Thyroid Regulatory Factors
Thyroid Regulatory Factors
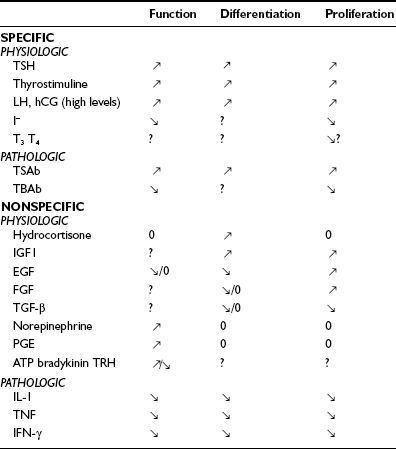
The two main factors that control the physiology of the thyroid after embryogenesis are the requirement for thyroid hormones and the supply of its main and specialized substrate iodide (Table 3-1). Thyroid hormone plasma levels and action are monitored by the hypothalamic supraoptic nuclei and by the thyrotrophs of the anterior lobe of the pituitary, where they exert a negative feedback through T3 receptor β. In normal rats, serum thyroid-stimulating hormone (TSH) levels are inversely related to thyrocyte sensitivity to TSH.1 The corresponding homeostatic control is expressed by TSH (thyrotropin). The TSH receptor is also stimulated by a new, different natural hormone cloned by homology, thyrostimuline. The physiologic role of this protein is unknown, but its level is not controlled by a thyroid hormone feedback, and it does not participate in the homeostatic control of the thyroid.2 Iodide supply is monitored in part indirectly as a substrate for the synthesis of thyroid hormones and therefore through its effects on the plasma level of thyroid hormones, but mainly in the thyroid itself, where it depresses various aspects of thyroid function and the response of the thyrocyte to TSH. These two major physiologic regulators control the function and size of the thyroid: TSH positively, iodide negatively.3–6 In mice embryo, other unknown factors control differentiation and organ growth which takes place normally until birth in the absence of TSH receptor.7,8 In humans, in whom birth occurs later in development, homozygous inactivating mutations of the TSH receptor in familial congenital hypothyroidism were found to be associated with a very hypoplastic thyroid gland.9 Although the thyroid contains receptors for thyroid hormones, and a direct effect of these hormones on thyrocytes would make sense,10 as yet little evidence has indicated that such control plays a role in physiology.11 However, expression of dominant-negative thyroid hormone receptors in mice represses PPARγ expression and induces thyroid tumors in thyroid.12 The role of increased TSH plasma level in this induction is not defined. Luteinizing hormone (LH) and human chorionic gonadotropin (hCG) at high levels activate the TSH receptor and thus directly stimulate the thyroid. This effect accounts for the depression of TSH levels and sometimes elevated thyroid activity at the beginning of pregnancy.13–15
Table 3-1
 , Stimulation;
, Stimulation;  , inhibition: 0, no effect; EGF, epidermal growth factor; FGF, fibroblast growth factor; hCG; human chorionic gonadotropin; IFN, interferon; IGF, insulin-like growth factor; IL, interleukin; LH, luteinizing hormone; PGF, prostaglandin F; T3, triiodothyronine; T4, thyroxine; TBAb, thyroid-blocking antibody; TGF, transforming growth factor; TRH, thyrotropin-releasing hormone; TSAb, thyroid-stimulating antibodies.
, inhibition: 0, no effect; EGF, epidermal growth factor; FGF, fibroblast growth factor; hCG; human chorionic gonadotropin; IFN, interferon; IGF, insulin-like growth factor; IL, interleukin; LH, luteinizing hormone; PGF, prostaglandin F; T3, triiodothyronine; T4, thyroxine; TBAb, thyroid-blocking antibody; TGF, transforming growth factor; TRH, thyrotropin-releasing hormone; TSAb, thyroid-stimulating antibodies.The thyroid gland is also influenced by various other nonspecific hormones.16 Hydrocortisone exerts a differentiating action in vitro.17 Estrogens affect the thyroid by unknown mechanisms, directly or indirectly, as exemplified in the menstrual cycle, in pregnancy, and in the higher prevalence of thyroid tumors and other pathologies in women. Growth hormone (GH) induces thyroid growth, but its effects are thought to be mediated by locally produced somatomedins (insulin-like growth factor 1 [IGF-1]). Nevertheless the presence of basal TSH levels might be a prerequisite for the growth-promoting action of IGF-1, because a GH replacement therapy did not increase thyroid size in patients deficient for both GH and TSH.18 Conversely, TSH does not induce the proliferation of human thyrocytes in the absence of IGF-1 or high levels of insulin. The anomalously low endemic goiter prevalence among pygmies living in iodine-deficient areas,19 a population genetically resistant to IGF-1, is also compatible with an in vivo permissive effect of IGF-1 and IGF-1 receptor on TSH mitogenic action. In dog and human thyroid primary cultures, the presence of insulin receptors strictly depends on TSH, suggesting that thyroid might be a more specific target of insulin than generally considered.20,21 Effects of locally secreted neurotransmitters and growth factors on thyrocytes have been demonstrated in vitro and sometimes in vivo, and the presence of some of these agents in the thyroid has been ascertained. The set of neurotransmitters acting on the thyrocyte and their effects vary from species to species.3,22 In human cells, well-defined, direct but short-lived responses to norepinephrine, ATP, adenosine, bradykinin, and thyrotropin-releasing hormone (TRH) have been observed.3,23,24 Growth-factor signaling cascades demonstrated in vitro can exert similar effects in vivo. In nude mice, the injection of epidermal growth factor (EGF) promotes DNA synthesis in thyroid and inhibits iodide uptake in xenotransplanted rat25 and human thyroid tissues.26 By contrast, the injection of fibroblast growth factor (FGF) induces a colloid goiter in mice, with no inhibition of iodide metabolism or thyroglobulin and thyroperoxidase mRNA accumulation.27 These effects are exact replicas of initial observations from the dog thyroid primary culture system28 and other thyroid primary culture systems.29–32 EGF and FGF have since been reported to be locally synthesized in the thyroid gland, as a possible response to thyroxine and TSH,33 respectively. Their exact role as autocrine and/or paracrine agents in the development, function, and pathology of the thyroid gland of different species has yet to be clarified.34,35 Transforming growth factor beta (TGF-β) constitutes another category of cytokines that are growth factors produced locally by thyrocytes and influence their proliferation and the action of mitogenic factors.34,36 TGF-β inhibits proliferation and prevents most of the effects of TSH and cAMP in human thyrocytes in vitro.37,38 TGF-β is synthesized as an inactive precursor which can be activated by different proteases produced by thyrocytes. TGF-β expression is up-regulated during TSH-induced thyroid hyperplasia in rats, suggesting an autocrine mechanism limiting goiter size.39 Also present in the thyroid are activin A and bone morphogenic peptide (BMP), which are related to TGF-β and inhibit thyrocyte proliferation in vitro.40 Unlike TGF-β, they are directly synthesized in an active form. Elements of a Wnt/β catenin signaling pathway (Wnt factors, Frizzled receptors, and disheveled isoforms) have been identified in human thyroid and thyroid cancer cell lines.41 The eventual role in vivo in humans of most of these factors remains to be proved and clarified.
In Pathology
Mutated, constitutively active TSH receptors and Gs proteins cause thyroid autonomous adenomas.42,43 Mutations which confer to the TSH receptor a higher sensitivity to LH/hCG cause hyperthyroidism in pregnancy.44,45 Pathologic extracellular signals play an important role in autoimmune thyroid disease. Thyroid-stimulating antibodies (TSAbs), which bind to the TSH receptor and activate it, reproduce the stimulatory effects of TSH on the function and growth of the tissue. Their effects on cAMP accumulation are slower and much more persistent than those of TSH.46 Their abnormal generation is responsible for the hyperthyroidism and goiter of Graves’ disease. Thyroid-blocking antibodies (TBAbs) also bind to the TSH receptor but do not activate it and hence behave as competitive inhibitors of the hormone. Such antibodies are responsible for some cases of hypothyroidism in thyroiditis. Both stimulating and inhibitory antibodies from mothers with positive sera induce transient hyperthyroidism or hypothyroidism in newborns.4 The existence of thyroid growth immunoglobulins has been hypothesized to explain the existence of Graves’ disease with weak hyperthyroidism and prominent goiter. The thyroid specificity of such immunoglobulins would imply that they recognize thyroid-specific targets. This hypothesis is now abandoned.47–49 Discrepancies between growth and functional stimulation may instead reflect kinetics or cell intrinsic factors. Local cytokines have been shown to influence, mostly negatively, the function, growth, and differentiation of thyrocytes in vitro and thyroid function in vivo. Because they are presumably secreted in loco in autoimmune thyroid diseases, these effects might play a role in the pathology of these diseases, but this notion has not yet been proved.3,16 Moreover, in selenium deficiency, secretion of TGF-β by macrophages has been implicated in the generation of thyroid fibrosis50 and the pathogenesis of thyroid failure in endemic cretinism. The overexpression of both FGF and FGF receptor 1 in thyrocytes from human multinodular goiter might explain their relative TSH independence.51 On the other hand, the subversion of tyrosine kinase pathways similar to those normally operated by local growth factors (i.e., the activation and thyrocyte expression of Ret52 and TRK,53 the overexpression of Met/HGF receptor, sometimes in association with hepatocyte growth factor (HGF),54 or erbB/EGF receptor in association with its ligand, TGF-α55) can be causally associated with TSH-independent thyroid papillary carcinomas. An autocrine loop involving IGF-2 and the insulin receptor isoform-A is also proposed to stimulate growth of some thyroid cancers.56 Thyroid cancer cells often escape growth inhibition by TGF-β.57
Regulatory Cascades
The Cyclic Adenosine Monophosphate Cascade
The cyclic adenosine monophosphate (cAMP) cascade in the thyroid corresponds, as far as it has been studied, to the canonic model of the β-adrenergic receptor cascade4 (Fig. 3-1). It is activated in the human thyrocyte by the TSH and the β-adrenergic and prostaglandin E receptors. These receptors are classical seven-transmembrane receptors controlling transducing guanosine triphosphate (GTP)-binding proteins. Activated G proteins belong to the Gs class and activate adenylyl cyclase; they are composed of a distinct αs subunit and nonspecific β and γ monomers. Activation of a G protein corresponds to its release of guanosine diphosphate (GDP) and binding of GTP and to its dissociation into αGTP and βγ dimers; αsGTP directly binds to and activates adenylyl cyclase. Inactivation of the G protein follows the spontaneous, more or less rapid hydrolysis of GTP to GDP by αs GTPase activity and the reassociation of αGDP with βγ. The effect of stimulation of the receptor by agonist binding is to increase the rate of GDP release and GTP binding, thus shifting the equilibrium of the cycle toward the αGTP active form. Unless constrained in a multiprotein complex, one receptor can consecutively activate several G proteins (hit-and-run model). A similar system negatively controls adenylyl cyclase through Gi. It is stimulated in the human thyroid by norepinephrine through α2-receptors and moderately by the TSH receptor. Adenosine at high concentrations directly inhibits adenylyl cyclase. The thyroid contains mainly three isoforms of adenylyl cyclase: III, VI, and IX.58 The cAMP generated by adenylyl cyclase binds to the regulatory subunit of protein kinase A (PKA) that is blocking the catalytic subunit and releases this now-active unit. The activated, released catalytic unit of protein kinase phosphorylates serines and threonines in the set of proteins containing accessible specific peptides that it recognizes. These phosphorylations, through more or less complex cascades, lead to the observed effects of the cascade. Two isoenzymes (I, II) of cAMP-dependent kinases have been found, the first of which is more sensitive to cAMP; as yet no clear specificity of action of these kinases has been demonstrated. In the case of the thyroid, this cascade is activated through specific receptors, by TSH in all species, and by norepinephrine receptors and prostaglandin E in humans, with widely different kinetics: prolonged for TSH and short lived (minutes) for norepinephrine and prostaglandins.59 Other neurotransmitters have been reported to activate the cascade in thyroid tissue, but not necessarily in the thyrocytes of the tissue.24 Besides PKA, cAMP activates EPAC (exchange protein directly activated by cAMP) or Rap guanine nucleotide exchange factor 1 (GEF1) and the less abundant GEF2, which activates the small G protein Rap.60 It does so in the thyroid.61 However, despite high expression of EPAC1 in thyrocytes and its further increase in response to TSH, all the physiologically relevant cAMP-dependent functions of TSH studied in dog thyroid cells, including acute regulation of cell functions (including thyroid hormone secretion) and delayed stimulation of differentiation expression and mitogenesis, are mediated only by PKA activation.61 The role of the cAMP/EPAC/Rap cascade in thyroid thus remains largely unknown. Activation of PKA inactivates small G proteins of the Rho family (RhoA, Rac1, and Cdc42), which reorganizes the actin cytoskeleton and could play an important role in stimulation of thyroid hormone secretion and induction of thyroid differentiation genes.62 Of the other known possible effectors of cAMP, cyclic guanosine monophosphate (cGMP)-dependent protein kinases are present in the thyroid but cyclic nucleotide–activated channels have not been looked for.
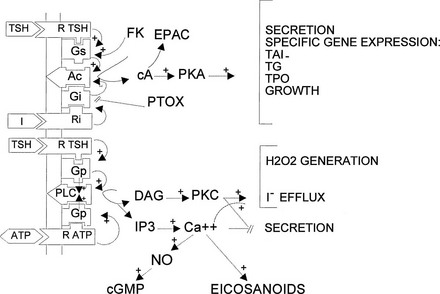
FIGURE 3-1 Regulatory cascades activated by thyroid-stimulating hormone (TSH) in human thyrocytes. In the human thyrocyte, H2O2 (H2O2) generation is activated only by the phosphatidylinositol 4,5-bisphosphate (PIP2) cascade—that is, by the Ca2+ (Ca++) and diacylglycerol (DAG) internal signals. In dog thyrocytes, it is activated also by the cyclic adenosine monophosphate (cAMP) cascade. In dog thyrocytes and FRTL-5 cells, TSH does not activate the PIP2 cascade at concentrations 100 times higher than those required to elicit its other effects. Ac, Adenylate cyclase; cA, 3′, 5′-cAMP; cGMP, 3′, 5′-cyclic guanosine monophosphate; FK, forskolin; Gi, guanosine triphosphate (GTP)-binding transducing protein inhibiting adenylate cyclase; Gq, GTP-binding transducing protein activating PIP2 phospholipase C; Gs, GTP-binding transducing protein activating adenylate cyclase; I, extracellular signal inhibiting adenylate cyclase (e.g., adenosine through A1 receptors); IP3, myoinositol 1,4,5-triphosphate; EPAC, cAMP-dependent Rap guanyl nucleotide exchange factor; PKA, cAMP-dependent protein kinases; PKC, protein kinase C; PLC, phospholipase C; PTOX, pertussis toxin; R ATP, ATP purinergic P2 receptor; R TSH, TSH receptor; Ri, receptor for extracellular inhibitory signal I; TAI, active transport of iodide; TG, thyroglobulin; TPO, thyroperoxidase.
In the thyrocyte, the cAMP cascade is controlled by several negative feedbacks, including the direct activation by phosphorylation of phosphodiesterases and the induction of several proteins inhibiting the cascade.63
The thyrocyte is very sensitive to small changes in cAMP concentrations; a mere doubling of this concentration is sufficient to cause maximal thyroglobulin endocytosis.64
The Calcium–Inositol 1,4,5-Triphosphate Cascade
The calcium (Ca2+)–inositol 1,4,5-triphosphate (IP3) cascade in the thyroid also corresponds, as far as has been studied, to the canonic model of the muscarinic or α1-adrenergic receptor–activated cascades. It is activated in the human thyrocyte by TSH—through the same receptors that stimulate adenylyl cyclase—and by ATP, bradykinin, and TRH through specific receptors. In this cascade, as in the cAMP pathway, the activated receptor causes the release of GDP and the binding of GTP by the GTP-binding transducing protein (Gq, G11, G12), and their dissociation into αq and βγ. In turn, αGTP then stimulates phospholipase C. The TSH receptor activates Gs and Gq, with a higher affinity for Gs.65,66 Phospholipase C hydrolyzes membrane phosphatidylinositol 4,5-bisphosphate (PIP2) into diacylglycerol and IP3. IP3 enhances calcium release from its intracellular stores, followed by an influx from the extracellular medium. The rise in free ionized intracellular Ca2+ leads to the activation of several proteins, including calmodulin. The latter protein in turn binds to target proteins and thus stimulates them (e.g., cyclic nucleotide phosphodiesterase and, most importantly, calmodulin-dependent protein kinases). These kinases phosphorylate a whole set of proteins exhibiting serines and threonines on their specific peptides and thus modulate them and cause many observable effects of this arm of the cascade. Calmodulin also activates constitutive nitric oxide (NO) synthase in thyrocytes. The generated NO itself enhances soluble guanylyl cyclase activity in thyrocytes and perhaps in other thyroid cells and thus increases cGMP accumulation.67 Nothing is yet known about the role of cGMP in the thyroid cell.
Diacylglycerol released from PIP2 activates protein kinase C, or rather the family of protein kinases C, which by phosphorylating serines or threonines in specific accessible peptides in target proteins causes the effects of the second arm of the cascade.68 It inhibits phospholipase C or its Gq, thus creating a negative feedback loop. In the human thyroid, the PIP2 cascade is stimulated through specific receptors by ATP, bradykinin, TRH, and TSH.24,66,69,70 The effects of bradykinin and TRH are very short lived. Acetylcholine, which is the main activator of this cascade in the dog thyrocyte, is inactive on the human cell, although it activates nonfollicular (presumably endothelial) cells in this tissue.24
Other Phospholipid-Linked Cascades
In dog thyroid cells and in a functional rat thyroid cell line (FRTL5), TSH activates PIP2 hydrolysis weakly and at concentrations several orders of magnitude higher than those required to enhance cAMP accumulation. Of course, these effects have little biological significance. However, at lower concentrations in dog cells, TSH increases the incorporation of labeled inositol and phosphate into phosphatidylinositol. Similar effects may exist in human cells, but they would be masked by the stimulation of the PIP2 cascade. They may reflect increased synthesis, perhaps coupled to and necessary for cell growth.71
Diacylglycerol can be generated by other cascades than the classic Ca2+-IP3 pathway. Activation of phosphatidylcholine phospholipase D takes place in dog thyroid cells stimulated by carbamylcholine. Because it is reproduced by phorbol esters (i.e., by stable analogs of diacylglycerol), it has been ascribed to phosphorylation of the enzyme by protein kinase C, which would represent a positive feedback loop.72 Although such mechanisms operate in many types of cells, their existence in human thyroid cells has not been demonstrated.73
Regulatory Cascades Controlled by Receptor Tyrosine Kinases
Many growth factors and hormones act on their target cells by receptors that contain one transmembrane segment. They interact with the extracellular domain and activate the intracellular domain, which phosphorylates proteins on their tyrosines. Receptor activation involves in some cases a dimerization and in others a conformational change of a preexisting dimer. The first step in activation is protein-tyrosine phosphorylation, followed by binding of various protein substrates on tyrosine phosphates containing segments of the receptor. Such binding through src homology domains (SH2) leads to phosphorylation of these proteins on their tyrosines. In turn, this phosphorylation causes sequential activation of the ras and raf proto-oncogenes, mitogen-activated protein (MAP) kinase kinase, MAP kinase, and so on, on the one hand, and phosphatidylinositol-3-kinase (PI-3-kinase), protein kinase B (PKB), and TOR (target of rapamycin) on the other hand. The set of proteins phosphorylated by a receptor defines the pattern of action of this receptor. In thyroids of various species, insulin, IGF-1, EGF, FGF, HGF, but not platelet-derived growth factor activate such cascades.74,75 In the human thyroid, effects of insulin, IGF-1, EGF, FGF, but not HGF have been demonstrated.3,21,51,76–78 TGF-β, acting through the serine threonine kinase activity of its receptors and its phosphorylated protein targets (Smad), inhibits proliferation and specific gene expression in human thyroid cells.37,38,79 TSH and cAMP do not activate either the MAPK-ERK nor the JUNK and p38 phosphorylation pathways in dog and human thyroids.75
Cross-Signaling Between the Cascades
Calcium, the intracellular signal generated by the PIP2 cascade, activates calmodulin-dependent cyclic nucleotide phosphodiesterases and thus inhibits cAMP accumulation and its cascade.80 This activity represents a negative cross-control between the PIP2 and the cAMP cascades. Activation of protein kinase C enhances the cAMP response to TSH and inhibits the prostaglandin E response, which suggests opposite effects on the TSH and prostaglandin receptors.81 No important effect of cAMP on the PIP2 cascade has been detected. On the other hand, stimulation of protein kinase C by phorbol esters inhibits EGF action.
Cross-signaling between the cyclic AMP pathway and growth factor–activated cascades have been observed in various cell types.82,83 In ovarian granulosa cells, FSH (through cAMP) activates MAP kinases and the PI3 kinase pathway.84 In FRTL5, but not in WRT cell lines, TSH (through cAMP) activates MAP kinase; in WRT cells but not in PCCl3 cells, TSH and cAMP activate PKB.85,86 Such cross signalings have not been observed in human or dog thyroid cells. Ras, MAPK, p38, Jun kinase and ERK5, as well as PI3 kinase and PKB, are not modulated by cAMP.74,75,87,88
Specific Control by Iodide
Iodide, the main substrate of the specific metabolism of the thyrocyte, is known to control the thyroid. Its main effects in vivo and in vitro are to decrease the response of the thyroid to TSH, to acutely inhibit its own oxidation (Wolff-Chaikoff effect), to reduce its trapping after a delay (adaptation to the Wolff-Chaikoff effect), and at high concentrations to inhibit thyroid hormone secretion (Fig. 3-2). The first effect is very sensitive inasmuch as small changes in iodine intake without any other changes (e.g., thyroid hormone levels) are sufficient to reset the thyroid system at different serum TSH levels, which suggests that in physiologic conditions, modulation of the thyroid response to TSH by iodide plays a major role in the negative feedback loop.5,89 Iodide in vitro has also been reported to inhibit a number of metabolic steps in thyroid cells.90,91 These actions might be direct or indirect as a result of an effect on an initial step of a regulatory cascade. Certainly, iodide inhibits the cAMP cascade at the level of Gs or cyclase and the Ca2+-PIP2 cascade at the level of Gq or phospholipase C; such effects can account for the inhibition of many metabolic steps controlled by these cascades.92,93 In one case in which this process has been studied in detail, the control of H2O2 generation (i.e., the limiting factor of iodide oxidation and thyroid hormone formation), iodide inhibited both the cAMP and the Ca2+-PIP2 cascades at their first step but also the effects of the generated intracellular signals cAMP, Ca2+, and diacylglycerol on H2O2 generation.94
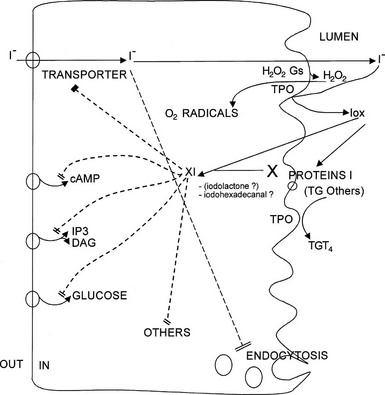
FIGURE 3-2 Effects of iodide on thyroid metabolism. All inhibitory effects of iodide, except in part the inhibition of secretion, are relieved by drugs that inhibit iodide trapping (e.g., perchlorate) or iodide oxidation (e.g., methimazole). Three possible mechanisms corresponding to this paradigm are outlined: generation of O2 radicals, iodination of target proteins, and synthesis of an XI compound. Any of these mechanisms could account for the various steps ascribed to XI inhibition by I− (indicated by slashes).
