[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 5
Mechanisms of Thyroid Hormone Action
Synthesis and Control of Secretion of Thyroid Hormones
Thyroid Hormone Action in the Periphery
Accumulation of Thyroid Hormone in Cells
Deiodinases in Thyroid Hormone Activation and Inactivation
Alternate Metabolic Fates of Thyroid Hormone: Inactive and Active Derivatives
History of the Mechanism of Action of Thyroid Hormones
Discovery of Thyroid Hormone Receptors and Their Mechanism of Action
Discovery That Thyroid Hormones Regulate Specific mRNAs and Gene Transcription
Discovery That TRs Bind Preferentially to Active Chromatin and Specific DNA Sequences
Thyroid Hormone Receptors: Mechanism
Receptor Structure and Function
Hormone Effects on Receptor Structure
LBD H12 Position and Coregulator Interactions
TR Antagonists: The Extension Hypothesis
Other Influences of H12 Rearrangement
Ligand Stabilizes LBD Structure
Structure and Function of TR Isoforms
TR Mutations in Disease: Structure-Function
Gene and Context-Specific Variations in TR Actions
The award of the 1909 Nobel Prize to Theodore Kocher for pioneering work on thyroid gland surgery lent timely recognition to the field of the thyroid gland and its actions, an area of study that was already several centuries in the making.1 Several Renaissance scholars, including Leonardo da Vinci, contributed to the early description of the thyroid gland and the realization that its swelling led to goiter. Later investigators linked goiter and cretinism to dietary deficiency of iodine (a key component of thyroid hormone) and uncovered links between altered thyroid gland function and apparently unconnected symptoms of hyperthyroidism (thyrotoxicosis or Graves’ disease) and hypothyroidism (or myxedema). Classic experiments of Murray in 1891 revealed that sheep thyroid gland extracts could be used to treat, and even cure, myxedema. Later searches for the active principle culminated in the isolation of thyroxine (T4; 3,5,3′,5′-tetra-iodo-l-thyronine) in 1914 by Kendall (who later, with others, received the Nobel Prize for discovering the therapeutic effects of the adrenal steroid cortisone on rheumatoid arthritis), the description of thyroxine structure and chemical synthesis by Harrington in the 1920s and the discovery of the major active form of thyroid hormone, triiodothyronine (T3; 3,5,3′-tri-iodo-l-thyronine), by Pitt-Rivers about 3 decades later. It is now known that thyroid hormones influence virtually every tissue and cell type; they play important roles in growth, development, and differentiation in fetal life and early childhood and act as master regulators of multiple metabolic processes in the adult. The past 50 years have witnessed great increases in our knowledge of thyroid hormone action, with the last 2 decades in particular producing very rapid advancements in understanding the functions of thyroid hormone receptors (TRs), members of the nuclear hormone receptor (NR) family. In this chapter, we review current knowledge of the mechanisms of thyroid hormone action.2–5
Synthesis and Control of Secretion of Thyroid Hormones
1. Spontaneous release of hormones from the hypothalamus and pituitary that stimulate thyroid hormone production
2. Feedback inhibition of production and release of these hypothalamic and pituitary hormones
3. Rates of intracellular conversion of T4 to T3
4. Metabolic destruction of both of these forms of the hormone6
Thyroid hormones are derivatives of tyrosine that are produced in the thyroid gland and secreted into the general circulation. Thyroid hormone synthesis occurs via iodination of tyrosine residues in a large protein called thyroglobulin, coupling of iodinated tyrosines, and enzymatic cleavage to liberate free thyroxine (T4). Recent human genetic linkage studies suggest that regulation of thyroid hormone production is complex, with up to eight different loci influencing biological set-point.7–9 Some of these probably correspond to known genes involved in synthesis of thyroid hormones and feedback inhibition of thyroid hormone production (see later discussion), but most of these loci are not identified.
T4 is the major form of thyroid hormone secreted from the gland, and much of this T4 is converted to T3 in peripheral tissues. Since T3 binds to TRs with higher affinity than T4, it is thought T3 is the major active form of thyroid hormone. Smaller amounts of alternate forms of thyroid hormone are also made in, and secreted by, the thyroid gland. These products are metabolites of thyroxine and include T3, reverse T3 (rT3), and others. Overall amounts of secreted T4 are much higher (more than 20-fold) than any of these alternate forms of thyroid hormone. The thyroid gland may also produce thyronamines (discussed in more detail later10), compounds that may constitute part of a completely distinct arm of the thyroid hormone signaling system; circulating levels of the most potent currently known thyronamine (3-iodothyronamine) appear higher than those of T3.
The negative feedback loop that controls thyroid hormone levels works through the hypothalamus and pituitary (hypothalamic-pituitary-thyroid [HPT] axis (Fig. 5-1). Thyrotropin-releasing hormone (TRH) is released from the hypothalamus and stimulates the synthesis and release of thyroid-stimulating hormone (TSH) by thyrotrophic cells in the pituitary gland. TSH is released into the circulation, where it stimulates the synthesis and release of T4 and to a lesser extent, T3. The circulating thyroid hormones block release of both TRH and TSH through repression of the transcription of genes that encode the TRH pre-pro-hormone and the α and β subunits of TSH and also by down-regulating TRH receptors in the pituitary. Together, these actions result in repression of hormone synthesis and release in response to high circulating levels of T4 and T3. This process helps maintain thyroid hormone levels within defined limits.
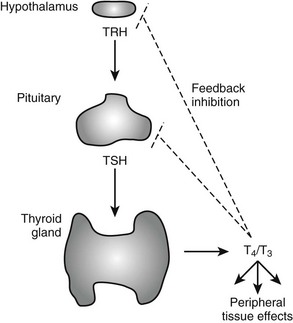
FIGURE 5-1 Regulation of thyroid hormone production in the hypothalamic-pituitary axis. Thyroid hormone is produced in the thyroid gland under control of thyroid-stimulating hormone (TSH). Circulating hormones exert multiple effects on different tissues in the periphery and also feed back to inhibit hypothalamic signals that stimulate TSH release and pituitary production of TSH, thereby maintaining thyroid hormone levels in defined limits.
Thyroid Hormone Action in the Periphery
Both major forms of thyroid hormone (T4 and T3) are transported in the circulation in complex with plasma proteins (Fig. 5-2). The ratio of total T4 to T3 in plasma is about 60:1. This is higher than the 20-fold ratio of T4 to T3 that is initially secreted by the thyroid gland because of greater plasma binding of T4 versus T3, resulting in greater clearance rates of T3. About 99.98% of total circulating T4 and 99.7% of T3 form noncovalent interactions with serum proteins, mostly to thyroxine-binding globulin but to a lesser extent to thyroxine-binding prealbumin, albumin and lipoproteins. The fact that less T3 circulates in complex with plasma proteins means that the ratio of free T4 to T3 is around three- to sixfold, with typical circulating free hormone levels around 20 picomolar (pM) T4 and 6 pM T3, respectively. The free fraction is biologically active and can enter target tissues. Thus, interactions with plasma binding proteins help to ensure even hormone delivery throughout the body.
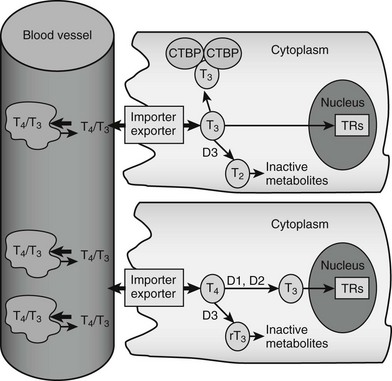
FIGURE 5-2 Peripheral actions of thyroid hormone. The figure summarizes possible fates of thyroid hormone in the periphery. Hormones are mostly transported as complexes with serum binding globulins with limited amounts of free T4 and T3. Hormones are actively taken up into cells by facilitated transport mechanisms, where they undergo several different fates. The upper cell represents possible intracellular fates of T3; it can be sequestered in complexes with CTBP dimers, undergo conversion to inactive metabolites, or enter the nucleus to interact with TRs. The lower cell represents fates of T4, which is either activated by metabolic modification to form T3 or converted to inactive metabolites. It should be noted that expression of D1 and D2 is under transcriptional control, making this step an important control point in thyroid hormone response.
Import and Export
Thyroid hormone entry into cells is mediated by specific transporters (see Fig. 5-2).11–13 While early models suggested that lipophilic thyroid hormone molecules enter cells by diffusion across the plasma membrane, and this may occur to some extent, it is now clear that T4 and T3 entry mostly involves facilitated transport, a form of passive diffusion that requires membrane transport proteins.
Accumulation of Thyroid Hormone in Cells
T3 binds with nanomolar affinity to an intracellular protein that was originally called cytoplasmic T3-binding protein (CTBP).14 This protein is found at high abundance in the kidney and, to a lesser extent, in liver. Because CTBP was later found to be homologous to mu-crystallin, a protein abundant in the lens of the kangaroo eye, the gene is now often called CRYM. Overexpression of CRYM in stable cell lines increases maximal cellular T3 binding capacity and decreases T3 efflux rates. However, these effects are coupled to reduced transcriptional responses to T3, suggesting that CRYM sequesters T3 in an inactive cytoplasmic complex, away from nuclear TRs.
CRYM is important for thyroid hormone action in vivo. Mice with a targeted CRYM gene deletion appear normal but exhibit moderate decreases in circulating T4 (25%) and T3 (13%) levels and perhaps more importantly, also exhibit extremely rapid rates of T3 entry into and escape from target tissues, presumably because there is no CRYM to sequester cytoplasmic hormone from export mechanisms. Human patients with a natural CRYM mutation that blocks its ability to bind T3 (K314T) are deaf, a known consequence of defective thyroid hormone signaling during development. Interestingly, CRYM requires NADPH for dimerization and T3 binding (Fig. 5-3). As NADPH levels are reflective of levels of cellular reducing power and anabolic capacity, it is conceivable that CRYM may sequester or release T3 in response to alterations in cellular metabolic status. This notion is attractive but not proven.
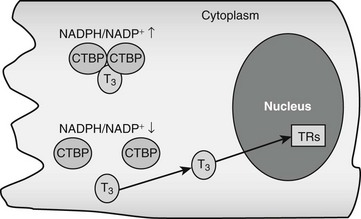
FIGURE 5-3 Thyroid hormone interactions with CTBP. Intracellular T3 can bind CTBP dimers and become sequestered in the cytoplasm. CTBP dimer formation is sensitive to intracellular NADPH/NADP+ concentrations such that free hormone will probably be released in response to reductions of NADPH levels.
There could be other T3-binding proteins in addition to CRYM and the nuclear TRs (which are described later). Radioisotope-labeled T3 interacts with proteins in the endoplasmic reticulum, mitochondrion, and nuclear envelope.2 These putative T3-binding proteins have not been identified, and the functional significance of these interactions is unknown.
Deiodinases in Thyroid Hormone Activation and Inactivation
The thyroid gland produces mostly thyroxine. Limited amounts of T3 and rT3 are secreted by the gland, but more than 80% of both these forms of thyroid hormone are produced in the periphery via actions of specific deiodinases, D1, D2, and D3.15,16 D1 and D2 remove the iodine group from the 5′ position of the outer thyronine ring. As T3 binds to TRs with higher affinity than T4, the actions of D1 and D2 deiodinases serve to increase thyroid hormone activity through generation of T3, and this represents a step-up in activity. Conversely, the action of D3 decreases hormone activity and represents a step down by removing the iodine group from the 5 position of the inner thyronine ring and converting T4 and T3 to rT3 and 3,3′-T2, respectively, neither of which interacts substantially with nuclear TRs at physiologic concentrations. Changes in deiodinase expression and activity are important control points in thyroid hormone signaling; they influence the production and availability of the biologically active form of thyroid hormone, T3.
D3 is important for clearance of plasma T3.17 It is highly expressed in human placenta, where it is thought to be important for protection of the developing fetus from effects of maternal thyroid hormones. D3 is also highly expressed in the brain and skin. Increased D3 levels may account for reductions of circulating thyroid hormone levels that are often observed in critically ill patients and states of inflammation. Finally, D3 is often overexpressed in vascular tumors, and this in turn can result in marked reductions in circulating thyroid hormone levels.
Alternate Metabolic Fates of Thyroid Hormone: Inactive and Active Derivatives
In addition to 5′ deiodination, T4 undergoes other modifications which are generally believed to inactivate the hormone.18 About 45% of circulating T4 is converted to rT3 and 35% to T3. T3 and rT3 undergo further deiodinations to create T4 derivatives with all possible combinations of iodinated and deiodinated inner and outer rings (3,3′-T2, 3,5-T2, 3′,5-T2, 3′-T1, 3-T1 and T0), none of which bind significantly to nuclear TRs. T4 and T3 can also be inactivated by glucuronidation in liver, followed by secretion into the bile or by sulfation in liver and kidney and excretion in the urine.
1. Deamination of the thyroid hormone alanine side group produces acetic acid derivatives of T4 (tetraiodoacetic acid, Tetrac) and T3 (triiodoacetic acid, Triac).19 Both are exclusive products of extrathyroidal metabolism; they are undetectable in the thyroid gland. Although they comprise about 2% of total circulating thyroid hormone, they are present at high levels in liver, where Triac represents about 14% of total thyroid hormones. Tetrac and Triac are excreted in the urine, suggesting that deamination may promote thyroid hormone clearance from the body. However, Triac binds to TRs with high affinity and acts as a potent activator. It also exhibits TR isoform preference (it preferentially binds the beta form of TR) and interacts weakly with CTBP/CRYM, suggesting that it may evade cytoplasmic sequestration by this protein. While these observations raise the issue that Triac has a distinct function from T3, this idea is not proven.
