Barbiturates constitute the oldest category of anticonvulsant medications that continue to be widely used in the management of epilepsy and other disorders. Their usefulness derives from a combination of efficacy, safety, and low cost. These virtues account for the fact that barbiturates likely remain among the most widely employed anticonvulsants in the world. Although phenobarbital (PB) is the major focus of this chapter, other drugs are reviewed, particularly mephobarbital, pentobarbital, and primidone. Mephobarbital (MPB) is a useful alternative to PB that is more widely employed in some other countries, such as Australia, than in the United States. Like primidone, it undergoes biotransformation to phenobarbital. Among the more sedative barbiturates, pentobarbital has been useful in the management of severe and persistent seizures that do not respond to more routine anticonvulsant therapy. Primidone (PRM) is not actually a barbiturate, as its pyrimidine ring contains only two carbonyl groups, while barbituric acid contains three carbonyl groups. Primidone is subject to biotransformation into phenobarbital and probably exerts most of its anticonvulsant effects in that form. Because the major antiepileptic effects and side effects can be ascribed to the phenobarbital metabolite and because of pharmacokinetic and pharmacodynamic similarities, it is quite appropriate to consider primidone in this chapter.
PHENOBARBITAL
Phenobarbital is 5-ethyl-5-phenyl substituted barbituric acid, with a molecular weight of 232.23. It is a weakly acidic substance with a pKa that is usually reported as 7.3 (1–4). The free acid is a white crystalline powder that has low aqueous and relatively low lipid solubility; however, the sodium salt, which is used for intravenous (IV) and intramuscular (IM) preparations, is freely soluble in slightly alkaline aqueous solutions.
Mechanisms of Action
Phenobarbital exhibits a wide spectrum of anticonvulsant activity, conferring protection to animals subjected either to electroshock or to chemically induced (pentylenetetrazol or bicuculline) experimental seizures (5,6). This spectrum is shared by most barbiturates and is consistent with their wide spectrum of activity in clinical seizure disorders.
Understanding of the antiepileptic activity of phenobarbital has been limited by the incomplete state of our understanding of the mechanisms of epilepsy. Current views suggest that phenobarbital modulates the postsynaptic effects of certain neurotransmitters. The modulation is thought to primarily affect the inhibitory substance gamma-aminobutyric acid (GABA).
Whether by these or other mechanisms, antiepileptic barbiturates appear to elevate the threshold to chemical or electrical induction of seizures in ways that differ from and are in some respects superior to those of phenytoin (7).
A large body of information has accumulated concerning the ability of barbiturates to depress physiologic excitation in the nervous system and enhance inhibition of synaptic transmission. Phenobarbital shares with pentobarbital (PnB) the capacity for selective postsynaptic augmentation of GABA-mediated inhibition and depression of glutamate- and quisqualate-mediated excitation in at least some central nervous system (CNS) regions (8–14). Barbiturate augmentation of GABA-stimulated postsynaptic inhibition appears to be due to activation of a subset (alpha-beta) of the GABAA-receptor gated chloride channels (13,15). Benzodiazepines and barbiturates work on the same GABA receptor, coupled to the chloride ionophore, but they bind to different allosteric sites (formed by different subunits of the pentameric GABA receptor) (16). However, benzodiazepines increase the frequency of opening of the chloride channel, while barbiturates prolong the opening of the channel (17). These postsynaptic effects are produced at clinically relevant concentrations (12,13).
The reduction of voltage-activated calcium currents may, in part, account for the sedative and anesthetic effects of barbiturates and may possibly play a role in the efficacy of pentobarbital and very high concentrations of phenobarbital in suppressing seizures (13). This is another potential mechanism whereby these agents may work in the setting of intractable status epilepticus with barbiturate coma and may represent one of the mechanisms for production of anesthesia (13).
Pharmacokinetics
Absorption. Phenobarbital is rapidly and nearly completely absorbed after oral or intramuscular (IM) administration to infants or children. For most children older than 6 months of age or adults, it is likely that peak serum concentrations of phenobarbital are achieved by 2 hours after oral and 2 to 4 hours after IM bolus administration of the usual age-appropriate maintenance doses. The bioavailability of most oral and parenteral formulations is essentially quantitative (85%–100%) through a wide range of doses in otherwise healthy children (>6 months of age) or adults. Rectally administered parenteral solutions of sodium phenobarbital are well absorbed at all ages, although the latency to peak concentration may be slightly longer and the bioavailability slightly lower than after IM administration (18).
Distribution. Phenobarbital disseminates into all body tissues. Only approximately 50% or less of circulating phenobarbital is bound to serum proteins in most patients whose ages are greater than 3 to 6 months. Equilibration of phenobarbital across the blood–brain barrier is relatively slow. Twelve to 60 minutes are required for maximal brain-to-plasma phenobarbital ratios in the adult mammalian brain after intravenous (IV) administration. These data suggest that (a) dosage of phenobarbital should be based on lean body mass to avoid overdosing obese individuals (19,20), and (b) sufficient time for maximal brain penetration should be allowed to occur after bolus administration of phenobarbital before administration of additional doses.
Phenobarbital readily crosses the placenta and is secreted in breast milk (21,22). Breast milk concentrations were 36% ± 20% and 41% ± 16% of maternal serum concentrations in two studies (23,24). The newborn infants of mothers treated with phenobarbital have levels equivalent to those of their mothers immediately after birth (23,25–28). Estimates of the apparent volume of distribution (Vd) of phenobarbital vary over nearly a four-fold range but are generally larger in infants and small children than in older individuals (29–31). The Vds for newborns and infants less than 4 months of age treated with IV phenobarbital average approximately 0.9 to 1.0 L/kg, independent of body weight, dose, gestational age, or occurrence of asphyxia (29,32–35). Older children and adults exhibit Vds that range from approximately 0.45 to 0.7 L/kg more or less irrespective of route of administration (36–38).
Metabolism and Elimination. Phenobarbital may be excreted unchanged or may undergo biotransformation before excretion. The most quantitatively important fates for phenobarbital metabolism include (a) aromatic hydroxylation to p-hydroxyphenobarbital (PBOH) and (b) N-glucosidation to 9-D-glucopyranosyl phenobarbital (PNG) (39,40). On average (with wide interindividual variation), approximately 20% to 30% of a daily dose of phenobarbital is converted to the pharmacologically unimportant PBOH. Hydroxylation of phenobarbital occurs by way of the cytochrome P450 system, the majority of which is converted by CYP2C9, and less so by CYP2C19 and CYP2E1. Genetic variations in different ethnicities exist, with 5% of Caucasian individuals and 20% of Asian individuals having poor CYP2C19 metabolism. The reverse occurs with poor metabolism by CYP2C9 being seen in 35% of Caucasian individuals and less than 10% of Asian and African American individuals. However, the degree to which genetic variations in CYP2C19 and CYP2C9 alter the metabolism of phenobarbital is not known (41). In most children and adults, about half of the PBOH is excreted unchanged and about half is excreted as a PBOH-glucuronide conjugate that is formed in the liver (42). Although phenobarbital is the classic inducer of hepatic microsomal metabolism, it does not appear to induce significant changes in its own metabolic rate or plasma clearance in humans, although slight effects may occasionally be detected (43,44).
