[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 1
The Principles, Enzymes, and Pathways of Human Steroidogenesis
Overview of the Human Steroidogenic Enzymes and Steroidogenesis
Overview of the Human Steroidogenic Enzymes and Steroidogenesis
1. The conversion of cholesterol to pregnenolone. Although viewed superficially as a single chemical transformation, the mobilization of cholesterol into the steroidogenic pathways is a complex event that serves as a key locus of regulation and also conventionally defines a tissue as “steroidogenic.” In humans, only the adrenal cortex, testicular Leydig cells, ovarian theca cells, trophoblast cells of the placenta, and specific glial and neuronal cells of the brain possess the capacity to cleave cholesterol into pregnenolone (the C21 precursor of all active steroid hormones) and isocaproaldehyde. The differences in how this process is regulated and in how pregnenolone is subsequently metabolized define the roles of the various steroidogenic cells and tissues in human physiology. Unlike peptide-secreting glands, steroidogenic cells do not store steroid hormones and intermediates, and it is the activation of this first step that enables the rapid production of steroids in response to hormonal and environmental stimuli.
2. The transformation of pregnenolone to active hormones, intermediates, and exported steroid derivatives. The repertoire of enzymes and cofactor proteins present in a given steroidogenic cell is responsible for the characteristic steroid profile of that cell type, and the coordinate regulation of their expression promotes the completion of all steps of a given pathway. Thus, these enzymes determine qualitatively what steroids are made, but since these steps are not kinetically limiting, it is step 1 that quantitatively regulates how much steroid is made at a given moment. Steroids diffuse into the bloodstream, although the sulfation of Δ5 steroids helps promote their binding to proteins and prolong their half-lives in the circulation.
3. Peripheral metabolism of hormones and precursors. Although not “steroidogenic” as defined earlier, some organs, such as the liver and skin, possess tremendous capacity to transform various steroids. For example, 70% to 80% of circulating testosterone in normally cycling women derives from the conversion of adrenal dehydroepiandrosterone (DHEA). Steroids can be activated in target tissues, such as the conversion of testosterone to dihydrotestosterone (DHT) in the prostate. In contrast, active androgens and estrogens are inactivated in the uterus and in other peripheral tissues.
4. Multiple layers of regulation. The regulation of steroidogenesis by trophic hormones, such as adrenocorticotropin (ACTH), angiotensin II, and luteinizing hormone (LH) is well known; less well appreciated are the multiple levels at which such regulation may occur. These include the transcriptional regulation of the genes encoding steroidogenic enzymes and co-factors; regulation of mitochondrial protein import; transfer of electrons from NADPH; post-translational modification of steroidogenic enzymes; and subcellular localization and/or targeting.
5. Catabolism and unproductive metabolism. A panoply of steroids can be isolated from human plasma and tissues, many of which have negligible biological activity. Most inactive byproducts derive from hepatic transformations (e.g., 6α/6β-hydroxylation of C19 steroids, 5β-reduction of C21 and C19 steroids, and 4-hydroxylation of estrogens), which promote renal excretion of these steroids.
6. More than one pathway to specific steroids. Particularly for the terminal steps of long pathways, such as the synthesis of DHT or estradiol, two or more routes may yield the same final product. Often the different routes utilize different enzymes, and these enzymes may be found in different tissues under different regulatory mechanisms. The relative importance of these pathways differ with age, sex, and physiologic state.
Steroids are molecules derived from the cyclopentanoperhydrophenanthrene four-ring hydrocarbon nucleus (Fig. 1-1). Most enzymes involved in steroid biosynthesis are either cytochrome P450s or hydroxysteroid dehydrogenases (Fig. 1-2). All of these reactions are functionally if not absolutely unidirectional, so the accumulation of products does not drive flux back to the precursor. All P450-mediated hydroxylations and carbon-carbon bond cleavage reactions are mechanistically and physiologically irreversible.1 Hydroxysteroid dehydrogenase reactions are mechanistically reversible and can run in either direction under certain conditions in vitro, but each hydroxysteroid dehydrogenase drives steroid flux predominantly in either the oxidative or reductive mode in vivo.2 However, two or more hydroxysteroid dehydrogenases drive the flux of a steroid pair in opposite directions, some favoring ketosteroid reduction and others favoring hydroxysteroid oxidation.
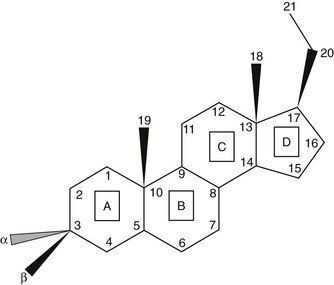
FIGURE 1-1 Cyclopentanoperhydrophenanthrene steroid nucleus. Steroid rings are identified with boxed capital letters, and carbon atoms are numbered. Substituents and hydrogens are labeled as α or β if they are positioned behind or in front of the plane of the page, respectively.
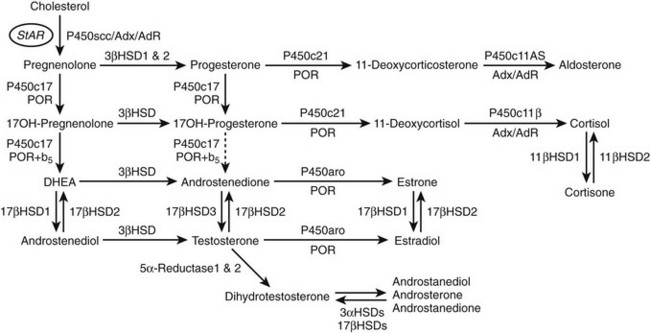
FIGURE 1-2 Major human steroidogenic pathways. Key enzymes and cofactor proteins are shown near arrows indicating chemical reactions. The StAR protein (oval) mobilizes cholesterol from the outer mitochondrial membrane to the inner mitochondrial membrane, where P450scc cleaves cholesterol to pregnenolone, the first committed intermediate in steroid biosynthesis. The steroids in the first column are Δ5-steroids, which constitute the preferred pathway to C19 steroids in human beings, and the dashed arrow indicates poor flux from 17α-hydroxyprogesterone to androstenedione. Steroids in the second column and farther right are Δ4-steroids, except the C18 estrogens (estrone and estradiol) and 5α-reduced steroids, including the potent androgen DHT and other androstanes (bottom row). Not all intermediate steroids, pathways, and enzymes are shown.
Cytochrome P450 Enzymes
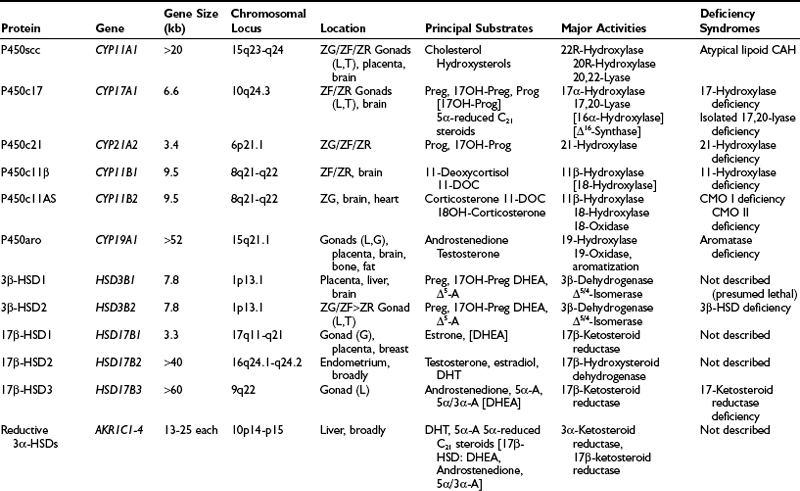
Mammalian cytochrome P450 enzymes fall into two broad classes, type 1 and type 2.3 Type 1 enzymes and their electron-transfer proteins reside in the mitochondria (Table 1-1) of eukaryotes; almost all bacterial P450s are also type 1 enzymes. Human type 1 P450 enzymes include the cholesterol side-chain cleavage enzyme P450scc; the two isozymes of 11-hydroxylase (i.e., P450c11β and P450c11AS); and two of the three principal enzymes in vitamin D metabolism (i.e., 1α-hydroxylase and 24-hydroxylase). Type 1 enzymes receive electrons from the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) via adrenodoxin, a small, soluble, iron-sulfur protein. Adrenodoxin does not oxidize NADPH directly, however, but receives the two electrons from NADPH via the flavoprotein adrenodoxin reductase (Fig. 1-3). Type 2 enzymes, in contrast, receive electrons from NADPH via the flavin adenine dinucleotide (FAD)-flavin mononucleotide (FMN) two-flavin protein, P450 oxidoreductase (POR). Type 2 enzymes are exclusively located in the smooth endoplasmic reticulum and constitute the majority of the human P450 enzymes.
Table 1-1
Intracellular Location of Steroidogenic Proteins
| Mitochondria | Cytoplasm | Endoplasmic Reticulum |
| P450scc | P450c17 | |
| P450c11β | P450c21 | |
| P450c11AS | P450aro | |
| Adrenodoxin reductase | P450-oxidoreductase | |
| Adrenodoxin | ||
| StAR | StAR | Cytochrome b5 |
| 3β-HSD1 and 2 | 3β-HSD1 and 2 | 3β-HSD1 and 2 |
| 17β-HSD1 | 17β-HSD1–3* | |
| Reductive 3α-HSDs includes 17β-HSD5 | Oxidative 3α-HSD 5α-Reductase 1 and 2 11β-HSD1 and 2 |
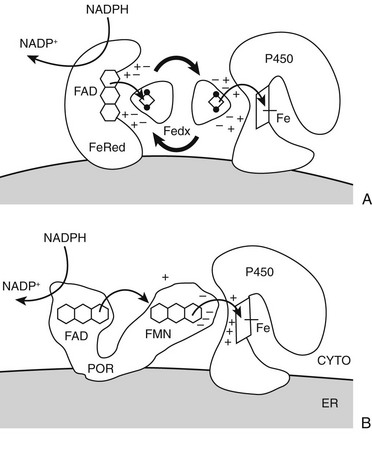
FIGURE 1-3 Electron transfer pathways for steroidogenic cytochrome P450 enzymes. A, In type 1 (mitochondrial) enzymes, the two electrons from the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) pass from the flavin (FAD) of ferredoxin (adrenodoxin) reductase (FeRed) to the iron-sulfur (Fe2S2, diamond with dots) cluster of ferredoxin (adrenodoxin, Fedx) and then to the heme of the P450 (square with iron atom [Fe]). Negatively-charged residues in Fedx (−) guide docking and electron transfer with positively-charged residues (+) in both Fedx and the P450. B, In type 2 (microsomal) enzymes, the flavoprotein P450-oxidoreductase (POR) receives electrons from NADPH to its FAD moiety, transfers electrons to its FMN moiety, and after a conformational rearrangement, directly transfers electrons from the FMN to the P450. Negative charges on POR (−) and positive charges (+) on the P450 guide the interaction as with the type 1 P450; phosphorylation and cytochrome b5 also regulate electron transfer and catalysis. Heme of P450 is indicated by square with iron atom (Fe).
P450 enzymes activate molecular oxygen using their heme center and electrons from NADPH. Substrate binding is required prior to heme reduction with one electron, which enables oxygen binding, the second one-electron transfer, and formation of the iron-oxygen complex which oxygenates the substrate. Thus, P450 reactions on steroids are limited to oxygen insertion (hydroxylation) reactions and, in a few notable cases, oxidative carbon-carbon bond cleavage reactions (Table 1-2).
Hydroxysteroid Dehydrogenases and Reductases
All hydroxysteroid dehydrogenases (HSDs) and related enzymes use nicotinamide cofactors either to reduce or to oxidize the steroid by two electrons through a hydride transfer mechanism.2 Most examples involve the conversion of a secondary alcohol to a ketone or vice versa, and in the case of the 3β-hydroxysteroid dehydrogenase/Δ5/4-isomerases, the dehydrogenation is accompanied by the isomerization of the adjacent carbon-carbon double bond from the Δ5 (between carbons 5 and 6) to the Δ4 positions (see Figs. 1-1 and 1-2). The human steroid 5α-reductases types 1 and 2, which are included with the HSDs for convenience, reduce olefinic carbon-carbon double bonds to the saturated state rather than acting on carbon centers bonded to oxygen.
The HSDs can be categorized according to either structural or functional classification schemes. Structurally, HSDs are members of either the short-chain dehydrogenase reductase (SDR) or aldo-keto reductase (AKR) families.4 The SDR enzymes are β-α-β proteins where up to seven parallel β-strands fan across the center of the molecule, forming the “Rossman fold” characteristic of oxidation/reduction enzymes that use nicotinamide cofactors. The AKR enzymes are soluble proteins that contain a beta-barrel or triosephosphate isomerase (TIM-barrel) motif in which eight parallel β-strands lie in a slanted circular distribution like the staves of a barrel. In both cases, the active site contains a critical tyrosine and lysine pair of residues involved in proton transfer from or to the steroid alcohol during catalysis. Functionally, HSDs act either as true dehydrogenases, using NAD+ as a cofactor to convert hydroxysteroids to ketosteroids, or as ketosteroid reductases, utilizing predominantly NADPH to reduce ketosteroids. Many HSDs catalyze either oxidation or reduction in vitro based on the pH and cofactor concentrations, but these enzymes, when expressed in intact mammalian cells, drive steroid flux primarily in one direction.4 These directional preferences derive primarily from the relative abundance of the oxidized and reduced form of cofactors and the relative affinity of each enzyme for NAD(H) versus NADP(H), because cofactor concentrations exceed steroid concentrations by many orders of magnitude.2,5 Consequently, the directional preference of some “reductive” enzymes can be reduced or reversed by depleting cells of NADPH or by mutations that impair NADPH binding.6
Acute Regulation of Steroidogenesis
Every time that a pulse of corticotrophin (adrenocorticotropic hormone [ACTH]) reaches the adrenal cortex, or a pulse of luteinizing hormone (LH) reaches the gonad, a subsequent pulse of steroid hormone production is observed within minutes. Although it has long been known that the loss of trophic hormones from the pituitary gland leads to adrenal and gonadal atrophy, the action of ACTH and LH to promote organ survival and to maintain steroidogenic capacity occurs at three distinct levels. First, as seen in long-term exposure to ACTH (e.g., in Cushing’s disease), ACTH promotes adrenal growth. This growth occurs primarily by ACTH stimulating the production of cyclic adenosine monophosphate (cAMP), which in turn promotes the synthesis of insulin-like growth factor 2 (IGF-2),7,8 basic fibroblast growth factor,9 and epidermal growth factor.10 Together, these growth factors stimulate adrenal cellular hypertrophy and hyperplasia. Second, ACTH acts long term through cAMP, and angiotensin II acts through the calcium/calmodulin pathway to promote the transcription of genes encoding various steroidogenic enzymes and electron-donating cofactor proteins. Third, ACTH fosters the increased flow of cholesterol into mitochondria, where it becomes substrate for the first and rate-limiting enzyme, P450scc. This acute response occurs within minutes and is inhibited by inhibitors of protein synthesis (e.g., puromycin or cycloheximide), indicating that a short-lived protein species mediates this process. Although other proteins are involved in the chronic replenishment of mitochondrial cholesterol, abundant biochemical, clinical, and genetic evidence implicates the steroidogenic acute regulatory protein (StAR) as this labile protein mediator.11
StAR is a 37-kilodalton (kD) phosphoprotein that is cleaved to a 30-kD form when it enters the mitochondrion. Overexpression of mouse StAR in mouse Leydig MA-10 cells increased their basal steroidogenic rate,12 and cotransfection of expression vectors for both StAR and the P450scc system in nonsteroidogenic COS-1 cells augmented pregnenolone synthesis above that obtained with the P450scc system alone.13 Mutations in StAR cause the most common form of congenital lipoid adrenal hyperplasia,13,14 in which very little steroid is made, and targeted disruption of the Star gene in the mouse causes a similar phenotype.15
The mechanism of StAR’s action is not known in detail.16 StAR acts exclusively on the outer mitochondrial membrane (OMM),17,18 and its activity in promoting steroidogenesis is proportional to its residency time on the OMM.18 When expressed in cytoplasm or added to mitochondria in vitro, both the 37-kD “precursor” and the 30-kD “mature form” of StAR are equally active, but StAR is inactive in the mitochondrial intramembranous space or matrix.18 Thus, it is StAR’s cellular localization, not its cleavage, that determines whether or not it is active. StAR has a sterol-binding pocket that accommodates a single molecule of cholesterol.19 The interaction of StAR with the OMM involves conformational changes20,21 that are necessary for StAR to accept and discharge cholesterol molecules. Although StAR can transfer cholesterol between synthetic membranes in vitro,22 suggesting that other protein molecules are not needed for its action, this activity can also be seen with the inactive mutant R182L, which causes lipoid CAH.23 Thus, StAR’s action to promote steroidogenesis is distinct from its cholesterol-transfer activity. StAR appears to interact with the peripheral benzodiazepine receptor (PBR)24 voltage-dependent anion channel 1 (VDAC1) and phosphate carrier protein,25 all proteins found on the outer mitochondrial membrane. Each molecule of StAR appears to be recycled, moving hundreds of molecules of cholesterol before the cleavage/inactivation event.26 Although StAR is required for the acute steroidogenic response, steroidogenesis will persist in the absence of StAR at about 14% of the StAR-induced rate,27 accounting for the steroidogenic capacity of tissues that lack StAR (e.g., the placenta and the brain).
Chronic Maintenance of the Steroidogenic Machinery
While the acute regulation of steroidogenesis is determined by access of cholesterol to the P450scc enzyme, which is mediated by StAR, P450scc is the enzymatic rate-limiting step in steroidogenesis. Thus the chronic regulation of steroidogenesis is quantitatively (how much) determined by P450scc gene expression28 and qualitatively (which steroids) determined by the expression of downstream enzymes. The episodic bursts of cAMP resulting from the binding of ACTH and LH to their respective receptors are necessary but not sufficient for the continued expression of the steroidogenic enzymes and the production of steroids. Patients with inactivating mutations in the ACTH receptor29 or LH receptor30 make negligible steroids from the affected glands. Conversely, activating mutations of the Gsα protein, which couples receptor binding to cAMP generation, and activating mutations of the LH receptor cause hypersecretion of steroids.31 Indeed, cAMP-responsive elements have been identified in the genes for most of the human steroidogenic P450 enzymes, but this mechanism alone does not allow for the diversity of steroid production observed in the various zones of the adrenal cortex, the gonads of both sexes, the placenta, and the brain.
Other transcription factors (e.g., AP-2, SP-1, SP-3, NF1C, NR4A1, NR4A2, GATA4, and GATA6) aid in defining the basal- and cAMP-stimulated transcription of each gene, which is also regulated in a tissue-specific manner by the regulatory elements unique to each gene. Among these factors, steroidogenic factor-1 (SF-1, NR5A1), an orphan nuclear receptor, coordinates the expression of steroidogenic enzymes in adrenal and gonadal cells.32 By contrast, steroidogenesis in the brain33 and placenta34,35 is independent of SF-1. Targeted disruption of SF-1 in the mouse not only disrupts steroid biosynthesis but also blocks the development of the adrenal glands, gonads, and the ventromedial hypothalamus in homozygous animals.36 Furthermore, SF-1 does not act in isolation, but its action is modified by other transcription factors (e.g., WT-1 and DAX-137) or by sumoylation and phosphorylation.38 The development of steroidogenic organs is intimately related to the capacity to produce steroids, and multiple factors acting on the genes for steroidogenic enzymes yield both common features and diversity among the steroidogenic tissues.
Most steroidogenic enzymes derive from a single mRNA species. The most prominent exception to this paradigm is aromatase, whose gene has four different promoters that enable vastly different regulation of expression of the same aromatase protein in many different tissues.39 Although different transcripts of several genes (including 17β-HSDs types 1, 2, and 3) have been described, the encoded proteins derived from “exon skipping” are inactive if translated.40
Human Steroidogenic P450s
Encoded by the CYP11A1 gene, P450scc consumes three equivalents of NADPH and molecular oxygen during the conversion of cholesterol to pregnenolone. Although the enzyme is named for the cleavage of the cholesterol side chain, this process consists of three discrete steps: (1) the 22-hydroxylation of cholesterol; (2) the 20-hydroxylation of 22(R)-hydroxycholesterol; and (3) the oxidative scission of the C20-C22 bond of 20(R), 22(R)-dihydroxycholesterol—the side-chain cleavage event. The enzyme will utilize free hydroxysterol intermediates as substrates for the side-chain cleavage reaction, a tool that is used experimentally because the hydroxysterols are much more soluble than cholesterol and because their access to P450scc is independent of StAR.13 In vivo, however, little of these free intermediates probably accumulate because their kcat/Km ratios are much higher than for cholesterol,41 and the high Kd for pregnenolone (about 3000 nM) drives product dissociation. This complex process is the rate-limiting step in steroidogenesis, with turnover numbers of only about 20 molecules of cholesterol per molecule P450scc per minute.41 P450scc will also cleave the side chain of other hydroxysterols (e.g., 7-dehydrocholesterol), and 20- and 22-hydroxylates vitamin D.42
The single human gene for P450scc43 encodes an mRNA of 2 kb.44 A 39-amino-acid mitochondrial leader peptide that targets P450scc to the mitochondria is then proteolytically removed to yield a 482-amino-acid protein. Forms of P450scc targeted to the endoplasmic reticulum are inactive,45 demonstrating that the mitochondrial environment is required for activity. Expression of P450scc is induced in the adrenal zona fasciculata/reticularis,46 testis,47 and ovary by cAMP; and in the zona glomerulosa by intracellular calcium/protein kinase C.48,49 In contrast, placental P450scc expression is constitutive50 and is caused at least in part by the LBP family of transcription factors.35,51 Side-chain cleavage activity and pregnenolone biosynthesis have been demonstrated in the rat and human brain52; and abundant P450scc expression is found in the rodent brain, especially in fetal life. Deletion of the gene for P450scc has been described in rabbits53 and mice,54 abrogating all steroidogenesis and thus proving that P450scc is the only enzyme that can convert cholesterol to pregnenolone. While homozygous mutations in P450scc are expected to be embryonic lethal by eliminating placental progesterone synthesis, a small number of patients has been described having P450scc mutations that typically retain partial enzymatic activity.55,56
P450c17
For investigators studying the enzymology and genetics of the steroidogenic pathways, P450c17 is especially interesting. Clinical observations showed that adrenal 17α-hydroxylase activity (reflected by serum cortisol concentrations) was fairly constant throughout life, whereas adrenal 17,20-lyase activity (reflected by serum DHEA and DHEAS concentrations) was low in early childhood but rose abruptly during adrenarche at ages 8 to 10 years.57,58 This dissociation between adrenal secretion of 17α-hydroxylase products (cortisol) and 17,20-lyase products (DHEA) suggested that distinct enzymes performed the two transformations, a hypothesis that was reinforced by the description of patients with putative isolated 17,20-lyase deficiency. Consequently, reports59 that the 17α-hydroxylase and 17,20-lyase activities of neonatal pig testes copurified were initially received with great skepticism. This controversy of “one enzyme or two” persisted until the cDNA for bovine P450c17 was cloned and shown to confer both 17-hydroxylase and 17,20-lyase activities when expressed in nonsteroidogenic COS-1 cells.60 The human genome has one gene for P450c17,61 which is expressed in the adrenals and gonads,62 and not two tissue-specific isozymes as had been thought. A single 2.1-kb mRNA species yields a 57-kD protein in these tissues, and mutations in this gene produce a spectrum of deficiencies in 17-hydroxysteroids and C19 steroids.
