Chapter 652 Diseases of Subcutaneous Tissue
Corticosteroid-Induced Atrophy
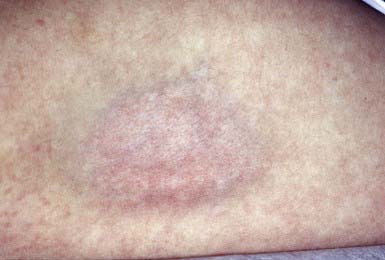
Intradermal injection of a corticosteroid can produce deep atrophy accompanied by surface pigmentary changes and telangiectasia (Fig. 652-1). These changes occur approximately 2 wk after injection and may last for months.
652.1 Panniculitis and Erythema Nodosum
Inflammation of fibrofatty subcutaneous tissue may primarily involve the fat lobule or, alternatively, the fibrous septum that compartmentalizes the fatty lobules. Lobular panniculitis that spares the subcutaneous vasculature includes post-steroid panniculitis, lupus erythematosus profundus, pancreatic panniculitis, α1-antitrypsin deficiency, subcutaneous fat necrosis of the newborn, sclerema neonatorum, cold panniculitis, subcutaneous sarcoidosis, and factitial panniculitis. Lobar panniculitis with vasculitis occurs in erythema induratum and, occasionally, as a feature of Crohn disease (Chapter 328.2). Inflammation predominantly within the septum, sparing the vasculature, may be seen in erythema nodosum (Table 652-1 and Fig. 652-2), necrobiosis lipoidica, progressive systemic sclerosis (Chapter 154), and subcutaneous granuloma annulare (Chapter 649). Septal panniculitis that includes inflammation of the vessels is found primarily in leukocytoclastic vasculitis and polyarteritis nodosa (Chapter 161).
Table 652-1 ETIOLOGY OF ERYTHEMA NODOSUM
VIRUSES
Epstein-Barr, hepatitis B, mumps
FUNGI
Coccidioidomycosis, histoplasmosis, blastomycosis, sporotrichosis
BACTERIA AND OTHER INFECTIOUS AGENTS
Group A streptococcus,* tuberculosis,* Yersinia, cat-scratch disease, leprosy, leptospirosis, tularemia, mycoplasma, Whipple disease, lymphogranuloma venereum, psittacosis, brucellosis
OTHER
Sarcoidosis, inflammatory bowel disease,* estrogen-containing oral contraceptives,* systemic lupus erythematosus, Behçet syndrome, severe acne, Hodgkin disease, lymphoma, sulfonamides, bromides, Sweet syndrome, pregnancy, idiopathic*
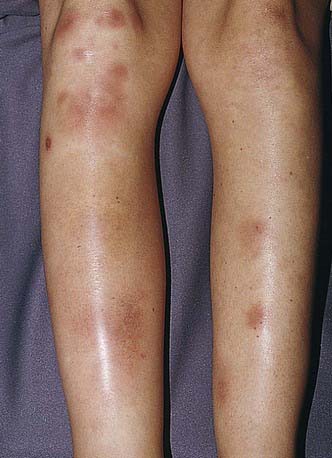
Figure 652-2 Tender red nodules with indistinct borders in a teenage girl with erythema nodosum.
(From Weston AL, Lane AT, Morelli JG: Color textbook of pediatric dermatology, ed 3, St Louis, 2002, Mosby, p 212.)
Erythema Nodosum
Erythema nodosum is a nodular, erythematous hypersensitivity reaction that typically appears with multiple lesions on the exterior surfaces of the arms and legs in the pretibial area (more common) and less often in other cutaneous areas containing subcutaneous fat. The lesions vary in size from 1 to 6 cm, are symmetric, and are oval with the longer axis parallel to the extremity. They initially appear bright or dull red but progress to a brown or purple; they are tense and painful and usually do not ulcerate (see Fig. 652-2). Initial lesions may resolve in 1-2 wk, but new lesions may continue to appear for 2-6 wk. Repeat episodes may occur weeks to months later. Prior to or immediately at the onset of lesions, there may be systemic manifestations that include fever, malaise, arthralgias (50-90%) and rheumatoid factor negative arthritis.
The etiology is unknown in 30-50% of pediatric cases of erythema nodosum; other etiologies are noted in Table 652-1. Group A streptococcal infection and inflammatory disorders (inflammatory bowel disease) are common etiologies in children; sarcoidosis should be considered in young adults.
Post-Steroid Panniculitis
Etiology/Pathogenesis
The mechanism of the inflammatory reaction in the fat in post-steroid panniculitis is unknown.
Lupus Erythematosus Profundus (Lupus Erythematosus Panniculitis)
Clinical Manifestations
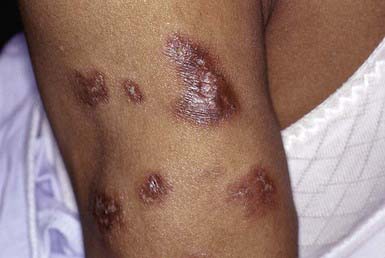
Lupus erythematosus profundus manifests as one to several firm, well-defined, purple plaques or nodules 1 to 3 cm in diameter, most commonly on the face, buttocks, or proximal extremities. This condition may occur in patients with systemic or discoid lupus erythematosus and may precede or follow the development of other cutaneous lesions. The overlying skin is usually normal but may be erythematous, atrophic, poikilodermatous, or hyperkeratotic (Fig. 652-3). Lesions may be painful and may ulcerate. On healing, a shallow depression generally remains or, rarely, soft pink areas of anetoderma result.