Phenobarbital is thought to be a substrate of P-glycoprotein, which, in some respects, varies according to MDR1 or ABCB1 polymorphisms, which are themselves quite variable across different ethnicities (41).
Phenobarbital has a long half-time of elimination. The two-standard-deviation range for half-time of elimination in children and adults is 24 to 140 hours, resulting in the capacity to eliminate between 11% and 50% of total body phenobarbital in 24 hours (43,45–49). Elimination half-time is longest in premature and full-term newborns, with various studies showing mean values of 100 to 200 hours with standard deviations of 30% to 80% (full range across these studies of 59 to greater than 400 hours) (31,33,50–53). Clearance may vary from day to day in individual babies (53); however, the rate of elimination may double by the second week of life and tends to continue to increase for the ensuing few weeks. Infants 6 weeks to 12 months old have the shortest mean half-times of elimination (30–75 hours) of any age group (31,35,54,55). At age 2 months, half-time of elimination is usually in the range of 39 to 55 hours. Children 1 to 15 years of age typically have half-time of elimination of 37 to 73 (68 ± 30) hours, while subjects 15 to 40 years of age have 53 to 141 (100 ± 20) hours half-time of elimination (30,56). Perinatal asphyxia may considerably decrease the clearance by newborns, probably due to the combination of renal and hepatic dysfunction (22,35,38,54,57–66). Clearance of unmetabolized phenobarbital may be higher at higher rates of urine formation (67–69) or in more alkaline urine, as both of these conditions reduce the rate of phenobarbital resorption in the distal nephron. Eight-fold increase in the rate of urine formation may increase phenobarbital clearance by three- to four-fold (70,71). This effect may be considerably enhanced by the alkalinization of urine with sodium bicarbonate (4).
Interactions With Other Drugs
Although some drugs affect phenobarbital kinetics, most of the pharmacokinetically important interactions encountered with the use of phenobarbital are those caused by the effects of phenobarbital on the kinetics of other drugs. The most common effect that phenobarbital has on the metabolism of other drugs is to increase their biotransformation, thereby increasing their clearance rate. Phenobarbital is the prototypical inducer of the hepatic mixed-function oxidase system that comprises, among other elements, the numerous isoenzymes of cytochrome P450 and of NADPH-cytochrome c reductase.
A list of drugs, that is not exhaustive, for which this effect may be clinically important is provided in Table 49.1 (44,72–96). In several instances, as indicated in Table 49.1, phenobarbital may result in increased concentrations of potentially toxic metabolites. The enhancement of potential valproate toxicity to the kidney and liver must be considered in cases in which these drugs are used in combination. Renal tubular injury may be exacerbated by the increased dose of valproate required in some patients taking phenobarbital (94,95), while the chance of hepatic injury may be increased because of phenobarbital-induced production of the “4-en” metabolite of valproate, which appears to be toxic to hepatocytes (97–100). Phenobarbital coadministration may increase the risk for valproate-induced hyperammonemic encephalopathy, perhaps by means of unfavorable effects on ammonia clearance (101).
TABLE 49.1
|
DRUGS SUBJECT TO KINETIC ALTERATION WHEN ADMINISTERED TO PATIENTS RECEIVING PHENOBARBITAL |
|
Shortened Half-Time of Elimination AND/OR Peak Levels |
|
Amidopyrine (44) Bishydroxycoumarin (74) Carbamazepine, 10,11-carbamazepine epoxide (75–77) Chloramphenicol (78) Cimetidine (81) Cyclosporine (82) Doxycycline (83) Ethylmorphine (84) Griseofulvin (87) Haloperidol (88) Hexobarbital (85) Mesoridazine (88) Methsuximide (91) Nortriptyline (92) Theophylline (93) Warfarin (96) |
*May result in increased levels of toxic metabolites.
In the past, it was thought that while phenobarbital induces the metabolism of phenytoin, the degree of that effect is seldom great enough to cause an adjustment of phenytoin dosage (102,103). More recent data suggests a more complex interaction that can result in either a decreased or increased serum level of phenytoin when administered concurrently with phenobarbital (104). As phenobarbital produces significant induction of CYP enzymes as a result of altered transcriptional regulation, there is increased clearance and reduced levels of multiple antiepileptic medications including valproic acid, carbamazepine, lamotrigine, felbamate, topiramate, tiagabine, ethosuximide, and zonisamide (41). Rufinamide clearance is also increased by phenobarbital (105). There is some evidence that clearance of lacosamide appears to be unaffected by concurrent administration with phenobarbital (104). However, there is conflicting evidence of a roughly 30% decrease in lacosamide levels when coadministered with phenobarbital (106).
Other important potential effects of comedication with phenobarbital include inadequate anticoagulation with warfarin (44,74,96), reduction of serum levels of exogenously administered prednisone or dexamethasone (107,108), or failure of oral contraception (109,110). Coadministration of phenobarbital with warfarin or other medications can be managed when the combination is essential. In such cases, care must be taken to adjust anticoagulants with any changes in or discontinuation of phenobarbital (111). The effect of phenobarbital on these various drugs may become manifest within days to weeks of initiation of comedication.
Less commonly, other drugs affect phenobarbital kinetics. The most important of these in everyday practice is the interaction of phenobarbital and valproic acid. Accumulation of phenobarbital occurs in most patients who are comedicated with phenobarbital and valproate, accompanied by a lower than expected serum level to dosage ratio of valproate. The rate and magnitude of these effects are variable but generally require lowering of phenobarbital and increasing of valproate dosages as compared to what might be expected with monotherapy (112). Elevation of phenobarbital levels may occur within days of initiation of valproate, but more typically the increase occurs slowly over a number of weeks.
