Chapter 559 Hypothyroidism
Hypothyroidism results from deficient production of thyroid hormone, either from a defect in the gland itself (primary hypothyroidism) or a result of reduced thyroid-stimulating hormone (TSH) stimulation (central or hypopituitary hypothyroidism; Table 559-1). The disorder may be manifested from birth (congenital) or acquired. When symptoms appear after a period of apparently normal thyroid function, the disorder may be truly acquired or might only appear so as a result of one of a variety of congenital defects in which the manifestation of the deficiency is delayed. The term cretinism, although often used synonymously with endemic iodine deficiency and congenital hypothyroidism, is to be avoided.
Table 559-1 ETIOLOGIC CLASSIFICATION OF CONGENITAL HYPOTHYROIDISM
PRIMARY HYPOTHYROIDISM
CENTRAL (HYPOPITUITARY) HYPOTHYROIDISM
ACTH, adrenocorticotropic hormone; FSH, follicle-stimulating hormone; LH, luteinizing hormone, TRH, thyroid-releasing hormone; TSH, thyroid-stimulating hormone.
Congenital Hypothyroidism
Etiology
Thyrotropin and Thyrotropin-Releasing Hormone Deficiency
Deficiency of TSH and hypothyroidism can occur in any of the conditions associated with developmental defects of the pituitary or hypothalamus (Chapter 551). More often in these conditions, the deficiency of TSH is secondary to a deficiency of thyrotropin-releasing hormone (TRH). TSH-deficient hypothyroidism is found in 1/30,000-50,000 infants; most screening programs are designed to detect primary hypothyroidism, so most of these cases are not detected by neonatal thyroid screening. The majority of affected infants have multiple pituitary deficiencies and present with hypoglycemia, persistent jaundice, and micropenis in association with septo-optic dysplasia, midline cleft lip, midface hypoplasia, and other midline facial anomalies.
Thyrotropin Hormone Unresponsiveness
Mild congenital hypothyroidism has been detected in newborn infants who subsequently proved to have type Ia pseudohypoparathyroidism. The molecular cause of resistance to TSH in these patients is the generalized impairment of cyclic adenosine monophosphate activation caused by genetic deficiency of the α subunit of the guanine nucleotide regulatory protein Gs (Chapter 566).
Thyroid Hormone Unresponsiveness
Iodine Exposure
Congenital hypothyroidism can result from fetal exposure to excessive iodides. Perinatal exposure can occur with the use of iodine antiseptic to prepare the skin for caesarian section or painting of the cervix before delivery. It has also been reported in infants born to mothers in Japan who consumed large quantities of iodine-rich seaweed. These conditions are transitory and must not be mistaken for the other forms of hypothyroidism. In the neonate, topical iodine-containing antiseptics used in nurseries and by surgeons can also cause transient congenital hypothyroidism, especially in low-birthweight infants, and can lead to abnormal results on neonatal screening tests. In older children, the usual sources of iodides are proprietary preparations used to treat asthma. In a few instances, the cause of hypothyroidism was amiodarone, an antiarrhythmic drug with high iodine content. In most of these instances, goiter is present (Chapter 561.3).
Clinical Manifestations
If congenital hypothyroidism goes undetected and untreated, these manifestations progress. Retardation of physical and mental development becomes greater during the following months, and by 3-6 mo of age the clinical picture is fully developed (Fig. 559-1). When there is only partial deficiency of thyroid hormone, the symptoms may be milder, the syndrome incomplete, and the onset delayed. Although breast milk contains significant amounts of thyroid hormones, particularly T3, it is inadequate to protect the breast-fed infant who has congenital hypothyroidism, and it has no effect on neonatal thyroid screening tests.
