The search for mutated genes in several hereditary forms of epilepsy affecting infants and children has resulted in the identification of epileptic channelopathies (ie, epilepsies resulting from mutations in ion channel genes). It is intuitively obvious to most readers that changes in ion channel proteins would lead to altered neuronal excitability. However, not so obvious is why a loss of the mutation function in a sodium channel would lead to epilepsy for those accustomed to managing seizures or even status epilepticus with a drug that blocks sodium channels crucial to fire an action potential. How does altered excitability in a neuronal population influence network excitability? We also evaluate how even genes that do not directly code for ion channels may participate in modifying cellular and network excitability as well as participate in processes that adversely influence neurodevelopment. We also explore how distinct mutations in the same gene may yield different syndromic phenotypes while mutations in many distinct genes may result in a similar syndrome. We also attempt to understand aspects of age-selective presentation or resolution of certain epilepsy syndromes, as well as the coexistence of epilepsy and neurodevelopmental disorders, where available information may support our explanation, formulated in part as speculative syntheses.
WHY HAVE INVESTIGATORS PURSUED A GENETIC STRATEGY FOCUSED ON RARE MENDELIAN FORMS OF EPILEPSY?
Only a small minority of seizure patients encountered in the clinic have family histories of epilepsy. Why focus on the even smaller group of patients in which pedigree analysis suggests inheritance of epilepsy resulting from effects of mutation in single genes? Investigators have chosen this approach partly out of recognition of the difficult nature of the problem. Seizures and epilepsy are complex phenomena. Seizures emerge suddenly from the brain’s interictal activity, often producing elaborate ictal behaviors and myriad consequences for development. During a seizure, normal neuronal firing patterns are replaced by synchronized rhythms that are excessively prominent and widespread. We know that the capacity to seize is latent in the normal brain—convulsions can be provoked in any child or adult as a symptom of metabolic disturbances (eg, hypoglycemia, hypocalcemia, uremia) or acute injury. What is different about the brains of people with epilepsy, who have seizures without such provocations? Furthermore, why do most syndromic forms of epilepsy exhibit distinctive developmental profiles, affecting individuals at particular periods of their lives? These are challenging questions, but we are gradually improving our understanding of these complex phenomena.
Genetics is an approach to these questions with unique advantages. Genetic research can lead to the identification of a responsible gene (or genes), even when no other biologic information about the cause of a disorder is available. So a successful genetic study offers the potential to “think outside the box” by identifying mechanisms previously completely unknown or unsuspected of being involved with a particular disease process (eg, the nicotinic receptor, KCNQ channel, and Lgi1 proteins, discussed later). Genetics is relatively unbiased by preconceptions born from prevailing models and currently available technical approaches. Such an approach is extremely valuable for scientists confronting a process as complex, heterogeneous, and poorly understood as epilepsy. Furthermore, even when the genetic form of a particular disorder is rare, there is a strong possibility that the mechanism revealed may be involved in the pathogenesis of more common forms of the disease.
Because of these potential advantages, over the last three decades efforts have been made to identify families in which epilepsy occurred in patterns suggesting Mendelian inheritance—that is, in patterns suggesting that the epileptic trait resulted from mutations in a single gene. In the last dozen years, with a burst of success aided by tools derived from the Human Genome Project, hundreds of such mutations have now been discovered (Table 3.1). Remarkably, nearly all of the mutations causing idiopathic epilepsy discovered so far lie in genes encoding ion channels, and the exceptions are primarily in genes for proteins that interact directly with channels and regulate their activity. Some types of hereditary epilepsy associated with other features, such as episodic or progressive motor dysfunction and cognitive impairment (ie, symptomatic or cryptogenic in traditional classification schemes), have also been found to be due to channel mutations. The primary and direct role of ion channels in the pathogenesis of Mendelian forms of idiopathic epilepsy has rapidly gone from hypothesis to irrevocable fact.
TABLE 3.1
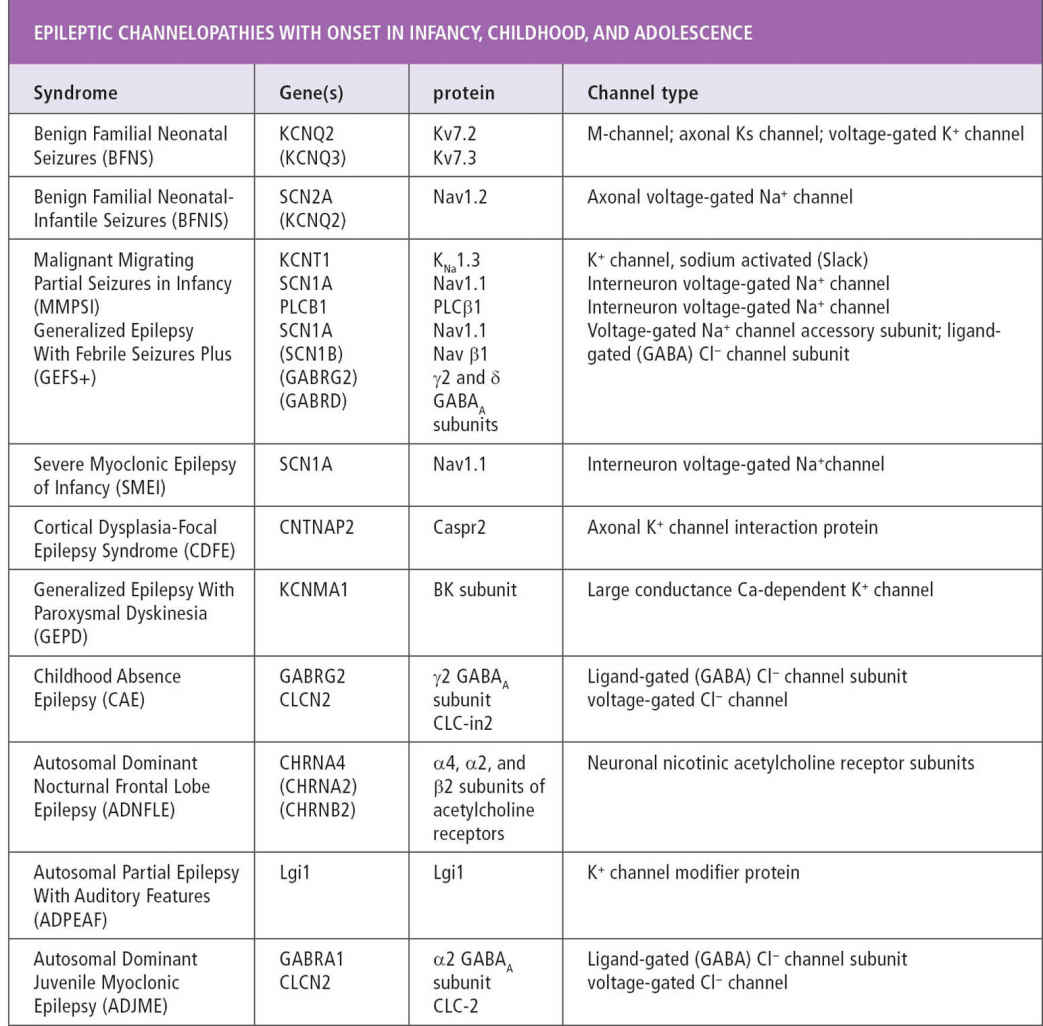
The forging of this robust conceptual link between epilepsy and channel genes occurred coincidentally with a period of revolutionary progress in our understanding of channels themselves coming from basic neuroscience. Investigators have begun to systematically explore the functions played by the newly identified “epilepsy genes.” They have used neurobiologic techniques to analyze with the requisite detail how mutations in particular channels alter the excitability of individual neurons and neural networks, ultimately resulting in epilepsy. These studies of the epileptic channelopathies are revealing important and previously unknown mechanisms controlling brain excitability at particular stages of development that represent novel targets for therapeutic drugs.
ION CHANNEL FUNCTION, STRUCTURE, DISTRIBUTION, AND DIVERSITY
What are ion channels? Ion channels are membrane proteins, tiny molecular machines about 10 nanometers in size, that allow ions to enter or leave the cell interior at very high rates—usually 106 to 107 ions per second per channel (1). These ion fluxes through individual channels cause transient, local changes in neuronal membrane potentials that represent the smallest units of brain electrical signaling. All neurons possess different classes of channels that are selectively permeable to the cations Na+, K+, and Ca+2 or the anion Cl−. Due to the activity of ATP-consuming pumps and cotransporters, Na+, Ca+2, and Cl− are usually more concentrated extracellularly and K+ more concentrated intracellularly. As a result, when channels permeable to Na+ or Ca+2 open, these ions enter the cell, depolarizing the membrane potential. When channels selective for K+ or Cl− open, the membrane is more hyperpolarized, due to K+ leaving or Cl− entering the cell interior. So, at the simplest level, Na+ and Ca+2 channel activity leads to increased excitation, and K+ and Cl− channel activity reduces excitation.
Although the function of an individual channel type may appear quite simple, groups of such channels can readily underlie complex, dynamic changes in the membrane potential, oscillations, and rhythmic activity in neurons and neuronal networks. Grouping even two channels capable of causing opposing changes in the membrane potential together in a neuronal membrane creates a mechanism for generating membrane potential oscillations. The propagated action potential, mediated by sequential opening of voltage-gated Na+ channels (depolarizing phase) and K+ channels (repolarizing and/or hyperpolarizing phase) grouped along nerve axons, is the best-studied example of such signaling.
We now know, however, that other channel groupings occur everywhere in nervous tissue. Particular channel types are deployed at specific locations, for specialized purposes. Indeed, humans possess several hundred genes encoding ion channel types that differ in key functional properties such as opening and closing rates (kinetics), the type of ions passed (ion selectivity), and the signals that bring about channel opening and closing (gating and modulation) (2). This allows different neurons to express distinct subsets of ion channels, and for different channel types expressed by a neuron to be localized at specific sites in the cell membrane with great precision. For example, some channels are expressed at synapses. Some channels are specifically expressed on dendrites, but others are found at particular locations along axons or at presynaptic terminals. Some channels are preferentially expressed on inhibitory or excitatory neurons within a brain region or on glia. Along with cell shape, cell-specific differences in ion channel expression contribute to each cell’s particular electrical “personality,” enabling neurons to perform distinctive functions in local networks and underlying the differences between the network properties of different brain regions. Importantly, the expression levels of many ion channels are not static during development—instead ion channel genes exhibit distinctive developmental profiles. One goal of current research is to improve the current fragmentary understanding of how the roles played by different ion channels change in parallel with the achievement of motor, sensory, and cognitive development milestones during infancy and childhood.
So, when we learn that an ion channel mutation causes seizures, this leads to functional questions at several levels. At the level of the channel molecule, we seek to learn how the mutation alters intrinsic channel properties such as kinetics, selectivity, gating, and modulation. At the cellular level, we want to know how the mutation affects channel abundance and distribution. At the local network level, we need to know how the mutation alters the rhythmic properties and synchronization of firing occurring in networks of neurons in particular brain regions, since it is at this level that the seizure phenotype is manifest. Finally, we seek to understand how the channels’ roles change during development, seeking clues from the clinical age of onset and natural history of the clinical syndrome.
Ion Channel Structure
Many channel mutations exert their effects by disrupting the normal structure of the encoded channel proteins. Here we introduce some key common aspects of channel structure/function relationships most relevant to the epileptic channelopathies; more detailed reviews are available elsewhere (3–6).
Cellular membranes are bilayers with polar surfaces facing the aqueous cytoplasmic and extracellular spaces and a ~50 angstrom thick, hydrophobic core (Figure 3.1A). Although seemingly thin and fluid, the oily core of biologic membranes represents a formidable barrier for ions, which are even tinier (~1 angstrom) and highly charged. Furthermore, ions in solution attract an admiring “entourage”—a surrounding shell of water molecules that are also repulsed by the hydrophobic core of the membrane. Thus, transport across the membrane requires each ion to be sequentially bound by the channel, dehydrated (stripped of surrounding water molecules), carried across the inhospitable membrane core, rehydrated, and released. Design constraints require all this to happen very quickly. It takes the electrical charge from about 10,000 ions to depolarize a 1 μm2 patch of membrane by 100 mV (the height of a typical action potential). For this to occur within a millisecond, ions must enter this small area of the membrane at a rate approaching 10 million/sec. Ion channels routinely achieve this amazing feat, but how?