2. Decarboxylation of thyroid hormone alanine side group produces amine derivatives (thyronamines) that are directly analogous to different iodinated forms of thyroid hormone.10 Thyronamines have been detected in the thyroid gland, central nervous system, fat, and other tissues. They are bound to transporter proteins in the circulation and, though completely quantitative analyses of circulating thyronamine levels have not been published, early estimates of their abundance suggest that they are present at concentrations that are at least similar to or greater than rT3 or T3.10 At the time of writing, an endocrine role for thyronamines has not been formally proven, but there is clear evidence of biological activity. Each thyronamine has been chemically synthesized and tested in cell culture in animal models; one derivative with a single iodine substituent at the 3 position of the inner thyronine ring (T1-amine) binds and activates a cell surface G-coupled protein receptor (trace amine–associated receptor 1; TAAR1) at physiologic concentrations (subnanomolar). T1-amine induces profound hypothermia, bradycardia, and reduced cardiac output in mice and alters metabolism in several species; it reduces metabolic rate and increases lipid utilization in a pattern that is reminiscent of sleep or hibernation. T1-amine may prove to be an important endocrine hormone, and it is interesting to suggest that T4 derivatives such as rT3 may be metabolic precursors of this compound.
Actions of Thyroid Hormone
Thyroid hormones exert profound influences on nearly all tissues (Fig. 5-4).2 Receptors for thyroid hormones (TRs) are expressed fairly ubiquitously, although well-defined target tissues such as liver and heart express higher levels of TRs than others, and there are differences in distributions of particular TR isoforms20 (described in more detail in a following discussion). Essentially, thyroid hormone is needed for proper growth and development of the fetus and children and exerts widespread influences on multiple aspects of metabolism in adults.
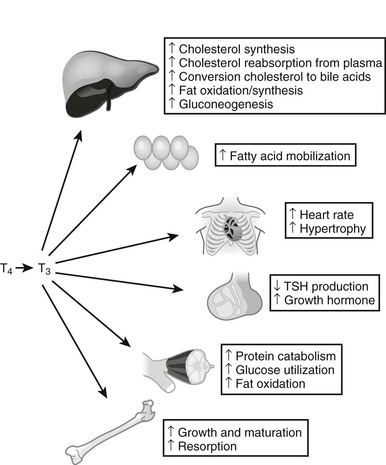
FIGURE 5-4 Tissue effects of thyroid hormones: summary of thyroid hormone effects on different tissues.
Analysis of gene expression patterns in mouse knockout models has confirmed that TRs mediate nearly all thyroid hormone responses, and that many of these effects involve changes in gene expression. While actual influences of thyroid hormone on gene expression are complex, there are important underlying principles.2 First, developmental defects that arise in cretinism and children born to hypothyroid mothers are not reversed by later hormone replacement, whereas adults with thyroid hormone imbalances exhibit metabolic disturbances that in most cases can be readily reversed by restoring TH levels to correct levels. This implies that hormone must trigger key developmental events in defined temporal windows, but thyroid hormone–regulated genes involved in metabolic regulation remain sensitive to alterations in hormone levels and continuously couple gene expression to thyroid status. Consideration of gene expression patterns, described in more detail later, reveals that some genes are induced in multiple tissues (D1 is an example), whereas other target genes are regulated in a manner that is highly tissue- and context-specific. Thus, thyroid hormone is a primary regulator of some genes, but hormone must cooperate with other factors to induce expression of other genes. Specific effects of thyroid hormone follow.
Basal Metabolic Rate
One of the most important effects of thyroid hormone involves changes in basal metabolic rate (BMR) in multiple tissues.21–24 Thyroid hormones stimulate oxygen consumption (indicative of enhanced metabolism) in multiple locations, including skeletal muscle, liver, kidney, and intestine. Increases in BMR are probably partly related to increased mitochondrial activity and number. Importantly, thyroid hormones induce expression of uncoupling proteins (UCPs), mitochondrial membrane proteins that dissipate the proton gradient in the absence of ATP synthesis, thereby converting potential energy to heat. This effect is probably a contributor to thyroid hormone–dependent increases in BMR.
Tissue Effects
Liver
Thyroid hormones exert multiple effects on the liver.2,25 They are potent mitogens in this tissue, especially in growing animals. Thyroid hormone also influences multiple metabolic processes. There is stimulation of fatty acid β-oxidation and gluconeogenesis, both key aspects of fasting response.26 However, thyroid hormone can also stimulate expression of enzymes involved in lipogenesis and generation of NADPH-reducing equivalents that are required for fat synthesis and protection against reactive oxygen species. Since it is thought that fat oxidation and synthesis do not occur simultaneously, these processes are probably separated spatially or temporally, and thyroid hormone must cooperate with other signaling mechanisms to regulate these effects.
Muscle2
Thyroid hormones promote muscle catabolism and increase skeletal muscle energy expenditure in adults. Thyroid hormone induces the insulin-sensitive glucose transporter in muscle and also promotes fat burning.30 These effects may help to sensitize muscle to insulin response. Accordingly, a human D2 gene polymorphism that reduces activity of the enzyme in muscle leads to a 20% reduction in insulin-dependent glucose disposal in humans and is correlated with insulin resistance in human populations that harbor this alteration.
Intestine
Thyroid hormone is required for normal maturation of the small intestine. TRα1 is required for proliferation of intestinal epithelial progenitor cells via induction of the β-catenin proto-oncogene.34
Systematic Analysis of Thyroid Hormone Target Genes
Many thyroid hormone–responsive genes involved in the responses described earlier are known.2 For example, in liver, thyroid hormone induces carnitine palmitoyl transferase 1a (that mediates the rate-limiting step in fatty acid oxidation); glucose-6-phosphatase and phosphoenolpyruvate carboxykinase (rate-limiting steps in gluconeogenesis); the NR coregulator PGC-1α, which is an important transcriptional coregulator that stimulates genes important for mitochondrial biogenesis, gluconeogenesis, and fat oxidation; CYP-7a1 (cholesterol-to–bile acid conversion); fatty acid synthetase; acetyl-CoA carboxylase; malic enzyme (increases fat synthesis); and spot 14, which is required for fat synthesis in some tissues and up-regulated in contexts in which carbohydrates are converted to fat. Many other hormone-responsive genes are probably still unknown.
The earliest systematic attempt to define thyroid hormone response was performed in the late 1970s. Use of two-dimensional (2D) gels to analyze extracts from radiolabeled methionine–pulsed cells revealed that about 1% of 1000 rat liver proteins change in response to altered thyroid hormone.36,37 There were inductions and repressions, and the studies began to define the “domain” of the thyroid hormone response. Recent advances in gene-profiling technology have permitted much more detailed descriptions of hormone-dependent alterations in gene profile. Presently there have been relatively few systematic genome-wide descriptions of thyroid hormone responses.25,26,38,39 An early study of thyroid hormone action in mouse liver revealed that about 1% of mouse genes change in response to altered thyroid hormone (hypothyroid versus hyperthyroid),25 confirming estimates from 2D gels. This study revealed target genes involved in carbohydrate, fat, and amino acid metabolism and unexpected thyroid hormone–regulated pathways; for example, genes involved in apoptosis are induced. The experiment also provided insights into patterns of thyroid hormone–responsive gene expression. Most regulated genes (>65%) in liver were repressed by thyroid hormone. This preponderance of negatively regulated genes is not seen in all tissues. Target genes are generally up-regulated by hormones in human muscle primary culture. Later studies of thyroid hormone patterns of gene expression in TR knockout mice provided insights into TR-specific effects and subtle differences in thyroid hormone–responsive gene expression and are discussed in more detail later in the chapter. Systematic screening of thyroid hormone–responsive genes should provide detailed descriptions of novel target genes and pathways in different tissues.
Thyroid Hormone–Regulated Micrornas
MicroRNAs (miRs) are small (18 to 25 nucleotides), noncoding RNAs that hybridize with coding mRNAs to inhibit translation and, in some cases, also promote mRNA degradation.40 The enormous roles these miRs play in regulation is just beginning to be appreciated. MiRs are produced by cleavage and processing of large primary transcripts, which often code for proteins. Thus, many of the influences that regulate expression of parental primary transcripts also regulate expression of the associated miR. While effects of thyroid hormone on miR expression have not been studied extensively, one miR (miR-208) lies within a noncoding region of the thyroid hormone–responsive human αMHC transcript (and is therefore up-regulated by thyroid hormone itself). MiR-208 is implicated in up-regulation of stress-dependent cardiac responses. It is likely that other thyroid hormone–responsive miRs exist, and they may play important roles in hormone effects on protein translation.
History of the Mechanism of Action of Thyroid Hormones
As the physiologic effects of thyroid hormones became known, investigators focused on roles of these hormones on stimulating the basal metabolic rate and on the liver.1,2 It was originally proposed that the hormones directly uncouple oxidative phosphorylation and that the primary site of action of thyroid hormones was at the mitochondria; here, investigators related actions of thyroid hormones to those of dinitrophenol. Later studies revealed other influences of thyroid hormones, including cholesterol reduction, that were hard to relate to mitochondrial actions. The notion that thyroid hormones directly uncouple mitochondrial oxidative phosphorylation did not provide a coherent unifying hypothesis to explain all actions of thyroid hormone.
In the late 1960s, Tata and colleagues conducted a number of measurements of the effects of thyroid hormones on the liver.41 He found that thyroid hormone–dependent increases in metabolic rate were blocked by actinomycin D, an inhibitor of RNA and, secondarily, protein synthesis. Also, there was an increased uptake of radiolabeled uridine into trichloric acid (TCA) precipitable material following administration of TH to animals, as well as increased activity of RNA polymerase, suggesting that TH stimulates RNA synthesis. Similar studies were being conducted with NR ligands such as with glucocorticoids, estrogens, and progestins, and there was great controversy as to whether these ligands acted through (1) transcriptional control, (2) precursor uptake, or (3) posttranscriptional control. The experiments did not address high specificity of TH responses, and as a result, controversy remained as to which of the three mechanisms applied to thyroid hormone action. Nevertheless, this study represented the first fundamentally correct proposal for the mechanism of thyroid hormone action.
Discovery of Thyroid Hormone Receptors and Their Mechanism of Action
TRs were identified in the 1970s.42,43 Following the discovery of several nuclear receptors, including the estrogen and glucocorticoid receptors, investigators in the thyroid hormone field utilized similar approaches to look for TRs. Specific binding of radiolabeled T3 to liver cells was initially discovered by Oppenheimer and colleagues using intact cells and later characterized further by Samuels and colleagues, who found that the mechanism of TR actions differed mechanistically from the steroid receptors, which translocate into the nucleus on ligand binding. For TRs, hormone could bind directly to nuclei, and the location of receptors did not alter with T3.
In the late 1970s and early 1980s, multiple aspects of the receptors were revealed.36,44 Analysis of properties of TRs revealed a single class of high-affinity binding sites with Kd values for T3 in the 0.1 nM range. Specific hormone-binding sites were observed in extracts of several responsive tissues, including liver, anterior pituitary, brain, and heart. Estimates of the number of binding sites per cell suggested that the proteins are rare; highly responsive tissues only contained about 10,000 specific hormone-binding sites per cell. Evidence that the receptor preparations correspond to physiologic hormone targets came from observations that affinities for different thyroid hormones parallel their biological potencies (Triac>T3>T4>rT3). Partial TR purification and photo-affinity labeling revealed two major nuclear hormone-binding proteins with molecular weights of 46 and 57 kD, which were later found to correspond to products of distinct genes—the TR α and β isoforms—described later in the chapter. TRs were also found to be associated with chromatin, and preferentially with active chromatin, suggesting that they influence gene expression through interactions with DNA.45–48
Discovery That TRs Bind Preferentially to Active Chromatin and Specific DNA Sequences
Studies during the 1970s and 1980s also set the stage for understanding the nature of TR binding sites and their relationship to active chromatin. GH genomic DNA was used for interaction studies with partially purified TR preparations, demonstrating that TRs bound to specific DNA sequences. These experiments were forerunners of the now familiar concept that receptors act by binding to specific thyroid hormone response elements (TREs). That the receptors could affect DNA was further evidenced by the finding that they induce bending of rat or human TR DNA binding sites. Subcellular fractionation revealed that TRs were associated with chromatin, and preferentially with active chromatin, suggesting a relationship between the receptors and transcriptionally active chromatin.45–48 These studies set the stage for definition of thyroid hormone response elements (TREs).
Cloning of TRs
TRs were cloned in 1986.49,50 They were identified as cellular homologs of a retroviral oncogene (cancer-causing gene), v-erbA,51 which causes erythroblastoma in chickens. Isolation of these cDNAs occurred after those for other members of the NR family, the glucocorticoid and estrogen receptors, which led to the realization that v-erbA was derived from an NR. Both of the v-erbA homologs bound thyroid hormone with high affinity and appropriate specificity. The Vennstrom group clone was most closely related to v-erbA and was originally designated as c-erbA, but it is now called thyroid hormone receptor α (TRα). As compared to TRα, v-erbA contains 17 different amino acid substitutions and a C-terminal truncation, which are now known to prevent the viral oncogene from binding hormone.49 The Evans group clone encoded a highly homologous but distinct 57-kD protein that was closely related to TRα and v-erbA but was the product of the THRB gene.50 The characterization of TR genes (THRA and THRB), which encode the two major TR isoforms (TRα1 and TRβ1) and several variants that arise from differential splicing (described later), helped to reveal that TRs belong to the NR family, which includes receptors for the steroid hormones, vitamins A and D, and other small lipophilic molecules.4
Thyroid Hormone Receptors: Mechanism
TRs work by altering gene expression in response to changes in thyroid hormone concentrations (mostly T3).2,38 This alteration in gene transcription profile is believed to account for most of the observed physiologic effects of thyroid hormones, although there are also actions of thyroid hormones that do not involve transcription (discussed later). Unlike the case with steroid receptors, TRs are active in the absence of hormone.
TR Action
Unlike classic models of hormone action in which unliganded receptor is inactive and hormone binding triggers its activity, TRs are transcriptionally active in the absence and in the presence of hormone (Fig. 5-5).52 Rather than activating the receptor, hormone binding alters the spectrum of its actions.