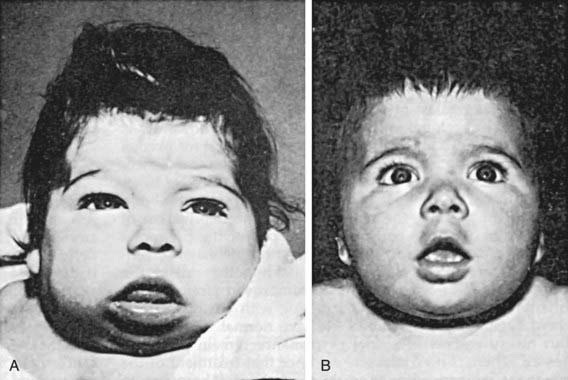
Laboratory Findings
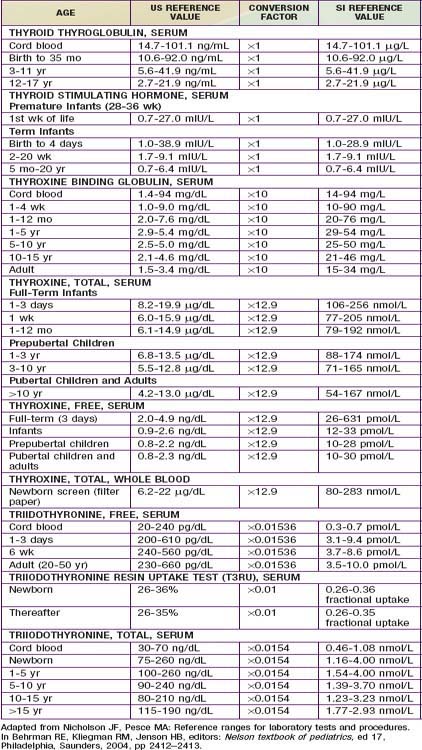
In developed countries, infants with congenital hypothyroidism are identified by newborn screening programs. Blood obtained by heel-prick between 2 and 5 days of life is placed on a filter paper card and sent to a central screening laboratory. Many newborn screening programs in North America and Europe measure levels of T4, followed by measurement of TSH when T4 is low. This approach identifies infants with primary hypothyroidism, some with hypothalamic or pituitary hypothyroidism, and infants with a delayed increase in TSH levels. Other neonatal screening programs in North America, Europe, Japan, Australia, and New Zealand are based on a primary measurement of TSH. This approach detects infants with primary hypothyroidism and can detect infants with subclinical hypothyroidism (normal T4, elevated TSH), but it misses infants with delayed TSH elevation and with hypothalamic or pituitary hypothyroidism. With any of these tests, special care should be given to the normal range of values for age of the patient, particularly in the 1st weeks of life (Table 559-2). Regardless of the approach used for screening, some infants escape detection because of technical or human errors; clinicians must maintain their vigilance for clinical manifestations of hypothyroidism.
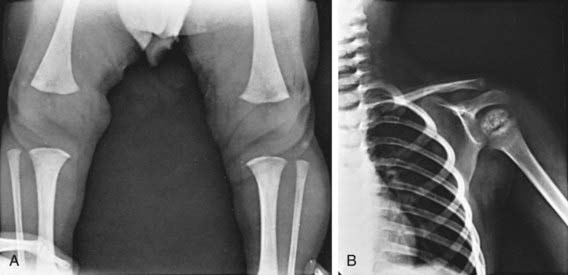
Retardation of osseous development can be shown radiographically at birth in about 60% of congenitally hypothyroid infants and indicates some deprivation of thyroid hormone during intrauterine life. The distal femoral epiphysis, normally present at birth, is often absent (Fig. 559-2A). In undetected and untreated patients, the discrepancy between chronologic age and osseous development increases. The epiphyses often have multiple foci of ossification (epiphyseal dysgenesis) (Fig. 559-2B); deformity (“beaking”) of the 12th thoracic or 1st or 2nd lumbar vertebra is common. Roentgenograms of the skull show large fontanels and wide sutures; intersutural (wormian) bones are common. The sella turcica is often enlarged and round; in rare instances, there may be erosion and thinning. Formation and eruption of teeth can be delayed. Cardiac enlargement or pericardial effusion may be present.
