Seizures are time-limited paroxysmal events that result from abnormal, involuntary rhythmic activity affecting small or large neuronal networks in the brain (1,2). The term seizure appears to have arisen from a notion that patients were possessed or seized by spirits in old times. The incidence of childhood epilepsy is approximately 80 cases per 100,000 persons per year (2). Incidence of first nonfebrile seizure in children is 25,000 to 40,000 per year in the United States (3). Although mortality directly related to seizure is low in these children, 45% to 50% have recurrence (4–6). Therefore, it is important to evaluate patients with seizures and events that look like seizures.
Common conditions that are mistaken for seizures are gastroesophageal (GE) reflux, apneic episodes, self-stimulating behavior, breath-holding spells, migraine, attention deficit hyperactivity disorder (ADHD), and sleep disorders (see Chapter 10).
Seizures can be classified based on etiologies and types. Etiologic classification includes provoked and unprovoked seizures. Provoked seizures are defined as seizures occurring in close temporal association with an acute systemic (immune or infectious), metabolic, or toxic insult or with an acute central nervous system (CNS) insult (7). Common causes of provoked seizures include metabolic disturbances such as hyponatremia, hypoglycemia and hyperglycemia, hypocalcemia, and acute neurologic conditions such as trauma, stroke, meningitis, and recreational drug or alcohol use or withdrawal. Unprovoked seizures can be classified based on etiologies as idiopathic (with particular clinical characteristics and specific electroencephalo graphic findings), symptomatic (due to metabolic or genetic disorders or structural abnormalities), or cryptogenic (unknown etiology) (7). The term cryptogenic suggests that a cause may be subsequently discovered. Classification of seizure types is based on the International League Against Epilepsy (ILAE) classification (see Section 3).
Epilepsy is defined conceptually as a disorder of the brain characterized by an enduring predisposition to generate epileptic seizures and by the neurobiologic, cognitive, psychological, and social consequences of this condition. The definition of epilepsy requires the occurrence of at least one epileptic seizure (8), while the practical definition is the occurrence of two or more unprovoked seizures (7). It is important to note that the occurrence of multiple seizures in a 24-hour period is considered as one seizure (7).
When a patient presents with a seizure or a seizure-like event, the goal is to identify whether the event was a seizure or not; if it was a seizure, to identify the type of seizure based on ILAE criteria, identify the etiology of seizure, and anticipate recurrence and the need for treatment.
A thorough history and physical exam are essential to identify critical elements during the evaluation of a seizure or event that appeared seizure-like, and these are the most important aspect of establishing the diagnosis of epilepsy. This chapter aims to recommend the initial evaluation of a patient presenting with seizure.
PATIENT EVALUATION
HISTORY
The following pertinent history should be elicited.
Age
Certain seizure types are age-dependent. Examples are infantile spasms, febrile seizures, benign epilepsy with centrotemporal spikes (BECTS), childhood absence seizures, juvenile absence seizures, and juvenile myoclonic epilepsy. Children are most vulnerable to develop epilepsy during the first year of life (9,10). Etiologies for status epilepticus differ based on age. During the first 2 years of life, status epilepticus is more common in neurologically normal children, whereas later on it is more common in neurologically abnormal children (11).
Preceding or Precipitating Events
Knowing the events immediately before a seizure episode is very important. Parents may not perceive that premonitory symptoms or auras are part of a seizure and may not volunteer this information to the physician. Most common manifestations of aura associated with temporal lobe seizures are autonomic. These include “funny feelings,” epigastric sensations, flushing, sweating, palpitations, nausea, or dizziness.
The presence of preceding events also helps to differentiate nonepileptic events, such as crying preceding breath-holding spells and feeding preceding reflux. Loud noises and stimuli can lead to hyperekplexia, which may look like seizure.
Certain seizures are provoked by specific stimuli and are called reflex seizures. Well-known precipitating factors are visual stimuli, flickering light, thinking, praxis, reading, somatosensory and proprioceptive stimuli, eating, music, hot water, and startle (12). Other nonspecific factors are also associated with induction of seizure. In a study of 1,677 patients with epilepsy, 53% reported one and 30% reported two or more precipitating factors. Most common factors were emotional stress, sleep deprivation, fatigue, and alcohol consumption. Patients with generalized seizures were more sensitive to sleep deprivation and flickering light than patients with localization-related epilepsy (13).
Timing of Event
Epilepsies associated with awakening seizures are usually primary generalized seizure disorders such as juvenile myoclonic epilepsy and absence seizures. Sleep epilepsies include focal or secondarily generalized seizure disorders such as BECTS, Landau–Kleffner, electrical status epilepticus in sleep (ESES), and frontal lobe seizures. Temporal lobe seizures are also more common during sleep. Complex partial seizures generalize more frequently during sleep. Seizures are more common during stages 1 and 2 sleep and least common during rapid eye movement (REM) sleep. Also, nonepileptic seizures do not occur out of sleep (14,15). Unprovoked seizures occurring out of sleep have a higher risk of recurrence, and the sleep state at the time of the first seizure correlates with successive seizures (16). Patients with seizures occurring in sleep have higher chances of having an abnormal electroencephalogram (EEG) (17). Knowing the child’s state immediately before and after the event is extremely important and frequently diagnostic.
The Event Itself
Have the parents and family members describe a chronological account of what they witnessed. Caregivers often use diagnostic terms such as “petit mal,” drawing conclusions. Often the exact description may allow the physician to determine whether an event was a seizure or not, and if a seizure, what type. Also, a majority of the time seizures are described as “shaking.” It is not reasonable to speculate that this means clonic activity because it could mean trembling associated with tonic seizure, clonic activity, or, simply, pelvic thrusting or bicycling motion. Witnesses should be asked to demonstrate the event if they can. A seizure may start in one part of the body and progress to the whole body with loss of consciousness. Therefore, it is important to ascertain how and where it started.
The history of forced eye closure during an event most likely suggests a nonepileptic event. The mouth is usually open during tonic seizure. Forced clenching of the mouth is associated with pseudoseizures (18). Gaze deviation can help determine the focal nature of the event and help in localization.
The duration of the event may allow differentiation of seizure from status epilepticus, which is treated differently and has a different prognosis. Status epilepticus has recently been operationally redefined as a seizure of 5 minutes or more of continuous clinical and/or electrographic seizure activity or recurrent seizure activity without return to baseline between seizures (19). This changes the definition of status epilepticus in terms of making the the practitioner focus on management from a treatment initiation perspective, in contrast to the older definition of 30 minutes or more, which tends to make the practitioner focus on the consequence of a prolonged seizure. In a study of 407 children presenting with a first unprovoked seizure, the authors reported that 50% had seizures lasting 5 minutes or longer, 29% 20 minutes, and 12% 30 minutes. Seizures of partial onset were more likely to be prolonged. Ninety-two percent of the seizures stopped spontaneously (20). The likelihood of a seizure stopping spontaneously decreases after 5 minutes and reaches a minimum at 15 minutes. Also, the duration of subsequent seizures correlates with the length of the first episode (20). It is not uncommon, however, to overestimate or underestimate the duration of the event.
It is important to ask patients about what they remember of the event. This may help to identify the exact duration of loss of consciousness or awareness, if any.
After the cessation of the event, it is important to know the patient’s condition. Malingering patients will be back to baseline immediately after a dramatic motor seizure. Others may show confusion, tiredness, sleepiness, or hemiparesis. Continued altered awareness may be due to postictal state or continued subclinical seizure.
Prior history of other paroxysmal events, similar events, other seizures, and febrile seizures is important. Recurrance risk of another seizure after a single unprovoked seizure is 40% to 52% (8). Recurrence risk after a second seizure increases to about 70%. High risk was seen if seizure recurred within 6 months of the first seizure (16). Patients with a first partial motor seizure before the age of 2 have a likely recurrence risk of 87% (21). In patients with newly diagnosed epilepsy, about 17% have seizures of more than one type (22). Such patients have a lower probability of remission of epilepsy (23). A total of 2% to 10% of patients with febrile seizures develop epilepsy. Complex febrile seizures are associated with an increased risk of subsequent development of epilepsy (4,24).