The mechanism of action of iodide on all the metabolic steps besides secretion fits the “XI” paradigm of Van Sande.95 These inhibitions are relieved by agents that block the trapping of iodide (e.g., perchlorate) or its oxidation (e.g., methimazole)—the Van Sande criteria. The effects are therefore ascribed to one or several postulated intracellular iodinated inhibitors called XI. The identity of such signals is still unproved. At various times, several candidates have been proposed for this role, such as thyroxine, iodinated eicosanoids (iodolactone),96 and more plausibly iodohexadecanal.97 The latter, the predominant iodinated lipid in the thyroid, can certainly account for the inhibition of adenylyl cyclase and of H2O2 generation.97–99 It should be emphasized that iodination of the various enzymes, as well as a catalytic role of iodide in the generation of O2 radicals (shown to be involved in the toxic effects of iodide), could account for the Van Sande criteria with no need for the XI paradigm.95,100 With regard to thyroid secretion, its inhibition by iodide in patients treated with antithyroid drugs suggests a direct, XI-independent effect.
In dogs in vivo, iodide at moderate doses decreases cell proliferation and the expression of TPO and NIS mRNA but not the synthesis or secretion of thyroid hormone. The down-regulation of NIS explains the adaptation to the Wolff-Chaikoff effect.101
The Thyroid-Stimulating Hormone Receptor
The β subunits of glycoprotein hormones, to which TSH belongs, are encoded by paralogous genes displaying substantial sequence similarity (Fig. 3-3). The corresponding receptors, FSHR, LH/CGR and TSHR, are members of the rhodopsin-like G protein–coupled receptor family. As such, TSHR contains a “serpentine” portion with seven transmembrane helices with many (but not all) of the sequence signatures typical of this receptor family. In addition, and a hallmark of the subfamily of glycoprotein hormone receptors, it contains a large (about 400 residues) aminoterminal ectodomain responsible for the high affinity and selective binding of TSH.102–104 The higher-sequence identity of the serpentine portions of glycoprotein hormone receptors (about 70%), when compared with the ectodomains (about 40%, see Fig. 3-3), suggested early that the former are interchangeable modules capable of activating the G proteins (mainly Gas) after specific binding of the individual hormones to the latter.4 Contrary to other rhodopsin-like GPCRs, binding of the hormones to their ectodomains can be observed with high affinity in the absence of the serpentine.105 The intramolecular transduction of the signal between these two portions of the receptors involves mechanisms specific to the glycoprotein hormone receptor family (see later). The relatively high sequence identity between the hormone-binding domains of the TSH and LH/CG receptors opens the possibility of spillover phenomena during normal or, even more so, molar or twin pregnancies, when hCG concentrations are several orders of magnitude higher than TSH. This provides an explanation to cases of pregnancy hyperthyroidism.15,15
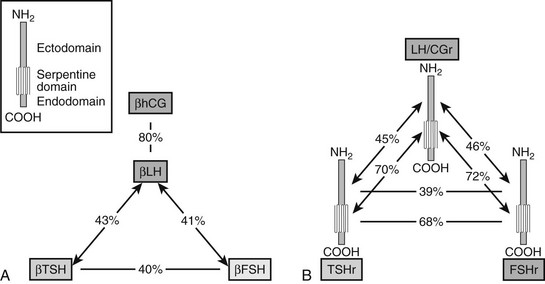
FIGURE 3-3 Both the glycoprotein hormone receptors (A) and the beta subunits of glycoprotein hormones (B) are encoded by paralogous genes. Sequence identities are indicated, separately for the ectodomains and serpentine domains of the three receptors, and for the β subunits of the four hormones. The pattern of shared similarities suggests coevolution of the hormones and the ectodomain of their receptors, resulting in generation of specificity barriers. The high similarity displayed by the serpentine portions of the receptors is compatible with a conserved mechanism of intramolecular signal transduction. (Data from Vassart G, Pardo L, Costagliola S: A molecular dissection of the glycoprotein hormone receptors, Trends Biochem Sci 29:119–126, 2004.)
The TSHR contains six sites for N-glycosylation, of which four have been shown to be effectively glycosylated.105 The functional role of the individual carbohydrate chains is still debated. It is likely that they contribute to the routing and stabilization of the receptor through the membrane system of the cell.
Alone among the glycoprotein hormone receptors, the TSHR undergoes cleavage of its ectodomain, severing it from the serpentine domain.106 This phenomenon has been related to the presence in the TSHR of a 50-amino-acid “insertion” for which there is no counterpart in the FSHR or LH/CGR. The initial cleavage step, by a metalloprotease, takes place around position 314 (within the 50-amino-acid insertion), followed by nibbling of the aminoterminal extremity of the serpentine-containing portion.107,108 The aminoterminal and serpentine portions of the resulting dimer remain bound to each other by disulphide bonds. The functional meaning of this TSHR-specific posttranslational modification remains unclear. Whereas all wild-type TSHR at the surface of thyrocytes seem to be in cleaved form, it has been shown that noncleavable mutant constructs are functionally undistinguishable from cleaved receptors when expressed in transfected cells.106 Noteworthy, when transiently or permanently transfected in nonthyroid cells, the wild-type human TSHR is present at the cell surface as a mixture of monomer and cleaved dimer. The cleavage and shedding of the ectodomain from a portion of the mature receptors has been related to the triggering or maintenance of autoimmunity in Graves’ disease.109,110
The TSH receptor is specifically inserted into the basolateral membrane of thyrocytes. This phenomenon involves signals encoded in the primary structure of the protein, as it is conserved when the receptor is expressed in the MDCK cell, a polarized cell of nonthyroid origin.111
The possibility that TSHR are present at the cell surface as “dimers of cleaved dimers” has recently been raised following demonstration that most rhodopsin-like GPCRs do dimerize112 (see following).
The TSH Receptor Gene
The gene coding for the human TSH receptor has been localized on the long arm of chromosome 14 (14q31).113,114 It spreads over more than 60 kb and is organized into 10 exons displaying an interesting correlation with the protein structure.104 The extracellular domain is encoded by a series of 9 exons, each of which corresponds to one or an integer number of leucine-rich repeat (LRR) motifs. The C-terminal half of the receptor containing the C-terminal part of the ectodomain and the serpentine domain is encoded by a single large exon. This finding is reminiscent of the fact that many G protein–coupled receptor genes are intronless. A likely evolutionary scenario derives from this gene organization115: the glycoprotein hormone receptor genes would have evolved from the condensation of an intronless classic G protein–coupled receptor with a mosaic gene encoding a protein with LRRs. Triplication of this ancestral gene and subsequent divergence led to the receptors for LH/CG, FSH, and TSH. The existence of 10 exons in both the TSH and FSH receptor genes (as opposed to the 11-exon LH/CG receptor), suggests the following phylogeny: one ancestral glycoprotein hormone receptor duplicating in the LH receptor and in the ancestor of TSH and FSH receptor, which lost one intron. The latter would later duplicate yielding the TSH and FSH receptors.
The gene promoter has been cloned and sequenced in humans and rats.115,116 It has characteristics of “housekeeping” genes in that it is GC-rich and devoid of TATA boxes; in the rat it was shown to drive transcription from multiple start sites.116
Expression of the TSH receptor gene is largely thyroid specific. Constructs made of a chloramphenicol acetyltransferase reporter gene under control of the 5′ flanking region of the rat gene show expression when transfected into FRTL5 cells and FRT cells but not into nonthyroid HeLa or a rat liver cell line (BRL) cells.116 However, TSH receptor mRNA has been clearly demonstrated in fat tissue of the guinea pig117 and following differentiation of preadipocytes into adipocytes.118,119 Functional significance in man of reports showing its presence in lymphocytes, extraocular tissue, and recently in cartilage and bone requires additional studies.109,120
Recently, functional expression of the TSH receptor has been demonstrated in pars tuberalis of the pituitary in quails and ependymal cells in mice, in relation with the control of photoperiodic behavior.118,119,121,122 No extrapolation of these studies to man has been attempted yet. Expression of the TSH receptor in thyroid cells is extremely robust. It is moderately up-regulated and down-regulated by TSH in vitro123 and down-regulated by iodide in vivo.101
Functional Aspects
Recognition of the Receptor by TSH
The three-dimensional structures are available for hCG and FSH,124–126 which allows accurate modelization of TSH on these templates. The crystal structure of the human FSHR-FSH complex127 has confirmed that the ectodomain of glycoprotein hormone receptors belongs to the family of proteins with LRRs, as was previously suggested by sequence analysis and homology modeling.128 The concave inner surface of the receptor (Fig. 3-4A) is an untwisted, noninclined β sheet formed by ten LRRs. Whereas the N-terminal portion of the β sheet (LRR1-7) is nearly flat, the C-terminal portion (LRR7-10) has the horseshoe-like curvature of LRR proteins. The crystal structure of part of the TSHR ectodomain in complex with a thyroid-stimulating autoantibody has recently been obtained.129 Notably, both the structure of the ectodomain of TSHR and the receptor-binding arrangements of the autoantibody are very similar to those reported for the FSHR-FSH complex. The ectodomain of glycoprotein hormone receptors also contains, downstream of the LRR region, a cysteine cluster domain of unknown structure (the hinge region), involved in receptor inhibition/activation and containing sites for tyrosine sulfation important for hormone binding (see later).
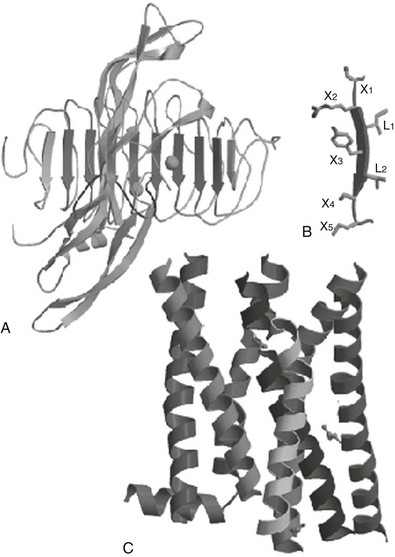
FIGURE 3-4 Schematic representation of the hormone receptor complex. A, General view of the follicle-stimulating hormone receptor (FSHR)-FSH crystal structure as a template to model the interaction between TSH and the TSH receptor 129. The concave inner surface of the receptor, formed by the β sheets of 10 leucine-rich repeats (LRRs), 2 through 9 (downward arrows), contact the middle section of the hormone molecule, both the C-terminal segment of the α subunit and the “seat-belt” segment of the β-subunit. B, Each LRR is composed of the X1-X2-L-X3-L-X4-X5 residues (where X is any amino acid, and L is usually Leu, Ile, or Val), forming the central X2-L-X3-L-X4, a typical β strand, while X1 and X5 are parts of the adjacent loops. C, Molecular model of the transmembrane domain of the TSH receptor, constructed from the crystal structure of bovine rhodopsin 123. The crystal structure of the β2-adrenergic receptor bound to the partial inverse agonist, carazolol, has been published.356 The structure of both rhodopsin and the β2 receptor are similar at the transmembrane domain. (Adapted from Caltabiano G, Campillo M, De Leener A et al: The specificity of binding of glycoprotein hormones to their receptors, Cell Mol Life Sci 65:2484–2492, 2008.)
Extensive amino acid substitutions by site-directed mutagenesis of the Xi residues in the LRR portion of the TSHR for their counterparts in the LH/CGR have been performed.130 Exchanging eight amino acids of the TSHR for the corresponding LH/CGR residues resulted in a mutant displaying a sensitivity to hCG matching that of the wild-type LH/CGR. Surprisingly, while gaining sensitivity to hCG, the mutant kept a normal sensitivity to TSH, making it a dual-specificity receptor. It is necessary to exchange 12 additional residues to fully transform it into a bona fide LH/CGR.130 From an evolutionary point of view, these observations indicate that nature has built recognition specificity of hormone-receptor couples on both attractive and repulsive residues, and that residues at different homologous positions have been selected to this result in the different receptors.
Inspection of electrostatic surface maps of models of the wild-type TSH and LH/CG receptors and some of the mutants is revealing in this respect.130,131 The LH/CGR displays an acidic groove in the middle of its horseshoe, extending to the lower part of it (corresponding to the C-terminal ends of the β strands). Generation of a similar distribution of charges in the dual-specificity and reverse-specificity TSHR mutants suggests that this is important for hCG recognition. A detailed modelization of the interactions between TSH and the ectodomain of its receptor has been realized.131
In addition to the hormone-specific interactions genetically encoded in the primary structure of glycoprotein hormones receptors and their ligands, the importance of nonhormone-specific ionic interactions has been demonstrated, involving sulfated tyrosine residues present in the ectodomains of all three receptors.132,133 Sulfation in the TSHR takes place on both tyrosine residues of a conserved Tyr-Asp-Tyr motif located close to the border between the ectodomain and the first transmembrane helix (Fig. 3-5). However, only sulfation of the first tyrosine of the motif seems to be functionally important,132 contributing crucially to the binding affinity without interfering with specificity. The functional role of this posttranslational modification of the TSH receptor has been confirmed by demonstration of profound hypothyroidism due to resistance to TSH in mice with inactivation of Tpst2, one of the enzymes responsible for tyrosine sulfation.134,135
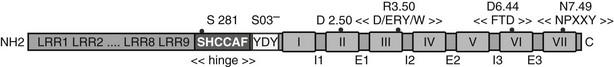
FIGURE 3-5 Linear representation of the TSH receptor. Sequence signatures common to all rhodopsin-like GPCRs and sequence signatures specific to the glycoprotein hormone receptor gene family are both implicated in activation of GPHRs. Key residues are indicated (dots) as well as conserved motifs: SO3− stands for posttranslational sulfation of the indicated tyrosine residues. The black boxes stand for transmembrane helices and I1-I3, E1-E3, for intracellular and extracellular loops, respectively. (Adapted from Vassart G, Pardo L, Costagliola S: A molecular dissection of the glycoprotein hormone receptors, Trends Biochem Sci 29:119–126, 2004.)
Activation of the Serpentine Portion of the TSH Receptor
Because it belongs to the rhodopsin-like GPCR family and displays many of the cognate signatures in primary structure, the serpentine portion of the TSH receptor is likely to share with rhodopsin common mechanisms of activation.136,137 However, sequence signatures characteristic of the serpentine portion of the glycoprotein hormone receptors suggest the existence of idiosyncrasies associated with specific mechanisms of activation (see Fig. 3-5). In addition, the TSHR has been found to be activated by a wide spectrum of gain-of-function mutations.138–140 Compilations of available data identify more than 30 residues, the mutation of which cause constitutive activation. Since many somatic mutations affecting a given residue have been found repeatedly, it is likely that we are close to a saturation map for spontaneous gain-of-function mutations. Attempts to translate this map into mechanisms of transition between inactive and active conformations of the receptor have been made, in the light of rhodopsin structural data. Three sequence patterns affected by gain-of-function mutations deserve special mention and may help in understanding how the TSH receptor is activated.
The first is centered on an aspartate in position 6.44 (Asp633) at the cytoplasmic side of transmembrane helix VI (TM-VI). When mutated to a variety of amino acids, the result is constitutive activation.138,141,142 This suggested that the gain of function resulted from the breakage of (a) bond(s) rather than the creation of novel interaction(s) by the mutated residue, and the main partner of Asp6.44 was identified as Asn7.49 in transmembrane helix VII. From a series of site-directed mutagenesis studies,141,143 it was tentatively concluded that in the inactive conformation of the TSH receptor, the side chain of Asp7.49 is normally “sequestered” by both Thr6.43 and Asp6.44, and that the active conformation(s) require(s) establishment of novel interaction(s) of N7.49 involving most probably Asp in position 2.50.
The second: Glutamate 3.49 and arginine 3.50 of the highly conserved “D/ERY/W” motif at the bottom of TM-III form an ionic lock with aspartate 6.30 at the cytoplasmic end of TM-VI. Disruption of this ionic lock (e.g., by mutations affecting Asp6.30) leads to constitutively active mutant receptors.143 Thus the movements of TM-III and TM-VI at the cytoplasmic side of the membrane (i.e., presumably an opening of the receptor) is necessary for receptor activation.144 The existence of this ionic lock has, however, been questioned recently, since it is not found in the determined structure of the β2-adrenergic receptor.136
The third: Serine 281 belongs to a GPHR-specific “YPSHCCAF” sequence signature located at the carboxyl-terminal end of the LRR portion in the ectodomain of the receptors (see Fig. 3-5). After mutation of this serine residue had been shown to activate the TSH receptor constitutively,145 this segment, sometimes referred to as the hinge motif, was shown to play an important role in activation of all three glycoprotein hormone receptors.146 The functional effect of substitutions of S281 in the TSHR likely results in a “loss of structure” locally, since the more destructuring the substitutions, the stronger the activation.146,147 This observation, together with results showing that mutation of specific residues in the extracellular loops of the TSHR cause constitutive activation,148 and the previous demonstration of activation of TSH receptor by extracellular trypsinization,149,150 led to the hypothesis that activation of the receptor could involve the rupture of an inhibitory interaction between the ectodomain and the serpentine domain (see later).145
Interaction Between the Ectodomain and the Serpentine Domain
Mutant TSHR constructs devoid of the ectodomain displayed a phenotype compatible with partial activation, thus confirming the inhibitory effect of the ectodomain on the serpentine portion already suggested by the partial activation of the receptor by a trypsin treatment. However, activation of cAMP production in cells transfected with truncated constructs was much lower than after full stimulation of the wild-type receptor by TSH or by mutation of Ser281.151 These observations led to the following model for activation of the TSHR (Fig. 3-6).151,152 In the resting state, the ectodomain would exert an inhibitory effect on the activity of an inherently noisy rhodopsin-like serpentine, qualifying pharmacologically as an inverse agonist of the serpentine. Upon activation, by binding of the hormone or secondary to mutation of S281 in the hinge region, the ectodomain would switch from inverse agonist to full agonist of the serpentine portion. The ability of the strongest S281 mutants to fully activate the receptor in the absence of hormone suggests that the ultimate agonist of the serpentine domain would be the “activated” ectodomain, with no need for an interaction between the hormone and the serpentine domain. This model in which the “real” agonist of the serpentine domain would be the activated ectodomain is confirmed by the identification of a monoclonal antibody recognizing the ectodomain and endowed with inverse agonistic activity.153,154 Recent data suggest that the activation step might be achieved by interaction of charged residues of the α subunit of the hormone with acidic residues of the hinge region.155
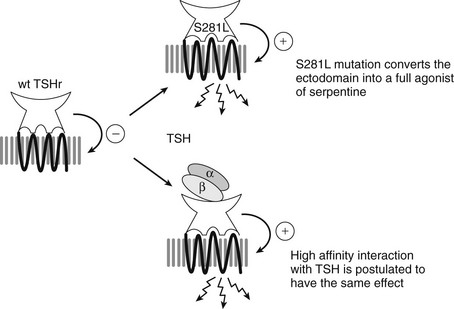
FIGURE 3-6 Model for activation of the thyrotropin receptor. Interactions between the ectodomain and the serpentine domains are implicated in the activation mechanism and functional specificity. The TSH receptor is represented with its ectodomain containing a concave, hormone-binding structure facing upwards, and a transmembrane serpentine portion. The basal state of the receptor is characterized by an inhibitory interaction between the ectodomain and the serpentine domain (indicated by the (−) sign). The ectodomain would function as a tethered inverse agonist of the serpentine portion. Mutation of Ser281 in the ectodomain into leucine switches the ectodomain from an inverse agonist into a full agonist of the serpentine domain (indicated by the (+) sign). Binding of TSH (indicated by αβ dimeric structure) to the ectodomain is proposed to have a similar effect, converting it into a full agonist of the serpentine portion. The serpentine portion in the basal state is represented as a compact, black structure. The fully activated serpentine portion is depicted as a relaxed gray structure with arrows indicating activation of Gαs.
Activation by Chorionic Gonadotropin
As indicated above, the sequence similarity between TSH and hCG, and between their receptors, allows for some degree of promiscuous stimulation of the TSH receptor by hCG during the first trimester of pregnancy, when hCG levels reach peak values. The inverse relation between TSH and hCG observed in most pregnant women is clear indication that their thyroid is subjected to the thyrotropic activity of hCG.15 Whereas this situation is usually associated with euthyroidism, thyrotoxicosis may develop in cases of excessive hCG production (as it occurs in twin pregnancies or hydatidiform moles), or in rare patients harboring a mutant TSH receptor with increased sensitivity to hCG.156
Of note, the bioactivity of human TSH (and of all glycoprotein hormones, including hCG) is lower than that of bovine TSH and of other nonprimate mammals. This is due to positive selection in higher primates of α subunits in which several key basic amino acids have been substituted.157 The observation that this phenomenon parallels the evolution of chorionic gonadotropin suggests that it may be related to protection against promiscuous stimulation of the TSH receptor by hCG during pregnancy.
Activation by Autoantibodies
Autoantibodies found in Graves’ disease and some types of idiopathic myxedema can stimulate (TSAb) or block (TSBAb) TSH receptor, respectively. Epitopes recognized by TSAbs are being identified from precise mapping of binding sites of murine or human monoclonal antibodies endowed with TSAb activity on the partial crystal structure of the ectodomain129 or models thereof.158 However, the actual mechanisms implicated in activation of the receptor by TSAbs (and by TSH) are still unknown. Although most TSAbs do compete with TSH for binding to the receptor,110,159 and despite similarity in interaction surfaces,129 the precise targets of the hormone and autoantibodies are likely to be different, at least in part. It has indeed been shown that sulfated tyrosine residues, which are important for TSH binding, are not implicated in recognition of TSH receptor by TSAbs.160 Also, contrary to TSH, most TSAbs from Graves’ patients display a delay in their ability to stimulate cAMP accumulation in transfected cells.46 The availability of TSAb preparations purified from individual patients should allow exploration of these issues in a direct fashion.161
Activation by Thyrostimulin
Thyrostimulin has been identified as a novel agonist of the TSH receptor. A glycoprotein hormone, it is composed of two subunits, coined α2 and β5, and activates the receptor with a lower EC50 than human TSH. It is produced by the pituitary in corticotrophs, but its physiologic significance remains mysterious.2,162–164
Down-Regulation of the TSH Receptor
Desensitization of some G protein–coupled receptors has been shown to involve phosphorylation of specific residues by G protein–receptor kinases (homologous desensitization) or PKA (heterologous desensitization) enzymes.165 When compared with other G protein–coupled receptors, the TSH receptor contains few phosphorylatable serine or threonine residues in its intracellular loops and C-terminal tail, which probably accounts for the limited desensitization observed after stimulation by TSH. Acute desensitization of the receptor in the presence of TSH, presumably by phosphorylation, is weak and delayed.166 Its internalization is rapidly followed by recycling to the cell surfaces, while the hormone is degraded in lysosomes.167 Similarly, a weak down-regulation, confounded by the long life of both TSHR mRNA and protein, takes place but has little functional role.123 On the other hand, in the presence of constant stimulation, cAMP accumulation is down-regulated mostly as a consequence of phosphodiesterase induction.168 The persistence of hyperthyroidism in patients submitted to constant TSH stimulations in TSH-secreting pituitary adenomas or TSAb stimulation in Graves’ disease testifies to the weakness of these negative regulations.