Acquired Hypothyroidism
Etiology
The most common cause of acquired hypothyroidism (Table 559-3) is chronic lymphocytic thyroiditis (Chapter 560). Autoimmune thyroid disease may be part of polyglandular syndromes; children with Down, Turner, and Klinefelter syndromes and celiac disease or diabetes are at higher risk for associated autoimmune thyroid disease (Chapter 560). In children with Down syndrome, anti-thyroid antibodies develop in approximately 30%, and subclinical or overt hypothyroidism occurs in approximately 15-20%. In girls with Turner syndrome, anti-thyroid antibodies develop in approximately 40%, and subclinical or overt hypothyroidism occurs in approximately 15-30%, rising with increasing age. In children with type 1 diabetes mellitus, approximately 20% develop anti-thyroid antibodies and 5% become hypothyroid. Additional autoimmune diseases with an increased risk of hypothyroidism include Sjögren syndrome, multiple sclerosis, pernicious anemia, Addison disease, and ovarian failure. Although typically seen in adolescence, it occurs as early as in the 1st yr of life. Williams syndrome is associated with subclinical hypothyroidism; this does not appear to be autoimmune, as anti-thyroid antibodies are negative.
Table 559-3 ETIOLOGIC CLASSIFICATION OF ACQUIRED HYPOTHYROIDISM
Protracted ingestion of medications containing iodides—for example, expectorants—can cause hypothyroidism, usually accompanied by goiter (Chapter 561). Amiodarone, a drug used for cardiac arrhythmias and consisting of 37% iodine by weight, causes hypothyroidism in about 20% of treated children. It affects thyroid function directly by its high iodine content as well as by inhibition of 5′-deiodinase, which converts T4 to T3. Children treated with this drug should have serial measurements of T4, T3, and TSH. Children with Graves’ disease treated with anti-thyroid drugs (methimazole or propylthiouracil) can develop hypothyroidism. Additional drugs that can produce hypothyroidism include lithium carbonate, interferon alpha, stavudine, thalidomide, valproate (subclinical), and aminoglutethimide.
Any hypothalamic or pituitary disease can cause acquired central hypothyroidism (Chapter 551). TSH deficiency may be the result of a hypothalamic-pituitary tumor (craniopharyngioma most common in children) or a result of treatment for the tumor. Other causes include cranial radiation, head trauma, or diseases infiltrating the pituitary gland, such as Langerhans cell histiocytosis.
Clinical Manifestations
Deceleration of growth is usually the first clinical manifestation, but this sign often goes unrecognized (Figs. 559-3 and 559-4). Goiter, which may be a presenting feature, typically is nontender and firm, with a rubbery consistency and a pebbly surface. Weight gain is mostly fluid retention (myxedema), not true obesity. Myxedematous changes of the skin, constipation, cold intolerance, decreased energy, and an increased need for sleep develop insidiously. Surprisingly, schoolwork and grades usually do not suffer, even in severely hypothyroid children. Additional features include bradycardia, muscle weakness or cramps, nerve entrapment, and ataxia. Osseous maturation is delayed, often strikingly, which is an indication of the duration of the hypothyroidism. Adolescents typically have delayed puberty; older adolescent girls manifest menometrorhhagia. Younger children might present with galactorrhea or pseudoprecocious puberty. Galactorrhea is a result of increased TRH stimulating prolactin secretion. The precocious puberty, characterized by breast development in girls and macro-orchidism in boys, is thought to be the result of abnormally high TSH concentrations binding to the FSH, receptor with subsequent stimulation.
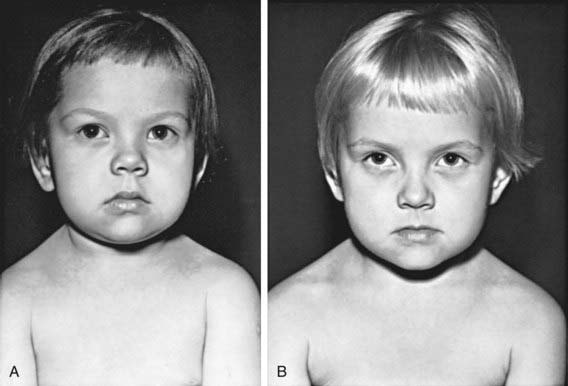
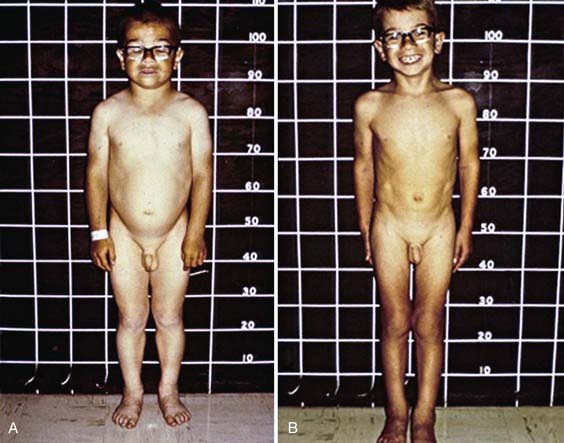
Some children have headaches and vision problems; they usually have hyperplastic enlargement of the pituitary gland, sometimes with suprasellar extension, after long-standing hypothyroidism; this condition, believed to be the result of thyrotroph hyperplasia, may be mistaken for a pituitary tumor (Chapter 551).Abnormal laboratory studies include hyponatremia, macrocytic anemia, hypercholesterolemia, and elevated CPK. Complications seen in severe hypothyroidism are noted in Table 559-4. All these changes return to normal with adequate replacement of T4.