Ion channel polypeptides traverse the membrane several times, and assemble into proteins shaped like bagels (or doughnuts) embedded at their midsection in the membrane (Figure 3.1). The surfaces of the channel that face the extracellular and intracellular aqueous solution are hydrophilic and may contain charged residues and sugars that attract ions, whereas the circumferential band touching the oily membrane core is made up of hydrophobic amino acid residues. Ions traverse the membrane through a centrally located pore (analogous to the doughnut’s hole). The channel pore is water-filled, except for a narrow segment, the “selectivity filter.” The selectivity filter possesses a row of sites where individual ions are briefly bound and released. The selectivity filter is equally accessible to ions from the intracellular and extracellular sides of the membrane. The net direction of flow is influenced by the fact that randomly moving ions are more likely to access the filter from the side where they are more highly concentrated, and less likely to return once released from the side where they are more dilute. In addition, once ions enter the channel pore, they come under the influence of the electrical potential difference between the two sides of the membrane—Na+, Ca2+, and K+ are attracted toward the negative side of the membrane and Cl– toward the positive side. The balance between these concentration and voltage-driven forces determines the direction of the net ion flow.
In addition to the pore-forming parts themselves, all ion channels have additional components that control the opening and closing of the ion pathway. Regulatory domains include “voltage-sensors” embedded in the membrane (Figure 3.1B). Alternatively, some channels have parts that bind and respond to intracellular second messengers and metabolites (eg, Ca2+, cAMP, ATP, or polyamines), and extracellular sites for binding neurotransmitters. The best understood regulatory mechanism is that of voltage-gating mechanisms common to Na+, K+, and Ca2+ channels, which has been revealed through detailed studies combining electrophysiology and x-ray crystallography. A series of positive charges clustered on one transmembrane segment (S4) cause it to move inward or outward, depending on which side of the membrane is more electronegative (Figure 3.1B). In the closed state (an inside-negative membrane potential), the S4 is pulled inward, forcing the pore-lining part of the channel into an angled position, so that the inner mouth of the pore is too small for ions to pass (Figure 3.1B, left). When the membrane is depolarized, the voltage sensor moves toward the extracellular space, pulling the pore-lining residues away from the center axis of the pore and opening the inner pore mouth sufficiently to allow ions to flow (Figure 3.1A and 3.1C).
Ion Channel Diversity and Nomenclature
Successive waves of technical advance over the last 20 years have left a wake of very confusing nomenclature for ion channel currents, proteins, and genes. First, electrophysiologic methods of improved sensitivity allowed previously overlooked subtypes in the channel currents to be recorded from neuronal membranes. This led to a new nomenclature subclassifying various voltage- and ligand-gated channel types based on their kinetics, pharmacology, and other functional properties. Next, the advent of methods for isolating mRNAs from neural cells led to the identification of a large number of cDNAs encoding channel subunits. Finally, the cloning of the genomes of humans and various model organisms led to the identification of many additional channel genes. The process of correlating the channels actually expressed in neurons with specific genes and proteins is typically slow and painstaking, in part because most native channels are heteromeric (ie, formed by the coassembly of the protein products of two or more different genes). Only rarely do channels appear to be homomeric (the term used by specialists to describe channels formed by multiple copies of the same subunit type). As a result, the exact subunit composition of most of the channels expressed in most brain neurons is not yet known—particularly in humans. The changes in channel composition and expression that occur during normal human development and in disease states are even less understood—with a few exceptions, such developmental changes have not yet been studied.
Although much remains to be learned about the function of particular channel genes, genomics has revealed how ion channel diversity has been generated, and, by implication, why. All human channels are derived from a very small number of ancestral genes, already present in the genomes of ancestral prokaryotes a billion years ago (1,2,7). Just as genes are subject to random mutations, it is now known that all genes are subject to random duplication, albeit very rarely, as the result of errors occurring during meiosis; indeed, the rate of such gene duplications is estimated to be one per hundred million years (8,9). Each gene duplication event creates a “surplus” gene that is relatively free to evolve new functional properties, since the original gene is still present and performing its essential functions. In the vast majority of cases, a duplicate gene does not evolve a novel useful function, but instead accumulates inactivating mutations over time and, eventually, disappears from the genome. Rarely, the duplicate gene evolves a new function that provides a survival advantage to the organism—in such cases, both duplicate genes become “fixed” in the genome. The related genes produced by duplication (called “paralogues”) can then themselves be subsequently duplicated. The human ion channel families are incredibly large—for example, one such family consists of 80 K channels, 10 voltage-gated Na channels, 11 voltage-gated Ca2+ channels, and an additional 42 cation channels of various classes, all derived from the same ancestral K+ channel (2)! The two other major families of channel genes, encoding voltage-gated Cl− channels and ligand-gated channels, were produced by the same process of duplication and subsequent functional divergence (10–12). Because a duplicated neuronal channel gene must provide a survival advantage to first establish itself in its species of origin and remain stably present over the course of evolutionary time, the gene’s survival alone is an indication that a duplicate gene has acquired a needed, novel role. That role may be restricted, for example, to particular neuronal cell types or to a particular stage of development. Many of these roles remain poorly understood. Indeed, analysis of the phenotypes produced by disease-causing mutations in individual channel genes is an important source for clues regarding these unknown functions.
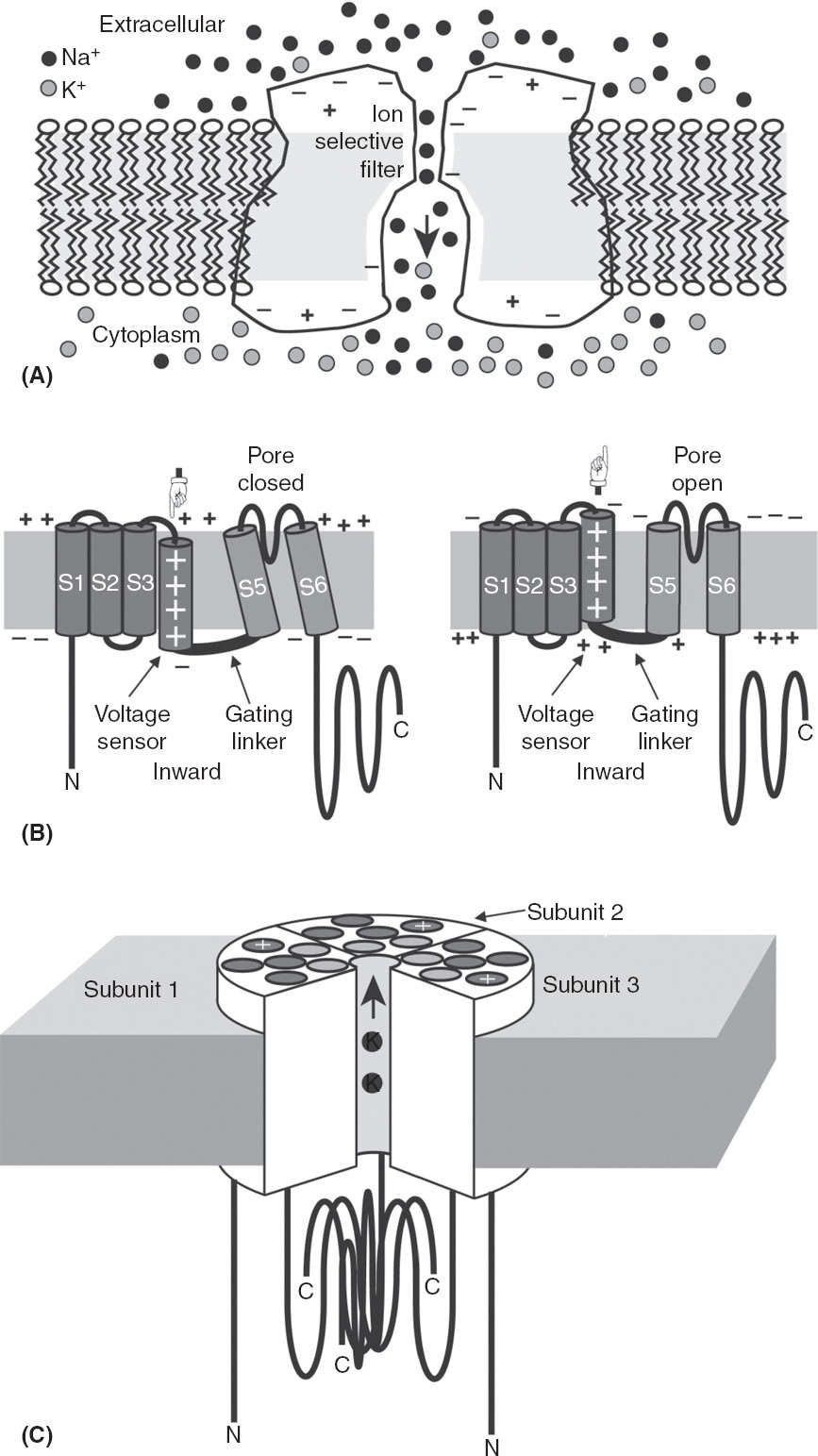
FIGURE 3.1 Structure and gating mechanism of representative ion channels. (A) Cartoon showing a cross-sectional view through the center of a model ion channel. The channel has hydrophobic surfaces facing the membrane lipid core (grey area) and hydrophilic surfaces with charged residues (indicated by +’s and –‘s) facing the internal and external solution and lining the centrally located transmembrane ion path. At the narrowest part of the pore, permeant ions bind preferentially to the “ion selective filter.” The channel depicted is selectively permeable to Na+ ions, which are shown moving through the channel from the extracellular solution to the cytoplasm. (B) Transmembrane topology, functional subdomains, and molecular movements of a voltage-gated K+ channel subunit. The subunit polypeptide has intracellular amino and carboxy terminal domains, and six segments (S1, S2 …) that traverse the membrane. S1–S4 form the voltage-sensor domain. S5–S6 and the loop that connects them, which is folded partly into the membrane, form the pore domain. The S4 domain possesses positively charged residues (indicated by +’s), and as a result undergoes molecular movements in response to changes in the transmembrane electrical potential, moving outward when the membrane is depolarized (indicated by pointers). Outward movement of the S4 domain pulls the pore at the cytoplasmic end of S5–S6, allowing ion flow (right panel). (C) Cartoon showing K+ channel subunits arranged around the central pore of an assembled channel—the fourth (front) subunit has been removed to reveal the ion path. Positions of transmembrane segments are indicated as in B (grey-S5 and S6; dark grey-S1 to S4).
EPILEPTIC CHANNELOPATHY SYNDROMES
A first expectation, based on earlier studies of channel defects in hyperexcitability disorders of skeletal muscle (eg, myotonia) and heart (long QT syndrome), would be that channel mutations leading to epilepsy would be associated with increases in Na+ and/or Ca+2 channel activity (since such activity leads to membrane depolarization) or with reductions in K+ or Cl− channel activity (since these currents prevent membrane depolarization) (13,14). As discussed later in this chapter, we have learned that this expectation is often, but not always, fulfilled. To facilitate further reading, we have provided the listing for each disorder in the National Library of Medicine’s online Mendelian Inheritance in Man database (www.ncbi.nlm.nih.gov/omim), which is frequently updated. As the diversity of genotypes producing a common phenotype is being recognized, the MIM entries have acquired a number following the abbreviation for the syndrome (for example, GEFS+ has entries ranging from 1 to 8).
Channel Mutations in Epilepsies With Onset in Neonates and Infants
Benign Familial Neonatal Seizures (BFNS, MIM 121200). Benign familial neonatal seizures (BFNS, also termed Benign Familial Neonatal Convulsions) is a syndrome characterized by recurrent, brief, generalized, or partial seizures that begin on about the fourth day of life and remit after 1 to 3 months (15). In the classic syndrome, first described by Rett and Teubel (16), infants otherwise grow and develop normally, without subsequent epilepsy, although affected persons carry a 10% to 16% risk of experiencing one or more seizures again later in life (15,17). In 1989, Leppert et al linked the disease gene to chromosome 20 (18). Later, investigators identified additional families linked to chromosome 8, a first example of genetic heterogeneity in epilepsy (19,20).
