Chapter 60 The Ketogenic Diet
The incidence of epilepsy in children and adolescents ranges from 50 to 100 per 100,000 [Hauser and Hesdorffer, 1980; Hauser, 1994]. Antiepileptic drugs (AEDs) are the primary treatment modality and provide good seizure control in most children. However, more than 25 percent of children with epilepsy have either intractable seizures or suffer treatment-limiting adverse medication effects [Pellock, 1993, 1995, 1996; Heller et al., 1993; Pellock and Pippenger, 1993; Patsalos and Duncan, 1993]. Only a limited number of these children benefit from surgical therapy.
Uncontrolled seizures pose a variety of risks to children, including higher rates of mortality, accidents, and injuries, a greater incidence of cognitive and psychiatric impairment, poorer self-esteem, higher levels of anxiety and depression, and social stigmatization or isolation [Fisher et al., 1996]. Thus, effective treatment to control seizures is fundamental to improving overall outcome in childhood epilepsy. The ketogenic diet has proven to be an effective treatment for many children with epilepsy [Wheless, 1995a, b; Lefever and Aronson, 2000; Levy and Cooper, 2003]. In this chapter, the history of the development of the diet, current understanding of the biochemistry of ketone body formation and its relation to the anticonvulsant effect of the diet, considerations related to patient selection, and diet efficacy, complications, advantages, and disadvantages are reviewed.
History
For centuries, fasting has been used to treat many diseases, including seizures; it was even used in biblical times [Bible, The King James Version, 1982; Livingston, 1972].
Contemporary accounts of fasting (Table 60-1) include a child who did not respond to the conventional treatment of the day [Hendricks, 1995; Freeman et al., 1994], which was a combination of bromides and phenobarbital [Lennox and Lennox, 1960]. This child’s seizures were managed with prayer and fasting, producing dramatic improvement in seizure control during the period of starvation. However, when the period of starvation was terminated, the seizures returned [Conklin, 1922; Freeman et al., 1994; Bridge, 1949]. The child’s uncle, a pediatrician, enlisted the aid of John Howland at Johns Hopkins Hospital to understand how starvation and fasting controlled seizures and how one could maintain the beneficial effects of fasting [Lennox and Lennox, 1960]. Concurrently, Wilder at the Mayo Clinic suggested that a diet high in fat and low in carbohydrates could maintain ketosis and its accompanying acidosis longer than fasting [Freeman et al., 1994; Schwartz et al., 1989b]. Wilder was also the first to coin the term ketogenic diet. (See Wheless [2004] for a review of the ketogenic diet history.) The beneficial effects of this diet were initially recorded by investigators from Johns Hopkins University, the Mayo Clinic, and Harvard University. Use of the ketogenic diet was included in almost every comprehensive textbook on epilepsy in childhood that appeared between 1941 and 1980 [Penfield and Erickson, 1941; Lennox, 1941; Bridge, 1949; Livingston, 1954, 1963, 1972; Lennox and Lennox, 1960; Keith, 1963; Livingston et al., 1977; Withrow, 1980; Bower, 1980].
Until 1938, the ketogenic diet was one of the few available therapies for epilepsy, but it fell into disfavor when researchers turned their attention to the development of new AEDs [Livingston and Pauli, 1975]. After the introduction of sodium valproate, it was believed that this branched-chain fatty acid would treat those children previously placed on the ketogenic diet and that the diet could no longer be justified [Aicardi, 1994]. Use of the ketogenic diet decreased greatly until it received national media attention [NBC Dateline, 1994; Hendricks, 1995; Schneider and Wagner, 1995; Charlie Foundation to Help Cure Pediatric Epilepsy, 1994a; Freeman et al., 1990, 1994, 2000]. After an almost two-decade hiatus, renewed public and scientific interest has led to a better understanding and increased worldwide use of the ketogenic diet [Kleinman, 2004; Stafstrom and Rho, 2004; Kossoff et al., 2009b; Kossoff and McGrogan, 2005a].
Efficacy
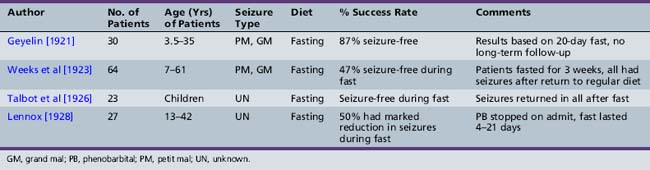
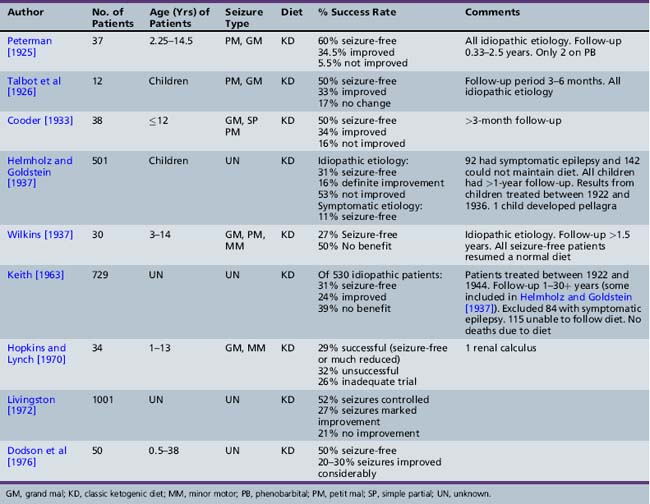
As presented in Table 60-2, initial reports began to appear in the 1920s and 1930s that documented the efficacy of the ketogenic diet [Geyelin, 1921; Wilder, 1921; Peterman, 1925; Helmholz, 1927; McQuarrie and Keith, 1927; Lennox and Cobb, 1928; Barborka, 1928a, 1929; Helmholz and Keith, 1930, 1932; Pulford, 1932; Fischer, 1935; Wilder and Pollack, 1935; Helmholz and Goldstein, 1937; Wilkins, 1937]. Over the next 60–70 years, many more clinical reports appeared (Table 60-3) [Keith, 1942; Prasad et al., 1996]. About one-third to one-half of children appeared to have had an excellent response to the diet, defined by cessation or marked reduction in seizure activity. About 10 percent of children become seizure-free after initiation of the ketogenic diet, and typically stop the diet after 2 years. Of those children who become seizure-free, a minority have recurrence after the diet is stopped [Martinez et al., 2007]. Risk factors that increase the likelihood of recurrence are epileptiform abnormalities on electroencephalograms (EEGs) obtained within 12 months of diet discontinuation, a focal abnormality on magnetic resonance imaging (MRI), lower initial seizure frequency, and tuberous sclerosis complex [Martinez et al., 2007]. When the diet has been discontinued, it has usually been due to lack of efficacy. It appears that younger children are more likely to have a more favorable response than older children [Freeman et al., 1998a].
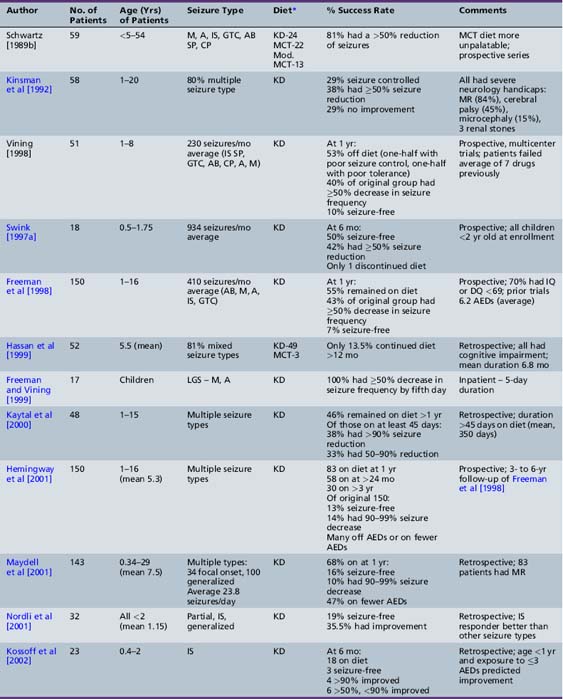
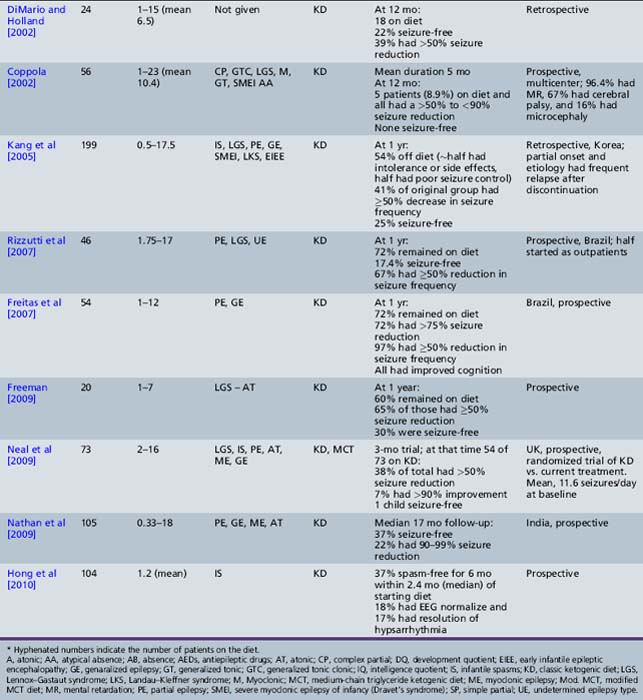
Recent studies attest to the efficacy and safety of the ketogenic diet in infants [Nordli et al., 2001; Kossoff et al., 2002; Klepper et al., 2002; Kang et al., 2005; Eun et al., 2006]. Nordli et al. retrospectively reviewed their experience with 32 infants who had been treated with the ketogenic diet, 17 of whom had infantile spasms [Nordli et al., 2001]. Most infants (71 percent) were able to maintain robust ketosis. The overall effectiveness of the diet in infants was similar to that reported for older children; 19.4 percent became seizure-free, and an additional 35.5 percent had a greater than 50 percent reduction in seizure frequency. The diet was particularly effective for children with infantile spasms. Kossoff et al. retrospectively reviewed their experience with the ketogenic diet as a treatment for infantile spasms during a 4-year period [Kossoff et al., 2002]. Twenty-three children, aged 5 months to 2 years, 39 percent of whom had symptomatic infantile spasms and 70 percent of whom had hypsarrhythmia, were started on the ketogenic diet. The children had been previously exposed to an average of 3.3 AEDs (range 0–7), and 74 percent had previously failed or relapsed after adrenocorticotropic hormone (ACTH) or steroids. The average time on the diet was 1.6 years, with 56 percent remaining on the diet at 12 months, 46 percent of whom were over 90 percent improved (three were seizure-free), and 100 percent were over 50 percent improved. Factors that predicted greater than 90 percent improvement at 12 months were age less than 1 year (p = 0.02) and exposure to ≤3 AEDs (p = 0.03). Improvement in development was related to seizure control. The same authors retrospectively reviewed their experience with the ketogenic diet as initial monotherapy for infantile spasms [Kossoff et al., 2008e]. At the end of 1 month, 8 of 13 (62 percent) infants were seizure-free (by parental report); however, the EEG normalization did not occur for 2–5 months. Time to spasm freedom was 6.5 days, suggesting that, if the ketogenic diet is used as initial treatment, a 2-week trial period is sufficient to judge efficacy for infantile spasms. In 2010, the same authors reported their 14-year experience with 104 consecutive infants prospectively started on the ketogenic diet [Hong et al., 2010]. Thirty-seven percent became spasm-free for at least a 6-month period, within a median 2.4 months of starting the ketogenic diet. A normal EEG was eventually obtained in 18 percent, and 17 percent demonstrated resolution of hypsarrhythmia. Eun et al. reviewed their experience treating 43 children with intractable infantile spasms (mean duration 13.4 months) with the ketogenic diet. Retrospective analysis revealed that the diet resulted in 53.5 percent of the patients being seizure-free; 81 percent had a greater than 50 percent reduction in seizure frequency after a mean duration of 9 months [Eun et al., 2006]. Complete seizure control was obtained within 4 weeks in 78.3 percent of the children who obtained seizure-freedom. The improvements in seizure control correlated with improvements in EEG findings. As part of the Korean multicentered experience, Kang et al. retrospectively reviewed efficacy in 49 infants treated with the ketogenic diet [Kang et al., 2005]. Only 1 of 14 children with severe myoclonic epilepsy of infancy was seizure-free at 12 months, but 56 percent had a greater than 50 percent seizure reduction. Carraballo et al. reported similar results, with 10 percent seizure-free, and 55 percent achieving a greater than 50 percent seizure reduction at 12 months [Carraballo et al., 2005]. In a small study (n = 3) of patients with a diagnosis of early infantile epileptic encephalothopy, none achieved seizure control [Kang et al., 2005].
A few early studies evaluated use of the ketogenic diet in adults (Table 60-4). Some revealed improved seizure control [Barborka, 1928b, 1930], but subsequent reports concluded that the diet is not particularly beneficial in adolescents and adults with epilepsy [Notkin, 1934; Lennox, 1941; Bridge, 1949; Livingston, 1954, 1963, 1972; Withrow, 1980; Mosek et al., 2009]. Reasons cited for this were poor dietary compliance, types of seizures seen in these age groups, and the developmental differences in the ability of the brain to use ketone bodies. As a result, past studies suggested that the optimum age of response to the diet may be in young children before the onset of adolescence. Recent studies have challenged the belief that older patients could not maintain therapeutic ketosis or comply with the rigors of the diet, and that the ketogenic diet was less efficacious in this age group. Sirven et al. [1999] reviewed the tolerability, efficacy, and adverse events in 11 adult patients prospectively begun on the classic ketogenic diet for refractory symptomatic epilepsy. All were on stable medication, and had not achieved seizure control with two or more medications; four had prior surgery. At eight months of follow-up, 1 patient was seizure-free, 5 patients had a greater than 50 percent decrease in seizure frequency, 1 patient had a less than 50 percent seizure decrease, and 4 patients discontinued the diet. Common adverse events included gastrointestinal complaints (100 percent), menstrual irregularities in all women, and increased serum cholesterol and cholesterol high-density lipoprotein (HDL) ratios. Subjective improvements in thinking and mood, without a decrease in AEDs, were reported in 7 patients, 1 without improved seizure control. Mady et al. reviewed their experience with 45 adolescents who had been on the ketogenic diet for an average duration of 1.2 years [Mady et al., 2003]. They found no evidence to support the belief that the diet was not efficacious and was too restrictive in this age group. Adolescents with multiple seizure types did best, and those with simple and complex partial seizures had the poorest response. The retention rate for motivated adolescents on the diet was not significantly different than reports in younger children.
Recent studies have critically re-examined the benefits of the diet in selected groups of patients (see Table 60-3). Schwartz et al. [1989a, 1989b] reported on the results of metabolic profiles and seizure control in 59 epileptic children receiving a normal diet or one of three forms of the ketogenic diet:
Patients fasted for 18 hours and then were placed on one of the diets and monitored for 6 weeks. All three diets produced a significant increase in total ketone body (aceto-acetate, β-hydroxybutyrate) levels that was most marked using the classical ketogenic diet [Freeman et al., 2000]. Ketone body concentrations reached a maximum in the afternoon and were frequently lower in the morning. Urinary ketones were measured and reflected changes noted in the serum. All three diets improved seizure control and none was superior to the others during the 6 weeks of observation. The first randomized trial of the classical and medium-chain triglyceride ketogenic diets was performed about 20 years later, and confirmed comparable efficacy over a longer follow-up period (1 year) [Neal et al., 2009]. Overall, these investigators found that 81 percent of patients had greater than a 45 percent reduction in seizures. Drowsiness, which occurred in 25 percent of patients during initiation of the diet, usually resolved. The medium-chain triglyceride diet was considered less palatable and associated with more side effects, including diarrhea and vomiting.