Dimerization
As do most rhodopsin-like GPCRs, the glycoprotein hormone receptors have been demonstrated to dimerize/oligomerize by a variety of experimental approaches, including FRET, BRET, and functional complementation of mutants.169–171 The physiologic significance of this phenomenon remains unknown, but it has been shown to be associated with allosteric properties of the dimers/oligomers. TSH binding to the TSHR displays strong negative cooperativity,169 which is classically considered to account for a shallow concentration action curve extending over more than two orders of magnitude.
Control of Thyroid Function
Iodide Transport
Iodide is actively transported by the iodide Na+/I− symporter (NIS) against an electrical gradient at the basal membrane of the thyrocyte and diffuses by a specialized channel (pendrin or another channel)172,173 from the cell to the lumen at the apical membrane. The opposite fluxes of iodide, from the lumen to the cell and from the cell to the outside, are generally considered to be passive and nonspecific. At least five types of control have been demonstrated.90,91,172
1. Rapid and transient stimulation of iodide efflux by TSH in vivo, which might reflect a general increase in membrane permeability. The cascade involved is not known.
2. Rapid activation of iodide apical efflux from the cell to the lumen by TSH. This effect, which contributes to the concentration of iodide at the site of its oxidation, is mediated—depending on the species—by Ca2+ and/or cAMP.172,174 In human cells, it is mainly controlled by Ca2+ and therefore by the TSH effect on phospholipase C.
3. Delayed increase in the capacity (Vmax) of the active iodide transport NIS in response to TSH. This effect is inhibited by inhibitors of RNA and protein synthesis and is due to activation of iodide transporter gene expression. This effect of TSH is reproduced by cAMP analogs in vitro and is therefore mediated by the cAMP cascade.6 Expression of mRNA is enhanced by TSH and cAMP and decreased by iodide.101,175 TSH enhancement of thyroid blood flow, more or less delayed depending on the species, also contributes to increase in the uptake of iodide.6 Blood flow is inversely related to iodine levels in the thyroid.176
4. Rapid inhibition by iodide of its own transport in vivo and in vitro. This inhibitory effect requires an intact transport and oxidation function—that is, it fulfills the criteria of an XI effect. After several hours, the capacity of the active transport mechanism is greatly impaired (adaptation to the Wolff-Chaikoff effect).91 The mechanism of the first effect is unknown but probably initially involves direct inhibition of the transport system itself (akin to the desensitization of a receptor), followed later by inhibition of NIS gene expression and NIS synthesis (akin to the down-regulation of a receptor).101
5. Inhibition by iodide of the thyroid blood flow. This effect does not fit the XI paradigm, since it takes place in patients treated with thyroperoxidase inhibitors.
Iodide Binding to Protein and Iodotyrosine Coupling
Iodide oxidation and binding to thyroglobulin and iodotyrosine coupling in iodothyronines are catalyzed by the same enzyme, thyroperoxidase, with H2O2 used as a substrate.177 The same regulations therefore apply to the two steps. H2O2 is generated by an NADPH-oxidase system constituted by two DUOX proteins.178,179 The system is very efficient in the basal state inasmuch as little of the iodide trapped can be chased by perchlorate in vivo. Also, in vitro the amount of iodine bound to proteins mainly depends on the iodide supply. Nevertheless, in human thyroid in vitro, stimulation of the iodination process takes place even at low concentrations of the anion, thus indicating that iodination is a second limiting step. Such stimulation is caused in all species by intracellular Ca2+ and is therefore a consequence of activation of the Ca2+-PIP2 cascade. In many species, phorbol esters and diacylglycerol, presumably through protein kinase C, also enhance iodination.180 It is striking that in a species such as the human, in which TSH activates the PIP2 cascade, cAMP inhibits H2O2 generation and iodination, whereas in a species (dog) in which TSH activates only the cAMP cascade, cAMP enhances iodination. Obviously, in the latter species, a supplementary cAMP control was necessary.180,181
Thyroperoxidase does not contain any obvious phosphorylation site in its intracellular tail. On the other hand, all the agents that activate iodination also activate H2O2 generation, and inhibition of H2O2 generation decreases iodination, which suggests that iodination is a substrate–driven process and that it is mainly controlled by iodide supply and H2O2 generation.180,182 Congruent with the relatively high Km of thyroperoxidase for H2O2, H2O2 is generated in disproportionate amounts with regard to the quantity of iodide oxidized. Negative control of iodination by iodide (the Wolff-Chaikoff effect) is accompanied and mostly explained by the inhibition of H2O2 generation. This effect of I− is relieved by perchlorate and methimazole and thus pertains to the XI paradigm.95,180
Iodotyrosine coupling to iodotyrosines is catalyzed by the same system and is therefore subject to the same regulations as iodination. However, coupling requires that suitable tyrosyl groups in thyroglobulin be iodinated (i.e., that the level of iodination of the protein be sufficient). In the case of severe iodine deficiency or when thyroglobulin exceeds the iodine available, insufficient iodination of each thyroglobulin molecule will preclude iodothyronine formation, whatever the activity of the H2O2 generating system and thyroperoxidase. On the other hand, when the iodotyrosines involved in the coupling are present, coupling is controlled by the H2O2 concentration but independent of iodide.177 In this case, H2O2 control has significance even at very low iodide concentrations.
Generation of H2O2 requires the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) as a coenzyme and is thus accompanied by NADPH oxidation. Limitation of the activity of the pentose phosphate pathway by NADP+ insures that NADPH oxidation for H2O2 generation causes stimulation of this pathway. Also, excess H2O2 leaking back into the thyrocyte is reduced mainly by catalase but also by glutathione (GSH) peroxidase, and the oxidized GSH (GSSG) produced is reduced by NADPH-linked GSH reductase. Thus both the generation of H2O2 and to some extent the disposal of excess H2O2 by pulling NADP reduction and the pentose pathway lead to activation of this pathway—historically one of the earliest and unexplained effects of TSH.6,180
Thyroid Hormone Secretion
Secretion of thyroid hormone requires endocytosis of human thyroglobulin, its hydrolysis, and the release of thyroid hormones from the cell. Thyroglobulin can be ingested by the thyrocyte by three mechanisms.6,183–185
In macropinocytosis, which is the first, pseudopods engulf clumps of thyroglobulin. In all species this process is triggered by acute activation of the camp/PKA cascade and therefore by TSH.186 Inactivation of the Rho small G proteins, resulting in microfilament depolarization and stress-fiber disruption, is probably involved in the process.62 Stimulation of macropinocytosis is preceded and accompanied by an enhancement of thyroglobulin exocytosis and thus of membrane to the apical surface.182,187,188 By micropinocytosis, the second process, small amounts of colloid fluid are ingested. This process does not appear to be greatly influenced by acute modulation of the regulatory cascades. It is enhanced in chronically stimulated thyroids and thyroid cells with induction of vesicle transport proteins Rab 5 and 7.189,190 It probably accounts for most of basal secretion. A third (hypothesized) process is receptor-mediated endocytosis; it would be enhanced in chronically stimulated thyroid cells.191–193 The protein involved could be megalin194 and/or asialoglycoprotein. This process probably accounts for transcytosis of thyroglobulin.195
Contrary to the last named, the first two processes are not specific for the protein. They can be distinguished by the fact that macropinocytosis is inhibited by microfilament and microtubule poisons and by lowering of the temperature (below 23° C). Whatever its mechanism, endocytosis is followed by lysosomal digestion, with complete hydrolysis of thyroglobulin. The main iodothyronine in thyroglobulin is thyroxine. However, during its secretion, a small fraction is deiodinated by type I 5-deiodinases (DIO1 and DIO2) to triiodothyronine (T3), thus increasing relative T3 (the active hormone) secretion.196
The free thyroid hormones are released by an unknown mechanism, which may be transport or exocytosis. The iodotyrosines are deiodinated by specific deiodinases and their iodide recirculated in the thyroid iodide compartments. Under acute stimulation, a release (spillover) of amino acids and iodide from the thyroid is observed. A mechanism for lysosome uptake of poorly iodinated thyroglobulin on N-acetylglucosamine receptors and recirculation to the lumen has been proposed. Under normal physiologic conditions, endocytosis is the limiting step of secretion, but after acute stimulation, hydrolysis might become limiting with the accumulation of colloid droplets. Secretion by macropinocytosis is triggered by activation of the cAMP cascade and inhibited by Ca2+ at two levels: cAMP accumulation and cAMP action. It is also inhibited in some thyroids by protein kinase C downstream from cAMP. Thus the PIP2 cascade negatively controls macropinocytosis.81
The thyroid also releases thyroglobulin. Inasmuch as this thyroglobulin was first demonstrated by its iodine, at least part of this thyroglobulin is iodinated; thus it must originate from the colloid lumen. Release is inhibited in vitro by various metabolic inhibitors and therefore corresponds to active secretion.187,197 The most plausible mechanism is transcytosis from the lumen to the thyrocyte lateral membranes.188 As for thyroid hormone, this secretion is enhanced by activation of the cAMP cascade and TSH and inhibited by Ca2+ and protein kinase C activation. Because thyroglobulin secretion does not require its iodination, it reflects the activation state of the gland regardless of the efficiency of thyroid hormone synthesis. Thyroglobulin serum levels and their increase after TSH stimulation constitute a very useful index of the functional state of the gland when thyroid hormone synthesis is impaired, as in iodine deficiency, congenital defects in iodine metabolism, treatment with antithyroid drugs, and the like.198 Regulated thyroglobulin secretion should not be confused with the release of this protein from thyroid tumors, which corresponds in large part to exocytosis of newly synthesized thyroglobulin in the extracellular space rather than in the nonexistent or disrupted follicular lumen. In inflammation or after even mild trauma, opening of the follicles can cause unregulated leakage of lumen thyroglobulin. Transcytosis or leakage from the lumen yields iodinated thyroglobulin, whereas newly synthesized exocytotic thyroglobulin is not iodinated.
Control of Thyroid-Specific Gene Expression
The so-called thyroid-specific genes encode proteins that are either found in the thyroid exclusively, like thyroglobulin and thyroperoxidase, or that, although being also found in a few additional tissues, are primarily involved in thyroid function, like TSH receptor and sodium/iodide symporter. The transcription of these genes in the thyroid appears to rely on the coordinated action of a master set of transcription factors that includes at least the homeodomain protein TTF-1 (also known as Nkx 2.1, or T/ebp), the paired-domain protein Pax 8, and perhaps also the forkhead-domain protein TTF-2 (also known as FoxE1).199,200 Loss-of-function mutant mice for TTF-1, Pax 8, or TTF-2 have been generated and allowed to identify a crucial role for these transcription factors in the development of the thyroid also. However, because none of these animals develop a normal mature thyroid, they could not be used to investigate the exact role of these key factors in the control of gene expression in the mature thyroid. Partial conditional inactivation of the TITF1 gene that encodes the TTF-1 protein was also achieved in the mouse,201but since inactivation remains only partial in this model, its use is limited in the detailed investigation of the consequences of the absence of TTF-1 on differentiated thyroid cell function. Most of the work concerning this last aspect has thus been conducted either in primary cultures of thyrocytes202 or in immortalized thyroid cell lines like FRTL-5 and PCCl3.203 Although the data gathered to date agree on most basic aspects, significative differences have sometimes been observed between primary versus immortalized cell models.204 Part of these discrepancies may result from the existence of occasional species-specific differences.205
The main regulator of thyroid function, the TSH signal, which is predominantly conveyed inside the cell by cAMP and PKA, up-regulates the expression of transcription factor Pax 8, both in primary cells206 and established cell lines.207 However, mice genetically deprived of TSH or of functional TSH receptor do not show reduced amounts of Pax 8 in their thyroids as compared to wild-type animals,8 suggesting that compensatory mechanisms may ensure an adequate production of this factor when thyroid development takes place in the absence of the normal physiologic stimulus. Besides this control on the amount of Pax 8 protein, there is no firm evidence that TSH or cAMP exert any other control at the level of the master thyroid transcription factors identified presently.204,208,209 The expression of several other transcription factors was shown to be up-regulated, often at least transiently, in response to TSH/cAMP in the thyroid, namely c-myc,210 c-fos,210 fos B, jun B, jun D,211 CREM,212 NGFI-B,213 and CHOP.214 A hypothetical role in the control exerted by TSH/cAMP on the expression of the thyroid-specific genes has been proposed for some of these factors,212,214 but no final link has yet been established.215 A recent report indicates that the dopamine and cAMP-regulated neuronal phosphoprotein DARPP-32 could play an essential role in this control.216
It is noteworthy that in addition to its control on the transcription of the individual thyroid-specific genes, which is detailed in a following discussion, TSH also regulates gene expression by acting at some posttranscriptional steps, as shown in the case of thyroglobulin.217 Finally, many effects of TSH and cAMP on gene expression (including on thyroid-specific genes such as thyroglobulin) might be rather indirect and depend in part on the profound modifications of cell morphology and cytoskeleton that result from PKA activation.31,62
TGF-β has been shown to down-regulate the expression of thyroid-specific genes.218,219 It seems to involve a reduction in the level of Pax 8 activity that is mediated by Smad proteins.219,220 In human thyroid primary cultures, TGF-β inhibits most effects of cAMP on gene expression.38 As above, this might be related in part to an inhibition of morphologic effects of TSH/cAMP. In all the species tested so far, EGF strongly represses thyroglobulin and thyroperoxidase gene expression as well as iodide transport.31,123,221–223 FGF has a similar action in some species including bovine.17 The mechanisms have not been explored. As recently quantified by SAGE analysis in the thyroid cell line PCCl3,224 exposure to a high dose of iodide also decreases the expression of most of the thyroid-specific genes within the thyrocyte.
Thyroglobulin
The regulatory DNA elements of the thyroglobulin gene have been characterized in several species.199,205,225 The proximal promoter, as defined in transfection experiments, extends over 200 base pairs and contains binding sites for transcription factors TTF-1, TTF-2, and Pax 8 (Fig. 3-7). An upstream enhancer element containing binding sites for TTF-1 has been identified in beef and man.226 In the latter, the enhancer region is longer and harbors additional binding sites for TTF-1 and cAMP responsive element–binding (CREB) protein.227 Both TTF-1 and Pax 8 proteins were individually shown to exert a major control on thyroglobulin gene transcription.228,229 By contrast, TTF-2 activity appears to be dispensable, because the thyroglobulin gene is expressed in cells devoid of TTF-2 protein.228 Synergism in the transcriptional activation of the gene by TTF-1 and Pax 8 appears to rely on a direct interaction between these two factors230 and on their coordinated action involving both the enhancer and proximal promoter sequences.231 It has also been proposed recently that the transcriptional coactivators p300232 and (or ?) TAZ, the transcriptional coactivator with PDZ-binding motif,233 are involved in this activation. The known thyroglobulin gene regulatory elements were shown to be sufficient to drive the thyroid-restricted expression of a linked gene in living mice.234 This thyroid-restricted expression likely results from the requirement for the simultaneous presence of both TTF-1 and Pax 8, which occurs in thyroid only. It is associated with the tissue-specific demethylation of thyroglobulin gene sequences.235 Demethylation of the DNA is supposed to relieve the constitutive silencing of the gene.236
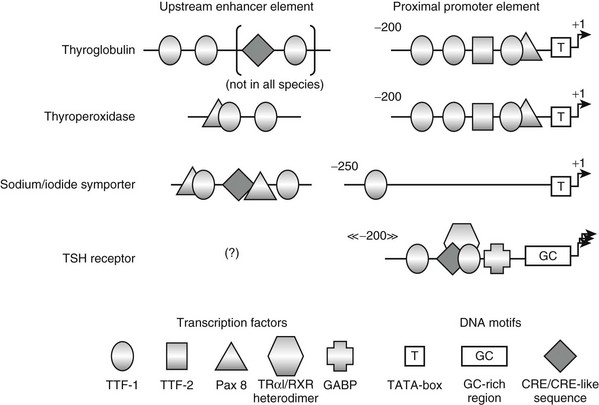
FIGURE 3-7 Schematic of the known regulatory elements of thyroglobulin, thyroperoxidase, sodium/iodide symporter, and TSH receptor genes. The organization of the proximal promoter and upstream enhancer elements of the different genes is depicted as determined in the species studied so far. Coordinates of the proximal promoters are in base pairs and refer to the transcription start site as +1. The positions of the upstream enhancer elements relative to the transcription start site are not indicated, since they vary extensively among the different species.
Thyroglobulin gene transcription has been shown to require the presence of circulating TSH in the adult rat237 and to be highly dependent on an elevated cAMP level in dog thyroid tissue slices,238 in primary cultured cells,222 and to a much lower extent in immortalized thyroid cell lines like FRTL-5.239 Although they are devoid of classical cAMP-responsive element (CRE), the proximal promoter sequences are essentially involved in this control, as indicated by the observation of TSH/cAMP-induced changes in their chromatin structure240 and their TSH/cAMP-dependent activity in transfection experiments.241 It has, however, been demonstrated recently that the onset of thyroglobulin gene expression during thyroid development takes place normally in mouse strains deprived of either circulating TSH or functional TSH receptors.7,8 This may be consistent with the observation that the thyroglobulin gene displays a low level of cAMP-independent transcription in primary cultured thyrocytes,222 which might depend on insulin, as observed in different culture models.242–244 In primary cultures of dog thyrocytes, the transcriptional activation of the thyroglobulin gene by cAMP after transient TSH withdrawal is also delayed as compared to that of the thyroperoxidase gene,222 and unlike thyroperoxidase transcription, it requires an active protein synthesis for sustained transcription.222 The increase in Pax 8 concentration consecutive to TSH/cAMP stimulation of the thyrocyte is not sufficient to account for the observed control on thyroglobulin gene transcription; TSH is still required for transcriptional activation even in cells expressing high levels of Pax 8 protein.207 Thus, besides TTF-1 and Pax 8, at least one additional, still unidentified, factor is likely to play a key role in the control of thyroglobulin gene expression, as also suggested by the observation that in the course of thyroid development, both TTF-1 and Pax 8 are present well before a thyroglobulin gene is expressed.
In addition to the full-length thyroglobulin mRNA, a shorter transcript accumulates in the rat thyroid in response to TSH stimulation.245 This transcript results from differential splicing and polyadenylation of the primary transcript and encodes a protein limited to the very N-terminal part of thyroglobulin. Because this truncated protein still contains a major hormonogenic site,246 it could suggest that in conditions in which the balance of thyroid metabolism would favor hormone synthesis over iodine storage (e.g., shortage of iodine), the rat thyrocyte would manufacture a shorter thyroglobulin with a preserved hormonogenic ability but lacking many of the nonhormonogenic tyrosines.
Thyroperoxidase
In the species studied so far, the architecture of the proximal promoter region of the thyroperoxidase gene strikingly resembles that of the corresponding region of the thyroglobulin gene247,248 (see Fig. 3-7). The upstream enhancer element also encompasses a pair of TTF-1 binding sites and contains an additional binding site for Pax 8, as compared to its counterpart in the thyroglobulin gene.249,250 Here again, the combination of the upstream enhancer and proximal promoter supports the synergistic action of TTF-1 and Pax 8 on gene transcription.231 The transcriptional coactivator p300 has also been reported to be involved in the activation of this promoter.251
Despite the existence of this high similarity, thyroperoxidase gene transcription is more tightly and more rapidly controlled by TSH and cAMP than that of the thyroglobulin gene in primary cultured thyrocytes and does not require a concomitant protein synthesis.222,252 Contrary to the thyroglobulin gene, the thyroperoxidase gene is not expressed in the absence of circulating TSH or functional TSH receptors in intact animals.8 On the other hand, the constitutive hyperactivation of the cAMP cascade leads to an increased expression of the gene as compared to the normal situation.253 In spite of their lack of a classical CRE, the proximal promoter sequences have been shown to mediate this TSH/cAMP control on transcription in transfection experiments.247 Exposure to a high dose of iodide reduces thyroperoxidase gene expression, as well as that of thyroglobulin, sodium/iodide symporter, and thyrotropin receptor genes in PCCl3 thyroid cells.224 Low doses of iodide also decrease thyroperoxidase gene expression in vivo, while the expression of thyroglobulin remains unaffected.101 Thus, apart from their basic dependence on the presence of the transcription factors TTF-1 and Pax 8, which insures their shared thyroid-restricted expression, the thyroperoxidase and thyroglobulin genes distinguish themselves significantly regarding the control of their transcription. It is worth mentioning in this context that a synergistic action of Pax8 and pRb, the retinoblastoma protein, appears to be required for thyroperoxidase promoter activation, whereas this is not the case for thyroglobulin promoter activation.254 It has also been postulated recently that the hormone-induced developmental activation of the thyroperoxidase gene involves the concerted action of TTF-2 and NF-1, both of which bind neighboring sequences in the gene promoter (see Fig. 3-7), resulting in the initial opening of the chromatin structure of the promoter.255
The existence of a major thyroperoxidase mRNA isoform has been detected in man.256 It appears to encode a protein devoid of its normal enzymatic activity.
Sodium/Iodide Symporter
Although the sodium/iodide symporter plays a key role in thyroid hormonogenesis, the expression of the corresponding gene is not restricted to the thyroid. Accordingly, the proximal promoter sequences identified so far do not exhibit a thyroid-specific activity in vitro,257,258 even if this activity may be marginally increased in the presence of TTF-1.259 The robust and appropriately controlled expression of this gene in the thyroid seems to be mediated essentially by the upstream enhancer element, which contains binding sites for both TTF-1 and Pax 8, and a cAMP-responsive element (CRE)-like DNA motif, which is involved in the control by TSH/cAMP173,260 (see Fig. 3-7). The cAMP-response element modulator (CREM) has recently been proposed to be involved in this control.261 As for the thyroperoxidase gene, TSH signaling is indispensable for sodium/iodide symporter gene transcriptional activation in vivo,7,8 and iodide down-regulates the expression of the gene.101,224 In addition, synergy between Pax8 and pRb appears to be required for the activation of both thyroperoxidase and sodium/iodide symporter promoters.254 A very similar control is thus exerted on the expression of both of these genes in the thyroid, despite the fact that their known regulatory regions bear only limited resemblances.