The two BFNC genes on chromosome 20 and 8 encode highly homologous potassium channel subunits, KCNQ2 and KCNQ3 (21–23). These neuronal channel subunits are relatives of a cardiac channel, KCNQ1, which previously was shown to be mutated in both dominantly and recessively inherited forms of the long QT syndrome (24,25). Four KCNQ1 subunits combine to form the channels in the heart. In neurons, some channels are KCNQ2 tetramers, but many are composed of a combination of KCNQ2 and KCNQ3 subunits (26–31). Some disease-causing missense mutations in KCNQ2 and KCNQ3 are associated with only modest reductions (20%–30%) in current magnitude (32). This may imply the critical dependence of neuronal excitability on the absolute magnitude of KCNQ2/KCNQ3 potassium channel activity, especially during the neonatal period. KCNQ channels underlie an extensively-studied potassium channel found in central and autonomic neurons, known as the M-channel (26). The name “M-channel” comes from the fact these channels are important effectors for acetylcholine acting via muscarinic receptors. Indeed, many neurotransmitters that activate intracellular G proteins increase neuronal excitability by reducing the openings of KCNQ channels (33). These channels open occasionally at the resting membrane potential and are slowly activated by membrane depolarization. Because of their slow kinetics, M-channel activation causes a period of membrane hyperpolarization after a cell receives excitatory input. As a result, neurons expressing M-channels tend to fire one action potential after receiving excitatory input, then become transiently quiescent. Inhibition of the M-channel by neurotransmitter receptors causes these neurons to become slightly depolarized and to fire multiple action potentials rhythmically after receiving excitatory inputs. Thus, M-channels may serve as a general brake on excess neuronal excitation, which can be selectively removed when the cell receives input from neurotransmitters such as acetylcholine.
KCNQ2 subunits are widely expressed in circuits implicated in generation of epileptic seizures (30,34,35). Interestingly, one conspicuous network localization of KCNQ2 expression is in the inhibitory neurons important for synchronization of the firing of excitatory neurons, including the reticular thalamic neurons and several populations of interneurons in the hippocampus and cortex (34). A second important localization of KCNQ2 and KCNQ3 is at the cellular level—at the spike initiation zone at the beginning of axons, including those of pyramidal cells in the hippocampus and neocortex (30,36). This localization may allow them to powerfully control the neuronal threshold, since each neuronal action potential originates in the proximal axon (37).
The near remission of BFNS may coincide with the ontogeny of more robust GABAergic inhibition accompanying the completion of transition of the chloride transporter from the neonatal form (NKCC1, which favors intracellular Cl− accumulation) to the mature form (KCC2, which favors Cl− efflux). To clarify the age-dependent etiology of BFNS, Okada and colleagues determined age-dependent functional switching of KCNQ channels, GABAergic, and glutamatergic transmission in rat hippocampal slices using multi-electrode arrays and specific inhibitors for those channels (38). KCNQ-channel inhibition shortened spike-frequency adaptation, but this stimulation was more predominant during P < 7 than P > 14. The authors suggested that the pathogenic mechanisms of age-dependent development and spontaneous remission of BFNS are, at least partially, associated with the interaction between age-dependent reduction of inhibitory KCNQ-channel activity and age-dependent functional switching of the GABAergic system from excitatory to inhibitory action in neonatal CNS.
Early Infantile Epileptic Encephalopathy (EIEE7, MIM 602235). A very severe form of early epileptic encephalopathy of the newborn, resembling Ohtahara syndrome (seizures in the first week of life accompanied by suppression-burst pattern in the EEG) and attributed to mutations in the KCNQ2 gene, has been described by Weckhuysen and colleagues. (39). Further description of KCNQ2 encephalopathy resembling the Ohtahara syndrome encountered in centers in Japan followed (40). These patients often exhibit tonic seizures in the first days of life with burst suppression EEG. In most of them, seizures were responsive to treatment with carbamazepine or other common AEDs such as topiramate, zonisamide, or even phenytoin. However, neurodevelopment was severely affected.
Unlike BFNS, in which autosomal dominant inheritance was common, this form of encephalopathy was consistently attributed to de novo mutations. Further, unlike BFNS, KCNQ3 mutations have not been identified in this syndrome. The KCNQ2 mutations found in EIEE patients were missense changes in highly conserved regions, whereas frameshift or nonsense mutations in KCNQ2 have been more frequently described in patients with BFNS (41,42). These findings support an intuitive understanding that an inherited mild loss of function mutations results in BFNS, while severe loss of function leads to EIEE. Not so obvious is the apparent contradiction between the relative ease of seizure control in this subset of EIEE cases and the poor developmental outcome.
More recent findings have challenged our hypothesis further. There have been recent reports of gain of function mutations associated with this gene in some patients (43,44). It would be expected that an increase in M current would dampen neuronal excitability, and this paradoxical finding seems to imply that the networks function optimally with a “Goldilocks-like” preference for the M current amplitude. It has been postulated by the authors that perhaps this effect somehow impacts interneurons more than pyramidal cells. Another interesting finding was that when a specific mutation (p.A294G, mutation located on the S6 segment of the Kv7.2 pore domain) encountered in a patient was induced in neurons, it resulted in a mislocalization of the heteromeric mutant channels to the somato-dendritic compartment in contrast to the normal axon initial segment localization (45). The authors concluded that disease severity is not necessarily a consequence of a strong inhibition of M current and that additional mechanisms such as abnormal subcellular distribution of Kv7 channels could be the determinant.
Ezogabine (retigabine) is a novel anticonvulsant with the ability to enhance M currents (Kv7.2/3 currents) (46,47). It was approved for clinical use in the United States in 2011. There was considerable enthusiasm to study it in these KCNQ2-related disorders, but reports of blue eye and skin discoloration in patients taking retigabine derailed further investigations. The discovery of a subset of patients with gain of function mutations raises some caution about the application of this concept even if an M-current enhancer with a cleaner safety profile were to be discovered.
Early Infantile Epileptic Encephalopathy (EIEE14, MIM 614959). This syndrome is more commonly identified as malignant migrating partial seizures of infancy. The diagnostic criteria for MMPSI suggested by Coppola et al included normal development before seizure onset, onset before age 6 months, migrating focal motor seizures at onset, multifocal seizures becoming intractable and treatment-resistant, profound psychomotor delay, and no identifiable etiology (48). No discernible inheritance pattern was encountered and MRIs early in the course of this disorder were unremarkable. Using the whole exome sequencing approach, de novo heterozygous mutations in the gene KCNT1 were identified as causative in 3 probands (49). KCNT1 (also known as SLACK) is a sodium-activated potassium (KNa) channel. KCNT1 is widely expressed in the nervous system. It can form homomeric channels as well as heteromeric channels by interacting with SLICK channel proteins coded by KCNT2. SLACK channel activity contributes to the slow hyperpolarization that follows repetitive firing (reminiscent of Kv7.2/3 channels). No KCNT2 mutations were identified in the MMPSI cohort studied by Barcia et al (49). KCNT1 regulates the rate of bursting and enhances the accuracy with which action potentials lock onto incoming stimuli (50). The KCNT1 protein represents the largest known potassium channel subunit. The C-terminal cytoplasmic domain interacts with a number of proteins, including the fragile X mental retardation protein (FMRP), which is a potent stimulator of KCNT1 channel activity (51). The demonstration that FMRP, defects in which cause a common form of intellectual disability, interacts with a membrane protein provides an intriguing link between ion channel rhythms and intellectual function. It is known that activation of SLACK channels contributes to the firing patterns of a wide variety of neurons.
The widening of genotypes associated with EIEE includes EIEE13, attributed to mutations in SCN8A, which codes for the Nav1.6 channels (52), while a range of phenotypes associated with SCN8A encephalopathy, including MMPSI, have been described (53). An interesting demonstration of the interplay among channels was demonstrated in a double mutant murine study in which heterozygous mutations in both SCN1A and SCN8A had been introduced (54). In this study, the SCN8A(med-jo/+) mice rescued the seizure severity of SCN1A(+/-). The SCN8A mutation also extended the life span of both SCN1A(+/-) and SCN1A(-/-) mice. The potential relevance of this finding to therapeutics is intriguing.
Benign Familial Neonatal-Infantile Seizures (BFNIS; MIM 607745). Shevell et al (55) and Kaplan and Lacey (56) first described families in which nonfebrile seizures, like those seen in BFNS but with a somewhat later onset ranging from the first week of life to 3.5 months, were inherited in an autosomal dominant pattern. This delayed onset led to the suspicion of a distinctive etiology, which was affirmed when the two BFNS loci on chromosomes 8 and 20 were excluded by genetic linkage analysis, and another location on chromosome 2, harboring a cluster of genes encoding several Na+ channel genes, was implicated (57–59). Screening of the chromosome 2 genes in BFNIS patients revealed mutations in SCN2A, encoding the Na+ channel subunit, Nav1.2.
Cell biologic studies using rodents indicate that Nav1.2 channels play an important, transient role on neuronal axons in the developing brain and peripheral nerve (60). In rats and mice, Nav1.2 is at birth the predominant sodium channel at the spike initiation zone in the proximal axon (ie, the axonal initial segment), but it is largely replaced at this location during the second and third postnatal week. A similar Nav1.2 to Nav1.6 switch occurs at the nodes of Ranvier of many newly myelinating fibers. Based on studies of other proteins, it seems likely that similar subunit switches occur in humans, but over a slower time period of several months (61). The observation that the channels mutated in BFNS and BFNIS are expressed in similar axonal locations during early development is certainly intriguing—as to why KCNQ2/3 mutations would produce an earlier onset than SCN2A mutations, that may be related to the findings of Okada et al (38) that the onset and remission of BFNS highlights the importance of KCNQ channels in providing inhibition before the switch in GABA-mediated currents from excitatory to inhibitory, with the ontogeny of the mature form of the Cl− transporter.
Generalized Epilepsy With Febrile Seizures Plus (GEFS+1, MIM 604233; GEFS+3, MIM 611277; GEFS+4, OMIM 609800; GEFS+6, MIM 612279; GEFS+7, MIM 613863; GEFS+8, MIM 613828), Severe Myoclonic Epilepsy of Infancy (SMEI; MIM 607208), Intractable Childhood Epilepsy With Generalized Tonic–Clonic Seizures (ICEGTCS; 607208). Febrile seizures occur in 3% to 5% of all children. When febrile seizures occur in isolation, between age 3 months and 5 years, in children who are otherwise developing normally, the risk of later epilepsy is low. For a minority of cases, however, febrile seizures herald epilepsy of a variety of forms and degrees of severity.
In a seminal clinical study, Scheffer and Berkovic (62) described an exceptionally large multigenerational family in which typical febrile seizures of infancy and early childhood were common, but a variety of nonfebrile seizure types also occurred. This spectrum of other seizure types represented in the pedigree included nonfebrile convulsions, absences, myoclonic seizures, atonic seizures, and severe myoclonic-astatic epilepsy with developmental delay. Scheffer and Berkovic argued that this heterogeneity represented variable phenotypes associated with the dominantly inherited trait, and introduced the phrase generalized epilepsy with febrile seizures plus (GEFS+) to describe the new seizure syndrome. They further noted that such phenotypic heterogeneity would make this syndrome difficult to detect in smaller families, obscuring its frequency in the clinic. They concluded that molecular studies of large pedigrees, such as the ones they had analyzed, might reveal the underlying genetic mechanism(s). Follow-up studies by the authors and others have confirmed all these predictions.
Mutations in four channel genes have thus far been identified in patients with GEFS+. By far the largest numbers of them are in SCN1A, while mutations in other channel genes have also been encountered. Several mutations have been found in SCN2A, SCN1B (encoding, a voltage-gated Na+ channel single-transmembrane accessory subunit), and GABRG2 and GABRD (encoding GABA receptor subunits).
The single mutation identified in Nav β1 (SCN1B), C121W, has been described in two Anglo-Australian pedigrees (63,64). C121 is located in the extracellular domain of Nav β1, where it forms a disulfide bridge that plays a critical role in the proper assembly of the extracellular domain into an immunoglobulin-like folding pattern (65). Nav β1 subunits have multiple roles, enhancing surface expression of the pore-forming α subunits, participating in protein–protein interactions within the extracellular space that are important for channel clustering at key sites such as the nodes of Ranvier, and enhancing rates of channel inactivation (66). Careful biochemical and electrophysiologic studies using coexpression with neuronal α subunits in mammalian cells have revealed two rather subtle effects of C121W (67). The mutation prevents protein–protein interactions mediated by the extracellular domain of Nav β1 and shifts inactivation to hyperpolarized potentials, thereby increasing the number of channels available for opening. The SCN1A mutations tend to represent loss of function mutations; as these channels are expressed in GABAergic interneurons, the net effect of principal cells is excitatory. The SCN2A channels are expressed in the axon initial segment of principal neurons, and thus gain of function mutations in this genes produce a net increase in excitability.
