[level-membership-for-pediatrics-category]
Chapter 462 The Inherited Pancytopenias
Inherited (“constitutional”) pancytopenia is defined as a decrease in marrow production of the 3 major hematopoietic lineages that occurs on an inherited basis, resulting in anemia, neutropenia, and thrombocytopenia. Any of these conditions (Table 462-1) can be transmitted as a simple mendelian disorder by mutant genes with inherited patterns of autosomal dominant, autosomal recessive, or X-linked types. Modifying genes and acquired factors may also be operative. Inherited pancytopenias account for approximately 30% of cases of pediatric marrow failure. Fanconi anemia is the most common of these disorders.
Fanconi Anemia
Clinical Manifestations
The most common anomaly in FA is hyperpigmentation of the trunk, neck, and intertriginous areas, as well as café-au-lait spots and vitiligo, alone or in combination (Fig. 462-1 and Table 462-2). Half the patients have short stature. Growth failure may be associated with abnormal growth hormone secretion or with hypothyroidism. Absence of radii and thumbs that are hypoplastic, supernumerary, bifid, or absent are common. Anomalies of the feet, congenital hip dislocation, and leg abnormalities are seen. A male patient with FA may have an underdeveloped penis; undescended, atrophic, or absence of the testes; and hypospadias or phimosis. Females can have malformations of the vagina, uterus, and ovary. Many patients have a FA “facies,” including microcephaly, small eyes, epicanthal folds, and abnormal shape, size, or positioning of the ears (see Fig. 462-1). Ectopic, pelvic, or horseshoe kidneys are detected by imaging and may show other organs as duplicated, hypoplastic, dysplastic, or absent kidneys. Cardiovascular and gastrointestinal malformations also occur. Approximately 10% of patients with FA are cognitively delayed.
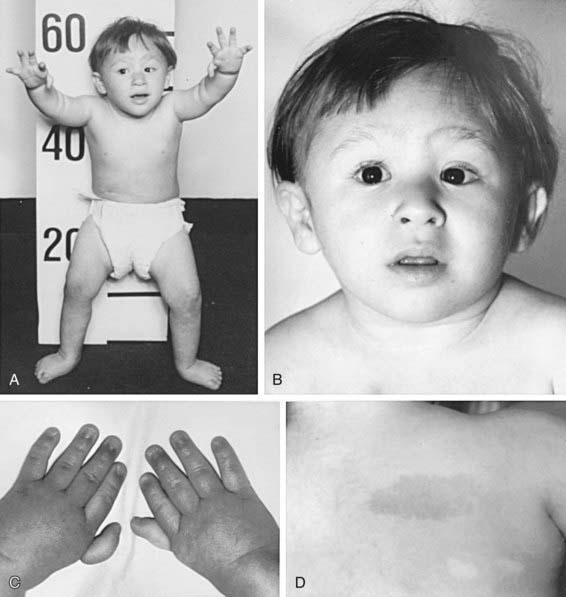
Table 462-2 CHARACTERISTIC PHYSICAL ANOMALIES IN FANCONI ANEMIA
| ANOMALY | APPROXIMATE FREQUENCY (% OF PATIENTS) |
|---|---|
| Skin pigment changes ± café-au-lait spots | 55 |
| Short stature | 51 |
| Upper limb abnormalities (thumbs, hands, radii, ulnas) | 43 |
| Hypogonadal and genital changes (mostly male) | 35 |
| Other skeletal findings (head/face, neck, spine) | 30 |
| Eye/lid/epicanthal fold anomalies | 23 |
| Renal malformations | 21 |
| Gastrointestinal/cardiopulmonary malformations | 11 |
| Hip, leg, foot, toe abnormalities | 10 |
| Ear anomalies (external and internal), deafness | 9 |
Treatment
Hematopoietic stem cell transplantation (HSCT; Chapter 129) is the only curative therapy for the hematologic abnormalities. Patients with FA <10 yr old who undergo transplantation using an HLA–identical sibling donor have a survival rate >80%. Survival rates are lower for patients undergoing the procedure when >10 yr. Preparative regimens are continuously evaluated, refined, and improved worldwide. For patients who do not have a matched sibling donor, a search for a matched unrelated donor (including a search of umbilical cord blood banks) might be initiated. Because of the heightened graft vs host response in patients with FA, the survival and cure rates have not been as good as those for matched sibling donor HSCT (≈ 30% survival). Molecular technology has led to preimplantation genetic diagnosis on parent-derived blastomeres to find an HLA-matched sibling donor without FA.