Adverse Effects
Experience has demonstrated that phenobarbital generally is a very safe and predictable medication. Nonetheless, it does produce various undesirable effects. The majority of these are reversible effects that can be tolerated but reduce the attractiveness of phenobarbital therapy. Serious side effects also occur, but they are rare. The most frequently encountered adverse characteristics of phenobarbital are (a) sedation, (b) disturbances of mood and behavior and possibly cognition, and (c) induction of hepatic metabolism, producing various effects on the disposition of a wide variety of other drugs (as discussed in preceding paragraphs). Exacerbation of seizures may possibly occur with weaning and discontinuation of phenobarbital maintenance. Serious allergic reactions may occur.
Sedation and Behavior. Although phenobarbital has the most favorable ratio of antiepileptic potency to sedative properties among the antiepileptic barbiturates, it is certainly more sedating than most other anticonvulsants (113). Drowsiness is most common at the initiation of therapy, afflicting as many as one-third of newly treated patients. Sedation may occur even at very low doses and may persist for several days, occasionally as long as several weeks (47). Sedation may return with dose increases, and there may be a dose-related increment in difficulty awakening in the morning or increase in the frequency with which a nap is required. However, many patients do not experience significant sedation after initially becoming accustomed to the medication, despite many-fold increases in dose (114,115). Patients receiving chronic phenobarbital therapy are least likely to feel drowsy if their serum level falls between 15 and 30 μg/mL, but there is considerable individual variation in tolerance. Some patients complain of little sedation with levels as high as 50 μg/mL, while others find levels of 10 to 15 μg/mL intolerable because of lethargy (116).
Mood Disturbance. Studies have shown that 30% to 42% of children with febrile seizures treated with phenobarbital prophylaxis experience deterioration in behavior, the majority having relatively low phenobarbital levels (i.e, less than 15 μg/mL). Hyperactivity, irritability, belligerence, intermittent agitation, disruptive and defiant behavior, insomnia, and uncharacteristic episodic sedation are among the most frequent troublesome manifestations—effects that are not related to dose or serum level (117–119).
Some older studies have failed to find a significant incidence of behavioral deterioration of children treated with phenobarbital (120,121). In a double-blind, placebo-controlled study of toddlers, the rate of hyperactivity was no different whether treated with phenobarbital or placebo. There were few significant phenobarbital-related side effects, and those that did occur (irritability and sleep disturbance) responded to dose reduction (122). The prospective, double-blind, randomized, crossover study of Young and coworkers (122) showed no significant worsening of behavior with either phenobarbital or mephobarbital. Drugs that are converted into phenobarbital, such as methylphenobarbital and primidone, are regarded by some as less likely to produce behavioral side effects than phenobarbital; however, these hypotheses have not been subjected to careful trials in children. One study of the treatment of childhood behavioral disturbances with phenobarbital or primidone has shown these drugs to be beneficial in 33% and 11% of children, respectively (123).
However, more recently, a meta-analysis conducted by Glauser (124) has shown that >40% of children with epilepsy treated with phenobarbital experienced a range of negative behavioral effects as well as depression.
Higher Cortical Function. Very early in what has been over a century of clinical use of phenobarbital, the intellectual function of many epileptic patients improved as their seizures came under better control with phenobarbital. With a wider choice of antiepileptic medications and other treatments now available, disturbances of cognition, especially attention and memory, and of skilled motor functions may occur in patients as a consequence of the medication rather than of the epilepsy. In an era of quite limited antiepileptic medication choices, Lennox (125) noted the additional toll that medications might take on epileptic patients with brain injuries, changes readily observed by patients, families, and teachers that were “often subtle and difficult to measure.” Nearly 60% of the epileptic patients treated with phenobarbital whom Lennox studied did not appear to have such difficulties.
Hillesmaa and coworkers (126) found a decreased rate of fetal head growth for human infants of mothers taking phenobarbital. Although some early studies documented changes in various measures of intelligence and learning in patients of various ages with some onset of epilepsy, no patterns of deterioration attributable to phenobarbital treatment were found. In individual cases, IQ measurements increased, while others decreased with phenobarbital treatment (127,128). Dosage in many of these early cases was lower than is now typical, and these studies did not address issues of adherence or attempt to relate intellectual dysfunction to drug levels. A subsequent study suggested, but did not prove, that the everyday intellectual performance of children on phenobarbital was deficient as compared to what might be expected given their performance on standardized tests of intellectual function (129). Schain and coworkers (130) found improvement in intelligence subtest scores, attentiveness, and impulse control in children who had their phenobarbital replaced with carbamazepine, although these children exhibited the simultaneous and potentially confounding variable of improved seizure control. Subtle but statistically significant lowering of performance- and full-scale IQ, verbal and nonverbal task subtest scores, and deterioration of behavior were found to be a consequence of phenobarbital as compared to valproate therapy of epilepsy in a double-blind crossover study (131). Similar results were obtained in another study (132), which also provided evidence on repeat testing for impairment of learning and cognitive development while on phenobarbital compared to valproate-treated or untreated control groups.
Several additional studies have aroused concern, especially in the setting of febrile seizure prophylaxis with phenobarbital. Hirtz and coworkers (133) and Farwell and colleagues (134) demonstrated that mean IQ scores of large cadres of such children were 5.2 to 8.4 points lower than anticipated, compared to untreated or placebo-treated control groups. A disparity of at least 5 points was shown to persist for as long as 6 months after phenobarbital prophylaxis had been discontinued in both studies. Various concerns have been raised about certain aspects of the design of these studies, including low enrollment rates of eligible children, incomplete testing of significant fractions of enrolled children, and particularly the intention-to-treat design. Thus, in the case of one study (135), some children in the placebo group actually received phenobarbital, while less than two-thirds of those in the phenobarbital-treated group received phenobarbital throughout the entire study period (2 years), and one-third of the treatment group had little if any phenobarbital exposure or low drug levels during follow-up. An earlier, smaller, but particularly well designed and executed study did not show any such significant effect of phenobarbital prophylaxis on infant developmental scales over an 8- to 12-month follow-up interval. However, Stanford-Binet Intelligence Scale assessment suggested that in some of these children there were negative effects of phenobarbital on performance of certain memory tasks that were drug concentration related, but the effect was not statistically significant (122). On reassessment of the IQ and academic achievement of 139 children between approximately 6 and 10 years of life, the Stanford–Binet IQ score difference between the treated and untreated groups diminished, but in the Wide Range Achievement Test, children that had been treated with phenobarbital had lower reading scores compared to those children in the placebo arm (136).