The discovery of a second GEFS+ locus on chromosome 2q (68,69), a region that harbors a cluster of three genes encoding sodium channel α subunits, led to a search for mutations within these genes in GEFS+ pedigrees. Sequencing of patient DNA revealed many different mutations in SCN1A and a single mutation in SCN2A, all resulting in single amino acid substitutions (missense mutations) in the resulting α subunit polypeptides (70–74). Interestingly, these studies have revealed a high frequency of partial seizures in members of some pedigrees that otherwise fit the original GEFS+ profile (64,73,74). The electrophysiologic properties of many of the mutant α subunits have now been studied fairly intensively in heterologous cells. A subset of the mutations show defective inactivation mechanisms, as observed first in studies of the C121W mutation in β1 discussed previously. For example, Lossin et al found three mutations that exhibited a persistent current during prolonged depolarizations due to an apparent failure of inactivation gating (75). However, other mutations seem to show reduced current amplitudes or changes in gating that are predicted to reduce current flow, without a defect in inactivation (76,77). Thus, only some GEFS+ mutations appear to be associated with enhanced channel activity similar to the mutations in the heart and skeletal muscle Na+ channels that cause disorders of hyperexcitability and excessive synchrony (myotonia, long QT syndrome) in those tissues (78,79).
This issue has been further complicated by genetic and clinical evidence that an additional epilepsy syndrome, severe myoclonic epilepsy of infancy (SMEI), represents a severe form of the GEFS+ spectrum (80–82). SMEI, first described by Dravet in 1978, is a progressive syndrome of refractory seizures and cognitive decline. After a normal early infancy, patients present with prolonged generalized or unilateral febrile seizures at 5 to 12 months of age. Other seizure types, including myoclonic, absence, partial, and atonic seizures, begin between 1 and 4 years of age. Psychomotor slowing becomes apparent during the second year of life, and the patient may also experience obtunded states associated with myoclonus, as well as develop ataxia, pyramidal dysfunction, and refractory generalized tonic–clonic and partial seizures. Intellectual outcome is generally poor. Although a family history of epilepsy had been previously recognized as common in SMEI, the nature of the seizure disorder in family members had not been systematically explored until recently. Singh et al (80) described 12 SMEI pedigrees within which other family members exhibited forms of epilepsy consistent with the GEFS+ spectrum. Subsequent screening of additional SMEI patients for mutations in SCN1A has identified more than 100 additional mutations. The majority of SMEI mutations are frame-shift, nonsense, or splice-donor mutations predicted to result in markedly aberrant, truncated forms of the channel protein. In contrast to the modest changes in gating seen with GEFS+ mutations causing single amino acid changes, the SMEI truncations would be expected to produce proteins incapable of conducting ions. Most recently, an additional clinical syndrome, intractable childhood epilepsy with generalized tonic–clonic seizures (ICEGTCS), which is quite similar to SMEI except that patients do not show myoclonic seizures, has also been shown to result from mutations in SCN1A (76,83).
These clinical and genetic observations are perplexing, and lead to many questions concerning genotype–phenotype relations in the GEFS+/SMEI patients. How could gain-of-function mutations (ie, the impaired inactivation seen in some GEFS+ patients) cause a mild syndrome, if loss-of-function mutations cause a severe phenotype (in SMEI and ICEGTCS). Cell biologic studies are beginning to shed light on this. In early studies, SCN1A was described as widely expressed by excitatory and inhibitory neurons, with Nav1.1 proteins observed on neuronal somata and proximal dendrites (84). Very recent experiments are revising this view, however. In the hippocampus, both the protein and mRNA are reportedly much more expressed in inhibitory neurons than in excitatory pyramidal cells, leading to the suggestion that the severe phenotype of null mutations reflects a simple loss of inhibition (85). Furthermore, careful immunohistochemical studies have revealed Nav1.1 channels clustered in the initial segments of some hippocampal neurons (86). The localization and relative importance of various subpopulations of Na+ channels is incompletely understood, but is evolving. Studies using transgenic mouse models that allow mutant channels to be studied in vivo have suggested that when some of the SCN1A mutations causing Dravet syndrome (DS, SMEI) are expressed in mice, there is a preferential localization to GABAergic interneurons (87,88). This has led to the explanation of this syndrome as an interneuronopathy, such that deficient Nav1.1 currents in GABAergic interneurons produced disinhibition of the principal neurons. Such an explanation is consistent with the observed exacerbation of seizures in DS patients by sodium channel antagonist medications such as carbamazepine or lamotrigine (89).
Beyond knock-in studies of human mutations in transgenic mice, Liu and colleagues from Dr. Parent’s laboratory have applied the induced pluripotent stem cell (iPSC) technology to derive forebrain-like pyramidal and bipolar-shaped neurons from patients with DS (90). Surprisingly, DS patient-derived neurons showed increased sodium currents in both bipolar and pyramidal neurons, with both types of cells displaying spontaneous bursting. Further, mimicking the clinical situation, there was no difference in the sodium currents between control neurons (also iPSC derived, from control patients) and DS patient-derived neurons when the currents were measured at 5 to 7 days, but the DS-derived cells had increased two-fold by 3 to 5 weeks in culture. Clearly, this is a very different finding from the transgenic mouse models, and is not readily consistent with the observation of seizure exacerbation by sodium-channel antagonists. The authors have speculated that increased excitability of both principal and inhibitory interneurons leads to hyperexcitability and synchronization to result in epilepsy and cognitive dysfunction. It is important to recognize that the iPSC studies are in neurons in culture, such as a hippocampal slice preparation from a transgenic mouse, rather than neurons in a network.
Screening of large clinical populations suggests, however, that mutations in SCN1A, SCN2A, and SCN1B account for only about 20% of familial cases of GEFS+ and a far lower percentage of cases of idiopathic generalized epilepsy (71,72). This implies that other genes may be involved, and the search has begun. Recently, mutations in the GABRG2 and GABRD genes that encode the γ2 and δ subunits of GABAA receptor were also identified as causes of GEFS+ (91,92). GABAA receptors are ligand-gated Cl− channels (93). GABA released at inhibitory synapses binds to sites on the extracellular side of the channel, leading to channel opening, Cl− influx, and generating a fast inhibitory postsynaptic potential (IPSP). There are 16 human genes for subunits of GABA receptors, α1–6, β1–4, γ1–3, δ, ε, and θ. Each GABA receptor is a pentameric complex, consisting of 2 α subunits, 2 β subunits, and a single subunit of class γ, δ, ε, or q. Although receptors containing a variety of different subunit combinations are represented in vivo, the most abundant and widely distributed form of the receptor is formed by α1, β2, and γ2 subunits. The known γ2 subunit mutations range widely in the severity of their effects on channel function and the associated clinical phenotype. One mutation (R43N), found in a pedigree with a relatively mild seizure phenotype (febrile seizures and childhood absence epilepsy), introduces a point mutation in the site for channel modulation by benzodiazepines such as diazepam and lorazepam (94). R43N mutant channels expressed in Xenopus oocytes exhibited only 10% reductions in GABA current amplitudes, though responsiveness to benzodiazepines was abolished completely. A second missense mutation (K289M) in an extracellular loop between the M2 and M3 transmembrane segments was associated with a ~90% reduction in GABA currents and a more severe phenotype of febrile seizures and afebrile generalized tonic–clonic seizures (91). Finally, a truncation mutation in GABRG2 was found in a patient with SMEI within a branch of a large GEFS+ pedigree with bilineal inheritance of epilepsy (95). When expressed in oocytes or cultured mammalian cells, this truncation mutation (Q351X) acted as a potent dominant negative, coassembling with normal GABA receptor subunits and preventing them from leaving the endoplasmic reticulum (95). Beyond these direct effects on GABA receptor functioning, three of the GEFS+ mutations have been shown to impair channel transport to the membrane when expressed in cells cultured and maintained at an elevated temperature (40°C); by contrast, wild-type channel trafficking did not show temperature dependence (96).
A fourth GABRG2 mutation was found in a small pedigree with febrile seizures and no other forms of epilepsy (97). The mutation described (R139G) was located in the extracellular portion of the subunit, near the location where benzodiazepines bind and thereby enhance channel openings. R139G had a slight effect on function, speeding the rate of channel closing after an opening elicited by GABA. Although the numbers of families is very small, so far there is a direct correlation between the severity of epilepsy and the severity of the effects on channel gating observed in the four known GABRG2 mutations.
A single GEFS+ pedigree with four affected patients has been linked to a mutation in GABRD. Channels including the d subunit encoded by GABRD are located at nonsynaptic sites on the soma and dendrites (98). Although δ-containing receptors are present at low density, they make important contributions to regulation of neuronal responsiveness because of their long-lasting openings (99).
Recessive Cortical Dysplasia-Focal Epilepsy Syndrome (CDFE; MIM #610042). An Amish family was described in which nine children were affected by a syndrome of cortical dysplasia, focal epilepsy, relative macrocephaly, and diminished deep-tendon reflexes (100). Intractable focal seizures began at 14 to 20 months of age. Subsequently, autism-like features, including language regression, hyperactivity, impulsive behavior, and mental retardation, developed in all children. Attempts to treat the refractory epilepsy by surgical removal of focal dysplasic cortical regions did not prevent the recurrence of seizures. Pathologic studies of dysplasias showed abnormal neurons, altered neuronal migration, and gliosis. All affected children were found to be homozygous for a mutation in CNTNAP2, encoding the protein contactin-associated protein-like 2 (Caspr2). Caspr2 is a single transmembrane protein possessing a large extracellular domain with multiple protein–protein interaction modules. Remarkably, Caspr2 is localized on axonal membranes at the axon initial segments and the nodes of Ranvier (101,102). Caspr2 is required for targeting the Kv1 subclass of voltage-gated potassium channels to these important axonal subdomains (103–105).
The Kv1 channel pore-forming subunits are encoded by KCNA genes. Six genes, KCNA1-6, are known to be expressed in the CNS, produced by gene duplication from a single ancient, ancestral gene. Indeed, these genes are the homologues of a single fly gene, called Shaker. Flies with mutations in Shaker are hyperactive when undisturbed and, if exposed to low levels of anesthetic vapors, develop rapid shaking movements of their wings, legs, and abdomen that prevent normal behavior (106). Recordings from Shaker flies show failure of action potentials propagated along neuronal axons to repolarize normally (107). In mammals, the role played by Kv1 channels on axons undergoes a developmental switch during myelination (104,108). Kv1 channels arrive near the node as it begins to form, and play a key role in axonal action potential repolarization during the early stages of myelination. Once myelination is complete, Kv1 channels have a diminished role at the nodes, because they are sequestered beneath the myelin sheath. However, they are also present on unmyelinated portions of the axon both proximally (ie, the initial segments) and distally (at presynaptic terminals), and in these locations contribute to control of initiation and neurotransmitter release (101,109). The basis for epilepsy based on recessive inheritance of Caspr2 mutations is not established, but the connection with axonal excitability is particularly provocative in view of the better established axonal roles of Nav and KCNQ subunits mutated in neonatal and infantile forms of epilepsy. This family study is also the first human example of recessive inheritance of epilepsy involving channel regulation. A large number of recessive epileptic channelopathies have been identified in inbred strains of laboratory mice (110); the relative lack of such mutations detected in human epilepsy reflects the uncommonness of consanguinity required for the rare pathogenic mutations to become symptomatic, and the difficulty in identifying mutations in small families.
Later research identified CNTNAP2 as an autism-susceptibility gene by linkage and gene-expression analyses, as well as demonstrated that CNTNAP2 was enriched in circuits important for language development (111). Studies using fMRI in children possessing at-risk alleles for CNTNAP2 revealed a diffuse pattern of left frontal lobe connectivity, compared to a more restricted pattern in nonrisk controls (112). CNTNAP2 protein has also been shown to play a role in the stabilization of new spines in dendrites in a study using CNTNAP2-/- knock-out mice (113), which may be related to the interaction of CNTNAP2 protein with the scaffold protein calcium/calmodulin-dependent serine protein kinase (CASK) (114). In hippocampal cultures, knockdown of CASK reduces spine density in neurites (115).
Axonal Hyperexcitability: An Emerging Theme in the Hereditary Epilepsies of Neonates and Infants. From the previous discussion, it is clear that recent evidence has linked KCNQ channels, Nav1.2, Nav1.1, and Caspr2 mutations to forms of epilepsy involving neonates and/or infants. In parallel, cell biologic studies have revealed that each of these proteins have major roles in regulation of intrinsic axonal excitability in the developing nervous system. It seems clear that in these early onset forms of epilepsy, disturbance of the ion channels that mediate the actual initiation and firing of action potentials is centrally important. It is not yet known why, for example, mutations in KCNQ2 should cause onset of seizures at an earlier age than mutations in Nav1.2. Further experiments should help to clarify the specific age-dependent phenotypes associated with various mutations and, in the process, identify the precise functions played by these channel types.