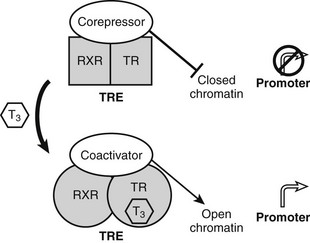
FIGURE 5-5 Thyroid hormone receptor action. Actions of unliganded and liganded TRs at positively regulated genes. The receptor binds DNA as a heterodimer with RXR in the absence and presence of the hormone. Unliganded TRs recruit corepressors, which repress gene transcription by condensing local chromatin structure and blocking coactivator binding. Hormone binding promotes a conformational change, which leads to exchange of corepressor for coactivators, which reverses effects of coactivators.
TRs bind constitutively to specific DNA sequences termed thyroid hormone response elements (TREs) in the proximal promoter of positively regulated target genes. As noted earlier, TRs function mostly via heterodimerization with another NR, RXR, which is a partner for many members of the NR family. RXR binds vitamin A derivatives and unsaturated fatty acids but is thought in many cases to be silent and unoccupied by ligands, when it forms complexes with TRs. Unliganded TRs repress transcription of positively regulated genes by recruiting corepressors such as the NR corepressor (N-CoR), silencing mediator of retinoid and thyroid hormone–responsive transcription (SMRT) and alien. Corepressors actively silence gene transcription by binding histone de-acetylases (mainly HDAC3) which condense local chromatin to prevent access of RNA polymerase II to target promoters.53 Hormone reverses these inhibitory effects and further stimulates gene expression by promoting corepressor release and subsequent recruitment of several different coactivator complexes which enhance transcription in different ways: ATP-dependent remodeling of local chromatin, catalysis of specific histone modifications that mark the genes for transcriptional activity, and enhancement of recruitment and processivity of the basal transcription machinery.
The converse process can also occur.54 Unliganded TRs stimulate gene transcription, and binding of T3 overcomes this process, resulting in repression of gene transcription. Generally, hormone also suppresses transcription below levels stimulated by the unliganded TRs. One model to explain this phenomenon is that coactivators and corepressors exert opposite actions at this type of gene, and there are several examples where so-called NR coactivators repress gene transcription, and corepressors act as coactivators that bind to the “corepressor binding site” (Fig. 5-6). However, several mechanisms of NR repression have been described, so there are other possibilities. For example, ligand-dependent activation of peroxisome proliferator–activated receptors (PPARs) and liver X receptors permits covalent attachment of lysine residue in the LBD to a large 100-amino-acid protein called small ubiquitin-like modifier (SUMO), which arrests corepressor complexes at target promoters (Fig. 5-7).54 This effect counters proinflammatory stimuli that promote corepressor release and gene induction and therefore acts as a repressive mechanism. Negative gene regulation may not involve direct receptor/DNA contacts.55 Although promoters of some genes that are repressed in response to ligand contain variant TREs, negative regulation may also involve TR interactions with heterologous DNA-bound transcription factors (such as AP-1 and CREB) instead of promoter DNA itself, as has been demonstrated for other NRs. Further investigation will be needed to clarify mechanisms of transcriptional repression at individual genes.
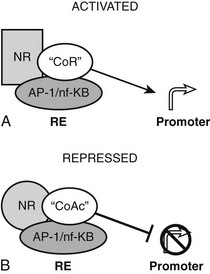
FIGURE 5-6 Transcriptional repression mechanisms. Transcriptional activation can occur by recruitment of a coactivator that binds to the corepressor binding surface. Transcriptional activation is mediated by a corepressor that binds the coactivator surface. In some cases, these proteins may be the same as coactivators and corepressors that mediate TR actions at classical TREs, with reversed functions.
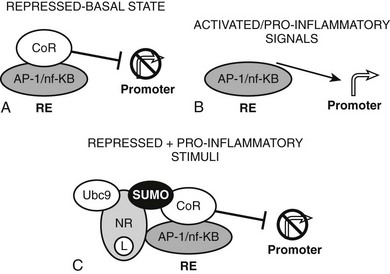
FIGURE 5-7 Repression by corepressor arrest. An alternative mode of transcriptional repression demonstrated for NRs at proinflammatory genes. A, Genes are constitutively repressed and B, activated by dismissal of corepressor complexes in response to proinflammatory signals. C, Liganded NRs repress transcription in the face of these proinflammatory stimuli by recruiting enzymes (including ubc9) which SUMOlate the liganded receptor LBD. SUMO modification, in turn, arrests the corepressor complex at the promoter.
Models of thyroid hormone response emphasize differences in TR complex formation in the absence of hormone and the presence of saturating levels of hormone. As discussed previously, physiologic levels of hormone do not saturate all receptors under these conditions,39 so thyroid hormone–responsive genes will probably be occupied by a mix of liganded and unliganded TRs, with different associated factors. The consequences of partial occupancy of DNA-bound receptors with hormone are far from clear; there could be active competition between liganded and unliganded TRs for the same sites in the same cells, stochastic choices between formation of liganded and unliganded TR-associated complexes in different cells, or other differences.
Receptor Structure and Function
TRs and other NRs are single polypeptide chains that adopt a common modular domain structure. Particular functions can be assigned to specific receptor domains (Fig. 5-8).2,4 Use of basic recombinant DNA technology to create short cDNA fragments corresponding to each domain and targeted mutagenesis coupled with expression and functional analysis revealed the role of each domain.
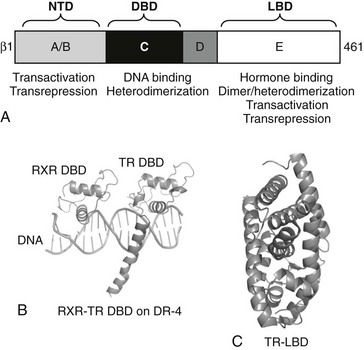
FIGURE 5-8 TR secondary and tertiary structure. Schematic representation of TR structures. A, The upper diagram represents TRβ secondary structure with domain positions marked. B, X-ray structures of the TR DBD on DNA in complex with RXR and the C, TR LBD. The two TR domain structures overlap in the “hinge” D-domain region.
DBD Structure
The TR DBD x-ray structure was solved in complex with that of its heterodimer partner RXR on a thyroid hormone receptor response element (TRE), a direct repeat of the consensus sequence AGGTCA spaced by four bases (DR-4 element, see Fig. 5-8).56,57 DNA contacts are mediated by a large α-helix in the first zinc finger, which docks into the major groove of DNA. This conserved region of the receptor contains the P-box, which dictates NR DNA element recognition specificity. Both the second zinc finger and a long carboxyl-terminal α-helix termed the C-terminal extension (CTE) also make DNA contacts. The second zinc finger interacts with the major groove, and the CTE interacts with the DNA minor groove and phosphate backbone. The organization of the CTE is unique to the TR, and these additional CTE-dependent DNA contacts may explain why TRs, unlike many other NRs, exhibit the capacity to bind to DNA as monomers (developed in following discussions).
RXR and TR DBDs interact with the DR-4 element in a head-to-tail manner, with RXR upstream. This polarity is dictated by the specific heterodimer contact surfaces, which involve multiple interactions between zinc finger regions of both receptors and between the outer face of the TR CTE and RXR. The 5′ location of RXR was already inferred by biochemical analysis58; introduction of the glucocorticoid receptor P-box sequence into TR permitted the RXR-TR complex to recognize a hybrid response element composed of one classic TR half-site (AGGTCA) and an equivalent glucocorticoid receptor response element half-site (TCTTGT) but only when the GR binding half-site is placed downstream in the 3′ position. This reflects that the TR preferentially associates with the downstream position.
RXR/TR pairs bind preferentially to DR-4 elements, whereas other RXR/NR pairs recognize direct AGGTCA repeats with different spacings.58 Comparison of RXR/TR structures with other RXR/nuclear receptor pairs reveals that the CTE sets this spacing preference.57 All NR CTEs fold across the core of the DBD, but different CTEs adopt different trajectories and occlude receptor binding to the wrong element. For RXR-TRs, spacings of less than 4 bp would not be permitted because of steric clashes between the CTE and the RXR DBD. Conversely, introduction of extra bases between the half-sites will impose geometric constraints that prevent proper engagement of the RXR-TR DBD heterodimer surface.
LBD Structure
Although there are now many publicly available NR LBD x-ray structures, TR (rat TRα) was the first structure solved in complex with native hormone (it was reported simultaneously with the structure of the liganded retinoic acid receptor, RAR).59–61 Strikingly, the ligand was buried in the core of the LBD; this was contrary to contemporary models which assumed that ligand would interact with an allosteric site on the receptor surface. The ligand-binding pocket is small and well tailored to cognate agonists such as T3 and Triac. This organization implies that admission of hormone into the core of the domain promotes its folding into an active conformation.
TR LBD structure strongly resembles other NRs.4,62 The domain is almost entirely helical, composed of 12 α-helices. The α-helices fold into three layers: two outer layers comprise three distinct helices, the central layer comprises two helices, with the additional space occupied by hormone. Several short helical loops and β sheets link the individual α-helices. N-terminal LBD helix (H1) constitutes a backbone that links helical layers. C-terminal helix (H12) adopts different positions in the presence and absence of hormone and dictates cofactor interactions that influence receptor activity.
Hinge Structure
The hinge domain (or D-domain) was first identified as a poorly conserved region that links the DBD and LBD. Original models suggested that it would behave as an unstructured peptide that facilitates rotation between the LBD and DBD in different contexts. However, TRβ LBD and DBD x-ray structures contain overlapping amino acids, and these reveal that residues that were originally assigned to the hinge actually comprise extensions of H1 of the LBD and the C-terminal portion of the DBD CTE.60,63 The true unstructured hinge is only 3 to 6 amino acids long. Presently it is not clear whether this short region is sufficient for true rotational flexibility between domains. Interestingly, the C-terminal helix of the DBD CTE region appears as a completely unstructured peptide in some LBD crystals but not others, and it has been suggested that this region of the receptor could either fold into a functional helix or unfold to improve rotational flexibility in different contexts.63
NTD Structure
There are no published studies of TR NTD structure. NR N-terminal domains (NTDs) are important for transcriptional response and integration of NR ligand signals with second messenger pathways, but this region is poorly conserved throughout the receptor family and not at all defined at the level of tertiary structure.64 Current models indicate that NR NTDs are composed of intrinsically unfolded domains; these types of proteins consist of multiple and potentially overlapping subregions that acquire active conformation on interaction with target cofactors.64
Full-Length Receptors
No high-resolution structures of TRs or RXR-TR fragments that make up more than one domain have yet been resolved. There are published low-resolution solution x-ray structures of liganded TR DBD-LBD dimers, and an unusual tetrameric form of unliganded TR DBD-LBD.65 However, these data provide sufficient resolution to determine likely shapes of the molecules, but not enough to define the trace of the protein backbone. Use of these low-resolution structures to dock high-resolution x-ray structural models of DBDs and LBDs suggests that in the dimer in solution, LBDs and DBDs are in close proximity to their counterparts in the neighboring subunit and that the DBDs are relatively distant from the LBDs, implying that the hinge is extended. In the solution tetramer, LBD pairs dock against each other with DBDs protruding in an X shape. Here, however, the DBDs are closer to the LBDs than they appear in the dimer, implying that there may be rearrangements in the hinge that are dependent on the oligomeric state of the LBD.
A recent structure of full-length liganded PPARγ in complex with liganded RXRα on a DNA element provides a guide to expected principles of organization of the full-length RXR-TR complex.66 The PPARγ-RXRα heterodimer binds to a DR-1 element, with PPARγ in the 5′ position, unlike the TR which occupies the 3′ position (Fig. 5-9). LBDs and DBDs adopt the expected conformation and engage in the expected heterodimer contacts predicted from LBD and DBD structures and mutational analysis, described more fully for the TRs later. However, the PPAR CTE portion of the hinge makes unexpected DNA contacts 5′ to the AGGTCA motif. Moreover, the complex is nonsymmetrical, the PPARγ LBD is close to the RXR DBD, and this facilitates unexpected heterodimer contacts which involve LBD surfaces around helix 3 and the β-sheet region and the surface of the RXR DBD on the opposite side of the molecule from the DNA-binding surface. The RXRα hinge appears unstructured and flexible. This permits the RXR LBD to adopt a more distant position from the DBD than the PPAR LBD and raises the possibility that the RXR hinge is a potential source of rotational flexibility in response-element recognition. Finally, NTDs of both receptors are disordered, in keeping with the prediction that these domains can only adopt appropriate conformation on contact with partners. It will be interesting to determine the structures of full-length TR/RXR to learn how TRs adapt to different elements and whether there are unexpected heterodimer contacts between LBDs and DBDs of these receptors.
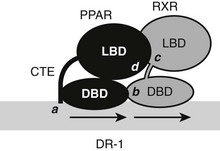
FIGURE 5-9 Schematic of full-length PPAR and RXR on a DR-1 element. Shown are expected and unexpected interaction points. The PPAR DBD CTE makes additional DNA contacts (a) outside of the consensus half-site. DBDs and LBDs are in predicted heterodimer contacts (b, c). Additionally, the PPAR LBD binds the RXR DBD (d). NTDs are unstructured and are not shown.
Variable TRE Sequence and Conformational Adaptation to DNA
Published liganded TR LBD x-ray structures are monomers.67 However, many NR LBDs have been crystallized as homodimers and heterodimers; there is a surface that binds both homo- and heterodimer partners which is large and comprises residues from helices 9, 10, 11, 7, and 8.68 Targeted mutagenesis of TRs and RXRs confirms that dimer and heterodimer formation is similar to this common interface, with a hotspot of particularly hydrophobic residues at the junction of H10 and H11 playing very important roles in binding.68
Most TREs are composed of DR-4 elements, but there is significant variation in sequence and arrangement of TREs (Fig. 5-10) which results in TR isoform and oligomer-specific differences in DNA recognition. TRs and RXR-TRs bind to direct repeats with different spacing, inverted palindromes (IP), and palindromes (pal).58,69 TRα- and TRβ-RXR heterodimers bind preferentially to DR-4 but also bind to IP-6 elements (where the DNA binding sites are in an inverted position with respect to each other, with a 6-nucleotide spacer) and palindromic (Pal) elements (where the DNA binding sites are arranged in a head-to-tail position with respect to each other, without spacer DNA). In contrast, TRβ homodimers bind strongly to IPs, weakly to consensus DR-4 elements, and not at all to Pal elements. TRα homodimers bind very weakly, if at all, to these TREs. In addition, TRβ homodimers bind strongly to a subset of variant DR-4 elements,70 implying that additional sequence differences can facilitate homodimer interactions. RXR-TRs are the major TR species in living cells, but TRβ homodimers can clearly modulate transcription from some TREs in cell culture and yeast.69 Moreover, blockade of RXR expression only inhibits T3 response at a subset of target genes.71 Both observations suggest that alternate oligomeric forms of TR can mediate thyroid hormone responses. The precise contributions of RXR-TRs versus other TR oligomers to T3 signals at different genes in physiologic conditions are not yet clear.
