BRAF
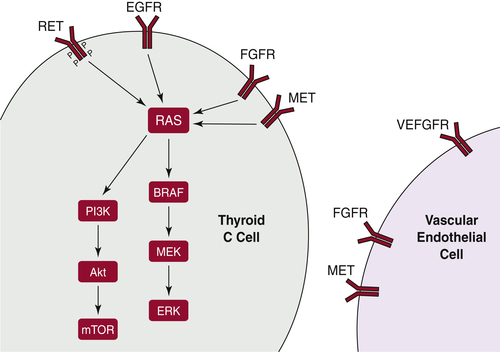
RAS
PI3K/Akt Pathway
PAX8/PPARγ
Medullary Thyroid Carcinoma
RET
Table 43-1
Most Common Mutations of the RET Gene Causing Hereditary Medullary Thyroid Carcinoma
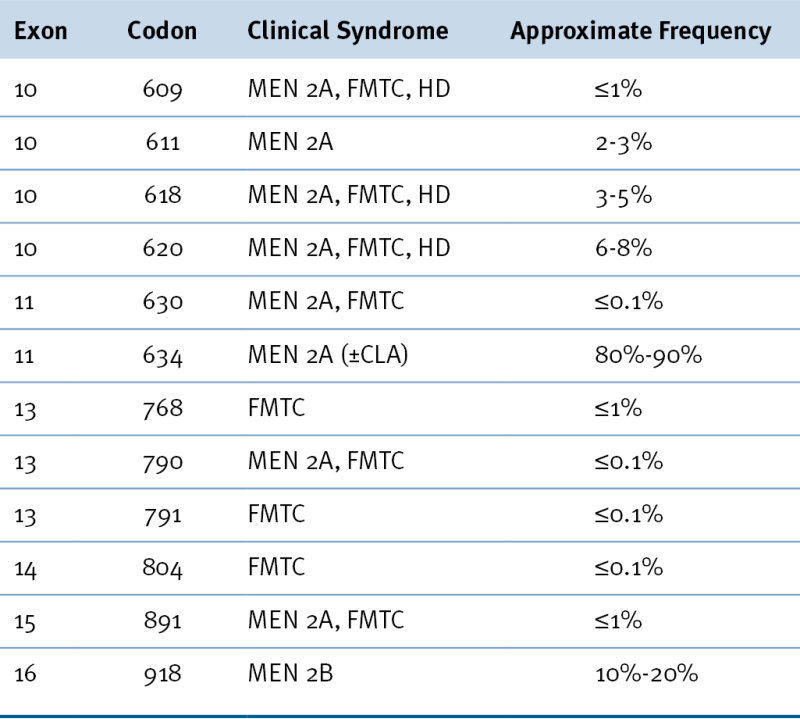
CLA, Cutaneous lichen amyloidosis; FMTC, familial medullary thyroid carcinoma; HD, Hirschsprung disease; MEN 2A, multiple endocrine neoplasia type 2A; MEN 2B, multiple endocrine neoplasia type 2B.
From Hu MI, Jimenez C, Cote G, et al. Medullary thyroid carcinoma. In: Braverman LE, Cooper DS, eds. Werner & Ingbar’s The Thyroid: A Fundamental and Clinical Text. 10th ed. Philadelphia, Pa: Wolters Kluwer; 2013:744-764.
RAS
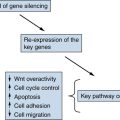
Other Molecular Mechanisms Active in Thyroid Carcinoma
Therapeutic Targeting
Future Directions
1. Global cancer statistics . CA Cancer J Clin . 2011 ; 61 : 69 – 90 .
2. Cancer statistics, 2012 . CA Cancer J Clin . 2012 ; 62 : 10 – 29 .
3. SEER Cancer Statistics Review, 1975-2007 . Bethesda, MD : National Cancer Institute ; 2010 .
4. Thyroid carcinoma . Lancet . 2003 ; 361 : 501 – 511 .
5. The GDNF family: signalling, biological functions and therapeutic value . Nat Rev Neurosci . 2002 ; 3 : 383 – 394 .
6. RET/PTC activation in human thyroid carcinomas . J Endocrinol Invest . 1995 ; 18 : 127 – 129 .
7. RET/PTC-induced cell growth is mediated in part by epidermal growth factor receptor (EGFR) activation: evidence for molecular and functional interactions between RET and EGFR . Cancer Res . 2008 ; 68 : 4183 – 4191 .