Table 559-4 PATHOGENESIS OF GENERAL COMPLICATIONS IN MANAGEMENT OF COMPLICATED HYPOTHYROIDISM
| COMPLICATION | PATHOGENESIS |
|---|---|
| Heart failure | Impaired ventricular systolic and diastolic functions and increased peripheral vascular resistance |
| Ventilatory failure | Blunted hypercapneic and hypoxic ventilatory drives |
| Hyponatremia | Impaired renal free water excretion and syndrome of inappropriate antidiuretic hormone secretion (SIADH) |
| Ileus | Bowel hypomotility |
| Medication sensitivity | Reduced clearance rate and increased sensitivity to sedative, analgesic, and anesthetic agents |
| Hypothermia and lack of febrile response to sepsis | Decreased calorigenesis |
| Delirium, dementia, seizure, stupor, and coma | Decreased central nervous system thyroid hormone actions, and encephalopathy due to hyponatremia and hypercapnia |
| Adrenal insufficiency | Associated intrinsic adrenal or pituitary disease, or reversible impairment of hypothalamic-pituitary-adrenal stress response |
| Coagulopathy | Acquired von Willebrand syndrome (type 1) and decreased factors VIII, VII, V, IX, and X |
From Roberts CG, Landenson PW: Hypothyroidism, Lancet 363:793–803, 2004.
Diagnostic Studies
Children with suspected hypothyroidism should undergo measurement of serum free T4 and TSH. Because the normal range for thyroid tests is slightly higher in children than adults, it is important to compare results to age-specific reference ranges. Measurement of antithyroglobulin and antiperoxidase (formerly, antimicrosomal) antibodies can pinpoint autoimmune thyroiditis as the cause. Generally, there is no indication for thyroid imaging. In cases with a goiter resulting from autoimmune thyroid disease, an ultrasound examination typically shows diffuse enlargement with scattered hypoechogenicity. Some children with acquired hypothyroidism and a goiter have a thyroid nodule discovered by palpation or neck sonography. Ultrasound examination is the most accurate method to follow nodule size and solid vs. cystic nature (Chapter 563.1). In children with a nodule and suppressed TSH, a radioactive iodine uptake and scan is indicated to determine if this is a “hot” or hyperfunctioning nodule. A bone age x-ray at diagnosis is useful, in that the degree of delay approximates duration and severity of hypothyroidism.
Treatment and Prognosis
In older children, after catch-up growth is complete, the growth rate provides a good index of the adequacy of therapy. Periodic bone age x-rays are useful to monitor treatment and future growth potential. In children with long-standing hypothyroidism, catch-up growth may be incomplete (see Fig. 559-4). During the first 18 mo of treatment, skeletal maturation often exceeds expected linear growth, resulting in a loss of about 7 cm of predicted adult height; the cause is unknown.
Al Taji E, Biebermann H, Limanova A, et al. Screening for mutations in transcription factors in a Czech cohort of 170 patients with congenital and early-onset hypothyroidism: identification of a novel PAX8 mutation in dominantly inherited early-onset non-autoimmune hypothyroidism. Eur J Endocrinol. 2007;156:521-529.
American Academy of PediatricsRose SR, Section on Endocrinology and Committee on Genetics, American Thyroid AssociationBrown RS, Public Health Committee, Lawson Wilkins Pediatric Endocrine SocietyFoley T, Kaplowitz PB, Kaye CI, Sundararajan S, Varma SK. Update of newborn screening and therapy for congenital hypothyroidism. Pediatrics. 2006;117:2290-2303.