Human P450c17 17-hydroxylates both pregnenolone and progesterone with approximately equal efficiency,63,64 but all other reactions show prominent differences between Δ4 and Δ5 substrates. The 17,20-lyase activity is about 50 times more efficient for the 17α-hydroxypregnenolone-to-DHEA reaction than for the 17α-hydroxyprogesterone-to-androstenedione reaction.63,64 Although the rate of the lyase reaction can be increased more than 10-fold by the addition of a molar excess of cytochrome b5,63–65 the Δ5 preference persists, and the lyase rate never quite reaches the rate of the hydroxylase reactions. In addition, human P450c17 16α-hydroxylates progesterone but not pregnenolone64; in the presence of cytochrome b5, it diverts about 10% of pregnenolone metabolism to a Δ16 andiene product63 that is also formed by this pathway in pigs and that acts as a pheromone in that species. Although experiments to study the chemistry of human P450c17 often require manipulations that could be considered nonphysiologic, the remarkable consistency for substrate preferences and kinetic constants observed for the modified, solubilized P450c17 expressed in Escherichia coli63,65 and native P450c17 expressed in yeast microsomes,64 or intact COS-1 cells,66 or that obtained from human tissues and cells,64,67 serve to verify these conclusions.
Given the diverse repertoire of reactions catalyzed by P450c17 in the classical pathways, it is not surprising that synthetic steroids such as dexamethasone68 and the enantiomer of progesterone,69 as well as planar drugs such as troglitazone,70 also bind to and inhibit P450c17. In addition, the 5α-reduced C21 steroids dihydroprogesterone (5α-pregnane-3,20-dione) and allopregnanolone (5α-pregnan-3α-ol-20-one) are excellent substrates for the 17α-hydroxylase activity of P450c1771 (Fig. 1-4A). Furthermore, 17α-hydroxylated allopregnanolone (5α-pregnane-3α,17α-diol-20-one; 17OH-Allo) is the most efficient substrate yet identified for the 17,20-lyase activity of human P450c17, and its cleavage to androsterone is minimally dependent on cytochrome b5,71 unlike 17α-hydroxypregnenolone metabolism to DHEA.63–65 The conversion of 17OH-Allo to androsterone by the 17,20-lyase activity of P450c17, first described in the testes of tammar wallaby pouch young,72 provides an alternative or “backdoor” pathway to DHT, by which DHT is produced without utilizing DHEA, androstenedione, and testosterone as intermediates73 (see Fig. 1-4B). Consequently, the presence of 5α-reductases in steroidogenic cells does not preclude the production of C19 steroids but rather paradoxically enhances the production of DHT by directing flux to 5α-reduced precursors of DHT.
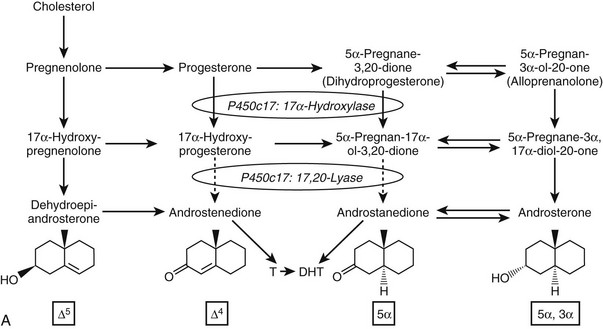
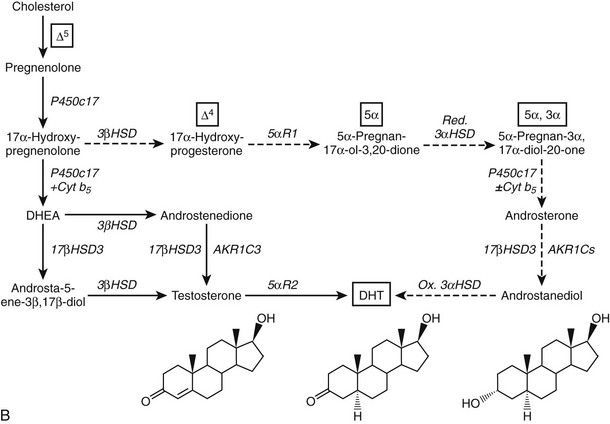
FIGURE 1-4 Reactions catalyzed by human P450c17 and pathways to C19 steroids. A, The four principal A/B-ring configurations of active endogenous steroids and their precursors: Δ5, Δ4, 5α, and 5α,3α (boxes and structures at bottom). Progesterone and 17α-hydroxyprogesterone can be 5α-reduced, and once the A-ring is saturated, these 5α-reduced steroids are substrates for reductive 3α-HSDs of the AKR1C family. Human P450c17 17α-hydroxylates all four classes of C21 steroids, but the 17,20-lyase activity is robust only with 17α-hydroxypregnenolone and 5α-pregnane-3α,17α-diol-20-one (Δ5– and 5α,3α-pathways, respectively). B, Two pathways to DHT using the different 17,20-lyase activities of human P450c17. In the conventional or Δ5-pathway (solid arrows), the 17,20-lyase activity of P450c17 requires cytochrome b5 to efficiently convert 17α-hydroxyprogesterone to DHEA, and testosterone is reduced in target tissues by 5α-reductase 2 (5αR2) to DHT. In the “backdoor” or 5α,3α-pathway (broken arrows), 5α-reduction by 5αR1 and 3α-reduction of C21 steroids occurs in the steroidogenic tissue prior to the 17,20-lyase reaction. In the best characterized pathway, 5α-pregnane-3α,17α-diol-20-one is cleaved to androsterone without requiring cytochrome b5 and reduced to androstanediol. Androstanediol is exported from the testis and metabolized to DHT by oxidative 3α-HSDs (Ox. 3α-HSD). Note that testosterone is not an intermediate in the backdoor pathway to DHT, that different isoforms of 5α-reductase appear to be involved in the two pathways, and that both reductive and oxidative 3αHSDs are required for the “backdoor” pathway. Structures of testosterone, DHT, and androstanediol are shown at bottom.
The backdoor pathway enables production of C19 steroids in the presence of abundant 3β-HSD activity, despite the poor 17,20-lyase activity of human P450c17 for 17α-hydroxyprogesterone, by using 17OH-Allo as the substrate for the 17,20-lyase reaction. The presence of 5α-reductase activity is a key requirement for the backdoor pathway. The best-studied example of 5α-reduction in a human steroidogenic tissue is the production of 5α-dihydroprogesterone in human corpus luteum by the type 1 enzyme.74 Human enzymes catalyze all of the other reactions required to complete this alternate route to DHT, and good evidence documents production of 5α-reduced androgens by the fetal adrenal, at least in some pathologic states. Consequently, it is possible that the backdoor pathway is the principal route to DHT in pathologic states in which 17α-hydroxyprogesterone accumulates, including 21-hydroxylase deficiency and P450 oxidoreductase deficiency (see the following). Androgen production by the backdoor pathway may explain why newborn girls with 21- and 11-hydroxlase deficiencies can be severely virilized, while those with 3β-HSD2 deficiency, whose adrenals cannot make 17α-hydroxyprogesterone, are minimally virilized.75 The fractional contributions of the conventional and backdoor pathways to DHT production during human sexual differentiation (at 8 to 12 weeks of gestation) and the expression of 5α-reductase in fetal adrenal and gonad tissues,76 however, are only beginning to be determined.
The chemistry of P450c17-mediated hydroxylations is believed to proceed via the common iron oxene species and “oxygen rebound” mechanism proposed for prototypical P450 hydroxylations.77 The mechanism of the 17,20-lyase reaction involving a carbon-carbon bond cleavage, however, is not known despite considerable study. The failure of hydrogen peroxide alone to support catalysis (as has been shown for some other P450-mediated deacylation reactions) and computer modeling studies suggest that the same heme-oxygen complex might participate in both hydroxylations and the 17,20-lyase reaction,78 but no conclusive evidence to exclude proposed mechanisms exist.
One consequence of the Δ5 preference of the human enzyme for the 17,20-lyase reaction is that most human C19 and C18 steroids derive from DHEA as an intermediate.67 This Δ5 preference allows for the phenomenon of adrenarche to occur in humans, an event that only takes place in large primates.79,80 However, Δ5-lyase activity is not sufficient for adrenarche to occur, because some monkeys (e.g., rhesus macaques) produce high amounts of DHEA throughout life, but most mammals (e.g., cattle, dogs, cats, etc.) never produce much DHEA.79 The biochemistry of P450c17, with its differential regulation of the 17α-hydroxylase and 17,20-lyase activities, provides clues to the genesis of this enigmatic process of adrenarche. P450c17 is a phosphoprotein, and phosphorylation selectively enhances the 17,20-lyase activity.81,82 It appears likely that the regulation of P450c17 phosphorylation, which is a dynamic balance between phosphorylation and dephosphorylation, plays an important role in adrenarche and pathologic hyperandrogenic states such as polycystic ovary syndrome.83 Whereas the kinase(s) responsible for P450c17 phosphorylation remain unknown, it is now apparent that the kinase activity is counterbalanced by protein phosphatase 2A, which in turn is regulated by cAMP via phosphoprotein SET.84 Cytochrome b5 also augments 17,20-lyase activity,64,82 and high expression of b5 in the zona reticularis of monkeys85 and humans86 suggests that the developmentally regulated expression of b5 might be a key event. The transcriptional regulation of cytochrome b5 in the adrenal is similar to that of P450c17,87 but mechanisms enabling zone-specific expression have not been elucidated. Finally, limiting steroid flux to the Δ5 pathway by lowering 3β-HSD activity in the zona reticularis (where most DHEA derives) potentiates the effect of increased 17,20-lyase activity.86,88
The initial description of 17α-hydroxylase deficiency was a case in which both 17α-hydroxylase and 17,20-lyase products were absent.89 When the gene for human P450c17 was cloned,61 patients with 17α-hydroxylase deficiency were found to harbor mutations in the CYP17A1 gene, and more than 40 mutations scattered throughout the CYP17A1 gene have been characterized,90 with mutations W406R and R362C being the most common and accounting for the high prevalence of 17α-hydroxylase deficiency in Brazil.91 The identification of CYP17A1 mutations causing apparent isolated 17,20-lyase deficiency is fraught with difficulty,92 but in the past decade, five cases of isolated 17,20-lyase deficiency caused by mutations in arginines 347 and 358 have been confirmed.93,94 Computer modeling studies demonstrate that R347H and R358Q neutralize positive charges in the redox-partner binding site.78,93 Biochemical studies confirm that mutations R347H and R358Q impair interactions of P450c17 with its electron donor POR and with cytochrome b5.95 Therefore, these cases of isolated 17,20-lyase deficiency are not caused by an inability of the mutant enzymes to bind the intermediate 17α-hydroxypregnenolone but rather are caused by subtle disturbances in interactions with redox partners.93,95 In contrast, mutation E305G has been shown to cause 17,20-lyase deficiency by selectively disrupting binding of 17α-hydroxypregnenolone and DHEA synthesis despite enhanced conversion of 17α-hydroxyprogesterone to androstenedione.96 This unusual variant of isolated 17,20-lyase deficiency provides further genetic evidence that the flux of androgens derived from conversion of 17α-hydroxyprogesterone to androstenedione in the minor Δ4 pathway is not sufficient to form normal male external genitalia. One of the first patients reported to have isolated 17,20 lyase deficiency was recently found to have a homozygous mutation in P450 oxidoreductase (G539R), further emphasizing the crucial role of efficient electron transfer in the 17,20 lyase reaction.97
P450c21
Microsomal P450c21 performs the 21-hydroxylation of the Δ4 steroids 17α-hydroxyprogesterone and progesterone, an essential step in the biosynthesis of both mineralocorticoids and glucocorticoids (see Fig. 1-2). The human P450c21 protein is found only in the adrenal glands; the extra-adrenal 21-hydroxylase activity found in other organs such as the liver and the aorta98 is not catalyzed by P450c2199 but appears to be catalyzed by CYP2C9, CYP3A4, and possibly CYP2C19 and other enzymes as well.100,101
The locus containing the CYP21 genes is among the most complex in the human genome and explains why 21-hydroxylase deficiency (affecting 1 of 14,000 live births) is one of the most common autosomal-recessive diseases. The CYP21A2 gene and the CYP21A1 pseudogene lie on chromosomal locus 6p21.1 in the midst of the human leukocyte antigen (HLA) locus. Because the HLA locus is highly recombinogenic, exchange between the CYP21A1 and CYP21A2 loci is common. Thus 85% of cases of 21-hydroxylase deficiency derive from micro- or macrogene conversion events where some or all of the CYP21A1 pseudogene replaces the corresponding area of the CYP21A2 gene, thus reducing the expression of the encoded P450c21 protein and/or impairing its activity.102 In addition, at least eight additional genes lie in this locus (Fig. 1-5), including the liver-specific C4A and C4B genes; the adrenal-specific “ZA” and “ZB” genes; and the ubiquitously expressed tenascin X or TNXB gene,103 the disruption of which is one cause of Ehlers-Danlos syndrome.104 Occasionally, a patient with 21-hydroxylase deficiency and Ehlers-Danlos syndrome will have a contiguous gene syndrome with tenascin X deficiency as well.105
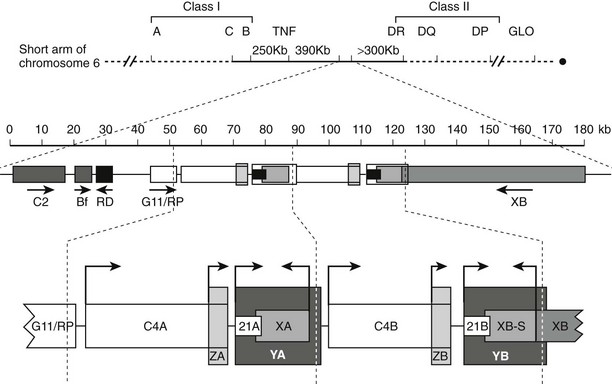
FIGURE 1-5 Genetic map of the human leukocyte antigen (HLA) locus containing the genes for P450c21. The top line shows the p21.1 region of chromosome 6, with the telomere to the left and the centromere to the right. Most HLA genes are found in the class I and class II regions; the class III region containing the CYP21 genes lies between these two. The second line shows the scale (in kb) for the diagram immediately below, showing (from left to right) the genes for complement factor C2, properdin factor Bf, and the RD and G11/RP genes of unknown function; arrows indicate transcriptional orientation. The bottom line shows the 21-hydroxylase locus on an expanded scale, including the C4A and C4B genes for the fourth component of complement, the “CYP21A” pseudogene (CYP21A1, 21A), and the active “CYP21B” gene (CYP21A2, 21B) that encodes P450c21. XA, YA, and YB are adrenal-specific transcripts that lack open reading frames. The XB gene encodes the extracellular matrix protein tenascin-X; XB-S encodes a truncated adrenal-specific form of the tenascin-X protein whose function is unknown. ZA and ZB are adrenal-specific transcripts that arise within the C4 genes and have open reading frames, but it is not known if they are translated into protein; however, the promoter elements of these transcripts are essential components of the CYP21A1 and CYP21A2 promoters. The arrows indicate transcriptional orientation. The vertical dotted lines designate the boundaries of the genetic duplication event that led to the presence of A and B regions.
Much less is known about the enzymology of P450c21 than of P450c17, but the available evidence suggests that unlike P450c17, P450c21 is not very sensitive to the abundance of POR or cytochrome b5. It is clear that genotype consistently predicts phenotype in very severe and very mild cases of 21-hydroxylase deficiency. In contrast, patients with P450c21 variants (e.g., the common Pro30Leu and Val281Leu mutations and less common mutations Arg339His and Pro453Ser), which have 20% to 50% of wild-type activity,102,106 can have various phenotypes, implying additional factors that can modify the clinical manifestations of 21-hydroxylase deficiency.
P450c11β and P450c11AS
The classical descriptions of distinct deficiencies in 11β-hydroxylase, 18-hydroxylase (also called corticosterone methyl oxidase I, or CMOI), and 18-oxidase (CMOII) suggested that three enzymes executed these three respective transformations.107,108 Analogous to the scenario for P450c17, a single enzyme109 and corresponding gene110 were found in bovine adrenals that possessed all three activities. In contrast, humans have two genes named CYP11B1 and CYP11B2111 that encode the mitochondrial enzymes 11β-hydroxylase (P450c11β) and aldosterone synthase (P450c11AS), respectively, and rats but not mice have three functional CYP11B genes.112 Although P450c11β and P450c11AS both possess 11β-hydroxylase activities, P450c11AS also performs the two oxygenations at C18 required for aldosterone biosynthesis.113,114 Mutations in CYP11B1 cause 11β-hydroxylase deficiency,115 whereas defects in CYP11B2 cause either CMOI or CMOII deficiencies.116 Severe defects can impair all P450c11AS activities, leading to the clinical phenotype of CMOI deficiency,113 whereas P450c11β provides 11β-hydroxylase activity in the zona fasciculata. Fortuitous site-directed mutagenesis experiments of nature have found amino acid substitutions such as Arg181Trp plus Val386Ala, which mainly impair 18-oxidase activity and lead to CMOII deficiency.117
The coding regions of the CYP11B1 and CYP11B2 genes share 93% amino acid identity and the same exonic gene structure found in all mitochondrial P450 genes.118 Despite the sequence similarities of these tandem genes, located within 40 kb on chromosome 8q24.3, the expression of P450c11AS is restricted to the adrenal zona glomerulosa, whereas P450c11β is found in the zona fasciculata and zona reticularis. The regulation of P450c11β is driven mainly by cAMP in response to ACTH, whereas P450c11AS expression derives from potassium and angiotensin II activation of the protein kinase C pathway.119 Thus, under normal circumstances, 18-hydroxylase and 18-oxidase activities are restricted to the zona glomerulosa, where 17-hydroxylase activity is low, limiting the repertoire of steroids that can undergo 18-oxygenation.
Although the organization of two highly homologous, adjacent CYP11B1 and CYP11B2 genes on chromosome 8 is reminiscent of the genetics of the CYP21A1 and CYP21A2, gene conversion in the CYP11B locus occurs rarely.120 Instead, a clinical entity called glucocorticoid remediable aldosteronism (GRA) arises when an unequal crossing over of the CYP11B1 and CYP11B2 genes creates a third, hybrid gene in which the ACTH-regulated promoter of CYP11B1 drives expression of a chimeric protein with aldosterone synthase activity.121,122 As a result, 18-hydroxylase and 18-oxidase activities are ectopically expressed in the zona fasciculata, leading to elevated renin-independent production of aldosterone, as well as 18-oxygenated metabolites of cortisol. The expression of this gene is suppressed by blunting ACTH production with glucocorticoids such as dexamethasone, which is used for diagnosis and treatment.123 The prevalence of GRA varies from nil to as high 2% of referred patients with hypertension.124
The genetics of GRA has assisted in the precise identification of residues in P450c11AS that enable 18-oxygenase activities. Residues 288, 296, 301, 302, 325, and perhaps most importantly, 320 are critical for 18-oxygenase activities.125,126 Therefore, crossovers 3′ to codon 320 do not enable aldosterone synthase activity. These key residues lie in or near the I-helix, which contains the catalytically important threonine residue implicated in oxygen activation for almost all P450s; thus, these mutations would be expected to alter active site geometry.
P450aro (Aromatase)
The oxidative demethylation of C19 steroids, mainly androstenedione and testosterone, consumes three equivalents of molecular oxygen and NADPH, yielding formic acid and C18 steroids with an aromatic A-ring, hence the common name for this enzyme, aromatase. As is the case for P450scc, each subsequent oxygenation proceeds with greater efficiency, aiding in the completion of this transformation that is essential for estrogen biosynthesis in all animals.127 The mechanism of this aromatization must account for the incorporation of the final oxygen atom from molecular oxygen into the formic acid byproduct. The weight of evidence favors a hydroxylation at C2 of 19-oxo-androstenedione, followed by an enzyme-assisted rearrangement and tautomerization of the intermediate dienone to the phenolic A-ring.128
P450aro is expressed in steroidogenic tissues (ovarian granuloma cells, placenta), in brain, and in nonsteroidogenic tissues, especially fat and bone.127 The CYP19A1 gene for P450aro spans over 75 kb129 and contains five different transcriptional start sites130 with individual promoters that permit the tissue-specific regulation of expression in diverse tissues. P450aro is a glycoprotein, but glycosylation per se does not appear to affect activity.
Studies of patients with aromatase deficiency confirm that biologically significant estrogen synthesis derives entirely from this enzyme,131,132 although dietary phytoestrogens can provide some estrogen action in mice with targeted deletion of the aromatase gene.133 Although very few cases of aromatase deficiency have been described, they are highly informative “knockouts of nature” that illustrate principles of fetoplacental steroidogenesis. In fetuses homozygous for aromatase deficiency, the principal manifestation results from its deficiency in the placenta,131 because ovarian steroidogenesis is quiescent during fetal life.134 The fetal adrenal makes large amounts of C19 steroids, principally DHEA-S, much of which is 16α-hydroxylated in the fetal liver before undergoing metabolism via steroid sulfatases, 3β-HSD1, aromatase, and 17β-HSD1 in the placenta to produce estriol, the characteristic estrogen of pregnancy. Although huge amounts of estriol and estradiol are produced by the fetoplacental unit, estrogens are not needed for fetal development, the maintenance of pregnancy, or the onset of parturition; all of these processes proceed normally in fetuses lacking StAR, P450c17, or aromatase, or even in fetuses wholly lacking adrenal glands because of mutations in SF-1 or DAX-1.135 However, in the absence of placental aromatase activity, androgenic C19 steroids derived from the fetal adrenal are passed into the maternal circulation, causing marked virilization of the mother.131
Furthermore, in pregnancies in which the mother has poorly treated 21-hydroxylase deficiency, maternal testosterone values can exceed 300 ng/dL (a midpubertal value for males), yet the fetus is not virilized136 because the maternal testosterone is efficiently metabolized to estradiol by placental aromatase. Thus, placental aromatase is a key enzyme in protecting the fetus and mother from unwanted androgen exposure. After birth, individuals with aromatase deficiency grow normally and continue linear growth after completion of puberty, with males producing normal amounts of testosterone. However, when treated with estrogens, aromatase-deficient subjects fuse their epiphyses and cease linear growth.137 These observations provide powerful evidence that bony maturation and epiphyseal fusion in children is mediated by estrogens, not androgens, even in males. These observations have led to the experimental use of aromatase inhibitors in various disorders of accelerated bone maturation.
