Chapter 74 The Human Genome
Fundamentals of Molecular Genetics
DNA consists of a pair of chains of a sugar-phosphate backbone linked by pyrimidine and purine bases to form a double helix (Fig. 74-1). The sugar in DNA is deoxyribose. The pyrimidines are cytosine (C) and thymine (T); the purines are guanine (G) and adenine (A). The bases are linked by hydrogen bonds such that A always pairs with T and G with C. Each strand of the double helix has polarity, with a free phosphate at one end (5′) and an unbonded hydroxyl on the sugar at the other end (3′). The two strands are oriented in opposite polarity in the double helix.
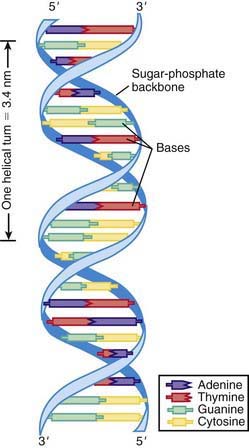
Figure 74-1 DNA double helix, with sugar-phosphate backbone and nitrogenous bases.
(From Jorde LB, Carey JC, Bamshad MJ, et al, editors: Medical genetics, ed 2, St Louis, 1999, Mosby, p 8.)
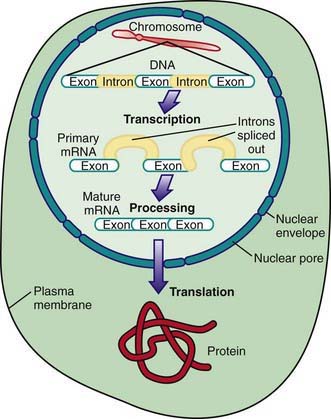
A prototypical gene consists of a regulatory region, segments called exons that encode the amino acid sequence of a protein, and intervening segments called introns (Fig. 74-2). Transcription starts at the promoter region and continues through the entire length of the gene to form mRNA. The introns are removed and the exons spliced together to form a mature message, which is exported to the cytoplasm. There the mRNA is bound to ribosomes and translated into protein.
Genetic Variation
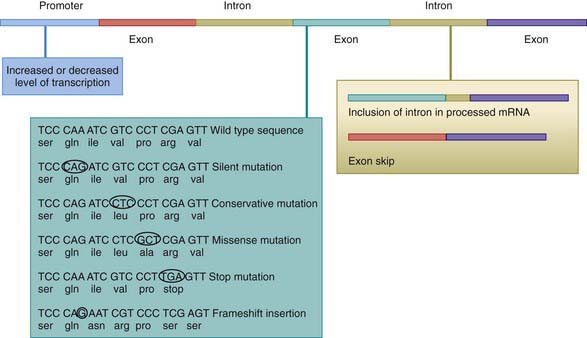
The process of producing protein from a gene is subject to disruption at multiple levels owing to alterations in the coding sequence (Fig. 74-3). Changes in the regulatory region can lead to altered gene expression, including increased or decreased rates of transcription, failure of gene activation, or activation of the gene at inappropriate times or in inappropriate cells. Changes in the coding sequence can lead to substitution of one amino acid for another (missense mutation or nonsynonymous) or creation of a stop codon in the place of an amino acid codon. Some single-base changes do not affect the amino acid (silent or wobble mutation or synonymous), because there may be several codons that correspond with a single amino acid. Amino acid substitutions can have a profound effect on protein function if the chemical properties of the substituted amino acid are markedly different from the usual one. Other substitutions can have a subtle or no effect on protein function, particularly if the substituted amino acid is chemically similar to the original one.
Mutations usually can be classified as causing a loss of function or a gain of function. Loss-of-function mutations cause a reduction in the level of protein function due to decreased expression or production of a protein that does not work as efficiently. In some cases, loss of protein function from one gene is sufficient to cause disease. Haploinsufficiency describes the situation in which maintenance of a normal phenotype requires the proteins produced by both copies of a gene, and a 50% decrease in gene function results in an abnormal phenotype. Hence, haploinsufficient phenotypes are by definition dominantly inherited. Loss-of-function mutations can also have a dominant negative effect when the abnormal protein product actively interferes with the function of the normal protein product. Both of these situations lead to diseases inherited in a dominant fashion (Chapter 75). In other cases, loss-of-function mutation must be present in both copies of a gene before an abnormal phenotype results. This situation typically results in diseases inherited in a recessive fashion (Chapter 75).
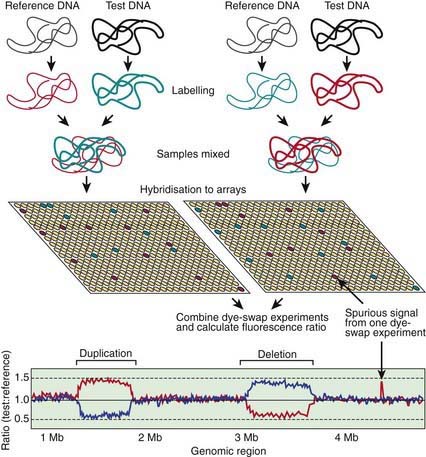
In some cases, changes in gene expression are caused by changes in the number of copies of a gene present in the genome (Fig. 74-4). Although some copy number variations (CNVs) are common and do not appear to cause or predispose to disease, others are clearly disease causing. Charcot-Marie-Tooth disease type 1A, the most common inherited form of chronic peripheral neuropathy of childhood, is caused by duplications of the gene for peripheral myelin protein 22, resulting in overexpression due to the existence of 3 active copies of this gene. Deletions of this same gene—leaving only one active copy—are responsible for a different disorder, hereditary neuropathy with liability to pressure palsies.