Teratogenicity. No prospective studies regarding antiepileptic drug (AED) teratogenicity had been completed until recently. The International Registry of Antiepileptic Drugs in Pregnancy (EURAP) showed a dose-dependent risk of malformations with exposure to a multitude of AEDs. Regarding phenobarbital specifically, at 1 year, 5.4% of the 166 infants born to mothers taking <150 mg of phenobarbital had congenital malformations, whereas 13.7% of the 51 infants born to mothers taking 150 mg or more of phenobarbital had congenital malformations. The malformations encompassed cardiac anomalies, neural tube defects, polydactyly, renal malformations, hypospadias, and others. The malformations occurred more frequently than in those mothers taking lamotrigine and carbamazepine, but less often than in those mothers taking valproic acid. A parental history of malformation was associated with malformations in the child irrespective of any treatment with antiepileptic drugs by roughly a factor of four (137).
Pregnancy registries have not had sufficient statistical power to make comparisons between one drug and another. It was noted, in multiple cohort studies that looked at cardiac malformations, neural tube defects, hypospadias, and orofacial clefts, that there was a higher proportion of cardiac malformations compared to the other three categories of malformations associated with phenobarbital use. The studies are not sufficient for use in a meta-analysis, however. In many studies, the rate of malformations appears to be dose-dependent. For instance, rates of malformation with use of any dose of phenobarbital were significantly higher than with lamotrigine doses of less than 300 mg per day, but at doses above 300 mg per day of lamotrigine, the differences were no longer significant (138).
The data on cognitive outcome in infants born to mothers with epilepsy who were prescribed phenobarbital is limited, and as such the practice parameter from the American Academy of Neurology and the American Epilepsy Society states that phenobarbital is only “possibly” associated with poor cognitive outcomes in male offspring of females with epilepsy compared to unexposed controls (139).
Dependence and Withdrawal. Prolonged administration of phenobarbital produces both habituation and dependence; therefore, significant withdrawal signs and symptoms may be provoked by abrupt discontinuation of the drug. Patients may experience anxiety, irritability, insomnia, mood disturbance, emotional lability, hyperexcitability, and tremulousness, various gastrointestinal disturbances, confusion, or delirium. Therefore, chronically administered phenobarbital should be withdrawn slowly to prevent these various reactions as well as withdrawal seizures. Seizures that occur during withdrawal of phenobarbital (whether the withdrawal is suggested by the physician or undertaken by the nonadherent patient) do not necessarily indicate that the drug remains therapeutically indispensable. In many cases, slower rates of withdrawal permit the drug to be withdrawn without seizure recurrence (114,135,140). A similar abstinence syndrome may occur in newborn infants of mothers treated with phenobarbital, given the ease with which phenobarbital crosses the placenta, rendering a level in the neonate that is close to that found in the mother (51). The abstinence syndrome of the newborn may persist for days to weeks and is likely to be better tolerated and of shorter duration in infants whose mothers received the usual antiepileptic doses of phenobarbital than in infants of phenobarbital-abusing mothers.
Overdosage. Intoxication with phenobarbital can occur because of dosing errors, coadministration of valproic acid, accidental ingestion, and suicide attempts. Several instances have been observed in which IV phenobarbital boluses have been administered in the emergency department to patients on chronic primidone therapy who have presented to the emergency department in status epilepticus, with failure to recognize that primidone is metabolized into phenobarbital. Rapid administration of a full loading dose of phenobarbital (20 mg/kg) to patients with phenobarbital levels of 30 to 40 μg/mL is particularly likely to prompt the development of pulmonary edema and respiratory failure. More frequently encountered are cases in which valproate is added to chronic phenobarbital therapy and patients developed progressive lethargy 3 to 5 weeks later with elevated phenobarbital levels. Inattention, drowsiness, and dysarthric slurring of speech that may resemble drunkenness are often exhibited by patients acutely intoxicated with phenobarbital. Curiously, plasma levels similar to those that produce such effects acutely may be tolerated without evident ill effects after the dose is slowly increased with chronic therapy. Other findings observed in patients that have toxic plasma concentrations (usually >40 μg/mL) include dizziness, constricted pupils, nystagmus, ataxia, or coma (generally with levels above 60 μg/mL).
Phenobarbital levels in excess of 80 μg/mL, although well tolerated in carefully monitored patients with appropriate cardiorespiratory intervention, are potentially lethal if such support is not provided. Such high levels, particularly if acutely achieved, may occasionally produce both cardiac and respiratory dysfunction (141). However, cardiac dysfunction is significantly less likely to occur with phenobarbital than with pentobarbital. Indeed, levels in excess of 130 μg/mL appear to be tolerated reasonably well by patients receiving appropriate intensive care support and requiring burst suppression for the management of intractable seizures.
Other Adverse Effects. Phenobarbital therapy has been associated with vitamin D-deficient osteomalacia (142). This may be the result of mixed-function oxidase induction, resulting in enhanced clearance of 25-hydroxy-cholicalciferol (143–145). The patients who are particularly vulnerable to “phenobarbital rickets” are those who have received many years of phenobarbital therapy, are poorly mobile, and have limited exposure to sunlight; diet also may play a role.
Patients on long-term therapy with older antiepileptic medications had improved bone mineral density with calcium and vitamin D supplementations, and there is data that adding risedronate could prevent new vertebral and nonvertebral fractures (146).
Such serious consequences as Stevens–Johnson syndrome, erythema multiforme, or toxic epidermal necrolysis are rare, but they do occur. Malaise, fatigue, fever, and eosinophilia typically accompany allergic rash, which may start centrally and spread to the face and extremities. Variable degrees of hepatic inflammation may accompany the hypersensitivity reaction in children, including fulminant fatal liver necrosis (147–149). Connective tissue disorders including Dupuytren contracture, Ledderhose syndrome (plantar fibromatosis), Peyronie disease, frozen shoulder, and aching joints have been associated with phenobarbital.
Clinical Use
Phenobarbital is indicated in the treatment of focal seizures with or without impairment of consciousness, as well as primary or secondarily generalized convulsive seizures (tonic, clonic, tonic–clonic) in all age groups. It is the drug of choice in the treatment of most forms of neonatal seizures and for prophylaxis of febrile seizures, and it is among the most valuable agents for the management of status epilepticus. Prior to the modern era of antiepileptic drug development, studies conducted in adults have shown that phenobarbital, primidone, phenytoin, and carbamazepine are equally effective in the management of generalized convulsive seizures (149), and although there may be differences in efficacy in treatment of focal seizures, they are slight. The cooperative VA study showed complete control of primary generalized tonic–clonic seizures in 43% of men receiving phenobarbital or phenytoin, 45% of those receiving primidone, and 48% of those receiving carbamazepine. Only 16% achieved complete control of focal or secondarily generalized seizures with phenobarbital, compared to 43% with carbamazepine (150). Other studies of adults have demonstrated similar results (151,152). One large noncrossover study of 3- to 14-year-old children with generalized tonic–clonic seizures showed a 22% rate of remission with phenobarbital monotherapy, compared to 34% for phenytoin, 40% for carbamazepine, and 16% for valproate (153). This study also showed that localization-related seizures with secondary generalization were completely controlled in only 3% of patients on phenobarbital, as compared to 21% with phenytoin, 25% with carbamazepine, and 4% with valproate (153).