Kinsman et al reviewed 59 patients with severe intractable epilepsy; 80 percent had multiple seizure types; and 88 percent were on multiple AEDs [Kinsman et al., 1992]. Improved seizure control occurred in 67 percent of patients; 64 percent were able to reduce the amount of AEDs they were taking; 36 percent became more alert; and 23 percent had better behavior. Comorbid neurologic conditions in this group of patients with refractory epilepsy included mental retardation (84 percent), microcephaly (15 percent), and cerebral palsy (45 percent). Seizure type did not predict success with the diet. Additionally, 75 percent of the improved patients continued the diet for at least 18 months, confirming the efficacy and palatability of the diet and willingness to continue administration of the diet on the part of patients and their families. This study confirmed the earlier work by Livingston, demonstrating that 52 percent of patients had complete control and an additional 27 percent had improved control [Livingston, 1972].
The first large prospective evaluation of the ketogenic diet was conducted in 150 consecutive children aged 1–16 years (mean age 5.3 years) [Freeman et al., 1998a]. The children were followed for a minimum of 1 year, had previously been on an average of 6.24 medications, and were on a mean of 1.97 medications at the diet’s initiation. Seventy percent of children had an intelligence or developmental quotient of <69. The children averaged 410 seizures per month before the ketogenic diet. At 6 months, 71 percent remained on the diet and 32 percent had a greater than 90 percent decrease in seizures. At 1 year, 55 percent remained on the diet, 7 percent were seizure-free, and 27 percent had a greater than 90 percent decrease in seizure frequency. There was no statistically significant difference in seizure control based on age, sex, or seizure type, although none of the patients had only partial seizures. Most of those discontinuing the diet did so because it was either insufficiently effective or too restrictive.
All of the prior reports of efficacy of the ketogenic diet described open-label trials. Only one randomized, blinded crossover study has been performed. This study evaluated the efficacy of ketogenic diet for atonic-myclonic or drop seizures associated with Lennox–Gastaut syndrome [Freeman and Vining, 1999; Freeman, 2009; Freeman et al., 2009b]. Twenty children, experiencing at least 15 atonic seizures per day, were fasted for 36 hours, and then received the ketogenic diet for 1 week, in conjunction with a solution of glucose or saccharin. They then crossed over to the other study arm for an additional week. Physicians and parents were blinded to the solution composition and level of ketosis. Fasting and the ketogenic diet resulted in a decrease in the numbers of atonic seizures, which was only partially reversed with the addition of the glucose solution. The improvement in seizure control seen even in the glucose arm may explain the effectiveness of the less restrictive diets (Atkins-like and low glycemic).
A single randomized controlled trial of the ketogenic diet has been performed in children aged 2–16 years [Neal et al., 2008a]. Children with various seizure types or epilepsy syndromes experiencing daily seizures were recruited. They were randomly assigned to receive the ketogenic diet immediately (n = 73), or after a 3-month delay, with no other changes in their treatment (n = 72, control group). Children were also randomly assigned to the classic or the MCT ketogenic diet. After 3 months, the percentage of patients with baseline numbers of seizures despite treatment was significantly lower (p <0.0001) in the diet group (62 percent) than in the controls (136.9 percent). The children in the diet group had a mean of 13.3 seizures per day at baseline, and after 3 months of treatment, 1 was seizure-free; 28 (38 percent) had a greater than 50 percent reduction in seizures and 5 (7 percent) had a greater than 90 percent reduction. Efficacy was the same for symptomatic generalized or symptomatic focal epilepsies. The most common adverse events were constipation, vomiting, lack of energy, and hunger.
The same authors reported on the 1-year follow-up of those children randomized to the classic or MCT versions of the ketogenic diet [Neal et al., 2009]. Of the 125 children who started the diets, data from 64 were available at 6 months, and from 47 at 12 months. At 12 months, there was no significant difference between groups in number achieving greater than 50 percent (17.8 vs. 22.2 percent) or 90 percent (9.6 vs. 9.7 percent) seizure reduction. There was no significant difference in tolerability of the diet, except increased reports of lack of energy after 3 months, and vomiting after 12 months, both in the classical group.
In recent years, two alternative diet regimens that are less restrictive and perhaps more palatable have emerged: the modified Atkins diet [Kossoff and Dorward, 2008a] and a low glycemic index treatment [Pfeifer et al., 2008] (Figure 60-1). No prospective, comparative trials have been performed with the three treatments to evaluate relative efficacy and side-effect profiles. The Atkins diet is less restrictive than the ketogenic diet. The modified Atkins diet is similar in fat composition to a 1:1 ketogenic ratio diet, with approximately 65 percent of the calories from fat sources; it can be started as an outpatient, with guidance from a nutritionist. Three out of 6 patients studied on this diet had a greater than 50 percent seizure reduction; 2 were seizure-free [Kossoff et al., 2003]. Subsequent studies in children, adolescents, and adults reveal that 45 percent have a 50–90 percent seizure reduction, and slightly over 25 percent have a greater than 90 percent seizure reduction (Table 60-5) [Kossoff et al., 2003, 2006, 2007, 2008b, 2008d; Kang et al., 2007b; Carrette et al., 2008; Ito et al., 2008; Weber et al., 2009; Porta et al., 2009; Kumada et al., 2010].
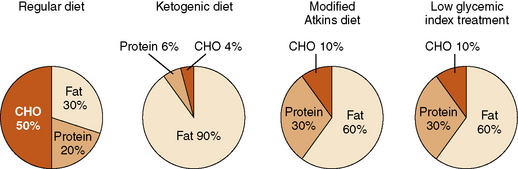
The low glycemic index treatment [Pfeifer et al., 2005; Pfeifer et al., 2008] includes approximately 20–30 percent of calories from protein and 60–70 percent from fat. Total carbohydrates are gradually decreased to 40–60 g/day (about 10 percent of calories), using foods with a low glycemic index [Foster-Powell et al., 2002]. A retrospective review of the efficacy of the low glycemic treatment demonstrated 50 percent of patients (n = 20) with refractory epilepsy experiencing a greater than 90 percent in seizure reduction [Pfeifer et al., 2005]. Of these 20 patients, 9 were transitioned from the ketogenic diet and 11 were initiated on the low glycemic treatment. Those with a greater than 90 percent seizure reduction had an average blood glucose level of 72.6 mg/dL. In a second retrospective review, 76 children on the low glycemic index diet – 42, 50, 54, 64, and 66 percent, respectively – achieved a greater than 50 percent seizure reduction from baseline seizure frequency at follow-up intervals of 1, 3, 6, 9, and 12 months [Muzykewicz et al., 2009]. Efficacy did not differ between partial onset and generalized seizure types. Increased efficacy was correlated with lower serum glucose levels at the 1- and 12-month follow-up visits. Side effects were minimal, with only three patients reporting transient lethargy. Children were placed on the low glycemic treatment for several reasons:
Mechanism of Action
While clinical reports support the efficacy of the ketogenic diet, several theories have emerged to explain the diet’s mechanism of action [Schwartzkroin, 1999]. Four major areas have been investigated: cerebral acidosis; water balance; the direct effect of ketones or lipids; and alteration in brain energy substrates. The importance of ketone body formation was recognized early in the search for the mechanism of action of the ketogenic diet [McQuarrie and Keith, 1927]. Initially, acidosis and partial dehydration were considered likely contributors to its success [Lennox, 1928; Bridge and Lob, 1931]. Even recently, the diet’s effects on water and electrolyte metabolism were thought responsible for its efficacy [Millichap et al., 1964]. Animal studies show no change in intracellular pH of the cerebral cortex on the ketogenic diet [Al-Mudallal et al., 1996; Harik et al., 1997]. Preliminary studies using MR spectroscopy have demonstrated that children on the diet do not develop alteration of brain water and electrolyte distribution [Seymour et al., 1999] or cerebral acidosis [Marks et al., 1997; Seymour et al., 1999]. This suggests that, although ketoacidosis occurs in these children, changes in brain pH may not be a critical factor. Animal models and human evidence suggest a prominent role of ketonemia [Uhlemann and Neims, 1972; Appleton and DeVivo, 1973, 1974; DeVivo et al., 1975; Al-Mudallal et al., 1995; Stafstrom, 1999a]. One study considered elevated plasma lipids a predictor of seizure control in children [Dekaban, 1966]. A subsequent analysis of blood metabolites in nine children on the ketogenic diet revealed that a rise in total serum arachidonate correlated with improved seizure control [Fraser et al., 2003]. Despite its long history of use and proven value, the exact mechanism of action of the diet remains unknown [Nordli and DeVivo, 1997].
Oxidation of Fatty Acids: Ketogenesis
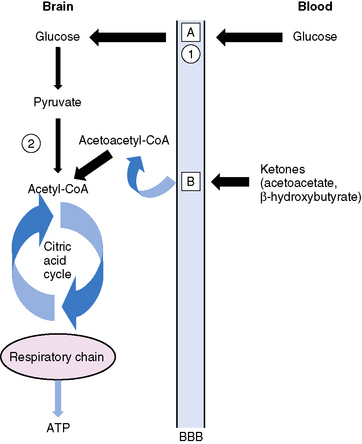
Ketonemia is essential for the ketogenic diet to work. Ketonemia occurs as a result of fatty-acid oxidation during fasting or while on the ketogenic diet [Hawkins and Biebuyck, 1980; DeVivo, 1980; Cahill, 1982; Mayes, 1996a, 1996b]. Fatty-acid biosynthesis (lipogenesis) takes place in the cytosol, whereas fatty-acid oxidation occurs in mitochondria and generates adenosine triphosphate (ATP) [Roe and Coates, 1995]. The oxidizable substrate may come from dietary sources (ketogenic diet) or from mobilization of peripheral adipose stores (starvation) (Figure 60-2). The brain does not directly use fatty acids but readily oxidizes ketone bodies [Hawkins and Biebuyck, 1979, 1980]. Increased fatty-acid oxidation leading to ketone body formation by the liver is characteristic of starvation or the ketogenic diet (Figure 60-3) [Mayes, 1996a]. Glucose, present in small concentrations, is necessary to facilitate ketone body metabolism. This is referred to as the permissive effect of glucose. Acetoacetate, a ketone body constituent, continually undergoes spontaneous decarboxylation to yield acetone that is volatilized in the lungs and gives the breath its characteristic odor.

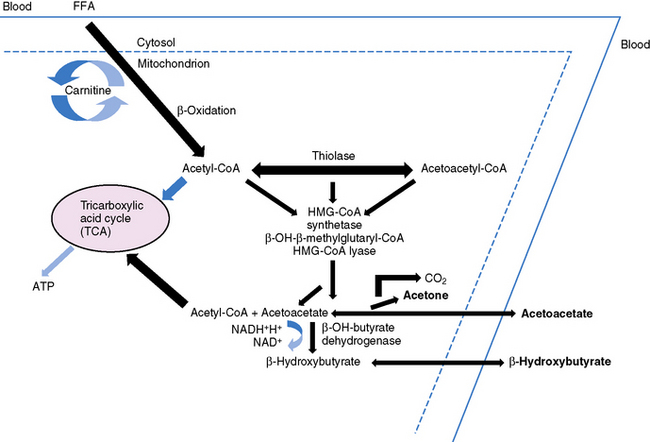
The liver is the only organ capable of synthesizing significant quantities of ketone bodies that are released into the blood (see Figure 60-3). The liver is equipped with an active enzymatic mechanism for the production of acetoacetate. Once formed, acetoacetate cannot be significantly metabolized back to fatty acids in the liver because it lacks the enzyme 3-oxoacid-CoA transferase. This accounts for the net production of ketone bodies by the liver. Ketone bodies are then transported to and oxidized in extrahepatic tissues proportionately to their concentration in the blood. Oxidation and brain influx rates of ketone bodies are proportional to their blood concentration of approximately 12 mmol/L. At this level, the oxidative machinery and uptake mechanisms of the cell are saturated [Mayes, 1996a].
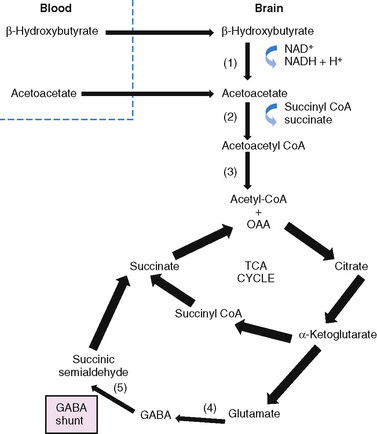
Glucose is the principal substrate for brain metabolism. Under certain conditions (e.g., fasting, ketogenic diet), the human brain uses ketone bodies for fuel [Owen et al., 1967]; the movement of ketone bodies into the brain is dependent on a monocarboxylic transport system [DeVivo, 1980]. Acetoacetate and β-hydroxybutyrate (BHB), the two constituents of ketone bodies, are metabolized primarily in the mitochondrial compartment. In the brain, the main pathway for the conversion of acetoacetate to acetoacetyl-CoA involves succinyl-CoA (Figure 60-4). Acetoacetyl-CoA is split to acetyl-CoA and oxidized in the tricarboxylic acid cycle. The enzymes that break down BHB and acetoacetate into acetyl CoA are regulated developmentally, with maximal expression early in life [Hawkins et al., 1971; Dahlquist et al., 1972; Nehlig, 1999]. This is consistent with the higher utilization of ketones by the brain in children compared to adults [Kraus et al., 1974]. Ketone bodies not only serve as a source of energy, but also contribute to the synthesis of the neurotransmitters, glutamate and gamma-aminobutyric acid (GABA), cerebral metabolic pathways normally dependent on glucose metabolism [Cremer, 1971; Sokoloff, 1973; DeLorey and Olsen, 1994; Rodwell, 1996; Cooper et al., 1996; Yudkoff et al., 2007; Lund et al., 2009]. Furthermore, fatty-acid oxidation increases brain ATP concentration [Mayes, 1996a, 1996b]. Elevation of brain ATP concentration has been verified in an animal model of the ketogenic diet [DeVivo et al., 1978; Nakazawa et al., 1983], suggesting that the ketogenic diet improves cerebral energetics. Increased alpha-ketoglutarate on the diet may also increase activity of the GABAA shunt [DeVivo, 1980; McGeer and McGeer, 1989; Peng et al., 1993]. Although whole-brain GABA levels are not changed in an animal model of chronic ketosis [Al-Mudallal et al., 1996; Harik et al., 1997], local changes in GABA concentration may occur [Rho et al., 1999b; Yudkoff et al., 2001a]. Two children studied with a new technology, two-dimensional double-quantum MR spectroscopy, showed low initial GABA levels that increased over time on the ketogenic diet [Wang et al., 2003]. The addition of acetoacetate or BHB to rat synaptosomes increased the rate of GABA formation by 35 and 43 percent, respectively [Erecinska et al., 1996]. The addition of β-hydroxybutyrate to cultured rat astrocytes suppresses GABA-transaminase in time- and dose-dependent manners [Suzuki et al., 2009]. This suppression of astrocytic GABA degradation may be another mechanism for increased GABA concentration. At the same time, the conversion of glutamate to aspartate, which has excitatory properties in the brain, is reduced [Erecinska et al., 1996; Yudkoff et al., 1997; Yudkoff et al., 2001b]. Thus, improved cerebral energetics, along with decreased excitatory (glutamatergic) and increased inhibitory (GABAergic) neurotransmission, may contribute to the efficacy of the ketogenic diet [Peyron et al., 1994; Greene et al., 2003].