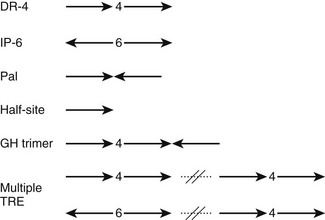
FIGURE 5-10 The diverse organization of TREs. The figure represents a schematic of orientations (arrows) and spacings (numbers between arrows) of AGGTCA half-sites observed in natural TREs. With multiple TREs, the orientations can differ between elements.
TRs and RXR-TRs must undergo significant conformational adaptation to bind different TREs.58 RXR-TR heterodimer and TR-TR homodimer formation involves the same interface at the junction of H10-H11, irrespective of TRE organization. Consideration of relative orientations of LBD and DBD required for binding to each TRE indicates that with DR-4 elements, the upstream LBD must rotate with respect to the DBD to form the heterodimer surface in a head-to-head fashion (Fig. 5-11). Since RXR-TRs preferetially bind DR-4 elements, it is likely that this rotation is provided by the upstream RXR. The TR DBD and LBD do not need to rotate for inverted palindromes, where TR-TR homodimer formation is strongest. Thus, RXR is probably responsible for flexible response-element recognition, and this may contribute to its function as a near-universal heterodimer partner for NRs. The unstructured appearance of the RXR hinge domain observed in the PPAR-RXR full-length structure mentioned earlier suggests that this could be one candidate for the source of flexibility.
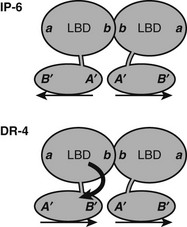
FIGURE 5-11 Predicted orientations of TR DBD and LBD on IP-6 and DR-4 elements. A need for swivel between domains at DR-4 elements? At an inverted palindrome, the LBDs and DBDs can bind in the same orientation, back to back, as indicated by positions of surfaces on DBDs (A’B”) and LBDs (a, b). At a direct repeat, the DBDs are in head-to-tail orientation, but the LBDs interact through the same surface that is utilized at the IP-6 element. Thus the LBD of the 5′ partner must rotate with respect to the 3′ partner. The source of this rotational flexibility is not clear, but the fact that RXR occupies this site in RXR-TR heterodimers suggests that the RXR hinge is one likely source.
Alternate TREs
There are examples where TRs may interact with DNA in other ways.58 Several response elements are composed of TRE half-sites that bind TR monomers. TRα and TRβ are unusual in the nuclear receptor family, with their high capacity to bind as monomers to AGGTCA half-sites in addition to more conventional interactions with DNA elements as dimers and heterodimers. In these cases, the recognition site is extended to include the core half-site and additional 5′ flanking sequences.
Natural TREs often contain multiple copies of half-sites. The spot 14 promoter contains two DR-4 elements, and the SERCa2 promoter contains a DR-4 element and two IP elements. In most cases, it is not known whether TRs recognize these elements as separate RXR-TR heterodimers or TR-TR homodimers or as larger assemblies, such as tetramers or heterotetramers. There is one clear example of the latter possibility. The rat growth-hormone promoter contains an unusual trimeric TRE that consists of a classic DR-4 element and a downstream half-site that forms a palindrome, the 3′ DR-4 half-site72 (Fig. 5-12). TRβ, but not TRα, forms homotrimers and heterotrimers with RXR that bind this element with high efficiency and cooperativity. Thus TRβ must contain additional subunit interaction surfaces, if it simultaneously engages in typical homo/heterodimer contacts and also binds another TRβ subunit in the 3′ position. TRβ0, a TR splice variant that lacks the NTD and is abundantly expressed in frogs, birds, and reptiles, binds this element with high affinity, suggesting that all surfaces are in the DBD-LBD region.
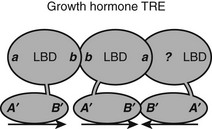
FIGURE 5-12 Predicted TR interactions with a complex TRE in the growth hormone promoter. The growth hormone TRE comprises three TRE half-sites arranged as a classic DR-4 element and a downstream half-site. The fact that there is cooperative binding suggests that the TR in the 3′ position in the DR-4 element must interact with both its upstream and downstream partner, although the mechanism for the latter interaction is not known.
There is significant variation in TRE half-site spacing and sequence in naturally occurring TREs that likely has consequences for TR activity.4,58 TRs preferentially recognize DR-4 and IP-6 elements but also bind similar elements with different spacings, including DR-3, DR-7, and IP-4. Actual half-site sequences diverge considerably from the classic AGGTCA half-site. In fact, negatively regulated genes sometimes contain weak TR binding sites that instead conform to the consensus TGGTTTGGGGTCCA.55 These influences will affect the affinity of the receptor for the element but may also influence receptor activity in other ways. DNA contact alters DBD structure, and a small subset of glucocorticoid-receptor DBD mutations that mimic allosteric switches which occur on DNA binding affect activity of AF-1 and AF-2 and change the response element–specific behavior of the receptor. One of these, a lysine residue at the base of the first zinc finger domain that contacts DNA, alters the activity of all NRs (including TR); it changes NRs from ligand-mediated repressors to ligand-mediated activators at promoters with AP-1 sites, where the receptor acts in the absence of direct DNA contact. Thus the nature of the DNA sequence element could alter both TR conformation and activity.
Hormone Effects on Receptor Structure
Presently there are few true unliganded NR LBD structures that permit direct comparison of liganded and unliganded states, and it is not clear how well such unliganded structures reflect the actual organization of unliganded receptors in the cell, where they are found in complexes with stabilizing protein partners.61 However, x-ray structural analysis, nuclear magnetic resonance, and hydrogen-deuterium exchange studies, which can reveal different aspects of receptor dynamics, and results of structure-directed mutagenesis approaches have revealed many interesting facts about the nature of these hormone-dependent alterations.
LBD H12 Position and Coregulator Interactions
The major known effect of hormone binding is stabilization of H12 in an active position in which it packs against the body of the LBD to form a surface called activation function 2 (AF-2)52,61 (Fig. 5-13). Comparisons of liganded TR and other liganded NR structures reveal that H12 is docked against the LBD in a similar position in all “active” liganded structures. Conversely, examination of unliganded structures and structures of NRs in complex with antagonists or partial agonists reveal that H12 can adopt various “inactive” or “partly active” conformations, all highly distinct from the active position of H12. This suggests that H12 may be unstable in the absence of an activating ligand. The idea that ligand binding stabilizes H12 has been confirmed by dynamics studies in which PPARγ H12 mobility was measured directly.73
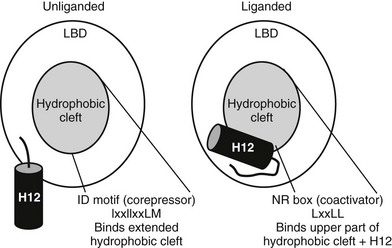
FIGURE 5-13 Helix 12 position and coregulator binding. TR C-terminal helix 12 is displaced away from the hydrophobic cleft without bound hormone, exposing a large hydrophobic cleft composed of residues form helices 3, 4, 5, and 6. This serves as interaction site for three-turn α-helical motifs from corepressors (IDs). With hormone, H12 packs over the lower part of the corepressor binding surface, blocking corepressor binding and creating a new binding site for coactivator NR boxes (LxxLL).
H12 position influences coregulator interactions.52 In the active position, H12 docks against the body of the LBD to form a surface-exposed hydrophobic cleft composed of residues from helices (H) 3, 4, 5, and 12. Saturation mutagenesis reveals that this surface corresponds to the hormone-dependent activation function, AF-2. Mutations in the AF-2 surface abolish LBD hormone-dependent transcriptional activity and interactions with coactivator proteins. AF-2 is not needed for heterodimer formation or for hormone binding, implying that these mutations in the AF-2 surface do not generally disrupt NR structure. Targeted mutation of LBD surface residues and examination of activities of the receptors in the unliganded state reveals that the corepressor binding surface overlaps with the upper part of AF-2 but also extends outside AF-2 and includes the region of the body of the LBD over which H12 is positioned in the active state. Thus, H12 is probably displaced in the absence of hormone, exposing an extended hydrophobic surface capable of corepressor binding, while hormone promotes H12 packing over the lower part of this surface, simultaneously inhibiting corepressor binding and completing the AF-2 surface.
TR Antagonists: The Extension Hypothesis
It has been possible to create TR antagonists based on x-ray structural analysis and knowledge of TR activation functions and interacting partners, outlined earlier.61 The “extension hypothesis” proposes that the design of agonist-like compounds with appropriately placed bulky extensions will prevent H12 from folding into the active conformation, thereby blocking coactivator binding (Fig. 5-14). Several competitive TR antagonists have been produced using the principles of this hypothesis. Moreover, later crystallization of estrogen-receptor LBDs in complex with the known antagonists tamoxifen and raloxifene revealed that both of these ligands possess extensions that displace H12.
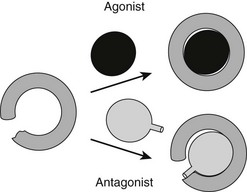
FIGURE 5-14 The extension hypothesis. Agonist binding promotes folding of the TR LBD around the ligand, thereby completing the coactivator binding surface shown in Fig. 5-13. Antagonists should comprise a hormone-like moiety that recognizes the hormone-binding pocket but would contain a bulky extension group that would prevent complete closing of the LBD and disturb the AF-2 surface.
Ligand Stabilizes LBD Structure
Hormone effects on TR LBD are probably more extensive than simple repositioning of H12.74–77 NR LBDs are somewhat mobile and unstable without ligand or association with cofactors, forming a so-called molten globular organization; hormone promotes widespread stabilization of domain structure.
The overall rearrangements that stabilize the domain may be functionally important. For TRs, homodimer interactions are inhibited by hormone in vitro, whereas RXR-TR LBD interactions are not. This implies that the homodimer and heterodimer interaction surfaces, though overlapping, are not identical, and that hormone promotes rearrangements in regions of the LBD surface that are specific for homodimer contact. Targeted mutagenesis suggests that conserved surface salt-bridge clusters within the TR H7-H8 region and H11 play a role in this effect.77 For other NRs, ligand binding leads to rearrangements in the same region of the LBD surface that expose a short peptide sequence that is a target for covalent attachment of SUMO peptides,54 an important event in some mechanisms of ligand-dependent repression, as described earlier. Finally, T3 promotes increased packing of H1 against the remaining LBD core (H2-H12 region). Remarkably, this can even be recapitulated in cis, using separate fragments for the H2-H12 and H1 region. Since H1 links the LBD to DBD, this event could conceivably communicate information about hormone binding to the DBD.
Mechanisms of Ligand Binding and Release
The ways ligand enters and leaves the buried TR hormone-binding pocket remain unknown.78,79 Hormone is completely enclosed within the domain, and entry/exit routes are not obvious from consideration of the structure alone. One possibility is that the major ligand entry and exit route lies under H12. In this model, referred to as the mousetrap model, H12 displacement in the absence of hormone opens the ligand entry route. Here, hormone-induced folding of H12 over the entry site traps ligand until H12 is displaced and the ligand is released. Interestingly, T4 displays a very high dissociation rate from TR LBDs, likely explaining low affinity for receptor. A crystal structure of the TR-LBD with T4 revealed that subtle rearrangements in side chains in the hormone-binding pocket create extra space to permit the receptor to accommodate the bulky 5′ iodine group, but that H12 packing is relatively inefficient. Inefficient H12 packing could increase T4 dissociation because it opens an exit route from the LBD.
Conformational Flexibility in The Hormone-Binding Pocket
A major surprise derived from recent x-ray and functional studies of TRs is that the TRβ LBD can adapt to bind a ligand that is much larger than classical thyroid hormones and still fold into an active conformation.76 A structure of TRβ with a synthetic agonist (GC-24) that contains a bulky 3′ phenyl extension reveals a different mechanism for adaptation from that described for T4 (Fig. 5-15). Here, helices 3 and 11 part to form an extension to the pocket.76 This phenomenon may be useful for drug design; GC-24 is highly TRβ selective, suggesting that it is possible to capitalize on TR isoform-specific differences in flexibility. The surprising implications of this finding are that TRs could bind and be activated by larger ligands than expected, although it is not clear whether natural ligands with appropriately placed extensions exist.
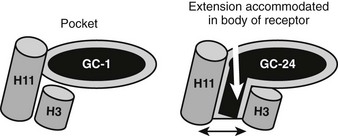
FIGURE 5-15 Plasticity of the TR hormone-binding pocket. Compounds such as T3, Triac, and GC-1 (an agonist that is the parental molecule for GC-24) are buried in the hormone-binding pocket, defined in the first TR x-ray structure. In this structure, H3 and H11 are linked. GC-24 resembles GC-1 but also contains an additional bulky 3′ hydrophobic group that should not be accommodated in the buried pocket. Here, an x-ray structure reveals that H3 and H11 part to form a hitherto unsuspected extension to the buried pocket. In spite of this significant rearrangement, the overall conformation of the LBD surface is mostly similar, and the AF-2 surface is active.
The reasons that the H3-H11 opens to extend the conventional hormone-binding pocket is not clear; one hypothesis is that the GC-24 extension is accommodated within a region of receptor that can open up to allow ligand exit under H12.76 There may be other regions of TR open to extend the pocket, as described earlier; some compounds with bulky 5′ extensions different from the GC-24 3′ extension can also act as agonists.