Redox Partner Proteins
The proteins collectively referred to as redox partners channel reducing equivalents from NADPH to the heme centers of P450 enzymes.3 Recent studies, however, suggest that these proteins act to promote catalysis by more than just their electron-transfer properties. Because of this, the precise nature of the interactions of the P450s with their redox partners is of considerable importance. Our understanding of these interactions has been greatly advanced by the x-ray crystal structures of these four proteins.
Adrenodoxin (Ferredoxin)
Adrenodoxin (Adx), also known as ferredoxin, is encoded by a gene on chromosome 11q22 that spans over 30 kb. Adx is a small (14 kD), soluble, Fe2S2 electron shuttle protein that resides either free in the mitochondrial matrix or is loosely bound to the inner mitochondrial membrane.138 Adx is expressed in many tissues, and its expression in steroidogenic tissues is induced by cAMP in parallel with P450scc.139
Bovine Adx consists of two domains,140 a core region and an interaction domain. The core region contains residues 1-55 and 91-end (bovine numbering), including the four cysteines whose sulfur atoms tether the Fe2S2 cluster to the protein. Residues 56 to 90 form the interaction domain, which is a hairpin containing a helix at its periphery that includes acidic residues critical for the interaction of Adx with P450scc141 (specifically, aspartates 72, 76, and 79, plus glutamate 73). The Fe2S2 cluster lies in a protuberance in the molecule at the junction of its two domains. The charged residues of Adx cluster in the interaction domain, giving the molecule a highly negatively charged surface above the Fe2S2 cluster (see Fig. 1-3A). This description of the Adx molecule concurs with earlier studies that showed that overlapping sets of negative charges on Adx drive Adx interactions with positive charges on both P450scc and adrenodoxin reductase (AdR).142 Because a preponderance of the evidence favors a model in which the same surface of Adx shuttles between AdR and the P450 to transport electrons,142,143 a model of how Adx interacts with AdR would approximate how mitochondrial P450s interact with Adx.
Adrenodoxin (Ferredoxin) Reductase
Like Adx, adrenodoxin reductase (AdR) is widely expressed in human tissues, but its expression is two orders of magnitude higher in steroidogenic tissues.144 The primary RNA transcript from the 11-kb AdR gene145 on chromosome 17q24-q25146 is alternatively spliced, generating two mRNA species that differ by only 18 bp,147 but only the protein encoded by the shorter mRNA is active in steroidogenesis.148 Unlike most steroidogenic genes, the promoter for AdR contains six copies of GGGCGGG sequences,145 which is the canonical binding site for the transcription factor SP-1 typically found in “housekeeping genes.” Accordingly, cAMP does not regulate transcription of the AdR gene, as is the case for Adx and P450scc,144 implying that AdR plays additional roles in human physiology beyond steroidogenesis. Given their essential roles in the conversion of cholesterol to pregnenolone, no null mutations in AdR or Adx have been described in humans, and impairment of the Drosophila AdR homologue dare causes developmental arrest and degeneration of the adult nervous system owing to the loss of ecdysteroid production.149
Bovine AdR also consists of two domains, each comprising a β-sheet core surrounded by α-helices.150 The NADP(H)-binding domain is a compact region composed of residues 106 to 331 (bovine numbering), whereas the more open FAD domain, formed by the remaining amino- and carboxy-terminal residues, binds the dinucleotide portion of FAD across a Rossman fold, with the redox-active flavin isoalloxazine ring abutting the NADP(H) domain. By analogy to related structures, including glutathione and thioredoxin reductases, the nicotinamide ring of NADPH is modeled to lie adjacent to the flavin ring in a position to transfer its two electrons to the FAD. Intramolecular electron transfer occurs in the cleft formed by the angled apposition of these two domains. Within this cleft, basic residues abound, including arginines 240 and 244, which are important for interactions with Adx.142,151 Hypothetical docking of the two structures suggests that the negative surface of Adx fits elegantly into the positive surface of AdR, even with NADP(H) bound.150 Basic residues are also critical for the interaction of P450scc with the negative surface charges on Adx,143 so that AdR-Adx docking is expected to share some key features with the mitochondrial P450-Adx interaction.
P450 Oxidoreductase
The flavoprotein P450 oxidoreductase (POR) is expressed widely in human tissues and serves as the sole electron-transfer protein for all microsomal P450s, including xenobiotic-metabolizing hepatic P450s, steroidogenic P450s, and P450s found in other tissues such as the kidney and brain. POR contains two lobes, one binding FAD and the other binding FMN, and a flexible amino terminus that tethers it to the endoplasmic reticulum. NADPH is bound to the cofactor-binding domain above the FAD in a β-sheet-rich FAD domain, and an α-helical connecting domain joins the FAD and the FMN domains.152 A disordered “hinge” of about 25 residues lies between the FMN domain and the connecting domain, suggesting that the FMN and FAD domains can move substantially relative to each other. In the x-ray structure of rat liver POR,152 the FMN and FAD lie at the base of a cleft formed by the butterfly-shaped apposition of the FAD and FMN domains, reminiscent of the electron transfer surface of AdR.150 It is not clear how the surface containing the FMN docks into the redox partner-binding surface of the P450, but the flexible hinge region on which the FMN domain resides suggests that the FMN domain can reorient itself significantly to accommodate docking to the P450 (see Fig. 1-3).3,64 The surface of the electron-donating FMN domain is dominated by acidic residues, whereas the redox-partner binding site of P450 enzymes contain numerous basic residues.
The crystal structure of the complex between the P450 and flavoprotein domains of the bacterial protein P450BM3 serves as a model of this flavoprotein-P450 interaction.153 Negative charges of the FMN domain guide interactions with positive charges on the P450. The FMN approaches no closer than 18 Å from the heme, similar to the 16 Å distance of FAD from the Fe2S2 cluster in the modeled AdR-Adx complex, and presumably similar to the distance of the heme from the Fe2S2 cluster in the P450-Adx complex.150 These distances are too far for electrons to “jump” directly to the heme; rather, electron transfer apparently uses the polypeptide chain as a conduit.153 Basic residues in the redox-partner binding surface are crucial for interactions with POR and for electron transfer,3,154 and these positive charges in human P450c17 are critical for maximal 17,20-lyase activity.78,95 Thus, these structures demonstrate several key principles of the electron transfer proteins involved in human steroidogenesis: NADPH and prosthetic groups lie at the interfaces of protein domains in which electron transfer occurs; the electron transfer surfaces are negatively charged to pair with positive charges on the P450s; the terminal electron transfer moiety (FMN domain or Adx) must be mobile or soluble to pass electrons on to the P450; and electrons flow from the FMN or Fe2S2 cluster along the adjacent polypeptide chain to the heme.
Cytochrome P450 Oxidoreductase Deficiency: A Disorder Affecting Multiple P450 Enzymes
Beginning with a clinical report in 1985,155 several patients have been described with clinical and hormonal findings suggesting partial deficiencies of both 17α-hydroxylase and 21-hydroxylase.156 Some of these individuals were born to mothers who had become virilized during pregnancy, suggesting fetoplacental aromatase deficiency, and many also had the Antley-Bixler congenital malformation syndrome characterized by craniosynostosis and radioulnar synostosis. About half of patients with Antley-Bixler syndrome have normal steroidogenesis and normal genitalia; these subjects have dominant gain-of-function mutations in the gene for fibroblast growth factor receptor type 2 (FGFR2); however, patients with Antley-Bixler syndrome who also have genital anomalies and disordered steroidogenesis do not have FGFR2 mutations.157 The initial report described three patients with Antley-Bixler syndrome, genital ambiguity, and hormonal findings suggestive of partial deficiencies of 17α-hydroxylase and 21-hydroxylase, as well as a fourth patient who was phenotypically normal but had a similar hormonal profile. All had recessive loss-of-function amino acid replacement mutations in POR.158 One of these patients was born to a woman who had become virilized during the pregnancy, suggesting partial fetoplacental aromatase deficiency.
In vitro biochemical assays of the recombinant mutant proteins showed that the mutations in the Antley-Bixler subjects had severely impaired but not totally absent activity, whereas the mutations found in the phenotypically normal subject with amenorrhea were less severe.158 Examination of the POR and FGFR2 genes in a series of 32 patients established that the recessive POR mutations and the dominant FGFR2 mutations segregate completely.159 To date, approximately 50 POR-deficient patients have been described.160 It appears unlikely that subjects will be found who are homozygous for null POR alleles, because knockouts of POR in mice cause embryonic lethality.161,162 On the other hand, liver-specific ablation of the Por gene in mice results in phenotypically and reproductively normal animals with profoundly impaired drug metabolism,163,164 because POR is required for the activities of all hepatic drug-metabolizing P450 enzymes. Consequently, it seems likely that patients with POR deficiency will also metabolize drugs poorly. Reports of Antley-Bixler syndrome in some infants of mothers who ingested fluconazole (an azole drug that inhibits the fungal P450 lanosterol-14-demethylase and also inhibits some human P450s) suggest that haploinsufficiency of POR may be a risk factor for teratogenic effects of some drugs, particularly those that inhibit P450s involved with embryogenesis.158
The human POR gene, located on chromosome 7, consists of 16 exons.160,165 The sequence of this gene in 842 normal persons from four ethnic groups revealed a high degree of polymorphism; most notably, the coding sequence variant A503V was found on ~28% of all alleles.166 This sequence variant reduces the 17α-hydroxylase and 17,20 lyase activities of P450c17 to ~60% of normal159,166 but has no measurable effect on the activities of P450c21167 or hepatic CYP1A2 or CYP2C19.168
Cytochrome b5
The small (12 to 17 kD) hemoprotein cytochrome b5 (b5) is found in many tissues (e.g., as a membrane-bound cytochrome in liver and as a soluble protein lacking the C-terminal membrane anchor in erythrocytes). Importantly, b5 is expressed in both the adrenals and gonads, where it can interact with P450c17; the adrenal expression is zone specific and may contribute to the genesis of adrenarche.85,86 Much evidence has shown that b5 can augment some activities of certain P450 enzymes, and the mechanism of this effect has been presumed to involve electron transfer from b5 to the P450 for the second electron during the P450 cycle.169 Although b5 can certainly receive electrons from flavoproteins like POR, the redox potentials of b5 and one-electron-reduced P450 are unfavorable for b5-to-P450 electron transfer. Indeed, some of the actions of b5 in experimental systems can be observed with apo-b5170 or Mn2+–b5 (which do not transfer electrons), including the stimulation of 17,20-lyase activity of human P450c17.63,64 These experiments suggest that b5 does not act alone as an electron donor but rather functions in concert with POR to somehow aid catalysis.
The soluble form of bovine b5 was one of the first proteins studied by x-ray crystallography, and a wealth of structural data for b5 have been acquired using molecular dynamics and nuclear magnetic resonance (NMR) spectroscopy for both the holo- and apo-b5.171,172 Analogous to Adx, b5 consists of two domains: (1) a heme-liganding core 1 domain (residues 40 to 65, bovine numbering) and (2) a structural core 2 domain, from which the C-terminal membrane-anchoring helix extends. The heme extends more to the periphery of b5 than does the Fe2S2 cluster of Adx, and the entire surface is dominated by negatively charged residues rather than just one cluster of negative charges near the heme. In addition, the core 1 domain acquires considerable conformational flexibility in apo-b5, whereas the core 2 domain remains folded as in holo-b5.172 Finally, the C-terminal membrane-spanning helix (exiting the core 2 domain) is required to stimulate the 17,20-lyase activity of human P450c17, but the signal peptide is not.173 Genetic and biochemical studies have implicated basic residues in P450c17, including R347, R358, and perhaps R449 and K89, as important for its interaction with b5,78,95,173 while E48 and E49 of b5 are required for high 17,20-lyase activity.174 The molecular details of how addition of b5 to the P450c17 POR complex augments 17,20-lyase activity, however, is not yet known.
Steroidogenic Dehydrogenases and Reductases
3β-Hydroxysteroid Dehydrogenase/Δ5-Δ4-Isomerases
Conversion of Δ5 steroids into their Δ4 congeners, a step required for the production of progestins, mineralocorticoids, glucocorticoids, and sex steroids, consists of two chemical transformations, both performed by the 3β-hydroxysteroid dehydrogenase/Δ5/4-isomerase (3β-HSD) enzymes. The first reaction is the oxidation of the 3β-hydroxyl group to the ketone, and during this process NAD+ is converted to NADH. The intermediate Δ5, 3-ketosteroid remains tightly bound to the enzyme with nascent NADH, and the presence of NADH in the cofactor binding site activates the enzyme’s second activity, the Δ5→Δ4-isomerase activity.175 Competition experiments have shown that the dehydrogenase and isomerase activities reside in a single active site,176 yet these enzymes are often referred to by their dehydrogenase activity alone.
Although rodents contain multiple 3β-HSD isoforms, only two active genes have been identified in humans. The type 1 enzyme (3β-HSD1) is expressed in the placenta, liver, brain, and some other tissues.177 This isoform is required for placental progesterone production during pregnancy, which may explain why a deficiency of 3β-HSD1 has never been described. In contrast, the type 2 enzyme (3β-HSD2) is by far the principal isoform in the adrenals and gonads.178 Deficiency of 3β-HSD2 causes the rare form of congenital adrenal hyperplasia known as 3β-HSD deficiency.75 The presence of the type 1 isozyme in these patients helps to explain the paradox of why 46,XX individuals born with severe 3β-HSD2 deficiency can virilize slightly in utero: The 3β-HSD block in the adrenal diverts Δ5-steroids away from cortisol and toward DHEA; extra-adrenal 3β-HSD1 enables testosterone synthesis despite 3β-HSD2 deficiency in the adrenal.
The types 1 and 2 enzymes share 93.5% amino acid identity, and all biochemical studies comparing the two enzymes yield very similar results. The enzymes are strongly inhibited by Δ4 products179 and by synthetic Δ4 steroids such as medroxyprogesterone acetate.68 Both enzymes have very similar affinities for the Δ5,17-ketosteroid pregnenolone, 17α-hydroxypregnenolone, and DHEA of about 5 µM68,176 and also convert the 17β-hydroxysteroid androsta-5-ene-3β,17β-diol to testosterone. The enzymes are primarily membrane bound and are found both in the microsomal and mitochondrial fractions during subcellular fractionation.176 Ultrastructural studies using immunogold labeling confirm that at least in bovine adrenal zona glomerulosa cells, 3β-HSD immunoreactivity is indeed found not only in mitochondria and the endoplasmic reticulum but also in the cytoplasm.180
Considerable evidence suggests that 3β-HSD activity is an important factor in regulating adrenal dehydroepiandrosterone sulfate (DHEA-S) production. The human fetal adrenal, which produces vast amounts of DHEA-S, contains little 3β-HSD immunoreactivity.181 Furthermore, the expression of 3β-HSD in the innermost regions of the adrenal cortex declines as the zona reticularis develops in childhood,86,182 and 3β-HSD immunoreactivity is low in the zona reticularis of the adult rhesus macaque85 and of humans.86 Thus the development of an adrenal cell type (reticularis) that is relatively deficient in 3β-HSD activity appears to be a necessary component of adrenarche, in which adrenal production of the Δ5 steroids DHEA and DHEA-S rises exponentially.183
17β-Hydroxysteroid Dehydrogenases
There are at least 14 human 17β-hydroxysteroid dehydrogenase (17β-HSD) isoforms; these isoforms vary widely in size, structure, substrate specificity, cofactor utilization, and physiologic functions.184 This section focuses on the human isoforms that possess significant, rather than gratuitous, 17β-HSD activity.
17β-HSD Type 1
The interconversion of estrone and estradiol by 17β-HSD1 has been studied more extensively than any other human steroidogenic enzyme, and in the late 1980s, three independent groups reported the cloning of its cDNA, the first of any human HSD.185 Located on chromosome 17q25186 adjacent to a pseudogene, the HSD17B1 gene187 encodes a 34-kD protein subunit that is expressed primarily in the placenta and in ovarian granulosa cells of developing follicles.186 The enzyme, which is active only as a dimer, accepts mainly estrogens such as estrone, although it also has low catalytic activity for the conversion of androstenedione to testosterone and DHEA to androsta-5-ene-3β,17β-diol.188 Although the enzyme can oxidize 17β-hydroxysteroids in the presence of NAD+ in vitro at a high pH, the enzyme functions in vivo to reduce estrone to estradiol and 16α-hydroxyestrone to estriol.188
Detailed kinetic analyses of 17β-HSD1 began in the late 1960s, and attempts to identify active-site residues using affinity labels and mechanism-based inactivators followed.189 Sequence alignments with other members of the SDR family identified a Tyr-X-X-X-Lys active-site motif in residues 155 to 159. These predictions were verified when the x-ray structure of 17β-HSD1 was solved.190 The structure demonstrates that cofactor lies across the β-sheet core of the protein in a Rossman fold characteristic of all SDR enzymes. Steroid appears to dangle from the top of the enzyme almost perpendicular to the cofactor, with a hydrophobic pocket holding the body of the steroid in place while the 3-hydroxyl forms hydrogen bonds with His 221 and Glu 282. At the place where the steroid and cofactor meet, Ser 142, Tyr 155, and Lys 159 help to form a proton-relay system that drives catalysis.
Because steroid flux to estrogens preferentially occurs via the aromatization of androstenedione to estrone, 17β-HSD1 appears to be required for the conversion of estrone to biologically active estradiol in the ovary and placenta.186 This role has not been proven unequivocally, because no cases of human 17β-HSD1 deficiency have been reported. Such a disease is theoretically compatible with life, because fetuses with aromatase deficiency and estrogen insensitivity (ER-α mutations) are viable.132 Nevertheless, this enzyme is probably critical for ovulation and may be important in the pathogenesis and progression of estrogen-dependent breast cancers.191
17β-HSD Type 2
In contrast to the “activating” role of 17β-HSD1 in the placenta and ovary, human endometrium was known to inactivate estradiol by the conversion to estrone. This activity, which was induced by progestins, was not 17β-HSD1, because 17β-HSD1 mRNA was not detected in the human uterus.186 Instead, a cDNA encoding microsomal HSD17B2 was cloned192 and found to be expressed in endometrium, placenta, and other tissues.193 In fact, 17β-HSD2 not only converts estradiol to estrone but also oxidizes testosterone and DHT to their inactive 17-ketosteroid homologues, androstenedione and 5α-androstanedione, respectively. The widespread tissue distribution and broad substrate specificity of 17β-HSD2 suggests that its role in human physiology is to protect tissues from excessive exposure to active steroid hormones by oxidation to inactive 17-ketosteroids.184 This role is again somewhat speculative, given that a human deficiency of this enzyme has not been described; but 17β-HSD2 is certainly the most active human inactivating (oxidizing) 17β-HSD that has been described to date. The type 2 enzyme also oxidizes 20α-dihydroprogesterone to progesterone, but this activity is low relative to its 17β-HSD activity.192
17β-HSD Type 3
Because 17β-HSD1 shows poor activity with C19 steroids, at least one other 17β-HSD enzyme capable of reducing androstenedione to testosterone was postulated to complete testosterone biosynthesis. Furthermore, reports of male pseudohermaphrodites lacking this putative androgenic “17-ketosteroid reductase” activity surfaced in the 1970s.194 When the large, complex gene for 17β-HSD3 was cloned, patients with “17-ketosteroid reductase deficiency” were found to harbor mutations in this HSD17B3 gene,195,196 proving the central role of this enzyme in male sexual differentiation and marking 17β-HSD3 as the only 17β-HSD enzyme whose role in human physiology is genetically established by a deficiency syndrome. Nonetheless, patients with 17β-HSD3 deficiency make small amounts of testosterone, suggesting that one or more additional human 17β-HSD enzymes convert androstenedione to testosterone. Similarly, the human ovary exports some testosterone despite an absence of 17β-HSD3 expression, and women with 17β-HSD3 deficiency produce normal amounts of androgens and estrogens.197
Unlike 17β-HSD1, which has been the subject of intense biochemical study, relatively little is known about 17β-HSD3 enzymology. This knowledge gap is at least in part caused by the very hydrophobic nature of the encoded 310-amino-acid protein, hampering the expression of this enzyme in bacteria. From experiments in transiently transfected HEK-293 cells, we know that 17β-HSD3 reduces all of the C19 17-ketosteroids that serve as precursors of testosterone and DHT in human beings, including DHEA, 5α-androstanedione, and androsterone.196 The conversion of DHEA to androsta-5-ene-3β,17β-diol by 17β-HSD3 may contribute significantly to testicular testosterone synthesis. Estrogens such as estrone are poor substrates for human 17β-HSD3.188
17β-HSD Types 4 and 5
Many additional 17β-HSD isoforms have been described in rodents and in humans, but the activities of these isoforms for steroids are generally poor. For example, the type 4 enzyme is a trifunctional protein located in peroxisomes,198 but its (oxidative) HSD activity toward estradiol is 106 times slower than its 3-hydroxyacyl-coenzyme A dehydrogenase activity.199 Deficiency of the type 4 enzyme causes Zellweger syndrome, in which bile acid synthesis is disturbed, but steroidogenesis is not affected.200 Thus, this enzyme has 17β-HSD activity as one of its repertoire of transformations, but steroidogenesis is not its principal physiologic function.
Unlike 17β-HSDs types 1 to 4, which are SDR enzymes, the type 5 enzyme is an AKR enzyme, which is expressed both in steroidogenic and non-steroidogenic tissues.201 There has been some confusion about the nature of the type 5 enzyme because of its multiple activities and inconsistent results from different laboratories. Originally described as hepatic 3α-HSD type 2 for its ability to reduce DHT to 3α-androstanediol,202 this protein was later found to also have 17β-HSD activity,201 including reducing androstenedione to testosterone.203 This enzyme, now known as AKR1C3 (see 3α-HSDs), may account for much of the extratesticular androstenedione-to-testosterone conversion, although its catalytic efficiency as a 17β-HSD is poor204 compared to its 20α-HSD activity with progesterone and 11-deoxycorticosterone205 or its prostaglandin dehydrogenase activity, reducing PGH2 to PGF2α.206 Nevertheless, AKR1C3 is more highly expressed in the human fetal adrenal during the time of sexual differentiation than 17β-HSD3207 and may participate in testosterone production, particularly in virilizing congenital adrenal hyperplasias.
Steroid 5α-Reductases
The conversion of testosterone to 5α-dihydrotestosterone (DHT) in target tissues was described in the 1960s,208 and studies using fibroblasts suggested that at least two human enzymes with different pH optima and genetics performed these transformations.209 These initial results were confirmed when the genes encoding the type 1210 and type 2211 enzymes were cloned, and patients with clinical 5α-reductase deficiency were found to have mutations in the SRD5A2 gene. The two isoforms are very hydrophobic 30-kD microsomal proteins that share 50% identity. The type 1 enzyme is limited to the nongenital skin and liver, and it is not expressed significantly in peripheral tissues of the fetus, which explains why a deficiency of the type 2 enzyme is not compensated for by the type 1 enzyme.212 The type 2 enzyme remains the predominant enzyme in genital skin, male accessory sex glands, and prostate, whereas the type 1 enzyme accounts for most of the hepatic 5α-reduction.