Clinical Studies
Ketonemia is necessary but not sufficient for ketogenic diet-induced seizure control [Vining, 1999b; Stafstrom and Spencer, 2000]. Urine ketones are the only readily available inexpensive approach to ketone assessment, and typically the desired range is 80–160 mmol/L. Seizure control correlates significantly (p = 0.03) with serum BHB levels greater than 4 mmol/L, although urine ketones of 160 mmol/L can be found when blood BHB levels exceed 2 mmol/L [Gilbert et al., 2000]. Although serum ketones, particularly BHB, are believed to reflect the immediate state of ketosis more accurately, it is not certain if it is the presence of these ketone bodies that produces the antiseizure effect. Brown et al. [1998] measured serum BHB levels during routine clinic visits and during illness in 12 children. Eight experienced an increase in seizures and decrease in BHB during illness, and 4 had no seizure increase and no lowering of BHB levels. This suggests that BHB levels are important for the antiseizure effect. Subsequently, Freeman et al. [1998] evaluated serum BHB levels of 35 children 3 months after diet initiation. There was a significant correlation between higher levels of BHB and seizure control. The mean serum BHB level in patients with greater than 90 percent seizure control was over 6 mM.
It is also accepted that the classic ketogenic diet produces the most ketone bodies [Schwartz et al., 1989a]. Despite the fact that ketone bodies partially replace glucose for cerebral metabolism [DeVivo, 1980; Haymond et al., 1983], cerebral glucose levels are unaltered [DeVivo et al., 1978; Al-Mudallal et al., 1995]. Ketones from the circulation are transported across the blood–brain barrier by facilitated diffusion, using a monocarboxylate transport system [Moore et al., 1976; Pellerin et al., 1998]. The efficacy of the diet in childhood and the apparent slightly lower efficacy in older children and adults may be due to maturational changes in this transport system [Persson et al., 1972; Kraus et al., 1974; Dodson et al., 1976; Dahlquist and Persson, 1976; DeVivo, 1980; Williamson, 1985]. A child’s ability to extract ketones from the blood into the brain is 4–5 times greater than that seen in adults [DeVivo, 1983]. However, even adult animals placed on the ketogenic diet can upregulate brain monocarboxylate transporter levels [Leino et al., 2001]. MR spectroscopy has documented the fact that both fasting and intravenous BHB infusion in humans increase brain BHB levels [Pan et al., 2000, 2001]. However, proton MR spectroscopy (1H-MRS) performed in occipital gray matter in five children on the ketogenic diet demonstrated a single resonance identified as acetone, and no detectable BHB or acetoacetate [Seymour et al., 1999]. Subsequent experiments in rats and mice showed that acetone is an anticonvulsant and that chronic administration may enhance its action [Likhodii et al., 2002, 2003; Rho et al., 2002]. MR spectroscopy has also been used to study changes in cerebral energetics induced by the ketogenic diet. Alteration of tricarboxylic acid cycle activity by ketosis, resulting in an increased adenosine triphosphate to adenosine diphosphate (ATP:ADP) ratio or greater cerebral energy, has been hypothesized to have an anticonvulsant effect [Masino and Geiger, 2008]. This hypothesis is supported by recent experiments in patients with Lennox–Gastaut syndrome that used MR spectroscopy (31P) to document improvement in cerebral energy metabolism on the ketogenic diet [Pan et al., 1999].
Additionally, during chronic ketosis, adaptive mechanisms are active that increase the cerebral extraction of ketone bodies [Gjedde and Crone, 1975]. These mechanisms may be why ketosis develops promptly within several days after initiation of the diet, but the anticonvulsant effect may be delayed for 1–2 weeks [Appleton and Devivo, 1973, 1974]. This observation suggests that ketosis per se is insufficient to explain the anticonvulsant effect. Once ketone bodies are extracted, it is postulated that there is a secondary biochemical change or a cascade of biochemical effects that has some form of anticonvulsant effect [Prasad et al., 1996].
The importance of ketosis was demonstrated by Huttenlocher in 1976. He studied two children with a prior history of myoclonic seizures, who were seizure-free on the ketogenic diet. After a 50-minute infusion of glucose, the first patient’s serum ketones decreased 67 percent, with no change in serum pH, and a seizure occurred. The second child’s EEG changed from normal to diffuse polyspike and slow-wave complexes accompanied by myoclonic jerks after 90 minutes of intravenous glucose. Another study, involving nine children with intractable atypical absence seizures treated with the MCT diet, noted a significant decrease in the mean number of epileptiform discharges in treated patients [Ross et al., 1985]. A rise in serum ketones was the only prominent early biochemical change. DeVivo et al. also reported two children suffering from seizures resulting from a glucose transporter type I defect who were treated with the ketogenic diet and achieved complete control of seizures and improvement in neurologic development [DeVivo et al., 1991]. These observations and others suggest a pivotal role for ketone bodies in providing an alternative energy source and in achieving a still unknown role in seizure control [DeVivo, 1983; Schwartz et al., 1989b; Huttenlocher, 1976; Livingston, 1954; Aicardi, 1992; DeVivo et al., 1978; Withrow, 1980; Millichap et al., 1964; Lamers et al., 1995].
Experimental studies
At the present time, there are several animal models used to study the effects of the ketogenic diet. (For review, see Stafstrom [1999a]; Bough et al. [2002]; Stafstrom and Bough [2003]; Bough and Stafstrom [2004]; Hartman et al. [2007]; Hartman et al. [2008]; Raffo et al. [2008].) In general, these animal models demonstrate that the ketogenic diet provides protection for partial-onset seizures with secondary generalization and generalized myoclonic, tonic, and tonic-clonic seizures. The mechanism appears to be a lowering of neuronal excitability and raising of seizure threshold [Thavendiranathan et al., 2003]. However, some studies have failed to show a protective effect of the ketogenic diet in models where animals were acutely challenged with a convulsant stimulus [Bough et al., 2000a, 2002; Thavendiranathan et al., 2000; Samala et al., 2008]. Differential efficacy in animal models undoubtedly relates to regional differences in protein phosphorylation and the concentration of GABA and other neurotransmitters, and differences between provoked and spontaneous seizures [Szot et al., 2001; Ziegler et al., 2002; Martillotti et al., 2006].
A limited number of animal studies have investigated the effect of age at diet onset versus efficacy. The diet provides a greater level of seizure protection to younger animals [Bough et al., 1999c; Rho et al., 1999a], but older animals also demonstrate an elevated seizure threshold [Appleton and DeVivo, 1974; Hori et al., 1997; Bough and Eagles, 1999b]. While it would seem intuitive that increasing ketonemia would correlate with efficacy of the ketogenic diet, experimental studies have not revealed a positive correlation between them [Bough et al., 1999a, 2000b; Likhodii et al., 2000]. However, rats that developed higher levels of ketosis also showed higher thresholds for seizure induction [Bough and Eagles, 1999b]. Caloric restriction can significantly influence seizure threshold and augment the effects of the ketogenic diet on seizure control [Bough et al., 2000a, 2002, 2003; Greene et al., 2001; Eagles et al., 2003; Mantis et al., 2004; Cheng et al., 2004]. A role for enhanced cerebral energetics in efficacy of the ketogenic diet was first proposed by DeVivo et al. [1978]. Bough et al. [2006] used gene expression profiling to confirm a coordinated upregulation of transcripts for genes encoding proteins involved in energy metabolism in adolescent rat hippocampus, including those specific to mitochondria, after maintenance on a ketogenic diet [Bough et al., 2006]. These data support the hypothesis that a ketogenic diet enhances brain metabolism. Several animal models provide evidence that the ketogenic diet can prevent epileptogenesis [Muller-Schwarze et al., 1999; Stafstrom et al., 1999b; Rho et al., 1999c, 2000; Su et al., 2000; Todorova et al., 2000; Bough et al., 2003; Kossoff and Rho, 2009a; Kossoff et al., 2009c] and mediate neuroprotection [Zieglar et al., 2003; Sullivan et al., 2004; Noh et al., 2005; Gasior et al., 2006; Maalouf et al., 2007; Kim and Rho, 2008; Maalouf et al., 2009]. These studies also support the clinical observation that children can be gradually weaned off the ketogenic diet, resume a normal diet, and not experience loss of seizure control.
Selection of Candidates for the Diet
Despite the fact that several thousand patients have been treated, there currently are no consensus criteria as to which patients are candidates for the diet [Maria et al., 1997] (see Tables 60-1 to 60-4). Many seizure types appear to respond to the ketogenic diet. In general, several groups of children are potential candidates for treatment. These include the following children:













Little is known about whether the ketogenic diet should be considered in patients who are being evaluated for epilepsy surgery. Forty-seven children with intractable epilepsy and surgically remediable focal malformations of cortical development were treated with the ketogenic diet [Jung et al., 2008b]. Those seizure-free at 3 months continued on the diet, while the rest underwent surgery. The seizure-free rate at 2 years was similar between the two groups, leading the authors to suggest that, in potentially surgically remediable epilepsy, a 3-month trial of the ketogenic diet may be an option. However, in those patients in whom a malignancy or vascular malformation is detected, surgery is preferable. The ketogenic diet may be the preferred initial therapy in children with seizures and specific metabolic defects or seizures associated with specific neurologic syndromes (see Box 60-1 and Figure 60-5) [DeVivo et al., 1973, 1991; Haas et al., 1986; Wijburg et al., 1992; Shafrir and Prensky, 1995; Melvin et al., 1996; Wexler et al., 1997; Bergqvist et al., 1999b; Weber et al., 2001; Klepper et al., 2002, 2004; Liebhaber et al., 2003; Busch et al., 2005; Kang et al., 2005, 2007a; Kossoff et al., 2005b; Lee et al, 2008; Brockmann, 2009, Joshi et al., 2009; Mantis et al., 2009 ? mouse model Rett?s syndrome; Nylen et al., 2009 ? mouse model SSADH; Seo et al., 2010]. These specific metabolic disorders are reviewed in other sections of this book.
For some patients, the ketogenic diet is contraindicated (Box 60-2). Children with acute intermittent porphyria should not be treated with the ketogenic diet because carbohydrate restriction can be harmful in this condition. The ketogenic diet may also worsen some mitochondrial diseases, pyruvate carboxylase deficiency, or organic acidurias [Demeritte et al., 1996b; Nordli, 2000; Kleinman, 2004; Kossoff et al., 2009b]. When indicated, metabolic screen, including urine amino and organic acids, serum amino acids, lactate, pyruvate, and carnitine profile, should be performed before starting the ketogenic diet [Wheless, 2001]. In addition, the combination of the use of the ketogenic diet with topiramate or zonisamide appears to be associated with an increased risk of acidosis.















Candidate Selection and the EEG
The predictive value of an EEG obtained before, during, or after initiation of the ketogenic diet is unknown. In a cohort of 29 children, 90 percent of children with normal pretreatment EEGs responded favorably to the diet, compared with only 12 percent with abnormal pretreatment EEGs [Eley, 1933]. Penfield and Erickson [1941] determined that those patients with abnormal hyperventilation studies were more likely to respond to the diet. Livingston [1954] reported on the EEG findings in 102 patients and found that those patients who did best on the diet had petit mal variants. Compared to other children with epilepsy, children with multifocal spikes did less well at 3 months, but showed no difference at 6 or 12 months [Vining et al., 1998].
Many studies, including the original works of Bridge [1949], Livingston [1954], and Keith [1963], have suggested improvement or normalization of the EEG after the diet has been implemented. Nellhaus [1971] correlated normalization of the EEG with the onset of ketosis, clinical improvement, and prognosis. Others have noted that EEG improvement may be seen with clinical improvement in patients with minor motor and petit mal seizures, but not in those with focal or grand mal seizures [Janaki et al., 1976]. Children with atypical absence seizures demonstrated a significant decrease in the number of epileptiform discharges at initiation of MCT therapy. However, their EEGs were unchanged from baseline at the end of a 10-week treatment period, despite two-thirds having a greater than 50 percent decrease in seizure frequency [Ross et al., 1985]. In one study, background EEG improvements correlated with a significant reduction in seizures [Demeritte et al., 1996a]. Kang et al. showed improvement in generalized epileptiform abnormalities more often than focal epileptiform discharges, and a correlation between seizure control and EEG improvement [Kang et al., 2005]. Carraballo also reported improvement in the EEG epileptiform abnormalities in children responding to the diet [Carraballo et al., 2005]. Correlation between EEG improvement and reduction in clinical seizures, and between improvement in interictal discharges and improvement in background, was variable in studies of ambulatory 24-hour EEGs [Freeman and Vining, 1999; Hallbook et al., 2007a; Remahl et al., 2008], perhaps because of differences in seizure type and patient population among these studies.
Initiation and Maintenance
A comprehensive monograph on the evaluation and management of patients being considered for placement on the ketogenic diet was published [Freeman et al., 1994] and subsequently revised [Freeman et al., 2000]. In addition, a videotape, “The Ketogenic Diet for Families, Dietitians, Nurses and Physicians,” and other resource material are available from The Charlie Foundation (www.charliefoundation.org). The following is a brief overview of how the diet can be implemented.
Prehospital evaluation
Once a child is considered a candidate for the diet, a screening evaluation by selected members of the team responsible for implementing the diet is initiated. This screening usually includes a comprehensive evaluation by the dietitians and nursing staff. The purpose of this evaluation is to educate the family and to assess their ability, on many levels, to carry out the rigorous and exacting program necessary to maximize the diet’s success. At the same time, the different types of meal plans and foods that the child can eat are discussed, along with their preparation. Issues involved in this evaluation are carefully considered and reviewed by Casey et al. [1999] and Freeman et al. [2000].
Hospitalization
The child is typically scheduled for elective admission to the hospital for initiation of the ketogenic diet (Box 60-3). However, in recent years, both retrospective [Vaisleib et al., 2004] and prospective studies [Rizzutti et al., 2007] have documented the feasibility of initiating the diet as an outpatient. The intense educational process afforded by inpatient initiation may be preferable for some families and ketogenic diet centers. Additionally, inpatient initiation allows observation of the child to gauge the initial response to and adverse effects of the diet. Parents are told to view this treatment as a 6–8-week trial. When effective, the ketogenic diet works quickly, typically within the first 1–2 weeks [Kossoff et al., 2008c]. The time to improvement is significantly quicker (mean 5 vs. 14 days, p <0.01) in those children who are fasted at onset, but there is no effect of initial fasting on long-term outcome. If there is no seizure reduction after 6–8 weeks, the ketogenic diet can be discontinued. During this time, patients must adhere strictly to the diet, with proof of persistent ketosis. The frequency of seizures usually decreases gradually, but reversal of the effect can occur rapidly. Weeks of hard work can be undone if a child eats a cookie or a piece of candy. If the seizures are improved, this usually is sufficient motivation for the parents to have the child adhere to the diet. If the child might have an underlying metabolic disease that could be exacerbated by the initial fast, an appropriate evaluation should be performed before initiating the diet. Valproate has been demonstrated experimentally to reduce fasting ketonemia [Turnbull et al., 1986; Thurston et al., 1983]. Clinically, valproate has not impaired the ability to initiate the ketogenic diet or achieve adequate ketosis, nor has it altered the diet’s side-effect profile or efficacy [Lyczkowski et al., 2005].