TR Interacting Proteins
Multiple TR interacting coactivators or corepressors have been identified (Fig. 5-16). The organization of these proteins is summarized briefly here. As already described, the liganded LBD recognizes coactivator NR box peptides (LxxLL), and the unliganded LBD binds IDs (CoRNR boxes, IxxIIxxLM). These motifs are often reiterated multiple times in the coregulator molecules. TRs exhibit clear preferences for certain coactivator and corepressor motifs (including the second NR box of the coactivator SRC2 [SRC2-2] and the first ID motif of N-CoR [ID3]).39,80,81 The structural basis for this preference is not clear but must involve sequences outside the box. The significance of the latter interaction was confirmed in a mouse knockout model in which the N-CoR exon overlapping ID3 was eliminated in liver; unliganded TR-dependent suppression of TR target genes was strongly affected.
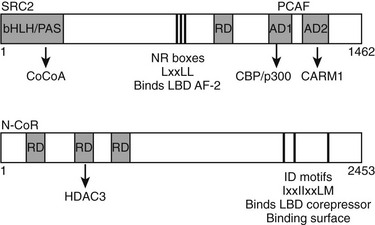
FIGURE 5-16 Schematic of coactivator and corepressor structure. Secondary structure of a representative coactivator (SRC2) and a corepressor (N-CoR) are shown. The positions of the NR interaction domains (inputs—NR boxes that bind TR AF-2 and IDs that bind the TR corepressor binding surface, respectively) are marked by thin black lines. The positions of independent domains that modulate gene expression (outputs) are also marked by gray boxes. These generally comprise binding sites for other factors, which are named beneath the diagram.
TRs and other NRs recruit multiple coactivators with different functions.82 Estrogen receptors recruit more than 60 different coactivators to target promoters, and it is likely that TRs will prove to bind a similar complement of proteins. The best known TR cofactors are the steroid-receptor coactivators (SRCs); these are implicated in chromatin modification, owing to their ability to recruit HATs such as p300 and pCAF and arginine methyltransferases such as CARM1. Other TR cofactors include the TRAP220/med1 subunit of the mediator complex, which contacts the basal transcription machinery, the metabolic coregulator PGC-1α, and many others. Some coactivators target TRs to protein degradation complexes, implying that TR turnover is an important component of transcriptional activation. Other coactivator complexes are implicated in modification of the basal transcription machinery that binds RNA polymerase, changes in RNA polymerase processivity, enzymatic modification of transcription factors, and alterations in RNA processing. Generally, individual coactivator complexes are recruited to target promoters sequentially with defined order and kinetics, implying that they must play different roles and function at different stages in transcriptional response.
As mentioned earlier, unliganded TRs interact with three known corepressors, N-CoR, SMRT, and alien.83 Like coactivators, these proteins form large complexes with auxiliary proteins; however, they serve to silence transcription. These auxiliary factors include HDACs, which condense local chromatin, enzymes which methylate DNA, a repressive promoter modification, and factors that target corepressors and associated factors to protein degradation complexes. The extent to which the three TR corepressors fulfill analogous functions is not clear. Targeted deletion of the N-CoR and SMRT genes leads to an embryonic-lethal phenotype, implying that one corepressor cannot compensate for the other.
There are examples of coactivators and corepressors that co-opt the classical binding mode of the other type of protein. Receptor-interacting protein 140 (RIP140) is a repressor with LxxLL motifs that binds to liganded nuclear receptors, including the TRs, and dampens their activity.84 This action is thought to repress NR activity in fat. RIP140 knockout mice are hypermetabolic and resistant to development of diet-induced obesity. Conversely, the adenoviral E1 protein is a coactivator that contains an ID/CoRnR box motif and binds unliganded TRs,85 and truncated versions of N-CoR that lack repression domains may fulfill a similar function.
Structure and Function of TR Isoforms
TRα1 and TRβ1 are similar in primary sequence (Fig. 5-17), structure, and mechanism of action, but they are not completely identical in function.2,86–88 Phenotypes of human patients with TRβ mutations that cause resistance to thyroid hormone syndrome (RTH) and mouse TR knockout models reveal that TRα and TRβ play distinct roles in T3 effects on different responses, genes, and tissues. These differential actions are often ascribed to differences in the receptor tissue distribution,20 but this is not proven. That TRα and TRβ may act differently to regulate some genes, which is also likely, also needs to be considered. In this regard, TRβ displays stronger activity at alternate inverted palindromic TREs than TRα, and this correlates with the extent of TR-TR homodimer binding to these elements. Moreover, TRs display different ligand preferences with TRβ binding to Triac, with about two- to threefold higher affinity than TRα. It is not clear whether these differences are functionally important, but the findings are suggestive of different mechanisms.
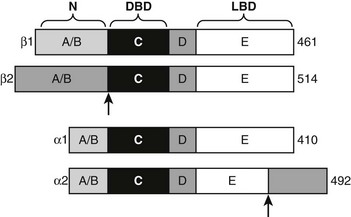
Alternate TR Splice Products
Both TR genes produce alternate protein products by combinations of differential exon splicing and promoter usage (see Fig. 5-17). The major differentially spliced THRA product is called TRα2. Here, the first 370 amino acids are identical to TRα, but an alternate C-terminus alters LBD sequences encoding H11 and H12, resulting in a TR that does not bind hormone. It is proposed that TRα2 can associate with wild-type TRs and block their actions in response to ligand (referred to as dominant inhibitor activity), especially in tissues where it is present in large amounts (e.g., brain, testis, kidney, and brown fat). TRα2 dampens T3 responses in transfection assays, and knockout mice that lack TRα2 display heightened thyroid hormone sensitivity in vivo. The mechanism of this dominant negative effect is unclear; many models of dominant negative activity require functional homodimer or heterodimer formation, but TRα2 appears to display reduced dimer formation through the altered LBD H10-H11 region that is reflected in reduced DNA binding of TRα2 relative to wild-type TRα.
Resistance to Thyroid Hormone
Inherited human TR mutations cause resistance to thyroid hormone syndrome (RTH).89 Symptoms of RTH, coupled with genetic analysis of TR mutations, yielded some of the first insights into differential actions of TRβ and TRα isoforms.
Patients with RTH exhibit reduced sensitivity to thyroid hormone.89 In most cases, circulating thyroid hormone levels are elevated to variable extents, but TSH levels (which should be suppressed by elevated hormone levels) are elevated (sometimes resulting in goiter) or normal. This is caused by failure of feedback inhibition of hormone synthesis through the hypothalamic-pituitary axis due to the reduced sensitivity of TRβ to thyroid hormone (see Fig. 5-1). Overall, the elevated thyroid hormone levels overcome the resistance and produce a mostly euthyroid state. (In some cases, a variant of the syndrome results in central but not peripheral resistance to thyroid hormone, where TSH levels are high, and there can be symptoms of frank hyperthyroidism.) However, there can also be a mix of hypo- and hyperthyroid phenotypes of variable clinical penetrance. Commonly there is tachycardia (hyperthyroid symptom) and attention deficit. To a lesser extent, there can be some mild mental retardation, skeletal malformations, and hearing abnormalities (hypothyroid manifestations). Most (>80%) RTH is caused by mutations in the LBD coding regions of the TRβ gene. Mutations in TRα have never been detected. The fact that there is elevated heart rate therefore suggested that normal TRα is responsible for this hyperthyroid effect and provided the first evidence for TR isoform–specific activities, later confirmed by gene knockout studies.
RTH can also be caused by mutations in other genes involved that play roles in thyroid hormone response. Cases have been described in which the target genes identified involve a thyroid hormone membrane transporter and enzymes required for transfer of an essential cofactor, selenium, to the active site of deiodinases.90
Analysis of TR Isoform Action in Mice
Functions of TR isoforms have been investigated in mouse models by targeted deletion of TR genes and knock-in of TR alleles with dominant negative activities.18,88,91,92
TR Knockouts
1. Mice that lack TRs (TRα0/0β−/−) fail to mount known thyroid hormone responses. This is interpreted as strong evidence that nuclear TRs mediate many known effects of thyroid hormones.
2. The mice exhibit clear developmental defects (such as dwarfism) and very high circulating thyroid hormone levels. Nevertheless, the overall phenotype is relatively mild compared to hypothyroid mice. This suggests that most harmful effects of hypothyroidism stem from unbalanced actions of unliganded TR isoforms, rather than lack of receptors per se, and emphasize the potent actions of unliganded TRs.
3. Deletions that target TRα or TRβ yield different phenotypes. TRα sets heart rate, in agreement with observed phenotypes of humans with RTH mutations. TRα is also involved in growth and development of the skeleton, muscle, and small intestine and plays an important role in body temperature regulation and setting metabolic rate. Conversely, TRβ plays major roles in regulation of serum cholesterol levels and feedback inhibition of thyroid hormone production and significant roles in regulating metabolic rate by directly inducing mitochondrial uncoupling proteins. Some genes respond selectively to particular TR isoforms. TRβ1 is needed for induction of spot 14, CYP7A1, Dio1, malic enzyme, and other liver-specific genes. Conversely, TRα1, but not TRβ1, is needed for induction of SERCa1 in muscle. Conceivably, differential actions could either reflect differences in TR tissue distributions or fundamental differences in mechanism of TRα1 and TRβ1. Evidence in favor of the latter possibility is that TRβ−/−TRα2−/− mice exhibit elevated expression of TRα1 in liver such that total liver T3-binding activity approaches wild-type levels, but this is not sufficient to rescue defects in T3 induction of CYP7A1.
4. Alternate spliced TR isoforms play specific roles. As expected from the tissue distribution of TRβ2, this form of TR mediates feedback inhibition of thyroid hormone production through the hypothalamic-pituitary axis. TRα2−/− mice exhibit features of hyperthyroidism, including elevated heart rate, weight loss, and elevated body temperature. Consideration of phenotypic differences of TRα0/0 (no TRα isoforms expressed) and TRα−/− mice (TRΔα1 and TRΔα2 expressed) reveal that some thyroid hormone responses are preserved in the intestine of the latter strain of mice.
TR Mutations in Disease: Structure-Function
Cancer
Cancer genome sequencing analysis has revealed that TR mutations occur frequently in cancer (including liver, kidney, thyroid gland, and breast).93 Presently, the significance of these mutations in cancer etiology is not completely clear, but it is well established that mutated TRs can cause cancer; as described earlier, v-erbA is a non-hormone-binding form of TRα that actively represses transcription and causes erythroblastoma.51 Moreover, mouse models which contain a targeted mutation of TRβ (knock-in) that resembles the human PV RTH mutant develop thyroid cancer at increased rates.
Gene and Context-Specific Variations in TR Actions
There are variations in the mechanism of thyroid hormone response at different target genes.2,38 Comparisons of gene expression patterns in livers of wild-type thyroidectomized and thyroid hormone–treated mice with similar TR−/− knockout mice reveals that most positively regulated genes adhere to the classic model of thyroid hormone action; transcription is suppressed by unliganded TRs and activated beyond basal levels with hormone. However, many genes deviate from this pattern (Fig. 5-18). Some are not suppressed by unliganded TRs but continue to be activated by the hormone, whereas others are suppressed by unliganded TRs, with hormone only partly relieving basal suppression. Similar variations are observed with negatively regulated genes.38 Some genes are activated by unliganded TRs with hormone-suppressing transcription below basal levels, while others are not activated by unliganded TRs but continue to be repressed by hormone. It is tempting to suggest that these variations in transcriptional response must reflect promoter-specific differences in corepressor/coactivator function or recruitment patterns, but this is not proven.
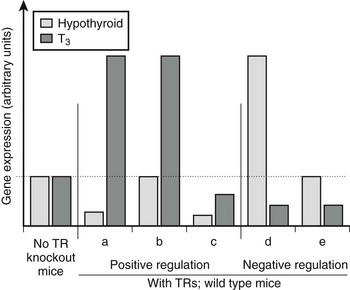
FIGURE 5-18 Differences in patterns of thyroid hormone regulation. Observed patterns of TR-responsive gene expression in knockout mice. Most positively regulated genes conform to pattern a; they are suppressed without ligand via corepressor recruitment and activated with ligand via coactivator recruitment. Other positively regulated genes differ and conform to patterns b and c; some lack basal repression, others are not activated beyond basal levels in response to hormone binding. Negatively regulated genes show similar variations. It is likely that different patterns correspond to variations in the mechanism of TR action at different genes.
Alternative Modes of TH Action
While most effects of thyroid hormone are mediated by nuclear TRs, there are some cases in which the hormones can exert rapid effects on cells that are unlikely to involve nuclear TRs.94,95 There are two possible mechanisms of these effects. It is now known that conventional nuclear TRs circulate in and out of the nucleus, and cytoplasmic TRs could mediate rapid effects of thyroid hormones. Alternatively, there may be alternate proteins that bind thyroid hormones and transduce thyroid hormone signals. Both mechanisms appear to be at play for thyroid hormones.