Shwachman-Diamond Syndrome
Etiology and Epidemiology
Shwachman-Diamond syndrome (SDS) is inherited in an autosomal recessive manner; it occurs in all racial and ethnic groups. The two essential diagnostic criteria are exocrine pancreatic insufficiency and variable hematologic cytopenias due to marrow failure (Chapter 341). Chromosomes are normal, and there is no increased chromosomal breakage after DEB testing of SDS lymphocytes.
Diagnosis
Mutational analysis for SBDS is definitive in 90% of cases. Pearson syndrome (Chapter 443), consisting of refractory sideroblastic anemia, cytoplasmic vacuolization of bone marrow precursors, metabolic acidosis, exocrine pancreatic insufficiency, and a diagnostic mitochondrial DNA mutation is similar to SDS, but the clinical course, morphologic features of the bone marrow, and gene mutation are different. Also, severe anemia requiring transfusion, rather than neutropenia, is present from birth to 1 yr of age. SDS shares some manifestations with Fanconi anemia, such as marrow dysfunction and growth failure, but patients with SDS are readily distinguished because of pancreatic insufficiency with fat malabsorption, fatty changes within the pancreatic body that can be visualized by imaging, characteristic skeletal abnormalities not seen in Fanconi anemia, and a normal chromosomal breakage study with DEB.
Treatment
Fat malabsorption responds to oral pancreatic enzyme replacement and supplemental fat-soluble vitamins, administered according to guidelines similar to those for cystic fibrosis (Chapter 395). A long-term plan should be initiated to monitor changes in peripheral blood counts that require corrective action and to look for early evidence of malignant myeloid transformation. The latter requires serial bone marrow aspirations for smears and cytogenetics and marrow biopsy. One recommendation is to perform marrow testing annually.
Dyskeratosis Congenita
Etiology and Epidemiology
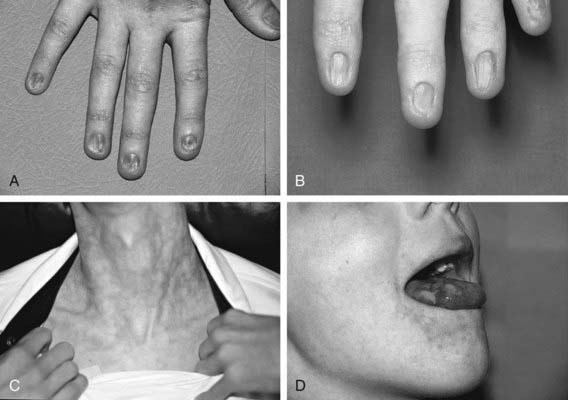
Dyskeratosis congenita (DC) is an inherited multisystem disorder characterized by mucocutaneous abnormalities, bone marrow failure, and a predisposition to cancer and MDS. The diagnostic mucocutaneous (ectodermal) triad is reticulate skin pigmentation of the upper body, mucosal leukoplakia, and nail dystrophy (Fig. 462-2). Skin and nail findings usually become apparent in the 1st 10 yr of life, whereas oral leukoplakia is seen later. These manifestations tend to progress as patients get older. Aplastic anemia occurs in approximately 50% of cases, usually in the 2nd decade of life. About 73% of patients with DC are male, a finding compatible with X-linked recessive inheritance. The remainder has either an autosomal dominant or autosomal recessive mode of inheritance.
Amegakaryocytic Thrombocytopenia
Diagnosis
If CAMT manifests beyond the neonatal period, marrow aspirate and biopsy will demonstrate deficient megakaryocytes and suggest the diagnosis; mutational analysis will confirm it. If CAMT occurs at birth or shortly after, it must be distinguished from other causes of inherited and acquired neonatal thrombocytopenia (Chapter 478). Thrombocytopenia with absent radii (TAR syndrome) is distinguished from CAMT because in TAR the radii are absent. CAMT blood lymphocytes do not show increased chromosomal breakage when exposed to DEB, distinguishing the disease it from FA.