Associated Events
Common events associated with generalized motor seizures are tongue biting, bowel or bladder incontinence, and respiratory attenuation. Tongue biting can be seen during the clonic phase of a seizure (18).
Irregular respiration and pauses in respirations are seen during clonic and tonic phases of generalized seizures. Cyanosis may be present with a seizure. Often peripheral cyanosis is not associated with central hypoxia. Malingering patients may not be able to demonstrate cyanosis. Respiratory involvement may also make seizures more frightening for the family, and early interventions may be needed for those at risk of prolonged convulsions.
Review of Systems
Fever, lethargy, headache, and photophobia may point toward meningitis and encephalitis as a cause of seizures. Diarrhea and vomiting leading to dehydration and electrolyte imbalance can provoke seizures. It is important to ask about diet, especially in infants and teenage females. In infants who are formula fed, if the formula is improperly mixed, there are chances of electrolyte imbalance leading to seizures. Teenage females should be asked questions regarding bulimia, “dieting,” and “diet pills” if suspected. Teenagers should also be asked about recreational drug and alcohol use. History of trauma should be evaluated. Other medical conditions such as diabetes and infections should be considered. Exposure to toxins and heavy metals should be suspected in a younger child.
In patients with “unknown cause” epilepsy, autoimmune antibodies are increasingly recognized as a cause of new-onset epilepsy, as in one series it has been found in almost 10% of patients. Thus, it should be taken into consideration when no other etiology can be determined, especially if comorbidities of personality change, sleep, or movement disorders are seen (25).
Medication
It is helpful to know whether the patient is taking medications that are known to decrease seizure threshold. Also, the history of hypoglycemic agents and diuretic use is helpful.
Birth History
Premature newborns who weigh less than 1.5 lb are at higher risk for brain injury leading to acute or remote symptomatic seizures. The differential diagnosis for neonatal seizures is broad (ie, structural, metabolic, genetic), but those arising from acute symptomatic causes (ie, hypoxic ischemic encephalopathy, transient metabolic disturbance, infection, stroke, or intracranial hemorrhage) are more common (26). In-utero exposure to infectious agents, recreational drugs, alcohol, and teratogenic medications can also cause structural brain abnormality. Meningitis, increased bilirubin levels, and respiratory problems occurring during the postnatal period are important to know for the same reason. Maternal history of oligohydramnios or polyhydramnios, fetal hiccups, or episodic hammering fetal movements may suggest neurological disorders in the fetus.
Developmental History
History of language, motor, or psychosocial delays, and regression and timing of regression should be evaluated. About 15% to 30% of patients with childhood-onset epilepsy have cerebral palsy or mental retardation and 11% to 39% of children with autistic spectrum disorders develop epilepsy (8,27,28). Patients with severe autism have a higher incidence of epilepsy (29). Developmental delays may be a hallmark of chromosomal disorders and associated epilepsy (30,31). Language regression and seizures may represent Landau–Kleffner syndrome. Global regression may be seen at the onset of ESES (27) (see Chapter 26). Children with developmental delay or mental retardation have a higher risk of intractable seizure. It is likely that these disorders represent widespread underlying CNS abnormalities (23).
Family History
In a study of patients with seizures, about 35% of patients reported seizures in first-degree relatives (32). Inherited epilepsies include autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE), BECTS, juvenile myoclonic epilepsy (JME), generalized epilepsy with febrile seizures plus syndrome, benign familial neonatal convulsions, benign familial infantile convulsions, and other idiopathic generalized epilepsy (33).
In a study looking at risk of epilepsy in adults with seizures, cryptogenic or idiopathic epilepsy with partial onset showed a higher occurrence rate in successive generations, whereas the occurrence rate for generalized epilepsy remained the same in all generations (34). In twin studies, there was concordance of occurrence of seizure in monozygotic and dizygotic twins and status epilepticus in monozygotic twins (32,35).
PHYSICAL EXAMINATION
General Examination
Head circumference should be measured. Presence of microcephaly could be secondary to congenital infections, hypoxia, or maternal diseases, and drug and toxin exposure. Cranial malformations such as lissencephaly, cortical dysplasia, and heterotropia are primary causes of microcephaly (36–38). Macrocephaly is associated with storage disorders and hydrocephalus (37).
Fundoscopic examination may show papilledema, suggesting increased intracranial pressure; or cherry-red spot, associated with storage diseases; or findings associated with neurocutaneous syndromes or congenital malformations.
The examiner should look for dysmorphic features. Microcephaly, craniosynostosis, deep-set eyes, hypertelorism, low-set ears, midface hypoplasia, depressed bridge of nose, microstomia or macrostomia, simian crease, and hand and foot abnormalities are some of the features that present in chromosomal disorders associated with seizures (30,31). Dysmorphic features such as cleft lip and cleft palate are known to be associated with cranial dysplasia. Abnormal hair patterning may suggest abnormalities in brain development before 18 weeks. Microcephaly with abnormal hair patterning may be seen with brain abnormality rather than craniosynostosis (39).
The examination of the skin is important; a number of neurocutaneous syndromes are associated with seizures. Incontentia pigmenti, linear nevus syndrome, hypomelanosis of Ito, tuberous sclerosis, Sturge–Weber syndrome, and neurofibromatosis are examples of these neurocutaneous syndromes (see Chapter 30). Skin examination can also reveal signs of physical abuse.
Nuchal rigidity and positive Kernig’s and Brudzinski’s signs suggest meningitis. Infants with open fontanel may not present with these signs, but will show a bulging or tense fontanel.
Organomegaly may be seen with metabolic storage diseases.
Neurological Examination
Determination of ongoing events should be the first step in the evaluation of a patient presenting with a history of seizure. The patient may present with altered sensorium, which could be due to ongoing seizure, postictal phenomena, or medication used. Dysarthria or visual changes may be present postictally. A paralysis, which may be partial or complete and usually occurring on just one side of the body, may suggest a Todd’s paralysis, which can last from half an hour to 36 hours, after which it resolves completely (NIH Website). The presence of Todd’s paralysis after a first unprovoked seizure is associated with increased risk of recurrence (16).
In patients suspected of having absence seizure, hyperventilate for at least 3 minutes, for this may precipitate a seizure.
Laboratory Studies
Basic Metabolic Panel
Seizures provoked by metabolic abnormalities in an otherwise normal child are rare. Therefore, basic tests should be done based on clinical judgment per American Academy of Neurology (AAN) guidelines for evaluation of first unprovoked seizure (3).
Lumbar Puncture
If there is clinical suspicion of meningitis or encephalitis, lumbar puncture should be done per AAN guidelines for evaluation of first unprovoked seizure.
Urine Toxicology
Routine urinalysis is not recommended, but if there is clinical suspicion of recreational drug use, a urine drug screen should be obtained.
Liver Function Tests
If there is a history suggestive of a potentially degenerative, metabolic, or storage disorder or there is anticipation of starting antiepilepsy drugs (AEDs), liver function tests should be performed.
Screening for Inherited Metabolic Disorders
These tests may not be obtained during the initial evaluation, but are very important in seizure evaluation and management and so are mentioned here.
About 200 inherited disorders are associated with seizures (40). There are clues such as certain seizure types, characteristic EEG patterns, and specific epilepsy syndromes that point to some of these disorders. Associated symptoms of mental retardation, developmental delays, hypotonia, macrocephaly and microcephaly, and failure to thrive may also suggest the possibility of a metabolic disorder. In patients with myoclonic seizures, spasms, early-onset tonic seizures, and infantile-onset hypomotor and versive seizures, inherited metabolic disorders should be suspected (40). Other characteristics of seizures associated with inherited disorders include onset in the first month of life, usually partial seizures with a migrant nature and successive appearance (41). West syndrome, early myoclonic encephalopathy, and Ohtahara syndrome are known to be associated with metabolic disorders (see Table 11.4).