8. Thyroid carcinomas in RET/PTC transgenic mice . Recent Results Cancer Res . 1998 ; 154 : 265 – 270 .
9. Oncogenic rearrangements of the NTRK1/NGF receptor . Cancer Lett . 2006 ; 232 : 90 – 98 .
10. Gene rearrangements in radiation-induced thyroid carcinogenesis . Med Pediatr Oncol . 2001 ; 36 : 574 – 582 .
11. RET/PTC rearrangements preferentially occurred in papillary thyroid cancer among atomic bomb survivors exposed to high radiation dose . Cancer Res . 2008 ; 68 : 7176 – 7182 .
12. Proximity of chromosomal loci that participate in radiation-induced rearrangements in human cells . Science . 2000 ; 290 : 138 – 141 .
13. BRAF mutation in papillary thyroid cancer and its value in tailoring initial treatment: a systematic review and meta-analysis . Medicine (Baltimore) . 2012 ; 91 : 274 – 286 .
14. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF . Cell . 2004 ; 116 : 855 – 867 .
15. Impact of mutational testing on the diagnosis and management of patients with cytologically indeterminate thyroid nodules: a prospective analysis of 1056 FNA samples . J Clin Endocrinol Metab . 2011 ; 96 : 3390 – 3397 .
16. BRAF(V600E) mutation and outcome of patients with papillary thyroid carcinoma: a 15-year median follow-up study . J Clin Endocrinol Metab . 2008 ; 93 : 3943 – 3949 .
17. Small-molecule MAPK inhibitors restore radioi47odine incorporation in mouse thyroid cancers with conditional BRAF activation . J Clin Invest . 2011 ; 121 : 4700 – 4711 .
18. Genome-wide alterations in gene methylation by the BRAF V600E mutation in papillary thyroid cancer cells . Endocr Relat Cancer . 2011 ; 18 : 687 – 697 .
19. RAS mutations are the predominant molecular alteration in poorly differentiated thyroid carcinomas and bear prognostic impact . J Clin Endocrinol Metab . 2009 ; 94 : 4735 – 4741 .
20. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1 . Cancer Res . 2009 ; 69 : 4885 – 4893 .
21. High frequency of ras oncogene activation in all stages of human thyroid tumorigenesis . Oncogene . 1989 ; 4 : 159 – 164 .
22. The biology and the genetics of Hurthle cell tumors of the thyroid . Endocr Relat Cancer . 2012 ; 19 : R131 – R147 .
23. RAS mutations are associated with aggressive tumor phenotypes and poor prognosis in thyroid cancer . J Clin Oncol . 2003 ; 21 : 3226 – 3235 .
24. Thyroid targeting of the N-ras(Gln61Lys) oncogene in transgenic mice results in follicular tumors that progress to poorly differentiated carcinomas . Oncogene . 2006 ; 25 : 5467 – 5474 .
25. Oncogenic Kras requires simultaneous PI3K signaling to induce ERK activation and transform thyroid epithelial cells in vivo . Cancer Res . 2009 ; 69 : 3689 – 3694 .
26. Incidence and clinical characteristics of thyroid cancer in prospective series of individuals with Cowden and Cowden-like syndrome characterized by germline PTEN, SDH, or KLLN alterations . J Clin Endocrinol Metab . 2011 ; 96 : E2063 – E2071 .
27. Lifetime cancer risks in individuals with germline PTEN mutations . Clin Cancer Res . 2012 ; 18 : 400 – 407 .
28. Phosphatidylinositol 3-kinase/Akt and RAS/RAF-mitogen-activated protein kinase pathway mutations in anaplastic thyroid cancer . J Clin Endocrinol Metab . 2008 ; 93 : 278 – 284 .