Selective Modulation
Excess TH production by the thyroid gland can produce beneficial effects, including reductions in serum LDL cholesterol and body fat.96 However, hyperthyroid patients also exhibit tachycardia (elevated heart rate) with atrial arrhythmias and can develop congestive heart failure. TH excess also causes muscle wasting, osteoporosis in postmenopausal women, and other symptoms, including fatigue, shortness of breath, and temperature sensitivity.97
Scientists have often suggested that TH derivatives could separate beneficial effects of TH from deleterious effects. Early human trials with dextrothyroxine (D-T4, the D-enantiomer of T4 that was contaminated also by small amounts of L-T4 that may have contributed to both beneficial and harmful effects) were performed as part of the coronary drug project (CDP) in the late 1960s to determine whether different compounds improved survival in men who had suffered one heart attack.98 This arm of the study was curtailed because of increased mortality, even though serum cholesterol was reduced. Other human trials involved Triac, which exhibited a small therapeutic window where beneficial effects on serum cholesterol are seen without increases in heart rate. Other groups identified synthetic TH analogs that reduce cholesterol without effects on heart, but these were not pursued for human use. In the last 10 to 15 years, improved understanding of TR structure and function and TH analog chemistry has led to creation of new, potent thyromimetics with clearer selective activities. Since analysis of RTH and mouse gene knockout models described previously revealed that TRα mediates tachycardia, several groups set out to create analogs that would selectively activate TRβ and promote the beneficial effects of TH, such as the lowering of serum cholesterol, while sparing the heart.96,99–101 Tests in animal models revealed that they also display liver uptake selectivity and vary in their abilities to accumulate in extrahepatic tissues, complicating interpretations of their profile of actions and side effects. Nevertheless, several compounds have shown promising effects on serum lipids and obesity in preclinical animal models and on serum lipids in early human clinical trials15 without obvious harmful effects on heart, muscle, or bone. In addition, some of the drugs cause rapid loss in body weight and white fat and potentially beneficial reductions in blood glucose. These compounds, and future derivatives, will improve treatments of thyroid hormone imbalances and could represent novel treatments for metabolic disease in euthyroid subjects.
References
1. Sawin, CT. The heritage of the thyroid: A brief history. In: Braverman LE, Utiger RD, Ingbar SH, Werner SC, eds. Werner and Ingbar’s The Thyroid: A Fundamental and Clinical Text. ed 9. Philadelphia: Lippincott Williams & Wilkins; 2000:3–7.
2. Yen, PM. Physiological and molecular basis of thyroid hormone action. Physiol Rev. 2001;81:1097–1142.
3. Zhang, J, Lazar, MA. The mechanism of action of thyroid hormones. Annu Rev Physiol. 2000;62:439–466.
4. Laudet, V, Gronemeyer, H. The Nuclear Receptor Facts Book. London: Academic Press; 2002.
5. Flamant, F, et al. International Union of Pharmacology. LIX. The pharmacology and classification of the nuclear receptor superfamily: thyroid hormone receptors. Pharmacol Rev. 2006;58:705–711.
6. Leonard, JL, Koehrle, J. Intracellular pathways of iodothyronine metabolism. In: Braverman LE, Utiger RD, Ingbar SH, et al, eds. Werner and Ingbar’s the thyroid: a fundamental and clinical text. ed 9. Philadelphia: Lippincott Williams & Wilkins; 2000:136–173.
7. Panicker, V, et al. A common variation in deiodinase 1 gene DIO1 is associated with the relative levels of free thyroxine and triiodothyronine. J Clin Endocrinol Metab. 2008;93:3075–3081.
8. Panicker, V, et al. Genetic loci linked to pituitary-thyroid axis set points: a genome-wide scan of a large twin cohort. J Clin Endocrinol Metab. 2008;93:3519–3523.
9. Sorensen, HG, et al. Identification and consequences of polymorphisms in the thyroid hormone receptor alpha and beta genes. Thyroid. 2008;18:1087–1094.
10. Scanlan, TS, et al. 3-Iodothyronamine is an endogenous and rapid-acting derivative of thyroid hormone. Nat Med. 2004;10:638–642.
11. Visser, WE, Friesema, EC, Jansen, J, et al. Thyroid hormone transport in and out of cells. Trends Endocrinol Metab. 2008;19:50–56.
12. Friesema, EC, et al. Effective cellular uptake and efflux of thyroid hormone by human monocarboxylate transporter 10. Mol Endocrinol. 2008;22:1357–1369.
13. Dumitrescu, AM, Refetoff, S. Novel biological and clinical aspects of thyroid hormone metabolism. Endocr Dev. 2007;10:127–139.
14. Suzuki, S, Mori, J, Hashizume, K. mu-crystallin, a NADPH-dependent T(3)-binding protein in cytosol. Trends Endocrinol Metab. 2007;18:286–289.
15. Berkenstam, A, et al. The thyroid hormone mimetic compound KB2115 lowers plasma LDL cholesterol and stimulates bile acid synthesis without cardiac effects in humans. Proc Natl Acad Sci U S A. 2008;105:663–667.
16. Gereben, B, et al. Cellular and Molecular Basis of Deiodinase-Regulated Thyroid Hormone Signaling. Endocr Rev. 2008.
17. Huang, SA, Bianco, AC. Reawakened interest in type III iodothyronine deiodinase in critical illness and injury. Nat Clin Pract Endocrinol Metab. 2008;4:148–155.
18. Moreno, M, et al. Metabolic effects of thyroid hormone derivatives. Thyroid. 2008;18:239–253.
19. Sherman, SI, et al. Augmented hepatic and skeletal thyromimetic effects of tiratricol in comparison with levothyroxine. J Clin Endocrinol Metab. 1997;82:2153–2158.
20. Bookout, AL, et al. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126:789–799.
21. Silva, JE. Thermogenic mechanisms and their hormonal regulation. Physiol Rev. 2006;86:435–464.
22. Silva, JE, Bianco, SD. Thyroid-adrenergic interactions: physiological and clinical implications. Thyroid. 2008;18:157–165.
23. Videla, LA, Fernandez, V, Tapia, G, et al. Thyroid hormone calorigenesis and mitochondrial redox signaling: upregulation of gene expression. Front Biosci. 2007;12:1220–1228.
24. Kim, B. Thyroid hormone as a determinant of energy expenditure and the basal metabolic rate. Thyroid. 2008;18:141–144.
25. Feng, X, Jiang, Y, Meltzer, P, et al. Thyroid hormone regulation of hepatic genes in vivo detected by complementary DNA microarray. Mol Endocrinol. 2000;14:947–955.
26. Weitzel, JM, et al. Hepatic gene expression patterns in thyroid hormone-treated hypothyroid rats. J Mol Endocrinol. 2003;31:291–303.
27. Obregon, MJ. Thyroid hormone and adipocyte differentiation. Thyroid. 2008;18:185–195.
28. Klein, I, Danzi, S. Thyroid disease and the heart. Circulation. 2007;116:1725–1735.
29. Wiersinga, WM. The role of thyroid hormone nuclear receptors in the heart: evidence from pharmacological approaches. Heart Fail Rev. 2008 Dec 19.
30. Crunkhorn, S, Patti, ME. Links between thyroid hormone action, oxidative metabolism, and diabetes risk? Thyroid. 2008;18:227–237.
31. Bassett, JH, Williams, GR. Critical role of the hypothalamic-pituitary-thyroid axis in bone. Bone. 2008;43:418–426.
32. Samuels, MH. Cognitive function in untreated hypothyroidism and hyperthyroidism. Curr Opin Endocrinol Diabetes Obes. 2008;15:429–433.
33. Williams, GR. Neurodevelopmental and neurophysiological actions of thyroid hormone. J Neuroendocrinol. 2008;20:784–794.
34. Plateroti, M, Kress, E, Mori, JI, et al. Thyroid hormone receptor alpha1 directly controls transcription of the beta-catenin gene in intestinal epithelial cells. Mol Cell Biol. 2006;26:3204–3214.
35. Doshi, DN, Blyumin, ML, Kimball, AB. Cutaneous manifestations of thyroid disease. Clin Dermatol. 2008;26:283–287.
36. Baxter, JD, et al. Thyroid hormone receptors and responses. Recent Prog Horm Res. 1979;35:97–153.
37. Ivarie, RD, Baxter, JD, Morris, JA. Interaction of thyroid and glucocorticoid hormones in rat pituitary tumor cells. Specificity and diversity of the responses analyzed by two-dimensional gel electrophoresis. J Biol Chem. 1981;256:4520–4528.
38. Yen, PM, Feng, X, Flamant, F, et al. Effects of ligand and thyroid hormone receptor isoforms on hepatic gene expression profiles of thyroid hormone receptor knockout mice. EMBO Rep. 2003;4:581–587.
39. Astapova, I, et al. The nuclear corepressor, NCoR, regulates thyroid hormone action in vivo. Proc Natl Acad Sci U S A. 2008;105:19544–19549.
40. van Rooij, E, et al. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–579.
41. Tata, JR, et al. The action of thyroid hormones at the cell level. Biochem J. 1963;86:408–428.
42. Oppenheimer, JH, Schwartz, HL, Surks, MI. Tissue differences in the concentration of triiodothyronine nuclear binding sites in the rat: liver, kidney, pituitary, heart, brain, spleen, and testis. Endocrinology. 1974;95:897–903.
43. Surks, MI, Koerner, DH, Oppenheimer, JH. In vitro binding of L-triiodothyronine to receptors in rat liver nuclei. Kinetics of binding, extraction properties, and lack of requirement for cytosol proteins. J Clin Invest. 1975;55:50–60.
44. Nelson, C, et al. Discrete cis-active genomic sequences dictate the pituitary cell type-specific expression of rat prolactin and growth hormone genes. Nature. 1986;322:557–562.
45. MacLeod, KN, Baxter, JD. DNA binding of thyroid hormone receptors. Biochem Biophys Res Commun. 1975;62:577–583.
46. Spindler, BJ, MacLeod, KM, Ring, J, et al. Thyroid hormone receptors. Binding characteristics and lack of hormonal dependency for nuclear localization. J Biol Chem. 1975;250:4113–4119.
47. Latham, KR, Ring, JC, Baxter, JD. Solubilized nuclear “receptors” for thyroid hormones. Physical characteristics and binding properties, evidence for multiple forms. J Biol Chem. 1976;251:7388–7397.
48. MacLeod, KM, Baxter, JD. Chromatin receptors for thyroid hormones. Interactions of the solubilized proteins with DNA. J Biol Chem. 1976;251:7380–7387.
49. Sap, J, et al. The c-erb-A protein is a high-affinity receptor for thyroid hormone. Nature. 1986;324:635–640.
50. Weinberger, C, et al. The c-erb-A gene encodes a thyroid hormone receptor. Nature. 1986;324:641–646.
51. Privalsky, ML. v-erb A, nuclear hormone receptors, and oncogenesis. Biochim Biophys Acta. 1992;1114:51–62.
52. Glass, CK, Rosenfeld, MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14:121–141.
53. Lazar, MA. Nuclear receptor corepressors. Nucl Recept Signal. 2003;1:e001.
54. Ricote, M, Glass, CK. PPARs and molecular mechanisms of transrepression. Biochim Biophys Acta. 2007;1771:926–935.
55. Shibusawa, N, Hollenberg, AN, Wondisford, FE. Thyroid hormone receptor DNA binding is required for both positive and negative gene regulation. J Biol Chem. 2003;278:732–738.
56. Rastinejad, F, Perlmann, T, Evans, RM, et al. Structural determinants of nuclear receptor assembly on DNA direct repeats. Nature. 1995;375:203–211.
57. Khorasanizadeh, S, Rastinejad, F. Nuclear-receptor interactions on DNA-response elements. Trends Biochem Sci. 2001;26:384–390.
58. Desvergne, B. How do thyroid hormone receptors bind to structurally diverse response elements? Mol Cell Endocrinol. 1994;100:125–131.
59. Wagner, RL, et al. A structural role for hormone in the thyroid hormone receptor. Nature. 1995;378:690–697.
60. Wagner, RL, et al. Hormone selectivity in thyroid hormone receptors. Mol Endocrinol. 2001;15:398–410.
61. Webb, P, et al. Design of thyroid hormone receptor antagonists from first principles. J Steroid Biochem Mol Biol. 2002;83:59–73.
62. Weatherman, RV, Fletterick, RJ, Scanlan, TS. Nuclear-receptor ligands and ligand-binding domains. Annu Rev Biochem. 1999;68:559–581.
63. Nascimento, AS, et al. Structural rearrangements in the thyroid hormone receptor hinge domain and their putative role in the receptor function. J Mol Biol. 2006;360:586–598.
64. Kumar, R, Thompson, EB. Transactivation functions of the N-terminal domains of nuclear hormone receptors: protein folding and coactivator interactions. Mol Endocrinol. 2003;17:1–10.
65. Figueira, AC, et al. Low-resolution structures of thyroid hormone receptor dimers and tetramers in solution. Biochemistry. 2007;46:1273–1283.
66. Chandra, V, et al. Structure of the intact PPAR-gamma-RXR-nuclear receptor complex on DNA. Nature. 2008;456:350–356.
67. Ribeiro, RC, et al. X-ray crystallographic and functional studies of thyroid hormone receptor. J Steroid Biochem Mol Biol. 1998;65:133–141.
68. Ribeiro, RC, et al. Definition of the surface in the thyroid hormone receptor ligand binding domain for association as homodimers and heterodimers with retinoid X receptor. J Biol Chem. 2001;276:14987–14995.
69. Velasco, LF, et al. Thyroid hormone response element organization dictates the composition of active receptor. J Biol Chem. 2007;282:12458–12466.
70. Wu, Y, Xu, B, Koenig, RJ. Thyroid hormone response element sequence and the recruitment of retinoid X receptors for thyroid hormone responsiveness. J Biol Chem. 2001;276:3929–3936.
71. Diallo, EM, Wilhelm, KG, Jr., Thompson, DL, et al. Variable RXR requirements for thyroid hormone responsiveness of endogenous genes. Mol Cell Endocrinol. 2007;264:149–156.
72. Mengeling, BJ, Lee, S, Privalsky, ML. Coactivator recruitment is enhanced by thyroid hormone receptor trimers. Mol Cell Endocrinol. 2008;280:47–62.
73. Kallenberger, BC, Love, JD, Chatterjee, VK, et al. A dynamic mechanism of nuclear receptor activation and its perturbation in a human disease. Nat Struct Biol. 2003;10:136–140.
74. Pissios, P, Tzameli, I, Kushner, P, et al. Dynamic stabilization of nuclear receptor ligand binding domains by hormone or corepressor binding. Mol Cell. 2000;6:245–253.
75. Pissios, P, Tzameli, I, Moore, DD. New insights into receptor ligand binding domains from a novel assembly assay. J Steroid Biochem Mol Biol. 2001;76:3–7.
76. Togashi, M, et al. Conformational adaptation of nuclear receptor ligand binding domains to agonists: potential for novel approaches to ligand design. J Steroid Biochem Mol Biol. 2005;93:127–137.