TABLE 11.1
|
EEG PATTERNS AND ASSOCIATED DISORDERS |
|
|
EEG Pattern |
Disorder |
|
Comblike rhythm Fast central spikes Rhythmic vertex positive spikes Vanishing EEG High amplitude 16–24 Hz activity Diminished spikes during sleep Marked photosensitivity |
Maple syrup urine disease Propionic academia Tay–Sachs disease Sialidosis (type I) Infantile NCL (type I) Infantile neuroaxonal dystrophy PME |
|
Burst suppression |
PME and NCL, particularly type II |
|
|
Neonatal citrullinemia Nonketotic hyperglycinemia Propionic academia Leigh disease D-Glyceric academia Molybdenum cofactor deficiency Menke’s syndrome Holocarboxylase synthetase deficiency Neonatal adrenoleukodystrophy |
|
Hypsarrhythmia |
Zellweger syndrome Neonatal adrenoleukodystrophy Neuroaxonal dystrophy Nonketotic hyperglycinemia Phenylketonuria Carbohydrate-deficient glycoprotein syndrome (type III) |
Source: From Ref. (40). Nordli DR Jr, De Vivo DC. Classification of infantile seizures: implications for identification and treatment of inborn errors of metabolism. J Child Neurol. 2002;17(Suppl 3):3S3–3S7; discussion 3S8.
EEG, electroencephalogram; NCL, neuronal ceroid lipofuscinosis; PME, progressive myeloclonic epilepsy.
EEGs showing burst suppression, hypsarrhythmia, and diffuse severe depression without obvious cause demand evaluation for inherited disorders (see Table 11.1 and Figure 11.1).
Serum amino acids and urine organic acids can be used as an initial screen in patients with the previously mentioned seizure types and syndromes. Cerebrospinal fluid (CSF) glucose, lactate, and amino acids are recommended for infantile seizures without clear etiology. It is important to obtain samples during the presentation of symptoms before treatment, because otherwise samples may be falsely normal or difficult to interpret (43) (see Tables 11.2 and 11.3) (41,43,44).
Disorders of carnitine and fatty acid oxidation acutely present with a history of decreased oral intake followed by increasing lethargy, obtundation or coma, and seizures. Other features include history of recurrent hypoglycemic, hypoketotic encephalopathy, potential developmental delay, and progressive lipid storage myopathy (see Tables 11.2 and 11.3).
Recurrent myoglobinuria, hypotonia, neuropathy, and progressive cardiomyopathy. Screening tests should include serum total and free carnitine, acylcarnitines, free fatty acids and ketones, urine organic acids, and acylglycines. A serum free fatty acid-to-ketone ratio of more than 2:1 suggests a block in fatty acid oxidation. Urine organic acid chain length and type will help to identify the site of the block. Confirmations can be obtained by specific cultured skin fibroblast tests (46).
A few cases of creatine metabolism defect and epilepsy have been reported. Associated symptoms include hypotonia, mental retardation, neurologic regression, and movement disorders. Initial tests should include serum and urine guanidinoacetic acid and creatine. Magnetic resonance (MR) spectroscopy also helps in the diagnosis (47).
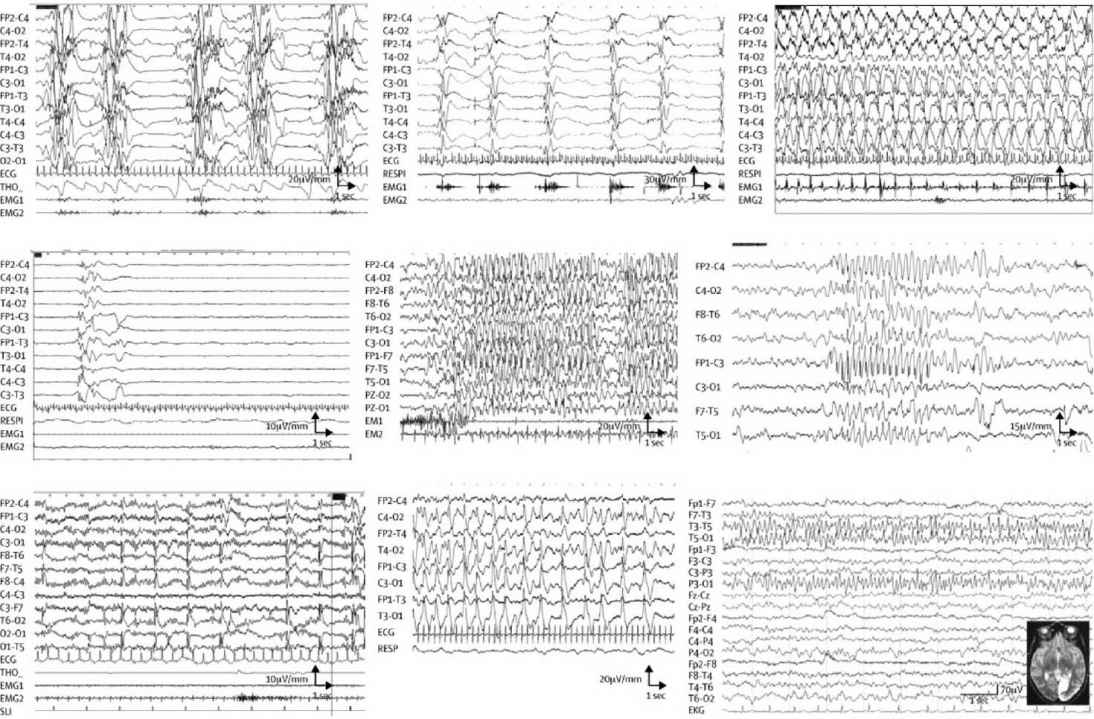
FIGURE 11.1 EEG in IEM according to etiology. (A) Interictal EEG in a 15-day-old baby with nonketotic hyperglycinemia, showing the characteristic suppression–bursts sequence (bursts of high-amplitude diffuse polyspikes separated by episodes of flat tracing). (B–D) EEG from a 3-month-old baby with pyridoxine-dependent epilepsy. (B) Epileptic spasms in clusters with high-amplitude slow complex and rhythmic waves. (C) High-amplitude rhythmic spike–waves predominate on the left hemisphere during a right clonic seizure. (D) After pyridoxine intravenous injection (100 mg), EEG shows flattening of the tracing. (E) Myoclonic status epilepticus in a 6-year-old girl with guanidinoacetate methyltransferase deficiency. Diffuse high-voltage spikes and spike–waves predominating on frontal or frontal-central areas; myoclonic jerks recorded on deltoid electromyography. (F) Atypical absence in an 11-year-old girl with glucose transporter-1 deficiency, with diffuse high-amplitude spike–waves. (G) Diffuse spikes, predominating on occipital areas triggered by slow photic stimulation in a 5-year-old girl with late-infantile neuronal ceroid lipofuscinosis. (H) 3-year-old girl with Alpers’ disease due to POLG1 mutations. Interictal EEG shows bilateral high-amplitude periodic sharp waves and spike–waves with left occipital predominance. (I) 16-year-old girl with MELAS and the classic mitochondrial 3243A→G mutation. EEG shows spike and spike–waves rhythmic discharges over the left posterior region, corresponding to the occipital area of hyperintense T2 signal.
MELAS, mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes.
Source: From Ref. (42). Dulac et al., Occasional seizures, epilepsy, and inborn errors of metabolism. Lancet Neurology. 2014;13(7):727–739.
CSF metabolites. Disorders affecting CNS energy metabolism, creatine synthesis, glucose transport, and neurotransmitter metabolism may present with hypotonia, temperature instability, developmental delay, ptosis, dystonia, and tremors along with seizures. These disorders may not be detected by routine screen for inherited metabolic disorders. If initial metabolic screen and CSF lactate and amino acids are normal in an infant with seizure without clear etiology, profiles of CSF metabolites, including biogenic amines, should be considered (48).
When gamma-aminobutyric acid (GABA)-related disorders are suspected—that is, GABA transaminase deficiency, succinic semialdehyde dehydrogenase deficiency (SSDD), glutamate decarboxylase, or GABA receptor defects—CSF GABA level should be measured. It is increased in patients with SSDD and vigabatrin treatment. SSDD can also be detected in lymphocytes and fibroblasts (49).