29. The PI3K-Akt-mTOR pathway in initiation and progression of thyroid tumors . Mol Cell Endocrinol . 2010 ; 321 : 20 – 28 .
30. Akt activation and localisation correlate with tumour invasion and oncogene expression in thyroid cancer . J Med Genet . 2004 ; 41 : 161 – 170 .
31. PAX8-PPARgamma1 fusion oncogene in human thyroid carcinoma . Science . 2000 ; 289 : 1357 – 1360 .
32. The paired box-8/peroxisome proliferator-activated receptor-gamma oncogene in thyroid tumorigenesis . Endocrinology . 2007 ; 148 : 932 – 935 .
33. PAX8-PPARgamma rearrangement in thyroid tumors: RT-PCR and immunohistochemical analyses . Am J Surg Pathol . 2002 ; 26 : 1016 – 1023 .
34. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A . Nature . 1993 ; 363 : 458 – 460 .
35. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC . Hum Mol Genet . 1993 ; 2 : 851 – 856 .
36. Activation of RET as a dominant transforming gene by germline mutations of MEN2A and MEN2B . Science . 1995 ; 267 : 381 – 383 .
37. Relevance of RET proto-oncogene mutations in sporadic medullary thyroid carcinoma . J Clin Endocrinol Metab . 1996 ; 81 : 3740 – 3745 .
38. Medullary thyroid cancer: management guidelines of the American Thyroid Association . Thyroid . 2009 ; 19 : 565 – 612 .
39. Prognostic significance of somatic RET oncogene mutations in sporadic medullary thyroid cancer: a 10-year follow-up study . J Clin Endocrinol Metab . 2008 ; 93 : 682 – 687 .
40. Prophylactic thyroidectomy in multiple endocrine neoplasia type 2A . N Engl J Med . 2005 ; 353 : 1105 – 1113 .
41. Somatic RAS mutations occur in a large proportion of sporadic RET-negative medullary thyroid carcinomas and extend to a previously unidentified exon . J Clin Endocrinol Metab . 2012 ; 97 : E2031 – E2035 .
42. Evidence of a low prevalence of RAS mutations in a large medullary thyroid cancer series . Thyroid . 2013 ; 23 : 50 – 57 .
43. Medullary thyroid carcinoma cell lines contain a self-renewing CD133+ population that is dependent on Ret proto-oncogene activity . J Clin Endocrinol Metab . 2010 ; 95 : 439 – 444 .
44. Increased expression of vascular endothelial growth factor and its receptors, VEGFR-1 and VEGFR-2, in medullary thyroid carcinoma . Thyroid . 2010 ; 20 : 863 – 871 .
45. Influence of the BRAF V600E mutation on expression of vascular endothelial growth factor in papillary thyroid cancer . J Clin Endocrinol Metab . 2006 ; 91 : 3667 – 3670 .
46. Expression of vascular endothelial growth factor (VEGF) and its receptors in thyroid carcinomas of follicular origin: a potential autocrine loop . Eur J Endocrinol . 2005 ; 153 : 701 – 709 .
47. Overexpression and activation of EGFR and VEGFR2 in medullary thyroid carcinomas is related to metastasis . Endocr Relat Cancer . 2010 ; 17 : 7 – 16 .
48. Expression of hepatocyte growth factor (HGF) and its receptor (MET) in medullary carcinoma of the thyroid . Endocr Pathol . 2000 ; 11 : 19 – 30 .
49. Over-expression of hepatocyte growth factor/scatter factor (HGF/SF) and the HGF/SF receptor (cMET) are associated with a high risk of metastasis and recurrence for children and young adults with papillary thyroid carcinoma . Clin Endocrinol (Oxf) . 2000 ; 53 : 635 – 644 .