Genotype-Phenotype Correlations in Genetic Disease
The long QT syndrome exemplifies a disorder with predictable associations between a patient’s genotype and his or her phenotype (Chapter 429.5). Long QT syndrome is genetically heterogeneous, meaning that mutations in several different genes can cause the same disorder. The risk for cardiac events (syncope, aborted cardiac arrest, or sudden death) is higher with long QT syndrome mutations involving the KCNQ1 gene (63%) or the KCNH2 gene (46%) than among subjects with mutations in the SCN5A gene (18%). In addition, those with mutations involving KCNQ1 experience most of their episodes during exercise and rarely during rest or sleep; those with KCNH2 and SCN5A mutations are more likely to have episodes during sleep or rest, and rarely during exercise. Therefore, mutations in specific genes (genotype) are correlated with specific manifestations (phenotype) of long QT syndrome. These types of relationships are commonly referred to as genotype-phenotype correlations.
Mutations in the fibrillin-1 gene associated with Marfan syndrome represent another example of predictable genotype-phenotype correlations (Chapter 693). Marfan syndrome is characterized by the combination of skeletal, ocular, and aortic manifestations, with the most devastating outcome being aortic root dissection and sudden death. Sixty-five exons make up the fibrillin-1 gene, and mutations have been found in almost all of these exons. The location of the mutation within the gene (genotype) might play a significant role in determining the severity of the condition (phenotype). Neonatal Marfan syndrome is caused by mutations in exons 24-27 and 31-32, whereas milder forms are caused by mutations in exons 59-65 and in exons 37 and 41.
Genotype-phenotype correlations have also been observed in cystic fibrosis (CF) (Chapter 395). Although pulmonary disease is the major cause of morbidity and mortality, CF is a multisystemic disorder that affects not only the epithelia of the respiratory tract but also the exocrine pancreas, intestine, male genital tract, hepatobiliary system, and exocrine sweat glands. CF is caused by mutations in the CF transmembrane conductance regulator (CFTR) gene. More than 1,600 different mutations have been identified. The most common is a deletion of three nucleotides that removes the amino acid phenylalanine (F) at the 508th position on the protein (ΔF508 mutation), which accounts for about 70% of all CF mutations and is associated with severe disease. The best genotype-phenotype correlations in CF are seen in the context of pancreatic function, with most common mutations being classified as either pancreatic sufficient or pancreatic insufficient. Persons with pancreatic sufficiency usually have either 1 or 2 pancreatic sufficient alleles, indicating that pancreatic sufficient alleles are dominant. In contrast, the genotype-phenotype correlation in pulmonary disease is much weaker, and persons with identical genotypes have wide variations in the severity of their pulmonary disease. This finding may be accounted for in part by genetic modifiers or environmental factors.
Human Genome Project
Availability of the entire human genomic sequence permits the study of large groups of genes, looking for patterns of gene expression or genome alteration. Microarrays have been developed that permit the expression of thousands of genes to be analyzed on a small glass chip. In some cases the patterns of gene expression provide signatures for particular disease states, such as cancer, or change in response to therapy (Fig. 74-5).

Ali-khan SE, Daar AS, Shuman C, et al. Whole genome scanning: resolving clinical diagnosis and management amidst complex data. Pediatr Res. 2009;66:357-363.
Alkan C, Kidd JM, Marques-Bonet T, et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet. 2009;41:1061-1067.
Ashley EA, Butte AJ, Wheeler MT, et al. Clinical assessment incorporating a personal genome. Lancet. 2010;375:1525-1535.
Bredenoord A, Braude P. Ethics of mitochondrial gene replacement: from bench to bedside. BMJ. 2011;342:87-89.
Christensen K, Murray JC. What genome-wide association studies can do for medicine. N Engl J Med. 2007;356:1094-1097.
Cordell HJ, Clayton DG. Genetic association studies. Lancet. 2005;366:1121-1130.
Feero WG, Guttmacher AE, Collins FS. Genomic medicine—an updated primer. N Engl J Med. 2010;362:2001-2011.
Goldstein DB. Common genetic variation and human traits. N Engl J Med. 2009;360:1696-1698.
Hamburg MA, Collins FS. The path to personalized medicine. N Engl J Med. 2010;363:301-304.
Hardy J, Singleton A. Genomewide association studies and human disease. N Engl J Med. 2009;360:1759-1768.
Hingorani AD, Shah T, Kumari M, et al. Translating genomics into improved healthcare. BMJ. 2010;341:1037-1042.
Lucassen A, Parker M. Confidentiality and sharing genetic information with relatives. Lancet. 2010;375:1507-1509.
McCarroll SA. Copy number variation and human genome maps. Nat Genet. 2010;42:365-366.
McGhee SA, McCabe ERB. Genome-wide testing: genomic medicine. Pediatr Res. 2006;60:243-244.
Moskowitz SA, Chmiel JF, Sternen DL, et al. CFTR-related disorders (2008) in GeneReviews at GeneTests: Medical Genetics Information Resource (database online). www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=cf#cf. Accessed March 9, 2009
Shaw CJ, Lupski JR. Implications of human genome architecture for rearrangement-based disorders: the genomic basis of disease. Hum Mol Genet. 2004;13:R57-R64.
Teare MD, Barrett JH. Genetic linkage studies. Lancet. 2005;366:1036-1044.
Veltman JA, Brunner HG. Understanding variable expressivity in microdeletion syndromes. Nat Genet. 2010;42:192-193.
Wain LV, Armour JAL, Tonin MD. Genomic copy number variation, human health, and disease. Lancet. 2009;374:340-350.