The day before hospital admission, the parents are asked to eliminate carbohydrates from the child’s diet. The child eats foods that contain only protein or fat. After dinner, the child begins fasting, with only noncaloric, noncaffeinated beverages given. On the first hospital day, an awake-and-sleeping EEG may be performed. Laboratory studies (complete blood count, platelet count, chemistry panel, and AED levels) are obtained, either before or at the time of admission. During this time, the child receives maintenance fluids and may have one caffeine-free diet drink per day. Blood glucose levels are checked every 6 hours. The dietitian uses this time for further review of meal plans and the child’s food preferences and eating habits with the parents [Lasser and Brush, 1973]. Recent studies [Vaisleib et al., 2004; Kim et al., 2004; Bergqvist et al., 2005; Kang et al., 2005; Kossoff et al., 2008c] have suggested no improvement in seizure control with initial fasting or fluid restriction, and fewer adverse events with a nonfasting, gradual initiation. On day one, the child is started on the ketogenic diet, using a 3:1 or 4:1 ratio (fat to protein plus carbohydrate), depending on the child’s age. A ketogenic eggnog is used for the initial feedings. The child receives three meals at one-third of the total calories, is advanced as tolerated to two-thirds of the total calories for three meals (day 2), and then eats his or her first full meal. Before discharge, the parents prepare their first ketogenic meal for their child under the supervision of the dietitian. At discharge, the parents have several meal plans for their child and are instructed to monitor the urine ketones on a daily basis and record all occurrences of seizures. Individual decisions are made during the hospital stay as to whether the AED regimen will be simplified. The child is given a prescription for sugar-free multivitamin, mineral, and calcium supplementation and instructed to begin this at home. Vitamin B supplements are given to prevent optic nerve dysfunction [Hoyt and Billson, 1979]. Vitamin D supplementation and calcium prevent osteomalacia while on the ketogenic diet [Hahn et al., 1979]. Inadvertent administration of carbohydrates may occur when intercurrent illnesses are treated. Parents and pediatricians must appreciate that decongestants, antipyretics, and antibiotics are often contained in carbohydrate-containing vehicles [Feldstein, 1996; Freeman et al., 2000; Lebel et al., 2001; McGhee and Katyal, 2001]. The child is then seen regularly for follow-up (Box 60-4). Special attention should be paid to the serum albumin and total protein concentration to make sure that the diet is providing enough protein. Cholesterol and triglyceride levels typically rise when the diet is started, but the diet may be continued unless the cholesterol level rises above 1000 mg/dL and consistently stays there. It is not known whether cholesterol-reducing drugs could be safely given while the child is maintained on the diet. It is not unusual to see minor elevations in the direct bilirubin. Decisions regarding withdrawal of AEDs depend on the child’s response to the diet and the family’s wishes. Early reduction (during diet initiation or the first month afterward) of AEDs appears to be safe and well tolerated; however, it offers no definite advantage compared with a late taper [Kossoff et al., 2004]. At follow-up, the dietitian also reviews the parental concerns about implementing the diet, and makes adjustments in the meal plan, as necessary, to maintain the child in maximum ketosis. If the child is in apparent maximum ketosis (i.e., urine ketones are more than 160 mg/dL) and seizures recur, this might be the result of caloric excess, which may be manifested by excessive weight gain. Lowering the total caloric and carbohydrate intake may help reduce the number of seizure recurrences. If the child is not in maximum ketosis, this may be due to a break in the diet (e.g., “cheating” or taking medicine with sugar) or a lower than 4:1 ratio of fat to protein and carbohydrate. In children who can be successfully withdrawn from AED therapy and are seizure-free for 2 years on the ketogenic diet (about 10 percent of treated children), an EEG is repeated and the ketogenic diet is slowly withdrawn over 1 year. Many of these families elect to continue a low-carbohydrate diet, concerned that seizures may recur.












Complications
It needs to be stressed to parents that the diet is a form of medical therapy, and as such, although it is relatively safe, it is not without side effects (Box 60-5 and Box 60-6) [Ballaban-Gil et al., 1996; Wheless, 2001; Kang et al., 2004]. However, only 6–17 percent of patients discontinue the diet for medical reasons [Thompson et al., 1998; Vining et al., 1998; Kang et al., 2004; Henderson et al., 2006]. If the child has an unrecognized metabolic defect, a catastrophic event could occur during the fasting phase (see Box 60-2). Other adverse events can occur during the initial hospital stay, and, if recognized, are usually easily treated (see Box 60-6). A number of adverse events can occur during the maintenance phase. Many can be prevented with close monitoring, vitamin supplementation, and anticipatory treatment. The most common side effect encountered is constipation, which can be reduced if a calcium supplement with magnesium is given. Many of the children who begin the diet are already prone to this problem because of limited mobility, hypotonia, or spasticity. Constipation can be treated with regular doses of polyethylene glycol, mineral oil, intermittent pediatric-dose enemas, or magnesium hydroxide. Renal stones have occurred in less than 5–10 percent of patients, and can usually be easily managed [Herzberg et al., 1990; Freeman et al., 1994, 2000; Casey et al., 1999; Chesney et al., 1999; Furth et al., 2000; Kaytal et al., 2000; Kielb et al., 2000; Maydell et al., 2001; Nordli et al., 2001; Kossoff et al., 2002; Sampath et al., 2007]. The risk of stone formation may be increased in younger patients (<3 years of age), and those with hypercalciuria and low urine volume [Furth et al., 2000; Sampath et al., 2007]. Children can be screened for hypercalciuria (urine creatinine: calcium), and fluid intake can be maximized to minimize risk of nephrolithiasis. Even children who develop renal calculi, however, may be able to remain on the ketogenic diet. Evaluation to determine the type of renal stone and the patient’s urinary calcium–creatinine ratio is necessary [Gillespie and Stapleton, 2004]. If renal stones appear to be due to hypercalciuria, then the patient can be treated with hydrochlorothiazide (2 mg/kg/day), given polycitra to alkalinize the urine, and monitored with repeated urinary studies and renal ultrasound. If the calcium–creatinine ratios normalize or dramatically improve and no additional renal stones occur, then the diet can be maintained. Oral potassium citrate as a preventative supplement results in urine alkalinization, decreasing the prevalence of kidney stones [Sampath et al., 2007]. Universal supplementation appears to be warranted.
Box 60-5 Possible Adverse Events During Initiation of the Ketogenic Diet
| Adverse Event | Monitoring/Treatment Strategy |
|---|---|
 Dehydration Dehydration |
 Encourage fluids (do not limit fluids to less than 75% of maintenance); intravenous fluids if necessary (without dextrose) Encourage fluids (do not limit fluids to less than 75% of maintenance); intravenous fluids if necessary (without dextrose) |
 Hypoglycemia Hypoglycemia |
 Check blood sugars every 6 hr until diet is initiated; if symptomatic or blood sugar <30 mg/dL, give orange juice. Screen for metabolic errors in advance Check blood sugars every 6 hr until diet is initiated; if symptomatic or blood sugar <30 mg/dL, give orange juice. Screen for metabolic errors in advance |
 Vomiting Vomiting |
 Intravenous fluids; give orange juice Intravenous fluids; give orange juice |
Box 60-6 Possible Adverse Events During Maintenance of the Ketogenic Diet
| Adverse Event | Monitoring/Treatment Strategy |
|---|---|
 Constipation Constipation |
 Polyethylene glycol, mineral oil, suppositories Polyethylene glycol, mineral oil, suppositories |
 Exacerbation of gastroesophageal reflux disease Exacerbation of gastroesophageal reflux disease |
 Medical management Medical management |
 Poor growth Poor growth |
 Check albumin, total protein Check albumin, total protein |
 Adequate protein Adequate protein |
|
 Monitor height, weight Monitor height, weight |
|
 Kidney stones Kidney stones |
 Urine dipstick for blood, renal ultrasonography Urine dipstick for blood, renal ultrasonography |
 Analyze stone (specific treatment) Analyze stone (specific treatment) |
|
 Increase fluids, alkalinize urine Increase fluids, alkalinize urine |
|
 Dyslipidemia, hyperlipidemia Dyslipidemia, hyperlipidemia |
 Check liver function tests, lipid profile; if sustained elevations of cholesterol/ triglycerides, adjust diet and/or treat Check liver function tests, lipid profile; if sustained elevations of cholesterol/ triglycerides, adjust diet and/or treat |
 Prolonged QT interval, cardiomyopathy Prolonged QT interval, cardiomyopathy |
 Electrocardiography, echocardiography, check selenium Electrocardiography, echocardiography, check selenium |
 Excessive bruising Excessive bruising |
 Complete blood count, platelet count Complete blood count, platelet count |
 Optic neuropathy Optic neuropathy |
 B vitamins B vitamins |
 Elevated very-long-chain fatty acids Elevated very-long-chain fatty acids |
 Check prior to ketogenic diet if clinically suspected Check prior to ketogenic diet if clinically suspected |
 Vitamin D deficiency, osteomalacia Vitamin D deficiency, osteomalacia |
 Vitamin D, calcium, bone mineral density/densitometry Vitamin D, calcium, bone mineral density/densitometry |
 Trace mineral deficiencies Trace mineral deficiencies |
 Copper, selenium, zinc Copper, selenium, zinc |
Acetazolamide, which is sometimes used to treat certain forms of seizures, should not be used during initiation of the diet for fear of producing symptomatic metabolic acidosis [Dodson et al., 1976]. Topiramate and zonisamide are weak carbonic anhydrase inhibitors, which, when combined with the ketogenic diet, predispose to metabolic acidosis. The ketogenic diet can be cautiously initiated in children on these drugs. However, the physician should be aware that, in some children, this may cause worsening of their metabolic acidosis, especially during ketogenic diet initiation, and may require adjustments in the drug dose, adjustments in the diet, or addition of polycitra [Takeoka et al., 2002]. To date, no increased risk of nephrolithiasis has been reported in patients on topiramate or zonisamide and the ketogenic diet [Kossoff et al., 2002; Takeoka et al., 2002]. All children on one of these medicines should be treated with increased hydration and monitoring of their urine calcium–creatinine ratio; if the latter is elevated, urine alkalinization should be carried out.
Fatigue occurs in many children during initiation of the diet but is prolonged in only a small number. Children with gastroesophageal reflux, especially if they have cerebral palsy, may have increased gastroesophageal reflux while on the diet. The high fat content decreases gastric emptying, which promotes gastroesophageal reflux. This problem usually can be managed medically [Jung et al., 2008a]. Use of the ketogenic diet in infants to treat their seizure disorders or selected metabolic disorders has rarely been associated with fat malabsorption and secondary failure to thrive due to physiologic pancreatic insufficiency [Goyens et al., 2002]. This has been corrected by alteration of the diet via use of MCT and pancreatic extract. Close attention to growth measurements, laboratory data, and medical supervision is indicated in infants on the ketogenic diet. Only short-term information on growth in a limited number of patients had previously been available [Couch et al., 1999; Kossoff et al., 2002; Williams et al., 2002; Liu et al., 2003]. A prospective cohort study of 237 children, with an average length of follow-up of 308 days, analyzed height and weight measurements over time on the ketogenic diet [Vining et al., 2002]. A small decrease in height scores was observed in the first 6 months, with bigger changes by 2 years. There was a drop in weight in the first 3 months, and after this, the weight remained constant in children who started the diet below the 50th percentile for their weight, while it continued to decrease in children starting above the 50th percentile. Very young children (0–2 years) grew poorly on the diet, while older children (7–10 years) grew almost normally. A subsequent prospective study of 75 children, followed for 12 months, examined growth in children on classical and MCT ketogenic diets [Neal et al., 2009]. Both weight and height percentiles decreased, and by 12 months, there was no difference in outcome between classical and MCT protocols, despite the increased protein in the latter diet.
Twenty-two children had growth parameters monitored for 1 year prior to initiation of the ketogenic diet, and then for 1 year on the diet [Spulber et al., 2009]. Additionally, serial measurements of serum ketones and insulin-like growth factor I (IGF-I) were performed. Weight and height percentiles, body mass index, and height velocity all decreased significantly (p <0.05). Height velocity correlated negatively with serum β-hydroxybutyric acid levels, and was most affected in those with pronounced ketosis. Serum IGF-I decreased immediately at diet initiation, but then reached stable levels at around 3 months after diet initiation.
Several laboratory abnormalities have been reported in children on the ketogenic diet, although none has been found to have clinical significance. Patients on the ketogenic diet are in a chronic acidotic state, putting them at risk for osteopenia. A single longitudinal study has evaluated the effect of the ketogenic diet on bone density, serum hydroxyvitamin D, and parathyroid hormone [Bergqvist et al., 2007, 2008]. At baseline, the children with intractable epilepsy had suboptimal growth status and poor bone health. A progressive loss of bone mineral content, resulting in osteopenia and osteoporosis, occurred with ketogenic diet treatment, despite improved serum vitamin D concentrations. Supplementation of vitamin D and calcium was not sufficient to prevent bone mineral content loss. Increased supplementation and periodic surveillance with dual energy x-ray absorptiometry may be needed to prevent or treat the loss of bone mineral content in children treated with the ketogenic diet [Bergqvist et al., 2008]. This compensated metabolic acidosis puts the young child, who becomes ill, at risk of becoming markedly acidotic, ketotic, or dehydrated. This can be addressed by forewarning parents to increase fluid intake during illness. Serum cholesterol and triglycerides may increase, especially during the first 6 months; they tend to plateau by 6 months, then decline and require routine monitoring [Dekaban et al., 1966; Bergqvist et al., 1998; Chesney et al., 1999; Vining, 1999a; Kang et al., 2004]. Adjustments to the diet (e.g., increased protein and polyunsaturated fat) can be made in children with significantly high cholesterol and triglyceride concentrations. Impaired neutrophil function was described in ketotic children, but only one child had severe, recurrent infections [Woody et al., 1989]. This finding has not been replicated. Slight to significant prolongation of the bleeding time and abnormal platelet aggregation, despite normal platelet count and partial thromboplastin time (PTT), was reported in six children [Berry-Kravis et al., 2001b]. Patients on the diet and undergoing surgery should be evaluated for symptoms of bleeding tendency. Elevated very-long-chain fatty acids were found in one center in 59 percent of patients on the diet, and subsequently raised concerns about confusing this side effect with peroxisomal disorders [Theda et al., 1993]. Changes in the serum carnitine concentrations have also been described. Some believe a carnitine supplement should be given routinely [Demeritte et al., 1996b]. However, a prospective study of 11 children showed no substantial change over 1 year after initiation of the ketogenic diet [Coppola et al., 2006]. Carnitine status should be monitored, although most children do not need supplementation [Berry-Kravis et al., 1998, 2001a; Lin et al., 1998]. Carnitine supplementation may have conflicting effects; carnitine deficiency may restrict fatty-acid beta-oxidation, and carnitine may also improve glucose transport into cells. Supplementation with magnesium, zinc, calcium, vitamin D, and B vitamins is recommended to avoid deficiency-related disease states [Hahn et al., 1979; Hoyt et al., 1979]. The ketogenic diet is deficient in several trace minerals, and if children are maintained on the diet for more than 2 years, these need to be supplemented [Goodwell et al., 1998; Chee et al., 1998; Bergqvist et al., 1999, 2003].
Serious complications of the ketogenic diet are rare and typically have only been described in single reports, including those of Fanconi’s renal tubular acidosis (in co-treatment with valproate) [Ballaban-Gil et al., 1996], severe hypoproteinemia [Ballaban-Gil et al., 1998], marked increase in liver function tests (in co-treatment with valproate) [Ballaban-Gil et al., 1998], cardiomyopathy [Ballaban-Gil, 1999; Best et al., 2000; Bergqvist et al., 2003; Kang et al., 2004; Bank et al., 2008], prolonged QTc [Best et al., 2000], acute hemorrhagic pancreatitis [Stewart et al., 2001], basal ganglia injury [Erickson et al., 2002], scurvy [Willmott and Bryan, 2008], lipoid pneumonia [Kang et al., 2005], and fatal propofol infusion syndrome [Baumeister et al., 2004].
A single retrospective chart review reports maintenance of efficacy and tolerability with long-term use of the ketogenic diet (duration of 6–12 years, n = 28) [Groesbeck et al., 2006]. However, side effects of decreased growth (23 out of 28), kidney stones (7 out of 28), and fractures (6 out of 28) occurred, and careful monitoring with strategies to minimize these complications is suggested.