50. Expression of fibroblast growth factors in thyroid cancer . J Clin Endocrinol Metab . 1995 ; 80 : 1006 – 1011 .
51. Biphasic effects of thyrotropin on invasion and growth of papillary and follicular thyroid cancer in vitro . Thyroid . 1995 ; 5 : 35 – 40 .
52. Thyrotrophin receptor signaling dependence of Braf-induced thyroid tumor initiation in mice . Proc Natl Acad Sci U S A . 2011 ; 108 : 1615 – 1620 .
53. Gene p53 mutations are restricted to poorly differentiated and undifferentiated carcinomas of the thyroid gland . J Clin Invest . 1993 ; 91 : 1753 – 1760 .
54. Unique association of p53 mutations with undifferentiated but not with differentiated carcinomas of the thyroid gland . Cancer Res . 1992 ; 52 : 1369 – 1371 .
55. p53 re-expression inhibits proliferation and restores differentiation of human thyroid anaplastic carcinoma cells . Oncogene . 1997 ; 14 : 729 – 740 .
56. Thyrocyte-specific inactivation of p53 and Pten results in anaplastic thyroid carcinomas faithfully recapitulating human tumors . Oncotarget . 2011 ; 2 : 1109 – 1126 .
57. Loss of p53 promotes anaplasia and local invasion in ret/PTC1-induced thyroid carcinomas . Am J Pathol . 2000 ; 157 : 671 – 677 .
58. Beta-catenin dysregulation in thyroid neoplasms: down-regulation, aberrant nuclear expression, and CTNNB1 exon 3 mutations are markers for aggressive tumor phenotypes and poor prognosis . Am J Pathol . 2001 ; 158 : 987 – 996 .
59. The beta-catenin axis integrates multiple signals downstream from RET/papillary thyroid carcinoma leading to cell proliferation . Cancer Res . 2009 ; 69 : 1867 – 1876 .
60. Extra-intestinal manifestations of familial adenomatous polyposis . Ann Surg Oncol . 2008 ; 15 : 2439 – 2450 .
61. Role of the Wnt pathway in thyroid cancer . Front Endocrinol (Lausanne) . 2012 ; 3 : 31 .
62. Targeted therapies for thyroid tumors . Mod Pathol . 2011 ; 24 ( suppl 2 ) : S44 – S52 .
63. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases . Cancer Res . 2002 ; 62 : 7284 – 7290 .
64. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial . J Clin Oncol . 2012 ; 30 ( 2 ) : 134 – 141 .
65. An international, double-blind, randomized, placebo-controlled phase III trial (EXAM) of cabozantinib (XL184) in medullary thyroid carcinoma (MTC) patients (pts) with documented RECIST progression at baseline . J Clin Oncol . 2012 ; 30 ( suppl ) : 5508 .
66. Motesanib diphosphate in progressive differentiated thyroid cancer . N Engl J Med . 2008 ; 359 : 31 – 42 .
67. Rationale and design of DECISION: a double-blind, randomized, placebo-controlled phase III trial evaluating the efficacy and safety of sorafenib in patients with locally advanced or metastatic radioactive iodine (RAI)-refractory, differentiated thyroid cancer . BMC Cancer . 2011 ; 11 : 349 .
68. Phase II trial of sorafenib in metastatic thyroid cancer . J Clin Oncol . 2009 ; 27 : 1675 – 1684 .
69. Phase II study of daily sunitinib in FDG-PET-positive, iodine-refractory differentiated thyroid cancer and metastatic medullary carcinoma of the thyroid with functional imaging correlation . Clin Cancer Res . 2010 ; 16 : 5260 – 5268 .
70. Inhibition of mutated, activated BRAF in metastatic melanoma . N Engl J Med . 2010 ; 363 : 809 – 819 .
71. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial . Lancet . 2012 ; 379 : 1893 – 1901 .
72. Reacquisition of RAI uptake in RAI-refractory metastatic thyroid cancers by pretreatment with the MEK inhibitor selumetinib . J Clin Oncol . 2012 ; 30 ( suppl ) : 5509 .
73. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations . N Engl J Med . 2012 ; 367 : 1694 – 1703 .