In nonketotic hyperglycinemia, CSF glycine is elevated. It is also elevated in patients who are receiving valproate treatment. It is decreased in defective serine synthesis (49).
Guanidinoacetate and creatine measurement can aid in diagnosis of arginine:glycine aminotransferase or guanidinoacetate methyletransferase deficiencies, and creatine transporter defect.
Measurement of tetrahydrobiopterin, neopterin, 7,8-dihydrobiopterin, homovanillic acid, 5-hydroxyin-doleacetic acid, and 3-O-methyldopa can help in diagnosis of tetrahydrobiopterin and biogenic amine metabolism.
TABLE 11.2
|
EPILEPSIES OF METABOLIC ORIGIN ACCORDING TO THEIR PATHOGENESIS |
|
|
|
Etiology |
|
Energy deficiency |
Hypoglycemia, glucose transporter-1 deficiency, respiratory chain deficiency, pyruvate dehydrogenase deficiency, Krebs cycle defects, creatine deficiencies |
|
Toxic effect |
Aminoacidopathies, organic acidurias, urea cycle defects, molybdenum cofactor deficiency, sulphite oxidase deficiency |
|
Impaired neuronal function |
Storage disorders |
|
Disturbance of neurotransmitter systems |
Nonketotic hyperglycinemia, atypical phenylketonuria, GABA transaminase deficiency, succinic semialdehyde dehydrogenase deficiency |
|
Associated brain malformations |
Peroxisomal disorders (Zellweger syndrome), respiratory chain deficiency, pyruvate dehydrogenase deficiency, O-glycosylation defects (congenital muscular dystrophies) |
|
Vitamin or cofactor dependency and vitamin transporter defects |
Biotinidase deficiency, pyridoxine-dependent and pyridoxal 5′-phosphate-dependent epilepsy (folinic-acid-responsive seizures), thiamine transporter deficiency, Menkes’ disease, folate transporter defect (FOLR1), dihydrofolate reductase deficiency |
|
Miscellaneous |
Serine biosynthesis deficiency |
Source: From Ref. (42). Dulac O, Plecko B, Gataullina S, Wolf NI. Occasional seizures, epilepsy, and inborn errors of metabolism. Lancet Neurology. 2014;13(7):727–739.
Pyridoxine-deficient and -responsive seizures are covered in another section (see Chapter 30).