Advantages and Disadvantages
Advantages
The ketogenic diet has several advantages when compared with other medical therapies. Anecdotal reports have indicated that many of the children who are maintained on the diet are able to have their AEDs decreased or withdrawn. This typically results in the child being more alert and exhibiting better behavior [Maydell et al., 2001; Nordli et al., 2001]. However, even children whose AEDs cannot be substantially lowered or withdrawn may have marked behavioral or cognitive improvements [Katyal et al., 2000]. This has occurred in children who have become seizure-free on the diet and had AEDs withdrawn, as well as in those whose seizures are only minimally improved and who have not had dramatic changes in their AED regimen.
Parents are concerned about cognitive side effects, and, although the newer AEDs tend to have fewer such side effects, there are none with the diet. A prospective pilot study assessed the effects of the ketogenic diet on development and behavior in 65 children with intractable epilepsy [Pulsifer et al., 2001]. Formal evaluation of development, behavior, and paternal stress using parental report measures was performed before diet initiation and 1 year later. At follow-up, mean developmental quotient showed statistically significant improvement (p <0.05), with significant (p <0.05) behavioral improvements in attention and social functioning. The observed developmental and behavioral improvements were not statistically related to reductions in seizure frequency or AEDs. Parental stress was essentially unchanged. This preliminary report supports the prior anecdotal observations of the beneficial effects of the diet on cognition and behavior. Hallbook et al. [2007b] evaluated 18 children with polysomnographic recordings, and quality of life, attention, and behavior scales at baseline, then after 3 and 12 months on the ketogenic diet in a prospective manner on stable AEDs. Eleven children continued with the ketogenic diet for 12 months. They showed improved sleep quality, a decrease in daytime sleep, and significant improvement in attention and quality of life. Experiments in rats have shown no adverse effect of the ketogenic diet on spatial learning and memory, the animal’s response to a novel environment, or a battery of behavioral tests [Hori et al., 1997; Bough et al., 2000b]. These reports of a positive cognitive effect of the diet in children and experimental models are in contrast to the single study of cognition in rats [Zhao et al., 2004] and one weight loss study in adults [Wing et al., 1995]. Adults on the ketogenic diet for weight loss performed more poorly on some neurocognitive tasks than did nontreated controls [Wing et al., 1995]. These concerns have not been raised by any study of human beings but should be pursued in experimental and clinical research arenas [Snead, 2004]. In addition, with the ketogenic diet, there appear to be no concerns about organ failure or toxicity.
Disadvantages
The most common reasons cited for discontinuation of the ketogenic diet are lack of efficacy, complications, noncompliance (especially in older children), and caregiver concerns [Lightstone et al., 2001]. The ketogenic diet is a strictly regulated medical diet that requires an epilepsy team for success. It requires active participation by the parents and children. The work required on the part of the parents to initiate and maintain this rigorous diet may be considered a disadvantage.
In the 1930s and 1940s, the cost of the diet was considered a major disadvantage. However, at that time the diet was compared with the cost of bromides or phenobarbital. Currently, the child with intractable epilepsy can incur costs of around $5000 per year or more for new AEDs. A retrospective study of medical costs in 15 children with intractable epilepsy who were placed on the ketogenic diet showed a saving of over $10,000 per child for the 1–2-year period after initiation of the diet, compared to the same time period prior to the diet [Mandel et al., 2002]. Successful maintenance on the ketogenic diet can provide a substantial financial benefit.
Many physicians perceive the diet as unpalatable and difficult to initiate and maintain. However, modern nutritional labeling requirements and the use of computers allow the dietitian to construct a palatable diet, keeping the patient’s food preferences in mind, and this permits a much more varied meal plan [Brake and Brake, 1997].
The Ketogenic Diet in the Twenty-First Century
More than 80 years have passed since the ketogenic diet was initially used, and many more therapies are now available for children with epilepsy. The ketogenic diet compares favorably with other new treatments that have been introduced to treat children with epilepsy [The Felbamate Study Group, 1993; George et al., 1994; The Vagus Nerve Stimulation Study Group, 1995; LeFever et al., 2000]. Studies on the newer AEDs, such as zonisamide, lamotrigine, tiagabine, gabapentin, topiramate, oxcarbazepine, lacosamide, pregabalin, and levetiracetam, typically indicate that only 3–10 percent of all intractable patients achieve complete relief of seizures. The question that remains unanswered is, “When, in the course of therapy for a child with intractable epilepsy, should the ketogenic diet be used?” Currently, it is not used until children have failed multiple medications and if they are not considered surgical candidates. However, in some epilepsy syndromes, the diet should be offered as a treatment strategy after failure of one or two drugs. If the child has an epilepsy syndrome that is often resistant to current therapy and if the seizures are not controlled on medication, the ketogenic diet should be mentioned as an alternative therapy at the time of the initial therapeutic discussions. This is especially true in children who are not good candidates for epilepsy surgery or whose parents do not wish their children to have epilepsy surgery. The renewed interest in the ketogenic diet has, once again, raised several research questions that, if answered, have the potential to improve our understanding of the neurochemistry of epilepsy and allow better treatment of all patients with epilepsy [Nordli and DeVivo, 1997; Prasad et al., 1996].
In addition to the need to gain a better understanding of the underlying neurochemical changes induced with the ketogenic diet, several other clinical questions remain. Specifically, for what seizure types or epilepsy syndromes does this diet have a higher chance of being a successful short- or long-term treatment? It would be helpful to have more long-term (i.e., 10–20 years) follow-up data on those patients who appear not to have a return of seizures after the diet is stopped [Keith, 1963].
Additional questions relate to the role of the ketogenic diet in treating less affected or normal children with epilepsy. How does the risk–benefit ratio of the ketogenic diet compare with other therapies? Finally, many adults with intractable epilepsy wish to know if there is any group of adults that could benefit from the ketogenic diet. The ketogenic diet might be useful in other childhood neurologic disorders, such as alternating hemiplegia of childhood or tumors of the nervous system [Nebeling et al., 1995]. There is an expanding experimental literature suggesting that the ketogenic diet may treat several neurologic conditions [Kossoff et al., 2009c; Santra et al., 2004; Murphy et al., 2004].
The ketogenic diet is a therapy that was first used at the beginning of the 20th century and appears still to have a definitive role in the treatment of childhood epilepsy into the 21st century. The preferred sequence of treatments for children with epilepsy will continue to need reassessment and redefinition. It will likely be several years before the exact roles for all current therapies are defined. Until then, it is critical that those taking care of children with seizures be aware of all the treatment options and refer children whose seizures are not controlled to centers specializing in the care of such children [The National Association of Epilepsy Centers, 1990]. For many such children, the ketogenic diet continues to represent a therapeutic alternative.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
. Videotapes on the ketogenic diet for families, dietitians, nurses, and physicians. The Charlie Foundation to Help Cure Pediatric Epilepsy, 501 10th Street, Santa Monica, CA 90402; Fax (310) 393-2347. (www.charliefoundation.org)
Freeman J.M., Kossoff E.H., Freeman J.B., et al. The Ketogenic Diet: A Treatment for Epilepsy in Children and Others, ed 4. New York: Demos; 2007. 386 Park Avenue South, Suite 301, New York City, NY 10016; Tel: (800) 532-8663
Brake D., Brake C. The ketogenic cookbook. Gilman, CT: Greider Pennycomer Press; 1996. Both authors are culinary arts graduate chefs whose children were on the ketogenic diet treatment program. George M. Greider, Pennycorner Press, Post Box 8, Gilman, CT 06336; Tel: (860) 873-3545; Fax: (860) 873-1311
Snyder D. Keto Kid. New York: Demos; 2007. 386 Park Avenue South, Suite 301, NYC, NY 10016. Tel. (800) 532-8663
Zupec-Kania B.A.. Ketogenic Diet: Parent’s Guide. Santa Monica, CA: The Charlie Foundation; 2009. www.charliefoundation.org.
The ketogenic diet. In: Kleinman R.E., editor. Pediatric Nutrition Handbook. ed 6. Elk Grove Village, IL: American Academy of Pediatrics; 2009:1021-1040. Policy of the American Academy of Pediatrics
Aicardi J. Clinics in developmental medicine 115/118: diseases of the nervous system in childhood. New York: Mac Keith; 1992.
Aicardi J. The international review of child neurology. Epilepsy in children, ed 2. New York: Raven Press; 1994.
Al-Mudallal A.S., LaManna J.C., Lust W.D., et al. Diet-induced ketosis does not cause cerebral acidosis. Epilepsia. 1996;37:258.
Al-Mudallal A.S., Levin B.E., Lust W.D., et al. Effects of unbalanced diets on cerebral glucose metabolism in the adult rat. Neurology. 1995;45:2261.
Appleton D.B., DeVivo D.C. An experimental model for the effect of ketogenic diet on epilepsy. Proc Aust Assoc Neurol. 1973;10:75.
Appleton D.B., DeVivo D.C. An animal model for the ketogenic diet. Epilepsia. 1974;15:211.
Ballaban-Gil K., Callahan C., O’Dell C., et al. Complications of the ketogenic diet. Ann Neurol. 1996;40:307.
Ballaban-Gil K.R. Cardiomyopathy associated with the ketogenic diet. Epilepsia. 1999;40(Suppl 7):129.
Bank I.M., Shemie S.D., Rosenblatt B., et al. Sudden cardiac death in association with the ketogenic diet. Pediatr Neurol. 2008;39:429.
Barborka C.J. The ketogenic diet. Proc Meet Mayo Clin. 1928;3:273.
Barborka C.J. Ketogenic diet treatment of epilepsy in adults. JAMA. 1928;91:73.
Barborka C.J. The ketogenic diet and its use. Med Clin North Am. 1929;12:1639.
Barborka C.J. Epilepsy in adults: results of treatment by ketogenic diet in one hundred cases. Arch Neurol Psychiatr. 1930;23:904.
Bastible C. The ketogenic treatment of epilepsy. Irish J Med Sci. 1931;2:506.
Baumeister F.A.M., Oberhoffer R., Liebhaber G.M., et al. Fatal propofol infusion syndrome in association with ketogenic diet. Neuropediatrics. 2004;35:250.
Bergqvist A.G.C., Chee C., Fink L., et al. Serial measurements of serum cholesterol and triglycerides in the ketogenic diet. Ann Neurol. 1998;44:565.
Bergqvist A.G.C., Chee C.M., Bettler J.E., et al. Zinc deficiency resulting from the ketogenic diet. Epilepsia. 1999;40(Suppl 7):133.
Bergqvist A.G.C., Chee C.M., Lutchka L.M., et al. Treatment of acquired epileptic aphasia with the ketogenic diet. J Child Neurol. 1999;14:696.
Bergqvist A.G.C., Chee C.M., Lutchka L., et al. Selenium deficiency associated with cardiomyopathy: a complication of the ketogenic diet. Epilepsia. 2003;44:618.
Bergqvist A.G.C., Schall J.I., Gallagher P.R., et al. Fasting versus gradual initiation of the ketogenic diet: a prospective, randomized clinical trial of efficacy. Epilepsia. 2005;45:1810.
Bergqvist A.G.C., Schall J.I., Stallings V.A. Vitamin D status in children with intractable epilepsy, and impact of the ketogenic diet. Epilepsia. 2007;48:66.
Bergqvist A.G.C., Schall J.I., Stallings V.A., et al. Progressive bone mineral content loss in children with intractable epilepsy treated with the ketogenic diet. AM J Clin Nutr. 2008;88:1678.
Berry-Kravis E., Booth G., Sanchez C., et al. The ketogenic diet: need for carnitine supplementation. Ann Neurol. 1998;44:566.
Berry-Kravis E., Booth G., Sanchez A.C., et al. Carnitine levels and the ketogenic diet. Epilepsia. 2001;42:1445.
Berry-Kravis E., Booth G., Taylor A., et al. Bruising and the ketogenic diet: evidence for diet-induced changes in platelet function. Ann Neurol. 2001;49:98.
Best T.H., Franz D.N., Gilbert D.L., et al. Cardiac complications in pediatric patients on the ketogenic diet. Neurol. 2000;54:2328.
Bible. The New King James Version Mark 9:14-29. Carmel, NY: Guideposts; 1982.
Bough K., Chen R., Eagles D. Path analysis shows that increasing ketogenic diet ratio but not beta-hydroxybutyrate, elevates seizure threshold in the rat. Dev Neurosci. 1999;21:400.
Bough K.J., Eagles D.A. A ketogenic diet increases the resistance to pentylenetetrazole-induced seizures in the rat. Epilepsia. 1999;40:138.
Bough K.J., Valiyil R., Han F.T., et al. Seizure resistance is dependent upon age and caloric restriction in rats fed a ketogenic diet. Epilepsy Res. 1999;35:21.
Bough K.J., Gudi K., Han F.T., et al. An anticonvulsant profile of the ketogenic diet in the rat. Epilepsy Res. 2002;50:313.
Bough K.J., Matthews P.J., Eagles D.A. A ketogenic diet has different effects upon seizures induced by maximal electroshock and by pentylenetetrazole infusion. Epilepsy Res. 2000;38:105.
Bough K.J., Yao S.G., Eagles D.A. Higher ketogenic diet ratios confer protection from seizures without neurotoxicity. Epilepsy Res. 2000;38:15.
Bough K.J., Schwartzkroin P.A., Rho J.M. Calorie restriction and ketogenic diet diminish neuronal excitability in rat dentate gyrus in vivo. Epilepsia. 2003;44:752.
Bough K.J., Stafstrom C.E. The ketogenic diet: scientific principles underlying its use. In: Rho J.M., Sankar R., Cavazos J.E., editors. Epilepsy: Scientific Foundations of Clinical Practice. New York City, NY: Marcel Dekker, Inc; 2004:263.
Bough K.J., Wetherington J., Hassel B., et al. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann Neurol. 2006;60:223.
Bower B.D.. Epilepsy in childhood and adolescence. Tyrer J.G., editor. Current status of modern therapy: the treatment of epilepsy, vol 5. Philadelphia: JB Lippincott, 1980.
Brake D., Brake C. The ketogenic cookbook. Gilman, Conn: Pennycorner Press; 1997.
Bridge E.M. Epilepsy and convulsive disorders in children. New York: McGraw-Hill; 1949.
Bridge E.M., Lob L.V. The mechanism of the ketogenic diet in epilepsy. Bull Johns Hopkins Hosp. 1931;48:373.
Brockmann K. The expanding phenotype of GLUT1 – deficiency syndrome. Brain Dev. 2009;31:545.
Brown L., Lutchka L.M., Bergqvist C., et al. Monitoring beta-hydroxybutyrate as an anti-convulsant level in the ketogenic diet. Epilepsia. 1998;39(Suppl 6):168.
Busch V., Gempel K., Hack A., et al. Treatment of glycogenosis type V with ketogenic diet. Ann Neurol. 2005;58:341.
Cahill G.F. Starvation. Trans Am Clin Climatol Assoc. 1982;94:1.
Carraballo R.H., Cersosimo R.O., Sakr D., et al. Ketogenic diet in patients with Dravet Syndrome. Epilepsia. 2005;46:1539.
Carrette E., Vonch K., Herdt V., et al. A pilot trial with modified Atkins diet in adult patients with refractory epilepsy. Clin Neurol Neurosurg. 2008;110:797.
Casey J.C., McGrogan J., Pillas D. The implementation and maintenance of the ketogenic diet in children. J Neurosci Nursing. 1999;31:294.
Charlie Foundation to Help Cure Pediatric Epilepsy. An Introduction to the ketogenic diet – a treatment for pediatric epilepsy. Videotape. Santa Monica, Calif: Charlie Foundation; 1994.
Chee C.M., Lutchka L., Brown L., et al. Ketogenic diet: unrecognized selenium deficiency. Epilepsia. 1998;39(Suppl 6):228.
Cheng C.M., Hicks K., Wang J., et al. Caloric restriction augments brain glutamic acid decarboxylase-65 and -67 expression. J Neurosci Res. 2004;77:270.
Chesney D., Brouhard B.H., Wyllie E., et al. Biochemical abnormalities of the ketogenic diet in children. Clin Pediatr. 1999;38:107.