Thyrotropin Receptor
The TSH receptor gene is also expressed in tissues other than the thyroid. Again, the promoter elements identified presently, which include binding sites for thyroid hormone receptor (TR)-α1/retinoid-X receptor (RXR) heterodimer,262 GA-binding protein (GABP),263 CREB protein,264 and TTF-1265 (see Fig. 3-7), do not display a clear thyroid-specific activity in transfection experiments, as could be expected. Contrary to the promoters described so far, the promoter of the TSH receptor gene does not contain a TATA-box motif but encompasses a GC-rich region preceding the multiple neighboring transcription start sites. Consistent with the presence of TTF-1 binding sites in the promoter region, the TSH receptor gene exhibits decreased activity in animals expressing reduced levels of TTF-1.266 No other regulatory DNA element specifically involved in the thyroid-specific expression of this gene has been identified as yet. On the other hand, DNA demethylation events in the promoter region have been observed in thyroid cells expressing the TSH receptor gene, as compared to nonexpressing cells.263
Control of TSH receptor gene expression has been studied in the FRTL5 cell line,267–269 the canine thyrocyte in primary culture,123 cultured human thyrocytes,270,271 and human thyroid cancer.272,273 The general conclusion emerging from these studies is the extreme robustness of TSH receptor gene expression as compared with the other markers of thyroid cell differentiation (thyroglobulin and thyroperoxidase). In the dog, levels of TSH receptor mRNA remain virtually unchanged in animals subjected for 28 days to hyperstimulation by TSH secondary to treatment with methimazole or to TSH withdrawal achieved by administration of thyroxine.123 In the same study, the effect of TSH or forskolin has been investigated in dog thyrocytes in primary culture. This experimental system has the advantage that the differentiation state of the cells can be manipulated at will: cAMP agonists maintain expression of the differentiated phenotype, whereas agents such as EGF, tetradecanoyl phorbol acetate (TPA), and serum lead to “dedifferentiation.”274 The results demonstrate that the dedifferentiating agents reduce accumulation of the receptor mRNA. However, contrary to what is observed with thyroglobulin and thyroperoxidase mRNA, the inhibition is never complete. TSH or forskolin are capable of promoting reaccumulation of the receptor mRNA, a maximum being reached after 20 hours. As with thyroglobulin but at variance with the thyroperoxidase gene, this stimulation requires ongoing protein synthesis.123 Chronic stimulation of cultured dog thyrocytes by TSH for several days does not lead to any important down-regulation in mRNA. Similar data have been obtained with human thyrocytes in primary culture.123,271 By contrast, negative regulation of receptor mRNA accumulation has been observed in immortal FRTL5 cells after treatment with TSH or TSAB.267,269 This difference from human and canine cells must probably be interpreted in the general framework of the other known differences in phenotype and regulatory behavior of this cell line as compared with primary cultured thyrocytes (see later).85 The presence in the promoter region of a CRE-like DNA motif which appears to be able to bind the CREB protein,264 a transcriptional activator directly activated by cAMP, as well as the CREM isoform ICER,212 a transcriptional repressor induced by cAMP, could explain both reported increase and decrease in gene expression following TSH stimulation, depending on the relative amounts of these factors (and likely of other CRE-binding proteins also) preexisting in the studied cells and the kinetics of the individual observations. Moreover, the binding site of the TRα1/RXR heterodimer identified in this promoter encompasses the CRE-like motif (see Fig. 3-7), which may add a further level of complexity depending on the availability of thyroid hormone in the experimental system. Recently, thyrotropin receptor mRNA was shown to be decreased in PCCl3 cells exposed to a high iodide concentration.224
The effect of malignant transformation on the amounts of TSH receptor mRNA has been studied in spontaneous tumors in humans,272,273 in a murine transgenic model of thyroid tumor promoted by expression of the simian virus-40 large T oncogene,275 and in FRTL5 cells transformed with v-ras.268 In the last two models, expression of the TSH receptor gene was suppressed: the tumor or cell growth became TSH independent. In the transgenic animal model, loss of TSH receptor mRNA seemed to take place gradually, with early tumors still displaying some TSH dependence for growth. In the human tumors, a spectrum of phenotypes was observed. As expected, anaplastic tumors had completely lost the receptor mRNA, as well as other markers of thyrocyte differentiation (thyroglobulin and thyroperoxidase). In papillary carcinoma, variable amounts of TSH receptor mRNA were invariably found,272 even in the tumors that had lost the capacity to express the thyroglobulin or thyroperoxidase genes.272 These data agree well with the observations of thyrocytes in primary culture: expression of the TSH receptor gene is robust, and it persists in the presence of agents (or after several steps in tumor progression) that promote extinction of the other markers of thyroid cell differentiation. This evidence leads to the conclusion that the basic marker of the thyroid phenotype is probably the TSH receptor itself, which makes sense: the gene encoding the sensor of TSH—the major regulator of thyroid function, growth, and differentiated phenotype—is virtually constitutive in thyrocytes. From a pragmatic viewpoint, these data provide a rationale for the common therapeutic practice of suppressing TSH secretion in patients with a differentiated thyroid tumor.276
Thyroid Oxidases
Two distinct genes, ThOX1 and ThOX2 (also known as DUOX-1 and DUOX-2), both significantly related to the gene encoding the phagocyte NADPH oxidase gp91Phox, are expressed essentially in the thyroid, but not exclusively, at least for ThOX2.179,277 In the dog, ThOX mRNAs accumulate in response to TSH/cAMP stimulation.179 This effect is much less apparent in man,179 and in the rat, conflicting results were obtained in vivo and in the established FRTL-5 cell line, respectively.277 The proximal promoter sequences of both human THOX1 and THOX2 genes have been delineated using a functional assay.278 These promoters do not exhibit a thyroid cell-restricted activity in vitro and are not controlled positively by TSH/cAMP, as could be expected. No known regulatory DNA motif could be identified within these promoter sequences. Notably, like the promoter of the TSH receptor gene, both of these promoters are devoid of a TATA-box motif. A recent report identified the ThOX-2 promoter as a target for either Pax8 or TTF-1,279 but another study indicated that this control is likely to involve regulatory sequences other than the ones identified and cloned so far.280
Recently, genes encoding proteins required for the functional maturation of the thyroid oxidases were identified in the close vicinity of both ThOX genes, and named DUOXA1 and DUOXA2, respectively.281 Within each ThOX-DUOXA pair, the two genes are arranged in a head-to-head configuration, the distance separating the putative transcription starts being in the range of only 100 bp. This raises the question as to whether the ThOX promoter sequences identified previously278 may also control transcription in the other direction.
Control of Growth and Differentiation
The thyroid is composed of thyrocytes (70%), endothelial cells (20%), and fibroblasts (10%) (proportions measured in dog thyroid). In a normal adult, the weight and composition of the tissue remain relatively constant. Because a low but significant proliferation is demonstrated in all types of cells, it must be assumed that the generation of new cells is balanced by a corresponding rate of cell death.3,282,283 The resulting turnover is on the order of 1 per 5 to 10 years for human thyrocytes (i.e., 6 to 8 renewals in adult life, as in other species).283 Normal cell population can therefore be modulated mainly at the level of proliferation and only secondarily by cell death. In growth situations—that is, either in normal development or after stimulation—the different cell types grow more or less in parallel, which implies a coordination between them.16,34,284,285 Because TSH receptors and iodine metabolism and signaling coexist only in the thyrocyte in thyroid, this cell, sole receiver of the physiologic information, must presumably control the other types of cells by paracrine factors such as VEGF, FGF, IGF-1, NO, and the like.16 The successful isolation of human thyroid endothelial cells will allow a more detailed study of these interactions.286 TSH has been demonstrated to up-regulate the production of vascular endothelial cell growth factor (VEGF) by human thyrocytes.287 It is interesting in this regard that the vascular support of the follicles reflects their activity, suggesting the concept of angiofollicular units.288,289
The Mitogenic Cascades
In the thyroid, at least three families of distinct mitogenic pathways have been well defined (Fig. 3-8): (1) the hormone receptor–adenylyl cyclase–cAMP-dependent protein kinase system, (2) the hormone receptor–tyrosine protein kinase pathways, and (3) the hormone receptor–phospholipase C cascade.3,290 The receptor–tyrosine kinase pathway may be subdivided into two branches; some growth factors, such as EGF, induce proliferation and repress differentiation expression, whereas others, such as FGF in dog cells or IGF-1 and insulin, are either mitogenic or are necessary for the proliferation effect of other factors without being mitogenic by themselves, but they do not inhibit differentiation expression.28,242 In human thyroid cells, IGF-1 is required for the mitogenic action of TSH or EGF, but by itself it only weakly stimulates proliferation.78 In dog and human thyrocytes in primary cultures, after induction of insulin receptors by TSH, physiologic concentrations of insulin permit the proliferative action of TSH.20,21 In FRTL5 and rat cells, and in mouse thyroid in vivo,291 IGF-1 is weakly mitogenic per se,292 whereas in pig thyroid cells, it produces a strong effect.293 Expression of both human IGF-1 and IGF-1R in mouse thyroid induces a mild goiter and a decrease in serum TSH level—that is, an increase in non-TSH-activated thyroid function.291 Similarly, relatively high serum IGF-1 levels in a human population are associated with higher goiter frequency.294
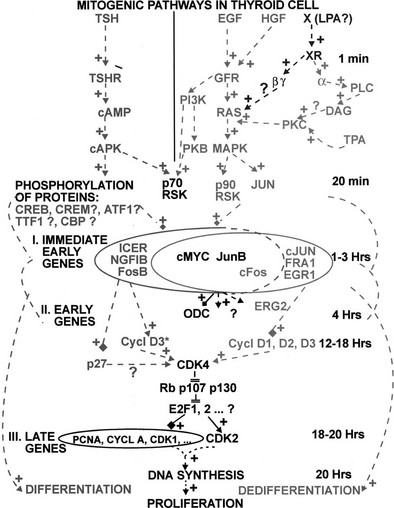
FIGURE 3-8 Mitogenic pathways in the thyroid. Data from the thyroid cell systems are integrated into the present general scheme of cell proliferation cascades. In the first line, known activators of various cascades in dog and human thyroid cells are shown. Various levels indicate a time sequence and postulated causal relationships from initial interaction of extracellular signal with its receptor to endpoints: proliferation and differentiation expression. In dog but not in human thyroid cells, acetylcholine through muscarinic receptors activates the phospholipase C cascade. cAPK, Cyclic adenosine monophosphate-dependent kinase; CDK, cyclin-dependent kinase; DAG, diacylglycerol; ——→+, stimulation; ——|+, inhibition; ——  +, induction; GFR, growth factor receptor; ODC, ornithine decarboxylase; PI3K, phosphatidylinositol 3-kinase; PKB, protein kinase B; PLC, phospholipase C; RSK, ribosomal S6 kinase.
+, induction; GFR, growth factor receptor; ODC, ornithine decarboxylase; PI3K, phosphatidylinositol 3-kinase; PKB, protein kinase B; PLC, phospholipase C; RSK, ribosomal S6 kinase.
It should be noted that TSH directly stimulates proliferation while maintaining the expression of differentiation. Differentiation expression, as evaluated by NIS or by thyroperoxidase and thyroglobulin mRNA content or nuclear transcription, is induced by TSH, forskolin, cholera toxin, and cAMP analogs.3 These effects are obtained in all the cells of a culture, as shown by in situ hybridization experiments.242 They are reversible; they can be obtained either after the arrest of proliferation or during the cell-division cycle.223,242 Moreover, the expression of differentiation, as measured by iodide transport, is stimulated by concentrations of TSH lower than those required for proliferation.78
All the proliferation effects of TSH are mimicked by nonspecific modulators of the cAMP cascade (e.g., cholera toxin and forskolin, which stimulate adenylate cyclase), cAMP analogs (which activate the cAMP-dependent protein kinases), and even synergistic pairs of cAMP analogs acting on the different sites of these two kinases.78,295,296 They are reproduced in vitro and in vivo by expression of the adenosine A2 receptor, which is constitutively activated by endogenous adenosine,253 and by constitutively active Gsα297 and cholera toxin.298 They are inhibited by antibodies blocking Gs.299 Inhibition of cAMP-dependent protein kinases (PKA) inhibits the proliferation and differentiation effects of cAMP.300,301 Stimulation of PKA by selective cAMP analogs that do not activate EPAC proteins is sufficient to fully mimic mitogenic effects of TSH and forskolin in dog thyrocytes.61 There is, therefore, no doubt that the mitogenic and differentiating effects of TSH are mainly and probably entirely mediated by cAMP-dependent protein kinases. A complementary role of the Rap guanyl nucleotide exchange factor EPAC and of Rap1 has been proposed in cell lines and in mice,302–304 but it is not observed in canine thyroid primary cultures.61 Permanent abolition of Gq expression in mouse thyroid leads as expected to an inhibition of iodide oxidation and thyroid hormone synthesis but also to hypotrophy of the gland.305 This suggests at least a necessary chronic permissive role of the Gq phospholipase C cascade. In dog thyroid cells, carbamylcholine acting through the Gq cascade has the same permissive effects on TSH mitogenic action as IGF-1, perhaps through activation of PKB.88,306
EGF also induces proliferation of thyroid cells from various species.3,16,78 However, the action of EGF is accompanied by a general and reversible loss of differentiation expression, assessed as described earlier.274 The effects of EGF on differentiation can be dissociated from its proliferative action. Indeed, they are obtained in cells that do not proliferate in the absence of insulin and in human cells, in which the proliferative effects are weaker, or in pig cells at concentrations lower than the mitogenic concentrations.3
Finally, the tumor-promoting phorbol esters, the pharmacologic probes of the protein kinase C system, and analogs of diacylglycerol also enhance the proliferation and inhibit the differentiation of thyroid cells. These effects are transient because of desensitization of the system by protein kinase C inactivation.3,78,307,308 Activation of the Gq/phospholipase C (PLC)/PKC cascade by a more physiologic agent, such as carbamylcholine, in dog thyroid cells does not reproduce all the effects of phorbol esters. In particular, prolonged stimulation of this cascade by carbamyl choline permits the cAMP-dependent mitogenesis of dog thyrocytes,306 but unlike phorbol esters, it does not induce proliferation in the presence of insulin.309 The Ras protooncogene is strongly activated by phorbol esters but very weakly by carbachol.88 Thus we cannot necessarily equate the effects of phorbol esters and prolonged stimulation of the PLC cascade. The dedifferentiating effects of phorbol esters do not require their mitogenic action either. Thus the effects of TSH, EGF, and phorbol esters on differentiation expression are largely independent of their mitogenic action.3
In several thyroid cell models, very high insulin concentrations are necessary for growth even in the presence of EGF. We now know that this prerequisite mainly reflects a requirement for IGF-1 receptor.3,76,292,310 It is interesting that in FRTL5 cells, as in cells from thyroid nodules, this requirement may disappear, perhaps because the cells secrete their own somatomedins and thus become autonomous with regard to these growth factors.76,311 By contrast, in primary cultures of normal dog and human thyrocytes, very low concentrations of insulin, acting on insulin receptors, are sufficient to support the mitogenic effects of TSH and cAMP when insulin receptors have been induced to high levels by TSH.20,21 This puzzling regulation, which is reminiscent of the induction of insulin receptors during the differentiation of adipocytes, suggests that thyroid might well be revealed as a more specific target of circulating insulin than hitherto recognized.
In the action of growth factors on receptor protein tyrosine kinase pathways, the effects on differentiation expression vary with the species and the factor involved: from stimulation (e.g., insulin, as well as IGF-2 in dog and FRTL5 cells),243 to an absence of effect,312 to transitory inhibition of differentiation during growth (FGF and HGF in dog cells),28,313 to full but reversible dedifferentiation effects (EGF in dog and human cells).77,274 Ret/PTC rearrangements, activating mutations of Ras, as well as oncogenic mutation of B-Raf, which are responsible for most differentiated carcinoma, constitutively activate the signaling cascades of growth factors.314–316
The kinetics of the induction of thymidine incorporation into nuclear DNA of dog thyroid cells is similar for TSH, forskolin, EGF, and TPA. Whatever the stimulant, a minimal delay of about 16 to 20 hours takes place before the beginning of labeling—that is, the beginning of DNA synthesis.317 This is the minimal amount of time required to prepare the necessary machinery. For the cAMP and EGF pathways, the stimulatory agent has to be present during this whole prereplicative period; any interruption in activation (e.g., by washing out the stimulatory forskolin) greatly delays the start of DNA synthesis.318 This limitation explains why norepinephrine and prostaglandin E, which also activate the cAMP cascade, do not induce growth and differentiation: the rapid desensitization of their receptors does not allow a sustained rise in cAMP levels.
The three main types of mitogenic cascade, specifically the growth factor–protein tyrosine kinase, phorbol ester–protein kinase C, and TSH-cAMP cascades, are fully distinct at the level of their primary intracellular signal and/or the first signal-activated protein kinase.3
Iodide actually inhibits the cAMP and the Ca2+-phosphatidylinositol cascades and in a more delayed and chronic effect decreases the sensitivity of the thyroid to the TSH growth response. These effects are relieved, according to the general paradigm of Van Sande, by perchlorate and methimazole95,319 in that they are mediated by a postulated organified derivative of iodide, iodohexadecanal.97
Steps in the Mitogenic Cascades
The phenomenology of EGF, TPA, and TSH proliferative action cells has been partially elucidated using dog thyroid primary cultures.3,85 The mechanisms of TSH/cAMP mitogenic effects have also been investigated using immortal rat thyroid cell lines (FRTL-5, WRT, and PC Cl3 cells).86 Whereas the signaling cascades involved in the action of growth factors and IGF-1 are likely to be well conserved in the different thyroid systems, as generally observed in the other cell types, the mechanistic logistics of cell-cycle regulation by cAMP has disappointingly turned out to strongly diverge in the various thyroid in vitro models.85 These divergences do not only reflect species differences.3 Among the apparently similar rat thyroid cell lines, or even among different subclones of FRTL-5 cells, major differences have been observed.85 For instance, the PI3 kinase/PKB cascade is activated by cAMP in WRT cells320 but inhibited by cAMP in PC Cl3 cells.321 The induction of c-Jun by TSH/cAMP in FRTL-5 cells and its repression by cAMP in WRT cells,322 as in dog323 and human thyrocytes, likely reflect major differences in upstream signaling cascades and should result in divergent expression of downstream target genes, such as cyclin D1. Cyclin D1 synthesis, an accepted endpoint of mitogenic cascades, is indeed induced by cAMP in FRTL-5 and PC Cl3 cells, but repressed by cAMP in dog and human thyroid primary cultures.85,324 The reasons for such discrepancies are unclear. Some signaling features, when they lead to selective proliferative advantages, might have been acquired during the establishment and continuous cultures of the cell lines and stabilized by subcloning. Many mechanisms demonstrated in the dog thyroid primary culture system so far apply to normal human thyrocytes, but much remains to be defined.85 In the following lines, we thus rely mostly on these systems.
Three biochemical aspects of the proliferative response occurring at different times of the prereplicative phase have been considered. The pattern of protein phosphorylation induced within minutes by TSH is reproduced by forskolin and cAMP analogs. It totally diverges from the phosphorylations induced by EGF and phorbol esters.325 EGF, HGF, and phorbol ester actions rapidly converge on the activation of Ras88 and the resulting activation of p42/p44 MAP kinases and p90RSK.74,75,87 PI-3-kinase and its effector enzyme PKB are activated for several hours only by insulin and IGF-1, the effect of EGF being short lived.74 This activity is therefore the one specific feature of insulin action and presumably the mechanism of the facilitating effect on mitogenesis. In dog thyrocytes, only HGF can trigger cell proliferation in the absence of insulin/IGF-1; this is explained by the fact that only this factor strongly activates both PI3 kinase and MAP kinase cascades.74 Only insulin, IGF-1 and HGF also markedly enhance general protein synthesis and induce cell hypertrophy.211 By contrast, TSH and cAMP are very unique as mitogens, since they do not activate Ras, the PI3kinase/PKB pathway, or any of different classes of MAP kinases in dog thyrocytes.74,75,87,88 TSH and cAMP also do not activate MAP kinases in human thyrocytes.75 The phosphorylation and activation of p70S6K and thus likely the mTOR cascade constitutes the only early convergence point of growth factor and cAMP-dependent mitogenic cascades.74,326 A recent study has demonstrated the crucial role of this cascade for TSH-elicited thyroid follicular hyperplasia in vivo in mice.327 Indeed, as found in dog thyroid primary cultures74 and PCCl3 cells (Blancquaert and Roger, unpublished), TSH stimulates in mice the mTOR/p70S6K axis without activating PKB, and a rapamycin derivative abrogates the hyperplastic (but interestingly, not the hypertrophic) responses to TSH.327 The cAMP-dependent mitogenesis and gene expression appears to require the phosphorylation by PKA and activity of CREB/CREM transcription factors.328,329
As in other types of cells, EGF and TPA first enhance c-fos and c-myc mRNA and protein concentrations in dog thyrocytes. On the other hand, TSH and forskolin strongly, but for a short period, enhance the c-myc mRNA concentration and with the same kinetics as the enhancement of the c-fos mRNA concentration by EGF/TPA. In fact, cAMP first enhances and then decreases c-myc expression. This second phenomenon is akin to what has been observed in the fibroblast, in which cAMP negatively regulates growth. As in fibroblasts, EGF and TPA enhance c-Jun, junB, junD, and egr1 expression. However, as in fibroblasts, activators of the cAMP cascade decrease c-Jun and egr1 expression. Therefore, c-Jun is not, as has been claimed, a gene whose expression is universally necessary for growth.211,323
The investigation of the pattern of proteins synthesized in response to the various proliferation stimuli has suggested very early that the proliferation of dog thyroid cells is controlled during G1 phase by at least two largely distinct cAMP-dependent or cAMP-independent pathways.330 Recent microarray analyses have confirmed and extended this concept in human thyrocytes.63,331 Nevertheless, the different mitogenic cascades are expected to finally modulate the level and activity of proteins that are the primary regulators of the cell-cycle machinery. As generally considered, mitogenic signals regulate the mammalian cell cycle by stimulating the accumulation of D-type cyclins and their assembly through an ill-defined mechanism, with their partners the cyclin-dependent kinases (CDKs) 4 and 6. These complexes operate in mid- to late G1 phase to promote progression through the restriction point and thus commit cells to replicate their genome.332 In the current model, this key decision depends on the initiation by cyclin D-CDK complexes of the phosphorylation of the growth/tumor suppressor protein pRb, which triggers the activation of transcription factors, including those of the E2F family, the synthesis of cyclin E and then cyclin A, and CDK2 activation by these cyclins. Activated CDK2 in turn further phosphorylates pRb and other substrates and initiates and organizes the progression through the DNA synthesis phase.332 The down-regulation of CDK inhibitors of the CIP/KIP family, including p27kip1, by mitogenic factors and/or their sequestration by cyclin D-CDK complexes participate to CDK2 activation, but their proposed role of adaptor and/or nuclear anchor for cyclin D-CDK complexes suggests positive influences on cell-cycle progression as well.333 These mechanisms have been well studied in dog thyroid cells324 (Fig. 3-9). As expected, the different mitogenic stimulations (TSH, cAMP, growth factors) require the activity of CDK4334 and converge on the inactivating phosphorylation of pRb and related proteins p107 and p130335 on the phosphorylation and nuclear translocation of CDK2, and on the induction of cyclin A and cdc2.336 These effects are dependent on insulin.335,337 What is strikingly different between the cascades is the mechanism of D-type cyclin-CDK4 activation. TSH, unlike all the other known mitogenic factors, does not induce the accumulation of D cyclins,338 but it paradoxically stimulates the expression of the CDK “inhibitor” p27kip1.339 However, the predominant cyclin, D3, is required for the proliferation stimulated by TSH but not in the proliferation of dog thyrocytes stimulated by EGF or HGF that induce cyclins D1 and D2, in addition to increasing cyclin D3 levels.338 The formation and the nuclear translocation of essential cyclin D3-CDK4 complexes depend on the synergistic interaction of TSH and insulin.337,338 These complexes are absent from cells stimulated by TSH or insulin alone. Paradoxically, in the absence of insulin, TSH inhibits the basal accumulation of cyclin D3.337 Conversely, insulin alone stimulates the required cyclin D3 accumulation, and it overcomes in large part the inhibition by TSH,337 but it is unable to assemble cyclin D3-CDK4 complexes in the absence of TSH. In the presence of insulin, TSH (cAMP) unmasks some epitopes of cyclin D3 and induces the assembly of cyclin D3-CDK4 complexes and their import into nuclei,337,338 where these complexes are anchored by their association with p27kip1.340,341 This also sequesters p27 away from CDK2 complexes,340 thus contributing to CDK2 activation. Moreover, cAMP exerts an additional crucial function in very late G1 phase to stimulate the enzymatic activity of cyclin D3-cdk4-p27 complexes, which involves the stimulation of the activating Thr172-phosphorylation of CDK4.324,342 TGF-β selectively inhibits the cAMP-dependent proliferation of dog thyrocytes by preventing the association of the cyclin D3-CDK4 complex with nuclear p27kip1 and the activating phosphorylation of CDK4.340,341
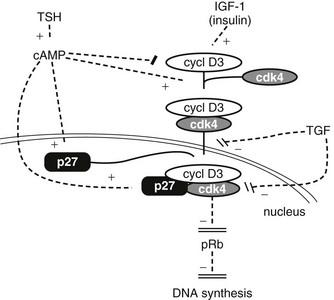
FIGURE 3-9 Targets of cell-cycle regulatory effects of TSH, insulin/IGF-1 and TGF-β, as demonstrated in the dog thyroid primary culture system. Diamond/rectangle arrowheads represent inductions/repressions; the other dashed arrows are activations (+) and inhibitions (−). TSH (cAMP) does not induce D cyclins, but it assembles and then activates the cyclin D3-CDK4-p27 holoenzyme by inducing the activating Thr172-phosphorylation of CDK4. IGF-1 and insulin allow the accumulation of the required cyclin D3. TGF-β inhibits the nuclear translocation of the cyclin D3-CDK4 complex, its association to p27, and its activation by TSH(cAMP). See text for full explanation.