Tests for mitochondrial disorders. Mitochondrial disorders causing epilepsy are rare. It is possible that this rarity is due to our inability to recognize these disorders. A review by DiMauro showed that seizures are associated with MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes), MERRF (myoclonus epilepsy with ragged red fibers), pyruvate dehydrogenase complex deficiencies, cytochrome c oxidase deficiencies, and maternally inherited Leigh’s disease (50). All the patients in the review presenting with these disorders and seizure had symptoms such as developmental delay and eventually developed classic symptoms for the disorder. Partial seizures are more common in patients with mitochondrial encephalopathies (51).
TABLE 11.3
|
EPILEPSIES OF METABOLIC ORIGIN ACCORDING TO MOST FREQUENT AGE AT ONSET |
|
|
|
Etiology |
|
Neonatal period |
Hypoglycemia, urea cycle defects, pyridoxine-dependent epilepsy (including folinic-acid-responsive seizures), pyridox(am)ine 5′-phosphate deficiency, nonketotic hyperglycinemia, holocarboxylase synthase deficiency, molybdenum cofactor deficiency, sulphite oxidase deficiency, organic acidurias, Zellweger syndrome, neonatal adrenoleukodystrophy, adenylosuccinate lyase deficiency, dihydrofolate reductase deficiency |
|
Infancy |
Sequelae of severe hypoglycemia, glucose transporter-1 deficiency, creatine deficiency, biotinidase deficiency, aminoacidopathies, organic acidurias, congenital disorders of glycosylation, pyridoxine-dependent epilepsy, infantile neuronal ceroid lipofuscinosis (CLN1), folate and thiamine transporter deficiencies, peroxisomal disorders, Menkes’ disease |
|
Toddlers |
Late infantile neuronal ceroid lipofuscinosis (CLN2), mitochondrial disorders including Alpers’ disease, lysosomal storage disorders, thiamine transporter deficiency, folate transporter deficiency |
|
School age and adolescence |
Mitochondrial disorders, juvenile form of neuronal ceroid lipofuscinosis (CLN3), progressive myoclonic encephalopathies, lysosomal storage disorders, thiamine transporter deficiency, Lafora’s disease, Gaucher’s disease, and Niemann-Pick type C disease |
Source: From Ref. (42). Dulac O, Plecko B, Gataullina S, Wolf NI. Occasional seizures, epilepsy, and inborn errors of metabolism. Lancet Neurology. 2014;13(7):727–739.
TABLE 11.4
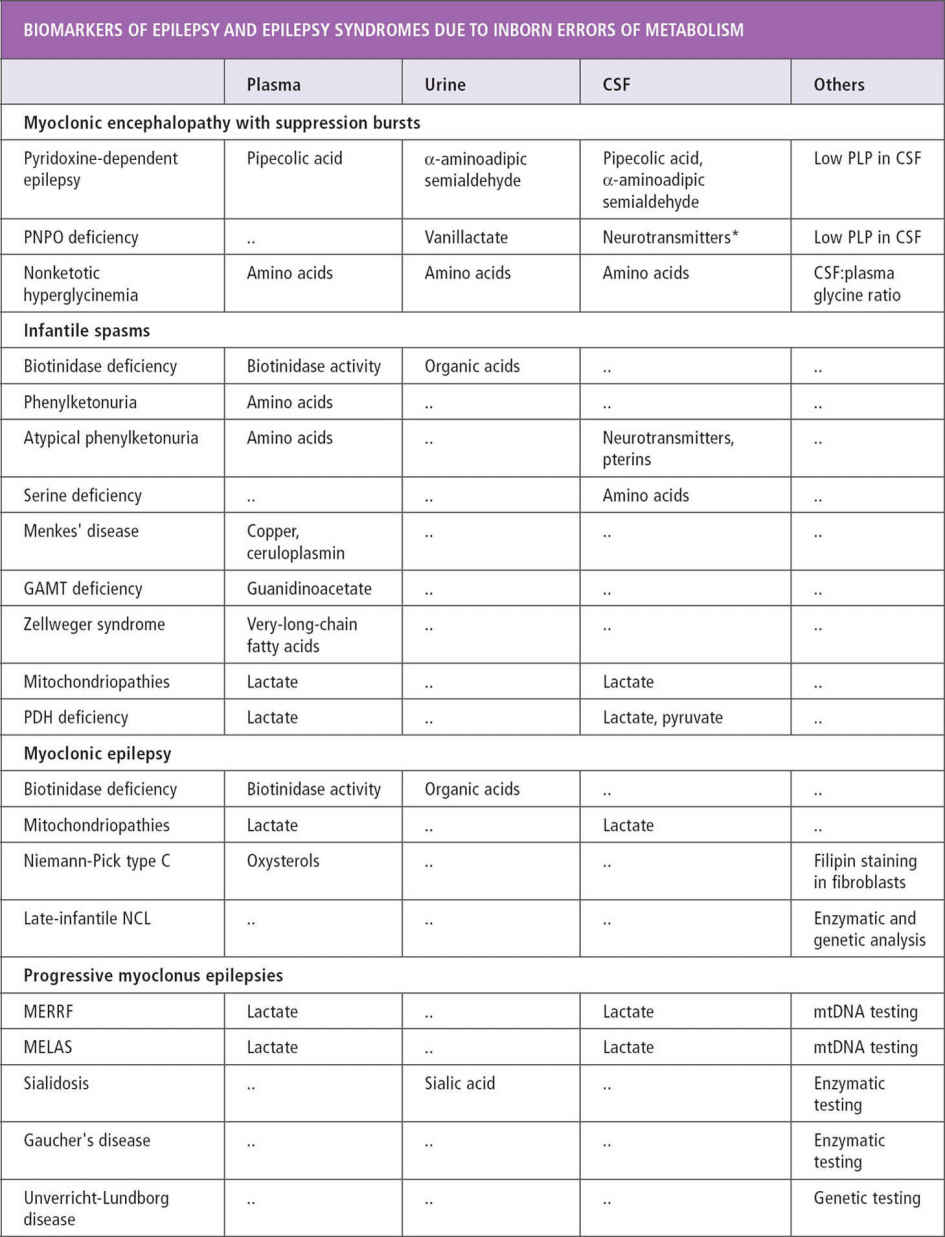
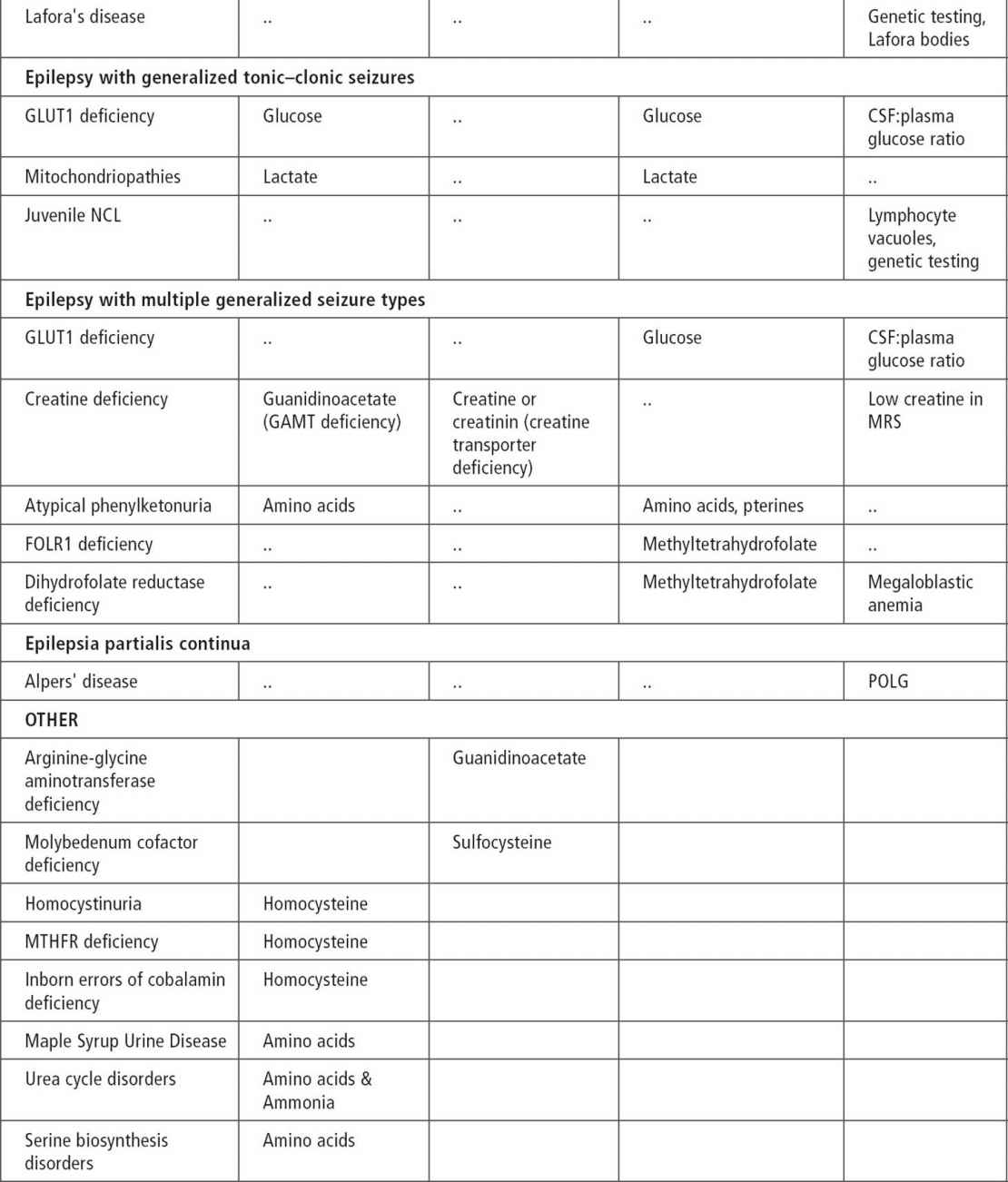
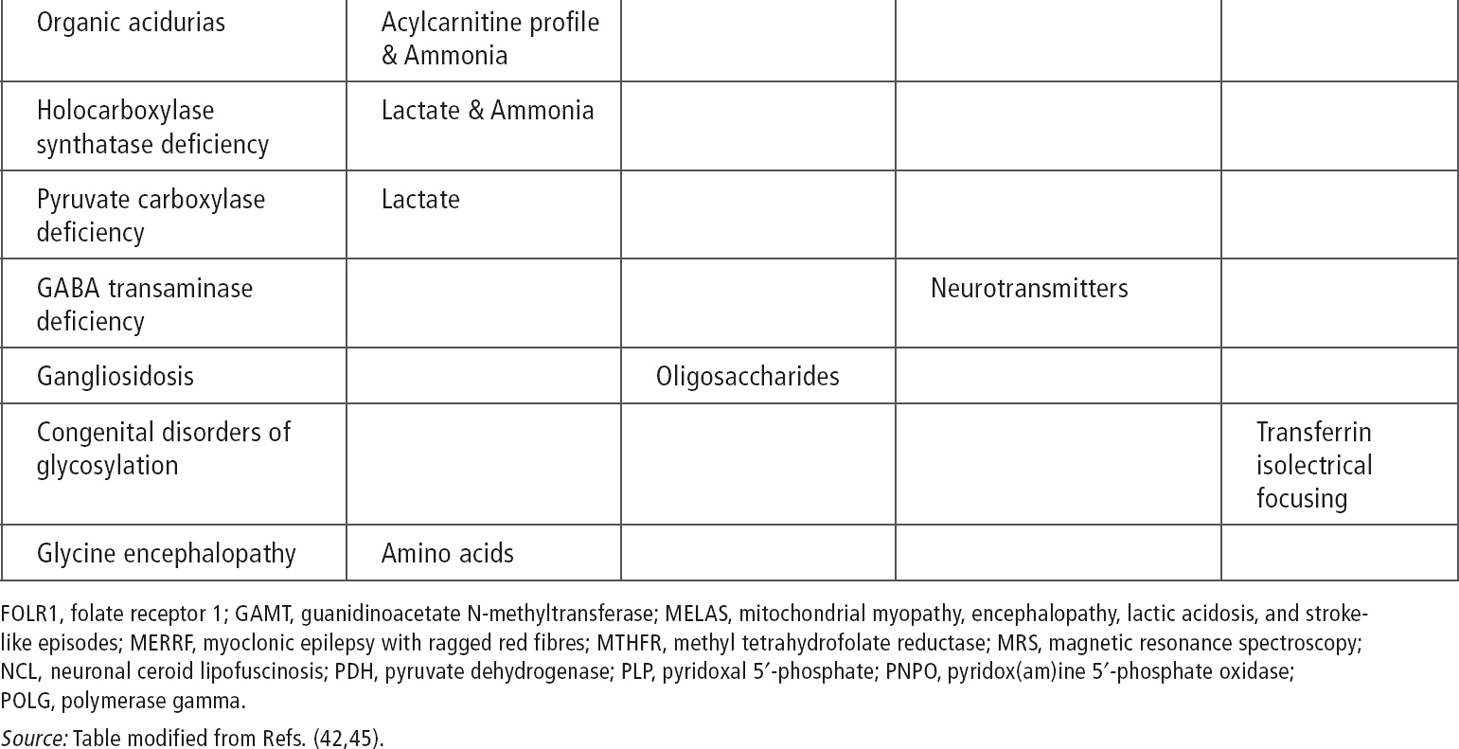
Ammonia, serum and CSF lactate, and urine organic acid can be used as an initial screen (43). In cases of intractable seizure and developmental delays, migraine, stroke-like episodes, or retinitis pigmentosa, serum testing and muscle biopsy should be considered.