Conklin H.W. Cause and treatment of epilepsy. J Am Osteo. 1922;22:11.
Cooder H.R. Epilepsy in children with particular reference to the ketogenic diet. Calif West Med. 1933;39:169.
Cooper J.R., Bloom F.E., Roth R.H. Amino acid transmitters. In: The biochemical basis of neuropharmacology. New York: Oxford Press; 1996.
Coppola G., Epifanio G., Auricchio G., et al. Plasma free carnitine in epilepsy children, adolescents and young adults treated with old and new anti-epileptic drugs with or without ketogenic diet. Brain Dev. 2006;28:358.
Coppola G., Veggiotti P., Cusmai R., et al. The Ketogenic diet in children, adolescents and young adults with refactory epilepsy: an Italian multicavtric experience. Epilepsy Res. 2002;48(3):221-227.
Couch S.C., Schwarzman F., Carroll J., et al. Growth and nutritional outcomes of children treated with the ketogenic diet. J Am Diet Assoc. 1999;99:1573.
Cremer J.E. Incorporation of label from D-beta-hydroxy[14C] butyrate and [3-14C] acetoacetate into amino acids in rat brain in vivo. Biochem J. 1971;122:135.
Dahlquist G., Persson U., Persson B. The activity of D-beta-hydroxybutyrate dehydrogenase in fetal, infant, and adult rat brain and the influence of starvation. Biol Neonate. 1972;20:40.
Dahlquist G., Persson B. The rate of cerebral utilization of glucose, ketone bodies, and oxygen: a comparative in vivo study of infant and adult rats. Pediatr Res. 1976;10:910.
Dekaban A.S. Plasma lipids in epileptic children treated with the high fat diet. Arch Neurol. 1966;15:177.
DeLorey T.M., Olsen R.W. GABA and glycine. In: Siegel G.J., Agranoff B.W., Albers R.W., et al, editors. Basic neurochemistry. New York: Raven Press, 1994.
Demeritte E.L., Coyne M., Ventimiglia J., et al. Electroencephalographic analysis in the ketogenic diet. Ann Neurol. 1996;40:305.
Demeritte E.L., Ventimiglia J., Coyne M., et al. Organic acid disorders and the ketogenic diet. Ann Neurol. 1996;40:305.
DeVivo D.C. The effects of ketone bodies on glucose utilization. In: Passoneau J.V., Lust W.D., Hawkins J.R., et al, editors. Cerebral metabolism and neural function. Baltimore: Williams & Wilkins; 1980:243.
DeVivo D.C. How to use other drugs (steroids) and the ketogenic diet. In: Morselli P.L., Pippenger C.E., Penry J.K., editors. Antiepileptic drug therapy in pediatrics. New York: Raven Press, 1983.
DeVivo D.C., Leckie M.P., Ferrendelli J.S., et al. Chronic ketosis and cerebral metabolism. Ann Neurol. 1978;3:331.
DeVivo D.C., Malas K.L., Leckie M.P. Starvation and seizures: observations on the electroconvulsive threshold and cerebral metabolism of the starved adult rat. Arch Neurol. 1975;32:755.
DeVivo D.C., Pagliara A.S., Prensky A.L. Ketotic hypoglycemia and the ketogenic diet. Neurology. 1973;23:640.
DeVivo D.C., Trifiletti R.R., Jacobson R.I., et al. Glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures and developmental delay. N Engl J Med. 1991;325:703.
DiMario F.J., Holland J. The ketogenic diet: a review of the experience at Connecticut Children’s Medical Center. Pediatr Neurol. 2002;26:288.
Dodson W.E., Prensky A.L., DeVivo D.C., et al. Management of seizure disorders. Selected aspects, part II. J Pediatr. 1976;89:695.
Eagles D.A., Boyd S.J., Kotak A., et al. Calorie restriction of a high-carbohydrate diet elevates the threshold of PTZ-induced seizures to values equal to those seen with a ketogenic diet. Epilepsy Res. 2003;54:41.
Eley R.C. Epilepsy. The value of encephalography in the selection of patients for treatment by the ketogenic diet. J Pediatr. 1933;3:359.
Erecinska M., Nelson D., Daikhin Y., et al. Regulation of GABA level in rat brain synaptosomes: fluxes through enzymes of the GABA shunt and effects of glutamate, calcium, and ketone bodies. J Neurochem. 1996;67:2325.
Erickson J.C., Jabbari B., Difazio M.P. Basal ganglia injury as a complication of the ketogenic diet. Mov Disord. 2002;18:448.
Eun S.H., Kang H.C., Kim D.W., et al. Ketogenic diet for treatment of infantile spasms. Brain Dev. 2006;28:566.
The Felbamate study group in Lennox-Gastaut syndrome. Efficacy of felbamate in childhood epileptic encephalopathy (Lennox-Gastaut syndrome). N Engl J Med. 1993;328:29.
Feldstein T.J. Carbohydrate and alcohol content of 200 oral liquid medications for use in patients receiving ketogenic diets. Pediatrics. 1996;97:506.
Fischer L. Epilepsy: its treatment by the use of the ketogenic diet versus drugs. Arch Pediatr. 1935;52:131.
Fisher R.S., Parks-Trusz S.L., Lehman C. Social issues in epilepsy. In: Shorvon S., Dreifuss F., Fish D., et al, editors. The treatment of epilepsy. Woburn, Mass: Blackwell Science, 1996.
Foster-Powell K., Holt S.H.A., Brand-Miller J.C. International table of glycemic index and glycemic load values: 2002. Am J Clin Nutr. 2002;76:5.
Fraser D.D., Witing S., Andrew R.D., et al. Elevated polyunsaturated fatty acids in blood obtained from children on the ketogenic diet. Neurol. 2003;60:1026.
Freeman J.M. The Ketogenic Diet: additional information from a crossover study. J. Child Neurol. 2009;24:509.
Freeman J.M., Freeman J.B., Kelly M.T. The Ketogenic Diet, ed 3. New York: Demos; 2000.
Freeman J.M., Kelly M.T., Freeman J.B. The Epilepsy diet treatment: an introduction to the ketogenic diet. New York: Demos; 1994.
Freeman J.M., Vining E.P.G. Seizures decrease rapidly after fasting: preliminary studies of the ketogenic diet. Arch Pediatr Adolesc Med. 1999;153:946.
Freeman J.M., Vining E.P.G., Kossoff E.H., et al. A blinded crossover study of the efficacy of the ketogenic diet. Epilepsia. 2009;50:322.
Freeman J.M., Vining E.P.G., Pillas D.J. Seizures and epilepsy in childhood: a guide for parents. Baltimore: Johns Hopkins University Press; 1990.
Freeman J.M., Vining E.P.G., Pillas D.J., et al. The efficacy of the ketogenic diet – 1998: A prospective evaluation of intervention in 150 children. Pediatrics. 1998;102:1358.
Freeman J.M., Vining E.P.G., Pyzik P.L., et al. Beta-hydroxybutyrate levels in blood correlate with seizure control in children on the ketogenic diet. Epilepsia. 1998;39(Suppl 6):167.
Freitas A., daPaz J.A., Casella E.B., et al. Ketogenic diet for the treatment of refractory epilepsy. Arq Neuropsiquiatr. 2007;65:381.
Furth S.L., Casey J.C., Pyzik P.L., et al. Risk factors for urolithiasis in children on the ketogenic diet. Pediatr Nephrol. 2000;15:125.
Gasior M., Rogawski M.A., Hartman A.L. Neuroprotective and disease modifying effects of the ketogenic diet. Behav Pharmaco. 2006;17:431.
George R., Salinsky M., Kuzniecky R., et al. Vagus nerve stimulation for treatment of partial seizures. 3. Long-term follow-up on the first 67 patients exiting a controlled study. Epilepsia. 1994;35:637.
Geyelin H.R. Fasting as a method for treating epilepsy. Med Rec. 1921;99:1037.
Gilbert D.L., Pyzik P.L., Freeman J.M. The ketogenic diet: Seizure control correlates better with serum beta-hydroxybutyrate than with urine ketones. J Child Neurol. 2000;15:787.
Gillespie R.S., Stapleton B.F. Nephrolithiasis in children. Pediatr Rev. 2004;25:131.
Gjedde A., Crone C. Induction processes in blood-brain transfer of ketone bodies during starvation. Am J Physiol. 1975;229:1165.
Goodwell W., Chez M.G., Buchanan C.P., et al. Bone marrow suppression from copper deficiency in 2 patients on the ketogenic diet. Ann Neurol. 1998;44:568.
Goyens P., DeLaet C., Ranguelov N., et al. Pitfalls of the ketogenic diet in a neonate. Pediatrics. 2002;109:1185.
Greene A.E., Todorova M.T., McGowan R., et al. Caloric restriction inhibits seizure susceptibility in epileptic EL mice by reducing blood glucose. Epilepsia. 2001;42:1371.
Greene A.E., Todorova M.T., Seyfried T.N. Perspectives on the metabolic management of epilepsy through dietary reduction of glucose and elevation of ketone bodies. J Neurochem. 2003;86:529.
Groesbeck D.K., Bluml R.M., Kossoff E.H. Long-term use of the ketogenic diet in the treatment of epilepsy. Devel Med Child Neurol. 2006;48:978.
Haas R.E., Rice M.A., Trauner D.A., et al. Therapeutic effects of a ketogenic diet in Rett syndrome. Am J Med Genet. 1986;1:225.
Hahn T.J., Halstead L.R., DeVivo D.C. Disordered mineral metabolism produced by ketogenic diet therapy. Calcif Tissue Int. 1979;28:17.
Hallbook T., Kohler S., Rosen I., et al. Effects of ketogenic diet on epileptiform activity in children with therapy resistant epilepsy. Epilepsy Res. 2007;77:134.
Hallbook T., Lundren J., Rosen I. Ketogenic diet improves sleep quality in children with therapy-resistant epilepsy. Epilepsia. 2007;48:59.
Harik S.I., Al-Mudallal A.S., LaManna J.C., et al. Ketogenic diet and the brain. Ann N Y Acad Sci. 1997;835:218.
Hartman A.L., Gasior M., Vining E.P.G., et al. The neuropharmacology of the ketogenic diet. Pediatr Neurol. 2007;36:281.
Hartman A.L., Lyle M., Rogawski M.A., et al. Efficacy of the ketogenic diet in the 6-Hz seizure test. Epilepsia. 2008;49:334.
Hassan A.M., Keene D.L., Whiting S.E., et al. Ketogenic diet in the treatment of refractory epilepsy in childhood. Pediatr Neurol. 1999;21(2):548.
Hauser W.A. The prevalence and incidence of convulsive disorders in children. Epilepsia. 1994;35:S1.
Hauser W.A., Hesdorffer D.H. Epilepsy: frequency, causes and consequences. New York: Demos Press; 1980.
Hawkins R.A., Biebuyck J.F. Ketone bodies are selectively used by individual brain regions. Science. 1979;205:325.
Hawkins R.A., Biebuyck J.F. Regional brain utilization of ketone bodies. In: Passoneau J.V., Lust W.D., Hawkins J.R., et al, editors. Cerebral metabolism and neural function. Baltimore: Williams & Wilkins; 1980:255.
Hawkins R.A., Williamson D.H., Krebs H.A. Ketone-body utilization by adult and suckling rat brain in vivo. Biochem J. 1971;122:13.
Haymond M.W., Howard C., Ben-Galim E., et al. Effects of ketosis on glucose flux in children and adults. Am J Physiol. 1983;245:E373.
Heller A.J., Stewart J., Hughes E., et al. Comparative efficacy and toxicity of phenobarbital, phenytoin, carbamazepine, and valproate in adults and children with newly diagnosed previously untreated epilepsy: a randomized long-term trial. Epilepsia. 1993;34:66.
Helmholz H.F. The treatment of epilepsy in childhood: five years experience with the ketogenic diet. JAMA. 1927;1927:88.
Helmholz H.F., Goldstein M. Results of 15 years’ experience with the ketogenic diet in the treatment of epilepsy in children. Proc Staff Meet Mayo Clinic. 1937;12:433.
Helmholz H.F., Keith H.M. Eight years’ experience with the ketogenic diet in the treatment of epilepsy. JAMA. 1930;95:707.
Helmholz H.F., Keith H.M. Ten years’ experience in the treatment of epilepsy with the ketogenic diet. Arch Neurol Psychiatr. 1932;29:808.
Hemingway C., Freeman J.M., Pillas D.J., et al. The ketogenic diet: a 3- to 6-year follow-up of 150 children enrolled prospectively. Pediatrics. 2001;108:898.
Henderson C.B., Filloux F.M., Alder S.C., et al. Efficacy of the ketogenic diet as a treatment option for epilepsy: meta-analysis. J Child Neurol. 2006;21:193.
Hendricks M.. High fat and seizure free. Johns Hopkins Mag. 1995(April):14.
Herzberg G.Z., Fivush B.A., Kinsman S.L., et al. Urolithiasis associated with the ketogenic diet. J Pediatr. 1990;117:743.
Hong A.M., Turner Z., Hamdy R.F., et al. Infantile spasms treated with the ketogenic diet: prospective single center experience in 104 consecutive infants. Epilepsia. 2010. (in press)
Hopkins I.J., Lynch B.C. Use of ketogenic diet in epilepsy in childhood. Aust Pediatr J. 1970;6:25.
Hori A., Tandon P., Holmes G.L., et al. Ketogenic diet: effects on expression of kindled seizures and behavior in adult rats. Epilepsia. 1997;38:750.
Hoyt C.S., Billson F.A. Optic neuropathy in ketogenic diet. Br J Ophthalmol. 1979;63:191.
Huttenlocher P.R. Ketonemia and seizures: metabolic and anticonvulsant effects of two ketogenic diets in childhood epilepsy. Pediatr Res. 1976;10:536.
Ito S., Oguni H., Ito Y., et al. Modified Atkins diet therapy for a case with glucose transporter type 1 deficiency syndrome. Brain Dev. 2008;30:226.
Janaki S., Rashid M.D., Gulati M.S., et al. A clinical electroencephalographic correlation of seizures on a ketogenic diet. Indian J Med Res. 1976;64:1057.
Joshi C.N., Greenberg C.R., Mhanni A.A., et al. Ketogenic Diet in Alpers-Huttenlocher syndrome. Pediatr Neurol. 2009;40:314.
Jung D.E., Chung J.Y., Kang H.C., et al. Improving tolerability of the ketogenic diet in patients with an abnormal endoscopic findings. Brain Dev. 2008;30:416.
Jung D.E., Kang H.C., Kim H.D. Long term outcome of the ketogenic diet for intractable childhood epilepsy with focal malformation of cortical development. Pediatrics. 2008;122:e330.
Kang H.C., Chung D.E., Kim D.W., et al. Early and late-onset complications of the ketogenic diet for intractable epilepsy. Epilepsia. 2004;45:1116.
Kang H.C., Kim Y.J., Kim D.W., et al. Efficacy and safety of the ketogenic diet for intractable childhood epilepsy; Korean multicentric experience. Epilepsia. 2005;46(2):272.
Kang H.C., Lee Y.M., Kim H.D., et al. Safe and effective use of the ketogenic diet in children with epilepsy and mitochondrial respiratory chain complex defects. Epilepsia. 2007;48:82.
Kang H.C., Lee H.S., You S.J., et al. Use of a modified Atkins diet in intractable childhood epilepsy. Epilepsia. 2007;48:182.
Kaytal N.G., Koehler A.N., McGhee B., et al. The ketogenic diet in refractory epilepsy: the experience of Children’s Hospital of Pittsburgh. Clin Pediatr. 2000;39:153.
Keith H.M. Results of treatment of recurring convulsive attacks of epilepsy. Am J Dis Child. 1942;74:140.
Keith H.M. Convulsive disorders in children: with reference to treatment with ketogenic diet. Boston: Little, Brown; 1963.