Although 5α-reductase activity is generally discussed in the context of male genital differentiation and androgen action, both isoenzymes reduce a variety of steroids in what are believed to be degradative pathways in human beings. In fact, progesterone, 17α-hydroxyprogesterone and related C21 steroids are the best substrates for both 5α-reductases, particularly the type 1; cortisol, cortisone, corticosterone, and related compounds are also good substrates.213 The 5α- (and 5β-) reduced steroids may be metabolized further and conjugated for excretion in the urine. Given the importance of 5α-reductase type 2 in prostate growth, inhibitors of the type 2 enzyme have been developed for the treatment of prostatic hyperplasia and the prevention of its recurrence after surgery.214 Finasteride selectively inhibits human 5α-reductase type 2, whereas dutasteride inhibits both the type 1 and type 2 isoenzymes. Both drugs are approved for treatment of prostatic hyperplasia in the United States.
Although the function of 5α-reductase type 2 is firmly established from the studies of male pseudohermaphrodites with this deficiency, the role of the type 1 isoform in human beings is less clear. Given the abundant expression of the type 1 isoform in liver and its high activity with C21 steroids, this enzyme has been ascribed a role of degrading circulating C21 steroids in preparation for excretion in the urine. However, disruption of Srd5a1 in mice results in delayed parturition, a defect that can be rescued with 5α-androstane-3α,17β-diol.215 In immature mice, 5α-reductase type 1 is expressed both in the ovary and in the Leydig cells, and this enzyme participates in the testicular synthesis of 5α-androstanediol via two pathways.216 Whether 5α-reductases are expressed in human adrenal or gonads in normal physiology or in pathologic states is not known.
3α-Hydroxysteroid Dehydrogenases
The four major human 3α-hydroxysteroid dehydrogenases (3α-HSDs) are AKR enzymes with reductive preferences that belong to the AKR1C family. The 3α-HSDs types 1, 2, 3, and 4 are trivial names for AKR1C4, 1C3, 1C2, and 1C1, respectively, which are located in tandem on chromosome 10p14-p15. Each enzyme has its characteristic tissue distribution217,218 and repertoire of catalytic activities.204 AKR1C3 also performs the 17β-HSD reaction with androstenedione and is known also as 17β-HSD5. All of the AKR1C isoforms catalyze additional reactions, such as the 20α-reduction of pregnanes. In the brain, 3α-HSDs reduce 5α-dihydroprogesterone to tetrahydroprogesterone (allopregnanolone), which is an allosteric activator of the GABAA receptor-chloride channel complex, with a nanomolar affinity.219 AKR1C4 is abundant in liver but has been found in adrenal and gonads; AKR1C3 was cloned from liver, prostate, and brain; AKR1C2 is found in the prostate and brain; and AKR1C1 is abundant in the uterus. Furthermore, the amino acid compositions of isozymes of the type 2 and 3 enzymes differ by a few residues. These minor differences in composition, however, cannot be neglected, because these differences might alter substrate utilization.
Recent studies have indicated an important role for 3α-HSDs in the nervous system. Antidepressant drugs in the selective serotonin reuptake inhibitor class (e.g., fluoxetine and paroxetine) directly lower the Km of rat brain type 2 3α-HSD for 5α-dihydroprogesterone by almost 10-fold,220 which explains why these drugs augment brain allopregnanolone concentrations and perhaps contribute to their antidepressant activity. In addition, x-ray crystallography has shown that the β subunit of the mammalian voltage-gated potassium channel is a tetrameric structure221 in which each subunit closely resembles the rat liver 3α-HSD (AKR1C9)222 and even contains bound NADP+ with high occupancy. Although the broader implications of this work are not yet known, these studies suggest a role of HSDs in coupling intracellular redox state to membrane excitation.
The 3α-HSDs differ from the 11β-HSDs, 3β-HSDs, and 17β-HSDs types 1 to 4 in several respects, because all reductive 3α-HSDs are AKR enzymes rather than SDR enzymes. As AKR enzymes, they function as monomers with a TIM-barrel structure; they bind cofactor with the nicotinamide ring draped across the mouth of the “barrel” rather than lying on a Rossman fold; and their kinetic mechanisms are highly ordered, with cofactor dissociation the final and rate-limiting step.223 Tight NADP(H) binding derives from interaction of Arg276 with the 2′-phosphate, and mutation of Arg276 eliminates a conformational change associated with tight binding224 and attenuates or reverses the preference for ketosteroid reduction in intact cells.6 As shown in the structure of AKR1C9,222 their active sites also contain tyrosine and lysine residues to facilitate proton transfer during catalysis, but these residues are distantly located in linear sequence rather than confined to the Tyr-X-X-X-Lys motif as in SDR enzymes.
In contrast to the reductive 3α-HSDs, the oxidative 3α-HSDs belong to the SDR family and show greatest similarity to the retinol dehydrogenase or cis-retinol/androgen dehydrogenase (RoDH/CRAD) subfamily.225 Although several of these RoDH/CRAD enzymes show some 3α-HSD activity, the most active enzyme appears to be RODH, the microsomal 3α-HSD, 3 (α→β)-hydroxysteroid epimerase, or formally, 17β-HSD6, whose cDNA was first cloned from prostate.226 This enzyme converts the inactive C19 steroid 5α-androstane-3α,17β-diol to DHT and thus may execute the final step in the backdoor pathway from 17α-hydroxyprogesterone to DHT via androsterone. However, prolonged incubation of 3α-hydroxysteroids with cells transfected with the cDNA for 17β-HSD6 or with microsomes containing the recombinant enzyme yields subsequent 3-ketosteroid metabolites, including both 3α- and 3β-hydroxysteroids and 17β-hydroxysteroids.227 Hence, this enzyme has complex catalytic flexibility and may serve a variety of biological functions.
11β-Hydroxysteroid Dehydrogenases
The 11β-hydroxysteroid dehydrogenases (11β-HSDs) regulate the bioactivity of endogenous and synthetic glucocorticoids, and a comparison of the types 1 and 2 enzymes exemplifies some key principles of HSD enzymology (Table 1-3). Both enzymes are hydrophobic, membrane-bound proteins that bind cortisol/cortisone and corticosterone/11-dehydrocorticosterone, but otherwise their properties and physiologic roles differ substantially228 (see Table 1-3). The type 2 enzyme shares only 21% sequence identity with 11β-HSD1, whereas 11β-HSD2 and 17β-HSD2 share 37% identity and favor steroid oxidation in vivo. Thus, 11β-HSD1 and 2 are only distantly related members of the SDR family, yet they perform opposite functions in specific tissues in human physiology and pharmacology.
Table 1-3
Comparison of 11β-Hydroxysteroid Dehydrogenases Types 1 and 2
| Property | Type 1 | Type 2 |
| Size | 34 kD | 41 kD |
| Orientation in ER | Luminal | Cytoplasmic |
| Expression | Liver, decidua, lung, gonad, pituitary, brain, fat, bone | Kidney, placenta, colon, salivary gland |
| Principal reaction | Reduction | Oxidation |
| Cofactor preference | NADPH via H6PDH | Cytoplasmic NAD+ |
| Substrate binding | Low affinity (Km 0.1-1 µM) | High affinity (Km 0.01-0.1 µM) |
| Inhibition by carbenoxolone | Moderate | Strong |
| Deficiency state | CRD (with H6PDH) | AME |
The 34-kD type 1 enzyme (11β-HSD1)229 is expressed in the liver, testis, lung, fat, and proximal convoluted tubule. The type 1 enzyme catalyzes both the oxidation of cortisol, using NADP+ as cofactor (Km 1 to 2 µM), and the reduction of cortisone, using NADPH cofactor (Km 0.1-1 µM), with cortisone reduction being the dominant reaction in transfected cells.230,231 Many synthetic glucocorticoids (e.g., prednisone and cortisone) are 11-ketosteroids that must be reduced to their 11β-hydroxy derivatives to attain biological activity, and these transformations are performed mainly in the liver by 11β-HSD1. In contrast, when recombinant 11β-HSD1 is studied in vitro, cortisol oxidation with NADP+ is most efficient, and cortisone reduction is only achieved if NADP+ is scrupulously removed with an enzymatic NADPH regeneration system.232,233 The net flux of steroid driven by 11β-HSD1 depends on the relative concentrations of available NADPH and NADP+, which usually favors reduction in cells, especially given the high Km of the enzyme for cortisol.233
The mechanism for the discrepancy between the prominent oxidative preference in vitro and the reductive dominance in vivo, however, is more complex and derives from the localization of 11β-HSD1 in the lumen of the endoplasmic reticulum.234 In this compartment, the ratio of NADPH to NADP+ is not maintained by the cytoplasmic NADP+-coupled dehydrogenases (mainly glucose-6-phosphate dehydrogenase) but by hexose-6-phosphate dehydrogenase (H6PDH). Indeed, the disorder cortisone reductase deficiency (CRD), which is characterized by high ratios of cortisone to cortisol and of their respective metabolites in blood and urine,235 is not caused by mutations in the coding regions of HSD11B1. Instead, CRD subjects most commonly harbor inactivating mutations in H6PDH236 which impair NADPH regeneration within the endoplasmic reticulum, where 11β-HSD1 resides. The genetics and pathophysiology of CRD provide an excellent example of the critical role of nicotinamide cofactors in HSD function and biology.
In contrast, the 41-kD type 2 enzyme237 catalyzes the oxidation of cortisol and corticosterone using NAD+, and although this enzyme has a high affinity for its steroid substrates (Km 0.01 to 0.1 µM),238 catalysis of the reductive reactions by 11β-HSD2 has not been conclusively demonstrated. Cortisol is a potent agonist at the mineralocorticoid (glucocorticoid type 2) receptor in the distal nephron, but its oxidized 11-keto derivative, cortisone, is not a mineralocorticoid. The reason cortisol does not act as a mineralocorticoid in vivo, even though cortisol concentrations can exceed aldosterone concentrations by three orders of magnitude, is because cortisol is enzymatically converted to cortisone in the cells lining the cortical and medullary collecting ducts. The type 2 enzyme inactivates the mineralocorticoid activity of cortisol in the kidney tubule,239 and inactivating mutations in the type 2 enzyme cause a syndrome of apparent mineralocorticoid excess240 (see also Chapter 13). The presence of the type 2 enzyme in the placenta241 also inactivates endogenous and synthetic corticosteroids such as prednisolone, allowing the use of these agents during pregnancy without affecting the fetus. In contrast, 9-fluorinated steroids such as dexamethasone are minimally inactivated by the type 2 enzyme, primarily because of a shift in the oxidation/reduction preference rather than a reduction in affinity for the enzyme.242 It is this resistance to inactivation by placental 11β-HSD2 that is essential for synthetic glucocorticoids to “cross the placenta” and to exert a pharmacologic effect on the fetus. Furthermore, the relatively high placental concentrations of NADP+ may also favor the oxidative action of 11β-HSD1, so that both placental enzymes protect the fetus from the high maternal concentrations of cortisol that occur during pregnancy.228
Pathways
Adrenal Steroidogenic Pathways
Diagrams of steroidogenic pathways, such as that shown in Fig. 1-2, typically combine the pathways from multiple cell types to give a comprehensive and inclusive illustration; however, such diagrams may be misleading, since dominant pathways by which specific steroids are synthesized differ in each steroidogenic cell type. The three major pathways of steroidogenesis in the human adrenal are shown in Fig. 1-6. The adrenal zona glomerulosa (ZG) is characterized by three features: its expression of AII receptors, its unique expression of P450c11AS, and its inability to express P450c17. This combination of factors permits the ZG to produce aldosterone under regulation by the renin/angiotensin system. By contrast, the adrenal zona fasciculata (ZF) does not express AII receptors or P450c11AS but instead expresses the melanocortin type 2 receptor (MC2R, the receptor for ACTH) and P450c11β, which cannot convert 18-hydroxycorticosterone to aldosterone and has minimal capacity to convert corticosterone to 18-hydroxycorticosterone.243 Both the ZG and ZF express P450c21, but the ZF also expresses P450c17, which allows aldosterone and cortisol synthesis, respectively, in these zones. The ZF, however, expresses very little cytochrome b586; consequently, P450c17 in the ZF catalyzes 17α-hydroxylation but very little 17,20 lyase activity. Thus the ZF produces two glucocorticoids, mainly cortisol and smaller amounts of corticosterone, under the influence of ACTH. Patients with severe mutations in P450c17 cannot synthesize cortisol but instead increase corticosterone production244 (as do rodent adrenals, which lack P450c17), explaining why they are not glucocorticoid deficient, despite the lack of cortisol (see Fig. 1-6). The adrenal zona reticularis (ZR) also expresses MC2R but very little P450c21 or P450c11β, and as a result, the ZR produces minimal amounts of cortisol. By contrast, the ZR expresses large amounts of cytochrome b5,86 maximizing the 17,20 lyase activity of P450c17,64 so that DHEA is produced and sulfated by SULT2A1 to DHEAS.245 The ZR expresses relatively little 3βHSD2, and the Km of 3βHSD2 is ~5 µM for pregnenolone and 17-hydroxypregnenolone,68 whereas the Km for both activities of P450c17 is ~1 µM,64 so that abundant DHEA is produced. As DHEA accumulates, small amounts are converted to androstenedione, and very small amounts of this androstenedione are converted to testosterone, probably by AKR1C3/17βHSD5. The pattern of steroid products secreted by each adrenal zone is determined by the enzymes produced in that zone and may be logically deduced from an understanding of their specific enzymatic properties.245
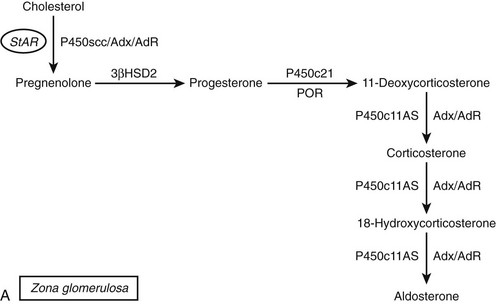
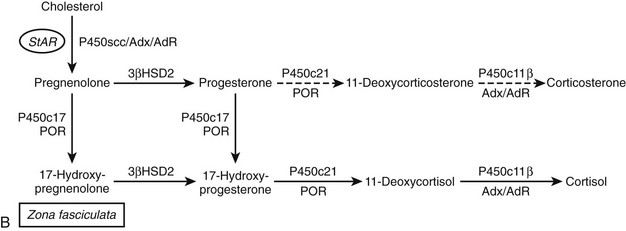
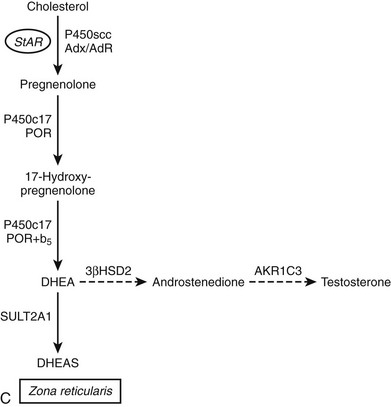
FIGURE 1-6 Major steroidogenic pathways of the human adrenal cortex. The conversion of cholesterol to pregnenolone is common to all three zones. A, In the zona glomerulosa, 3βHSD2 converts pregnenolone to progesterone. P450c17 is absent, but P450c21 produces 11-deoxycorticosterone, which is a substrate for P450c11AS (aldosterone synthase), the enzyme catalyzing 11-hydroxylation and two 18-oxygenations to complete aldosterone synthesis. B, The zona fasciculata expresses P450c17 but little cytochrome b5, so pregnenolone is hydroxylated to 17α-hydroxypregnenolone but not cleaved to DHEA. Instead, 3βHSD2 yields 17α-hydroxyprogesterone, the preferred substrate for P450c21, which produces 11-deoxycortisol. P450c11β, which is unique to the zona fasciculata, completes the synthesis of cortisol. Corticosterone is normally a minor product (dashed arrows) derived from a parallel pathway without the action of P450c17. C, The zona reticularis has high P450c17 and cytochrome b5 but low 3βHSD2, so pregnenolone is sequentially oxidized to 17α-hydroxypregnenolone and then DHEA. SULT2A1 sulfates DHEA, and DHEAS is exported to the circulation.
Gonadal Steroidogenic Pathways
Testicular synthesis of testosterone follows a pathway that is similar to C19 steroid production in the adrenal zona reticularis, with the notable exceptions that Leydig cells express abundant 3βHSD2 and 17βHSD3 but no SULT2A1, and the stimulus for steroidogenesis is transduced by the luteinizing hormone (LH) receptor rather than MC2R. Consequently, DHEA produced under LH stimulation is not sulfated but readily converted to androstenediol and androstenedione and then testosterone (Fig. 1-7). As in the adrenal, the principal pathway to C19 steroids is via Δ5 steroids to DHEA; the Δ4 pathway from 17OHP to androstenedione makes a minimal contribution.67,96 By contrast, ovarian steroidogenesis is more complex, as the enzymatic steps are partitioned between the granulosa and theca cells, which surround the oocyte and form a follicle. In addition, the patterns of steroidogenesis vary during the cycle, directed mainly to estradiol in the follicular phase and to progesterone in the luteal phase (see Fig. 1-7). The key point in ovarian steroidogenesis is that granulosa cells do not express P450c17. Thus, in general, steroidogenesis is initiated in granulosa cells under the influence of LH, which, via cAMP, stimulates the expression of P450scc.28 Pregnenolone and progesterone from granulosa cells diffuse into adjacent theca cells, where they can be acted upon by P450c17 and 3βHSD2 to produce androstenedione. Small amounts of this androstenedione are secreted or converted to testosterone (probably by AKR1C3/17βHSD5), but most androstenedione returns to the granulosa cells, where it is converted to estrone and then to estradiol by P450aro and 17βHSD1, respectively. As with the three zones of the adrenal, the patterns of gonadal steroidogenesis are dictated by the cell-specific expression of specific steroidogenic enzymes.
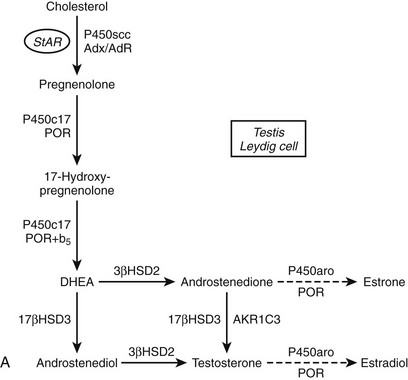
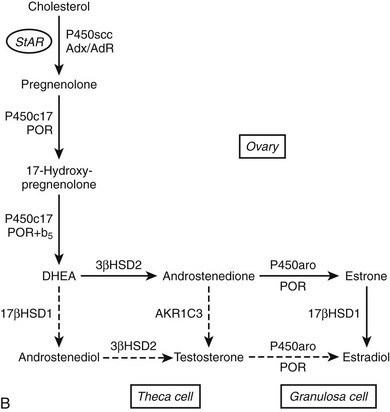
FIGURE 1-7 Major steroidogenic pathways of the gonads. Both the Leydig cell of the testis and the theca cell of the ovary convert cholesterol to DHEA and have the capacity to synthesize androstenedione and testosterone. The major difference is their enzymatic machinery is abundant 17βHSD3 in the Leydig cells (A), which efficiently completes testosterone synthesis in the testis, whereas androstenedione is the major C19 product of the theca cell (B). The granulosa cells of the ovary contain abundant aromatase and 17βHSD1, which complete the biosynthesis of estradiol. Minor pathways are shown with dashed arrows.
References
1. Hall, PF. Cytochromes P450 and the regulation of steroid synthesis. Steroids. 1986;48:133–196.
2. Agarwal, AK, Auchus, RJ. Minireview: cellular redox state regulates hydroxysteroid dehydrogenase activity and intracellular hormone potency. Endocrinology. 2005;146:2531–2538.
3. Miller, WL. Regulation of steroidogenesis by electron transfer. Endocrinology. 2005;146:2544–2550.
4. Penning, TM. Molecular endocrinology of hydroxysteroid dehydrogenases. Endocr Rev. 1997;18:281–305.
5. Sherbet, DP, Papari-Zareei, M, Khan, N, et al. Cofactors, redox state, and directional preferences of hydroxysteroid dehydrogenases. Mol Cell Endocrinol. 2007;265–266:83–88.
6. Papari-Zareei, M, Brandmaier, A, Auchus, RJ. Arginine 276 controls the directional preference of AKR1C9 (rat liver 3α-hydroxysteroid dehydrogenase) in human embryonic kidney 293 cells. Endocrinology. 2006;147:100–107.
7. Voutilainen, R, Miller, WL. Coordinate tropic hormone regulation of mRNAs for insulin-like growth factor II and the cholesterol side-chain cleavage enzyme, P450scc, in human steroidogenic tissues. Proc Natl Acad Sci USA. 1987;84:1590–1594.
8. Mesiano, S, Mellon, SH, Jaffe, RB. Mitogenic action, regulation, and localization of insulin-like growth factors in the human fetal adrenal gland. J Clin Endocrinol Metab. 1993;76:968–976.
9. Mesiano, S, Mellon, SH, Gospodarowicz, D, et al. Basic fibroblast growth factor expression is regulated by corticotropin in the human fetal adrenal: a model for adrenal growth regulation. Proc Natl Acad Sci USA. 1991;88:5428–5432.
10. Coulter, CL, Read, LC, Carr, BR, et al. A role for epidermal growth factor in the morphological and functional maturation of the adrenal gland in the fetal rhesus monkey in vivo. J Clin Endocrinol Metab. 1996;81:1254–1260.
11. Stocco, DM, Clark, BJ. Regulation of the acute production of steroids in steroidogenic cells. Endocr Rev. 1996;17:221–244.
12. Clark, BJ, Wells, J, King, SR, et al. The purification, cloning and expression of a novel luteinizing hormone-induced mitochondrial protein in MA-10 mouse Leydig tumor cells. Characterization of the steroidogenic acute regulatory protein (StAR). J Biol Chem. 1994;269:28314–28322.
13. Lin, D, Sugawara, T, Strauss, JF, III., et al. Role of steroidogenic acute regulatory protein in adrenal and gonadal steroidogenesis. Science. 1995;267:1828–1831.
14. Bose, HS, Sugawara, T, Strauss, JF, III., et al. The pathophysiology and genetics of congenital lipoid adrenal hyperplasia. N Engl J Med. 1996;335:1870–1878.
15. Caron, K, Soo, S-C, Wetsel, W, et al. Targeted disruption of the mouse gene encoding steroidogenic acute regulatory protein provides insights into congenital lipoid adrenal hyperplasia. Proc Natl Acad Sci USA. 1997;94:11540–11545.
16. Miller, WL. StAR search: What we know about how the steroidogenic acute regulatory protein mediates mitochondrial cholesterol import. Mol Endocrinol. 2007;21:589–601.
17. Arakane, F, Sugawara, T, Nishino, H, et al. Steroidogenic acute regulatory protein (StAR) retains activity in the absence of its mitochondrial targeting sequence: implications for the mechanism of StAR action. Proc Natl Acad Sci USA. 1996;93:13731–13736.