At the time of the writing of this book, testing with comprehensive epilepsy panels are available for the following:
• Adenylosuccinate lyase deficiency
• Alpers syndrome (Alpers-Huttenlocher syndrome)
• Angelman syndrome (AS)
• Angelman-like syndrome, X-linked syndromic mental retardation (Christianson type)
• Atypical Rett syndrome
• Autoimmune epilepsy
• Benign familial neonatal seizures (BFNS)
• Benign familial neonatal-infantile seizures (BFNIS)
• Creatine deficiency syndromes
• Dravet syndrome
• Early-onset epileptic encephalopathy and/or infantile spasms
• Epilepsy and mental retardation limited to females
• Epilepsy with variable learning and behavioral disorders
• Familial hemiplegic migraine (sometimes a/w seizures)
• Generalized epilepsy with febrile seizures plus (GEFS+)
• Glucose transporter type I deficiency syndrome
• Juvenile myoclonic epilepsy
• Lafora disease
• Microcephaly with early-onset intractable seizures and developmental delay (MCSZ)
• Mowat–Wilson syndrome
• Neuronal ceroid lipofuscinoses (NCL)
• Neuronal migration disorders
• Nocturnal frontal lobe epilepsy, autosomal dominant
• Ohtahara syndrome
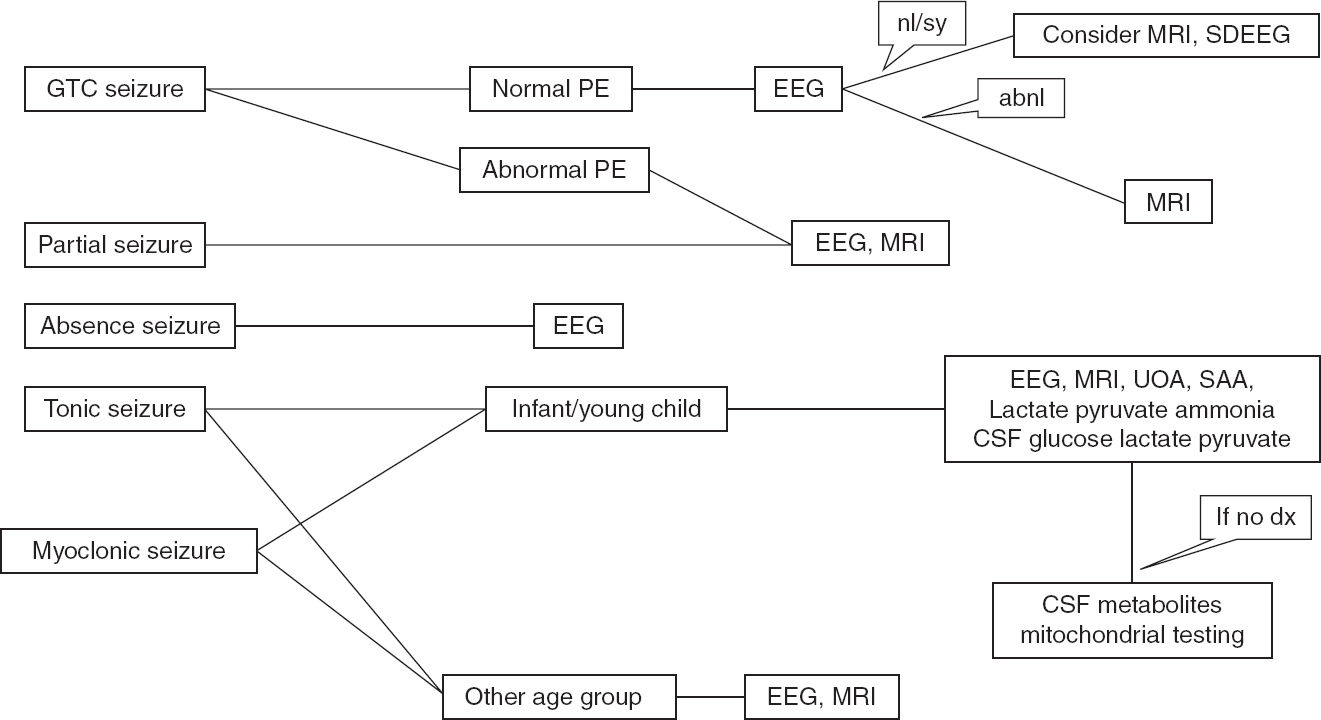
FIGURE 11.2 Recommended algorithm based on seizure types.
abnl, abnormal; GTC, generalized tonic clonic; nl/sy, normal or benign syndromic; PE, physical examination; SAA, serum amino acid; SDEEG, sleep-deprived EEG; UAO, urine organic acid.
• Partial epilepsy with auditory features, autosomal dominant
• Progressive myoclonic epilepsy
• Pyridoxine dependent seizures
• Rett syndrome
• Severe myoclonic epilepsy of infancy
• Tuberous sclerosis
• Unverricht–Lundborg disease (Baltic myoclonus)
• West syndrome
[level-membership-for-pediatrics-category]
Chromosomal Studies
Chromosomal abnormalities with high association of seizures include 4p-syndrome, Miller–Dieker syndrome, Angelman syndrome, inversion duplication 15 syndrome, terminal deletions of chromosome 1q and 1p, and ring chromosomes 14 and 20. Patients presenting with specific phenotypes and EEG abnormalities should have genetic testing done as discussed in Chapters 7 and 8 (30,31,52).
EEG
EEG remains the gold-standard test for evaluation of seizures, especially when an ictus is captured during the study. It can help to differentiate nonepileptic events from seizure. It can also help to identify an epileptic syndrome, because epilepsy syndromes are classified based on specific patterns of abnormality on EEG. It helps to provide long-term prognosis and identify any need to do further testing. It influences the management by helping to select AEDs. Certain metabolic disorders have classic EEG patterns (see Table 11.1 and Figure 11.1).
In a study of patients with unprovoked seizures, EEG abnormalities were associated with increased recurrence. Abnormal EEGs were more common in patients with partial seizures than with generalized seizures (53). A study by Yoshinaga et al showed that more than 20% of seizures were classified incorrectly or missed when only history was used to identify paroxysmal events (54). Detection without EEG, using clinical evaluation to identify neonatal seizures, is approximately 50% accurate for events detected at bedside, but clinical evaluation cannot help identify subclinical and/or nonconvulsive seizures.
Recommendations regarding timing for the EEG testing are controversial. In a study by King et al, an EEG performed within the first 24 hours had a higher diagnostic yield (55). In a study done at Virginia Commonwealth University when EEGs were done in the emergency room in patients presenting with a seizure, 80% of patients with an abnormal EEG developed recurrence (56). EEG abnormalities are the best predictors of recurrence in neurologically normal children (3).
Activation methods such as hyperventilation and photic stimulation increase the yield of EEG and are recommended (57). Even though Gilbert et al showed that sleep deprivation did not increase EEG abnormalities (58), a number of other studies have shown that sleep recordings can increase the yield of EEG (3,15,53,57,59).