α1-Antitrypsin Deficiency
Etiology/Pathogenesis
Individuals with α1-antitrypsin deficiency have severe homozygous deficiency or, rarely, a partial deficiency of the protease inhibitor α1-antitrypsin, which inhibits trypsin activity and the activity of elastase, serine proteases, collagenase, factor VIII, and kallikrein (Chapter 385). Panniculitis occurs with the Z subtype.
Clinical Manifestations
Cellulitis-like areas or tender, red nodules occur on the trunk or proximal extremities (Chapter 385). Nodules tend to ulcerate spontaneously and discharge an oily yellow fluid. Panniculitis may be associated with other manifestations of the disease, such as panacinar emphysema, noninfectious hepatitis, cirrhosis, persistent cutaneous vasculitis, cold contact urticaria, and acquired angioedema. Diagnosis can be substantiated by a decreased level of serum α1-antitrypsin activity.
Subcutaneous Fat Necrosis
Clinical Manifestations
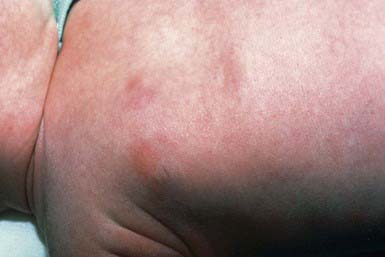
This inflammatory disorder of adipose tissue occurs primarily in the first 4 wk of life in full-term or post-term infants. Typical lesions are asymptomatic, rubbery to firm, erythematous to violaceous plaques or nodules on the cheeks, buttocks, back, thighs, or upper arms (Fig. 652-4). Lesions may be focal or extensive and are generally asymptomatic, although they may be tender during the acute phase. Uncomplicated lesions involute spontaneously within weeks to months, usually without scarring or atrophy. Calcium deposition may occasionally occur within areas of fat necrosis, which may sometimes result in rupture and drainage of liquid material. A rare but potentially life-threatening complication is hypercalcemia. It manifests at 1-6 mo of age as lethargy, poor feeding, vomiting, failure to thrive, irritability, seizures, shortening of the QT interval on electrocardiography, or renal failure. The origin of the hypercalcemia is unknown.
Chilblains (Pernio)
Clinical Manifestations
The condition is characterized by localized symmetric erythematous to purplish edematous plaques and nodules in areas exposed to cold, typically acral areas (distal hands and feet, ears, face; Chapter 69). Lesions develop 12-24 hr after cold exposure and may be associated with itching, burning, or pain. Blister formation and ulceration are rare.
Centers for Disease Control and Prevention. Outbreak of erythema nodosum of unknown cause—New Mexico, November 2007–January 2008. MMWR. 2009;58:1347-1351.
Fraga J, Garcia-Diez A. Lupus erythematosus panniculitis. Dermatol Clin. 2008;26:453-463.
Fregonese L, Stolk J. Hereditary alpa-1-antitrypsin deficiency and its clinical consequences. Orphanet J Rare Dis. 2008;19:16.
Kerstan A, Goebeier M, Schmidt E, et al. Lupus erythematosus profundus in an 8-year old child. J Eur Acad Dermatol Venereol. 2007;21:132-133.
Kwon EJ, Emanuel PO, Gribetz CH, et al. Poststeroid panniculitis. J Cutan Pathol. 2007;34(Suppl 1):64-67.
Lombardi G, Cabana R, Bollani L, et al. Effectiveness of pamidronate in severe neonatal hypercalcemia caused by subcutaneous fat necrosis: a case report. Eur J Pediatr. 2009;168:625-627.
Narváez J, Bianchi MM, Santo P, et al. Pancreatitis, panniculitis and polyarthritis. Semin Arthritis Rheum. 2008;47:1814-1819.
Quesada-Cortes A, Campos-Munoz L, Daiz-Diaz RM, et al. Cold panniculitis. Dermatol Clin. 2008;26:458-459.
Sanmartin O, Requena C, Requena L. Factitial panniculitis. Dermatol Clin. 2008;26:519-527. viii
Simon TD, Soep J, Hollister JR. Pernio in pediatrics. Pediatrics. 2005;116:e472-e475.
Zeb A, Darmstadt GL. Sclerema neonatorum: a review of nomenclature, clinical presentation, histological features, differential diagnosis and management. J Perinatol. 2008;28:453-460.
652.2 Lipodystrophy
Partial Lipodystrophy
There are 3 forms of familial partial lipodystrophy (FPLD):
AKT2 and ZMPSTE24 mutations are newly recognized causes of partial lipodystrophy.
Generalized Lipodystrophy
Generalized lipodystrophy may also be congenital or acquired.
Congenital generalized lipodystrophy is seen in 3 forms:
Type 3 (CAV1) is autosomal recessive and caused by mutations in the caveolin 1 gene.
Garg A, Agarwal AK. Lipodystrophies: disorders of adipose tissue biology. Biochim Biophys Acta. 2009;1791:507-513.
Pope E, Janson A, Khambalia A, et al. Childhood acquired lipodystrophy: a retrospective study. J am Acad Dermatol. 2006;55:947-950.
Serrao VV, Feio AB. Localized abdominal idiopathic lipodystrophy. Dermatol Online. 2008;15:15.