18. Bose, H, Lingappa, VR, Miller, WL. Rapid regulation of steroidogenesis by mitochondrial protein import. Nature. 2002;417:87–91.
19. Tsujishita, Y, Hurley, JH. Structure and lipid transport mechanism of a StAR-related domain. Nat Struct Biol. 2000;7:408–414.
20. Bose, HS, Whittal, RM, Baldwin, MA, et al. The active form of the steroidogenic acute regulatory protein, StAR, appears to be a molten globule. Proc Natl Acad Sci U S A. 1999;96:7250–7255.
21. Baker, BY, Yaworsky, DC, Miller, WL. A pH-dependent molten globule transition is required for activity of the steroidogenic acute regulatory protein, StAR. J Biol Chem. 2005;280:41753–41760.
22. Tuckey, RC, Headlam, MJ, Bose, HS, et al. Transfer of cholesterol between phospholipid vesicles mediated by the steroidogenic acute regulatory protein (StAR). J Biol Chem. 2002;277:47123–47128.
23. Baker, BY, Epand, RF, Epand, RM, et al. Cholesterol binding does not predict activity of the steroidogenic acute regulatory protein, StAR. J Biol Chem. 2007;282:10223–10232.
24. Hauet, T, Yao, ZX, Bose, HS, et al. Peripheral-type benzodiazepine receptor-mediated action of steroidogenic acute regulatory protein on cholesterol entry into Leydig cell mitochondria. Mol Endocrinol. 2005;19:540–554.
25. Bose, M, Whittal, RM, Miller, WL, et al. Steroidogenic activity of StAR requires contact with mitochondrial VDAC1 and phosphate carrier protein. J Biol Chem. 2008;283:8837–8845.
26. Artemenko, IP, Zhao, D, Hales, DB, et al. Mitochondrial processing of newly synthesized steroidogenic acute regulatory protein (StAR), but not total StAR, mediates cholesterol transfer to cytochrome P450 side chain cleavage enzyme in adrenal cells. J Biol Chem. 2001;276:46583–46596.
27. Miller, WL, Strauss, JF, III. Molecular pathology and mechanism of action of the steroidogenic acute regulatory protein, StAR. J Steroid Biochem Mol Biol. 1999;69:131–141.
28. Voutilainen, R, Tapanainen, J, Chung, B, et al. Hormonal regulation of P450scc (20,22-desmolase) and P450c17 (17α-hydroxylase/17,20-lyase) in cultured human granulosa cells. J Clin Endocrinol Metab. 1986;63:202–207.
29. Tsigos, C, Arai, K, Hung, W, et al. Hereditary isolated glucocorticoid deficiency is associated with abnormalities of the adrenocorticotropin receptor gene. J Clin Invest. 1993;92:2458–2461.
30. Martens, JWM, Verhoef-Post, M, Abelin, N, et al. A homozygous mutation in the luteinizing hormone receptor causes partial Leydig cell hypoplasia: correlation between receptor activity and phenotype. Mol Endocrinol. 1998;12:775–784.
31. Shenker, A. G protein-coupled receptor structure and function: the impact of disease-causing mutations. Baillieres Clin Endocrinol Metab. 1995;9:427–451.
32. Parker, KL, Schimmer, BP. Steroidogenic factor 1:A key determinant of endocrine development and function. Endocr Rev. 1997;18:361–377.
33. Zhang, P, Rodriguez, H, Mellon, SH. Transcriptional regulation of P450scc gene expression in neural and in steroidogenic cells: implications for regulation of neurosteroidogenesis. Mol Endocrinol. 1995;9:1571–1582.
34. Huang, N, Miller, WL. Cloning of factors related to HIV-inducible LBP proteins that regulate steroidogenic factor-1-independent human placental transcription of the cholesterol side-chain cleavage enzyme, P450scc. J Biol Chem. 2000;275:2852–2858.
35. Henderson, YC, Frederick, MJ, Wang, MT, et al. LBP-1b, LBP-9, and LBP-32/MGR detected in syncytiotrophoblasts from first-trimester human placental tissue and their transcriptional regulation. DNA Cell Biol. 2008;27:71–79.
36. Luo, X, Ikeda, Y, Parker, KL. A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell. 1994;77:481–490.
37. Nachtigal, MW, Hirokawa, Y, Enyeart-VanHouten, DL, et al. Wilms’ tumor 1 and Dax-1 modulate the orphan nuclear receptor SF-1 in sex-specific gene expression. Cell. 1998;93:445–454.
38. Hammer, GD, Krylova, I, Zhang, Y, et al. Phosphorylation of the nuclear receptor SF-1 modulates cofactor recruitment: integration of hormone signaling in reproduction and stress. Mol Cell. 1999;3:521–526.
39. Simpson, ER, Zhao, Y, Agarwal, VR, et al. Aromatase expression in health and disease. Recent Prog Horm Res. 1997;52:185–214.
40. Labrie, Y, Durocher, F, Lachance, Y, et al. The human type II 17β-hydroxysteroid dehydrogenase gene encodes two alternatively spliced mRNA species. DNA Cell Biol. 1995;14:849–861.
41. Tuckey, RC, Cameron, KJ. Catalytic properties of cytochrome P-450scc purified from the human placenta: comparison to bovine cytochrome P-450scc. Arch Biochem Biophys. 1993;1163:185–194.
42. Guryev, O, Carvalho, RA, Usanov, S, et al. A pathway for the metabolism of vitamin D3:unique hydroxylated metabolites formed during catalysis with cytochrome P450scc (CYP11A1). Proc Natl Acad Sci U S A. 2003;100:14754–14759.
43. Morohashi, K, Sogawa, K, Omura, T, et al. Gene structure of human cytochrome P-450(scc), cholesterol desmolase. J Biochem. 1987;101:879–887.
44. Chung, B, Matteson, KJ, Voutilainen, R, et al. Human cholesterol side-chain cleavage enzyme, P450scc: cDNA cloning, assignment of the gene to chromosome 15, and expression in the placenta. Proc Natl Acad Sci USA. 1986;83:8962–8966.
45. Black, SM, Harikrishna, JA, Szklarz, GD, et al. The mitochondrial environment is required for activity of the cholesterol side-chain cleavage enzyme, cytochrome P450scc. Proc Natl Acad Sci USA. 1994;91:7247–7251.
46. John, ME, John, MC, Boggaram, V, et al. Transcriptional regulation of steroid hydroxylase genes by corticotropin. Proc Natl Acad Sci USA. 1986;83:4715–4719.
47. Mellon, SH, Vaisse, C. cAMP regulates P450scc gene expression by a cycloheximide-insensitive mechanism in cultured mouse Leydig MA-10 cells. Proc Natl Acad Sci USA. 1989;86:7775–7779.
48. Barrett, PQ, Bollag, WB, Isales, CM, et al. The role of calcium in angiotensin II-mediated aldosterone secretion. Endocr Rev. 1989;10:496–518.
49. Moore, CCD, Brentano, ST, Miller, WL. Human P450scc gene transcription is induced by cyclic AMP and repressed by 12-O-tetradecanolyphorbol-13-acetate and A23187 by independent cis-elements. Mol Cell Biol. 1990;10:6013–6023.
50. Moore, CCD, Hum, DW, Miller, WL. Identification of positive and negative placental-specific basal elements, a transcriptional repressor, and a cAMP response element in the human gene for P450scc. Mol Endocrinol. 1992;6:2045–2058.
51. Huang, N, Miller, WL. LBP proteins modulate SF1-independent expression of P450scc in human placental JEG-3 cells. Mol Endocrinol. 2005;19:409–420.
52. Baulieu, EE. Neurosteroids: of the nervous system, by the nervous system, for the nervous system. Recent Prog Horm Res. 1997;52:1–32.
53. Yang, X, Iwamoto, K, Wang, M, et al. Inherited congenital adrenal hyperplasia in the rabbit is caused by a deletion in the gene encoding cytochrome P450 cholesterol side-chain cleavage enzyme. Endocrinology. 1993;132:1977–1982.
54. Hu, MC, Hsu, NC, El Hadj, NB, et al. Steroid deficiency syndromes in mice with targeted disruption of Cyp11a1. Mol Endocrinol. 2002;16:1943–1950.
55. Tajima, T, Fujieda, K, Kouda, N, et al. Heterozygous mutation in the cholesterol side chain cleavage enzyme (P450scc) gene in a patient with 46,XY sex reversal and adrenal insufficiency. J Clin Endocrinol Metab. 2001;86:3820–3825.
56. Kim, CJ, Lin, L, Huang, N, et al. Severe combined adrenal and gonadal deficiency caused by novel mutations in the cholesterol side chain cleavage enzyme, P450scc. J Clin Endocrinol Metab. 2008;93:696–702.
57. Apter, D, Pakarinen, A, Hammond, GL, et al. Adrenocortical function in puberty. Acta Paediatr Scand. 1979;68:599–604.
58. Orentreich, N, Brind, JL, Rizer, RL, et al. Age changes and sex differences in serum dehydroepiandrosterone sulfate concentrations throughout adulthood. J Clin Endocrinol Metab. 1984;59:551–555.
59. Nakajin, S, Shively, JE, Yuan, P, et al. Microsomal cytochrome P450 from neonatal pig testis: two enzymatic activities (17α-hydroxylase and C17,20-lyase) associated with one protein. Biochemistry. 1981;20:4037–4042.
60. Zuber, MX, Simpson, ER, Waterman, MR. Expression of bovine 17α-hydroxylase cytochrome P450 cDNA in non-steroidogenic (COS-1) cells. Science. 1986;234:1258–1261.
61. Picado-Leonard, J, Miller, WL. Cloning and sequence of the human gene encoding P450c17 (steroid 17α-hydroxylase/17,20 lyase): Similarity to the gene for P450c21. DNA. 1987;6:439–448.
62. Chung, BC, Picado-Leonard, J, Haniu, M, et al. Cytochrome P450c17 (steroid 17α-hydroxylase/17,20 lyase): cloning of human adrenal and testis cDNAs indicates the same gene is expressed in both tissues. Proc Natl Acad Sci U S A. 1987;84:407–411.
63. Lee-Robichaud, P, Wright, JN, Akhtar, ME, et al. Modulation of the activity of human 17α-hydroxylase-17,20-lyase (CYP17) by cytochrome b5:endocrinological and mechanistic implications. Biochem J. 1995;308:901–908.
64. Auchus, RJ, Lee, TC, Miller, WL. Cytochrome b5 augments the 17,20 lyase activity of human P450c17 without direct electron transfer. J Biol Chem. 1998;273:3158–3165.
65. Katagiri, M, Kagawa, N, Waterman, MR. The role of cytochrome b5 in the biosynthesis of androgens by human P450c17. Arch Biochem Biophys. 1995;317:343–347.
66. Lin, D, Black, SM, Nagahama, Y, et al. Steroid 17α-hydroxylase and 17,20 lyase activities of P450c17: contributions of serine106 and P450 reductase. Endocrinology. 1993;132:2498–2506.
67. Flück, CE, Miller, WL, Auchus, RJ. The 17,20-lyase activity of cytochrome P450c17 from human fetal testis favors the Δ5 steroidogenic pathway. J Clin Endocrinol Metab. 2003;88:3762–3766.
68. Lee, TC, Miller, WL, Auchus, RJ. Medroxyprogesterone acetate and dexamethasone are competitive inhibitors of different human steroidogenic enzymes. J Clin Endocrinol Metab. 1999;84:2104–2110.
69. Auchus, RJ, Kumar, AS, Boswell, CA, et al. The enantiomer of progesterone (ent-progesterone) is a competitive inhibitor of human cytochromes P450c17 and P450c21. Arch Biochem Biophys. 2003;409:134–144.
70. Arlt, W, Auchus, RJ, Miller, WL. Thiazolidinediones but not metformin directly inhibit the steroidogenic enzymes P450c17 and 3β-hydroxysteroid dehydrogenase. J Biol Chem. 2001;276:16767–16771.
71. Gupta, MK, Guryev, OL, Auchus, RJ. 5α-reduced C21 steroids are substrates for human cytochrome P450c17. Arch Biochem Biophys. 2003;418:151–160.
72. Wilson, JD, Auchus, RJ, Leihy, MW, et al. 5α-androstane-3α,17β-diol is formed in tammar wallaby pouch young testes by a pathway involving 5α-pregnane-3α,17α-diol-20-one as a key intermediate. Endocrinology. 2003;144:575–580.
73. Auchus, RJ. The backdoor pathway to dihydrotestosterone. Trends Endocrinol Metab. 2004;15:432–438.
74. Milewich, L, Mendonca, BB, Arnhold, I, et al. Women with steroid 5α-reductase 2 deficiency have normal concentrations of plasma 5α-dihydroprogesterone during the luteal phase. J Clin Endocrinol Metab. 1995;80:3136–3139.
75. Moisan, AM, Ricketts, ML, Tardy, V, et al. New insight into the molecular basis of 3β-hydroxysteroid dehydrogenase deficiency: identification of eight mutations in the HSD3B2 gene in eleven patients from seven new families and comparison of the functional properties of twenty-five mutant enzymes. J Clin Endocrinol Metab. 1999;84:4410–4425.
76. Hanley, NA, Arlt, W. The human fetal adrenal cortex and the window of sexual differentiation. Trends Endocrinol Metab. 2006;17:391–397.
77. Ortiz de Montellano, PR. Oxygen activation and reactivity. In: Ortiz de Montellano PR, ed. Cytochrome P-450:Structure, Mechanism, and Biochemistry. ed 2. New York: Plenum; 1995:245–303.
78. Auchus, RJ, Miller, WL. Molecular modeling of human P450c17 (17α-hydroxylase/17,20-lyase): insights into reaction mechanisms and effects of mutations. Mol Endocrinol. 1999;13:1169–1182.
79. Cutler, GB, Glenn, M, Bush, M, et al. Adrenarche: a survey of rodents, domestic animals and primates. Endocrinology. 1978;103:2112–2118.
80. Arlt, W, Martens, JW, Song, M, et al. Molecular evolution of adrenarche: structural and functional analysis of P450c17 from four primate species. Endocrinology. 2002;143:4665–4672.
81. Zhang, L, Rodriguez, H, Ohno, S, et al. Serine phosphorylation of human P450c17 increases 17,20 lyase activity: implications for adrenarche and for the polycystic ovary syndrome. Proc Natl Acad Sci USA. 1995;92:10619–10623.
82. Pandey, AV, Miller, WL. Regulation of 17,20 lyase activity by cytochrome b5 and by serine phosphorylation of P450c17. J Biol Chem. 2005;280:13265–13271.
83. Bremer, AA, Miller, WL. The serine phosphorylation hypothesis of polycystic ovary syndrome: a unifying mechanism for hyperandrogenemia and insulin resistance. Fertil Steril. 2008;89:1039–1048.
84. Pandey, AV, Mellon, SH, Miller, WL. Protein phosphatase 2A and phosphoprotein SET regulate androgen production by P450c17. J Biol Chem. 2003;278:2837–2844.
85. Nguyen, AD, Mapes, SM, Corbin, CJ, et al. Morphological adrenarche in rhesus macaques: development of the zona reticularis is concurrent with fetal zone regression in the early neonatal period. J Endocrinol. 2008;199:367–378.
86. Suzuki, T, Sasano, H, Takeyama, J, et al. Developmental changes in steroidogenic enzymes in human postnatal adrenal cortex: immunohistochemical studies. Clin Endocrinol (Oxf). 2000;53:739–747.
87. Huang, N, Miller, WL. Regulation of cytochrome b5 gene transcription by Sp3, GATA-6, and steroidogenic factor 1 in human adrenal NCI-H295A cells. Mol Endocrinol. 2005;19:2020–2034.
88. Gell, JS, Carr, BR, Sasano, H, et al. Adrenarche results from development of a 3β-hydroxysteroid dehydrogenase-deficient adrenal reticularis. J Clin Endocrinol Metab. 1998;83:3695–3701.
89. Biglieri, EG, Herron, MA, Brust, N. 17-hydroxylation deficiency in man. J Clin Invest. 1966;45:1946–1954.
90. Auchus, RJ. The genetics, pathophysiology, and management of human deficiencies of P450c17. Endocrinol Metab Clin North Am. 2001;30:101–119.
91. Costa-Santos, M, Kater, CE, Auchus, RJ. Two prevalent CYP17 mutations and genotype/phenotype correlations in 24 Brazilian patients with 17-hydroxylase deficiency. J Clin Endocrinol Metab. 2004;89:49–60.
92. Gupta, MK, Geller, DH, Auchus, RJ. Pitfalls in characterizing P450c17 mutations associated with isolated 17,20-lyase deficiency. J Clin Endocrinol Metab. 2001;86:4416–4423.
93. Geller, DH, Auchus, RJ, Mendonça, BB, et al. The genetic and functional basis of isolated 17,20 lyase deficiency. Nature Genet. 1997;17:201–205.
94. Van Den Akker, EL, Koper, JW, Boehmer, AL, et al. Differential inhibition of 17α-hydroxylase and 17,20-lyase activities by three novel missense CYP17 mutations identified in patients with P450c17 deficiency. J Clin Endocrinol Metab. 2002;87:5714–5721.
95. Geller, DH, Auchus, RJ, Miller, WL. P450c17 mutations R347H and R358Q selectively disrupt 17,20-lyase activity by disrupting interactions with P450 oxidoreductase and cytochrome b5. Mol Endocrinol. 1999;13:167–175.
96. Sherbet, DP, Tiosano, D, Kwist, KM, et al. CYP17 mutation E305G causes isolated 17,20-lyase deficiency by selectively altering substrate binding. J Biol Chem. 2003;278:48563–48569.
97. Hershkovitz, E, Parvari, R, Wudy, SA, et al. Homozygous mutation G539R in the gene for P450 oxidoreductase in a family previously diagnosed as having 17,20-lyase deficiency. J Clin Endocrinol Metab. 2008;93:3584–3588.
98. Winkel, CA, Casey, ML, Worley, RJ, et al. Extraadrenal steroid 21-hydroxylase activity in a woman with congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. 1983;56:104–107.
99. Mellon, SH, Miller, WL. Extra-adrenal steroid 21-hydroxylation is not mediated by P450c21. J Clin Invest. 1989;84:1497–1502.
100. Yamazaki, H, Shimada, T. Progesterone and testosterone hydroxylation by cytochromes P450 2C19, 2C9, and 3A4 in human liver microsomes. Arch Biochem Biophys. 1997;346:161–169.
101. Gomes, LG, Huang, N, Agrawal, V, et al. Extraadrenal 21-hydroxylation by CYP2C19 and CYP3A4:Effect on 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2009;94:89–95.
102. White, PC, Speiser, PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev. 2000;21:245–291.
103. Bristow, J, Tee, MK, Gitelman, SE, et al. Tenascin-X. A novel extracellular matrix protein encoded by the human XB gene overlapping P450c21B. J Cell Biol. 1993;122:265–278.
104. Schalkwijk, J, Zweers, MC, Steijlen, PM, et al. A recessive form of the Ehlers-Danlos syndrome caused by tenascin-X deficiency. N Engl J Med. 2001;345:1167–1175.
105. Burch, GH, Gong, Y, Liu, W, et al. Tenascin-X deficiency is associated with Ehlers-Danlos syndrome. Nature Genet. 1997;17:104–108.
106. Helmburg, A, Tusie-Luna, M, Tabarelli, M, et al. R339H and P453S: CYP21 mutations associated with nonclassic steroid 21-hydroxylase deficiency that are not apparent gene conversion. Mol Endocrinol. 1992;6:1318–1322.
107. Ulick, S. Diagnosis and nomenclature of the disorders of the terminal portion of aldosterone biosynthetic pathway. J Clin Endocrinol Metab. 1976;43:92–96.
108. Veldhuis, JD, Kulin, HE, Santen, RJ, et al. Inborn error in the terminal step of aldosterone biosynthesis: corticosterone methyl oxidase type II deficiency in a North American pedigree. N Engl J Med. 1980;303:118–121.
109. Yanagibashi, K, Haniu, M, Shively, JE, et al. The synthesis of aldosterone by the adrenal cortex: two zones (fasciculata and glomerulosa) possess one enzyme for 11-, 18-hydroxylation, and aldehyde synthesis. J Biol Chem. 1986;261:3556–3562.
110. Morohashi, K, Yoshioka, H, Gotoh, O, et al. Molecular cloning and nucleotide sequence of DNA of mitochondrial P-450(11) of bovine adrenal cortex. J Biochem. 1987;102:559–568.
111. Mornet, E, Dupont, J, Vitek, A, et al. Characterization of two genes encoding human steroid 11β-hydroxylase (P45011β). J Biol Chem. 1989;264:20961–20967.
112. Mellon, SH, Bair, SR, Monis, H. P450c11B3 mRNA, transcribed from a third P450c11 gene, is expressed in a tissue-specific, developmentally and hormonally regulated fashion in the rodent adrenal, and encodes a protein with both 11-hydroxylase and 18-hydroxylase activities. J Biol Chem. 1995;270:1643–1649.
113. Carnow, KM, Tusie-Lung, M, Pascoe, L, et al. The product of the CYP11B2 gene is required for aldosterone biosynthesis in the human adrenal cortex. Mol Endocrinol. 1991;5:1513–1522.
114. Kawamoto, T, Mitsuuchi, Y, Toda, K, et al. Role of steroid 11β-hydroxylase and 18-hydroxylase in the biosynthesis of glucocorticoids and mineralocorticoids in humans. Proc Natl Acad Sci USA. 1992;89:1458–1462.
115. White, PC, Dupont, J, New, MI, et al. A mutation in CYP11B1 (Arg 448→His) associated with steroid 11β-hydroxylase deficiency in Jews of Moroccan origin. J Clin Invest. 1991;87:1664–1667.
116. White, PC, Curnow, KM, Pascoe, L. Disorders of steroid 11β-hydroxylase isozymes. Endocr Rev. 1994;15:421–438.
117. Pascoe, L, Curnow, K, Slutsker, L, et al. Mutations in the human CYP11B2 (aldosterone synthase) gene causing corticosterone methyloxidase II deficiency. Proc Natl Acad Sci USA. 1992;89:4996–5000.
118. Fu, GK, Portale, AA, Miller, WL. Complete structure of the human gene for the vitamin D 1α-hydroxyase, P450c1α. DNA Cell Biol. 1997;16:1499–1507.
119. Clyne, CD, Zhang, Y, Slutsker, L, et al. Angiotensin II and potassium regulate human CYP11B2 transcription through common cis-elements. Mol Endocrinol. 1997;11:638–649.
120. Fardella, CE, Hum, DW, Rodriguez, H, et al. Gene conversion in the CYP11B2 gene encoding aldosterone synthase (P450c11AS) is associated with, but does not cause, the syndrome of corticosterone methyl oxidase II deficiency. J Clin Endocrinol Metab. 1996;81:321–326.
121. Lifton, R, Dluhy, RG, Powers, M, et al. A chimaeric 11β-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature. 1992;335:262–265.