Kielb S., Koo H.P., Bloom D.A., et al. Nephrolithiasis associated with the ketogenic diet. J Urol. 2000;164:464.
Kim D.W., Kang H.C., Park J.C., et al. Benefits of the nonfasting ketogenic diet compared with the initial fasting ketogenic diet. Pediatrics. 2004;114:1627.
Kim D.Y., Rho J.M. The ketogenic diet and epilepsy. Curr Opin Clin Nutr Metab Care. 2008;11:113.
Kinsman S.L., Vining E.P.G., Quaskey S.A., et al. Efficacy of the ketogenic diet for intractable seizure disorders: review of 58 cases. Epilepsia. 1992;33:1132.
Kleinman R.E., editor. The Ketogenic Diet. Pediatric Nutrition Handbook. ed 5. Chicago, IL: American Academy of Pediatrics; 2004:773.
Klepper J., Diefenbach S., Kohlschutter A., et al. Effects of the ketogenic diet in the glucose transporter 1 deficiency syndrome. Prostaglandins Leukot Essent Fatty Acids. 2004;70:321.
Klepper J., Leiendecker B., Bredahl R., et al. Introduction of a ketogenic diet in young infants. J Inherit Metab Dis. 2002;25:449.
Kossoff E.H., Dorward J.L. The modified Atkins diet. Epilepsia. 2008;49:37.
Kossoff E.H., Dorward J.L., Molinero M.R., et al. The modified Atkins diet: a potential treatment for developing countries. Epilepsia. 2008;49:1646.
Kossoff E.H., Laux L.C., Blackford R., et al. When do seizures usually improve with the ketogenic diet? Epilepsia. 2008;49:329.
Kossoff E.H., Rowley H., Sinha S.R., et al. A prospective study of the modified Atkins diet for intractable epilepsy in adults. Epilepsia. 2008;49:316.
Kossoff E.H., Hedderick E.F., Turner Z., et al. A case control evaluation of the ketogenic diet versus ACTH for new-onset infantile spasms. Epilepsia. 2008;49:1504.
Kossoff E.H., Krauss G.L., McGrogan J.R., et al. Efficacy of the Atkins diet as therapy for intractable epilepsy. Neurol. 2003;61:1789.
Kossoff E.H., McGrogan. Worldwide use of the ketogenic diet. Epilepsia. 2005;46:280.
Kossoff E.H., Thiele E.A., Pfeifer H.H., et al. Tuberous Sclerosis complex and the ketogenic diet. Epilepsia. 2005;46:1684.
Kossoff E.H., McGrogan, Bluml R.M., et al. A modified Atkins diet is effective for the treatment of intractable pediatric epilepsy. Epilepsia. 2006;47:421.
Kossoff E.H., Pyzik P.L., McGrogan J.R., et al. Efficacy of the ketogenic diet for infantile spasms. Pediatrics. 2002;109:780.
Kossoff E.H., Pyzik P.L., McGrogan J.R., et al. The impact of early versus late anticonvulsant reduction after ketogenic diet initiation. Epilepsy Behav. 2004;5:499.
Kossoff E.H., Rho J.M. Ketogenic diets: evidence for short and long term efficacy. Neurotherapeutics. 2009;6:406.
Kossoff E.H., Zupec-Kania B.A., Amark P.E., et al. Optimal clinical management of children receiving the ketogenic diet: recommendations of the international ketogenic diet study group. Epilepsia. 2009;50:304.
Kossoff E.H., Zupec-Kania B.A., Rho J.M. Ketogenic Diets: An update for child neurologists. J Child Neurol. 2009;24:979.
Kossoff E.H., Turner Z., Bluml R.M., et al. A randomized, crossover comparison of daily carbohydrate limits using the modified Atkins diet. Epilepsy Behav. 2007;10:432.
Kraus H., Schlenker S., Schwedesky D. Developmental changes of cerebral ketone body utilization in human infants. Hoppe Seylers Z Physiol Chem. 1974;355:164.
Kumuda T., Miyojima T., Kimura A.N., et al. Modified Atkins diet for the treatment of nonconvulsive status epilepticus in children. J Child Neurol. 2010;25:485.
Lamers K.J.B., Gabreels F.J.M., Renier W.O., et al. Fasting studies in cerebrospinal fluid and blood in children with epilepsy of unknown origin. Epilepsy Res. 1995;21:59.
Lasser J.L., Brush M.K. An improved ketogenic diet for treatment of epilepsy. J Am Diet Assoc. 1973;62:281.
Lebel D., Morin C., Laberge M., et al. The carbohydrate and caloric content of concomitant medications for children with epilepsy on the ketogenic diet. Can J Neurol Sci. 2001;28:232.
Lee Y.M., Kang H.C., Lee J.S., et al. Mitochondrial respiratory chain defects: underlying etiology in various epileptic conditions. Epilepsia. 2008;49:685.
Lefevre F., Aronson N. Ketogenic diet for the treatment of refractory epilepsy in children: a systematic review of efficacy. Pediatrics. 105(4), 2000. Available at http://www.pediatrics.org/cgi/content/full/105/4/e46
Leino R.L., Gerhart D.Z., Duelli R., et al. Diet-induced ketosis increases monocarboxylate transporter (MCT1) levels in rat brain. Neurochem Int. 2001;38:519.
Lennox W.G. Ketogenic diet in the treatment of epilepsy. N Engl J Med. 1928;199:74.
Lennox W.G. Science and seizures: new light on epilepsy and migraine. New York: Harper; 1941.
Lennox W.G., Cobb S. Studies in epilepsy. VIII, the clinical effect of fasting. Arch Neurol Psychiatr. 1928;20:771.
Lennox W.G., Lennox M.A.. Epilepsy and related disorders. vol 2,. Boston: Little, Brown; 1960.
Levy R., Cooper P. Ketogenic diet for epilepsy. Cochrane Database Syst Rev. 3, 2003. CD001903
Liebhaber G.M., Riemann E., Baumeister F.A. Ketogenic diet in Rett syndrome. J Child Neurol. 2003;18:74.
Lightstone L., Shinnar S., Callahan C.M., et al. Reasons for failure of the ketogenic diet. J Neurosci Nurs. 2001;36:292.
Likhodii S.S., Burnham W.M. Ketogenic diet: does acetone stop seizures? Med Sci Monit. 2002;8:HY19.
Likhodii S.S., Musa K., Mendonca A., et al. Dietary fat, ketosis, and seizure resistance in rats on the ketogenic diet. Epilepsia. 2000;41:1400.
Likhodii S.S., Serbanescu I., Cortez M., et al. Anticonvulsant properties of acetone, a brain ketone elevated by the ketogenic diet. Ann Neurol. 2003;54:219.
Lin M., Mitchell W.G., Chen L.S., et al. Symptomatic carnitine depletion is uncommon in children on ketogenic diet. Epilepsia. 1998;39(Suppl 6):168.
Liu Y.M., William S., Basualdo-Hammond C., et al. A prospective study: growth and nutritional status of children treated with the ketogenic diet. J Am Diet Assoc. 2003;103:707.
Livingston S. The diagnosis and treatment of convulsive disorders in children. Springfield, Ill: Charles C Thomas; 1954.
Livingston S. Living with epileptic seizures. Springfield, Ill: Charles C Thomas; 1963.
Livingston S. Comprehensive management of epilepsy in infancy, childhood and adolescence. Springfield, Ill: Charles C Thomas; 1972.
Livingston S., Pauli L.L. Ketogenic diet and epilepsy. Dev Med Child Neurol. 1975;17:818.
Livingston S., Pauli L.L., Pruce I. Ketogenic diet in the treatment of childhood epilepsy. Dev Med Child Neurol. 1977;19:833.
Lund T.M., Risa O., Sonnewald U., et al. Availability of neurostransmitter glutamate is diminished when B-hydroxybutyrate replaces glucose in cultured neurons. J Neurochem. 2009;110:80.
Lyczkowski D.A., Pfeifer H.H., Ghosh S., et al. Safety and tolerability of the ketogenic diet in pediatric epilepsy: effects of valproate combination therapy. Epilepsia. 2005;46:1533.
Maalouf M., Rho J.M., Mattson M.P. The neuroproductive properties of calorie restriction, the ketogenic diet and ketone bodies. Brain Res Rev. 2009;59:293.
Maalouf M., Sullivan P.G., Davis L., et al. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neurosci. 2007;145:256.
Mady M.A., Kossoff E.H., McGregor A.L., et al. The ketogenic diet: adolescents can do it, too. Epilepsia. 2003;44(6):847-851.
Mandel A., Ballew M., Pina-Garza J.E., et al. Medical costs are reduced when children with intractable epilepsy are successfully treated with the ketogenic diet. J Am Diet Assoc. 2002;102:396.
Mantis J.G., Centeno N.A., Todorova M., et al. Management of multifactorial idiopathic epilepsy in EL mice with caloric restriction and the ketogenic diet: role of glucose and ketone bodies. Nutr Metab. 2004;1:11.
Maria B.L., Borum P.R., Friedman T., et al. The ketogenic diet. In: Maria B.L., editor. Consensus in child neurology. Hamilton, Ontario: Decker Periodicals, 1997.
Marks W.J., Vermathen P., Sum J.M., et al. Brain pH in humans treated with the ketogenic diet. Neurology. 1997;48:A111.
Martillotti J., Weinshenker D., Liles L.C., et al. A ketogenic diet and knockout of the norepinephrine transporter both reduce seizure severity in mice. Epilepsy Res. 2006;68:207.
Martinez C.C., Pyzik P.L., Kossoff E.H. Discontinuing the ketogenic diet in seizure free children: recurrence and risk factors. Epilepsia. 2007;48:187.
Masino S.A., Geiger J.D. Are purines mediators of the anticonvulsant/neuroprotective effects of ketogenic diets? Trends Neurosci. 2008;31:273.
Mantis J.G., Fritz C.L., Marsh J., et al. Improvement in motor and exploratory behavior in Rett Syndrome mice with restricted ketogenic and standard diets. Epilepsy Behav. 2009;15:133.
Maydell B.V., Wyllie E., Akhtar N., et al. Efficacy of the ketogenic diet in focal versus generalized seizures. Pediatr Neurol. 2001;25:208.
Mayes P.A. Integration of metabolism and the provision of tissue fuels. In Murray R.K., Granner D.K., Mayes P.A., et al, editors: Harper’s Biochemistry, ed 24, Stamford, Conn: Appleton & Lange, 1996.
Mayes P.A. Oxidation of fatty acids: ketogenesis. In Murray R.K., Granner D.K., Mayes P.A., et al, editors: Harper’s Biochemistry, ed 24, Stamford, Conn: Appleton & Lange, 1996.
McGeer P.L., McGeer E.G. Amino acid neurotransmitters. In: Siegel G.J., Agranoff B.W., Albers R.W., et al, editors. Basic neurochemistry. New York: Raven Press, 1989.
McGhee B., Katyal N. Avoid unnecessary drug-related carbohydrates for patients consuming the ketogenic diet. J Am Diet Assoc. 2001;101:87.
McQuarrie I., Keith H.M. Epilepsy in children. Am J Dis Child. 1927;34:1013.
Melvin J.J., Legido A., Faerber E.N., et al. Pyruvate dehydrogenase complex deficiency associated with brain dysgenesis: response of lactic acidemia to a ketogenic diet. Neurology. 1996;46:A114.
Millichap J.G., Jones J.D., Rudis B.P. Mechanism of anticonvulsant action of ketogenic diet. Am J Dis Child. 1964;107:593.
Moore T., Lione A., Sugden M., et al. Hydroxybutyrate transport in rat brain: developmental and dietary modification. Am J Physiol. 1976;230:619.
Mosek A., Natour H., Neufeld M.Y., et al. Ketogenic diet treatment in adults with refractory epilepsy: A prospective pilot study. Seizure. 2009;18:30.
Muller-Schwarze A.B., Tandon P., Liu Z., et al. Ketogenic diet reduces spontaneous seizures and mossy fiber sprouting in the kainic acid model. Neuro Report. 1999;10:1517.
Murphy P., Likhodiis S., Nylen K., et al. The antidepressant properties of the ketogenic diet. Biol Psychiatry. 2004;56:981.
Muzykewicz D.A., Lyczkowski D.A., Memon N., et al. Efficacy safety and tolerability of the low glycemic index treatment in pediatric epilepsy. Epilepsia. 2009;50:1118.
The National Association of Epilepsy Centers. The National Association of Epilepsy Centers Guidelines for diagnosis and treatment in specialized epilepsy centers. Epilepsia. 1990;31:1.
Nathan J.K., Purandare A.S., Parekh Z.B., et al. Ketogenic diet in Indian children with uncontrolled epilepsy. Indian Pediatr. 2009;46:669.
Nakazawa M., Kodama S., Matsuo T. Effects of ketogenic diet on electroconvulsive threshold and brain contents of adenosine nucleotides. Brain Dev. 1983;5:375.
NBC Dateline. The ketogenic diet. October 26. 1994.
Neal E.G., Chaffe H.M., Schwartz R.H., et al. The ketogenic diet for the treatment of childhood epilepsy: a randomized controlled trial. Lancet Neurol. 2008;7:500.
Neal E.G., Chaffe H.M., Schwartz R.H., et al. A randomized trial of classical and medium chain triglyceride ketogenic diets in the treatment of childhood epilepsy. Epilepsia. 2009;50:1109.
Nebeling L.C., Miraldi F., Shurin S.B., et al. Effects of a ketogenic diet on tumor metabolism and nutritional status in pediatric oncology patients: two case reports. J Am Coll Nutr. 1995;14:202.
Nehlig A. Age-dependent pathways of brain energy metabolism: the suckling rat, a natural model of the ketogenic diet. Epilepsy Res. 1999;37:211.
Nellhaus G. The ketogenic diet reconsidered: correlation with EEG. Neurology. 1971;21:424.
Noh H.S., Kang S.S., Kim D.W., et al. Ketogenic diet increases caldindin-D28k in the hippocampi of male ICR mice with kainic acid seizures. Epilepsy Res. 2005;65:153.
Nordli D.R. The ketogenic diet: uses and abuses. Neurol. 2000;58(12, Suppl 7):S21.
Nordli D.R.Jr, DeVivo D.C. The ketogenic diet revisited: back to the future. Epilepsia. 1997;38:743.
Nordli D.R., Kuroda M.M., Carroll J., et al. Experience with the ketogenic diet in infants. Pediatrics. 2001;108:129.
Notkin J. Epilepsy: treatment of institutionalized adult patients with a ketogenic diet. Arch Neurol Psychiatr. 1934;31:787.
Nylen K., Velazquez J.L., Sayed V., et al. The effects of a ketogenic diet on ATP concentrations and the number of hippocampal mitochondria in Aldh5a1 (-/-) mice. Biochim Biophys Acta. 2009;1790:208.
Owen O.E., Morgan A.P., Kemp H.G., et al. Brain metabolism during fasting. J Clin Invest. 1967;46:1589.
Pan J., Bebin E., Chu W., et al. Ketosis and epilepsy: 31P spectroscopic imaging at 4.1T. Epilepsia. 1999;40:703.
Pan J.W., Rothman D.L., Behar K.L., et al. Human brain beta-hydroxybutyrate and lactate increase in fasting-induced ketosis. J Cereb Blood Flow Metab. 2000;20:1502.
Pan J.W., Telang F.W., Lee J.H., et al. Measurement of beta-hydroxybutyrate in acute hyperketonemia in human brain. J Neurochem. 2001;79:539.
Patsalos P.N., Duncan J.S. Antiepileptic drugs: a review of clinically significant drug interactions. Drug Safety. 1993;9:156.
Pellerin L., Pellegri G., Martin J.-L., et al. Expression of monocarboxylate transporter mRNAs in mouse brain: support for a distinct role of lactate as an energy substrate for the neonatal vs. Adult brain. Proc Natl Acad Sci USA. 1998;95:3990.