Plasma concentrations of phenobarbital required for control of generalized tonic–clonic seizures may be bimodally distributed. Schmidt (154) found that the majority of responding patients achieved control at phenobarbital levels of 18 ± 10 μg/mL but that a significant minority achieved control at levels of 38 ± 6 μg/mL. One-third of patients considered to have intractable focal seizures (eg, with phenobarbital levels less than 20 μg/mL) were found to improve significantly if “adequate” phenobarbital or primidone levels were achieved (ie, levels high enough to achieve control without intolerable side effects). An additional 13.5% in one study, and 16% in another study, responded if either of these drugs at higher concentrations was combined with phenytoin or carbamazepine (155,156).
For the management of children under 1 year of age who present with focal or generalized convulsive seizures excepting infantile spasms, phenobarbital represents a treatment option because of its relative ease of administration, reliable kinetics, wide therapeutic window, and safety as compared to phenytoin or valproate. It is less frequently chosen in older children because of sedative qualities and potential effects on behavior. Nonetheless, it should still be considered for older children whose seizures remain resistant to other treatments. The use of phenobarbital as part of polytherapy for such resistant seizures may introduce difficulties because of sedative effects and induction of hepatic enzymes. One study showed that one-third of patients receiving combinations containing phenobarbital had improved seizure control when phenobarbital was eliminated from the regimen (157).
Initiation of Phenobarbital Therapy. Phenobarbital loading can be achieved by IV or oral administration. IV loading typically requires administration of 15 to 20 mg/kg for newborns or very young infants and 10 to 20 mg/kg for older infants and children. The drug may be administered as a single dose or in two doses divided by a few hours (34,158,159). A number of different approaches to oral loading have been described. Administration of 6 to 8 mg/kg/day for 2 days followed by an age-appropriate daily maintenance dose will quickly render plasma levels of at least 10 μg/mL (151,160). Bourgeois (161) demonstrated that the phenobarbital dose could be increased over 4 days as total daily doses of 3, 3.5, 4, and 5 mg/kg/day on successive days to achieve, in a linear fashion and without significant interim sedation, a serum level of approximately 20 μg/mL at 96 hours. The maintenance dose must be adjusted thereafter to prevent toxic accumulation of phenobarbital. Without some form of initial loading, as many as 30 days may be necessary to achieve maximal steady-state phenobarbital concentrations (162). Obese adolescent patients may have a volume of distribution of 0.5 L/kg or less, and suitable adjustment in their loading dose must be considered in some cases.
Daily maintenance dose requirements for children are higher, on a weight basis, than those for adults, averaging 2 to 4 mg/kg/day (158,163). To maintain plasma levels of 10 to 25 μg/mL, Rossi (164) recommended oral maintenance doses of 4.79 ± 1.3 mg/kg in infants aged 2 to 12 months, 3.5 ± 0.99 mg/kg in children aged 1 to 3 years, and 2.31 ± 0.74 mg/kg in those 3 to 6.5 years. The data of others would suggest that for children older than 3 years who weigh less than 40 kg, doses of 1.5 to 3 mg/kg/day are appropriate, whereas doses no greater than 1 to 1.5 mg/kg/day may maintain satisfactory levels for adolescents and adults who weigh more than 40 kg (151,160). The phenobarbital dose may be administered entirely at bedtime or divided throughout the day, depending on the degree of sleep disturbance and susceptibility to behavioral abnormalities or sedative effects. Once-daily administration is beneficial to some but not all patients.
Neonatal Seizures. Phenobarbital is the most widely employed drug for the management of neonatal seizures. This practice reflects familiarity with the agent and widely shared confidence in the efficacy and safety of phenobarbital in neonates rather than any well-established evidence for the superiority of this agent over other anticonvulsants. The choice of phenobarbital may be based more on the potential risks associated with other drugs than on any demonstrated superiority of phenobarbital (165–168). The second most commonly employed agent, phenytoin, carries risks for tissue injury if tissues are infiltrated at the site of IV line placement or for adverse cardiac effects if the rate of administration is excessive, problems that are less frequently encountered with fosphenytoin. However, nonlinear kinetics make oral administration of phenytoin a problem in very small infants. Phenobarbital and phenytoin were found to be equally effective, though incompletely so, when used for neonatal seizures, with less than half of infants achieving seizure control with either drug used as monotherapy (169).
The initiation of phenobarbital therapy for seizures in newborns should start with IV administration of a loading dose of 16 to 20 mg/kg delivered as a single bolus or as two divided boluses. Volume of distribution of an administered bolus of phenobarbital to a neonate has been variously estimated at 0.81 to 0.97 L/kg, with 15% to 20% deviation of such mean values, and does not vary as a result of gestational age of the newborn infant (170,171). From a practical vantage point, a volume of distribution of approximately 1.0 L/kg can safely be presumed in most cases, with a loading dose of 20 mg/kg resulting in a peak serum level of 20 μg/mL. Plasma protein binding of phenobarbital averages 22% to 25% in neonates, approximately half the value that is anticipated in older children and adults (172).
Successful control of neonatal seizures is seldom achieved with serum levels less than 16 μg/mL; initial loading should attempt to achieve serum levels ranging from 15 to 25 μg/mL. Electrographic monitoring has shown that many clinically responding newborns show persistence of electrographic seizures after routine loading doses (173). Although the significance of electrographic activity in the newborn without clinical seizures remains uncertain, infants with recalcitrant clinical seizures require additional loading boluses delivered as 10 mg/kg at intervals of one to several hours. At plasma concentrations of at least 40 μg/mL, as many as 77% to 85% of neonates respond (171,172). Serum levels as high as 60 to 80 μg/mL appear to be tolerated by most newborns, although such high levels may necessitate greater degrees of cardiorespiratory support, compromise clinical examination, and interfere with feeding. Svenningson and coworkers (174) found that plasma phenobarbital concentrations in excess of 50 μg/mL in newborns were associated with slowing of the heart rate to less than 100 beats per minute. This is a potentially serious matter, because the neonate lacks the reflexive capacity to alter stroke volume in compensation for bradycardia.