Other Genetic Syndromes
Down Syndrome
Down syndrome (trisomy 21; Chapter 76) has a unique association with aberrant hematologic findings. In addition to the propensity for acute lymphoblastic and myeloblastic leukemias, especially acute megakaryoblastic leukemia, a few patients with Down syndrome have been reported as having pancytopenia due to aplastic anemia.
Reticular Dysgenesis
Reticular dysgenesis (Chapter 120) is an immunologic deficiency syndrome coupled with congenital agranulocytosis. The mode of inheritance is probably autosomal recessive, but an X-linked mode is also possible in some cases. The disorder is a variant of severe combined immune deficiency in which cellular and humoral immunity are absent and severe lymphopenia and neutropenia are also seen. Anemia and thrombocytopenia may also be present. Bone marrow specimens are hypocellular, with markedly reduced myeloid and lymphoid elements. The only curative therapy is HSCT.
Unclassified Inherited Bone Marrow Failure Syndromes
Unclassified inherited bone marrow failure syndromes are heterogeneous disorders that may be either atypical presentations of identifiable diseases or new syndromes. Characterized by various cytopenias, with or without physical manifestations, they do not fit into a classic genetic bone marrow failure disease because all features may not be evident at the time of presentation. Compared with classic disorders (presentation ≈ 1 mo of age), infants with unclassified disorders present later (≈ 9 mo) and manifest single or multilineage cytopenia, aplastic anemia, myelodysplasia, or malignancy with variable expression of malformations. Criteria for the diagnosis are seen in Table 462-3. With follow-up, some may demonstrate typical physical features of known syndromes, such as Shwachman-Diamond syndrome, although without obvious mutations in the SBDS gene.
Table 462-3 CANADIAN INHERITED MARROW FAILURE REGISTRY CRITERIA FOR UNCLASSIFIED INHERITED BONE MARROW FAILURE SYNDROMES
FULFILLS CRITERIA 1 AND 2:
FULFILLS AT LEAST 2 OF THE FOLLOWING:
FULFILLS AT LEAST 1 OF THE FOLLOWING:
* The Canadian Inherited Marrow Failure Registry diagnostic guidelines for selected syndromes were adapted from the literature and are available at www.sickkids.ca/cimfr.
† Cytopenia was defined as follows: neutropenia, neutrophil count of <1.5 × 109/L; thrombocytopenia, platelet count of <150 × 109/L; anemia, hemoglobin concentration of <2 standard deviations below mean, adjusted for age.
‡ Hemoglobinopathies with ineffective erythropoiesis and high hemoglobin F should be excluded by clinical or laboratory testing.
Modified from Teo JT, Klaassen R, Fernandez CV, et al: Clinical and genetic analysis of unclassifiable inherited bone marrow failure syndromes. Pediatrics 22:e139–e148, 2008.
Alter BP. Diagnosis, genetics and management of inherited bone marrow failure syndromes. Hematol Am Soc Hematol Educ Program. 2007;2007:29-39.
Alter BP, Giri N, Savage SA, et al. Cancer in dyskeratosis congenita. Blood. 2009;113:6549-6557.
Bagby GC. Constitutional marrow failure. guest editor. Semin Hematol. 2006;43:145-203.
Burroughs L, Woolfrey A, Shimamura A. Shwachman-Diamond syndrome: a review of the clinical presentation, molecular pathogenesis, diagnosis, and treatment. Hematol Oncol Clin North Am. 2009;23:233-248.
D’Andrea AD. Susceptibility pathways in Fanconi’s anemia and breast cancer. N Engl J Med. 2010;362:1909-1918.
De Winter JP, Joenje H. The genetic and molecular basis of Fanconi anemia. Mutat Res. 2008;668:11-19.
Dokal I, Vulliamy T. Inherited aplastic anemias/bone marrow failure syndromes. Blood Rev. 2008;22:141-153.
Freedman MH. Inherited forms of bone marrow failure. In: Hoffman R, Benz EJ, Shattil SJ, et al, editors. Hematology: basic principles and practice. ed 5. Philadelphia: Churchill Livingstone, Elsevier; 2009:319-357.
Ganapathi KA, Shimamura A. Ribosomal dysfunction and inherited marrow failure. Br J Haematol. 2008;141:376-387.