Channel Protein Interactions With Processes Beyond Neuronal Excitability. The other important emerging theme is the association of the interaction of ion-channel proteins with processes involved in neural plasticity, as exemplified by the previously mentioned interaction of the KCNT1 (SLACK) channel with the FRMP protein, and the role of CNTNAP2 in mediating plasticity in language-specific regions of the forebrain by stabilization of spines and synapses.
Channel Mutations in Epilepsies With Onset in Childhood and Adolescence
Autosomal Dominant Nocturnal Frontal Lobe Epilepsy (ADNFLE; MIM #600513). In 1994, Scheffer et al described a new epilepsy syndrome characterized by clusters of brief nocturnal motor seizures (116,117). The attacks began in childhood (median age, 8) and persisted throughout life, often misdiagnosed as night terrors, nightmares, hysteria, or paroxysmal nocturnal dystonia. Video EEG recordings revealed that the nocturnal attacks were epileptic seizures heralded by frontally predominant sharp and slow wave activity (117), usually arising from stage 2 nonREM sleep. In spite of improved understanding of the disorder, clinical differentiation from parasomnias and other paroxysmal disorders of sleep can be challenging and may require video-EEG monitoring and polysomnography (118,119).
Initial analysis of ADNFLE pedigrees suggested autosomal dominant inheritance with incomplete (~70%) penetrance. Subsequent molecular genetic studies have revealed mutations in genes for three subunits of neuronal nicotinic acetylcholine receptors (nAchRs) in affected individuals: CHRNA4 on chromosome 20, CNRNB2 on chromosome 1, and CHRNA2 on 8 (120–124).
Central nAchRs are pentameric cation channels, closely related to the well-studied nAchRs at skeletal muscle endplates, but are composed of α and β subunits expressed only (or at least, primarily) by neurons. Binding of acetylcholine to sites on α subunits leads to channel opening and membrane depolarization. Although there are distinct genes for seven neuronal nAchR α subunits and 3 β subunits, heteromeric channels formed by coassembly of α4 and β2 subunits appear to be the most abundant form expressed in the brain. The subunits share a common basic structure, with a large, extracellular amino-terminal domain and four transmembrane α-helices. The second (M2) helix lines the transmembrane ion pathway and is believed to undergo a twisting conformational change after acetylcholine binding that allows ions to flow.
The ADNFLE mutations in α4 or β2 described so far all result in single amino acid changes at key positions within the pore-lining helices, and might therefore be expected to cause changes in gating or ion conductance properties. Indeed, electrophysiologic experiments using the mutant subunits expressed in experimental cells such as Xenopus oocytes or mammalian cultured cells have revealed marked, albeit complex, changes in these properties (121,125,126). The α4 S248F mutant AchR responses showed faster desensitization, slower recovery from desensitization, less inward rectification, and virtually no Ca+2 permeability as compared to wild-type α4/β2 AChRs (126). Although these effects would all tend to reduce channel activity, this mutation also caused use-dependent upregulation of activity and was more strongly activated by low levels of agonist, effects that might result in enhanced activity in vivo. Studies of the two known CHRNB2 mutants show differing effects: the V287M mutation shows normal kinetics but 10-fold increased Ach sensitivity (123), whereas the V287L mutation shows normal Ach sensitivity but 30-fold slower kinetics (122). The α2 mutation, I279N, is in the first transmembrane segment (M1) that links the extracellular ligand-binding domain to the pore-lining segment (M1). Recordings of transfected cells expressing either the mutant or the wild-type receptor showed that I279N markedly increases the receptor sensitivity to acetylcholine (124).
Immunolocalization studies show that the α4 subunit is widely expressed and is localized on neuronal somata and dendrites in cortical and subcortical structures, and is particularly highly expressed by dopaminergic neurons in the substantia nigra (127). Labarca et al generated CHRNA mutant mice harboring a point mutation in the pore near the position of the ADNFLE mutations (128). This mutation caused 30-fold increased Ach sensitivity and is partially activated by choline at levels that are normally present in cerebrospinal fluid. Mice exhibited early lethality and loss of nigral dopaminergic neurons. Investigators generated two lines of transgenic mice expressing different ADNFLE mutations, and found, paradoxically, a 20-fold increase in nicotine-induced GABAergic inhibitory postsynaptic currents in cortical neurons, with no change in excitatory currents (129).
How could increased inhibition lead to seizures? Because cortical GABAergic interneurons project widely to large numbers of neighboring pyramidal cells, they are powerful synchronizers of cortical output. Neurons in basal cholinergic nuclei project to the thalamocortical circuits that mediate transitions between sleep and wakefulness; acetylcholine release in these circuits contributes to arousal and alertness (130). Excessive cholinergic responsiveness during a critical stage of desynchronization of this circuit appears to be sufficient to cause seizures. The frontal lobe predominance of the semiology and epileptic discharges in ADNFLE patients is not explained by the distribution patterns of the receptors, which are widespread, but may reflect the importance of the physiologic contribution to the critical arousal transition, disrupted in the disorder made by the frontal lobes. Indeed, the hypothesis that the release of acetyl choline is reduced during sleep initially, but the increase in acetyl choline release during rapid-eye-movement (REM) sleep may mediate seizures in the channels with gain of function, was tested by using a nicotine patch to desensitize those receptors by continuous tonic nicotinergic activation (131).
The list of mutations associated with ADNFLE has expanded, with the identification of a distinct set of mutations in the KCNT1 (SLACK) gene in four different families (132). Affected patients exhibited a severe form of ADNFLE with earlier onset and significant psychiatric comorbidities. The same investigators also found KCNT1 mutations in sporadic nocturnal frontal lobe epilepsy. The mutations in KCNT1 in ADNFLE, like in MMPSI, are gain of function mutations. An intriguing in vitro study using a Xenopus laevis expression system demonstrating a reduction in the SLACK currents in quinidine has raised tantalizing therapeutic possibilities (133).
Autosomal Dominant Juvenile Myoclonic Epilepsy (JME, EJM1, MIM 254770). Juvenile myoclonic epilepsy is characterized by the onset, usually during adolescence, of bilateral single or repetitive myoclonic jerks, occurring most commonly upon morning awakening (134). A majority of patients have at least one generalized tonic–clonic seizure, and about 15% have absences. A characteristic EEG finding is frontally predominant bilateral 4 to 6 Hz polyspike–wave discharges. JME is not uncommon, and represents approximately 4% to 6% of all patients with epilepsy (134). Nevertheless, identifying genes for JME has been particularly difficult, likely due to the genetic heterogeneity and complexity underlying the syndrome. Recently, mutations in genes for a GABA receptor subunit and a subtype of voltage-gated Cl− channel were identified.
Cossette et al ascertained a large family in which epilepsy, with characteristic clinical and electroencephalographic features of common JME, was inherited in an autosomal dominant pattern (135). A missense mutation was found in GABRA1, resulting in an A322D change in a conserved site in the third transmembrane helix of the GABAα1 subunit. Two subunits are present in each pentameric GABA receptor; each of these subunits contributes both to a binding site for GABA and to the transmembrane pore. Peak currents mediated by receptors including the A322D GABAα1 mutants were only 10% of those of wild-type receptors, and this weak response required 200-fold higher concentrations of GABA. Accordingly, the epileptic phenotype would be expected to be associated with greatly reduced responsiveness to inhibitory neurotransmission. It is unclear why this mutation results in epilepsy of the JME type.
A genomewide linkage analysis of 130 families with idiopathic generalized epilepsy (IGE) identified a susceptibility locus on chromosome 3q (136). The gene CLCN2, encoding a voltage-gated Cl− channel protein (called ClC-2 or CLC-2), was identified within the linked region and sequenced for mutations in samples from 46 families (137). Three mutations were identified among these families, one in a small family with five patients with JME. CLC-2, like other members of the chloride channel family, forms dimeric channel complexes. Each chloride channel subunit possesses its own transmembrane ion path, and gating of the two pores within each channel is regulated by allosteric interactions within the protein (10). The JME mutation identified in the single family produced a truncated form of the protein that was unable to conduct ions itself or coassemble with wild-type subunits.
In 2008, Medina and colleagues found mutations in EFHC1 (Myoclonin1) in 9% of consecutive cases with JME from Mexico and Honduras as well as in 3% of clinic patients from Japan, a stronger association than has been seen with other genes in this syndrome (138). A subsequent report by many authors from that study suggested that the association might have been private, since they did not find those mutations in a wider Hispanic population (139). Indeed, both heterozygous and homozygous EFHC1-deficient mice developed spontaneous myoclonus as well as enhanced sensitivity to chemoconvulsants (140). However, the association may be highly dependent also on the genetic background. This possibility was highlighted by a population study that found the implicated JME haplotypes in many unaffected individuals, especially of West African ancestry (141). These findings are highly reminiscent of a classic paper by Schauwecker and Steward, who demonstrated the importance of genetic background in seizure-induced excitotoxic vulnerability and the pitfalls in experiments involving targeted gene knockouts (142). The reader should be reminded of the importance of this work after nearly two decades, and the lessons for human studies as eloquently expressed in a commentary by Pal and Helbig (143).
Epilepsy With Grand Mal Seizures on Awakening (EGMA, MIM #607628) and Childhood Absence Epilepsy (CAE; MIM #600131). JME overlaps with another syndrome, termed “awakening epilepsy” or “epilepsy with grand mal seizures on awakening” (EGMA). Patients with EGMA have difficulty falling asleep, abnormal sleep architecture, and the vast majority of their seizures occur clustered within the first minutes after awakening from sleep (irrespective of the time of day) (144). A CLCN2 mutation encoding a nonfunctional channel was identified in a large family with EGMA (137).
A subset of JME and EGMA patients experience absence spells. A third CLCN2 mutation was found to segregate in a small family in which three children had typical childhood absence epilepsy (CAE) (137). This CAE-linked mutation resulted in a single amino acid change (G715E); mutant channels were reported to be functional but were reported to exhibit altered voltage-dependence. This finding has been contradicted by two subsequent investigations showing activity of the mutant indistinguishable from wild type, further weakening the modest genetic evidence provided by the small family studied (10). A more recent survey of 55 families with IGE failed to identify additional CLCN2 mutations (145). At present, the strongest evidence linking CLCN2 to generalized epilepsy is provided by the large EGMA pedigree.
Autosomal Dominant Partial Epilepsy With Auditory Features (ADPEAF, MIM #600512). In 1995, Ottman et al described a large family in which partial seizures associated with unusual auditory auras were inherited in an autosomal dominant pattern linked to a gene on chromosome 10 (146). Several additional families with this distinctive syndrome were subsequently identified (147,148). The age of seizure onset was typically at 9 to 12 years of age. Seizures are heralded by a variety of stereotyped, but in nearly all cases nonverbal, auditory hallucinations (147). Mutations were subsequently identified in the gene Lgi1, a gene of unknown function previously shown to be disrupted in some patients with primary brain tumors (149). This set in motion efforts to understand the function of the Lgi1 protein in brain development, auditory processing, and epileptogenesis.
At present, evidence supporting two conflicting models has been described. One set of experiments shows that Lgi1 is physically associated with and enhances the activity of Kv1 family voltage-gated potassium channels whose pore-forming subunits are encoded by the KCNA genes (150). In this model, Lgi1 is a cytosolic protein that binds to the intracellular portion of channels that contain both pore-forming subunits and previously described channel intracellular accessory units, called β subunits. The β1 subunit causes Kv1 channels to close after a period of milliseconds of membrane depolarization. This time and voltage-dependent closing, called inactivation, is similar to the fast inactivation of sodium channels that helps terminate each action potential. However, inactivation of K+ channels will generally tend to increase neuronal excitability. Wild type Lgi1 proteins antagonize this inactivation, prolonging channel opening. Mutations derived from ADPEAF patients, which produce truncated proteins, all abolish the ability of the Lgi1 protein to prolong the K+ channel openings. This model is consistent with previously discussed findings that loss-of-function KCNA1 mutations cause epilepsy in some patients with episodic ataxia-1, and mutations in Caspr2 disrupt Kv1 channel targeting in the recessive focal epilepsy.
The alternative, and very different, model is based on the recent report that Lgi1 protein is packaged in secretory vesicles and released into the synaptic cleft, where it binds tightly to a cluster of proteins linked to postsynaptic glutamate receptors (151,152). Lgi1 binds directly to ADAM22, and is thereby linked to PSD-95 and stargazin, all of which are components of the excitatory postsynaptic density. Application of soluble Lgi1 proteins to brain slices in vitro results in enhancement of AMPA-type glutamate receptor currents. Lgi1 applied to neurons grown in dissociated culture increases the number of AMPA receptors at the cell surface. Mutations in stargazin and ADAM22 that are capable of disrupting the protein complex are associated with epilepsy in mice (although human mutations in these genes are unknown) (153–155). Effects of Lgi1 mutations on this complex and the implications for receptor function are incompletely known, but the implication is that mutations would tend to reduce postsynaptic excitatory currents.