Newborns respond to an initial loading dosage of 20 mg/kg, a serum level of 20 μg/mL, and oral or IV total daily maintenance dose of 2.25 to 4 mg/kg. Although doses of 5 mg/kg/day are widely recommended, the continuation of such a dose through the first few weeks of life generally results in accumulation to serum levels significantly in excess of 20 mg/dL (34,53,175). Plasma clearance of phenobarbital usually increases after 1 to 2 weeks of life and may require modification of the maintenance dosage in some but not all cases.
Status Epilepticus. The ease and relative safety of administration, wide therapeutic window, and long duration of action combine to make phenobarbital a particularly attractive choice in the treatment of status epilepticus. Negative aspects of the use of this drug include the sedative effects, and the possible provocation of respiratory depression, hypotension, or pulmonary edema. On the other hand, phenobarbital can be administered more rapidly and in higher concentration than phenytoin, and it can be administered intramuscularly if necessary. The benzodiazepines (IM midazolam, IV lorazepam, or IV diazepam) are the first-line treatments of choice for status epilepticus and are supported by multiple randomized controlled trials. The second-line choices, though none are preferred over the other based on evidence, will be one out of three medications: fosphenytoin, valproate, and levetiracetam, with the choice of IV phenobarbital being an option both for first- and second-line therapy for status epilepticus if the previous options are not available (176).
The rate of administration of sequential boluses of phenobarbital determines the risk for cardiopulmonary complications, which is lower for phenobarbital than for pentobarbital or too rapidly administered phenytoin. In general, the rate of IV administration of phenobarbital for treatment of status epilepticus should be 2 mg/kg/min for children who weigh less than 40 kg. The rate should be 100 mg/min for children and adults who weigh more than 40 kg. Slower rates may be required in special cases, such as in patients with acute cardiac disease (eg, tricyclic overdose). In general, respiratory depression is not seen below plasma levels of 60 μg/mL, and hypotension may not arise as a complication of phenobarbital until after even higher levels are achieved. Pulmonary edema is an uncommon complication and usually requires very massive phenobarbital bolusing over short time intervals to high levels. The most widely accepted practice for phenobarbital administration in treatment of status epilepticus in the phenobarbital-naive patient is to administer a total loading dose of 20 mg/kg. This quite reliably produces a plasma level close to 20 μg/mL. It is clear that the administration of only a partial loading dose (eg, 10 mg/kg) to the anticonvulsant-naive patient usually is inadequate. Seizures may well diminish or stop, but they often recur as the phenobarbital becomes distributed throughout the body. Respiratory support is usually required as the plasma level rises above 50 to 70 μg/mL, and vasopressor support may be required above levels of 70 to 90 μg/mL, whether as the result of phenobarbital, of the causative illness, or both.
Febrile Seizures. From a historical perspective, phenobarbital has been the most commonly employed prophylactic agent for prevention of febrile seizures. As febrile seizures are generally without significant immediate or long-term serious medical consequences, there has been significant momentum away from providing prophylactic treatment. Enthusiasm for prophylactic treatment has diminished because (a) approximately two-thirds of children have just one febrile seizure, (b) recurrent febrile seizures have exceedingly low risk for untoward consequences, (c) phenobarbital and other agents introduce a risk for various drug-related side effects (117), and (d) it has been difficult to provide convincing proof that prophylaxis is effective. Although there is some evidence supporting the effectiveness of treatment with phenobarbital, sodium valproate, or benzodiazepines, it is not conclusive proof.
Phenobarbital is usually judged a safer choice than valproate for administration to very young children, and long-term prophylaxis with benzodiazepines poses unacceptable problems with sedation and tachyphylaxis. However, phenobarbital is not without risk, as the death of one child has been ascribed to the use of phenobarbital for prophylaxis against febrile seizures (147). Furthermore, the efficacy of phenobarbital as prophylactic therapy has recently come into question (177).
There is evidence to suggest that if phenobarbital prophylaxis is to be effective, steady-state serum concentrations of 16 to 30 μg/mL are required (178). This study demonstrated a 4% risk for recurrence in children with such levels compared to approximately 20% rates of recurrence for untreated children and for those with phenobarbital levels of 8 to 15 μg/mL. Several studies have failed to demonstrate a difference in outcome between groups of children at risk for febrile seizures who received either phenobarbital or valproate for prophylaxis compared to children who received no prophylactic anticonvulsant medication (134,179,180). However, these studies did not control for the important element of adherence by assessing serum phenobarbital levels at time of recurrence. Adherence with phenobarbital prophylactic regimens for febrile seizures is notoriously low (181). Herranz and coworkers (182)found that 20% of children treated with phenobarbital prophylaxis for febrile seizures had recurrences at mean levels of 16.4 ± 2.8 μg/mL, compared to 88% of those treated with primidone (mean phenobarbital levels 14.1 ± 3.7 μg/mL) and 92% of those treated with valproate (mean levels 35.2 ± 5.9 μg/mL). Side effects were experienced by 7% of those on phenobarbital, 53% of those on primidone, and 45% of those on valproate, although most side effects were tolerable.
Phenobarbital is discontinued gradually in most cases. This is based on evidence that dependence on phenobarbital results in provocation of seizures if the rate of decline of phenobarbital levels is too rapid. This presumption has been placed in question in one study of patients with focal complex seizures that appeared to demonstrate that the risk for seizures was not dependent on rate of withdrawal but on the achievement of a concentration below 15 to 20 μg/mL (183).
In more recent history, an extensive review found no clinically significant or important benefits from treatment with any of several antiepileptic or antipyretic drugs for children with febrile seizures. As such, phenobarbital is not widely used in the treatment of febrile seizures given the potential of both physical and cognitive side effects (184).
OTHER BARBITURATES
The N-methylbarbiturates (methylphenobarbital and metharbital) and the anesthetic pentobarbital have all been used as anticonvulsants.
Methylphenobarbital
Methylphenobarbital (Mebaral®, MBL), although infrequently used in the United States, is widely employed in some countries, such as Australia. A considerable portion of the administered dose is rapidly cleared as the R-enantiomer; therefore, the dose of racemic methylphenobarbital should be approximately twice the phenobarbital dose required to achieve satisfactory clinical effects (185–187). The single oral dose half-time of elimination for the R-enantiomer has been estimated at 7.5 ± 1.7 hours, compared to 69.8 ± 19.7 hours for the S-enantiomer and 98.0 ± 19.7 hours for the phenobarbital metabolite, with most of the circulating phenobarbital being derived from the S-enantiomer (188), unless the patient has CYP2C19 deficiency, in which case a much larger portion of the phenobarbital metabolite is derived from the R-enantiomer.