122. Pascoe, L, Curnow, K, Slutsker, L, et al. Glucocorticoid-suppressible hyperaldosteronism results from hybrid genes created by unequal crossover between CYP11B1 and CYP11B2. Proc Natl Acad Sci USA. 1992;89:8327–8331.
123. Litchfield, WR, New, MI, Coolidge, C, et al. Evaluation of the dexamethasone suppression test for the diagnosis of glucocorticoid-remediable aldosteronism. J Clin Endocrinol Metab. 1997;82:3570–3573.
124. Fardella, CE, Mosso, L, Gómez-Sánchez, C, et al. Primary hyperaldosteronism in essential hypertensives: prevalence, biochemical profile, and molecular biology. J Clin Endocrinol Metab. 2000;85:1863–1867.
125. Bottner, B, Denner, K, Bernhardt, R. Conferring aldosterone synthesis to human CYP11B1 by replacing key amino acid residues with CYP11B2-specific ones. Eur J Biochem. 1998;252:458–466.
126. Curnow, KM, Mulatero, P, Emeric-Blanchouin, N, et al. The amino acid substitutions Ser288Gly and Val320Ala convert the cortisol producing enzyme, CYP11B1, into an aldosterone producing enzyme [letter]. Nat Struct Biol. 1997;4:32–35.
127. Simpson, ER, Mahendroo, MS, Means, GD, et al. Aromatase cytochrome P450, the enzyme responsible for estrogen biosynthesis. Endocr Rev. 1994;15:342–355.
128. Beusen, DD, Carrell, HL, Covey, DF. Metabolism of 19-methyl-substituted steroids by human placental aromatase. Biochemistry. 1987;26:7833–7841.
129. Mahendroo, MS, Means, GD, Mendelson, CR, et al. Tissue-specific expression of human P450 arom: the promoter responsible in adipose tissue is different from that utilized in placenta. J Biol Chem. 1991;266:11276.
130. Simpson, ER, Mahendroo, MS, Means, GD, et al. Tissue-specific promoters regulate aromatase cytochrome P450 expression. Clin Chem. 1993;39:317.
131. Conte, FA, Grumbach, MM, Ito, Y, et al. A syndrome of female pseudohermaphrodism, hypergonadotropic hypogonadism, and multicystic ovaries associated with missense mutations in the gene encoding aromatase (P450arom). J Clin Endocrinol Metab. 1994;78:1287–1292.
132. Grumbach, MM, Auchus, RJ. Estrogen: consequences and implications of human mutations in synthesis and action. J Clin Endocrinol Metab. 1999;84:4677–4694.
133. Fisher, CR, Graves, KH, Parlow, AF, et al. Characterization of mice deficient in aromatase (ArKO) because of targeted disruption of the cyp19 gene. Proc Natl Acad Sci U S A. 1998;95:6965–6970.
134. Voutilainen, R, Miller, WL. Developmental expression of genes for the steroidogenic enzymes P450scc (20,22 desmolase), P450c17 (17α-hydroxylase/17,20 lyase) and P450c21 (21-hydroxylase) in the human fetus. J Clin Endocrinol Metab. 1986;63:1145–1150.
135. Miller, WL. Steroid hormone biosynthesis and actions in the materno-feto-placental unit. Clin Perinatol. 1998;25:799–817.
136. Lo, JC, Schwitzgebel, VM, Tyrrell, JB, et al. Normal female infants born of mothers with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 1999;84:930–936.
137. Morishima, A, Grumbach, MM, Simpson, ER, et al. Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens. J Clin Endocrinol Metab. 1995;80:3689–3698.
138. Hanukoglu, I, Suh, BS, Himmelhoch, S, et al. Induction and mitochondrial localization of cytochrome P450scc system enzymes in normal and transformed ovarian granulosa cells. J Cell Biol. 1990;111:1373–1381.
139. Voutilainen, R, Picado-Leonard, J, DiBlasio, AM, et al. Hormonal and developmental regulation of human adrenodoxin mRNA in steroidogenic tissues. J Clin Endocrinol Metab. 1988;66:383–388.
140. Müller, A, Müller, JJ, Müller, YA, et al. New aspects of electron transfer revealed by the crystal structure of a truncated bovine adrenodoxin, Adx(4-108). Structure. 1998;6:269–280.
141. Coghlan, VM, Vickery, LE. Site-specific mutations in human ferredoxin that affect binding to ferredoxin reductase and cytochrome P450scc. J Biol Chem. 1991;266:18606–18612.
142. Vickery, LE. Molecular recognition and electron transfer in mitochondrial steroid hydroxylase systems. Steroids. 1997;62:124–127.
143. Wada, A, Waterman, MR. Identification by site-directed mutagenesis of two lysine residues in cholesterol side chain cleavage cytochrome P450 that are essential for adrenodoxin binding. J Biol Chem. 1992;267:22877–22882.
144. Brentano, ST, Black, SM, Lin, D, et al. cAMP post-transcriptionally diminishes the abundance of adrenodoxin reductase mRNA. Proc Natl Acad Sci USA. 1992;89:4099–4103.
145. Lin, D, Shi, Y, Miller, WL. Cloning and sequence of the human adrenodoxin reductase gene. Proc Natl Acad Sci USA. 1990;87:8516–8520.
146. Sparkes, RS, Klisak, I, Miller, WL. Regional mapping of genes encoding human steroidogenic enzymes: P450scc to 15q23-q24, adrenodoxin to 11q22; adrenodoxin reductase to 17q24-q25; and P450c17 to 10q24-q25. DNA Cell Biol. 1991;10:359–365.
147. Solish, SB, Picado-Leonard, J, Morel, Y, et al. Human adrenodoxin reductase: two mRNAs encoded by a single gene of chromosome 17cen→q25 are expressed in steroidogenic tissues. Proc Natl Acad Sci USA. 1988;71:7104–7108.
148. Brandt, ME, Vickery, LE. Expression and characterization of human mitochondrial ferredoxin reductase in Escherichia coli. Arch Biochem Biophys. 1992;294:735–740.
149. Freeman, MR, Dobritsa, A, Gaines, P, et al. The dare gene: steroid hormone production, olfactory behavior, and neural degeneration in Drosophila. Development. 1999;126:4591–4602.
150. Ziegler, GA, Vonrhein, C, Hanukoglu, I, et al. The structure of adrenodoxin reductase of mitochondrial P450 systems: electron transfer for steroid biosynthesis. J Mol Biol. 1999;289:981–990.
151. Brandt, ME, Vickery, LE. Charge pair interactions stabilizing ferredoxin-ferredoxin reductase complexes. Identification by complementary site-specific mutations. J Biol Chem. 1993;268:17126–17130.
152. Wang, M, Roberts, DL, Paschke, R, et al. Three-dimensional structure of NADPH-cytochrome P450 reductase: prototype for FMN- and FAD-containing enzymes. Proc Natl Acad Sci USA. 1997;94:8411–8416.
153. Sevrioukova, I, Huiying, L, Zhong, H, et al. Structure of a cytochrome P450-redox partner electron-transfer complex. Proceedings of the National Academy of Sciences (USA). 1999;96:1863–1868.
154. Hasemann, CA, Kurumbail, RG, Boddupalli, SS, et al. Structure and function of cytochromes P450: a comparative analysis of three crystal structures. Structure. 1995;2:41–62.
155. Peterson, RE, Imperato-McGinley, J, Gautier, T, et al. Male pseudohermaphroditism due to multiple defects in steroid-biosynthetic microsomal mixed-function oxidases. A new variant of congenital adrenal hyperplasia. N Engl J Med. 1985;313:1182–1191.
156. Shackleton, C, Malunowicz, E. Apparent pregnene hydroxylation deficiency (APHD): seeking the parentage of an orphan metabolome. Steroids. 2003;68:707–717.
157. Reardon, W, Smith, A, Honour, JW, et al. Evidence for digenic inheritance in some cases of Antley-Bixler syndrome? J Med Genet. 2000;37:26–32.
158. Flück, CE, Tajima, T, Pandey, AV, et al. Mutant P450 oxidoreductase causes disordered steroidogenesis with and without Antley-Bixler syndrome. Nat Genet. 2004;36:228–230.
159. Huang, N, Pandey, AV, Agrawal, V, et al. Diversity and function of mutations in P450 oxidoreductase in patients with Antley-Bixler syndrome and disordered steroidogenesis. Am J Hum Genet. 2005;76:729–749.
160. Scott, RR, Miller, WL. Genetic and clinical features of P450 oxidoreductase deficiency. Horm Res. 2008;69:266–275.
161. Shen, AL, O’Leary, KA, Kasper, CB. Association of multiple developmental defects and embryonic lethality with loss of microsomal NADPH-cytochrome P450 oxidoreductase. J Biol Chem. 2002;277:6536–6541.
162. Otto, DM, Henderson, CJ, Carrie, D, et al. Identification of novel roles of the cytochrome P450 system in early embryogenesis: effects on vasculogenesis and retinoic acid homeostasis. Mol Cell Biol. 2003;23:6103–6116.
163. Henderson, CJ, Otto, DM, Carrie, D, et al. Inactivation of the hepatic cytochrome P450 system by conditional deletion of hepatic cytochrome P450 reductase. J Biol Chem. 2003;278:13480–13486.
164. Gu, J, Weng, Y, Zhang, QY, et al. Liver-specific deletion of the NADPH-cytochrome P450 reductase gene: impact on plasma cholesterol homeostasis and the function and regulation of microsomal cytochrome P450 and heme oxygenase. J Biol Chem. 2003;278:25895–25901.
165. Scott, RR, Gomes, LG, Huang, N, et al. Apparent manifesting heterozygosity in P450 oxidoreductase deficiency and its effect on coexisting 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2007;92:2318–2322.
166. Huang, N, Agrawal, V, Giacomini, KM, et al. Genetics of P450 oxidoreductase: sequence variation in 842 individuals of four ethnicities and activities of 15 missense mutations. Proc Natl Acad Sci U S A. 2008;105:1733–1738.
167. Gomes, LG, Huang, N, Agrawal, V, et al. The common P450 oxidoreductase variant A503V is not a modifier gene for 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2008;93:2913–2916.
168. Agrawal, V, Huang, N, Miller, WL. Pharmacogenetics of P450 oxidoreductase: effect of sequence variants on activities of CYP1A2 and CYP2C19. Pharmacogenet Genomics. 2008;18:569–576.
169. Bridges, A, Gruenke, L, Chang, YT, et al. Identification of the binding site on cytochrome P450 2B4 for cytochrome b5 and cytochrome P450 reductase. J Biol Chem. 1998;273:17036–17049.
170. Yamazaki, H, Johnson, WW, Ueng, YF, et al. Lack of electron transfer from cytochrome b5 in stimulation of catalytic activities of cytochrome P450 3A4. Characterization of a reconstituted cytochrome P450 3A4/NADPH-cytochrome P450 reductase system and studies with apo-cytochrome b5. J Biol Chem. 1996;271:27438–27444.
171. Muskett, FW, Kelly, GP, Whitford, D. The solution structure of bovine ferricytochrome b5 determined using heteronuclear NMR methods. J Mol Biol. 1996;258:172–189.
172. Falzone, CJ, Mayer, MR, Whiteman, EL, et al. Design challenges for hemoproteins: the solution structure of apocytochrome b5. Biochemistry. 1996;35:6519–6526.
173. Lee-Robichaud, P, Kaderbhai, MA, Kaderbhai, N, et al. Interaction of human CYP17 (P-45017α, 17α-hydroxylase-17,20-lyase) with cytochrome b5: importance of the orientation of the hydrophobic domain of cytochrome b5. Biochem J. 1997;321:857–863.
174. Naffin-Olivos, JL, Auchus, RJ. Human cytochrome b5 requires residues E48 and E49 to stimulate the 17, 20-lyase activity of cytochrome P450c17. Biochemistry. 2006;46:100–108.
175. Thomas, JL, Frieden, C, Nash, WE, et al. An NADH-induced conformational change that mediates the sequential 3β-hydroxysteroid dehydrogenase/isomerase activities is supported by affinity labeling and the time-dependent activation of isomerase. J Biol Chem. 1995;270:21003–21008.
176. Thomas, JL, Myers, RP, Strickler, RC. Human placental 3β-hydroxy-5-ene-steroid dehydrogenase and steroid 5/4-ene-isomerase: purification from mitochondria and kinetic profiles, biophysical characterization of the purified mitochondrial and microsomal enzymes. J Steroid Biochem. 1989;33:209–217.
177. Luu-The, V, Lechance, Y, Labrie, C, et al. Full length cDNA structure and deduced amino acid sequence of human 3β-hydroxy-5-ene steroid dehydrogenase. Mol Endocrinol. 1989;3:1310–1312.
178. Rhéaume, E, Lachance, Y, Zhao, HL, et al. Structure and expression of a new complementary DNA encoding the almost exclusive 3β-hydroxysteroid dehydrogenase/Δ5-Δ4-isomerase in human adrenals and gonads. Mol Endocrinol. 1991;5:1147–1157.
179. Thomas, JL, Berko, EA, Faustino, A, et al. Human placental 3β-hydroxy-5-ene-steroid dehydrogenase and steroid 5/4-ene-isomerase: purification from microsomes, substrate kinetics, and inhibition by product steroids. J Steroid Biochem. 1988;31:785–793.
180. Cherradi, N, Rossier, MF, Vallotton, MB, et al. Submitochondrial distribution of three key steroidogenic proteins (steroidogenic acute regulatory protein and cytochrome P450scc and 3β-hydroxysteroid dehydrogenase isomerase enzymes) upon stimulation by intracellular calcium in adrenal glomerulosa cells. J Biol Chem. 1997;272:7899–7907.
181. Mesiano, S, Coulter, CL, Jaffe, RB. Localization of cytochrome P450 cholesterol side-chain cleavage, cytochrome P450 17α-hydroxylase/17,20 lyase, and 3β-hydroxysteroid dehydrogenase-isomerase steroidogenic enzymes in human and rhesus monkey fetal adrenal glands: reappraisal of functional zonation. J Clin Endocrinol Metab. 1993;77:1184–1189.
182. Endoh, A, Kristiansen, SB, Casson, PR, et al. The zona reticularis is the site of biosynthesis of dehydroepiandrosterone and dehydroepiandrosterone sulfate in the adult human adrenal cortex resulting from its low expression of 3β-hydroxysteroid dehydrogenase. J Clin Endocrinol Metab. 1996;81:3558–3565.
183. Remer, T, Boye, KR, Hartmann, MF, et al. Urinary markers of adrenarche: reference values in healthy subjects, aged 3–18 years. J Clin Endocrinol Metab. 2005;90:2015–2021.
184. Peltoketo, H, Luu-The, V, Simard, J, et al. 17β-hydroxysteroid dehydrogenase (HSD)/17-ketosteroid reductase (KSR) family; nomenclature and main characteristics of the 17HSD/KSR enzymes. J Mol Endocrinol. 1999;23:1–11.
185. Peltoketo, H, Isomaa, V, Mäenlavsta, O, et al. Complete amino acid sequence of human placental 17β-hydroxysteroid dehydrogenase deduced from cDNA. FEBS Lett. 1988;239:73–77.
186. Tremblay, Y, Ringler, GE, Morel, Y, et al. Regulation of the gene for estrogenic 17-ketosteroid reductase lying on chromosome 17cen→q25. J Biol Chem. 1989;264:20458–20462.
187. Luu-The, V, Labrie, C, Simard, J, et al. Structure of two in tandem human 17β-hydroxysteroid dehydrogenase genes. Mol Endocrinol. 1990;4:268–275.
188. Luu-The, V, Zhang, Y, Poirier, D, et al. Characteristics of human types 1, 2 and 3 17β-hydroxysteroid dehydrogenase activities: oxidation/reduction and inhibition. J Steroid Biochem Mol Biol. 1995;55:581–587.
189. Auchus, RJ, Covey, DF, Bork, V, et al. Solid-state NMR observation of cysteine and lysine Michael adducts of inactivated estradiol dehydrogenase. J Biol Chem. 1988;263:11640–11645.
190. Ghosh, D, Pletnev, VZ, Zhu, DW, et al. Structure of human estrogenic 17β-hydroxysteroid dehydrogenase at 2.20 Å resolution. Structure. 1995;3:503–513.
191. Sasano, H, Frost, AR, Saitoh, R, et al. Aromatase and 17β-hydroxysteroid dehydrogenase type 1 in human breast carcinoma. J Clin Endocrinol Metab. 1996;81:4042–4046.
192. Wu, L, Einstein, M, Geissler, WM, et al. Expression cloning and characterization of human 17β−hydrosteroid dehydrogenase Type 2, a microsomal enzyme possessing 20α-hydroxysteroid dehydrogenase activity. J Biol Chem. 1993;268:12964–12969.
193. Casey, ML, MacDonald, PC, Andersson, S. 17β-Hydroxysteroid dehydrogenase type 2:chromosomal assignment and progestin regulation of gene expression in human endometrium. Journal of Clinical Investigation. 1994;94:2135–2141.
194. Saez, JM, De Peretti, E, Morera, AM, et al. Familial male pseudohermaphroditism with gynecomastia due to a testicular 17-ketosteroid reductase defect. I. Studies in vivo. J Clin Endocrinol Metab. 1971;32:604–610.
195. Geissler, WM, Davis, DL, Wu, L, et al. Male pseudohermaphroditism caused by mutations of testicular 17β-hydroxysteroid dehydrogenase 3. Nature Genet. 1994;7:34–39.
196. Moghrabi, N, Hughes, IA, Dunaif, A, et al. Deleterious missense mutations and silent polymorphism in the human 17β-hydroxysteroid dehydrogenase 3 gene (HSD17B3). J Clin Endocrinol Metab. 1998;83:2855–2860.
197. Mendonca, BB, Arnhold, IJ, Bloise, W, et al. 17β-hydroxysteroid dehydrogenase 3 deficiency in women. J Clin Endocrinol Metab. 1999;84:802–804.
198. Adamski, J, Normand, T, Leenders, F, et al. Molecular cloning of a novel widely expressed human 80 kDa 17β-hydroxysteroid dehydrogenase IV. Biochemical Journal. 1995;311:437–443.
199. Qin, YM, Poutanen, MH, Helander, HM, et al. Peroxisomal multifunctional enzyme of beta-oxidation metabolizing D-3-hydroxyacyl-CoA esters in rat liver: molecular cloning, expression and characterization. Biochem J. 1997;321:21–28.
200. van Grunsven, EG, van Berkel, E, Ijlst, L, et al. Peroxisomal D-hydroxyacyl-CoA dehydrogenase deficiency: resolution of the enzyme defect and its molecular basis in bifunctional protein deficiency. Proc Natl Acad Sci U S A. 1998;95:2128–2133.
201. Lin, HK, Jez, JM, Schlegel, BP, et al. Expression and characterization of recombinant type 2 3α-hydroxysteroid dehydrogenase (HSD) from human prostate: demonstration of bifunctional 3α/17β-HSD activity and cellular distribution. Molecular Endocrinology. 1997;11:1971–1984.
202. Deyashiki, Y, Ogasawara, A, Nakayama, T, et al. Molecular cloning of two human liver 3α-hydroxysteroid/dihydrodiol dehydrogenase isoenzymes that are identical with chlordecone reductase and bile-acid binder. Biochem J. 1994;299:545–552.
203. El-Alfy, M, Luu-The, V, Huang, XF, et al. Localization of type 5 17β-hydroxysteroid dehydrogenase, 3beta-hydroxysteroid dehydrogenase, and androgen receptor in the human prostate by in situ hybridization and immunocytochemistry. Endocrinology. 1999;140:1481–1491.
204. Penning, TM, Burczynski, ME, Jez, JM, et al. Human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem J. 2000;351:67–77.
205. Sharma, KK, Lindqvist, A, Zhou, XJ, et al. Deoxycorticosterone inactivation by AKR1C3 in human mineralocorticoid target tissues. Mol Cell Endocrinol. 2006;248:79–86.
206. Byrns, MC, Steckelbroeck, S, Penning, TM. An indomethacin analogue, N-(4-chlorobenzoyl)-melatonin, is a selective inhibitor of aldo-keto reductase 1C3 (type 2 3α-HSD, type 5 17β-HSD, and prostaglandin F synthase), a potential target for the treatment of hormone dependent and hormone independent malignancies. Biochem Pharmacol. 2008;75:484–493.
207. Goto, M, Piper Hanley, K, Marcos, J, et al. In humans, early cortisol biosynthesis provides a mechanism to safeguard female sexual development. J Clin Invest. 2006;116:953–960.
208. Bruchovsky, N, Wilson, JD. The intranuclear binding of testosterone and 5α-androstan-17β-ol-3-one by rat prostate. J Biol Chem. 1968;243:5953–5960.
209. Moore, RJ, Wilson, JD. Steroid 5α-reductase in cultured human fibroblasts. Biochemical and genetic evidence for two distinct enzyme activities. J Biol Chem. 1976;251:5895–5900.
210. Jenkins, EP, Andersson, S, Imperato-McGinley, J, et al. Genetic and pharmacologic evidence for more than one human steroid 5α−reductase. J Clin Invest. 1992;89:293–300.
211. Andersson, S, Berman, DM, Jenkins, EP, et al. Deletion of a steroid 5α-reductase 2 gene in male pseudohermaphroditism. Nature. 1991;354:159–161.
212. Thigpen, AE, Silver, RI, Guileyardo, JM, et al. Tissue distribution and ontogeny of steroid 5α−reductase isozyme expression. J Clin Invest. 1993;92:903–910.
213. Frederiksen, DW, Wilson, JD. Partial characterization of the nuclear reduced nicotinamide adenine dinucleotide phosphate: Δ4–3-ketosteroid 5α-oxidoreductase of rat prostate. J Biol Chem. 1971;246:2584–2593.
214. McConnell, JD, Bruskewitz, R, Walsh, P, et al. The effect of finasteride on the risk of acute urinary retention and the need for surgical treatment among men with benign prostatic hyperplasia. Finasteride Long-Term Efficacy and Safety Study Group. N Engl J Med. 1998;338:557–563.
215. Mahendroo, MS, Cala, KM, Russell, DW. 5α-reduced androgens play a key role in murine parturition. Mol Endocrinol. 1996;10:380–392.
216. Mahendroo, MS, Wilson, JD, Richardson, JA, et al. Steroid 5α-reductase 1 promotes 5α-androstane-3α,17β-diol synthesis in immature mouse testes by two pathways. Mol Cell Endocrinol. 2004;222:113–120.
217. Khanna, M, Qin, KN, Wang, RW, et al. Substrate specificity, gene structure and tissue-specific distribution of multiple human 3α-hydroxysteroid dehydrogenases. J Biol Chem. 1995;270:20162–20168.
218. Nishizawa, M, Nakajima, T, Yasuda, K, et al. Close kinship of human 20α-hydroxysteroid dehydrogenase gene with three aldo-keto reductase genes. Genes Cells. 2000;5:111–125.
219. Rupprecht, R, Hauser, CA, Trapp, T, et al. Neurosteroids: molecular mechanisms of action and psychopharmacological significance. J Steroid Biochem Mol Biol. 1996;56:163–168.