How can the two proposed functional roles of Lgi1 be reconciled? Immunoblots of proteins separated by SDS gel electrophoresis reveal two forms of Lgi1 that differ by 3–5 kDa, indicating that the protein is either alternatively spliced or posttranslationally processed (156). Moreover, the larger form localizes to the cytoplasm, whereas the smaller is found in a microsomal fraction containing secretory vesicles. Although it is not clear which form is most important for epilepsy, hippocampal and cerebellar cortex areas (with particularly high densities of AMPA-type glutamatergic synapses) showed predominantly the smaller, presumed secreted form. The highest percentage of the cytosolic, high molecular weight form was present in the temporal neocortex (156). This favors the idea that the cytoplasmic interaction with the potassium channel contributes to the auditory auras and seizures in ADPEAF, but additional studies are needed.
Generalized Epilepsy With Paroxysmal Dyskinesias (MIM #609446). Clinical studies have noted patients, sometimes in familial groups, with a combination of generalized epilepsy and paroxysmal dyskinesia (157,158). Du et al (159) described an unusual family with 16 affected individuals in five consecutive generations, strongly indicating dominant inheritance. Seven had paroxysmal nonkinesigenic dyskinesias, four developed epilepsy only, and five exhibited both symptoms. The symptom onset age ranged broadly from 3 to 15 years, with one patient developing seizures before 6 months. After establishing linkage to chromosome 10, the investigators identified a mutation in KCNMA1, encoding a pore-forming α subunit of a type of calcium-activated potassium channel, called BK. The name BK (“Big-K” or “Maxi-K”) is based on the fact that these pass ions at higher rates than other K+ channel types. BK channels are activated synergistically by membrane depolarization and elevated intracellular Ca2+ (1). Their opening rates are very fast, enabling them to speed repolarization of action potentials, shortening the sodium channel refractory period and thereby facilitating rapid neuronal bursting. The pathogenic mutation in BK identified increases in channel activity (160). Interestingly, a knockout mutation in a BK accessory subunit has also recently been shown to cause epilepsy in transgenic mice (161). Careful electrophysiologic analysis of these mice indicates that this mutation also increases the activity of BK, leading to increased neuronal firing. The detailed biophysical mechanism whereby excitability is increased in the mice is intriguingly indirect. First, the enhanced BK activity and associated shortened action potential duration cause a reduction in Ca2+ entry during each spike. This diminishes the activation of a second type of Ca2+-activated K+ channel, known as SK (“small conductance K+ channel”). Despite their name, SK channels very potently limit excitability due to their long-lasting openings. So preventing SK activation (by increasing BK openings) leads to markedly increased cellular excitability. In effect, the cell must balance BK and SK activity precisely to maintain the desired overall level of excitability. This elegant electrophysiologic analysis again illustrates the importance of transgenic mouse models to current studies of the epileptic channelopathies, and begins to hint at how very indirect the effects of a mutation can be.
Conclusions and Implications for Therapeutics
Although the first identification of a channel gene mutation in epilepsy occurred only in 1994, many such mutations have now been identified in 15 different channel subunits and channel-associated proteins. What do these efforts reveal about the causes of epilepsy and potential for novel treatments? One remarkable result is that many of the mutations have been found in subunits of voltage-dependent Na+ channels and GABA receptors, important targets of the majority of currently approved drugs, all developed through in vivo pharmacologic screening. Many (but not all) Na+ channel mutations in GEFS+, for example, result in increased Na+ channel activity; by contrast, drugs like phenytoin, carbamazepine, and lamotrigine act by blocking Na+ channel activity. Similarly, in autosomal dominant JME and some cases of GEFS+, mutations in GABA receptor subunits that reduce activity are implicated, whereas benzodiazepines and barbiturates potentiate GABA receptor activity. This is satisfying, but is also potentially a cause for concern. If genetics were to only lead us back to the therapeutic targets we know about, it would be informative but of little practical use. In this regard, the stories of the identification of the KCNQ channel mutations in BFNS, nicotinic acetylcholine receptor mutations in ADNFLE, and Lgi1 mutations in ADPEAF are exciting and reassuring counterexamples. The neuronal KCNQ channels and the functions of Lgi1 regulating K+channels and glutamate receptors were unknown before these mutations led to the recent mechanistic studies discussed previously. It seems possible that a new class of KCNQ opener drugs may be developed, with patterns of clinical usefulness that are quite different from available agents. The Lgi1-dependent augmentation of K+ channel activity is another potential target for future drug development. As neurobiologic studies allow us to understand the mechanistic pathways through which individual mutations lead to seizures, additional targets will be revealed beyond the mutated gene itself. An example of this is the unexpected observation that nicotinic receptor mutations in ADNFLE may cause seizures through enhanced inhibition during arousal from deep sleep. Interestingly, low doses of picrotoxin, a GABA antagonist that is strongly convulsant at higher doses, blocked epileptiform EEG activity and spontaneous seizures in transgenic mice bearing ADNFLE mutations (129). The effect of quinidine in limiting gain of function mutations in KCNT1 was mentioned earlier (133)
Although many aspects of genotype–phenotype relationships remain poorly understood, recent years have brought considerable progress, encouraging optimism that the pathogenic mechanisms in these disorders can be understood in detail. Examples of such progress are the identification of the role of KCNQ channels in setting thresholds by targeting the axonal spike initiation zone, the discovery that SCN1A subunits are preferentially expressed on inhibitory interneurons, and the analysis showing how KCNM1 mutations increasing the activity of BK channels can paradoxically increase neuronal excitability (30,85,162). However, it remains unclear whether, in GEFS+ families, the variable phenotype seen within pedigrees may reflect differences in genetic background between individuals, environmental exposures, or both. The spectrum of phenotypes associated with SCN1A mutations, ranging from normal gene carriers or patients with only FS in GEFS+ pedigrees to SMEI patients with severe refractory seizures and progressive motor and cognitive decline, is impressive and requires further study. It remains unclear whether the more severe phenotypes in GEFS+ pedigrees and SMEI should be seen as a part of the GEFS+ spectrum, since they are almost certainly associated with secondary changes in brain structure and function (eg, cell death, network reorganization, changes in the levels of expression of other channels, and signaling proteins) that do not occur in mildly affected individuals. Finally, as pathogenic mechanisms in these Mendelian forms of epilepsy become clearer, efforts will focus on understanding to what extent these mechanisms contribute to the common, non-Mendelian forms of epilepsy. Already, the epileptic channelopathies fit poorly with our traditional classification of the epilepsies as idiopathic (ie, hereditary, with normal function except seizures) versus symptomatic (associated with cognitive, motor, and other deficits). Not only can different mutations in the same channel cause varying degrees of impairment, the same mutation can have variable effects within a pedigree.
Finally, there is increasing recognition of the importance of comorbidities in epilepsy, especially the neurodevelopmental disabilities. The mechanistic connections between epilepsy and these disorders seem extensive (163). The connections between these problems at a molecular level are beginning to be unraveled as described earlier in the interaction between KCNT1 channel protein and FMRP, the role of CNTNAP2 in both ion channel function and neural plasticity, thus heralding a new era in our understanding of the different facets of epileptic disorders.
REFERENCES
1. Hille B. Ionic Channels of Excitable Membranes. 3rd ed. Sunderland, MA: Sinauer; 2001.
2. Yu FH, Catterall WA, Vgl-Chanome T. A protein superfamily specialized for electrical signaling and ionic homeostasis. Sci STKE. 2004;2004:re15.
3. Tombola F, Pathak MM, Isacoff EY. How far will you go to sense voltage? Neuron. 2005;48:719–725.
4. Madden DR. The structure and function of glutamate receptor ion channels. Nat Rev Neurosci. 2002;3:91–101.
5. Long SB, Campbell EB, Mackinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 2005;309:903–908.
6. Jiang Y, Lee A, Chen J, et al. X-ray structure of a voltage-dependent K+ channel. Nature. 2003;423:33–41.
7. Okamura Y, Nishino A, Murata Y, et al. Comprehensive analysis of the ascidian genome reveals novel insights into the molecular evolution of ion channel genes. Physiol Genomics. 2005;22:269–282.
8. Zhang J. Evolution by gene duplication: an update. Trends Evolution and Ecology. 2003;18:292–298.
9. Lynch M, Conery JS. The origins of genome complexity. Science. 2003;302:1401–1404.
10. Jentsch TJ, Poet M, Fuhrmann JC, et al. Physiological functions of CLC cl- channels gleaned from human genetic disease and mouse models. Ann Rev Physiol. 2005;67:779–807.
11. Mohler H. GABA(A) receptor diversity and pharmacology. Cell Tissue Res. 2006;326:505–516.
12. Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Curr Opin Pharmacol. 2007;7:39–47.
13. Ptacek L. The familial periodic paralyses and nondystrophic myotonias. Am J Med. 1998;105:58–70.
14. Cooper EC, Jan LY. Ion channel genes and human neurological disease: recent progress, prospects, and challenges. Proc Natl Acad Sci U S A. 1999;96:4759–4766.
15. Ronen GM, Rosales TO, Connolly M, et al. Seizure characteristics in chromosome 20 benign familial neonatal convulsions. Neurology. 1993;43:1355–1360.
16. Rett A, Teubel R. Neugeborenenkrampfe im Rahmen einer epileptisch belasteten Familie. Wien Klin Wochenschr. 1964;76:609–613.
17. Zimprich F, Ronen GM, Stogmann W, et al. Andreas Rett and benign familial neonatal convulsions revisited. Neurology. 2006;67:864–866.
18. Leppert M, Anderson VE, Quattlebaum T, et al. Benign familial neonatal convulsions linked to genetic markers on chromosome 20. Nature. 1989;337:647–648.
19. Lewis TB, Leach RJ, Ward K, et al. Genetic heterogeneity in benign familial neonatal convulsions: identification of a new locus on chromosome 8q. Am J Hum Genet. 1993;53:670–675.
20. Steinlein O, Schuster V, Fischer C, et al. Benign familial neonatal convulsions: confirmation of genetic heterogeneity and further evidence for a second locus on chromosome 8q. Hum Genet. 1995;95:411–415.
21. Singh NA, Charlier C, Stauffer D, et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet. 1998;18:25–29.
22. Charlier C, Singh NA, Ryan SG, et al. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat Genet. 1998;18:53–55.
23. Biervert C, Schroeder BC, Kubisch C, et al. A potassium channel mutation in neonatal human epilepsy. Science. 1998;279:403–406.
24. Neyroud N, Tesson F, Denjoy I, et al. A novel mutation in the potassium channel gene KVLQT1 causes the cardioauditory syndrome. Nat Genet. 1997;15:186–189.
25. Wang Q, Curran ME, Splawski I, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23.
26. Wang H-S, Pan Z, Shi W, et al. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the m-channel. Science. 1998;282:1890–1893.
27. Hadley JK, Passmore GM, Tatulian L, et al. Stoichiometry of expressed KCNQ2/KCNQ3 potassium channels and subunit composition of native ganglionic m channels deduced from block by tetraethylammonium. J Neurosci. 2003;23:5012–5019.
28. Martire M, Castaldo P, D’Amico M, et al. M channels containing KCNQ2 subunits modulate norepinephrine, aspartate, and GABA release from hippocampal nerve terminals. J Neurosci. 2004;24:592–597.
29. Shen W, Hamilton SE, Nathanson NM, et al. Cholinergic suppression of KCNQ channel currents enhances excitability of striatal medium spiny neurons. J Neurosci. 2005;25:7449–7458.
30. Pan Z, Kao T, Horvath Z, et al. A common ankyrin-g-based mechanism retains KCNQ and Nav channels at electrically active domains of the axon. J Neurosci. 2006;26:2599–2613.
31. Schwarz JR, Glassmeier G, Cooper EC, et al. KCNQ channels mediate iks, a slow K+ current regulating excitability in the rat node of Ranvier. J Physiol. 2006;573:17–34.
32. Schroeder BC, Kubisch C, Stein V, et al. Moderate loss of function of cyclic-amp-modulated KCNQ2 KCNQ3 K+ channels causes epilepsy. Nature. 1998;396:687–690.