Atwell TD, Lteif AN, Brown DL, et al. Neonatal thyroid function after administration of IV iodinated contrast agent to 21 pregnant patients. AJR Am J Roentgenol. 2008;191:268-271.
Balhara B, Misra M, Levitsky LL. Clinical monitoring guidelines for congenital hypothyroidism: laboratory outcome data in the first year of life. J Pediatr. 2011;158:532-537.
Bizhanova A, Kopp A. Minireview: The sodium-iodide symporter NIS and pendrin in iodide homeostasis of the thyroid. Endocrinology. 2009;150:1084-1090.
Bonomi M, Busnelli M, Beck-Peccoz P, et al. A family with complete resistance to thyrotropin-releasing hormone. N Engl J Med. 2009;360:731-734.
Carre A, Szinnai G, Castanet M, et al. Five new TTF1/NKX2.1 mutations in brain-lung-thyroid syndrome: Rescue by PAX8 syngerism I one case. Hum Mol Genet. 2009;18:2266-2276.
Dimitropoulos A, Molinari L, Etter K, et al. Children with congenital hypothyroidism: long-term intellectual outcome after early high-dose treatment. Pediatr Res. 2009;65:242-248.
Eugene D, Djemli A, VanVliet G. Sexual dimorphism of thyroid function in newborns with congenital hypothyroidism. J Clin Endocrinol Metab. 2005;90:2696-2700.
Gu YH, Kato T, Harada S, et al. Time trend and geographic distribution of treated patients with congenital hypothyroidism relative to the number of available endocrinologists in Japan. J Pediatr. 2010;157:153-157.
Harris KB, Pass KA. Increase in congenital hypothyroidism in New York State and in the United States. Mol Genet Metab. 2007;91:268.
Heuer H, Visser T. Minireview: pathophysiological importance of thyroid hormone transporters. Endocrinology. 2009;150:1078-1083.
Hinton CF, Harris KB, Borgfeld L, et al. Trends in incidence rates of congenital hypothyroidism related to select demographic factors: data from the United States, California, Massachusetts, New York, and Texas. Pediatrics. 2010;125:S37-S47.
Jones JH, Gellen B, Paterson WF, et al. Effect of high versus low initial doses of L-thyroxine for congenital hypothyroidism on thyroid function and somatic growth. Arch Dis Child. 2008;93:940-944.
Kumar J, Gordillo R, Kaskel FJ, et al. Increased prevalence of renal and urinary tract anomalies in children with congenital hypothyroidism. J Pediatr. 2009;154:263-266.
LaFranchi SH, Austin J. How should we be treating children with congenital hypothyroidism? J Pediatr Endocrinol Metab. 2007;20:559-571.
Leonardi D, Polizzotti N, Carta A, et al. Longitudinal study of thyroid function in children with mild hyperthyrotropinemia at neonatal screening for congenital hypothyroidism. J Clin Endocrinol Metab. 2008;93:2679.
Maruo Y, Takahashi H, Soeda I, et al. Transient congenital hypothyroidism caused by biallelic mutations of the dual oxidase 2 gene in Japanese patients detected by a neonatal screening program. J Clin Endocrinol Metab. 2008;93:4261-4267.
Mathai S, Cutfield WS, Gunn AJ, et al. A novel therapeutic paradigm to treat congenital hypothyroidism. Clin Endocrinol (Oxf). 2008;69:142-147.
Moreno JC, Klootwijk W, van Toor H, et al. Mutations in the iodotyrosine deiodinase gene and hypothyroidism. N Engl J Med. 2008;358:1811-1818.
Mouat F, Evans HM, Cutfield WS, et al. Massive hepatic hemangioendothelioma and consumptive hypothyroidism. J Pediatr Endocrinol Metab. 2008;21:701-703.
Narumi S, Muroya K, Abe Y, et al. TSHR mutations as a cause of congenital hypothyroidism in Japan: a population-based genetic epidemiology study. J Clin Endocrinol Metab. 2009;94:1317-1323.
Olney RS, Grosse SD, Vogt RFJr. Prevalence of congenital hypothyroidism—current trends and future directions: workshop summary. Pediatrics. 2010;125:S31-S36.