Geddis AE. Congenital amegakaryocytic thrombocytopenia and thrombocytopenia with absent radii. Hematol Oncol Clin North Am. 2009;23:321-331.
Green AM, Kupfer GM. Fanconi anemia. Hematol Oncol Clin North Am. 2009;23:193-214.
Kerr EN, Ellis L, Dupuis A, et al. The behavioral phenotype of school-age children with Shwachman-Diamond syndrome indicates neurocognitive dysfunction with loss of Shwachman-Bodian-Diamond syndrome gene function. J Pediatr. 2010;156:433-438.
Teo JT, Klaassen R, Fernandez CV, et al. Clinical and genetic analysis of unclassifiable inherited bone marrow failure syndromes. Pediatrics. 2008;122:e139-e148.
Vaz F, Hanenberg H, Schuster B, et al. Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat Genet. 2010;42:406-409.
Walne AJ, Dokal I. Advances in the understanding of dyskeratosis congenita. Br J Haematol. 2009;145:164-172.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 462 The Inherited Pancytopenias
Inherited (“constitutional”) pancytopenia is defined as a decrease in marrow production of the 3 major hematopoietic lineages that occurs on an inherited basis, resulting in anemia, neutropenia, and thrombocytopenia. Any of these conditions (Table 462-1) can be transmitted as a simple mendelian disorder by mutant genes with inherited patterns of autosomal dominant, autosomal recessive, or X-linked types. Modifying genes and acquired factors may also be operative. Inherited pancytopenias account for approximately 30% of cases of pediatric marrow failure. Fanconi anemia is the most common of these disorders.
Fanconi Anemia
Clinical Manifestations
The most common anomaly in FA is hyperpigmentation of the trunk, neck, and intertriginous areas, as well as café-au-lait spots and vitiligo, alone or in combination (Fig. 462-1 and Table 462-2). Half the patients have short stature. Growth failure may be associated with abnormal growth hormone secretion or with hypothyroidism. Absence of radii and thumbs that are hypoplastic, supernumerary, bifid, or absent are common. Anomalies of the feet, congenital hip dislocation, and leg abnormalities are seen. A male patient with FA may have an underdeveloped penis; undescended, atrophic, or absence of the testes; and hypospadias or phimosis. Females can have malformations of the vagina, uterus, and ovary. Many patients have a FA “facies,” including microcephaly, small eyes, epicanthal folds, and abnormal shape, size, or positioning of the ears (see Fig. 462-1). Ectopic, pelvic, or horseshoe kidneys are detected by imaging and may show other organs as duplicated, hypoplastic, dysplastic, or absent kidneys. Cardiovascular and gastrointestinal malformations also occur. Approximately 10% of patients with FA are cognitively delayed.
Table 462-2 CHARACTERISTIC PHYSICAL ANOMALIES IN FANCONI ANEMIA
| ANOMALY | APPROXIMATE FREQUENCY (% OF PATIENTS) |
|---|---|
| Skin pigment changes ± café-au-lait spots | 55 |
| Short stature | 51 |
| Upper limb abnormalities (thumbs, hands, radii, ulnas) | 43 |
| Hypogonadal and genital changes (mostly male) | 35 |
| Other skeletal findings (head/face, neck, spine) | 30 |
| Eye/lid/epicanthal fold anomalies | 23 |
| Renal malformations | 21 |
| Gastrointestinal/cardiopulmonary malformations | 11 |
| Hip, leg, foot, toe abnormalities | 10 |
| Ear anomalies (external and internal), deafness | 9 |
Treatment
Hematopoietic stem cell transplantation (HSCT; Chapter 129) is the only curative therapy for the hematologic abnormalities. Patients with FA <10 yr old who undergo transplantation using an HLA–identical sibling donor have a survival rate >80%. Survival rates are lower for patients undergoing the procedure when >10 yr. Preparative regimens are continuously evaluated, refined, and improved worldwide. For patients who do not have a matched sibling donor, a search for a matched unrelated donor (including a search of umbilical cord blood banks) might be initiated. Because of the heightened graft vs host response in patients with FA, the survival and cure rates have not been as good as those for matched sibling donor HSCT (≈ 30% survival). Molecular technology has led to preimplantation genetic diagnosis on parent-derived blastomeres to find an HLA-matched sibling donor without FA.