77. Togashi, M, Nguyen, P, Fletterick, R, et al. Rearrangements in thyroid hormone receptor charge clusters that stabilize bound 3,5′,5-triiodo-L-thyronine and inhibit homodimer formation. J Biol Chem. 2005;280:25665–25673.
78. Martinez, L, et al. Molecular dynamics simulations reveal multiple pathways of ligand dissociation from thyroid hormone receptors. Biophys J. 2005;89:2011–2023.
79. Martinez, L, Webb, P, Polikarpov, I, et al. Molecular dynamics simulations of ligand dissociation from thyroid hormone receptors: evidence of the likeliest escape pathway and its implications for the design of novel ligands. J Med Chem. 2006;49:23–26.
80. Moore, JM, et al. Quantitative proteomics of the thyroid hormone receptor-coregulator interactions. J Biol Chem. 2004;279:27584–27590.
81. Webb, P, et al. The nuclear receptor corepressor (N-CoR) contains three isoleucine motifs (I/LXXII) that serve as receptor interaction domains (IDs). Mol Endocrinol. 2000;14:1976–1985.
82. Lonard, DM, O’Malley, BW. Nuclear receptor coregulators: judges, juries, and executioners of cellular regulation. Mol Cell. 2007;27:691–700.
83. Privalsky, ML. The role of corepressors in transcriptional regulation by nuclear hormone receptors. Annu Rev Physiol. 2004;66:315–360.
84. White, R, et al. Role of RIP140 in metabolic tissues: connections to disease. FEBS Lett. 2008;582:39–45.
85. Meng, X, et al. E1A and a nuclear receptor corepressor splice variant (N-CoRI) are thyroid hormone receptor coactivators that bind in the corepressor mode. Proc Natl Acad Sci U S A. 2005;102:6267–6272.
86. Lazar, MA. Thyroid hormone receptors: multiple forms, multiple possibilities. Endocr Rev. 1993;14:184–193.
87. O’Shea, PJ, Williams, GR. Insight into the physiological actions of thyroid hormone receptors from genetically modified mice. J Endocrinol. 2002;175:553–570.
88. Forrest, D, Vennstrom, B. Functions of thyroid hormone receptors in mice. Thyroid. 2000;10:41–52.
89. Yen, PM. Molecular basis of resistance to thyroid hormone. Trends Endocrinol Metab. 2003;14:327–333.
90. Refetoff, S, Dumitrescu, AM. Syndromes of reduced sensitivity to thyroid hormone: genetic defects in hormone receptors, cell transporters and deiodination. Best Pract Res Clin Endocrinol Metab. 2007;21:277–305.
91. Liu, YY, et al. A mutant thyroid hormone receptor alpha antagonizes peroxisome proliferator-activated receptor alpha signaling in vivo and impairs fatty acid oxidation. Endocrinology. 2007;148:1206–1217.
92. Sjogren, M, et al. Hypermetabolism in mice caused by the central action of an unliganded thyroid hormone receptor alpha1. EMBO J. 2007;26:4535–4545.
93. Yen, PM, Cheng, SY. Germline and somatic thyroid hormone receptor mutations in man. J Endocrinol Invest. 2003;26:780–787.
94. Davis, PJ, Leonard, JL, Davis, FB. Mechanisms of nongenomic actions of thyroid hormone. Front Neuroendocrinol. 2008;29:211–218.
95. Furuya, F, Lu, C, Guigon, CJ, et al. Nongenomic activation of phosphatidylinositol 3-kinase signaling by thyroid hormone receptors. Steroids. 2009;74:628–634.
96. Webb, P. Selective activators of thyroid hormone receptors. Expert Opin Investig Drugs. 2004;13:489–500.
97. Biondi, B, Cooper, DS. The clinical significance of subclinical thyroid dysfunction. Endocr Rev. 2008;29:76–131.
98. The coronary drug project. Findings leading to further modifications of its protocol with respect to dextrothyroxine. The coronary drug project research group. JAMA. 1972;220:996–1008.
99. Baxter, JD, et al. Selective modulation of thyroid hormone receptor action. J Steroid Biochem Mol Biol. 2001;76:31–42.
100. Baxter, JD, Webb, P, Grover, G, et al. Selective activation of thyroid hormone signaling pathways by GC-1: a new approach to controlling cholesterol and body weight. Trends Endocrinol Metab. 2004;15:154–157.
101. Scanlan, TS. Sobetirome: a case history of bench-to-clinic drug discovery and development. Heart Fail Rev. 2008 Nov 11.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 5
Mechanisms of Thyroid Hormone Action
Synthesis and Control of Secretion of Thyroid Hormones
Thyroid Hormone Action in the Periphery
Accumulation of Thyroid Hormone in Cells
Deiodinases in Thyroid Hormone Activation and Inactivation
Alternate Metabolic Fates of Thyroid Hormone: Inactive and Active Derivatives
History of the Mechanism of Action of Thyroid Hormones
Discovery of Thyroid Hormone Receptors and Their Mechanism of Action
Discovery That Thyroid Hormones Regulate Specific mRNAs and Gene Transcription
Discovery That TRs Bind Preferentially to Active Chromatin and Specific DNA Sequences
Thyroid Hormone Receptors: Mechanism
Receptor Structure and Function
Hormone Effects on Receptor Structure
LBD H12 Position and Coregulator Interactions
TR Antagonists: The Extension Hypothesis
Other Influences of H12 Rearrangement
Ligand Stabilizes LBD Structure
Structure and Function of TR Isoforms
TR Mutations in Disease: Structure-Function
Gene and Context-Specific Variations in TR Actions
The award of the 1909 Nobel Prize to Theodore Kocher for pioneering work on thyroid gland surgery lent timely recognition to the field of the thyroid gland and its actions, an area of study that was already several centuries in the making.1 Several Renaissance scholars, including Leonardo da Vinci, contributed to the early description of the thyroid gland and the realization that its swelling led to goiter. Later investigators linked goiter and cretinism to dietary deficiency of iodine (a key component of thyroid hormone) and uncovered links between altered thyroid gland function and apparently unconnected symptoms of hyperthyroidism (thyrotoxicosis or Graves’ disease) and hypothyroidism (or myxedema). Classic experiments of Murray in 1891 revealed that sheep thyroid gland extracts could be used to treat, and even cure, myxedema. Later searches for the active principle culminated in the isolation of thyroxine (T4; 3,5,3′,5′-tetra-iodo-l-thyronine) in 1914 by Kendall (who later, with others, received the Nobel Prize for discovering the therapeutic effects of the adrenal steroid cortisone on rheumatoid arthritis), the description of thyroxine structure and chemical synthesis by Harrington in the 1920s and the discovery of the major active form of thyroid hormone, triiodothyronine (T3; 3,5,3′-tri-iodo-l-thyronine), by Pitt-Rivers about 3 decades later. It is now known that thyroid hormones influence virtually every tissue and cell type; they play important roles in growth, development, and differentiation in fetal life and early childhood and act as master regulators of multiple metabolic processes in the adult. The past 50 years have witnessed great increases in our knowledge of thyroid hormone action, with the last 2 decades in particular producing very rapid advancements in understanding the functions of thyroid hormone receptors (TRs), members of the nuclear hormone receptor (NR) family. In this chapter, we review current knowledge of the mechanisms of thyroid hormone action.2–5
Synthesis and Control of Secretion of Thyroid Hormones
1. Spontaneous release of hormones from the hypothalamus and pituitary that stimulate thyroid hormone production
2. Feedback inhibition of production and release of these hypothalamic and pituitary hormones
3. Rates of intracellular conversion of T4 to T3
4. Metabolic destruction of both of these forms of the hormone6
Thyroid hormones are derivatives of tyrosine that are produced in the thyroid gland and secreted into the general circulation. Thyroid hormone synthesis occurs via iodination of tyrosine residues in a large protein called thyroglobulin, coupling of iodinated tyrosines, and enzymatic cleavage to liberate free thyroxine (T4). Recent human genetic linkage studies suggest that regulation of thyroid hormone production is complex, with up to eight different loci influencing biological set-point.7–9 Some of these probably correspond to known genes involved in synthesis of thyroid hormones and feedback inhibition of thyroid hormone production (see later discussion), but most of these loci are not identified.
T4 is the major form of thyroid hormone secreted from the gland, and much of this T4 is converted to T3 in peripheral tissues. Since T3 binds to TRs with higher affinity than T4, it is thought T3 is the major active form of thyroid hormone. Smaller amounts of alternate forms of thyroid hormone are also made in, and secreted by, the thyroid gland. These products are metabolites of thyroxine and include T3, reverse T3 (rT3), and others. Overall amounts of secreted T4 are much higher (more than 20-fold) than any of these alternate forms of thyroid hormone. The thyroid gland may also produce thyronamines (discussed in more detail later10), compounds that may constitute part of a completely distinct arm of the thyroid hormone signaling system; circulating levels of the most potent currently known thyronamine (3-iodothyronamine) appear higher than those of T3.
The negative feedback loop that controls thyroid hormone levels works through the hypothalamus and pituitary (hypothalamic-pituitary-thyroid [HPT] axis (Fig. 5-1). Thyrotropin-releasing hormone (TRH) is released from the hypothalamus and stimulates the synthesis and release of thyroid-stimulating hormone (TSH) by thyrotrophic cells in the pituitary gland. TSH is released into the circulation, where it stimulates the synthesis and release of T4 and to a lesser extent, T3. The circulating thyroid hormones block release of both TRH and TSH through repression of the transcription of genes that encode the TRH pre-pro-hormone and the α and β subunits of TSH and also by down-regulating TRH receptors in the pituitary. Together, these actions result in repression of hormone synthesis and release in response to high circulating levels of T4 and T3. This process helps maintain thyroid hormone levels within defined limits.
FIGURE 5-1 Regulation of thyroid hormone production in the hypothalamic-pituitary axis. Thyroid hormone is produced in the thyroid gland under control of thyroid-stimulating hormone (TSH). Circulating hormones exert multiple effects on different tissues in the periphery and also feed back to inhibit hypothalamic signals that stimulate TSH release and pituitary production of TSH, thereby maintaining thyroid hormone levels in defined limits.
Thyroid Hormone Action in the Periphery
Both major forms of thyroid hormone (T4 and T3) are transported in the circulation in complex with plasma proteins (Fig. 5-2). The ratio of total T4 to T3 in plasma is about 60:1. This is higher than the 20-fold ratio of T4 to T3 that is initially secreted by the thyroid gland because of greater plasma binding of T4 versus T3, resulting in greater clearance rates of T3. About 99.98% of total circulating T4 and 99.7% of T3 form noncovalent interactions with serum proteins, mostly to thyroxine-binding globulin but to a lesser extent to thyroxine-binding prealbumin, albumin and lipoproteins. The fact that less T3 circulates in complex with plasma proteins means that the ratio of free T4 to T3 is around three- to sixfold, with typical circulating free hormone levels around 20 picomolar (pM) T4 and 6 pM T3, respectively. The free fraction is biologically active and can enter target tissues. Thus, interactions with plasma binding proteins help to ensure even hormone delivery throughout the body.
FIGURE 5-2 Peripheral actions of thyroid hormone. The figure summarizes possible fates of thyroid hormone in the periphery. Hormones are mostly transported as complexes with serum binding globulins with limited amounts of free T4 and T3. Hormones are actively taken up into cells by facilitated transport mechanisms, where they undergo several different fates. The upper cell represents possible intracellular fates of T3; it can be sequestered in complexes with CTBP dimers, undergo conversion to inactive metabolites, or enter the nucleus to interact with TRs. The lower cell represents fates of T4, which is either activated by metabolic modification to form T3 or converted to inactive metabolites. It should be noted that expression of D1 and D2 is under transcriptional control, making this step an important control point in thyroid hormone response.
Import and Export
Thyroid hormone entry into cells is mediated by specific transporters (see Fig. 5-2).11–13 While early models suggested that lipophilic thyroid hormone molecules enter cells by diffusion across the plasma membrane, and this may occur to some extent, it is now clear that T4 and T3 entry mostly involves facilitated transport, a form of passive diffusion that requires membrane transport proteins.
Accumulation of Thyroid Hormone in Cells
T3 binds with nanomolar affinity to an intracellular protein that was originally called cytoplasmic T3-binding protein (CTBP).14 This protein is found at high abundance in the kidney and, to a lesser extent, in liver. Because CTBP was later found to be homologous to mu-crystallin, a protein abundant in the lens of the kangaroo eye, the gene is now often called CRYM. Overexpression of CRYM in stable cell lines increases maximal cellular T3 binding capacity and decreases T3 efflux rates. However, these effects are coupled to reduced transcriptional responses to T3, suggesting that CRYM sequesters T3 in an inactive cytoplasmic complex, away from nuclear TRs.
CRYM is important for thyroid hormone action in vivo. Mice with a targeted CRYM gene deletion appear normal but exhibit moderate decreases in circulating T4 (25%) and T3 (13%) levels and perhaps more importantly, also exhibit extremely rapid rates of T3 entry into and escape from target tissues, presumably because there is no CRYM to sequester cytoplasmic hormone from export mechanisms. Human patients with a natural CRYM mutation that blocks its ability to bind T3 (K314T) are deaf, a known consequence of defective thyroid hormone signaling during development. Interestingly, CRYM requires NADPH for dimerization and T3 binding (Fig. 5-3). As NADPH levels are reflective of levels of cellular reducing power and anabolic capacity, it is conceivable that CRYM may sequester or release T3 in response to alterations in cellular metabolic status. This notion is attractive but not proven.
FIGURE 5-3 Thyroid hormone interactions with CTBP. Intracellular T3 can bind CTBP dimers and become sequestered in the cytoplasm. CTBP dimer formation is sensitive to intracellular NADPH/NADP+ concentrations such that free hormone will probably be released in response to reductions of NADPH levels.
There could be other T3-binding proteins in addition to CRYM and the nuclear TRs (which are described later). Radioisotope-labeled T3 interacts with proteins in the endoplasmic reticulum, mitochondrion, and nuclear envelope.2 These putative T3-binding proteins have not been identified, and the functional significance of these interactions is unknown.