33. Brown D. M-currents: an update. Trends in neuroscience. 1988;11:294–299.
34. Cooper EC, Harrington E, Jan YN, et al. M-channel KCNQ2 subunits are localized to key sites for control of neuronal network oscillations and synchronization in mouse brain. J Neurosci. 2001;21:9529–9540.
35. Cooper EC, Aldape KD, Abosch A, et al. Colocalization and coassembly of two human brain m-type potassium channel subunits that are mutated in epilepsy. Proc Natl Acad Sci USA. 2000;97:4914–4919.
36. Devaux JJ, Kleopa KA, Cooper EC, et al. KCNQ2 is a nodal K+ channel. J Neurosci. 2004;24:1236–1244.
37. Stuart G, Spruston N, Sakmann B, et al. Action potential initiation and backpropagation in neurons of the mammalian CNS. Trends Neurosci. 1997;20:125–131.
38. Okada M1, Zhu G, Hirose S, et al. Age-dependent modulation of hippocampal excitability by KCNQ-channels. Epilepsy Res. 2003;53:81–94.
39. Weckhuysen S, Mandelstam S, Suls A, et al. KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann Neurol. 2012;71(1):15–25.
40. Kato M, Yamagata T, Kubota M, et al. Clinical spectrum of early onset epileptic encephalopathies caused by KCNQ2 mutation. Epilepsia. 2013;54(7):1282–1287.
41. Singh NA, Westenskow P, Charlier C, et al; For BFNC Physician Consortium. KCNQ2 and KCNQ3 potassium channel genes in benign familial neonatal convulsions: expansion of the functional and mutation spectrum. Brain. 2003;126(Pt 12):2726–2737.
42. Steinlein OK, Conrad C, Weidner B. Benign familial neonatal convulsions: always benign? Epilepsy Res. 2007;73:245–249.
43. Miceli F, Soldovieri MV, Ambrosino P, et al. Early-onset epileptic encephalopathy caused by gain-of-function mutations in the voltage sensor of kv7.2 and kv7.3 potassium channel subunits. J Neurosci. 2015;3:3782–3793.
44. Devaux J, Abidi A, Roubertie A, et al. A kv7.2 mutation associated with early onset epileptic encephalopathy with suppression-burst enhances kv7/m channel activity. Epilepsia. 2016;57:e87–e93.
45. Abidi A, Devaux JJ, Molinari F, et al. A recurrent KCNQ2 pore mutation causing early onset epileptic encephalopathy has a moderate effect on M current but alters subcellular localization of kv7 channels. Neurobiol Dis. 2015;80:80–92.
46. Gunthorpe MJ, Large CH, Sankar R. The mechanism of action of retigabine (ezogabine), a first-in-class K+ channel opener for the treatment of epilepsy. Epilepsia. 2012;53:412–424.
47. Large CH, Sokal DM, Nehlig A, et al. The spectrum of anticonvulsant efficacy of retigabine (ezogabine) in animal models: implications for clinical use. Epilepsia. 2012;53(3):425–436.
48. Coppola G, Plouin P, Chiron C, et al. Migrating partial seizures in infancy, a malignant disorder with developmental arrest. Epilepsia. 1995;36:1017–1024.
49. Barcia G, Fleming MR, Deligniere A, et al. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet. 2012;44(11):1255–1259.
50. Bhattacharjee A, Kaczmarek LK. For K+ channels, Na+ is the new Ca2+. Trends Neurosci. 2005;28:422–428.
51. Brown MR, Kronengold J, Gazula VR, et al. Fragile X mental retardation protein controls gating of the sodium-activated potassium channel slack. Nat Neurosci. 2010;13(7):819–821.
52. Ohba C, Kato M, Takahashi S, et al. Early onset epileptic encephalopathy caused by de novo SCN8A mutations. Epilepsia. 2014;55(7):994–1000.
53. Larsen J, Carvill GL, Gardella E, et al. EuroEPINOMICS RES Consortium CRP. The phenotypic spectrum of SCN8A encephalopathy. Neurology. 2015;84(5):480–489.
54. Martin MS, Tang B, Papale LA, et al. The voltage-gated sodium channel SCN8A is a genetic modifier of severe myoclonic epilepsy of infancy. Hum Mol Genet. 2007;16:2892–2899.
55. Shevell MI, Sinclair DB, Metrakos K. Benign familial neonatal seizures: clinical and electroencephalographic characteristics. Pediatr Neurol. 1986;2:272–275.
56. Kaplan RE, Lacey DJ. Benign familial neonatal-infantile seizures. Am J Med Genet. 1983;16:595–599.
57. Lewis TB, Shevell MI, Andermann E, et al. Evidence of a third locus for benign familial convulsions. J Child Neurol. 1996;11:211–214.
58. Malacarne M, Gennaro E, Madia F, et al. Benign familial infantile convulsions: mapping of a novel locus on chromosome 2q24 and evidence for genetic heterogeneity. Am J Hum Genet. 2001;68:1521–1526.
59. Leppert M, McMahon WM, Quattlebaum TG, et al. Searching for human epilepsy genes: a progress report. Brain Pathol. 1993;3:357–369.
60. Boiko T, Rasband MN, Levinson SR, et al. Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron. 2001;30:91–104.
61. Dzhala VI, Talos DM, Sdrulla DA, et al. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11:1205–1213.
62. Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain. 1997;120:479–490.
63. Wallace RH, Wang DW, Singh R, et al. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel β1 subunit gene SCN1B. Nat Genet. 1998;19:366–370.
64. Wallace RH, Scheffer IE, Parasivam G, et al. Generalized epilepsy with febrile seizures plus: mutation of the sodium channel subunit SCN1B. Neurology. 2002;58:1426–1429.
65. McCormick KA, Isom LL, Ragsdale D, et al. Molecular determinants of Na+ channel function in the extracellular domain of the β1 subunit. J Biol Chem. 1998;273:3954–3962.
66. Isom LL. Sodium channel β subunits: anything but auxiliary. Neuroscientist. 2001;7:42–54.
67. Meadows LS, Malhotra J, Loukas A, et al. Functional and biochemical analysis of a sodium channel β1 subunit mutation responsible for generalized epilepsy with febrile seizures plus type 1. J Neurosci. 2002;22:10699–10709.
68. Baulac S, Gourfinkel-An I, Picard F, et al. A second locus for familial generalized epilepsy with febrile seizures plus maps to chromosome 2q21-q33. Am J Hum Genet. 1999;65:1078–1085.
69. Moulard B, Guipponi M, Chaigne D, et al. Identification of a new locus for generalized epilepsy with febrile seizures plus (GEFS+) on chromosome 2q24-q33. Am J Hum Genet. 1999;65:1396–1400.
70. Escayg A, Macdonald BT, Meisler MH, et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nature Genetics. 2000;24:343–345.
71. Wallace RH, Scheffer IE, Barnett S, et al. Neuronal sodium-channel alpha1-subunit mutations in generalized epilepsy with febrile seizures plus. Am J Hum Genet. 2001;68:859–865.
72. Escayg A, Heils A, Macdonald BT, et al. A novel SCN1A mutation associated with generalized epilepsy with febrile seizures plus—and prevalence of variants in patients with epilepsy. Am J Hum Genet. 2001;68:866–873.
73. Abou-Khalil B, Ge Q, Desai R, et al. Partial and generalized epilepsy with febrile seizures plus and a novel SCN1A mutation. Neurology. 2001;57:2265–2272.
74. Sugawara T, Mazaki-Miyazaki E, Ito M, et al. Nav1.1 mutations cause febrile seizures associated with afebrile partial seizures. Neurology. 2001;57:703–705.
75. Lossin C, Wang DW, Rhodes TH, et al. Molecular basis of an inherited epilepsy. Neuron. 2002;34:877–884.
76. Vanoye CG, Lossin C, Rhodes TH, et al. Single-channel properties of human Nav1.1 and mechanism of channel dysfunction in SCN1A-associated epilepsy. J Gen Physiol. 2006;127:1–14.
77. Barela AJ, Waddy SP, Lickfett JG, et al. An epilepsy mutation in the sodium channel SCN1A that decreases channel excitability. J Neurosci. 2006;26:2714–2723.
78. Bennett PB, Yazawa K, Makita N, et al. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685.
79. Cannon SC, Brown RH Jr, Corey DP. A sodium channel defect in hyperkalemic periodic paralysis: potassium-induced failure of inactivation. Neuron. 1991;6:619–626.
80. Singh R, Andermann E, Whitehouse WP, et al. Severe myoclonic epilepsy of infancy: extended spectrum of GEFS+? Epilepsia. 2001;42:837–844.
81. Claes L, del-Favero J, Ceulemans B, et al. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. 2001;68:1327–1332.
82. Sugawara T, Mazaki-Miyazaki E, Fukushima K, et al. Frequent mutations of SCN1A in severe myoclonic epilepsy in infancy. Neurology. 2002;58:1122–1124.
83. Fujiwara T, Sugawara T, Mazaki-Miyazaki E, et al. Mutations of sodium channel alpha subunit type 1 (SCN1A) in intractable childhood epilepsies with frequent generalized tonic–clonic seizures. Brain. 2003;126:531–546.
84. Westenbroek RE, Merrick DK, Catterall WA. Differential subcellular localization of the RI and RII Na+ channel subtypes in central neurons. Neuron. 1989;3:695–704.
85. Yu FH, Mantegazza M, Westenbroek RE, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9:1142–1149.
86. van Wart A, Trimmer JS, Matthews G. Polarized distribution of ion channels within microdomains of the axon initial segment. J Comp Neurol. 2007;500:339–352.
87. Yu FH, Mantegazza M, Westenbroek RE, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9:1142–1149.
88. Ogiwara I, Miyamoto H, Morita N, et al. Na(v)1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an SCN1A gene mutation. J Neurosci. 2007;27:5903–5914.
89. Guerrini R, Dravet C, Genton P, et al. Lamotrigine and seizure aggravation in severe myoclonic epilepsy. Epilepsia. 1998;39:508–512.
90. Liu Y, Lopez-Santiago LF, Yuan Y, et al. Dravet syndrome patient-derived neurons suggest a novel epilepsy mechanism. Ann Neurol. 2013;74(1):128–139.
91. Baulac S, Huberfeld G, Gourfinkel-An I, et al. First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat Genet. 2001;28:46–48.
92. Dibbens LM, Feng HJ, Richards MC, et al. GABRD encoding a protein for extra- or peri-synaptic GABA(A) receptors is a susceptibility locus for generalized epilepsies. Hum Mol Genet. 2004;13:1315–1319.
93. Moss SJ, Smart TG. Constructing inhibitory synapses. Nat Rev Neurosci. 2001;2:240–250.
94. Wallace RH, Marini C, Petrou S, et al. Mutant GABA(A) receptor γ2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet. 2001;28:49–52.
95. Harkin LA, Bowser DN, Dibbens LM, et al. Truncation of the gaba(a)-receptor γ2 subunit in a family with generalized epilepsy with febrile seizures plus. Am J Hum Genet. 2002;70:530–536.
96. Kang JQ, Shen W, Macdonald RL. Why does fever trigger febrile seizures? GABA(A) receptor γ2 subunit mutations associated with idiopathic generalized epilepsies have temperature-dependent trafficking deficiencies. J Neurosci. 2006;26:2590–2597.
97. Audenaert D, Schwartz E, Claeys KG, et al. A novel GABRG2 mutation associated with febrile seizures. Neurology. 2006;67:687–690.
98. Nusser Z, Sieghart W, Somogyi P. Segregation of different GABA(A) receptors to synaptic and extrasynaptic membranes of cerebellar granule cells. J Neurosci. 1998;18:1693–1703.
99. Mortensen M, Smart TG. Extrasynaptic αβ subunit GABA(A) receptors on rat hippocampal pyramidal neurons. J Physiol. 2006;577:841–856.
100. Strauss KA, Puffenberger EG, Huentelman MJ, et al. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl J Med. 2006;354:1370–1377.
101. Inda MC, Defelipe J, Munoz A. Voltage-gated ion channels in the axon initial segment of human cortical pyramidal cells and their relationship with chandelier cells. Proc Natl Acad Sci USA. 2006;103:2920–2925.
102. Poliak S, Gollan L, Martinez R, et al. Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with k+ channels. Neuron. 1999;24:1037–1047.
103. Poliak S, Gollan L, Salomon D, et al. Localization of caspr2 in myelinated nerves depends on axon-glia interactions and the generation of barriers along the axon. J Neurosci. 2001;21:7568–7575.
104. Vabnick I, Trimmer JS, Schwarz TL, et al. Dynamic potassium channel distributions during axonal development prevent aberrant firing patterns. J Neurosci. 1999;19:747–758.