The investigation of cell-cycle regulatory proteins has thus clearly established that both CDK4 activation and pRb phosphorylation result from distinct but complementary actions of TSH and insulin, rather than from their interaction at an earlier step of the signaling cascades306,337 (see Fig. 3-9). Together with the fact that the necessary increase of cell mass before division depends on insulin/IGF-1 but not TSH,211 these observations provide a molecular basis for the well-established physiologic concept that in the regulation of normal thyroid cell proliferation, TSH is the “decisional” mitotic trigger, while locally produced IGF-1 and/or circulating insulin are supporting “permissive” factors.3 Of note, in all these experiments, the facilitative action of insulin can be replaced by activation of the Gq/PLC cascade by carbamylcholine.306
Studies of protein phosphorylation, proto-oncogene expression, and cell-cycle regulatory proteins in dog thyrocytes allow discrimination between two models of cAMP action on proliferation in this system: a direct effect on the thyrocyte or an indirect effect through the secretion and autocrine action of another growth factor. If the effect of TSH through cAMP involved such an autocrine loop, one would expect to find faster kinetics of action of the growth factor and at least some common parts in the patterns of protein phosphorylation and protein synthesis induced by cAMP and the growth factor. The results do not support such a hypothesis, at least for the growth factors tested3 (see Fig. 3-8). Nor do the data on cAMP action in the dog and human thyrocyte systems support a major role for various mechanisms involving cross-signaling of cAMP with growth factor pathways, as claimed in rat thyroid cell line studies (reviewed and discussed in85). Indeed, in primary cultures of normal human thyrocytes, EGF+serum increases cyclin D1 and p21 accumulation, and it stimulates the assembly and activity of cyclin D1-CDK4-p21. By contrast, TSH (cAMP) represses cyclin D1 and p21, but it stimulates the activating phosphorylation of CDK4 and the pRb-kinase activity of preexisting cyclin D3-CDK4 complexes.343 Cyclin D1 and cyclin D3 are thus differentially used in the distinct mitogenic stimulations by growth factors and TSH and potentially in hyperproliferative diseases generated by the overactivation of their respective signaling pathways.
The validity of these concepts in vivo has been established by using transgenic mice models. The expression in thyroid of oncogene E7 of HPV16, which sequestrates pRb protein, leads to thyroid growth and euthyroid goiter. Expression in the thyroid of the adenosine A2 receptor, which behaves as a constitutive activator of adenylyl cyclase, induces thyroid growth, goitrogenesis, and hyperthyroidism.253 Similar, albeit weaker, phenotypes are obtained in mice expressing constitutive Gs (the G protein activating adenylyl cyclase)297 or cholera toxin.298 On the contrary, the expression in thyroid of a dominant negative CREB provokes a marked thyroid hypotrophy, suggesting the crucial role of CREB and its activating phosphorylation by PKA.329 By contrast, transgenic mice overexpressing both human IGF-1 and IGF-1 receptor in their thyroid (TgIGF-1–TgIGF-IR) develop only a mild thyroid hyperplasia and respond to a goitrogenic effect of antithyroid drugs while maintaining a comparatively low serum TSH level. This indicates some autonomy of these thyroids, as in acromegalic patients, and a much greater sensitivity to endogenous TSH.291 Recently, thyrocyte-specific deficiency of Gq/G11 (the G proteins activating PLCb) in mice was shown to impair not only the TSH-stimulated iodine-organification and thyroid hormone synthesis, but also TSH-dependent development of goiter.305 It remains to be defined whether this impaired follicular cell hyperplasia could result in part from the lack of induction of VEGF and angiogenesis305 which normally accompany goitrogenesis. Nevertheless, the phenotype of these mice underscores the role in TSH-dependent goitrogenesis of PLC, which is activated by TSH but even more strongly by neurotransmitters. Noteworthy, section of inferior laryngeal nerve in rats was similarly found to impair both thyroid function and growth stimulated by TSH.344 Moreover, activation of Gq /PLC by carbamylcholine can facilitate cAMP-dependent mitogenesis in dog thyrocytes cultured without insulin or IGF-1.306 On the other hand, expression of Ret (which is a constitutive growth factor receptor) in papillary thyroid carcinoma (PTC) leads to growth, cancer, and hypothyroidism.345,346
Proliferation an Differentiation (Fig. 3-10)
The incompatibility at the cell level of a proliferation and differentiation program is commonly accepted in biology. In general, cells with a high proliferative capacity are poorly differentiated, and during development, such cells lose this capacity as they progressively differentiate. Some cells even lose all potential to divide when reaching their full differentiation, a phenomenon called terminal differentiation. Conversely, in tumor cells, proliferation and differentiation expression are inversely related. Activation of Ras and p42/p44 MAP kinases, induction of c-Jun, sustained expression of c-myc, induction of cyclin D1 and down-regulation of p27kip1 all have been shown to be causatively associated not only with proliferation but also with loss of differentiation in a large variety of systems, sometimes independently of proliferation effects. It is therefore not surprising that in thyroid cells, the general mitogenic agents and pathways, phorbol esters and the protein kinase C pathway, EGF, and in calf and porcine cells, FGF and the protein tyrosine kinase pathway, induce both proliferation and the loss of differentiation expression. The effects of the cAMP cascade are in striking contrast with this general concept. Indeed, TSH and cAMP induce proliferation of dog thyrocytes while maintaining differentiation expression; both proliferation and differentiation programs can be triggered by TSH in the same cells at the same time.242 This situation is by no means unique, because neuroblasts in the cell cycle may simultaneously differentiate. It is tempting to relate this apparent paradox to the unique characteristics of the cAMP-dependent mitogenic pathway, such as the lack of activation (or even the inhibition) of the Ras/MAP kinase/c-Jun/cyclin D1 cascade, as demonstrated in dog and human thyrocytes. For instance, if one generalization could be made about proto-oncogenes, it is the dedifferentiating role of c-myc. A rapid and dramatic decrease in c-myc mRNA by antisense myc sequences induces differentiation of a variety of cell types. It is therefore striking that in the case of the thyrocyte, in which activation of the cAMP cascade leads to both proliferation and differentiation, the kinetics of the c-myc gene appears to be tightly controlled. After a first phase of 1 hour of higher levels of c-myc mRNA, c-myc expression is decreased below control levels. In this second phase, cAMP decreases c-myc mRNA levels, as it does in proliferation-inhibited fibroblasts. It even depresses EGF-induced expression. The first phase could be necessary for proliferation, whereas the second phase could reflect stimulation of differentiation by TSH.210,347 The specific involvement of cyclin D3 in the cAMP-dependent mitogenic stimulation of dog and human thyrocytes is also interesting in this context.343 We have recently shown that the differential utilization of cyclin D1 or cyclin D3 affects the site specificity of the pRb-kinase of CDK4, including in dog and human thyrocytes.343,348 In addition to inhibiting E2F-dependent gene transcription related to cell-cycle progression, pRb plays positive roles in the induction of tissue-specific gene expression by directly interacting with a variety of transcription factors, including Pax8 in thyroid cells.254 Whether the selective utilization of cyclin D3 in the TSH cascade, associated with a more restricted pRb-kinase activity, could allow the preservation of some differentiation-related functions of pRb thus remains to be examined. Indeed, unlike cyclins D1 and D2, cyclin D3 is highly expressed in several quiescent tissues in vivo, and its expression is not only stimulated by mitogenic factors but also induced during several differentiation processes associated with a repression of cyclin D1.349
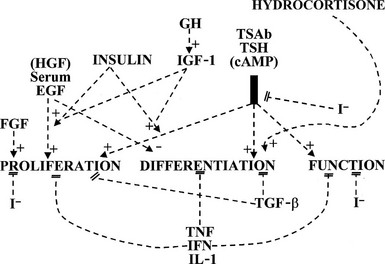
FIGURE 3-10 Main controls of the principal biological variables of the human thyrocyte. EGF, Epidermal growth factor; FGF, fibroblast growth factor; GH, growth hormone; HGF, hepatocyte growth factor; I−, iodide; IGF-1, insulin-like growth factor; IFN, interferon; IL-1, interleukin-1; TGF-β, transforming growth factor β; TNF, tumor necrosis factor; TSAb, thyroid-stimulating immunoglobulins; +, positive control (stimulation); −, negative control (inhibition).
We now consider the distinct cAMP-dependent mitogenic pathway, which appears to be adjuncted to the more general mechanisms used by growth factors, as pertaining to the specialized differentiation program of thyroid cells.82 In dog thyrocytes, the proliferation in response to serum or growth factors specifically extinguishes their capacity to respond to TSH/cAMP as a mitogenic stimulus.350 Similarly, in less differentiated thyroid cancers generated by the subversion of growth factor mechanisms, the TSH dependence of growth is generally found to be lost, and in various cell lines derived from thyroid carcinomas, cAMP and PKA activation even inhibit CDK4 activity and cell-cycle progression.351
Because the cell renewal rate is very low in the thyroid (once every 8 years in adults), the role of apoptosis is also very low. However, under different circumstances, the apoptotic role can greatly increase, such as after the arrest of an important stimulation in vitro352 and in vivo.353–355
References
1. Moeller, LC, Alonso, M, Liao, X, et al. Pituitary-thyroid setpoint and thyrotropin receptor expression in consomic rats. Endocrinology. 2007;148:4727–4733.
2. Nakabayashi, K, Matsumi, H, Bhalla, A, et al. Thyrostimulin, a heterodimer of two new human glycoprotein hormone subunits, activates the thyroid-stimulating hormone receptor. J Clin Invest. 2002;109:1445–1452.
3. Dumont, JE, Lamy, F, Roger, PP, et al. Physiological and pathological regulation of thyroid cell proliferation and differentiation by thyrotropin and other factors. Physiol Rev. 1992;72:667–697.
4. Vassart, G, Dumont, JE. The thyrotropin receptor and the regulation of thyrocyte function and growth. Endocr Rev. 1992;13:596–611.
5. Brabant, G, Bergmann, P, Kirsch, CM, et al. Early adaptation of thyrotropin and thyroglobulin secretion to experimentally decreased iodine supply in man. Metabolism. 1992;41:1093–1096.
6. Dumont, JE. The action of thyrotropin on thyroid metabolism. Vitam Horm. 1971;29:287–412.
7. Marians, RC, Ng, L, Blair, HC, et al. Defining thyrotropin-dependent and -independent steps of thyroid hormone synthesis by using thyrotropin receptor-null mice. Proc Natl Acad Sci U S A. 2002;99:15776–15781.
8. Postiglione, MP, Parlato, R, Rodriguez-Mallon, A, et al. Role of the thyroid-stimulating hormone receptor signaling in development and differentiation of the thyroid gland. Proc Natl Acad Sci U S A. 2002;99:15462–15467.
9. Abramowicz, MJ, Duprez, L, Parma, J, et al. Familial congenital hypothyroidism due to inactivating mutation of the thyrotropin receptor causing profound hypoplasia of the thyroid gland. J Clin Invest. 1997;99:3018–3024.
10. Toyoda, N, Nishikawa, M, Horimoto, M. Synergistic effect of thyroid hormone and thyrotropin on iodothyronine 5′-adenosinase in FRTL-5 rat thyroid cells. Endocrinology. 1990;127:1199–1205.
11. Paire, A, Bernier-Valentin, F, Rabilloud, R, et al. Expression of alpha- and beta-subunits and activity of Na+ K+ ATPase in pig thyroid cells in primary culture: modulation by thyrotropin and thyroid hormones. Mol Cell Endocrinol. 1998;146:93–101.
12. Ying, H, Suzuki, H, Zhao, L, et al. Mutant thyroid hormone receptor beta represses the expression and transcriptional activity of peroxisome proliferator-activated receptor gamma during thyroid carcinogenesis. Cancer Res. 2003;63:5274–5280.
13. Glinoer, D, de Nayer, P, Bourdoux, P, et al. Regulation of maternal thyroid during pregnancy. J Clin Endocrinol Metab. 1990;71:276–287.
14. Hershman, JM, Lee, HY, Sugawara, M, et al. Human chorionic gonadotropin stimulates iodide uptake, adenylate cyclase, and deoxyribonucleic acid synthesis in cultured rat thyroid cells. J Clin Endocrinol Metab. 1988;67:74–79.
15. Glinoer, D. The regulation of thyroid function in pregnancy: pathways of endocrine adaptation from physiology to pathology. Endocr Rev. 1997;18:404–433.
16. Dumont, JE, Maenhaut, C, Pirson, I, et al. Growth factors controlling the thyroid gland. Baillieres Clin Endocrinol Metab. 1991;5:727–754.
17. Gerard, CM, Roger, PP, Dumont, JE. Thyroglobulin gene expression as a differentiation marker in primary cultures of calf thyroid cells. Mol Cell Endocrinol. 1989;61:23–35.
18. Cheung, NW, Lou, JC, Boyages, SC. Growth hormone does not increase thyroid size in the absence of thyrotropin: a study in adults with hypopituitarism. J Clin Endocrinol Metab. 1996;81:1179–1183.
19. Dormitzer, PR, Ellison, PT, Bode, HH. Anomalously low endemic goiter prevalence among Efe pygmies. Am J Phys Anthropol. 1989;78:527–531.
20. Burikhanov, R, Coulonval, K, Pirson, I, et al. Thyrotropin via cyclic AMP induces insulin receptor expression and insulin co-stimulation of growth and amplifies insulin and insulin-like growth factor signaling pathways in dog thyroid epithelial cells. J Biol Chem. 1996;271:29400–29406.
21. Van Keymeulen, A, Dumont, JE, Roger, PP. TSH induces insulin receptors that mediate insulin costimulation of growth in normal human thyroid cells. Biochem Biophys Res Commun. 2000;279:202–207.
22. Ahrén, B. Regulatory peptides in the thyroid gland—a review on their localization and function. Acta Endocrinol (Copenh). 1991;124:225–232.
23. Raspé, E, Laurent, E, Andry, G, et al. ATP, bradykinine, TRH and TSH activate the Ca2+-phophatidyl inositol cascade of human thyrocytes in primary culture. Mol Cell Endocrinol. 1991;81:175–183.
24. Raspé, E, Andry, G, Dumont, JE. Adenosine triphosphate, bradykinin, and thyrotropin-releasing hormone regulate the intracellular Ca2+ concentration and the 45Ca2+ efflux of human thyrocytes in primary culture. J Cell Physiol. 1989;140:608–614.
25. Ozawa, S, Spaulding, SW. Epidermal growth factor inhibits radioiodine uptake but stimulates deoxyribonucleic acid synthesis in newborn rat thyroids grown in nude mice. Endocrinology. 1990;127:604–612.
26. Paschke, R, Eck, T, Herfurth, J, et al. Stimulation of proliferation and inhibition of function of xenotransplanted human thyroid tissue by epidermal growth factor. J Endocrinol Invest. 1995;18:359–363.
27. De Vito, WJ, Chanoine, JP, Alex, S, et al. Effect of in vivo administration of recombinant acidic fibroblast growth factor on thyroid function in the rat: induction of colloid goiter. Endocrinology. 1992;131:729–735.
28. Roger, PP, Dumont, JE. Factors controlling proliferation and differentiation of canine thyroid cells cultured in reduced serum conditions: effects of thyrotropin, cyclic AMP and growth factors. Mol Cell Endocrinol. 1984;36:79–93.
29. Westermark, K, Karlsson, FA, Westermark, B. Epidermal growth factor modulates thyroid growth and function in culture. Endocrinology. 1983;112:1680–1686.
30. Eggo, MC, King, WJ, Black, EG, et al. Functional human thyroid cells and their insulin-like growth factor-binding proteins: regulation by thyrotropin, cyclic 3′,5′ adenosine monophosphate, and growth factors. J Clin Endocrinol Metab. 1996;81:3056–3062.
31. Lamy, F, Taton, M, Dumont, JE, et al. Control of protein synthesis by thyrotropin and epidermal growth factor in human thyrocytes: role of morphological changes. Mol Cell Endocrinol. 1990;73:195.
32. Kraiem, Z, Sadeh, O, Yosef, M, et al. Mutual antagonistic interactions between the thyrotropin (adenosine 3′,5′-monophosphate) and protein kinase C/epidermal growth factor (tyrosine kinase) pathways in cell proliferation and differentiation of cultured human thyroid follicles. Endocrinology. 1995;136:585–590.
33. Becks, GP, Logan, A, Phillips, ID, et al. Increase of basic fibroblast growth factor (FGF) and FGF receptor messenger RNA during rat thyroid hyperplasia: temporal changes and cellular distribution. J Endocrinol. 1994;142:325–338.
34. Bidey, SP, Hill, DJ, Eggo, MC. Growth factors and goitrogenesis. J Endocrinol. 1999;160:321–332.
35. Derwahl, M, Broecker, M, Kraiem, Z. Clinical review 101: thyrotropin may not be the dominant growth factor in benign and malignant thyroid tumors. J Clin Endocrinol Metab. 1999;84:829–834.
36. Roger, PP. Thyrotropin-dependent transforming growth factor beta expression in thyroid gland [comment]. Eur J Endocrinol. 1996;134:269–271.
37. Grubeck-Loebenstein, B, Buchan, G, Sadeghi, R, et al. Transforming growth factor beta regulates thyroid growth. J Clin Invest. 1989;83:764–770.
38. Taton, M, Lamy, F, Roger, PP, et al. General inhibition by transforming growth factor beta1 of thyrotropin and cAMP responses in human thyroid cells in primary culture. Mol Cell Endocrinol. 1993;95:13–21.
39. Logan, A, Smith, C, Becks, GP, et al. Enhanced expression of transforming growth factor-beta 1 during thyroid hyperplasia in rats. J Endocrinol. 1994;141:45–57.
40. Franzen, A, Piek, E, Westermark, B, et al. Expression of transforming growth factor-beta1, activin A, and their receptors in thyroid follicle cells: negative regulation of thyrocyte growth and function. Endocrinology. 1999;140:4300–4310.
41. Helmbrecht, K, Kispert, A, von Wasielewski, R, et al. Identification of a Wnt/beta-catenin signaling pathway in human thyroid cells. Endocrinology. 2001;142:5261–5266.
42. Lyons, J, Landis, CA, Harsh, G, et al. Two G protein oncogenes in human endocrine tumors. Science. 1990;249:655–659.
43. Parma, J, Duprez, L, Vansande, J, et al. Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature. 1993;365:649–651.
44. Vasseur, C, Rodien, P, Beau, I, et al. A chorionic gonadotropin-sensitive mutation in the follicle-stimulating hormone receptor as a cause of familial gestational spontaneous ovarian hyperstimulation syndrome. N Engl J Med. 2003;349:753–759.
45. Smits, G, Campillo, M, Govaerts, C, et al. Glycoprotein hormone receptors: determinants in leucine-rich repeats responsible for ligand specificity. EMBO J. 2003;22:2692–2703.
46. Van Sande, J, Costa, MJ, Massart, C, et al. Kinetics of thyrotropin-stimulating hormone (TSH) and thyroid-stimulating antibody binding and action on the TSH receptor in intact TSH receptor-expressing CHO cells. J Clin Endocrinol Metab. 2003;88:5366–5374.
47. Dumont, JE, Roger, PP, Ludgate, M. Assays for thyroid growth immunoglobulins and their clinical implications: methods, concepts and misconceptions. Endocr Rev. 1987;8:448–452.
48. Zakarija, M, Jin, S, McKenzie, JM. Evidence supporting the identity in Graves’ disease of thyroid-stimulating antibody and thyroid growth-promoting immunoglobulin G as assayed in FRTL-5 cells. J Clin Invest. 1988;81:879–884.