220. Griffin, LD, Mellon, SH. Selective serotonin reuptake inhibitors directly alter activity of neurosteroidogenic enzymes. Proc Natl Acad Sci U S A. 1999;96:13512–13517.
221. Gulbis, JM, Mann, S, MacKinnon, R. Structure of a voltage-dependent K+ channel beta subunit. Cell. 1999;97:943–952.
222. Hoog, SS, Pawlowski, JE, Alzari, PM, et al. Three-dimensional structure of rat liver 3α- hydroxysteroid/dihydrodiol dehydrogenase: a member of the aldo-keto reductase superfamily. Proc Natl Acad Sci U S A. 1994;91:2517–2521.
223. Cooper, WC, Jin, Y, Penning, TM. Elucidation of a complete kinetic mechanism for a mammalian hydroxysteroid dehydrogenase (HSD) and identification of all enzyme forms on the reaction coordinate: the example of rat liver 3α-HSD (AKR1C9). J Biol Chem. 2007;282:33484–33493.
224. Ratnam, K, Ma, H, Penning, TM. The arginine 276 anchor for NADP(H) dictates fluorescence kinetic transients in 3α-hydroxysteroid dehydrogenase, a representative aldo-keto reductase. Biochemistry. 1999;38:7856–7864.
225. Napoli, JL. 17β-Hydroxysteroid dehydrogenase type 9 and other short-chain dehydrogenases/reductases that catalyze retinoid, 17β- and 3α-hydroxysteroid metabolism. Mol Cell Endocrinol. 2001;171:103–109.
226. Biswas, MG, Russell, DW. Expression cloning and characterization of oxidative 17β- and 3α-hydroxysteroid dehydrogenases from rat and human prostate. J Biol Chem. 1997;272:15959–15966.
227. Chetyrkin, SV, Hu, J, Gough, WH, et al. Further characterization of human microsomal 3α-hydroxysteroid dehydrogenase. Arch Biochem Biophys. 2001;386:1–10.
228. White, PC, Mune, T, Agarwal, AK. 11β-hydroxysteroid dehydrogenase and the syndrome of apparent mineralocorticoid excess. Endocr Rev. 1997;18:135–156.
229. Tannin, GM, Agarwal, AK, Monder, C, et al. The human gene for 11β-hydroxysteroid dehydrogenase. Structure, tissue distribution, and chromosomal localization. J Biol Chem. 1991;266:16653–16658.
230. Agarwal, AK, Monder, C, Eckstein, B, et al. Cloning and expression of rat cDNA encoding corticosteroid 11β-dehydrogenase. J Biol Chem. 1989;264:18939–18943.
231. Moore, CCD, Mellon, SH, Murai, J, et al. Structure and function of the hepatic form of 11β-hydroxysteroid dehydrogenase in the squirrel monkey, an animal model of glucocorticoid resistance. Endocrinology. 1993;133:368–375.
232. Agarwal, AK, Tusie-Luna, M-T, Monder, C, et al. Expression of 11β-hydroxysteroid dehydrogenase using recombinant vaccinia virus. Mol Endocrinol. 1990;4:1827–1832.
233. Walker, EA, Clark, AM, Hewison, M, et al. Functional expression, characterization, and purification of the catalytic domain of human 11β-hydroxysteroid dehydrogenase type 1. J Biol Chem. 2001;276:21343–21350.
234. Odermatt, A, Arnold, P, Stauffer, A, et al. The N-terminal anchor sequences of 11β-hydroxysteroid dehydrogenases determine their orientation in the endoplasmic reticulum membrane. J Biol Chem. 1999;274:28762–28770.
235. Jamieson, A, Wallace, AM, Andrew, R, et al. Apparent cortisone reductase deficiency: a functional defect in 11β-hydroxysteroid dehydrogenase type 1. J Clin Endocrinol Metab. 1999;84:3570–3574.
236. Lavery, GG, Walker, EA, Tiganescu, A, et al. Steroid biomarkers and genetic studies reveal inactivating mutations in hexose-6-phosphate dehydrogenase in patients with cortisone reductase deficiency. J Clin Endocrinol Metab. 2008;93:3827–3832.
237. Albiston, AL, Obeyesekere, VR, Smith, RE, et al. Cloning and tissue distribution of the human 11β-hydroxysteroid dehydrogenase Type II enzyme. Mol Cell Endocrinol. 1994;105:R11–R17.
238. Brown, RW, Chapman, KE, Edwards, CRW, et al. Human placental 11β-hydroxysteroid dehydrogenase: evidence for and partial purification of a distinct NAD+-dependent isoform. Endocrinology. 1993;132:2614–2621.
239. Funder, JW, Pearce, PT, Smith, R, et al. Mineralocorticoid action: target tissue specificity is enzyme, not receptor, mediated. Science. 1988;242:583–585.
240. Mune, T, Rogerson, FM, Nikkila, H, et al. Human hypertension caused by mutations in the kidney isozyme of 11β-hydroxysteroid dehydrogenase. Nature Genet. 1995;10:394–399.
241. Krozowski, Z, MaGuire, JA, Stein-Oakley, AN, et al. Immunohistochemical localization of the 11β-hydroxysteroid dehydrogenase type II enzyme in human kidney and placenta. J Clin Endocrinol Metab. 1995;80:2203–2209.
242. Li, KX, Obeyesekere, VR, Krozowski, ZS, et al. Oxoreductase and dehydrogenase activities of the human and rat 11β-hydroxysteroid dehydrogenase type 2 enzyme. Endocrinology. 1997;138:2948–2952.
243. Mulatero, P, Curnow, KM, Aupetit-Faisant, B, et al. Recombinant CYP11B genes encode enzymes that can catalyze conversion of 11-deoxycortisol to cortisol, 18-hydroxycortisol, and 18-oxocortisol. J Clin Endocrinol Metab. 1998;83:3996–4001.
244. Kater, CE, Biglieri, EG. Disorders of steroid 17α-hydroxylase deficiency. Endocrinol Metab Clin North Am. 1994;23:341–357.
245. Auchus, RJ, Rainey, WE. Adrenarche: physiology, biochemistry and human disease. Clin Endocrinol (Oxf). 2004;60:288–296.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 1
The Principles, Enzymes, and Pathways of Human Steroidogenesis
Overview of the Human Steroidogenic Enzymes and Steroidogenesis
Overview of the Human Steroidogenic Enzymes and Steroidogenesis
1. The conversion of cholesterol to pregnenolone. Although viewed superficially as a single chemical transformation, the mobilization of cholesterol into the steroidogenic pathways is a complex event that serves as a key locus of regulation and also conventionally defines a tissue as “steroidogenic.” In humans, only the adrenal cortex, testicular Leydig cells, ovarian theca cells, trophoblast cells of the placenta, and specific glial and neuronal cells of the brain possess the capacity to cleave cholesterol into pregnenolone (the C21 precursor of all active steroid hormones) and isocaproaldehyde. The differences in how this process is regulated and in how pregnenolone is subsequently metabolized define the roles of the various steroidogenic cells and tissues in human physiology. Unlike peptide-secreting glands, steroidogenic cells do not store steroid hormones and intermediates, and it is the activation of this first step that enables the rapid production of steroids in response to hormonal and environmental stimuli.
2. The transformation of pregnenolone to active hormones, intermediates, and exported steroid derivatives. The repertoire of enzymes and cofactor proteins present in a given steroidogenic cell is responsible for the characteristic steroid profile of that cell type, and the coordinate regulation of their expression promotes the completion of all steps of a given pathway. Thus, these enzymes determine qualitatively what steroids are made, but since these steps are not kinetically limiting, it is step 1 that quantitatively regulates how much steroid is made at a given moment. Steroids diffuse into the bloodstream, although the sulfation of Δ5 steroids helps promote their binding to proteins and prolong their half-lives in the circulation.
3. Peripheral metabolism of hormones and precursors. Although not “steroidogenic” as defined earlier, some organs, such as the liver and skin, possess tremendous capacity to transform various steroids. For example, 70% to 80% of circulating testosterone in normally cycling women derives from the conversion of adrenal dehydroepiandrosterone (DHEA). Steroids can be activated in target tissues, such as the conversion of testosterone to dihydrotestosterone (DHT) in the prostate. In contrast, active androgens and estrogens are inactivated in the uterus and in other peripheral tissues.
4. Multiple layers of regulation. The regulation of steroidogenesis by trophic hormones, such as adrenocorticotropin (ACTH), angiotensin II, and luteinizing hormone (LH) is well known; less well appreciated are the multiple levels at which such regulation may occur. These include the transcriptional regulation of the genes encoding steroidogenic enzymes and co-factors; regulation of mitochondrial protein import; transfer of electrons from NADPH; post-translational modification of steroidogenic enzymes; and subcellular localization and/or targeting.
5. Catabolism and unproductive metabolism. A panoply of steroids can be isolated from human plasma and tissues, many of which have negligible biological activity. Most inactive byproducts derive from hepatic transformations (e.g., 6α/6β-hydroxylation of C19 steroids, 5β-reduction of C21 and C19 steroids, and 4-hydroxylation of estrogens), which promote renal excretion of these steroids.
6. More than one pathway to specific steroids. Particularly for the terminal steps of long pathways, such as the synthesis of DHT or estradiol, two or more routes may yield the same final product. Often the different routes utilize different enzymes, and these enzymes may be found in different tissues under different regulatory mechanisms. The relative importance of these pathways differ with age, sex, and physiologic state.
Steroids are molecules derived from the cyclopentanoperhydrophenanthrene four-ring hydrocarbon nucleus (Fig. 1-1). Most enzymes involved in steroid biosynthesis are either cytochrome P450s or hydroxysteroid dehydrogenases (Fig. 1-2). All of these reactions are functionally if not absolutely unidirectional, so the accumulation of products does not drive flux back to the precursor. All P450-mediated hydroxylations and carbon-carbon bond cleavage reactions are mechanistically and physiologically irreversible.1 Hydroxysteroid dehydrogenase reactions are mechanistically reversible and can run in either direction under certain conditions in vitro, but each hydroxysteroid dehydrogenase drives steroid flux predominantly in either the oxidative or reductive mode in vivo.2 However, two or more hydroxysteroid dehydrogenases drive the flux of a steroid pair in opposite directions, some favoring ketosteroid reduction and others favoring hydroxysteroid oxidation.
FIGURE 1-1 Cyclopentanoperhydrophenanthrene steroid nucleus. Steroid rings are identified with boxed capital letters, and carbon atoms are numbered. Substituents and hydrogens are labeled as α or β if they are positioned behind or in front of the plane of the page, respectively.
FIGURE 1-2 Major human steroidogenic pathways. Key enzymes and cofactor proteins are shown near arrows indicating chemical reactions. The StAR protein (oval) mobilizes cholesterol from the outer mitochondrial membrane to the inner mitochondrial membrane, where P450scc cleaves cholesterol to pregnenolone, the first committed intermediate in steroid biosynthesis. The steroids in the first column are Δ5-steroids, which constitute the preferred pathway to C19 steroids in human beings, and the dashed arrow indicates poor flux from 17α-hydroxyprogesterone to androstenedione. Steroids in the second column and farther right are Δ4-steroids, except the C18 estrogens (estrone and estradiol) and 5α-reduced steroids, including the potent androgen DHT and other androstanes (bottom row). Not all intermediate steroids, pathways, and enzymes are shown.
Cytochrome P450 Enzymes
Mammalian cytochrome P450 enzymes fall into two broad classes, type 1 and type 2.3 Type 1 enzymes and their electron-transfer proteins reside in the mitochondria (Table 1-1) of eukaryotes; almost all bacterial P450s are also type 1 enzymes. Human type 1 P450 enzymes include the cholesterol side-chain cleavage enzyme P450scc; the two isozymes of 11-hydroxylase (i.e., P450c11β and P450c11AS); and two of the three principal enzymes in vitamin D metabolism (i.e., 1α-hydroxylase and 24-hydroxylase). Type 1 enzymes receive electrons from the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) via adrenodoxin, a small, soluble, iron-sulfur protein. Adrenodoxin does not oxidize NADPH directly, however, but receives the two electrons from NADPH via the flavoprotein adrenodoxin reductase (Fig. 1-3). Type 2 enzymes, in contrast, receive electrons from NADPH via the flavin adenine dinucleotide (FAD)-flavin mononucleotide (FMN) two-flavin protein, P450 oxidoreductase (POR). Type 2 enzymes are exclusively located in the smooth endoplasmic reticulum and constitute the majority of the human P450 enzymes.
Table 1-1
Intracellular Location of Steroidogenic Proteins
| Mitochondria | Cytoplasm | Endoplasmic Reticulum |
| P450scc | P450c17 | |
| P450c11β | P450c21 | |
| P450c11AS | P450aro | |
| Adrenodoxin reductase | P450-oxidoreductase | |
| Adrenodoxin | ||
| StAR | StAR | Cytochrome b5 |
| 3β-HSD1 and 2 | 3β-HSD1 and 2 | 3β-HSD1 and 2 |
| 17β-HSD1 | 17β-HSD1–3* | |
| Reductive 3α-HSDs includes 17β-HSD5 | Oxidative 3α-HSD 5α-Reductase 1 and 2 11β-HSD1 and 2 |
FIGURE 1-3 Electron transfer pathways for steroidogenic cytochrome P450 enzymes. A, In type 1 (mitochondrial) enzymes, the two electrons from the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) pass from the flavin (FAD) of ferredoxin (adrenodoxin) reductase (FeRed) to the iron-sulfur (Fe2S2, diamond with dots) cluster of ferredoxin (adrenodoxin, Fedx) and then to the heme of the P450 (square with iron atom [Fe]). Negatively-charged residues in Fedx (−) guide docking and electron transfer with positively-charged residues (+) in both Fedx and the P450. B, In type 2 (microsomal) enzymes, the flavoprotein P450-oxidoreductase (POR) receives electrons from NADPH to its FAD moiety, transfers electrons to its FMN moiety, and after a conformational rearrangement, directly transfers electrons from the FMN to the P450. Negative charges on POR (−) and positive charges (+) on the P450 guide the interaction as with the type 1 P450; phosphorylation and cytochrome b5 also regulate electron transfer and catalysis. Heme of P450 is indicated by square with iron atom (Fe).
P450 enzymes activate molecular oxygen using their heme center and electrons from NADPH. Substrate binding is required prior to heme reduction with one electron, which enables oxygen binding, the second one-electron transfer, and formation of the iron-oxygen complex which oxygenates the substrate. Thus, P450 reactions on steroids are limited to oxygen insertion (hydroxylation) reactions and, in a few notable cases, oxidative carbon-carbon bond cleavage reactions (Table 1-2).
Hydroxysteroid Dehydrogenases and Reductases
All hydroxysteroid dehydrogenases (HSDs) and related enzymes use nicotinamide cofactors either to reduce or to oxidize the steroid by two electrons through a hydride transfer mechanism.2 Most examples involve the conversion of a secondary alcohol to a ketone or vice versa, and in the case of the 3β-hydroxysteroid dehydrogenase/Δ5/4-isomerases, the dehydrogenation is accompanied by the isomerization of the adjacent carbon-carbon double bond from the Δ5 (between carbons 5 and 6) to the Δ4 positions (see Figs. 1-1 and 1-2). The human steroid 5α-reductases types 1 and 2, which are included with the HSDs for convenience, reduce olefinic carbon-carbon double bonds to the saturated state rather than acting on carbon centers bonded to oxygen.
The HSDs can be categorized according to either structural or functional classification schemes. Structurally, HSDs are members of either the short-chain dehydrogenase reductase (SDR) or aldo-keto reductase (AKR) families.4 The SDR enzymes are β-α-β proteins where up to seven parallel β-strands fan across the center of the molecule, forming the “Rossman fold” characteristic of oxidation/reduction enzymes that use nicotinamide cofactors. The AKR enzymes are soluble proteins that contain a beta-barrel or triosephosphate isomerase (TIM-barrel) motif in which eight parallel β-strands lie in a slanted circular distribution like the staves of a barrel. In both cases, the active site contains a critical tyrosine and lysine pair of residues involved in proton transfer from or to the steroid alcohol during catalysis. Functionally, HSDs act either as true dehydrogenases, using NAD+ as a cofactor to convert hydroxysteroids to ketosteroids, or as ketosteroid reductases, utilizing predominantly NADPH to reduce ketosteroids. Many HSDs catalyze either oxidation or reduction in vitro based on the pH and cofactor concentrations, but these enzymes, when expressed in intact mammalian cells, drive steroid flux primarily in one direction.4 These directional preferences derive primarily from the relative abundance of the oxidized and reduced form of cofactors and the relative affinity of each enzyme for NAD(H) versus NADP(H), because cofactor concentrations exceed steroid concentrations by many orders of magnitude.2,5 Consequently, the directional preference of some “reductive” enzymes can be reduced or reversed by depleting cells of NADPH or by mutations that impair NADPH binding.6
Acute Regulation of Steroidogenesis
Every time that a pulse of corticotrophin (adrenocorticotropic hormone [ACTH]) reaches the adrenal cortex, or a pulse of luteinizing hormone (LH) reaches the gonad, a subsequent pulse of steroid hormone production is observed within minutes. Although it has long been known that the loss of trophic hormones from the pituitary gland leads to adrenal and gonadal atrophy, the action of ACTH and LH to promote organ survival and to maintain steroidogenic capacity occurs at three distinct levels. First, as seen in long-term exposure to ACTH (e.g., in Cushing’s disease), ACTH promotes adrenal growth. This growth occurs primarily by ACTH stimulating the production of cyclic adenosine monophosphate (cAMP), which in turn promotes the synthesis of insulin-like growth factor 2 (IGF-2),7,8 basic fibroblast growth factor,9 and epidermal growth factor.10 Together, these growth factors stimulate adrenal cellular hypertrophy and hyperplasia. Second, ACTH acts long term through cAMP, and angiotensin II acts through the calcium/calmodulin pathway to promote the transcription of genes encoding various steroidogenic enzymes and electron-donating cofactor proteins. Third, ACTH fosters the increased flow of cholesterol into mitochondria, where it becomes substrate for the first and rate-limiting enzyme, P450scc. This acute response occurs within minutes and is inhibited by inhibitors of protein synthesis (e.g., puromycin or cycloheximide), indicating that a short-lived protein species mediates this process. Although other proteins are involved in the chronic replenishment of mitochondrial cholesterol, abundant biochemical, clinical, and genetic evidence implicates the steroidogenic acute regulatory protein (StAR) as this labile protein mediator.11
StAR is a 37-kilodalton (kD) phosphoprotein that is cleaved to a 30-kD form when it enters the mitochondrion. Overexpression of mouse StAR in mouse Leydig MA-10 cells increased their basal steroidogenic rate,12 and cotransfection of expression vectors for both StAR and the P450scc system in nonsteroidogenic COS-1 cells augmented pregnenolone synthesis above that obtained with the P450scc system alone.13 Mutations in StAR cause the most common form of congenital lipoid adrenal hyperplasia,13,14 in which very little steroid is made, and targeted disruption of the Star gene in the mouse causes a similar phenotype.15
The mechanism of StAR’s action is not known in detail.16 StAR acts exclusively on the outer mitochondrial membrane (OMM),17,18 and its activity in promoting steroidogenesis is proportional to its residency time on the OMM.18 When expressed in cytoplasm or added to mitochondria in vitro, both the 37-kD “precursor” and the 30-kD “mature form” of StAR are equally active, but StAR is inactive in the mitochondrial intramembranous space or matrix.18 Thus, it is StAR’s cellular localization, not its cleavage, that determines whether or not it is active. StAR has a sterol-binding pocket that accommodates a single molecule of cholesterol.19 The interaction of StAR with the OMM involves conformational changes20,21 that are necessary for StAR to accept and discharge cholesterol molecules. Although StAR can transfer cholesterol between synthetic membranes in vitro,22 suggesting that other protein molecules are not needed for its action, this activity can also be seen with the inactive mutant R182L, which causes lipoid CAH.23 Thus, StAR’s action to promote steroidogenesis is distinct from its cholesterol-transfer activity. StAR appears to interact with the peripheral benzodiazepine receptor (PBR)24 voltage-dependent anion channel 1 (VDAC1) and phosphate carrier protein,25 all proteins found on the outer mitochondrial membrane. Each molecule of StAR appears to be recycled, moving hundreds of molecules of cholesterol before the cleavage/inactivation event.26 Although StAR is required for the acute steroidogenic response, steroidogenesis will persist in the absence of StAR at about 14% of the StAR-induced rate,27 accounting for the steroidogenic capacity of tissues that lack StAR (e.g., the placenta and the brain).
Chronic Maintenance of the Steroidogenic Machinery
While the acute regulation of steroidogenesis is determined by access of cholesterol to the P450scc enzyme, which is mediated by StAR, P450scc is the enzymatic rate-limiting step in steroidogenesis. Thus the chronic regulation of steroidogenesis is quantitatively (how much) determined by P450scc gene expression28 and qualitatively (which steroids) determined by the expression of downstream enzymes. The episodic bursts of cAMP resulting from the binding of ACTH and LH to their respective receptors are necessary but not sufficient for the continued expression of the steroidogenic enzymes and the production of steroids. Patients with inactivating mutations in the ACTH receptor29 or LH receptor30 make negligible steroids from the affected glands. Conversely, activating mutations of the Gsα protein, which couples receptor binding to cAMP generation, and activating mutations of the LH receptor cause hypersecretion of steroids.31 Indeed, cAMP-responsive elements have been identified in the genes for most of the human steroidogenic P450 enzymes, but this mechanism alone does not allow for the diversity of steroid production observed in the various zones of the adrenal cortex, the gonads of both sexes, the placenta, and the brain.
Other transcription factors (e.g., AP-2, SP-1, SP-3, NF1C, NR4A1, NR4A2, GATA4, and GATA6) aid in defining the basal- and cAMP-stimulated transcription of each gene, which is also regulated in a tissue-specific manner by the regulatory elements unique to each gene. Among these factors, steroidogenic factor-1 (SF-1, NR5A1), an orphan nuclear receptor, coordinates the expression of steroidogenic enzymes in adrenal and gonadal cells.32 By contrast, steroidogenesis in the brain33 and placenta34,35 is independent of SF-1. Targeted disruption of SF-1 in the mouse not only disrupts steroid biosynthesis but also blocks the development of the adrenal glands, gonads, and the ventromedial hypothalamus in homozygous animals.36 Furthermore, SF-1 does not act in isolation, but its action is modified by other transcription factors (e.g., WT-1 and DAX-137) or by sumoylation and phosphorylation.38 The development of steroidogenic organs is intimately related to the capacity to produce steroids, and multiple factors acting on the genes for steroidogenic enzymes yield both common features and diversity among the steroidogenic tissues.