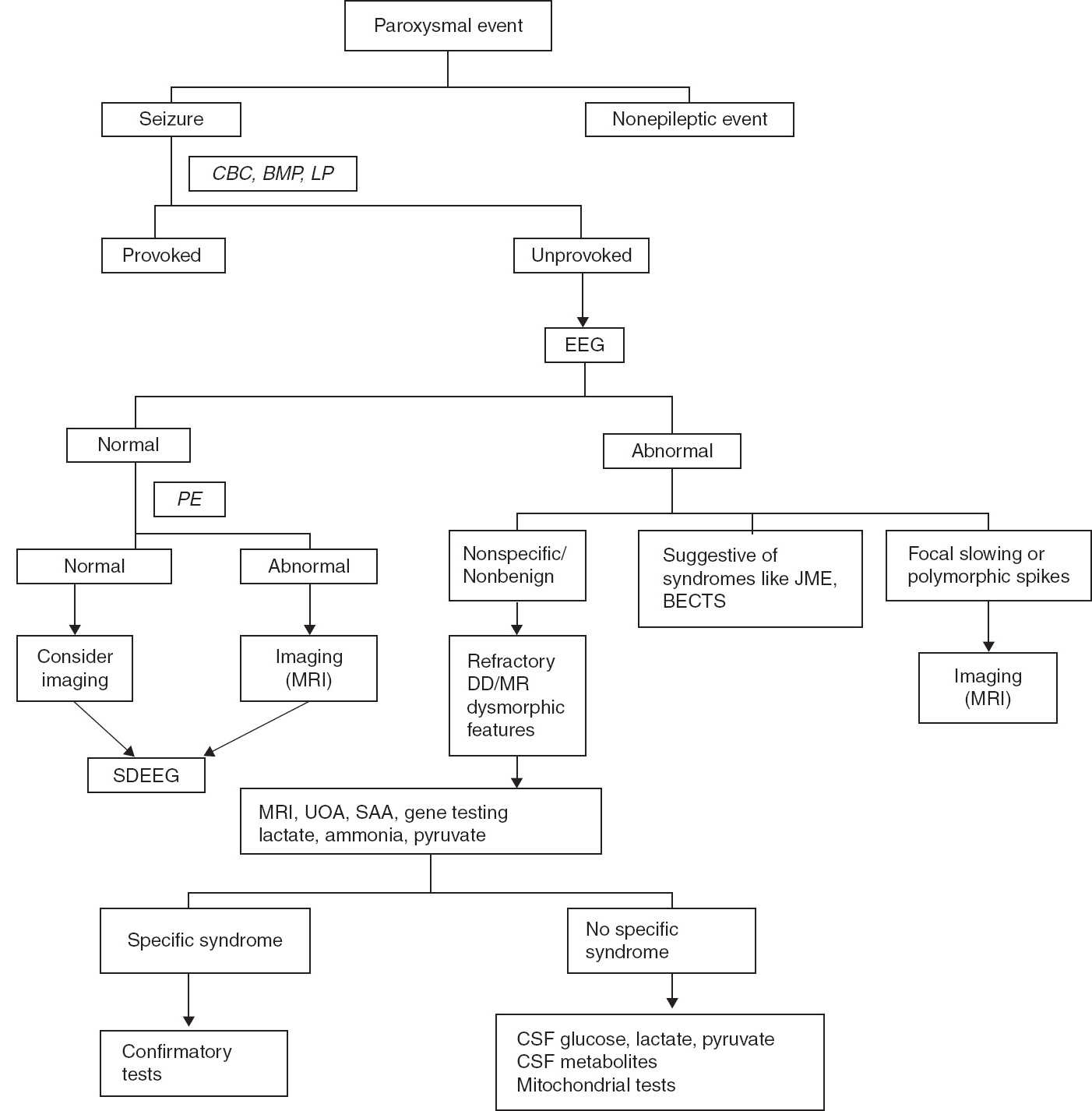
FIGURE 11.3 Recommended algorithm for seizure evaluation.
BECTS, benign epilepsy with centrotemporal spikes; BMP, basic metabolic panel; CBC, complete blood count; DD, developmental delay; JME, juvenile myoclonic epilepsy; LP, lumbar puncture; MR, mental retardation; PE, physical examination; SAA, serum amino acid; SDEEG, sleep-deprived EEG; UOA, urine organic acid.
Thus, EEG is recommended to evaluate paroxysmal events. In patients with altered consciousness and recurrent seizures, emergent EEG is recommended. Hyperventilation and sleep deprivation increase the yield of EEG. When possible, an EEG should be obtained immediately after seizure presentation. A normal or suspicious EEG should be followed by sleep-deprived EEG.
Imaging
In a study by Shinnar et al (60), only 1% of children with a first unprovoked seizure had neuroimaging abnormalities requiring acute intervention. All these children had indications for neuroimaging such as an abnormal neurologic examination or prolonged seizure. The most common abnormalities were focal encephalomalacia and cerebral dysgenesis. Suggested risk factors for abnormal imaging were partial seizures, abnormal neurological examination, and abnormal EEG (61). In children with a brief tonic–clonic seizure, normal examination, and normal EEG, 10% of imaging studies were abnormal (60).
Imaging is recommended for seizure evaluation (3). Emergent imaging studies are required in case of prolonged seizure, prolonged postictal phase, and postictal hemiparesis to rule out stroke and other structural abnormalities (3). Outside of the acute situation, magnetic resonance imaging is a superior study because it can identify subtle cortical abnormalities (3,61).
CONCLUSION
Seizures are frightening events both for the families and the children in whom they occur and require precautions associated with driving, water activities, heights, and work environment. The diagnosis has immense psychosocial impact on both the family and the patient. Thus, the first seizure or similar event should be evaluated carefully to make an accurate diagnosis, the recurrence risk should be explained to the family, and appropriate treatment decisions should be made. Recommended algorithms for seizure evaluation are shown in Figures 11.2 and 11.3.
REFERENCES
1. Shneker BF, Fountain NB. Epilepsy. Dis Mon. 2003;49(7):426–478.
2. Tamber MS, Mountz JM. Advances in the diagnosis and treatment of epilepsy. Semin Nucl Med. 2012;42(6):371–386.
3. Hirtz D, Ashwal S, Berg A, et al. Practice parameter: evaluating a first nonfebrile seizure in children: report of the Quality Standards Subcommittee of the American Academy of Neurology, the Child Neurology Society, and the American Epilepsy Society. Neurology. 2000;55(5):616–623.
4. Shinnar S, Berg AT, Moshe SL, et al. The risk of seizure recurrence after a first unprovoked afebrile seizure in childhood: an extended follow-up. Pediatrics. 1996;98(2 Pt 1):216–225.
5. Shinnar S, Berg AT, Moshe SL, et al. Risk of seizure recurrence following a first unprovoked seizure in childhood: a prospective study. Pediatrics. 1990;85(6):1076–1085.
6. Shinnar S, O’Dell C, Berg AT. Mortality following a first unprovoked seizure in children: a prospective study. Neurology. 2005;64(5):880–882.
7. ILAE Commission Report. The epidemiology of the epilepsies: future directions (International League against Epilepsy). Epilepsia. 1997;38(5):614–618.
8. Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014;55(4):475–482.
9. Hauser WA, Annegers JF, Kurland LT. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935–1984. Epilepsia. 1993;34(3):453–468.
10. Shinnar S, Pellock JM. Update on the epidemiology and prognosis of pediatric epilepsy. J Child Neurol. 2002;17 (Suppl 1):S4–S17.
11. Haut SR, Legatt AD, O’Dell C, et al. Seizure lateralization during EEG monitoring in patients with bilateral foci: the cluster effect. Epilepsia. 1997;38(8):937–940.
12. Ferlazzo E, Zifkia BG, Andermann E, Andermann F. Cortical triggers in generalized reflex seizures and epilepsies. Brain. 2005;128:700–710.
13. Nakken KO, Solaas MH, Kjeldsen MJ, et al. Which seizure-precipitating factors do patients with epilepsy most frequently report? Epilepsy Behav. 2005;685(1):85–89.
14. Nordli DR, Bazil CW, Scheuer ML, Pedley TA. Recognition and classification of seizures in infants. Epilepsia. 1997;38(5):553–560.
15. Mendez M, Radtke RA. Interactions between sleep and epilepsy. J Clin Neurophysiol. 2001;18(2):106–127.
16. Shinnar S, Berg AT, O’Dell C, et al. Predictors of multiple seizures in a cohort of children prospectively followed from the time of their first unprovoked seizure. Ann Neurol. 2000;48(2):140–147.
17. Shinnar S, Berg AT, Ptachewich Y, Alemany M. Sleep state and the risk of seizure recurrence following a first unprovoked seizure in childhood. Neurology. 1993;43(4):701–706.
18. Detoledo JC, Ramsay RE. Patterns of involvement of facial muscles during epileptic and nonepileptic events: review of 654 events. Neurology. 1996;47(3):621–625.
19. Brophy GM, Bell R, Claassen J, et al. for Neurocritical Care Society Status Epilepticus Guideline Writing Committee. Guidelines for the evaluation and management of status epilepticus. Neurocrit Care. 2012;17(1):3–23.