Methylphenobarbital clearance is almost entirely by biotransformation with urinary excretion of the major metabolites, which are phenobarbital and para-hydroxymethyl phenobarbital (as a phenolic glucuronide conjugate) (187,189,190). Phenobarbital is the only pharmacologically important biotransformation product. The capacity to generate this product may increase with chronic therapy because of increased rate of methylphenobarbital clearance with faster rates of appearance and higher peak levels of phenobarbital. Naive subjects may excrete less than 11% of their methylphenobarbital dose as phenobarbital, while subjects exposed to methylphenobarbital or phenobarbital may increase that amount to more than 50% (189). Because phenobarbital has a smaller volume of distribution and is cleared more slowly than methylphenobarbital, plasma phenobarbital levels may accumulate over time to much higher values than simultaneous serum methylphenobarbital values. Many or most of the drug interactions experienced with chronic methylphenobarbital therapy are thought to be due to phenobarbital, and any interaction or side effect that has been described for phenobarbital can occur with methylphenobarbital therapy.
The experimental and clinical spectrum of methylphenobarbital is similar to that of phenobarbital (191). It may be administered once or twice daily. Because of the tendency for phenobarbital levels to accumulate to higher serum levels than methylphenobarbital levels over the long term and the greater availability of phenobarbital level determinations, most clinicians follow-up only the phenobarbital level in patients taking methylphenobarbital. Doses that are calculated to produce therapeutic steady-state phenobarbital levels may result in unacceptable drowsiness during the initial phases of therapy, and therefore adherence may be poor. Starting with half the expected dose and accelerating the dose over 1 to 2 weeks avoids this problem but delays the achievement of the desired steady-state peak phenobarbital concentrations to as long as 4 to 5 weeks after initiation of therapy. Full initial doses may be started in patients who have recently been treated with phenobarbital or other “inducing” anticonvulsants. At steady state, phenobarbital concentration is generally 7 to 10 times greater than the total (R + S) methylphenobarbital concentration in serum.
At the current time, methylphenobarbital is no longer available in the United States or the United Kingdom, but it is still employed in various other countries in epilepsy treatment.
Pentobarbital
Pentobarbital (PnB) is a 5-ethyl-5 (1-methylbutyl) barbiturate that is clinically employed as a sodium salt (Nembutal®). It is a short-acting barbiturate. The half-time of elimination in adults ranges from 18 to 50 hours and is dose dependent. The partition coefficient for pentobarbital is approximately 11 times greater than that for phenobarbital. This reflects much faster lipid solubility, accounting for shorter latency in onset of activity, shorter duration of action, and faster metabolic degradation. The superior lipid solubility may be, in part, responsible for the fact that pentobarbital is much more potently sedative and hypnotic than phenobarbital. Acutely achieved blood concentrations of 0.5 to 3 μg/mL produce approximately the same degree of sedation as phenobarbital levels of 5 to 40 μg/mL. Pentobarbital levels >10 to 18 μg/mL usually induce coma. When pentobarbital is employed as a constant infusion of 0.3 to 4.0 mg/kg/hr after bolus administration of 15 mg/kg over 1 hour, the serum half-time of elimination ranges from approximately 11 to 23 hours in adults (192).
Pentobarbital is indicated for the treatment of status epilepticus that is unresponsive to such first-line therapies as benzodiazepines, fosphenytoin, or phenobarbital. The aim of pentobarbital therapy is to produce coma with suppression-burst pattern on EEG; therefore, the sedative properties of this drug are not deleterious. On the other hand, pentobarbital is more likely than phenobarbital to have negative effects on cardiac contractility and to require the addition of cardiotonic medications to support blood pressure and perfusion when employed in the usual doses. Particular caution should be used in the management of patients who have cardiac disease and those whose seizures are the result of hypoxic-ischemic injury or tricyclic overdose, because the negative effects on cardiac function may be particularly deleterious in such settings. Barbiturate anesthesia (with pentobarbital more commonly than thiopental or methohexital) is considered by many neurologists to be the ultimate form of therapy for status epilepticus, which has proved intractable to the usual combinations and dosages of short- and long-acting anticonvulsants, including phenobarbital (193,194). Treatment with pentobarbital has not been proven to be superior to the use of very high doses of phenobarbital to achieve burst suppression or control of seizures.
Lowenstein and associates (192) reviewed their results with eight retrospectively studied and six prospectively enrolled patients treated with pentobarbital, thiopental, or methohexital anesthesia; only one of these patients was a child. Pentobarbital loading with 15 mg/kg over 1 hour to the prospective group resulted in prompt cessation of seizures in patients treated with this protocol. Pentobarbital was infused at rates that varied from 0.3 to 4.0 mg/kg/hr, and additional boluses were administered as needed of 5 mg/kg (maximum of 30 mg/kg of bolus drug in the first 12 hours) to achieve and maintain burst suppression. The median peak serum pentobarbital level for patients treated in this fashion was 10.8 μg/mL (range 6.5–21.2 μg/mL). Treatment resulted in a favorable outcome for three of these six adults, whereas one had a poor outcome and two had indeterminate outcomes. Only two of these six required vasopressors. Significant drop in blood pressure occurred within a few hours of initiation of therapy in 9 of the 14 patients of the entire group reported by these investigators.
Lowenstein and colleagues stopped pentobarbital after approximately 12 hours of treatment, restarting therapy if seizures recurred. Duration of infusion ranged (in the prospective group) from 11 to 77 hours. For those patients who recovered function after cessation of anesthesia, brainstem functions returned in 6 to 24 hours, and various forms of motor activity returned in 1 to 72 hours. Some possible withdrawal phenomena occurred, including repetitive twitching of the extremities, resembling activity observed in some cases of barbiturate overdose and was difficult to distinguish on a clinical basis from seizure activity (192,195,196). Patients receiving particularly high doses of pentobarbital were found in some instances to have profound weakness and areflexia that persisted for as long as 2 weeks (including as many as 5 days of complete paralysis after cessation of infusion), despite much more rapid recovery of alertness and intellectual interactiveness. This effect may have been the result of pentobarbital-induced dysfunction of peripheral nerve function or release of neurotransmitter in the peripheral synaptic cleft, as has been observed experimentally (197,198). These transient forms of dysfunction may introduce confusion into attempts to assess patients for “brain death” or to estimate prognosis.