Pellock J.M. Seizures and epilepsy in infancy and childhood. Neurol Clin. 1993;3:755.
Pellock J.M. Antiepileptic drug therapy in the United States: a review of clinical studies and unmet needs. Neurology. 1995;45:S17.
Pellock J.M. Utilization of new antiepileptic drugs in children. Epilepsia. 1996;37:S66.
Pellock J.M., Pippenger C.E. Adverse effects of antiepileptic drugs. In: Dodson W.E., Pellock J.M., editors. Pediatric epilepsy: diagnosis and therapy. New York: Demos, 1993.
Penfield W., Erickson T.C. Epilepsy and cerebral localization. A study of the mechanism, treatment and prevention of epileptic seizures. Baltimore: Charles C Thomas; 1941.
Peng L., Hertz L., Huang R., et al. Utilization of glutamine and of TCA cycle constituents as precursors for transmitter glutamate and GABA. Dev Neurosci. 1993;15:367.
Persson B., Settergress G., Dahlquist G. Cerebral arterio-venous difference of acetoacetate and D-beta-hydroxybutyrate in children. Acta Paediatr Scand. 1972;61:273.
Peterman M.G. The ketogenic diet in epilepsy. JAMA. 1925;84:1979.
Peyron R., LeBars D., Cinotti L., et al. Effects of GABAA receptor activation on brain glucose metabolism in normal subjects and temporal lobe epilepsy (TLE) patients. A positron emission tomography (PET) study. Part I: brain glucose metabolism is increased after GABAA receptor activation. Epilepsy Res. 1994;19:45.
Pfeifer H.H., Lyczkowski D.A., Thiele E.A. Low glycemic index treatment implementation and new insights into efficacy. Epilepsia. 2008;49:42.
Pfeifer H.H., Thiele E.A. Low glycemic index treatment: a liberalized ketogenic diet for treatment of intractable epilepsy. Neurol. 2005;65:1810.
Porta N., Vallee L., Boutry E., et al. Comparison of seizure reduction and serum fatty acid levels after receiving the ketogenic and modified Atkins diet. Seizure. 2009;18:359.
Prasad A.N., Stafstrom C.F., Holmes G.L. Alternative epilepsy therapies: the ketogenic diet, immunoglobulins, and steroids. Epilepsia. 1996;37:S81.
Pulford D.S. The present status of the ketogenic diet. Ann Int Med. 1932;6:795.
Pulsifer M.B., Gordon J.M., Brandt J., et al. Effects of ketogenic diet on development and behavior: preliminary report of a prospective study. Devel Med Child Neurol. 2001;43:301.
Raffo E., Francois J., Ferrandon A., et al. Calorie-restricted ketogenic diet increases thresholds to all patterns of pentylenetetrazol-induced seizures: critical importance of electroclinical assessment. Epilepsia. 2008;49:320.
Remahl S., Dahlin M.G., Amark P.E. Influence of the ketogenic diet on 24 hour electroencephalogram in children with epilepsy. Pediatr Neurol. 2008;38:38.
Rho J., Kim D., Robbins C., et al. Age-dependent differences in flurothyl seizure sensitivity in mice treated with a ketogenic diet. Epilepsy Res. 1999;37:233.
Rho J.M., Shin D.H., Robbins C.A., et al. GABA levels are increased in the hippocampus of mature mice fed a ketogenic diet. Epilepsia. 1999;40(Suppl 7):161.
Rho J.M., Szot P., Tempel B.L., et al. Developmental seizure susceptibility of Kv1.1 potassium channel knockout mice. Dev Neurosci. 1999;21:320.
Rho J.M., Anderson G.D., Donevan S.D., et al. Acetoacetate, acetone, and dibenzylamine (a contaminant in L-(+)-beta-hydroxybutyrate) exhibit direct anticonvulsant actions in vivo. Epilepsia. 2002;43:358.
Rho J.M., Robbins C.A., Wenzel J., et al. An experimental ketogenic diet promotes long-term survival and reduces synaptic reorganization in the hippocampus of epileptic Kv1.1 null mutant mice. Epilepsia. 2000;41(Suppl 7):34.
Rizzutti S., Ramos A.M.F., Muszkat M., et al. Is hospitalization really necessary during the introduction of the ketogenic diet? J Child Neurol. 2007;22:33.
Rodwell V.W. Conversion of amino acids to specialized product. In Murray R.K., Granner D.K., Mayes P.A., et al, editors: Harper’s Biochemistry, ed 24, Stamford, Conn: Appleton & Lange, 1996.
Roe C.R., Coates P.M. Mitochondrial fatty acid oxidation disorders. In: Scriver C.R., Beaudet a.l., Sly W.S., et al, editors. The metabolic and molecular basis of inherited disease. New York: McGraw-Hill; 1995:1501.
Ross D.L., Swaiman K.F., Torres F., et al. Early biochemical and EEG correlates of the ketogenic diet in children with atypical absence epilepsy. Pediatr Neurol. 1985;1:104.
Samala R., Willis S., Borges K. Anticonvulsant profile of a balanced ketogenic diet in acute mouse seizure models. Epilepsy Res. 2008;81:119.
Sampath A., Kossoff E.H., Furth S.L., et al. Kidney stones and the ketogenic diet: risk factors and prevention. J Child Neurol. 2007;22:375.
Santra S., Gilkerson R.W., Davidson M., et al. Ketogenic treatment reduces deleted mitochondrial DNAs in cultured human cells. Ann Neurol. 2004;56:662.
Schneider K.S., Wagner J.. Recipe for hope. People Magazine. 1995(April 17):54.
Schwartz R.M., Boyes S., Aynsley-Green A. Metabolic effects of three ketogenic diets in the treatment of severe epilepsy. Dev Med Child Neurol. 1989;31:152.
Schwartz R.H., Eaton J., Bower B.D., et al. Ketogenic diets in the treatment of epilepsy: Short term clinical effects. Dev Med Child Neurol. 1989;31:145.
Schwartzkroin P.A. Mechanisms underlying the anti-epileptic efficacy of the ketogenic diet. Epilepsy Res. 1999;37:171.
Seo J.H., Lee Y.M., Lee J.S., et al. A case of Ohtahara syndrome with mitochondrial respiratory chain complex I deficiency. Brain Dev. 2010;32:253.
Seymour K.J., Blum S., Sutherling J., et al. Identification of cerebral acetone by 1H-MRS in patients with epilepsy controlled by ketogenic diet. MAGMA. 1999;8:33.
Shafrir Y., Prensky A.L. Acquired epileptiform opercular syndrome: a second case report, review of the literature, and comparison to the Landau-Kleffner syndrome. Epilepsia. 1995;36:1050.
Sirven J., Whedon B., Caplan D., et al. The ketogenic diet for intractable epilepsy in adults: preliminary results. Epilepsia. 1999;40:1721.
Snead O.C. The ketogenic diet: a cautionary note. Pediatr Res. 2004;55:368.
Sokoloff L. Metabolism of ketone bodies by the brain. Ann Rev Med. 1973;24:271.
Spulber G., Spulber S., Hagenas L., et al. Growth dependance on insulin like growth factor I during the ketogenic diet. Epilepsia. 2009;50:297.
Stafstrom C.E. Animal models of the ketogenic diet: what have we learned, what can we learn? Epilepsy Res. 1999;37:241.
Stafstrom C.E., Wang C., Jensen F.E. Electrophysiological observations in hippocampal slices from rats treated with the ketogenic diet. Dev Neurosci. 1999;21:393.
Stafstrom C.E., Bough K.J. The ketogenic diet for the treatment of epilepsy: a challenge for nutritional neuroscientists. Nutr Neurosci. 2003;6:67.
Stafstrom C.E., Rho J.M., editors. Epilepsy and the Ketogenic Diet. Totawa, NJ: Humana Press, Inc, 2004.
Stafstrom C.E., Spencer S. The ketogenic diet: a therapy in search of an explanation. Neurol. 2000;54:282.
Stewart W.A., Gordon K., Camfield P. Acute pancreatitis causing death in a child on the ketogenic diet. J Child Neurol. 2001;16:682.
Su S.W., Cilio M.R., Sogawa Y., et al. Timing of the ketogenic diet initiation in an experimental epilepsy model. Dev Brain Res. 2000;125:131.
Sullivan P.G., Rippy N.A., Dorenbos K., et al. The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Ann Neurol. 2004;55:576.
Suzuki Y., Takahashi H., Fukuda M., et al. β-hydroxybutyrate alters GABA- transaminase activity in cultured astrocytes. Brain Res. 2009;1268:17.
Swink T., Vining E., Casey, et al. Efficacy of the ketogenic diet in children under 2 years of age. Epilepsia. 1997;38:26.
Szot P., Weinshenker D., Rho J.M., et al. Norepinephrine is required for the anticonvulsant effect of the ketogenic diet. Dev Brain Res. 2001;129:211.
Takeoka M., Riviello J.J., Pfeifer H., et al. Concomitant treatment with Topiramate and ketogenic diet in pediatric epilepsy. Epilepsia. 2002;43:1072.
Talbot F.B., Metcalf K., Moriarty M. The ketogenic diet in the treatment of idiopathic epilepsy. Am J Dis Child. 1926;32:316.
Thavendiranathan P., Chow C., Cunnane S., et al. The effect of the ‘classic’ ketogenic diet on animal seizure models. Brain Res. 2003;959:206.
Thavendiranathan P., Mendonca A., Dell C., et al. The MCT ketogenic diet: effects on animal seizure models. Exp Neurol. 2000;161:696.
Theda C., Woody R.C., Naidu S., et al. Increased very long chain fatty acids in patients on a ketogenic diet: a cause of diagnostic confusion. J Pediatr. 1993;122:724.
Thompson J.A., Van Orman C.B., Peterson P. Ketogenic diet: long-term efficacy and patient tolerance in the treatment of efficacy and patient tolerance in the treatment of intractable pediatric epilepsy. Epilepsia. 1998;39(Suppl 6):167.
Thurston J.H., Carroll J.E., Dodson W.E., et al. Chronic valproate administration reduces fasting ketonemia in children. Neurology. 1983;33:1348.
Todorova M.T., Tandon P., Madore R.A., et al. The ketogenic diet inhibits epileptogenesis in EL mice: a genetic model for idiopathic epilepsy. Epilepsia. 2000;41:933.
Turnbull D.M., Dick D.J., Wilson L., et al. Valproate causes metabolic disturbance in normal man. J Neurol Neurosurg Psychiatr. 1986;49:405.
Uhlemann E.R., Neims A.H. Anticonvulsant properties of the ketogenic diet in mice. J Pharmacol Exp Ther. 1972;180:231.
The Vagus Nerve Stimulation Study Group. A randomized controlled trial of chronic vagus nerve stimulation for treatment of medically intractable seizures. Neurology. 1995;45:224.
Vaisleib I.I., Buchhalter J.R., Zupanc M.L. Ketogenic diet: outpatient initiation, without fluid, or caloric restriction. Pediatr Neurol. 2004;31:198.
Vining E.P.G., Freeman J.M., Kwiterovich P., et al. The ketogenic diet induces dyslipidemia. Epilepsia. 1999;40(Suppl 7):122.
Vining E.P.G. Clinical efficacy of the ketogenic diet. Epilepsy Res. 1999;37:181.
Vining E.P.G., Freeman J.M., Ballaban-Gil K., et al. Multi-center study of the efficacy of the ketogenic diet. Arch Neurol. 1998;55:1433.
Vininig E.P.G., Pyzik P., McGrogan J., et al. Growth of children on the ketogenic diet. Dev Med Child Neurol. 2002;44:796.
Wang Z.J., Bergqvist C., Hunter J.V., et al. In vivo measurement of brain metabolites using two-dimensional double-quantum MR spectroscopy-exploration of GABA levels in a ketogenic diet. Magn Reson Med. 2003;49:615.
Weber S., Molgaard C., Taudorf K., et al. Modified Atkins diet to children and adolescents with medical intractable epilepsy. Seizure. 2009;18:237.
Weber T.A., Antognetti R., Stacpoole P.W. Caveats when considering ketogenic diets for the treatment of pyruvate dehydrogenase complex deficiency. J Pediatr. 2001;138:390.
Weeks D.F., Renner D.S., Allen F.M., et al. Observations on fasting and diets in the treatment of epilepsy. J Metab Res. 1923;3:317.
Wexler I.D., Hemalatha S.G., McConnell J., et al. Outcome of pyruvate dehydrogenase deficiency treated with ketogenic diet. Neurol. 1997;49:1655.
Wheless J.W. The epilepsy diet treatment: an introduction to the ketogenic diet, book review. J Child Neurol. 1995;10:489.
Wheless J.W. The ketogenic diet: fa(c)t or fiction. J Child Neurol. 1995;10:419.
Wheless J.W. The ketogenic diet: an effective medical therapy with side-effects. J Child Neurol. 2001;16:633.
Wheless J.W. History and origin of the ketogenic diet. In: Stafstrom C.E., Rho J.M., editors. Epilepsy and the Ketogenic Diet. Totawa, NJ: Humana Press, Inc; 2004:31.
Wijburg F.A., Barth P.G., Bindoff L.A., et al. Leigh syndrome associated with a deficiency of the pyruvate dehydrogenase complex: results of treatment with a ketogenic diet. Neuropediatrics. 1992;23:147.
Wilder R.M. The effects of ketonuria on the course of epilepsy. Bull Mayo Clin. 1921;2:307.
Wilder R.M., Pollack H. Ketosis and the ketogenic diet: their application to treatment of epilepsy and infections of the urinary tract. Int Clin. 1935;1:1.
Wilkins L. Epilepsy in childhood: III. Results with the ketogenic diet. J Pediatr. 1937;10:341.
Williams S., Basualdo-Hammond C., Curtis R., et al. Growth retardation in children with epilepsy on the ketogenic diet: a retrospective chart review. J Am Diet Assoc. 2002;102:405.
Williamson D.H. Ketone body metabolism during development. Fed Proc. 1985;44:2342.
Willmott N.S., Bryan R.A.E. Case report: scurvy in child with epilepsy on a ketogenic diet with oral complications. Eur Arch Paediatr Dent. 2008;9:148.
Wing R., Vazquez J., Ryan C. Cognitive effects of ketogenic weight-reducing diets. Int J Obes. 1995;19:811.
Withrow C.D. Antiepileptic drugs, the ketogenic diet: mechanism of anticonvulsant action. In: Glaser G.H., Penry J.K., Woodbury D.M., editors. Antiepileptic drugs: mechanism of action. New York: Raven, 1980.
Woody R.C., Steele R.W., Knapple W., et al. Impaired neutrophil function in children with seizures treated with the ketogenic diet. J Pediatr. 1989;115:427.
Yudkoff M., Daikhin Y., Nissim I., et al. Effects of ketone bodies on astrocyte amino acid metabolism. J Neurochem. 1997;69:682.
Yudkoff M., Daikhin Y., Nissim I., et al. Brain amino acid metabolism and ketosis. J Neurosci Res. 2001;66:272.
Yudkoff M., Daikhin Y., Nissim I., et al. Ketogenic diet, amino acid metabolism, and seizure control. J Neurosci Res. 2001;66:931.
Yudkoff M., Daikhin Y., Melo T.M., et al. The ketogenic diet and brain metabolism of amino acids: relationship to the anticonvulsant effect. Annu Rev Nutr. 2007;27:415.
Zhao Q., Stafstrom C.E., Fu D.D., et al. Detrimental effects of the ketogenic diet on cognitive function in rats. Pediatr Res. 2004;55:498.
Ziegler D.R., Araujo E., Rotta L., et al. A ketogenic diet increases protein phosphorylation in brain slices of rats. J Nutr. 2002;132:483.
Zieglar D.R., Ribeiro L.C., Hagenn M., et al. Ketogenic diet increases glutathione preoxidase activity in rat hippocampus. Neurochem Res. 2003;28:1793.