Most steroidogenic enzymes derive from a single mRNA species. The most prominent exception to this paradigm is aromatase, whose gene has four different promoters that enable vastly different regulation of expression of the same aromatase protein in many different tissues.39 Although different transcripts of several genes (including 17β-HSDs types 1, 2, and 3) have been described, the encoded proteins derived from “exon skipping” are inactive if translated.40
Human Steroidogenic P450s
Encoded by the CYP11A1 gene, P450scc consumes three equivalents of NADPH and molecular oxygen during the conversion of cholesterol to pregnenolone. Although the enzyme is named for the cleavage of the cholesterol side chain, this process consists of three discrete steps: (1) the 22-hydroxylation of cholesterol; (2) the 20-hydroxylation of 22(R)-hydroxycholesterol; and (3) the oxidative scission of the C20-C22 bond of 20(R), 22(R)-dihydroxycholesterol—the side-chain cleavage event. The enzyme will utilize free hydroxysterol intermediates as substrates for the side-chain cleavage reaction, a tool that is used experimentally because the hydroxysterols are much more soluble than cholesterol and because their access to P450scc is independent of StAR.13 In vivo, however, little of these free intermediates probably accumulate because their kcat/Km ratios are much higher than for cholesterol,41 and the high Kd for pregnenolone (about 3000 nM) drives product dissociation. This complex process is the rate-limiting step in steroidogenesis, with turnover numbers of only about 20 molecules of cholesterol per molecule P450scc per minute.41 P450scc will also cleave the side chain of other hydroxysterols (e.g., 7-dehydrocholesterol), and 20- and 22-hydroxylates vitamin D.42
The single human gene for P450scc43 encodes an mRNA of 2 kb.44 A 39-amino-acid mitochondrial leader peptide that targets P450scc to the mitochondria is then proteolytically removed to yield a 482-amino-acid protein. Forms of P450scc targeted to the endoplasmic reticulum are inactive,45 demonstrating that the mitochondrial environment is required for activity. Expression of P450scc is induced in the adrenal zona fasciculata/reticularis,46 testis,47 and ovary by cAMP; and in the zona glomerulosa by intracellular calcium/protein kinase C.48,49 In contrast, placental P450scc expression is constitutive50 and is caused at least in part by the LBP family of transcription factors.35,51 Side-chain cleavage activity and pregnenolone biosynthesis have been demonstrated in the rat and human brain52; and abundant P450scc expression is found in the rodent brain, especially in fetal life. Deletion of the gene for P450scc has been described in rabbits53 and mice,54 abrogating all steroidogenesis and thus proving that P450scc is the only enzyme that can convert cholesterol to pregnenolone. While homozygous mutations in P450scc are expected to be embryonic lethal by eliminating placental progesterone synthesis, a small number of patients has been described having P450scc mutations that typically retain partial enzymatic activity.55,56
P450c17
For investigators studying the enzymology and genetics of the steroidogenic pathways, P450c17 is especially interesting. Clinical observations showed that adrenal 17α-hydroxylase activity (reflected by serum cortisol concentrations) was fairly constant throughout life, whereas adrenal 17,20-lyase activity (reflected by serum DHEA and DHEAS concentrations) was low in early childhood but rose abruptly during adrenarche at ages 8 to 10 years.57,58 This dissociation between adrenal secretion of 17α-hydroxylase products (cortisol) and 17,20-lyase products (DHEA) suggested that distinct enzymes performed the two transformations, a hypothesis that was reinforced by the description of patients with putative isolated 17,20-lyase deficiency. Consequently, reports59 that the 17α-hydroxylase and 17,20-lyase activities of neonatal pig testes copurified were initially received with great skepticism. This controversy of “one enzyme or two” persisted until the cDNA for bovine P450c17 was cloned and shown to confer both 17-hydroxylase and 17,20-lyase activities when expressed in nonsteroidogenic COS-1 cells.60 The human genome has one gene for P450c17,61 which is expressed in the adrenals and gonads,62 and not two tissue-specific isozymes as had been thought. A single 2.1-kb mRNA species yields a 57-kD protein in these tissues, and mutations in this gene produce a spectrum of deficiencies in 17-hydroxysteroids and C19 steroids.
Human P450c17 17-hydroxylates both pregnenolone and progesterone with approximately equal efficiency,63,64 but all other reactions show prominent differences between Δ4 and Δ5 substrates. The 17,20-lyase activity is about 50 times more efficient for the 17α-hydroxypregnenolone-to-DHEA reaction than for the 17α-hydroxyprogesterone-to-androstenedione reaction.63,64 Although the rate of the lyase reaction can be increased more than 10-fold by the addition of a molar excess of cytochrome b5,63–65 the Δ5 preference persists, and the lyase rate never quite reaches the rate of the hydroxylase reactions. In addition, human P450c17 16α-hydroxylates progesterone but not pregnenolone64; in the presence of cytochrome b5, it diverts about 10% of pregnenolone metabolism to a Δ16 andiene product63 that is also formed by this pathway in pigs and that acts as a pheromone in that species. Although experiments to study the chemistry of human P450c17 often require manipulations that could be considered nonphysiologic, the remarkable consistency for substrate preferences and kinetic constants observed for the modified, solubilized P450c17 expressed in Escherichia coli63,65 and native P450c17 expressed in yeast microsomes,64 or intact COS-1 cells,66 or that obtained from human tissues and cells,64,67 serve to verify these conclusions.
Given the diverse repertoire of reactions catalyzed by P450c17 in the classical pathways, it is not surprising that synthetic steroids such as dexamethasone68 and the enantiomer of progesterone,69 as well as planar drugs such as troglitazone,70 also bind to and inhibit P450c17. In addition, the 5α-reduced C21 steroids dihydroprogesterone (5α-pregnane-3,20-dione) and allopregnanolone (5α-pregnan-3α-ol-20-one) are excellent substrates for the 17α-hydroxylase activity of P450c1771 (Fig. 1-4A). Furthermore, 17α-hydroxylated allopregnanolone (5α-pregnane-3α,17α-diol-20-one; 17OH-Allo) is the most efficient substrate yet identified for the 17,20-lyase activity of human P450c17, and its cleavage to androsterone is minimally dependent on cytochrome b5,71 unlike 17α-hydroxypregnenolone metabolism to DHEA.63–65 The conversion of 17OH-Allo to androsterone by the 17,20-lyase activity of P450c17, first described in the testes of tammar wallaby pouch young,72 provides an alternative or “backdoor” pathway to DHT, by which DHT is produced without utilizing DHEA, androstenedione, and testosterone as intermediates73 (see Fig. 1-4B). Consequently, the presence of 5α-reductases in steroidogenic cells does not preclude the production of C19 steroids but rather paradoxically enhances the production of DHT by directing flux to 5α-reduced precursors of DHT.
FIGURE 1-4 Reactions catalyzed by human P450c17 and pathways to C19 steroids. A, The four principal A/B-ring configurations of active endogenous steroids and their precursors: Δ5, Δ4, 5α, and 5α,3α (boxes and structures at bottom). Progesterone and 17α-hydroxyprogesterone can be 5α-reduced, and once the A-ring is saturated, these 5α-reduced steroids are substrates for reductive 3α-HSDs of the AKR1C family. Human P450c17 17α-hydroxylates all four classes of C21 steroids, but the 17,20-lyase activity is robust only with 17α-hydroxypregnenolone and 5α-pregnane-3α,17α-diol-20-one (Δ5– and 5α,3α-pathways, respectively). B, Two pathways to DHT using the different 17,20-lyase activities of human P450c17. In the conventional or Δ5-pathway (solid arrows), the 17,20-lyase activity of P450c17 requires cytochrome b5 to efficiently convert 17α-hydroxyprogesterone to DHEA, and testosterone is reduced in target tissues by 5α-reductase 2 (5αR2) to DHT. In the “backdoor” or 5α,3α-pathway (broken arrows), 5α-reduction by 5αR1 and 3α-reduction of C21 steroids occurs in the steroidogenic tissue prior to the 17,20-lyase reaction. In the best characterized pathway, 5α-pregnane-3α,17α-diol-20-one is cleaved to androsterone without requiring cytochrome b5 and reduced to androstanediol. Androstanediol is exported from the testis and metabolized to DHT by oxidative 3α-HSDs (Ox. 3α-HSD). Note that testosterone is not an intermediate in the backdoor pathway to DHT, that different isoforms of 5α-reductase appear to be involved in the two pathways, and that both reductive and oxidative 3αHSDs are required for the “backdoor” pathway. Structures of testosterone, DHT, and androstanediol are shown at bottom.
The backdoor pathway enables production of C19 steroids in the presence of abundant 3β-HSD activity, despite the poor 17,20-lyase activity of human P450c17 for 17α-hydroxyprogesterone, by using 17OH-Allo as the substrate for the 17,20-lyase reaction. The presence of 5α-reductase activity is a key requirement for the backdoor pathway. The best-studied example of 5α-reduction in a human steroidogenic tissue is the production of 5α-dihydroprogesterone in human corpus luteum by the type 1 enzyme.74 Human enzymes catalyze all of the other reactions required to complete this alternate route to DHT, and good evidence documents production of 5α-reduced androgens by the fetal adrenal, at least in some pathologic states. Consequently, it is possible that the backdoor pathway is the principal route to DHT in pathologic states in which 17α-hydroxyprogesterone accumulates, including 21-hydroxylase deficiency and P450 oxidoreductase deficiency (see the following). Androgen production by the backdoor pathway may explain why newborn girls with 21- and 11-hydroxlase deficiencies can be severely virilized, while those with 3β-HSD2 deficiency, whose adrenals cannot make 17α-hydroxyprogesterone, are minimally virilized.75 The fractional contributions of the conventional and backdoor pathways to DHT production during human sexual differentiation (at 8 to 12 weeks of gestation) and the expression of 5α-reductase in fetal adrenal and gonad tissues,76 however, are only beginning to be determined.
The chemistry of P450c17-mediated hydroxylations is believed to proceed via the common iron oxene species and “oxygen rebound” mechanism proposed for prototypical P450 hydroxylations.77 The mechanism of the 17,20-lyase reaction involving a carbon-carbon bond cleavage, however, is not known despite considerable study. The failure of hydrogen peroxide alone to support catalysis (as has been shown for some other P450-mediated deacylation reactions) and computer modeling studies suggest that the same heme-oxygen complex might participate in both hydroxylations and the 17,20-lyase reaction,78 but no conclusive evidence to exclude proposed mechanisms exist.
One consequence of the Δ5 preference of the human enzyme for the 17,20-lyase reaction is that most human C19 and C18 steroids derive from DHEA as an intermediate.67 This Δ5 preference allows for the phenomenon of adrenarche to occur in humans, an event that only takes place in large primates.79,80 However, Δ5-lyase activity is not sufficient for adrenarche to occur, because some monkeys (e.g., rhesus macaques) produce high amounts of DHEA throughout life, but most mammals (e.g., cattle, dogs, cats, etc.) never produce much DHEA.79 The biochemistry of P450c17, with its differential regulation of the 17α-hydroxylase and 17,20-lyase activities, provides clues to the genesis of this enigmatic process of adrenarche. P450c17 is a phosphoprotein, and phosphorylation selectively enhances the 17,20-lyase activity.81,82 It appears likely that the regulation of P450c17 phosphorylation, which is a dynamic balance between phosphorylation and dephosphorylation, plays an important role in adrenarche and pathologic hyperandrogenic states such as polycystic ovary syndrome.83 Whereas the kinase(s) responsible for P450c17 phosphorylation remain unknown, it is now apparent that the kinase activity is counterbalanced by protein phosphatase 2A, which in turn is regulated by cAMP via phosphoprotein SET.84 Cytochrome b5 also augments 17,20-lyase activity,64,82 and high expression of b5 in the zona reticularis of monkeys85 and humans86 suggests that the developmentally regulated expression of b5 might be a key event. The transcriptional regulation of cytochrome b5 in the adrenal is similar to that of P450c17,87 but mechanisms enabling zone-specific expression have not been elucidated. Finally, limiting steroid flux to the Δ5 pathway by lowering 3β-HSD activity in the zona reticularis (where most DHEA derives) potentiates the effect of increased 17,20-lyase activity.86,88
The initial description of 17α-hydroxylase deficiency was a case in which both 17α-hydroxylase and 17,20-lyase products were absent.89 When the gene for human P450c17 was cloned,61 patients with 17α-hydroxylase deficiency were found to harbor mutations in the CYP17A1 gene, and more than 40 mutations scattered throughout the CYP17A1 gene have been characterized,90 with mutations W406R and R362C being the most common and accounting for the high prevalence of 17α-hydroxylase deficiency in Brazil.91 The identification of CYP17A1 mutations causing apparent isolated 17,20-lyase deficiency is fraught with difficulty,92 but in the past decade, five cases of isolated 17,20-lyase deficiency caused by mutations in arginines 347 and 358 have been confirmed.93,94 Computer modeling studies demonstrate that R347H and R358Q neutralize positive charges in the redox-partner binding site.78,93 Biochemical studies confirm that mutations R347H and R358Q impair interactions of P450c17 with its electron donor POR and with cytochrome b5.95 Therefore, these cases of isolated 17,20-lyase deficiency are not caused by an inability of the mutant enzymes to bind the intermediate 17α-hydroxypregnenolone but rather are caused by subtle disturbances in interactions with redox partners.93,95 In contrast, mutation E305G has been shown to cause 17,20-lyase deficiency by selectively disrupting binding of 17α-hydroxypregnenolone and DHEA synthesis despite enhanced conversion of 17α-hydroxyprogesterone to androstenedione.96 This unusual variant of isolated 17,20-lyase deficiency provides further genetic evidence that the flux of androgens derived from conversion of 17α-hydroxyprogesterone to androstenedione in the minor Δ4 pathway is not sufficient to form normal male external genitalia. One of the first patients reported to have isolated 17,20 lyase deficiency was recently found to have a homozygous mutation in P450 oxidoreductase (G539R), further emphasizing the crucial role of efficient electron transfer in the 17,20 lyase reaction.97
P450c21
Microsomal P450c21 performs the 21-hydroxylation of the Δ4 steroids 17α-hydroxyprogesterone and progesterone, an essential step in the biosynthesis of both mineralocorticoids and glucocorticoids (see Fig. 1-2). The human P450c21 protein is found only in the adrenal glands; the extra-adrenal 21-hydroxylase activity found in other organs such as the liver and the aorta98 is not catalyzed by P450c2199 but appears to be catalyzed by CYP2C9, CYP3A4, and possibly CYP2C19 and other enzymes as well.100,101
The locus containing the CYP21 genes is among the most complex in the human genome and explains why 21-hydroxylase deficiency (affecting 1 of 14,000 live births) is one of the most common autosomal-recessive diseases. The CYP21A2 gene and the CYP21A1 pseudogene lie on chromosomal locus 6p21.1 in the midst of the human leukocyte antigen (HLA) locus. Because the HLA locus is highly recombinogenic, exchange between the CYP21A1 and CYP21A2 loci is common. Thus 85% of cases of 21-hydroxylase deficiency derive from micro- or macrogene conversion events where some or all of the CYP21A1 pseudogene replaces the corresponding area of the CYP21A2 gene, thus reducing the expression of the encoded P450c21 protein and/or impairing its activity.102 In addition, at least eight additional genes lie in this locus (Fig. 1-5), including the liver-specific C4A and C4B genes; the adrenal-specific “ZA” and “ZB” genes; and the ubiquitously expressed tenascin X or TNXB gene,103 the disruption of which is one cause of Ehlers-Danlos syndrome.104 Occasionally, a patient with 21-hydroxylase deficiency and Ehlers-Danlos syndrome will have a contiguous gene syndrome with tenascin X deficiency as well.105
FIGURE 1-5 Genetic map of the human leukocyte antigen (HLA) locus containing the genes for P450c21. The top line shows the p21.1 region of chromosome 6, with the telomere to the left and the centromere to the right. Most HLA genes are found in the class I and class II regions; the class III region containing the CYP21 genes lies between these two. The second line shows the scale (in kb) for the diagram immediately below, showing (from left to right) the genes for complement factor C2, properdin factor Bf, and the RD and G11/RP genes of unknown function; arrows indicate transcriptional orientation. The bottom line shows the 21-hydroxylase locus on an expanded scale, including the C4A and C4B genes for the fourth component of complement, the “CYP21A” pseudogene (CYP21A1, 21A), and the active “CYP21B” gene (CYP21A2, 21B) that encodes P450c21. XA, YA, and YB are adrenal-specific transcripts that lack open reading frames. The XB gene encodes the extracellular matrix protein tenascin-X; XB-S encodes a truncated adrenal-specific form of the tenascin-X protein whose function is unknown. ZA and ZB are adrenal-specific transcripts that arise within the C4 genes and have open reading frames, but it is not known if they are translated into protein; however, the promoter elements of these transcripts are essential components of the CYP21A1 and CYP21A2 promoters. The arrows indicate transcriptional orientation. The vertical dotted lines designate the boundaries of the genetic duplication event that led to the presence of A and B regions.
Much less is known about the enzymology of P450c21 than of P450c17, but the available evidence suggests that unlike P450c17, P450c21 is not very sensitive to the abundance of POR or cytochrome b5. It is clear that genotype consistently predicts phenotype in very severe and very mild cases of 21-hydroxylase deficiency. In contrast, patients with P450c21 variants (e.g., the common Pro30Leu and Val281Leu mutations and less common mutations Arg339His and Pro453Ser), which have 20% to 50% of wild-type activity,102,106 can have various phenotypes, implying additional factors that can modify the clinical manifestations of 21-hydroxylase deficiency.
P450c11β and P450c11AS
The classical descriptions of distinct deficiencies in 11β-hydroxylase, 18-hydroxylase (also called corticosterone methyl oxidase I, or CMOI), and 18-oxidase (CMOII) suggested that three enzymes executed these three respective transformations.107,108 Analogous to the scenario for P450c17, a single enzyme109 and corresponding gene110 were found in bovine adrenals that possessed all three activities. In contrast, humans have two genes named CYP11B1 and CYP11B2111 that encode the mitochondrial enzymes 11β-hydroxylase (P450c11β) and aldosterone synthase (P450c11AS), respectively, and rats but not mice have three functional CYP11B genes.112 Although P450c11β and P450c11AS both possess 11β-hydroxylase activities, P450c11AS also performs the two oxygenations at C18 required for aldosterone biosynthesis.113,114 Mutations in CYP11B1 cause 11β-hydroxylase deficiency,115 whereas defects in CYP11B2 cause either CMOI or CMOII deficiencies.116 Severe defects can impair all P450c11AS activities, leading to the clinical phenotype of CMOI deficiency,113 whereas P450c11β provides 11β-hydroxylase activity in the zona fasciculata. Fortuitous site-directed mutagenesis experiments of nature have found amino acid substitutions such as Arg181Trp plus Val386Ala, which mainly impair 18-oxidase activity and lead to CMOII deficiency.117
The coding regions of the CYP11B1 and CYP11B2 genes share 93% amino acid identity and the same exonic gene structure found in all mitochondrial P450 genes.118 Despite the sequence similarities of these tandem genes, located within 40 kb on chromosome 8q24.3, the expression of P450c11AS is restricted to the adrenal zona glomerulosa, whereas P450c11β is found in the zona fasciculata and zona reticularis. The regulation of P450c11β is driven mainly by cAMP in response to ACTH, whereas P450c11AS expression derives from potassium and angiotensin II activation of the protein kinase C pathway.119 Thus, under normal circumstances, 18-hydroxylase and 18-oxidase activities are restricted to the zona glomerulosa, where 17-hydroxylase activity is low, limiting the repertoire of steroids that can undergo 18-oxygenation.
Although the organization of two highly homologous, adjacent CYP11B1 and CYP11B2 genes on chromosome 8 is reminiscent of the genetics of the CYP21A1 and CYP21A2, gene conversion in the CYP11B locus occurs rarely.120 Instead, a clinical entity called glucocorticoid remediable aldosteronism (GRA) arises when an unequal crossing over of the CYP11B1 and CYP11B2 genes creates a third, hybrid gene in which the ACTH-regulated promoter of CYP11B1 drives expression of a chimeric protein with aldosterone synthase activity.121,122 As a result, 18-hydroxylase and 18-oxidase activities are ectopically expressed in the zona fasciculata, leading to elevated renin-independent production of aldosterone, as well as 18-oxygenated metabolites of cortisol. The expression of this gene is suppressed by blunting ACTH production with glucocorticoids such as dexamethasone, which is used for diagnosis and treatment.123 The prevalence of GRA varies from nil to as high 2% of referred patients with hypertension.124
The genetics of GRA has assisted in the precise identification of residues in P450c11AS that enable 18-oxygenase activities. Residues 288, 296, 301, 302, 325, and perhaps most importantly, 320 are critical for 18-oxygenase activities.125,126 Therefore, crossovers 3′ to codon 320 do not enable aldosterone synthase activity. These key residues lie in or near the I-helix, which contains the catalytically important threonine residue implicated in oxygen activation for almost all P450s; thus, these mutations would be expected to alter active site geometry.
P450aro (Aromatase)
The oxidative demethylation of C19 steroids, mainly androstenedione and testosterone, consumes three equivalents of molecular oxygen and NADPH, yielding formic acid and C18 steroids with an aromatic A-ring, hence the common name for this enzyme, aromatase. As is the case for P450scc, each subsequent oxygenation proceeds with greater efficiency, aiding in the completion of this transformation that is essential for estrogen biosynthesis in all animals.127 The mechanism of this aromatization must account for the incorporation of the final oxygen atom from molecular oxygen into the formic acid byproduct. The weight of evidence favors a hydroxylation at C2 of 19-oxo-androstenedione, followed by an enzyme-assisted rearrangement and tautomerization of the intermediate dienone to the phenolic A-ring.128
P450aro is expressed in steroidogenic tissues (ovarian granuloma cells, placenta), in brain, and in nonsteroidogenic tissues, especially fat and bone.127 The CYP19A1 gene for P450aro spans over 75 kb129 and contains five different transcriptional start sites130 with individual promoters that permit the tissue-specific regulation of expression in diverse tissues. P450aro is a glycoprotein, but glycosylation per se does not appear to affect activity.
Studies of patients with aromatase deficiency confirm that biologically significant estrogen synthesis derives entirely from this enzyme,131,132 although dietary phytoestrogens can provide some estrogen action in mice with targeted deletion of the aromatase gene.133 Although very few cases of aromatase deficiency have been described, they are highly informative “knockouts of nature” that illustrate principles of fetoplacental steroidogenesis. In fetuses homozygous for aromatase deficiency, the principal manifestation results from its deficiency in the placenta,131 because ovarian steroidogenesis is quiescent during fetal life.134 The fetal adrenal makes large amounts of C19 steroids, principally DHEA-S, much of which is 16α-hydroxylated in the fetal liver before undergoing metabolism via steroid sulfatases, 3β-HSD1, aromatase, and 17β-HSD1 in the placenta to produce estriol, the characteristic estrogen of pregnancy. Although huge amounts of estriol and estradiol are produced by the fetoplacental unit, estrogens are not needed for fetal development, the maintenance of pregnancy, or the onset of parturition; all of these processes proceed normally in fetuses lacking StAR, P450c17, or aromatase, or even in fetuses wholly lacking adrenal glands because of mutations in SF-1 or DAX-1.135 However, in the absence of placental aromatase activity, androgenic C19 steroids derived from the fetal adrenal are passed into the maternal circulation, causing marked virilization of the mother.131
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