49. Zakarija, M, McKenzie, JM. Do thyroid growth-promoting immunoglobulins exist? J Clin Endocrinol Metab. 1990;70:308–310.
50. Contempré, B, Le Moine, O, Dumont, JE, et al. Selenium deficiency and thyroid fibrosis. A key role for macrophages and transforming growth factor beta (TGF-beta). Mol Cell Endocrinol. 1996;124:7–15.
51. Thompson, SD, Franklyn, JA, Watkinson, JC, et al. Fibroblast growth factors 1 and 2 and fibroblast growth factor receptor 1 are elevated in thyroid hyperplasia. J Clin Endocrinol Metab. 1998;83:1336–1341.
52. Fusco, A, Santoro, M, Grieco, M, et al. RET/PTC activation in human thyroid carcinomas. J Endocrinol Invest. 1995;18:127–129.
53. Pierotti, MA, Bongarzone, I, Borrello, MG, et al. Rearrangements of TRK proto-oncogene in papillary thyroid carcinomas. J Endocrinol Invest. 1995;18:130–133.
54. Trovato, M, Villari, D, Bartolone, L, et al. Expression of the hepatocyte growth factor and c-met in normal thyroid, non-neoplastic, and neoplastic nodules. Thyroid. 1998;8:125–131.
55. Aasland, R, Akslen, LA, Varhaug, JE, et al. Co-expression of the genes encoding transforming growth factor-alpha and its receptor in papillary carcinomas of the thyroid. Int J Cancer. 1990;46:382–387.
56. Vella, V, Pandini, G, Sciacca, L, et al. A novel autocrine loop involving IGF-II and the insulin receptor isoform-A stimulates growth of thyroid cancer. J Clin Endocrinol Metab. 2002;87:245–254.
57. Blaydes, JP, Schlumberger, M, Wynford-Thomas, D, et al. Interaction between p53 and TGF beta 1 in control of epithelial cell proliferation. Oncogene. 1995;10:307–317.
58. Vanvooren, V, Allgeier, A, Cosson, E, et al. Expression of multiple adenylyl cyclase isoforms in human and dog thyroid. Mol Cell Endocrinol. 2000;170:185–196.
59. Van Sande, J, Mockel, J, Boeynaems, JM, et al. Regulation of cyclic nucleotide and prostaglandin formation in human thyroid tissues and in autonomous nodules. J Clin Endocrinol Metab. 1980;50:776–785.
60. de Rooij, J, Zwartkruis, FJT, Verheijen, MHG, et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477.
61. Dremier, S, Milenkovic, M, Blancquaert, S, et al. Cyclic adenosine 3′,5′-monophosphate (cAMP)-dependent protein kinases, but not exchange proteins directly activated by cAMP (Epac), mediate thyrotropin/cAMP-dependent regulation of thyroid cells. Endocrinology. 2007;148:4612–4622.
62. Fortemaison, N, Blancquaert, S, Dumont, JE, et al. Differential involvement of the actin cytoskeleton in differentiation and mitogenesis of thyroid cells: inactivation of Rho proteins contributes to cyclic adenosine monophosphate-dependent gene expression but prevents mitogenesis. Endocrinology. 2005;146:5485–5495.
63. van Staveren, WC, Solis, DW, Delys, L, et al. Gene expression in human thyrocytes and autonomous adenomas reveals suppression of negative feedbacks in tumorigenesis. Proc Natl Acad Sci U S A. 2006;103:413–418.
64. Ketelbant-Balasse, P, Van Sande, J, Neve, P, et al. Time sequence of 3′,5′-cyclic AMP accumulation and ultrastructural changes in dog thyroid slices after acute stimulation by TSH. Horm Metab Res. 1976;8:212–215.
65. Cleator, JH, Ravenell, R, Kurtz, DT, et al. A dominant negative Galphas mutant that prevents thyroid-stimulating hormone receptor activation of cAMP production and inositol 1,4,5-trisphosphate turnover: competition by different G proteins for activation by a common receptor. J Biol Chem. 2004;279:36601–36607.
66. Van Sande, J, Dequanter, D, Lothaire, P, et al. Thyrotropin stimulates the generation of inositol 1,4,5-trisphosphate in human thyroid cells. J Clin Endocrinol Metab. 2006;91:1099–1107.
67. Esteves, R, Van Sande, J, Dumont, JE. Nitric oxide as a signal in thyroid. Mol Cell Endocrinol. 1992;90:R1–R3.
68. Munari-Silem, Y, Audebet, C, Rousset, B. Protein kinase C in pig thyroid cells: activation, translocation and endogenous substrate phosphorylating activity in response to phorbol esters. Mol Cell Endocrinol. 1987;54:81–90.
69. Van Sande, J, Raspe, E, Perret, J, et al. Thyrotropin activates both the cyclic AMP and the PIP2 cascades in CHO cells expressing the human cDNA of TSH receptor. Mol Cell Endocrinol. 1990;74:R1–R6.
70. Laurent, E, Mockel, J, Van Sande, J, et al. Dual activation by thyrotropin of the phospholipase C and cAMP cascades in human thyroid. Mol Cell Endocrinol. 1987;52:273–278.
71. Mockel, J, Laurent, E, Lejeune, C, et al. Thyrotropin does not activate the phosphatidylinositol bisphosphate hydrolyzing phospholipase C in the dog thyroid. Mol Cell Biol. 1991;82:221–227.
72. Mockel, J, Lejeune, C, Dumont, JE. Relative contribution of phosphoinositides and phosphatidylcholine hydrolysis to the actions of carbamylcholine, thyrotropin, and phorbol esters on dog thyroid slices: regulation of cytidine monophosphate-phosphatidic acid accumulation and phospholipase-D activity. II. Actions of phorbol esters. Endocrinology. 1994;135:2497–2503.
73. Lejeune, C, Mockel, J, Dumont, JE. Relative contribution of phosphoinositides and phosphatidylcholine hydrolysis to the actions of carbamylcholine, thyrotropin (TSH), and phorbol esters on dog thyroid slices: regulation of cytidine monophosphate-phosphatidic acid accumulation and phospholipase-D activity. I. Actions of carbamylcholine, calcium ionophores, and TSH. Endocrinology. 1994;135:2488–2496.
74. Coulonval, K, Vandeput, F, Stein, RC, et al. Phosphatidylinositol 3-kinase, protein kinase B and ribosomal S6 kinases in the stimulation of thyroid epithelial cell proliferation by cAMP and growth factors in the presence of insulin. Biochem J. 2000;348:351–358.
75. Vandeput, F, Perpete, S, Coulonval, K, et al. Role of the different mitogen-activated protein kinase subfamilies in the stimulation of dog and human thyroid epithelial cell proliferation by cyclic adenosine 5′-monophosphate and growth factors. Endocrinology. 2003;144:1341–1349.
76. Williams, DW, Williams, ED, Wynford-Thomas, D. Evidence for autocrine production of IGF-1 in human thyroid adenomas. Mol Cell Endocrinol. 1989;61:139–147.
77. Errick, JE, Ing, KW, Eggo, MC, et al. Growth and differentiation in cultured human thyroid cells: effects of epidermal growth factor and thyrotropin. In Vitro Cell Dev Biol. 1986;22:28–36.
78. Roger, PP, Taton, M, Van Sande, J, et al. Mitogenic effects of thyrotropin and adenosine 3′,5′-monophosphate in differentiated normal human thyroid cells in vitro. J Clin Endocrinol Metab. 1988;66:1158–1165.
79. Heldin, NE, Bergström, D, Hermansson, A, et al. Lack of responsiveness to TGF-β1 in a thyroid carcinoma cell line with functional type I and type II TGF-β receptors and Smad proteins, suggests a novel mechanism for TGF-β insensitivity in carcinoma cells. Mol Cell Endocrinol. 1999;153:79–90.
80. Dumont, JE, Miot, F, Erneux, C, et al. Negative regulation of cyclic AMP levels by activation of cyclic nucleotide phosphodiesterases: the example of the dog thyroid. Adv Cyclic Nucleotide Res. 1984;16:325–336.
81. Mockel, J, Van Sande, J, Decoster, C, et al. Tumor promoters as probes of protein kinase C in dog thyroid cell: inhibition of the primary effects of carbamylcholine and reproduction of some distal effects. Metabolism. 1987;36:137–143.
82. Roger, PP, Reuse, S, Maenhaut, C, et al. Multiple facets of the modulation of growth by cAMP. Vitam Horm. 1995;51:59–191.
83. Stork, PJ, Schmitt, JM. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol. 2002;12:258–266.
84. Richards, JS. New signaling pathways for hormones and cyclic adenosine 3′,5′-monophosphate action in endocrine cells. Mol Endocrinol. 2001;15:209–218.
85. Kimura, T, Van Keymeulen, A, Golstein, J, et al. Regulation of thyroid cell proliferation by TSH and other factors: a critical evaluation of in vitro models. Endocr Rev. 2001;22:631–656.
86. Rivas, M, Santisteban, P. TSH-activated signaling pathways in thyroid tumorigenesis. Mol Cell Endocrinol. 2003;213:31–45.
87. Lamy, F, Wilkin, F, Baptist, M, et al. Phosphorylation of mitogen-activated protein kinases is involved in the epidermal growth factor and phorbol ester, but not in the thyrotropin/cAMP, thyroid mitogenic pathways. J Biol Chem. 1993;268:8398–8401.
88. Van Keymeulen, A, Roger, PP, Dumont, JE, et al. TSH and cAMP do not signal mitogenesis through Ras activation. Biochem Biophys Res Commun. 2000;273:154–158.
89. Bray, GA. Increased sensitivity of the thyroid in iodine-depleted rats to the goitrogenic effects of thyrotropin. J Clin Invest. 1968;47:1640–1647.
90. Wolff, J. Congenital goiter with defective iodide transport. Endocr Rev. 1983;4:240.
91. Wolff, J. Iodide goiter and the pharmacologic effects of excess iodide. Am J Med. 1969;47:101–124.
92. Cochaux, P, Van Sande, J, Swillens, S, et al. Iodide-induced inhibition of adenylate cyclase activity in horse and dog thyroid. Eur J Biochem. 1987;170:435–442.
93. Laurent, E, Mockel, J, Takazawa, K, et al. Stimulation of generation of inositol phosphates by carbamylcholine and its inhibition by phorbol esters and iodide in dog thyroid cells. Biochem J. 1989;263:795–801.
94. Corvilain, B, Laurent, E, Lecomte, M, et al. Role of the cyclic adenosine 3′,5′-monophosphate and the phosphatidylinositol-Ca2+ cascades in mediating the effects of thyrotropin and iodide on hormone synthesis and secretion in human thyroid slices. J Clin Endocrinol Metab. 1994;79:152.
95. Van Sande, J, Grenier, G, Willems, C, et al. Inhibition by iodide of the activation of the thyroid cyclic 3′,5′-AMP system. Endocrinology. 1975;96:781–786.
96. Dugrillon, A, Bechtner, G, Uedelhoven, WM, et al. Evidence that an iodolactone mediates the inhibitory effect of iodine on thyroid cell proliferation but not on adenosine 3′,5′-monophosphate formation. Endocrinology. 1990;127:337–343.
97. Panneels, V, Macours, P, Van den, BH, et al. Biosynthesis and metabolism of 2-iodohexadecanal in cultured dog thyroid cells. J Biol Chem. 1996;271:23006–23014.
98. Panneels, V, Van Sande, J, Van den, BH, et al. Inhibition of human thyroid adenylyl cyclase by 2-iodoaldehydes. Mol Cell Endocrinol. 1994;106:41–50.
99. Panneels, V, Van den, BH, Jacoby, C, et al. Inhibition of H2O2 production by iodoaldehydes in cultured dog thyroid cells. Mol Cell Endocrinol. 1994;102:167–176.
100. Many, MC, Mestdagh, C, Van Den Hove, MF, et al. In vitro study of acute toxic effects of high iodide doses in human thyroid follicles. Endocrinology. 1992;131:621–630.
101. Uyttersprot, N, Pelgrims, N, Carrasco, N, et al. Moderate doses of iodide in vivo inhibit cell proliferation and the expression of thyroperoxidase and Na+/I− symporter mRNAs in dog thyroid. Mol Cell Endocrinol. 1997;131:195–203.
102. Dias, JA, Van Roey, P. Structural biology of human follitropin and its receptor. Arch Med Res. 2001;32:510–519.
103. Ascoli, M, Fanelli, F, Segaloff, DL. The lutropin/chorionic gonadotropin receptor, a 2002 perspective. Endocr Rev. 2002;23:141–174.
104. Szkudlinski, MW, Fremont, V, Ronin, C, et al. Thyroid-stimulating hormone and thyroid-stimulating hormone receptor structure-function relationships. Physiol Rev. 2002;82:473–502.
105. Cornelis, S, Uttenweiler-Joseph, S, Panneels, V, et al. Purification and characterization of a soluble bioactive amino-terminal extracellular domain of the human thyrotropin receptor. Biochemistry. 2001;40:9860–9869.
106. Rapoport, B, Chazenbalk, GD, Jaume, JC, et al. The thyrotropin (TSH) receptor: interaction with TSH and autoantibodies. Endocr Rev. 1998;19:673–716.
107. de Bernard, S, Misrahi, M, Huet, JC, et al. Sequential cleavage and excision of a segment of the thyrotropin receptor ectodomain. J Biol Chem. 1999;274:101–107.
108. Couet, J, Sar, S, Jolivet, A, et al. Shedding of human thyrotropin receptor ectodomain: involvement of a matrix metalloprotease. J Biol Chem. 1996;271:4545–4552.
109. Mizutori, Y, Chen, CR, Latrofa, F, et al. Evidence that shed thyrotropin receptor A subunits drive affinity maturation of autoantibodies causing Graves’ disease. J Clin Endocrinol Metab. 2009;94:927–935.
110. Rapoport, B, McLachlan, SM. The thyrotropin receptor in Graves’ disease. Thyroid. 2007;17:911–922.
111. Beau, I, Misrahi, M, Gross, B, et al. Basolateral localization and transcytosis of gonadotropin and thyrotropin receptors expressed in Madin-Darby canine kidney cells. J Biol Chem. 1997;272:5241–5248.
112. Angers, S, Salahpour, A, Bouvier, M. Dimerization: An emerging concept for G protein–coupled receptor ontogeny and function. Annu Rev Pharmacol Toxicol. 2002;42:409–435.
113. Libert, F, Passage, E, Lefort, A, et al. Localization of human thyrotropin receptor gene to chromosome region 14q3 by in situ hybridization. Cytogenet Cell Genet. 1990;54:82–83.
114. Rousseau-Merck, MF, Misrahi, M, Loosfelt, H, et al. Assignment of the human thyroid stimulating hormone receptor (TSHR) gene to chromosome 14q31. Genomics. 1990;8:233–236.
115. Gross, B, Misrahi, M, Sar, S, et al. Composite structure of the human thyrotropin receptor gene. Biochem Biophys Res Commun. 1991;177:679–687.
116. Ikuyama, S, Niller, HH, Shimura, H, et al. Characterization of the 5′-flanking region of the rat thyrotropin receptor gene. Mol Endocrinol. 1992;6:793–804.
117. Roselli-Rehfuss, L, Robbins, LS, Cone, RD. Thyrotropin receptor messenger ribonucleic acid is expressed in most brown and white adipose tissues in the guinea pig. Endocrinology. 1992;130:1857–1861.
118. Bell, A, Gagnon, A, Grunder, L, et al. Functional TSH receptor in human abdominal preadipocytes and orbital fibroblasts. Am J Physiol Cell Physiol. 2000;279:C335–C340.
119. Crisp, MS, Lane, C, Halliwell, M, et al. Thyrotropin receptor transcripts in human adipose tissue. J Clin Endocrinol Metab. 1997;82:2003–2005.
120. Bassett, JH, Williams, AJ, Murphy, E, et al. A lack of thyroid hormones rather than excess thyrotropin causes abnormal skeletal development in hypothyroidism. Mol Endocrinol. 2008;22:501–512.
121. Nakao, N, Ono, H, Yamamura, T, et al. Thyrotrophin in the pars tuberalis triggers photoperiodic response. Nature. 2008;452:317–322.
122. Ono, H, Hoshino, Y, Yasuo, S, et al. Involvement of thyrotropin in photoperiodic signal transduction in mice. Proc Natl Acad Sci U S A. 2008;105:18238–18242.
123. Maenhaut, C, Brabant, G, Vassart, G, et al. In vitro and in vivo regulation of thyrotropin receptor mRNA levels in dog and human thyroid cells. J Biol Chem. 1992;267:3000–3007.
124. Lapthorn, AJ, Harris, DC, Littlejohn, A, et al. Crystal structure of human chorionic gonadotropin. Nature. 1994;369:455–461.
125. Wu, H, Lustbader, JW, Liu, Y, et al. Structure of human chorionic gonadotropin at 2.6-angstrom resolution from MAD analysis of the selenomethionyl protein. Structure. 1994;2:545–558.
126. Fox, KM, Dias, JA, Van Roey, P. Three-dimensional structure of human follicle-stimulating hormone. Mol Endocrinol. 2001;15:378–389.
127. Fan, QR, Hendrickson, WA. Structure of human follicle-stimulating hormone in complex with its receptor. Nature. 2005;433:269–277.
128. Kajava, AV, Vassart, G, Wodak, SJ. Modeling of the 3-dimensional structure of proteins with the typical leucine-rich repeats. Structure. 1995;3:867–877.
129. Sanders, J, Chirgadze, DY, Sanders, P, et al. Crystal structure of the TSH receptor in complex with a thyroid-stimulating autoantibody. Thyroid. 2007;17:395–410.
130. Smits, G, Govaerts, C, Nubourgh, I, et al. Lysine 183 and glutamic acid 157 of the TSH receptor: two interacting residues with a key role in determining specificity toward TSH and human CG. Mol Endocrinol. 2002;16:722–735.
131. Caltabiano, G, Campillo, M, De Leener, A, et al. The specificity of binding of glycoprotein hormones to their receptors. Cell Mol Life Sci. 2008;65:2484–2492.
132. Costagliola, S, Panneels, V, Bonomi, M, et al. Tyrosine sulfation is required for agonist recognition by glycoprotein hormone receptors. EMBO J. 2002;21:504–513.
133. Bonomi, M, Busnelli, M, Persani, L, et al. Structural differences in the hinge region of the glycoprotein hormone receptors: evidence from the sulfated tyrosine residues. Mol Endocrinol. 2006;20:3351–3363.
134. Westmuckett, AD, Hoffhines, AJ, Borghei, A, et al. Early postnatal pulmonary failure and primary hypothyroidism in mice with combined TPST-1 and TPST-2 deficiency. Gen Comp Endocrinol. 2008;156:145–153.
135. Sasaki, N, Hosoda, Y, Nagata, A, et al. A mutation in Tpst2 encoding tyrosylprotein sulfotransferase causes dwarfism associated with hypothyroidism. Mol Endocrinol. 2007;21:1713–1721.
136. Palczewski, K, Kumasaka, T, Hori, T, et al. Crystal structure of rhodopsin: a G protein–coupled receptor. Science. 2000;289:739–745.
137. Ridge, KD, Abdulaev, NG, Sousa, M, et al. Phototransduction: crystal clear. Trends Biochem Sci. 2003;28:479–487.
138. Parma, J, Duprez, L, Van Sande, J, et al. Diversity and prevalence of somatic mutations in the thyrotropin receptor and Gs alpha genes as a cause of toxic thyroid adenomas. J Clin Endocrinol Metab. 1997;82:2695–2701.
139. Refetoff, S, Dumont, JE, Vassart, G. Thyroid disorders. In: Scriver CR, Sly WS, Childs B, et al, eds. The Metabolic and Molecular Bases of Inherited Diseases. New York: McGraw-Hill; 2001:4029–4076.
140. Duprez, L, Parma, J, Van Sande, J, et al. Germline mutations in the thyrotropin receptor gene cause non-autoimmune autosomal dominant hyperthyroidism. Nat Genet. 1994;7:396–401.
141. Govaerts, C, Lefort, A, Costagliola, S, et al. A conserved Asn in transmembrane helix 7 is an on/off switch in the activation of the thyrotropin receptor. J Biol Chem. 2001;276:22991–22999.
142. Neumann, S, Krause, G, Chey, S, et al. A free carboxylate oxygen in the side chain of position 674 in transmembrane domain 7 is necessary for TSH receptor activation. Mol Endocrinol. 2001;15:1294–1305.
143. Claeysen, S, Govaerts, C, Lefort, A, et al. A conserved Asn in TM7 of the thyrotropin receptor is a common requirement for activation by both mutations and its natural agonist. FEBS Lett. 2002;517:195–200.
144. Ballesteros, JA, Jensen, AD, Liapakis, G, et al. Activation of the beta(2)-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J Biol Chem. 2001;276:29171–29177.
145. Duprez, L, Parma, J, Costagliola, S, et al. Constitutive activation of the TSH receptor by spontaneous mutations affecting the N-terminal extracellular domain. FEBS Lett. 1997;409:469–474.
146. Nakabayashi, K, Kudo, M, Kobilka, B, et al. Activation of the luteinizing hormone receptor following substitution of Ser-277 with selective hydrophobic residues in the ectodomain hinge region. J Biol Chem. 2000;275:30264–30271.
147. Ho, SC, Van Sande, J, Lefort, A, et al. Effects of mutations involving the highly conserved S281HCC motif in the extracellular domain of the thyrotropin (TSH) receptor on TSH binding and constitutive activity. Endocrinology. 2001;142:2760–2767.
148. Parma, J, Van Sande, J, Swillens, S, et al. Somatic mutations causing constitutive activity of the thyrotropin receptor are the major cause of hyperfunctioning thyroid adenomas: identification of additional mutations activating both the cyclic adenosine 3′,5′-monophosphate and inositol phosphate-Ca2+ cascades. Mol Endocrinol. 1995;9:725–733.
149. Van Sande, J, Massart, C, Costagliola, S, et al. Specific activation of the thyrotropin receptor by trypsin. Mol Cell Endocrinol. 1996;119:161–168.
150. Chen, CR, Chazenbalk, GD, McLachlan, SM, et al. Evidence that the C terminus of the A subunit suppresses thyrotropin receptor constitutive activity. Endocrinology. 2003;144:3821–3827.
151. Vlaeminck-Guillem, V, Ho, SC, Rodien, P, et al. Activation of the cAMP pathway by the TSH receptor involves switching of the ectodomain from a tethered inverse agonist to an agonist. Mol Endocrinol. 2002;16:736–746.
152. Vassart, G, Pardo, L, Costagliola, S. A molecular dissection of the glycoprotein hormone receptors. Trends Biochem Sci. 2004;29(3):119–126.
153. Chen, CR, McLachlan, SM, Rapoport, B. Suppression of thyrotropin receptor constitutive activity by a monoclonal antibody with inverse agonist activity. Endocrinology. 2007;148:2375–2382.