Deiodinases in Thyroid Hormone Activation and Inactivation
The thyroid gland produces mostly thyroxine. Limited amounts of T3 and rT3 are secreted by the gland, but more than 80% of both these forms of thyroid hormone are produced in the periphery via actions of specific deiodinases, D1, D2, and D3.15,16 D1 and D2 remove the iodine group from the 5′ position of the outer thyronine ring. As T3 binds to TRs with higher affinity than T4, the actions of D1 and D2 deiodinases serve to increase thyroid hormone activity through generation of T3, and this represents a step-up in activity. Conversely, the action of D3 decreases hormone activity and represents a step down by removing the iodine group from the 5 position of the inner thyronine ring and converting T4 and T3 to rT3 and 3,3′-T2, respectively, neither of which interacts substantially with nuclear TRs at physiologic concentrations. Changes in deiodinase expression and activity are important control points in thyroid hormone signaling; they influence the production and availability of the biologically active form of thyroid hormone, T3.
D3 is important for clearance of plasma T3.17 It is highly expressed in human placenta, where it is thought to be important for protection of the developing fetus from effects of maternal thyroid hormones. D3 is also highly expressed in the brain and skin. Increased D3 levels may account for reductions of circulating thyroid hormone levels that are often observed in critically ill patients and states of inflammation. Finally, D3 is often overexpressed in vascular tumors, and this in turn can result in marked reductions in circulating thyroid hormone levels.
Alternate Metabolic Fates of Thyroid Hormone: Inactive and Active Derivatives
In addition to 5′ deiodination, T4 undergoes other modifications which are generally believed to inactivate the hormone.18 About 45% of circulating T4 is converted to rT3 and 35% to T3. T3 and rT3 undergo further deiodinations to create T4 derivatives with all possible combinations of iodinated and deiodinated inner and outer rings (3,3′-T2, 3,5-T2, 3′,5-T2, 3′-T1, 3-T1 and T0), none of which bind significantly to nuclear TRs. T4 and T3 can also be inactivated by glucuronidation in liver, followed by secretion into the bile or by sulfation in liver and kidney and excretion in the urine.
1. Deamination of the thyroid hormone alanine side group produces acetic acid derivatives of T4 (tetraiodoacetic acid, Tetrac) and T3 (triiodoacetic acid, Triac).19 Both are exclusive products of extrathyroidal metabolism; they are undetectable in the thyroid gland. Although they comprise about 2% of total circulating thyroid hormone, they are present at high levels in liver, where Triac represents about 14% of total thyroid hormones. Tetrac and Triac are excreted in the urine, suggesting that deamination may promote thyroid hormone clearance from the body. However, Triac binds to TRs with high affinity and acts as a potent activator. It also exhibits TR isoform preference (it preferentially binds the beta form of TR) and interacts weakly with CTBP/CRYM, suggesting that it may evade cytoplasmic sequestration by this protein. While these observations raise the issue that Triac has a distinct function from T3, this idea is not proven.
2. Decarboxylation of thyroid hormone alanine side group produces amine derivatives (thyronamines) that are directly analogous to different iodinated forms of thyroid hormone.10 Thyronamines have been detected in the thyroid gland, central nervous system, fat, and other tissues. They are bound to transporter proteins in the circulation and, though completely quantitative analyses of circulating thyronamine levels have not been published, early estimates of their abundance suggest that they are present at concentrations that are at least similar to or greater than rT3 or T3.10 At the time of writing, an endocrine role for thyronamines has not been formally proven, but there is clear evidence of biological activity. Each thyronamine has been chemically synthesized and tested in cell culture in animal models; one derivative with a single iodine substituent at the 3 position of the inner thyronine ring (T1-amine) binds and activates a cell surface G-coupled protein receptor (trace amine–associated receptor 1; TAAR1) at physiologic concentrations (subnanomolar). T1-amine induces profound hypothermia, bradycardia, and reduced cardiac output in mice and alters metabolism in several species; it reduces metabolic rate and increases lipid utilization in a pattern that is reminiscent of sleep or hibernation. T1-amine may prove to be an important endocrine hormone, and it is interesting to suggest that T4 derivatives such as rT3 may be metabolic precursors of this compound.
Actions of Thyroid Hormone
Thyroid hormones exert profound influences on nearly all tissues (Fig. 5-4).2 Receptors for thyroid hormones (TRs) are expressed fairly ubiquitously, although well-defined target tissues such as liver and heart express higher levels of TRs than others, and there are differences in distributions of particular TR isoforms20 (described in more detail in a following discussion). Essentially, thyroid hormone is needed for proper growth and development of the fetus and children and exerts widespread influences on multiple aspects of metabolism in adults.
FIGURE 5-4 Tissue effects of thyroid hormones: summary of thyroid hormone effects on different tissues.
Analysis of gene expression patterns in mouse knockout models has confirmed that TRs mediate nearly all thyroid hormone responses, and that many of these effects involve changes in gene expression. While actual influences of thyroid hormone on gene expression are complex, there are important underlying principles.2 First, developmental defects that arise in cretinism and children born to hypothyroid mothers are not reversed by later hormone replacement, whereas adults with thyroid hormone imbalances exhibit metabolic disturbances that in most cases can be readily reversed by restoring TH levels to correct levels. This implies that hormone must trigger key developmental events in defined temporal windows, but thyroid hormone–regulated genes involved in metabolic regulation remain sensitive to alterations in hormone levels and continuously couple gene expression to thyroid status. Consideration of gene expression patterns, described in more detail later, reveals that some genes are induced in multiple tissues (D1 is an example), whereas other target genes are regulated in a manner that is highly tissue- and context-specific. Thus, thyroid hormone is a primary regulator of some genes, but hormone must cooperate with other factors to induce expression of other genes. Specific effects of thyroid hormone follow.
Basal Metabolic Rate
One of the most important effects of thyroid hormone involves changes in basal metabolic rate (BMR) in multiple tissues.21–24 Thyroid hormones stimulate oxygen consumption (indicative of enhanced metabolism) in multiple locations, including skeletal muscle, liver, kidney, and intestine. Increases in BMR are probably partly related to increased mitochondrial activity and number. Importantly, thyroid hormones induce expression of uncoupling proteins (UCPs), mitochondrial membrane proteins that dissipate the proton gradient in the absence of ATP synthesis, thereby converting potential energy to heat. This effect is probably a contributor to thyroid hormone–dependent increases in BMR.
Tissue Effects
Liver
Thyroid hormones exert multiple effects on the liver.2,25 They are potent mitogens in this tissue, especially in growing animals. Thyroid hormone also influences multiple metabolic processes. There is stimulation of fatty acid β-oxidation and gluconeogenesis, both key aspects of fasting response.26 However, thyroid hormone can also stimulate expression of enzymes involved in lipogenesis and generation of NADPH-reducing equivalents that are required for fat synthesis and protection against reactive oxygen species. Since it is thought that fat oxidation and synthesis do not occur simultaneously, these processes are probably separated spatially or temporally, and thyroid hormone must cooperate with other signaling mechanisms to regulate these effects.
Muscle2
Thyroid hormones promote muscle catabolism and increase skeletal muscle energy expenditure in adults. Thyroid hormone induces the insulin-sensitive glucose transporter in muscle and also promotes fat burning.30 These effects may help to sensitize muscle to insulin response. Accordingly, a human D2 gene polymorphism that reduces activity of the enzyme in muscle leads to a 20% reduction in insulin-dependent glucose disposal in humans and is correlated with insulin resistance in human populations that harbor this alteration.
Intestine
Thyroid hormone is required for normal maturation of the small intestine. TRα1 is required for proliferation of intestinal epithelial progenitor cells via induction of the β-catenin proto-oncogene.34
Systematic Analysis of Thyroid Hormone Target Genes
Many thyroid hormone–responsive genes involved in the responses described earlier are known.2 For example, in liver, thyroid hormone induces carnitine palmitoyl transferase 1a (that mediates the rate-limiting step in fatty acid oxidation); glucose-6-phosphatase and phosphoenolpyruvate carboxykinase (rate-limiting steps in gluconeogenesis); the NR coregulator PGC-1α, which is an important transcriptional coregulator that stimulates genes important for mitochondrial biogenesis, gluconeogenesis, and fat oxidation; CYP-7a1 (cholesterol-to–bile acid conversion); fatty acid synthetase; acetyl-CoA carboxylase; malic enzyme (increases fat synthesis); and spot 14, which is required for fat synthesis in some tissues and up-regulated in contexts in which carbohydrates are converted to fat. Many other hormone-responsive genes are probably still unknown.
The earliest systematic attempt to define thyroid hormone response was performed in the late 1970s. Use of two-dimensional (2D) gels to analyze extracts from radiolabeled methionine–pulsed cells revealed that about 1% of 1000 rat liver proteins change in response to altered thyroid hormone.36,37 There were inductions and repressions, and the studies began to define the “domain” of the thyroid hormone response. Recent advances in gene-profiling technology have permitted much more detailed descriptions of hormone-dependent alterations in gene profile. Presently there have been relatively few systematic genome-wide descriptions of thyroid hormone responses.25,26,38,39 An early study of thyroid hormone action in mouse liver revealed that about 1% of mouse genes change in response to altered thyroid hormone (hypothyroid versus hyperthyroid),25 confirming estimates from 2D gels. This study revealed target genes involved in carbohydrate, fat, and amino acid metabolism and unexpected thyroid hormone–regulated pathways; for example, genes involved in apoptosis are induced. The experiment also provided insights into patterns of thyroid hormone–responsive gene expression. Most regulated genes (>65%) in liver were repressed by thyroid hormone. This preponderance of negatively regulated genes is not seen in all tissues. Target genes are generally up-regulated by hormones in human muscle primary culture. Later studies of thyroid hormone patterns of gene expression in TR knockout mice provided insights into TR-specific effects and subtle differences in thyroid hormone–responsive gene expression and are discussed in more detail later in the chapter. Systematic screening of thyroid hormone–responsive genes should provide detailed descriptions of novel target genes and pathways in different tissues.
Thyroid Hormone–Regulated Micrornas
MicroRNAs (miRs) are small (18 to 25 nucleotides), noncoding RNAs that hybridize with coding mRNAs to inhibit translation and, in some cases, also promote mRNA degradation.40 The enormous roles these miRs play in regulation is just beginning to be appreciated. MiRs are produced by cleavage and processing of large primary transcripts, which often code for proteins. Thus, many of the influences that regulate expression of parental primary transcripts also regulate expression of the associated miR. While effects of thyroid hormone on miR expression have not been studied extensively, one miR (miR-208) lies within a noncoding region of the thyroid hormone–responsive human αMHC transcript (and is therefore up-regulated by thyroid hormone itself). MiR-208 is implicated in up-regulation of stress-dependent cardiac responses. It is likely that other thyroid hormone–responsive miRs exist, and they may play important roles in hormone effects on protein translation.
History of the Mechanism of Action of Thyroid Hormones
As the physiologic effects of thyroid hormones became known, investigators focused on roles of these hormones on stimulating the basal metabolic rate and on the liver.1,2 It was originally proposed that the hormones directly uncouple oxidative phosphorylation and that the primary site of action of thyroid hormones was at the mitochondria; here, investigators related actions of thyroid hormones to those of dinitrophenol. Later studies revealed other influences of thyroid hormones, including cholesterol reduction, that were hard to relate to mitochondrial actions. The notion that thyroid hormones directly uncouple mitochondrial oxidative phosphorylation did not provide a coherent unifying hypothesis to explain all actions of thyroid hormone.
In the late 1960s, Tata and colleagues conducted a number of measurements of the effects of thyroid hormones on the liver.41 He found that thyroid hormone–dependent increases in metabolic rate were blocked by actinomycin D, an inhibitor of RNA and, secondarily, protein synthesis. Also, there was an increased uptake of radiolabeled uridine into trichloric acid (TCA) precipitable material following administration of TH to animals, as well as increased activity of RNA polymerase, suggesting that TH stimulates RNA synthesis. Similar studies were being conducted with NR ligands such as with glucocorticoids, estrogens, and progestins, and there was great controversy as to whether these ligands acted through (1) transcriptional control, (2) precursor uptake, or (3) posttranscriptional control. The experiments did not address high specificity of TH responses, and as a result, controversy remained as to which of the three mechanisms applied to thyroid hormone action. Nevertheless, this study represented the first fundamentally correct proposal for the mechanism of thyroid hormone action.
Discovery of Thyroid Hormone Receptors and Their Mechanism of Action
TRs were identified in the 1970s.42,43 Following the discovery of several nuclear receptors, including the estrogen and glucocorticoid receptors, investigators in the thyroid hormone field utilized similar approaches to look for TRs. Specific binding of radiolabeled T3 to liver cells was initially discovered by Oppenheimer and colleagues using intact cells and later characterized further by Samuels and colleagues, who found that the mechanism of TR actions differed mechanistically from the steroid receptors, which translocate into the nucleus on ligand binding. For TRs, hormone could bind directly to nuclei, and the location of receptors did not alter with T3.
In the late 1970s and early 1980s, multiple aspects of the receptors were revealed.36,44 Analysis of properties of TRs revealed a single class of high-affinity binding sites with Kd values for T3 in the 0.1 nM range. Specific hormone-binding sites were observed in extracts of several responsive tissues, including liver, anterior pituitary, brain, and heart. Estimates of the number of binding sites per cell suggested that the proteins are rare; highly responsive tissues only contained about 10,000 specific hormone-binding sites per cell. Evidence that the receptor preparations correspond to physiologic hormone targets came from observations that affinities for different thyroid hormones parallel their biological potencies (Triac>T3>T4>rT3). Partial TR purification and photo-affinity labeling revealed two major nuclear hormone-binding proteins with molecular weights of 46 and 57 kD, which were later found to correspond to products of distinct genes—the TR α and β isoforms—described later in the chapter. TRs were also found to be associated with chromatin, and preferentially with active chromatin, suggesting that they influence gene expression through interactions with DNA.45–48