20. Shinnar S, Berg AT, Moshe SL, Shinnar R. How long do new-onset seizures in children last? Ann Neurol. 2001;49(5):659–667.
21. Fenichel GM. Paroxysmal Disorders. Clinical Pediatric Neurology. 6th ed. 2009:18–19.
22. Berg AT, Shinnar S, Levy SR, Testa FM. Newly diagnosed epilepsy in children: presentation at diagnosis. Epilepsia. 1999;40(4):445–452.
23. Oskoui M, Webster RI, Zhang X, Shevell MI. Factors predictive of outcome in childhood epilepsy. J Child Neurol. 2005;20(11):898–904.
24. Shinnar S, Glauser TA. Febrile seizures. J Child Neurol. 2002;17(Suppl 1):S44–S52.
25. Suleiman J, Wright S, Gill D, et al. Autoantibodies to neuronal antigens in children with new onset seizures classified according to the revised ILAE organization of seizures and epilepsies. Epilepsia. 2013;54(6):2091–2100.
26. Glass HC. Neonatal seizures: advances in mechanisms and management. Clin Perinatol. 2014;41(1):177–190.
27. Ballaban-Gil K, Tuchman R. Epilepsy and epileptiform EEG: association with autism and language disorders. Ment Retard Dev Disabil Res Rev. 2000;6(4):300–308.
28. Filipek PA, Accardo PJ, Ashwal S, et al. Practice parameter: screening and diagnosis of autism (Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Child Neurology Society). Neurology. 2000;55(4):468–479.
29. Danielsson S, Gillberg IC, Billstedt E, et al. Epilepsy in young adults with autism: a prospective population-based follow-up study of 120 individuals diagnosed in childhood. Epilepsia. 2005;46(6):918–923.
30. Hachiya Y, Arai H, Hayashi M, et al. Autonomic dysfunction in cases of spinal muscular atrophy type 1 with long survival. Brain & Dev. 2005;27(8):574–578.
31. Schinzel A, Niedrist D. Chromosome imbalances associated with epilepsy. Am J Med Genet. 2001;106(2):119–124.
32. Miller LL, Pellock JM, Boggs JG, et al. Epilepsy and seizure occurrence in a population-based sample of Virginian twins and their families. Epilepsy Res. 1999;34(2–3):135–143.
33. Callenbach PM, van den Maagdenberg AM, Frants RR, Brouwer OF. Clinical and genetic aspects of idiopathic epilepsies in childhood. Eur J Paediatr Neurol. 2005;9(2):91–103.
34. Ottman R, Lee JH, Risch N, et al. Clinical indicators of genetic susceptibility to epilepsy. Epilepsia. 1996;37(4):353–361.
35. Corey LA, Pellock JM, Boggs JG, et al. Evidence for a genetic predisposition for status epilepticus. Neurology. 1998;50(2):558–560.
36. Barkovich AJ, Kuzniecky RI, Jackson GD, et al. Classification system for malformations of cortical development: update 2001. Neurology. 2001;57(12):2168–2178.
37. Glass RB, Fernbach SK, Norton KI, et al. The infant skull: a vault of information. Radiographics. 2004;24(2):507–522.
38. Sztrha L, Dawodu A, Gururaj A, Johansen JG. Microcephaly associated with abnormal gyral pattern. Neuropediatrics. 2004;35(6):346–352.
39. Smith DW, Gong BT. Scalp-hair patterning: its origin and significance relative to early brain and upper facial development. Teratology. 1974;9(1):17–34.
40. Nordli DR, de Vivo DC. Classification of infantile seizures: implications for identification and treatment of inborn errors of metabolism. J Child Neurol. 2002;17(Suppl 3):3S3–3S7; discussion 3S8.
41. Vigevano F, Bartuli A. Infantile epileptic syndromes and metabolic etiologies. J Child Neurol. 2002;17(Suppl 3):3S9–3S13; discussion 3S14.
42. Dulac O, Plecko B, Gataullina S, Wolf NI. Occasional seizures, epilepsy, and inborn errors of metabolism. Lancet Neurol. 2014;13(7):727–739.
43. Buist NR, Dulac O, Bottiglieri T, et al. for Amalfi Group. Metabolic evaluation of infantile epilepsy: summary recommendations of the Amalfi Group. J Child Neurol. 2002;17(Suppl 3):3S98–3S102.
44. Leary LD, Nordli DR, Jr, de Vivo DC. Epilepsy in the setting of inherited metabolic and mitochondrial disorders. In: Wyllie E, Gupta A, Lachhwani DK. The Treatment of Epilepsy: Principles and Practice. Philadelphia, PA: Lippincott, Williams, & Wilkins; 2006.
45. Patel J, Mercimek-Mahmutoglu S. Epileptic encephalopathy in childhood: a stepwise approach for identification of underlying genetic causes. Indian J Pediatr. 2016:1–11.
46. Tein I. Role of carnitine and fatty acid oxidation and its defects in infantile epilepsy. J Child Neurol. 2002;17(Suppl 3):3S57–3S82; discussion 3S82–3S83.
47. Leuzzi V. Inborn errors of creatine metabolism and epilepsy: clinical features, diagnosis, and treatment. J Child Neurol. 2002;17(Suppl 3):3S89–3S97; discussion 3S97.
48. Hyland K, Arnold LA. Value of lumbar puncture in the diagnosis of infantile epilepsy and folinic acid-responsive seizures. J Child Neurol. 2002;17(Suppl 3):3S48–3S55; discussion 3S56.
49. Jaeken J. Genetic disorders of gamma-aminobutyric acid, glycine, and serine as causes of epilepsy. J Child Neurol. 2002;17(Suppl 3):3S84–3S87; discussion 3S88.
50. DiMauro S, Andreu AL, de Vivo DC. Mitochondrial disorders. J Child Neurol. 2002;17(Suppl 3):3S35–3S45; discussion 3S46–3S47.
51. Canafoglia L, Franceschetti S, Antozzi C, et al. Epileptic phenotypes associated with mitochondrial disorders. Neurology. 2001;56(10):1340–1346.
52. Reuber M, Fernández G, Bauer J, Singh DD, Elger CE. Interictal EEG abnormalities in patients with psychogenic nonepileptic seizures. Epilepsia. 2002;43(9):1013–1020.
53. Shinnar S, Kang H, Berg AT, et al. EEG abnormalities in children with a first unprovoked seizure. Epilepsia. 1994;35(3):471–476.
54. Yoshinaga H, Hattori J, Ohta H, et al. Utility of the scalp-recorded ictal EEG in childhood epilepsy. Epilepsia. 2001;42(6):772–777.
55. King MA, Newton MR, Jackson GD, et al. Epileptology of the first-seizure presentation: a clinical, electroencephalographic, and magnetic resonance imaging study of 300 consecutive patients. Lancet. 1998;352(9133): 1007–1011.
56. Alehan FK, Morton LD, Pellock JM. Utility of electroencephalography in the pediatric emergency department. J Child Neurol. 2001;16(7):484–487.
57. Jan MM. Assessment of the utility of paediatric electroencephalography. Seizure. 2002;11(2):99–103.
58. Gilbert DL, Deroos S, Bare MA. Does sleep or sleep deprivation increase epileptiform discharges in pediatric electroencephalograms? Pediatrics. 2004;114(3):658–662.
59. Flink R, Pederson B, Guekhi AB, et al. Guidelines for the use of EEG methodology in the diagnosis of epilepsy (International League against Epilepsy: Commission Report). Commission on European Affairs: subcommission on European Guidelines. Acta Neurol Scand. 2002;106(1):1–7.
60. Shinnar S, O’Dell C, Mitnick R, et al. Neuroimaging abnormalities in children with an apparent first unprovoked seizure. Epilepsy Res. 2001;43(3):261–269.
61. Berg AT, Testa FM, Levy SR, Shinnar S. Neuroimaging in children with newly diagnosed epilepsy: a community-based study. Pediatrics. 2000;106(3):527–532.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
[/not-level-membership-for-pediatrics-category]