154. Chen, CR, McLachlan, SM, Rapoport, B. Identification of key amino acid residues in a thyrotropin receptor monoclonal antibody epitope provides insight into its inverse agonist and antagonist properties. Endocrinology. 2008;149:3427–3434.
155. Kleinau, G, Krause, G. Thyrotropin and homologous glycoprotein hormone receptors: structural and functional aspects of extracellular signaling mechanisms. Endocr Rev. 2009.
156. Rodien, P, Bremont, C, Samson, ML, et al. Familial gestational hyperthyroidism caused by a mutant thyrotropin receptor hypersensitive to human chorionic gonadotropin. N Engl J Med. 1998;339:1823–1826.
157. Szkudlinski, MW, Teh, NG, Grossmann, M, et al. Engineering human glycoprotein hormone superactive analogues. Nat Biotechnol. 1996;14:1257–1263.
158. Costagliola, S, Bonomi, M, Morgenthaler, NG, et al. Delineation of the discontinuous-conformational epitope of a monoclonal antibody displaying full in vitro and in vivo thyrotropin activity. Mol Endocrinol. 2004;18:3020–3034.
159. Costagliola, S, Morgenthaler, NG, Hoermann, R, et al. Second generation assay for thyrotropin receptor antibodies has superior diagnostic sensitivity for Graves’ disease. J Clin Endocrinol Metab. 1999;84:90–97.
160. Costagliola, S, Franssen, JDF, Bonomi, M, et al. Generation of a mouse monoclonal TSH receptor antibody with stimulating activity. Biochem Biophys Res Commun. 2002;299:891–896.
161. Morgenthaler, NG, Minich, WB, Willnich, M, et al. Affinity purification and diagnostic use of TSH receptor autoantibodies from human serum. Mol Cell Endocrinol. 2003;212:73–79.
162. Okada, SL, Ellsworth, JL, Durnam, DM, et al. A glycoprotein hormone expressed in corticotrophs exhibits unique binding properties on thyroid-stimulating hormone receptor. Mol Endocrinol. 2006;20:414–425.
163. Nagasaki, H, Wang, Z, Jackson, VR, et al. Differential expression of the thyrostimulin subunits, glycoprotein alpha2 and beta5 in the rat pituitary. J Mol Endocrinol. 2006;37:39–50.
164. Dos, SS, Bardet, C, Bertrand, S, et al. Distinct expression patterns of glycoprotein hormone-α2 (GPA2) and -β5 (GPB5) in a basal chordate suggest independent developmental functions. Endocrinology. 2009.
165. Raymond, JR, Hnatowich, M, Lefkowitz, RJ, et al. Adrenergic receptors. Models for regulation of signal transduction processes. Hypertension. 1990;15:119–131.
166. Delbeke, D, Van Sande, J, Swillens, S, et al. Cooling enhances adenosine 3′,5′ monophosphate accumulation in thyrotropin stimulated dog thyroid slices. Metabolism. 1982;31:797–804.
167. Baratti-Elbaz, C, Ghinea, N, Lahuna, O, et al. Internalization and recycling pathways of the thyrotropin receptor. Mol Endocrinol. 1999;13:1751–1765.
168. Persani, L, Lania, A, Alberti, L, et al. Induction of specific phosphodiesterase isoforms by constitutive activation of the cAMP pathway in autonomous thyroid adenomas. J Clin Endocrinol Metab. 2000;85:2872–2878.
169. Urizar, E, Montanelli, L, Loy, T, et al. Glycoprotein hormone receptors: link between receptor homodimerization and negative cooperativity. EMBO J. 2005;24:1954–1964.
170. Tao, YX, Johnson, NB, Segaloff, DL. Constitutive and agonist-dependent self-association of the cell surface human lutropin receptor. J Biol Chem. 2004;279:5904–5914.
171. Latif, R, Graves, P, Davies, TF. Ligand-dependent inhibition of oligomerization at the human thyrotropin receptor. J Biol Chem. 2002;277:45059–45067.
172. Nilsson, M, Björkman, U, Ekholm, R, et al. Polarized efflux of iodide in porcine thyrocytes occurs via a cAMP-regulated iodide channel in the apical plasma membrane. Acta Endocrinol (Copenh). 1992;126:67–74.
173. Rodriguez, AM, Perron, B, Lacroix, L, et al. Identification and characterization of a putative human iodide transporter located at the apical membrane of thyrocytes. J Clin Endocrinol Metab. 2002;87:3500–3503.
174. Raspé, E, Dumont, JE. Control of the dog thyrocyte plasma membrane iodide permeability by the Ca2+-phosphatidylinositol and adenosine 3′,5′-monophosphate cascades. Endocrinology. 1994;135:986–995.
175. Saito, T, Endo, T, Kawaguchi, A, et al. Increased expression of the Na+/I− symporter in cultured human thyroid cells exposed to thyrotropin and in Graves’ thyroid tissue. J Clin Endocrinol Metab. 1997;82:3331–3336.
176. Arntzenius, AB, Smit, LJ, Schipper, J. Inverse relation between iodine intake and thyroid blood flow: color Doppler flow imaging in euthyroid humans. J Clin Endocrinol Metab. 1991;73:1051–1055.
177. Nunez, J, Pommier, J. Formation of thyroid hormones. Vitam Horm. 1982;39:175–229.
178. Dupuy, C, Ohayon, R, Valent, A, et al. Purification of a novel flavoprotein involved in the thyroid NADPH oxidase. Cloning of the porcine and human cDNAs. J Biol Chem. 1999;274:37265–37269.
179. De Deken, X, Wang, D, Many, MC, et al. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. J Biol Chem. 2000;275:23227–23233.
180. Corvilain, B, Van Sande, J, Laurent, E, et al. The H2O2-generating system modulates protein iodination and the activity of the pentose phosphate pathway in dog thyroid. Endocrinology. 1991;128:779–785.
181. Björkman, U, Ekholm, R. Hydrogen peroxide generation and its regulation in FRTL-5 and porcine thyroid cells. Endocrinology. 1992;130:393–399.
182. Björkman, U, Ekholm, R. Accelerated exocytosis and H2O2 generation in isolated thyroid follicles enhance protein iodination. Endocrinology. 1988;122:488–494.
183. Dumont, JE, Boeynaems, JM, de Coster, C, et al. Biochemical mechanisms in the control of thyroid function and growth. Adv Cyclic Nucleotide Res. 1978;9:723.
184. Bernier-Valentin, F, Kostrouch, Z, Rabilloud, R, et al. Analysis of the thyroglobulin internationalization process using in vitro reconstituted thyroid follicles: evidence for a coated vesicle-dependent endocytic pathway. Endocrinology. 1991;129:2194–2201.
185. Deshpande, V, Venkatesh, SG. Thyroglobulin, the prothyroid hormone: chemistry, synthesis and degradation. Biochim Biophys Acta. 1999;1430:157–178.
186. Deery, WJ, Heath, JP. Phagocytosis induced by thyrotropin in cultured thyroid cells is associated with myosin light chain dephosphorylation and stress fiber disruption. J Cell Biol. 1993;122:21–37.
187. Chambard, M, Depetris, D, Gruffat, D, et al. Thyrotropin regulation of apical and basal exocytosis of thyroglobulin by porcine thyroid monolayers. J Mol Endocrinol. 1990;4:193–199.
188. Herzog, V. Pathways of endocytosis in thyroid follicle cells. Int Rev Cytol. 1984;91:107–139.
189. Croizet-Berger, K, Daumerie, C, Couvreur, M, et al. The endocytic catalysts, Rab5a and Rab7, are tandem regulators of thyroid hormone production. Proc Natl Acad Sci U S A. 2002;99:8277–8282.
190. Rocmans, PA, Ketelbant-Balasse, P, Dumont, JE, et al. Hormonal secretion by hyperactive thyroid cells is not secondary to apical phagocytosis. Endocrinology. 1978;103:1834–1848.
191. Van Den Hove, MF, Couvreur, M, De Visscher, M. A new mechanism for the reabsorption of thyroid iodoproteins: selective fluid pinocytosis. Eur J Biochem. 1982;122:415–422.
192. Lemansky, P, Herzog, V. Endocytosis of thyroglobulin is not mediated by mannose-6-phosphate receptors in thyrocytes. Evidence for low-affinity-binding sites operating in the uptake of thyroglobulin. Eur J Biochem. 1992;209:111–119.
193. Marino, M, McCluskey, RT. Role of thyroglobulin endocytic pathways in the control of thyroid hormone release. Am J Physiol Cell Physiol. 2000;279:C1295–C1306.
194. Marino, M, Zheng, G, McCluskey, RT. Megalin (gp330) is an endocytic receptor for thyroglobulin on cultured fisher rat thyroid cells. J Biol Chem. 1999;274:12898–12904.
195. Lisi, S, Chiovato, L, Pinchera, A, et al. Impaired thyroglobulin (Tg) secretion by FRTL-5 cells transfected with soluble receptor associated protein (RAP): evidence for a role of RAP in the Tg biosynthetic pathway. J Endocrinol Invest. 2003;26:1105–1110.
196. Laurberg, P. Mechanisms governing the relative proportions of thyroxine and 3,5,3′-triiodothyronine in thyroid secretion. Metabolism. 1984;33:379–392.
197. Unger, J, Boeynaems, JM, Van Herle, A, et al. In vitro nonbutanol-extractable iodine release in dog thyroid. Endocrinology. 1979;105:225–231.
198. Van Herle, AJ, Vassart, G, Dumont, JE. Control of thyroglobulin synthesis and secretion. (First of two parts). N Engl J Med. 1979;301:239–249.
199. Damante, G, Tell, G, Di Lauro, R. A unique combination of transcription factors controls differentiation of thyroid cells. Prog Nucleic Acid Res Mol Biol. 2001;66:307–356.
200. Damante, G, DiLauro, R. Thyroid-specific gene-expression. Biochim Biophys Acta. 1994;1218:255–266.
201. Kusakabe, T, Kawaguchi, A, Hoshi, N, et al. Thyroid-specific enhancer-binding protein/NKX2.1 is required for the maintenance of ordered architecture and function of the differentiated thyroid. Mol Endocrinol. 2006;20:1796–1809.
202. Roger, PP, Christophe, D, Dumont, JE, et al. The dog thyroid primary culture system: a model of the regulation of function, growth and differentiation expression by cAMP and other well-defined signaling cascades. European J Endocrinol. 1997;137:579–598.
203. Medina, DL, Suzuki, K, Pietrarelli, M, et al. Role of insulin and serum on thyrotropin regulation of thyroid transcription factor-1 and Pax-8 genes expression in FRTL-5 thyroid cells. Thyroid. 2000;10:295–303.
204. Pouillon, V, Pichon, B, Donda, A, et al. TTF-2 does not appear to be a key mediator of the effect of cyclic AMP on thyroglobulin gene transcription in primary cultured dog thyrocytes. Biochem Biophys Res Commun. 1998;242:327–331.
205. Donda, A, Javaux, F, Van Renterghem, P, et al. Human, bovine, canine and rat thyroglobulin promoter sequences display species-specific differences in an in vitro study. Mol Cell Endocrinol. 1993;90:R23–R26.
206. Van Renterghem, P, Vassart, G, Christophe, D. Pax 8 expression in primary cultured dog thyrocyte is increased by cyclic AMP. Biochim Biophys Acta. 1996;1307:97–103.
207. Mascia, A, Nitsch, L, Di Lauro, R, et al. Hormonal control of the transcription factor Pax8 and its role in the regulation of thyroglobulin gene expression in thyroid cells. J Endocrinol. 2002;172:163–176.
208. Van Renterghem, P, Dremier, S, Vassart, G, et al. Study of TTF-1 gene expression in dog thyrocytes in primary culture. Mol Cell Endocrinol. 1995;112:83–93.
209. Zannini, M, Acebron, A, DeFelice, M, et al. Mapping and functional role of phosphorylation sites in the thyroid transcription factor-1 (TTF-1). J Biol Chem. 1996;271:2249–2254.
210. Reuse, S, Maenhaut, C, Dumont, JE. Regulation of protooncogenes c-fos and c-myc expressions by protein tyrosine kinase, protein kinase C, and cyclic AMP mitogenic pathways in dog primary thyrocytes: a positive and negative control by cyclic AMP on c-myc expression. Exp Cell Res. 1990;189:33–40.
211. Deleu, S, Pirson, I, Coulonval, K, et al. IGF-1 or insulin, and the TSH cyclic AMP cascade separately control dog and human thyroid cell growth and DNA synthesis, and complement each other in inducing mitogenesis. Mol Cell Endocrinol. 1999;149:41–51.
212. Lalli, E, Sassonecorsi, P. Thyroid-stimulating hormone (TSH)-directed induction of the Crem gene in the thyroid gland participates in the long-term desensitization of the TSH receptor. Proc Natl Acad Sci U S A. 1995;92:9633–9637.
213. Pichon, B, Jimenez-Cervantes, C, Pirson, I, et al. Induction of nerve growth factor-induced gene-B (NGFI-B) as an early event in the cyclic adenosine monophosphate response of dog thyrocytes in primary culture. Endocrinology. 1996;137:4691–4698.
214. Pomerance, M, Carapau, D, Chantoux, F, et al. CCAAT/enhancer-binding protein-homologous protein expression and transcriptional activity are regulated by 3′,5′-cyclic adenosine monophosphate in thyroid cells. Mol Endocrinol. 2003;17:2283–2294.
215. Pichon, B, Vassart, G, Christophe, D. A canonical nerve growth factor-induced gene-B response element appears not to be involved in the cyclic adenosine monophosphate-dependent expression of differentiation in thyrocytes. Mol Cell Endocrinol. 1999;154:21–27.
216. Garcia-Jimenez, C, Zaballos, MA, Santisteban, P. DARPP-32 (dopamine and 3′,5′-cyclic adenosine monophosphate-regulated neuronal phosphoprotein) is essential for the maintenance of thyroid differentiation. Mol Endocrinol. 2005;19:3060–3072.
217. Davies, E, Dumont, JE, Vassart, G. Thyrotropin-stimulated recruitment of free monoribosomes on to membrane-bound thyroglobulin synthesizing polyribosomes. Biochem J. 1978;172:227–231.
218. Colletta, G, Cirafici, AM, Dicarlo, A. Dual effect of transforming growth factor-beta on rat thyroid cells: inhibition of thyrotropin-induced proliferation and reduction of thyroid-specific differentiation markers. Cancer Res. 1989;49:3457–3462.
219. Nicolussi, A, D’Inzeo, S, Santulli, M, et al. TGF-beta control of rat thyroid follicular cells differentiation. Mol Cell Endocrinol. 2003;207:1–11.
220. Costamagna, E, Garcia, B, Santisteban, P. The functional interaction between the paired domain transcription factor Pax8 and Smad3 is involved in transforming growth factor-beta repression of the sodium/iodide symporter gene. J Biol Chem. 2004;279:3439–3446.
221. Roger, PP, Van Heuverswyn, B, Lambert, C, et al. Antagonistic effects of thyrotropin and epidermal growth factor on thyroglobulin mRNA level in cultured thyroid cells. Eur J Biochem. 1985;152:239–245.
222. Gerard, CM, Lefort, A, Christophe, D, et al. Control of thyroperoxidase and thyroglobulin transcription by cAMP: evidence for distinct regulatory mechanisms. Mol Endocrinol. 1989;3:2110–2118.
223. Pohl, V, Abramowicz, M, Vassart, G, et al. Thyroperoxidase mRNA in quiescent and proliferating thyroid epithelial cells: expression and subcellular localization studied by in situ hybridization. Eur J Cell Biol. 1993;62:94–104.
224. Leoni, SG, Galante, PA, Ricarte-Filho, JC, et al. Differential gene expression analysis of iodide-treated rat thyroid follicular cell line PCCl3. Genomics. 2008;91:356–366.
225. Blackwood, L, Onions, DE, Argyle, DJ. Characterization of the feline thyroglobulin promoter. Domest Anim Endocrinol. 2001;20:185–201.
226. Christophe-Hobertus, C, Christophe, D. Two binding sites for thyroid transcription factor 1 (TTF-1) determine the activity of the bovine thyroglobulin gene upstream enhancer element. Mol Cell Endocrinol. 1999;149:79–84.
227. Berg, V, Vassart, G, Christophe, D. A zinc-dependent DNA-binding activity co-operates with cAMP-responsive-element-binding protein to activate the human thyroglobulin enhancer. Biochem J. 1997;323:349–357.
228. Mascia, A, DeFelice, M, Lipardi, C, et al. Transfection of TTF-1 gene induces thyroglobulin gene expression in undifferentiated FRT cells. Biochim Biophys Acta. 1997;1354:171–181.
229. di Magliano, MP, Di Lauro, R, Zannini, M. Pax8 has a key role in thyroid cell differentiation. Proc Natl Acad Sci U S A. 2000;97:13144–13149.
230. Di Palma, T, Nitsch, R, Mascia, A, et al. The paired domain-containing factor Pax8 and the homeodomain-containing factor TTF-1 directly interact and synergistically activate transcription. J Biol Chem. 2003;278:3395–3402.
231. Miccadei, S, De Leo, R, Zammarchi, E, et al. The synergistic activity of thyroid transcription factor 1 and Pax8 relies on the promoter/enhancer interplay. Mol Endocrinol. 2002;16:837–846.
232. Grasberger, H, Ringkananont, U, Lefrancois, P, et al. Thyroid transcription factor 1 rescues PAX8/p300 synergism impaired by a natural PAX8 paired domain mutation with dominant negative activity. Mol Endocrinol. 2005;19:1779–1791.
233. Di Palma, T, D’Andrea, B, Liguori, GL, et al. TAZ is a coactivator for Pax8 and TTF-1, two transcription factors involved in thyroid differentiation. Exp Cell Res. 2009;315:162–175.
234. Ledent, C, Parmentier, M, Vassart, G. Tissue-specific expression and methylation of a thyroglobulin-chloramphenicol acetyltransferase fusion gene in transgenic mice. Proc Natl Acad Sci U S A. 1990;87:6176–6180.
235. Libert, F, Vassart, G, Christophe, D. Methylation and expression of the human thyroglobulin gene. Biochim Biophys Acta. 1986;134:1109–1113.
236. Pichon, B, Christophe-Hobertus, C, Vassart, G, et al. Unmethylated thyroglobulin promoter may be repressed by methylation of flanking DNA sequences. Biochem J. 1994;298:537–541.
237. Van Heuverswyn, B, Streydio, C, Brocas, H, et al. Thyrotropin controls transcription of the thyroglobulin gene. Proc Natl Acad Sci U S A. 1984;81:5941–5945.
238. Van Heuverswyn, B, Leriche, A, Van Sande, J, et al. Transcriptional control of thyroglobulin gene expression by cyclic AMP. FEBS Lett. 1985;188:192–196.
239. Avvedimento, VE, Tramontano, D, Ursini, MV. The level of thyroglobulin mRNA is regulated by TSH both in vitro and in vivo. Biochem Biophys Res Commun. 1984;122:472–477.
240. Hansen, C, Gerard, C, Vassart, G, et al. Thyroid-specific and cAMP-dependent hypersensitive regions in thyroglobulin gene chromatin. Eur J Biochem. 1988;178:387–393.
241. Christophe, D, Gérard, C, Juvenal, G, et al. Identification of a cAMP-responsive region in thyroglobulin gene promoter. Mol Cell Endocrinol. 1989;64:5–18.
242. Pohl, V, Roger, PP, Christophe, D, et al. Differentiation expression during proliferative activity induced through different pathways: in situ hybridization study of thyroglobulin gene expression in thyroid epithelial cells. J Cell Biol. 1990;111:663–672.
243. Ortiz, L, Zannini, M, Di Lauro, R, et al. Transcriptional control of the forkhead thyroid transcription factor TTF-2 by thyrotropin, insulin, and insulin-like growth factor I. J Biol Chem. 1997;272:23334–23339.
244. Fayet, G, Hovsepian, S. Isolation of a normal human thyroid cell line: hormonal requirement for thyroglobulin regulation. Thyroid. 2002;12:539–546.
245. Graves, PN, Davies, TF. A second thyroglobulin messenger RNA species (rTg-2) in rat thyrocytes. Mol Endocrinol. 1990;4:155–161.
246. Mercken, L, Simons, MJ, Vassart, G. The 5′-end of bovine thyroglobulin mRNA encodes a hormonogenic peptides. FEBS Lett. 1982;149:285–287.
247. Abramowicz, MJ, Vassart, G, Christophe, D. Functional study of the human thyroid peroxidase gene promoter. Eur J Biochem. 1992;203:467–473.
248. Francis-Lang, H, Price, M, Polycarpou-Schwarz, M, et al. Cell-type-specific expression of the rat thyroperoxidase promoter indicates common mechanism for thyroid-specific gene expression. Mol Cell Biol. 1992;12:576–588.
249. Mizuno, K, Gonzalez, FJ, Kimura, S. Thyroid-specific enhancer-binding protein (T/EBP): cDNA cloning, functional characterization, and structural identity with thyroid transcription factor TTF-1. Mol Cell Biol. 1991;11:4927–4933.
250. Esposito, C, Miccadei, S, Saiardi, A, et al. PAX 8 activates the enhancer of the human thyroperoxidase gene. Biochem J. 1998;331:37–40.
251. De Leo, R, Miccadei, S, Zammarchi, E, et al. Role for p300 in Pax 8 induction of thyroperoxidase gene expression. J Biol Chem. 2000;275:34100–34105.
252. Gérard, C, Lefort, A, Libert, F, et al. Transcriptional regulation of the thyroperoxidase gene by thyrotropin and forskolin. Mol Cell Endocrinol. 1988;60:239–242.
253. Ledent, C, Dumont, JE, Vassart, G, et al. Thyroid expression of an A2 adenosine receptor transgene induces thyroid hyperplasia and hyperthyroidism. EMBO J. 1992;11:537–542.
254. Miccadei, S, Provenzano, C, Mojzisek, M, et al. Retinoblastoma protein acts as Pax 8 transcriptional coactivator. Oncogene. 2005;24:6993–7001.
255. Cuesta, I, Zaret, KS, Santisteban, P. The forkhead factor FoxE1 binds to the thyroperoxidase promoter during thyroid cell differentiation and modifies compacted chromatin structure. Mol Cell Biol. 2007;27:7302–7314.
256. Niccoli, P, Fayadat, L, Panneels, V, et al. Human thyroperoxidase in its alternatively spliced form (TPO2) is enzymatically inactive and exhibits changes in intracellular processing and trafficking. J Biol Chem. 1997;272:29487–29492.
257. Tong, Q, Ryu, KY, Jhiang, SM. Promoter characterization of the rat Na+/I− symporter gene. Biochem Biophys Res Commun. 1997;239:34–41.
258. Behr, M, Schmitt, TL, Espinoza, CR, et al. Cloning of a functional promoter of the human sodium/iodide-symporter gene. Biochem J. 1998;331:359–363.
259. Endo, T, Kaneshige, M, Nakazato, M, et al. Thyroid transcription factor-1 activates the promoter activity of rat thyroid Na+/I− symporter gene. Mol Endocrinol. 1997;11:1747–1755.