Pardo V, Rubio IG, Knobel M, et al. Phenotypic variation among four family members with congenital hypothyroidism caused by two distinct thyroglobulin gene mutations. Thyroid. 2008;18:783-786.
Partsch CJ, Riepe FG, Krone N, et al. Initially elevated TSH and congenital hypothyroidism due to a homozygous mutation of the TSH beta subunit gene: case report and review of the literature. Exp Clin Endocrinol Diabetes. 2006;114:227-234.
Selva KA, Harper A, Downs A, et al. Neurodevelopmental outcomes in congenital hypothyroidism: comparison of initial T4 dose and time to reach target T4 and TSH. J Pediatr. 2005;147:775-780.
Trueba SS, Auge J, Mattei G, et al. PAX8, TITF1, and FOXE1 gene expression patterns during human development: new insight into human thyroid development and thyroid dysgenesis–associated malformations. J Clin Endocrinol Metab. 2005;90:455-462.
van der Sluijs Veer L, Kempers MJ, Last BF, et al. Quality of life, developmental milestones, and self-esteem of young adults with congenital hypothyroidism diagnosed by neonatal screening. J Clin Endocrinol Metab. 2008;93:2654-2661.
Woo HC, Lizarda A, Tucker R, et al. Congenital hypothyroidism with a delayed thyroid-stimulating hormone elevation in very premature infants: incidence and growth and developmental outcomes. J Pediatr. 2011;158:538-542.
Bilimoria KY, Pescovitz OH, DiMeglio LA. Autoimmune thyroid dysfunction in children with type 1 diabetes mellitus: screening guidelines based on a retrospective analysis. J Pediatr Endocrinol Metab. 2003;16:1111-1117.
Cambiaso P, Orazi C, Digilio MC, et al. Thyroid morphology and subclinical hypothyroidism in children and adolescents with Williams syndrome. J Pediatr. 2007;150:62-65.
Cooper DS. Thyroxine monotherapy after thyroidectomy. JAMA. 2008;299:817-818.
DeBoer MD, LaFranchi S. Differential presentation for children with autoimmune thyroiditis discovered because of symptom development or screening. J Pediatr Endocrinol Metab. 2008;21:753-761.
de Vries L, Bulvick S, Phillip M. Chronic autoimmune thyroiditis in children and adolescents: at presentation and during long-term follow-up. Arch Dis Child. 2009;94:33-37.
El-Mansoury M, Bryman I, Berntorp K, et al. Hypothyroidism is common in Turner syndrome: results of a five-year follow-up. J Clin Endocrinol Metab. 2005;90:2131-2135.
Indolfi G, Stagi S, Bartolini E, et al. Thyroid function and anti-thyroid autoantibodies in untreated children with vertically acquired chronic hepatitis C virus infection. Clin Endocrinol. 2008;68:117-121.
Ishiguro H, Yasuda Y, Tomita Y, et al. Long-term follow-up of thyroid function in patients who receive bone marrow transplantation during childhood and adolescence. J Clin Endocrinol Metab. 2004;89:5981-5986.
Mikati MA, Tarabay H, Khaul A, et al. Risk factors for development of subclinical hypothyroidism during valproic acid therapy. J Pediatr. 2007;151:178-181.
Popova G, Paterson WF, Brown A, et al. Hashimoto’s thyroiditis in Down’s syndrome: clinical presentation and evolution. Horm Res. 2008;70:278-284.
Remuzzi G, Garattini S. Elimination of iodine-deficiency disorders in Tibet. Lancet. 2008;371:1980-1981.
Teng W, Shan Z, Teng X, et al. Effect of iodine in take on thyroid diseases in China. N Engl J Med. 2006;354:2783-2792.
van Trotsenburg AS, Vulsma T, van Rozenburg-Marres SL, et al. The effect of thyroxine treatment started in the neonatal period on development and growth of two-year-old Down syndrome children: a randomized clinical trial. J Clin Endocrinol Metab. 2005;90:3304-3311.
Wasniewska M, Salerno M, Cassio A, et al. Elevated TSH levels normalize or remain unchanged in the majority of children with subclinical hypothyroidism. Eur J Endocrinol. 2009;160:417-421.