105. Rasband MN, Park EW, Zhen D, et al. Clustering of neuronal potassium channels is independent of their interaction with PSD-95. J Cell Biol. 2002;159:663–672.
106. Kaplan WD, Trout WE. The behavior of four neurological mutants of sophila. Genetics. 1969;61:399–409.
107. Tanouye MA, Ferrus A, Fujita SC. Abnormal action potentials associated with the shaker complex locus of drosophila. Proc Natl Acad Sci USA. 1981;78:6548–6552.
108. Rasband MN, Shrager P. Ion channel sequestration in central nervous system axons. J Physiol. 2000;525(Pt 1):63–73.
109. Geiger JR, Jonas P. Dynamic control of presynapticca2+ inflow by fast-inactivating K+ channels in hippocampal mossy fiber boutons. Neuron. 2000;28:927–939.
110. Noebels JL. The biology of epilepsy genes. Annu Rev Neurosci. 2003;26:599–625.
111. Alarcón M1, Abrahams BS, Stone JL, et al. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am J Hum Genet. 2008;82:150–159.
112. Scott-van Zeeland AA, Abrahams BS, Alvarez-Retuerto AI, et al. Altered functional connectivity in frontal lobe circuits is associated with variation in the autism risk gene CNTNAP2. Sci Transl Med. 2010;2(56):56ra80.
113. Gdalyahu A, Lazaro M, Penagarikano O, et al. The autism related protein contactin-associated protein-like 2 (CNTNAP2) stabilizes new spines: an in vivo mouse study. PLoS One. 2015;10:e0125633.
114. Horresh I, Poliak S, Grant S, et al. Multiple molecular interactions determine the clustering of caspr2 and kv1 channels in myelinated axons. J Neurosci. 2008;28:14213–14222.
115. Chao HW, Hong CJ, Huang TN, et al. Sumoylation of the MAGUK protein CASK regulates dendritic spinogenesis. J Cell Biol. 2008;182:141–155.
116. Scheffer IE, Bhatia KP, Lopes-Cendes I, et al. Autosomal dominant frontal epilepsy misdiagnosed as sleep disorder. Lancet. 1994;343:515–517.
117. Scheffer IE, Bhatia KP, Lopes-Cendes I, et al. Autosomal dominant nocturnal frontal lobe epilepsy. A distinctive clinical disorder. Brain. 1995;118(Pt 1):61–73.
118. Derry CP, Duncan JS, Berkovic SF. Paroxysmal motor disorders of sleep: the clinical spectrum and differentiation from epilepsy. Epilepsia. 2006;47:1775–1791.
119. Provini F, Plazzi G, Tinuper P, et al. Nocturnal frontal lobe epilepsy, a clinical and polygraphic overview of 100 consecutive cases. Brain. 1999;122(Pt 6):1017–1031.
120. Steinlein OK, Mulley JC, Propping P, et al. A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 1995;11:201–203.
121. Steinlein OK, Magnusson A, Stoodt J, et al. An insertion mutation of the CHRNA4 gene in a family with autosomal dominant nocturnal frontal lobe epilepsy. Hum Mol Genet. 1997;6:943–947.
122. de Fusco M, Becchetti A, Patrignani A, et al. The nicotinic receptor β2 subunit is mutant in nocturnal frontal lobe epilepsy. Nat Genet. 2000;26:275–276.
123. Phillips HA, Favre I, Kirkpatrick M, et al. CHRNB2 is the second acetylcholine receptor subunit associated with autosomal dominant nocturnal frontal lobe epilepsy. Am J Hum Genet. 2001;68:225–231.
124. Aridon P, Marini C, di Resta C, et al. Increased sensitivity of the neuronal nicotinic receptor alpha 2 subunit causes familial epilepsy with nocturnal wandering and ictal fear. Am J Hum Genet. 2006;79:342–350.
125. Bertrand S, Weiland S, Berkovic SF, et al. Properties of neuronal nicotinic acetylcholine receptor mutants from humans suffering from autosomal dominant nocturnal frontal lobe epilepsy. Br J Pharmacol. 1998;125:751–760.
126. Kuryatov A, Gerzanich V, Nelson M, et al. Mutation causing autosomal dominant nocturnal frontal lobe epilepsy alters Ca2+ permeability, conductance, and gating of human α4β2 nicotinic acetylcholine receptors. J Neurosci. 1997;17:9035–9047.
127. Arroyo-Jimenez MM, Bourgeois JP, Marubio LM, et al. Ultrastructural localization of the alpha4-subunit of the neuronal acetylcholine nicotinic receptor in the rat substantia nigra. J Neurosci. 1999;19:6475–6487.
128. Labarca C, Schwarz J, Deshpande P, et al. Point mutant mice with hypersensitive alpha 4 nicotinic receptors show dopaminergic deficits and increased anxiety. Proc Natl Acad Sci USA. 2001;98:2786–2791.
129. Klaassen A, Glykys J, Maguire J, et al. Seizures and enhanced cortical GABAergic inhibition in two mouse models of human autosomal dominant nocturnal frontal lobe epilepsy. Proc Natl Acad Sci USA. 2006;103:19152–19157.
130. McCormick DA. Bal T. Sleep and arousal: thalamocortical mechanisms. Annu Rev Neurosci. 1997;20:185–215.
131. Willoughby JO, Pope KJ, Eaton V. Nicotine as an antiepileptic agent in ADNFLE: an n-of-one study. Epilepsia. 2003;44:1238–1240.
132. Heron SE, Smith KR, Bahlo M, et al. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 2012;44(11):1188–1190.
133. Milligan CJ, Li M, Gazina EV, et al. KCNT1 gain of function in 2 epilepsy phenotypes is reversed by quinidine. Ann Neurol. 2014;75(4):581–590.
134. Janz D. Epilepsy with impulsive petit mal (juvenile myoclonic epilepsy). Acta Neurol Scand. 1985;72:449–459.
135. Cossette P, Liu L, Brisebois K, et al. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet. 2002;31:184–189.
136. Sander T, Schulz H, Saar K, et al. Genome search for susceptibility loci of common idiopathic generalised epilepsies. Hum Mol Genet. 2000;9:1465–1472.
137. Haug K, Warnstedt M, Alekov AK, et al. Mutations in CLCN2 encoding a voltage-gated chloride channel are associated with idiopathic generalized epilepsies. Nat Genet. 2003;33:527–532.
138. Medina MT, Suzuki T. Alonso T, et al. Novel mutations in myoclonin1/efhc1 in sporadic and familial juvenile myoclonic epilepsy. Neurology. 2008;70:2137–2144.
139. Bai D, Bailey JN, Duron RM, et al. DNA variants in coding region of EFHC1: SNPs do not associate with juvenile myoclonic epilepsy. Epilepsia. 2009;50:1184–1190.
140. Suzuki T, Miyamoto H, Nakahari T, et al. EFHC1 deficiency causes spontaneous myoclonus and increased seizure susceptibility. Hum Mol Genet. 2009;18(6): 1099–1109.
141. Subaran RL, Conte JM, Stewart WC, et al. Pathogenic EFHC1 mutations are tolerated in healthy individuals dependent on reported ancestry. Epilepsia. 2015;56:188–194.
142. Schauwecker PE, Steward O. Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. Proc Natl Acad Sci U S A. 1997;94:4103–4108.
143. Pal D, Helbig I. Commentary: pathogenic EFHC1 mutations are tolerated in healthy individuals dependent on reported ancestry. Epilepsia. 2015;56(2):195–196.
144. Janz D. Epilepsy with grand mal on awakening and sleep-waking cycle. Clin Neurophysiol. 2000;111(Suppl 2):S103–110.
145. Marini C, Scheffer IE, Crossland KM, et al. Genetic architecture of idiopathic generalized epilepsy: clinical genetic analysis of 55 multiplex families. Epilepsia. 2004;45:467–478.
146. Ottman R, Risch N, Hauser WA, et al. Localization of a gene for partial epilepsy to chromosome 10q. Nat Genet. 1995;10:56–60.
147. Winawer MR, Boneschi FM, Barker-Cummings C, et al. Four new families with autosomal dominant partial epilepsy with auditory features: clinical description and linkage to chromosome 10q24. Epilepsia. 2002;43:60–67.
148. Poza JJ, Saenz A, Martinez-Gil A, et al. Autosomal dominant lateral temporal epilepsy: clinical and genetic study of a large basque pedigree linked to chromosome 10q. Ann Neurol. 1999;45:182–188.
149. Kalachikov S, Evgrafov O, Ross B, et al. Mutations in Lgi1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet. 2002;30:335–341.
150. Schulte U, Thumfart JO, Klocker N, et al. The epilepsy-linked Lgi1 protein assembles into presynaptic kv1 channels and inhibits inactivation by kvβ1. Neuron. 2006;49:697–706.
151. Fukata Y, Adesnik H, Iwanaga T, et al. Epilepsy-related ligand/receptor complex Lgi1 and Adam22 regulate synaptic transmission. Science. 2006;313:1792–1795.
152. Snyder SH. Neuroscience. Adam finds an exciting mate. Science. 2006;313:1744–1745.
153. Letts VA, Felix R, Biddlecome GH, et al. The mouse stargazer gene encodes a neuronal Ca2+-channel gamma subunit. Nat Genet. 1998;19:340–347.
154. Chen L, Chetkovich DM, Petralia RS, et al. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408:936–943.
155. Sagane K, Hayakawa K, Kai J, et al. Ataxia and peripheral nerve hypomyelination in Adam22-deficient mice. BMC Neurosci. 2005;6:33.
156. Furlan S, Roncaroli F, Forner F, et al. The Lgi1/epitempin gene encodes two protein isoforms differentially expressed in human brain. J Neurochem. 2006;98:985–991.
157. Cuenca-Leon E, Cormand B, Thomson T, et al. Paroxysmal kinesigenic dyskinesia and generalized seizures: clinical and genetic analysis in a Spanish pedigree. Neuropediatrics. 2002;33:288–293.
158. Guerrini R, Sanchez-Carpintero R, Deonna T, et al. Early-onset absence epilepsy and paroxysmal dyskinesia. Epilepsia. 2002;43:1224–1229.
159. Du W, Bautista JF, Yang H, et al. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nature Genet. 2005;37:733–738.
160. Diez-Sampedro A, Silverman WR, Bautista JF, et al. Mechanism of increased open probability by a mutation of the BK channel. J Neurophysiol. 2006;96:1507–1516.
161. Wang B, Rothberg BS, Brenner R. Mechanism of β4 subunit modulation of BK channels. J Gen Physiol. 2006;127:449–465.
162. Brenner R, Chen QH, Vilaythong A, et al. BK channel β4 subunit reduces dentate gyrus excitability and protects against temporal lobe seizures. Nat Neurosci. 2005;8:1752–1759.
163. Mazarati A, Sankar R. Common mechanisms underlying epileptogenesis and the comorbidities of epilepsy. In: Holmes GL, Noebels JL, eds. Epilepsy: The Biology of a Spectrum Disorder. Cold Spring Harb Perspect Med. New York, NY: Cold Spring Harbor; 2016:361–378.
Related posts:
 Epilepsy Febrile Seizure Plus (GEFS+)Douglas R. Nordli, Jr. and John M. Pellock
Epilepsy Febrile Seizure Plus (GEFS+)Douglas R. Nordli, Jr. and John M. Pellock
 Dominant Focal EpilepsiesSarah Weckhuysen, Mary Kurian, Fabienne Picard, and Stephanie Baulac
and PrimidoneStephen Fulton and Amy McGregor
Dominant Focal EpilepsiesSarah Weckhuysen, Mary Kurian, Fabienne Picard, and Stephanie Baulac
and PrimidoneStephen Fulton and Amy McGregor
 Self-Limited EpilepsiesDouglas R. Nordli, Jr., Colin D. Ferrie, and Chrysostomos P. Panayiotopoulos
Self-Limited EpilepsiesDouglas R. Nordli, Jr., Colin D. Ferrie, and Chrysostomos P. Panayiotopoulos
 and Pharmacologic Consequences of SeizuresShilpa D. Kadam and Michael V. Johnston
and Pharmacologic Consequences of SeizuresShilpa D. Kadam and Michael V. Johnston
 Presurgical Functional MappingAndrew C. Papanicolaou, Roozbeh Rezaie, Shalini Narayana, Marina Kilintari, Asim F. Choudhri, Frederick A. Boop, and James W. Wheless
Presurgical Functional MappingAndrew C. Papanicolaou, Roozbeh Rezaie, Shalini Narayana, Marina Kilintari, Asim F. Choudhri, Frederick A. Boop, and James W. Wheless