260. Taki, K, Kogai, T, Kanamoto, Y, et al. A thyroid-specific far-upstream enhancer in the human sodium/iodide symporter gene requires Pax-8 binding and cyclic adenosine 3′,5′-monophosphate response element-like sequence binding proteins for full activity and is differentially regulated in normal and thyroid cancer cells. Mol Endocrinol. 2002;16:2266–2282.
261. Fenton, MS, Marion, KM, Hershman, JM. Identification of cyclic adenosine 3′,5′-monophosphate response element modulator as an activator of the human sodium/iodide symporter upstream enhancer. Endocrinology. 2008;149:2592–2606.
262. Saiardi, A, Falasca, P, Civitareale, D. The thyroid-hormone inhibits the thyrotropin receptor promoter activity: evidence for a short loop regulation. Biochem Biophys Res Commun. 1994;205:230–237.
263. Yokomori, N, Tawata, M, Saito, T, et al. Regulation of the rat thyrotropin receptor gene by the methylation-sensitive transcription factor GA binding protein. Mol Endocrinol. 1998;12:1241–1249.
264. Saiardi, A, Falasca, P, Civitareale, D. Synergistic transcriptional activation of the thyrotropin receptor promoter by cyclic AMP-responsive-element-binding protein and thyroid transcription factor-1. Biochem J. 1995;310:491–496.
265. Civitareale, D, Castelli, MP, Falasca, P, et al. Thyroid transcription factor-1 activates the promoter of the thyrotropin receptor gene. Mol Endocrinol. 1993;7:1589–1595.
266. Moeller, LC, Kimura, S, Kusakabe, T, et al. Hypothyroidism in thyroid transcription factor 1 haploinsufficiency is caused by reduced expression of the thyroid-stimulating hormone receptor. Mol Endocrinol. 2003;17:2295–2302.
267. Saji, M, Akamizu, T, Sanchez, M. Regulation of thyrotropin receptor gene expression in rat FRTL-5 thyroid cells. Endocrinology. 1992;130:520–533.
268. Berlingieri, MT, Akamizu, T, Fusco, A. Thyrotropin receptor gene expression in oncogene-transfected rat thyroid cells: correlation between transformation, loss of thyrotropin-dependent growth, and loss of thyrotropin receptor gene expression. Biochem Biophys Res Commun. 1990;173:172–178.
269. Akamizu, T, Ikuyama, S, Saji, M, et al. Cloning, chromosomal assignment, and regulation of the rat thyrotropin receptor: expression of the gene is regulated by thyrotropin, agents that increase cAMP levels, and thyroid autoantibodies. Proc Natl Acad Sci U S A. 1990;87:5677–5681.
270. Kung, AW, Collison, K, Banga, JP, et al. Effect of Graves’ IgG on gene transcription in human thyroid cell cultures. Thyroglobulin gene activation. FEBS Lett. 1988;232:12–16.
271. Huber, GK, Weinstein, SP, Graves, PN, et al. The positive regulation of human thyrotropin (TSH) receptor messenger ribonucleic acid by recombinant human TSH is at the intranuclear level. Endocrinology. 1992;130:2858–2864.
272. Brabant, G, Maenhaut, C, Kohrle, J, et al. Human thyrotropin receptor gene: expression in thyroid tumors and correlation to markers of thyroid differentiation and dedifferentiation. Mol Cell Endocrinol. 1991;82:R7–R12.
273. Ohta, K, Endo, T, Onaya, T. The mRNA levels of thyrotropin receptor, thyroglobulin and thyroid peroxidase in neoplastic human thyroid tissues. Biochem Biophys Res Commun. 1991;174:1148–1153.
274. Roger, PP, Van Heuverswyn, B, Lambert, C, et al. Antagonistic effects of thyrotropin and epidermal growth factor on thyroglobulin mRNA level in cultured thyroid cells. Eur J Biochem. 1985;152:239–245.
275. Ledent, C, Dumont, JE, Vassart, G, et al. Thyroid adenocarcinomas secondary to tissue-specific expression of Simian virus-40 large T-antigen in transgenic mice. Endocrinology. 1991;129:1391–1401.
276. Mazzaferri, EL. Papillary and follicular thyroid cancer: a selective approach to diagnosis and treatment. Annu Rev Med. 1981;32:73–91.
277. Dupuy, C, Pomerance, M, Ohayon, R, et al. Thyroid oxidase (THOX2) gene expression in the rat thyroid cell line FRTL-5. Biochem Biophys Res Commun. 2000;277:287–292.
278. Pachucki, J, Wang, D, Christophe, D, et al. Structural and functional characterization of the two human ThOX/Duox genes and their 5’-flanking regions. Mol Cell Endocrinol. 2004;214:53–62.
279. D’Andrea, B, Iacone, R, Di Palma, T, et al. Functional inactivation of the transcription factor Pax8 through oligomerization chain reaction. Mol Endocrinol. 2006;20:1810–1824.
280. Christophe-Hobertus, C, Christophe, D. Human thyroid oxidases genes promoter activity in thyrocytes does not appear to be functionally dependent on thyroid transcription factor-1 or Pax8. Mol Cell Endocrinol. 2007;264:157–163.
281. Grasberger, H, Refetoff, S. Identification of the maturation factor for dual oxidase. Evolution of an eukaryotic operon equivalent. J Biol Chem. 2006;281:18269–18272.
282. Christov, K. Cell population kinetics and DNA content during thyroid carcinogenesis. Cell Tissue Kinet. 1985;18:119–131.
283. Coclet, J, Foureau, F, Ketelbant, P, et al. Cell population kinetics in dog and human adult thyroid. Clin Endocrinol (Oxf). 1989;31:655–665.
284. Smeds, S, Wollman, SH. 3H-thymidine labeling of endothelial cells in thyroid arteries, veins, and lymphatics during thyroid stimulation. Lab Invest. 1983;48:285–291.
285. Many, MC, Denef, JF, Haumont, S. Precocity of the endothelial proliferation during a course of rapid goitrogenesis. Acta Endocrinol (Copenh). 1984;105:487–491.
286. Patel, VA, Logan, A, Watkinson, JC, et al. Isolation and characterization of human thyroid endothelial cells. Am J Physiol Endocrinol Metab. 2003;284:E168–E176.
287. Sato, K, Yamazaki, K, Shizume, K, et al. Stimulation by thyroid-stimulating hormone and Grave’s immunoglobulin G of vascular endothelial growth factor mRNA expression in human thyroid follicles in vitro and flt mRNA expression in the rat thyroid in vivo. J Clin Invest. 1995;96:1295–1302.
288. Gerard, CM, Many, MC, Daumerie, C, et al. Structural changes in the angiofollicular units between active and hypofunctioning follicles align with differences in the epithelial expression of newly discovered proteins involved in iodine transport and organification. J Clin Endocrinol Metab. 2002;87:1291–1299.
289. Gerard, AC, Xhenseval, V, Colin, IM, et al. Evidence for coordinated changes between vascular endothelial growth factor and nitric oxide synthase III immunoreactivity, the functional status of the thyroid follicles, and the microvascular bed during chronic stimulation by low iodine and propylthiouracil in old mice. Eur J Endocrinol. 2000;142:651–660.
290. Takasu, N, Komiya, I, Nagasawa, Y, et al. Stimulation of porcine thyroid cell alkalinization and growth by EGF, phorbol ester, and diacylglycerol. Am J Physiol. 1990;258:E445–E450.
291. Clement, S, Refetoff, S, Robaye, B, et al. Low TSH requirement and goiter in transgenic mice overexpressing IGF-I and IGF-I receptor in the thyroid gland. Endocrinology. 2001;142:5131–5139.
292. Tramontano, D, Cushing, GW, Moses, AC, et al. Insulin-like growth factor-I stimulates the growth of rat thyroid cells in culture and synergizes the stimulation of DNA synthesis induced by TSH and Graves’ IgG. Endocrinology. 1986;119:940–942.
293. Saji, M, Tsushima, T, Isozaki, O, et al. Interaction of insulin-like growth factor I with porcine thyroid cells cultured in monolayer. Endocrinology. 1987;121:749–756.
294. Volzke, H, Friedrich, N, Schipf, S, et al. Association between serum insulin-like growth factor-I levels and thyroid disorders in a population-based study. J Clin Endocrinol Metab. 2007;92:4039–4045.
295. Roger, PP, Servais, P, Dumont, JE. Stimulation by thyrotropin and cyclic AMP of the proliferation of quiescent canine thyroid cells cultured in a defined medium containing insulin. FEBS Lett. 1983;157:323–329.
296. Van Sande, J, Lefort, A, Beebe, S, et al. Pairs of cyclic AMP analogs, that are specifically synergistic for type I and type II cAMP-dependent protein kinases, mimic thyrotropin effects on the function, differentiation expression and mitogenesis of dog thyroid cells. Eur J Biochem. 1989;183:699–708.
297. Michiels, FM, Caillou, B, Talbot, M, et al. Oncogenic potential of guanine nucleotide stimulatory factor alpha subunit in thyroid glands of transgenic mice. Proc Natl Acad Sci U S A. 1994;91:10488–10492.
298. Zeiger, MA, Saji, M, Gusev, Y, et al. Thyroid-specific expression of cholera toxin A1 subunit causes thyroid hyperplasia and hyperthyroidism in transgenic mice. Endocrinology. 1997;138:3133–3140.
299. Meinkoth, JL, Goldsmith, PK, Spiegel, AM, et al. Inhibition of thyrotropin-induced DNA synthesis in thyroid follicular cells by microinjection of an antibody to the stimulatory G protein of adenylate cyclase, Gs. J Biol Chem. 1992;267:13239–13245.
300. Kupperman, E, Wen, W, Meinkoth, JL. Inhibition of thyrotropin-stimulated DNA synthesis by microinjection of inhibitors of cellular Ras and cyclic AMP-dependent protein kinase. Mol Cell Biol. 1993;13:4477–4484.
301. Dremier, S, Pohl, V, Poteet-Smith, C, et al. Activation of cyclic AMP-dependent kinase is required but may not be sufficient to mimic cyclic AMP-dependent DNA synthesis and thyroglobulin expression in dog thyroid cells. Mol Cell Biol. 1997;17:6717–6726.
302. Saavedra, AP, Tsygankova, OM, Prendergast, GV, et al. Role of cAMP, PKA and Rap1A in thyroid follicular cell survival. Oncogene. 2002;21:778–788.
303. Ribeiro-Neto, F, Urbani, J, Lemee, N, et al. On the mitogenic properties of Rap1b: cAMP-induced G(1)/S entry requires activated and phosphorylated Rap1b. Proc Natl Acad Sci U S A. 2002;99:5418–5423.
304. Hochbaum, D, Hong, K, Barila, G, et al. Epac, in synergy with cAMP-dependent protein kinase (PKA), is required for cAMP-mediated mitogenesis. J Biol Chem. 2008;283:4464–4468.
305. Kero, J, Ahmed, K, Wettschureck, N, et al. Thyrocyte-specific Gq/G11 deficiency impairs thyroid function and prevents goiter development. J Clin Invest. 2007;117:2399–2407.
306. Van Keymeulen, A, Deleu, S, Bartek, J, et al. Respective roles of carbamylcholine and cyclic adenosine monophosphate in their synergistic regulation of cell cycle in thyroid primary cultures. Endocrinology. 2001;142:1251–1259.
307. Bacharach, LK, Eggo, MC, Mak, WW, et al. Phorbol esters stimulate growth and inhibit differentiation in cultured thyroid cells. Endocrinology. 1985;116:1603–1609.
308. Roger, PP, Reuse, S, Servais, P, et al. Stimulation of cell proliferation and inhibition of differentiated expression by tumor-promoting phorbol esters in dog thyroid cells in primary culture. Cancer Res. 1986;46:898.
309. Raspe, E, Reuse, S, Roger, PP, et al. Lack of correlation between the activation of the Ca2+-phosphatidylinositol cascade and the regulation of DNA synthesis in the dog thyrocyte. Exp Cell Res. 1992;198:17–26.
310. Ollis, CA, Hill, DJ, Munro, DS. A role for insulin-like growth factor-I in the regulation of human thyroid cell growth by thyrotropin. J Endocrinol. 1989;123:495–500.
311. Maciel, RMB, Mores, AC, Villone, G, et al. Demonstration of the production and physiological role of insulin-like growth factor II in rat thyroid follicular cells in culture. J Clin Invest. 1988;82:1546–1553.
312. De Vita, G, Berlingieri, MT, Visconti, R, et al. Akt/protein kinase B promotes survival and hormone-independent proliferation of thyroid cells in the absence of dedifferentiating and transforming effects. Cancer Res. 2000;60:3916–3920.
313. Dremier, S, Taton, M, Coulonval, K, et al. Mitogenic, dedifferentiating, and scattering effects of hepatocyte growth factor on dog thyroid cells. Endocrinology. 1994;135:135–140.
314. Gire, V, Marshall, CJ, Wynford-Thomas, D. Activation of mitogen-activated protein kinase is necessary but not sufficient for proliferation of human thyroid epithelial cells induced by mutant Ras. Oncogene. 1999;18:4819–4832.
315. Melillo, RM, Santoro, M, Ong, SH, et al. Docking protein FRS2 links the protein tyrosine kinase RET and its oncogenic forms with the mitogen-activated protein kinase signaling cascade. Mol Cell Biol. 2001;21:4177–4187.
316. Kimura, ET, Nikiforova, MN, Zhu, Z, et al. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003;63:1454–1457.
317. Roger, PP, Servais, P, Dumont, JE. Induction of DNA synthesis in dog thyrocytes in primary culture: synergistic effects of thyrotropin and cyclic AMP with epidermal growth factor and insulin. J Cell Physiol. 1987;130:58–67.
318. Roger, PP, Servais, P, Dumont, JE. Regulation of dog thyroid epithelial cell cycle by forskolin, an adenylate cyclase activator. Exp Cell Res. 1987;172:282–292.
319. Becks, GP, Eggo, MC, Burrow, GN. Organic iodide inhibits deoxyribonucleic acid synthesis and growth in FRTL5 cells. Endocrinology. 1988;123:545–550.
320. Tsygankova, OM, Saavedra, A, Rebhun, JF, et al. Coordinated regulation of Rap1 and thyroid differentiation by cyclic AMP and protein kinase A. Mol Cell Biol. 2001;21:1921–1929.
321. Lou, LG, Urbani, J, Ribeiro-Neto, F, et al. cAMP inhibition of Akt is mediated by activated and phosphorylated Rap1b. J Biol Chem. 2002;277:32799–32806.
322. Tominaga, T, Dela Cruz, J, Burrow, GN, et al. Divergent patterns of immediate early gene expression in response to thyroid-stimulating hormone and insulin-like growth factor I in Wistar rat thyrocytes. Endocrinology. 1994;135:1212–1219.
323. Reuse, S, Pirson, I, Dumont, JE. Differential regulation of protooncogenes c-Jun and Jun D expressions by protein tyrosine kinase, protein kinase C, and cyclic-AMP mitogenic pathways in dog primary thyrocytes: TSH and cyclic-AMP induce proliferation but downregulate c-Jun expression. Exp Cell Res. 1991;196:210–215.
324. Bockstaele, L, Kooken, H, Libert, F, et al. Regulated activating Thr172 phosphorylation of cyclin-dependent kinase 4(CDK4): its relationship with cyclins and CDK “inhibitors,”. Mol Cell Biol. 2006;26:5070–5085.
325. Contor, L, Lamy, F, Lecocq, R, et al. Differential protein phosphorylation in induction of thyroid cell proliferation by thyrotropin, epidermal growth factor, or phorbol ester. Mol Cell Biol. 1988;8:2494–2503.
326. Cass, LA, Meinkoth, JL. Differential effects of cyclic adenosine 3′,5′-monophosphate on p70 ribosomal S6 kinase. Endocrinology. 1998;139:1991–1998.
327. Brewer, C, Yeager, N, Di Cristofano, A. Thyroid-stimulating hormone initiated proliferative signals converge in vivo on the mTOR kinase without activating AKT. Cancer Res. 2007;67:8002–8006.
328. Uyttersprot, N, Costagliola, S, Dumont, JE, et al. Requirement for cAMP-response element (CRE) binding protein/CRE modulator transcription factors in thyrotropin-induced proliferation of dog thyroid cells in primary culture. Eur J Biochem. 1999;259:370–378.
329. Nguyen, LQ, Kopp, P, Martinson, F, et al. A dominant negative CREB (cAMP response element-binding protein) isoform inhibits thyrocyte growth, thyroid-specific gene expression, differentiation, and function. Mol Endocrinol. 2000;14:1448–1461.
330. Lamy, F, Roger, PP, Lecocq, R, et al. Protein synthesis during induction of DNA replication in thyroid epithelial cells: evidence for late markers of distinct mitogenic pathways. J Cell Physiol. 1989;138:568–578.
331. Hebrant, A, van Staveren, WC, Delys, L, et al. Long-term EGF/serum-treated human thyrocytes mimic papillary thyroid carcinomas with regard to gene expression. Exp Cell Res. 2007;313:3276–3284.
332. Bartek, J, Bartkova, J, Lukas, J. The retinoblastoma protein pathway and the restriction point. Curr Opin Cell Biol. 1996;8:805–814.
333. Sherr, CJ, Roberts, JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512.
334. Lukas, J, Bartkova, J, Bartek, J. Convergence of mitogenic signalling cascades from diverse classes of receptors at the cyclin D-cyclin-dependent kinase-pRb-controlled G(1) checkpoint. Mol Cell Biol. 1996;16:6917–6925.
335. Coulonval, K, Maenhaut, C, Dumont, JE, et al. Phosphorylation of the three Rb protein family members is a common step of the cAMP-, the growth factor, and the phorbol ester-mitogenic cascades but is not necessary for the hypertrophy induced by insulin. Exp Cell Res. 1997;233:395–398.
336. Baptist, M, Lamy, F, Gannon, J, et al. Expression and subcellular localization of CDK2 and cdc2 kinases and their common partner cyclin A in thyroid epithelial cells: comparison of cyclic AMP-dependent and -independent cell cycles. J Cell Physiol. 1996;166:256–273.
337. Van Keymeulen, A, Bartek, J, Dumont, JE, et al. Cyclin D3 accumulation and activity integrate and rank the comitogenic pathways of thyrotropin and insulin in thyrocytes in primary culture. Oncogene. 1999;18:7351–7359.
338. Depoortere, F, Van Keymeulen, A, Lukas, J, et al. A requirement for cyclin D3-cyclin-dependent kinase (CDK)-4 assembly in the cyclic adenosine monophosphate-dependent proliferation of thyrocytes. J Cell Biol. 1998;140:1427–1439.
339. Depoortere, F, Dumont, JE, Roger, PP. Paradoxical accumulation of the cyclin-dependent kinase inhibitor p27kip1 during the cAMP-dependent mitogenic stimulation of thyroid epithelial cells. J Cell Sci. 1996;109:1759–1764.
340. Depoortere, F, Pirson, I, Bartek, J, et al. Transforming growth factor beta(1) selectively inhibits the cyclic AMP-dependent proliferation of primary thyroid epithelial cells by preventing the association of cyclin D3-CDK4 with nuclear p27(kip1). Mol Biol Cell. 2000;11:1061–1076.
341. Coulonval, K, Bockstaele, L, Paternot, S, et al. The cyclin D3-CDK4-p27(kip1) holoenzyme in thyroid epithelial cells: activation by TSH, inhibition by TGFbeta, and phosphorylations of its subunits demonstrated by two-dimensional gel electrophoresis. Exp Cell Res. 2003;291:135–149.
342. Paternot, S, Coulonval, K, Dumont, JE, et al. Cyclic AMP-dependent phosphorylation of cyclin D3-bound CDK4 determines the passage through the cell cycle restriction point in thyroid epithelial cells. J Biol Chem. 2003;278:26533–26540.
343. Paternot, S, Dumont, JE, Roger, PP. Differential utilization of cyclin D1 and cyclin D3 in the distinct mitogenic stimulations by growth factors and TSH of human thyrocytes in primary culture. Mol Endocrinol. 2006;20:3279–3292.
344. Romeo, HE, Diaz, MC, Ceppi, J, et al. Effect of inferior laryngeal nerve section on thyroid function in rats. Endocrinology. 1988;122:2527–2532.
345. Jhiang, SM, Sagartz, JE, Tong, Q, et al. Targeted expression of the ret/PTC1 oncogene induces papillary thyroid carcinomas. Endocrinology. 1996;137:375–378.
346. Powell, DJ, Jr., Russell, J, Nibu, K, et al. The RET/PTC3 oncogene: metastatic solid-type papillary carcinomas in murine thyroids. Cancer Res. 1998;58:5523–5528.
347. Pirson, I, Coulonval, K, Lamy, F, et al. c-myc expression is controlled by the mitogenic cAMP-cascade in thyrocytes. J Cell Physiol. 1996;168:59–70.
348. Paternot, S, Arsenijevic, T, Coulonval, K, et al. Distinct specificities of pRb phosphorylation by CDK4 activated by cyclin D1 or cyclin D3: differential involvement in the distinct mitogenic modes of thyroid epithelial cells. Cell Cycle. 2006;5:61–70.
349. Bartkova, J, Lukas, J, Strauss, M, et al. Cyclin D3: requirement for G1/S transition and high abundance in quiescent tissues suggest a dual role in proliferation and differentiation. Oncogene. 1998;17:1027–1037.
350. Roger, PP, Baptist, M, Dumont, JE. A mechanism generating heterogeneity in thyroid epithelial cells: suppression of the thyrotropin/cAMP-dependent mitogenic pathway after cell division induced by cAMP-independent factors. J Cell Biol. 1992;117:383–393.
351. Rocha, AS, Paternot, S, Coulonval, K, et al. Cyclic AMP inhibits the proliferation of thyroid carcinoma cell lines through regulation of CDK4 phosphorylation. Mol Biol Cell. 2008;19:4814–4825.
352. Dremier, S, Golstein, J, Mosselmans, R, et al. Apoptosis in dog thyroid cells. Biochem Biophys Res Commun. 1994;200:52–58.
353. Rognoni, JB, Penel, C, Golstein, J, et al. Cell kinetics of thyroid epithelial cells during hyperplastic goiter involution. J Endocrinol. 1987;114:483–490.
354. Riesco, JM, Juanes, JA, Carretero, J, et al. Cell proliferation and apoptosis of thyroid follicular cells are involved in the involution of experimental non-tumoral hyperplastic goiter. Anat Embryol (Berl). 1998;198:439–450.
355. Tamura, M, Kimura, H, Koji, T, et al. Role of apoptosis of thyrocytes in a rat model of goiter. A possible involvement of Fas system. Endocrinology. 1998;139:3643–3646.
356. Rasmussen, SG, Choi, HJ, Rosenbaum, DM, et al. Crystal structure of the human beta2 adrenergic G protein–coupled receptor. Nature. 2007;450:383–387.