Primidone
Primidone (PRM), 5-ethyldihydro-5-phenyl-4,6 (1H, 5H) pyrimidinedione or 2-desoxy-phenobarbital, was synthesized in 1949 and was shown to be effective in the treatment of major motor epilepsy shortly thereafter (199). It is therefore the third oldest anticonvulsant that continues in regular use (200). It has just two carbonyl substituents of the pyrimidine ring rather than the three that are characteristic of barbiturates. Hepatic biotransformation renders two main metabolites of PRM: phenylethylmalonamide (PEMA) and phenobarbital. The identification of phenobarbital as a metabolite raised questions as to the importance of the other substances in control of seizures, a subject that remains controversial. Although it has remained difficult to clearly demonstrate the contribution that primidone and PEMA make to clinical management of epilepsy independent of the effects of phenobarbital, there are both experimental and clinical data that support the view that they are pharmacodynamically important.
Anticonvulsant Activity
The fact that primidone has anticonvulsant properties is suggested by several lines of evidence, including the facts that (a) administration of primidone lowers the plasma level of phenobarbital required to protect animals against experimental forms of seizure, and (b) protection against seizures is afforded in the few hours after a single primidone dose is administered before the achievement of significant levels of PEMA or phenobarbital (5,6,201–203). The potency of primidone is equal to or possibly superior to that of phenobarbital in the prevention of electroshock-induced seizures, but it has much less activity against chemically induced (pentylenetetrazol or bicuculline) seizures (6,200,201,203).
Baumel and coworkers (202) roughly estimated the potency (ED50 for prevention of maximal electroshock seizures) of primidone to be 25% more and that of PEMA to be 90% less than that of phenobarbital. They also found that primidone had no detectable antichemoconvulsant activity, while PEMA is approximately 0.025 as potent as phenobarbital for prevention of that form of experimental seizure. Other researchers in a variety of animal lines have found somewhat different potency ratios of these compounds (5,203,204), but primidone and phenobarbital generally have had similar antielectroshock potency, while PEMA has been 12- to 18-fold less potent. Experimental evidence from mice evaluated on the rotorod (5) suggests that primidone is approximately 2.5 times less neurotoxic than phenobarbital and that the two substances are synergistic in seizure control, possibly exerting different mechanisms of action. Bourgeois and coworkers established that a ratio of 1:1 brain concentration of primidone to phenobarbital achieved the best therapeutic index (ratio of therapeutic efficacy to toxicity) in control of seizures (5). A therapeutic range for primidone trough concentration of 3 to 12 μg/mL has been suggested (205), but somewhat higher levels may be tolerated. Although PEMA also has anticonvulsant properties, the potency appears to be too low to contribute much to the antiepileptic effects or toxicity of primidone at the usually encountered plasma concentrations of clinical practice (161). Monitoring of serum PEMA levels in patients treated with primidone has no practical value.
Pharmacokinetics
Peak serum primidone levels are achieved in approximately 3 hours in adults and 4 to 6 hours in children after single-dose ingestion of tablets in doses of 12.5 to 20 mg/kg (206). Brand-name tablets probably have nearly complete bioavailability, with 72% to 100% of a single oral dose excreted in urine as primidone or its metabolites (206–208). Absorption of generic preparations may be less reliable (209). Absorption of primidone tablets is reduced by concurrent acetazolamide administration (210). Volume of distribution of primidone and of PEMA after single oral dose has been estimated at 0.86 L/kg and 0.69 L/kg, respectively. There is less than 10% plasma protein binding of either primidone or of PEMA (211,212).
Biotransformation of primidone is very complicated. PEMA and phenobarbital are the two major metabolites with antiepileptic potency. It has been difficult to estimate the relative contribution of PEMA and phenobarbital to the pharmacodynamic properties of primidone, including both antiepileptic potency and toxicity, although these properties have been studied carefully in animals (5,6). Zavadil and Gallagher (213) showed that, on average, approximately 65% of a single IV primidone dose is excreted unchanged in the urine within 5 days of administration to adults, while 7% is excreted as PEMA and 2% is excreted as phenobarbital. The rate of conversion of primidone to phenobarbital is much slower than that to PEMA. Under steady-state conditions of chronic administration, approximately 25% of monotherapeutically administered primidone can be relied on to be converted to phenobarbital (214). The bioconversion of primidone to phenobarbital shows age-related variation. In general, the more complete transformation of primidone to phenobarbital by children aged 0.5 to 6.5 years often results in disappointingly low primidone to phenobarbital serum ratios, although there is considerable individual variability (215,216).
Primidone and its various metabolites are primarily (at least 75%–77%) renally excreted (213). The half-time for elimination of orally administered primidone ranges from 10 to 15 hours in adults on monotherapy, but comedication with antiepileptic inducers of hepatic biotransformation enzymes lowers this time to 6.5 to 8.3 hours (207,210,213,217). On average, half-time of elimination for primidone in children ranges from 4.5 to 11 hours, lower with phenytoin comedication than with primidone monotherapy (206). The hepatic immaturity of newborns accounts for half-times of primidone elimination that vary from 8 to 80 hours.
In one study of steady-state primidone monotherapy, the average trough serum concentration to total daily dose of primidone for primidone, PEMA, and phenobarbital were 0.78 ± 0.25, 0.64 ± 0.39, and 1.47 ± 0.53, respectively (218). At steady state, phenobarbital levels are almost always higher than primidone levels. Because phenobarbital is probably responsible for many of the toxic effects of primidone, especially sedation, attempts have been made to diminish the biotransformation of primidone to phenobarbital. As noted previously, a brain ratio of 1:1 for primidone and phenobarbital concentrations has been established experimentally as ideal for the achievement of maximal therapeutic efficacy with minimal toxicity (5). Both nicotinamide and isoniazid have been tried for this purpose, but gastrointestinal, hepatic, and other possible toxicities have limited the usefulness of such approaches (219–221). In most clinical situations, primidone levels are of relatively little value as management tools. They may be useful in two situations, however: (a) assigning a cause for possible drug side effects or (b) establishing that the conversion rate of primidone to phenobarbital is unfavorably rapid and therefore that the use of the more expensive and inconvenient-to-administer primidone is poorly justified.
Drug Interactions
Coadministration of primidone and some other major antiepileptic drugs to adults decreases excretion of unchanged primidone after a single dose by more than one-third. This is the result of four-fold increase in the conversion rate to PEMA and 50% increase in conversion to phenobarbital. Interactions with various anticonvulsants may result in considerable changes in the ratios of primidone, PEMA, and phenobarbital. Phenytoin is the most potent accelerator of primidone biotransformation, while carbamazepine has a lesser effect (210,215,222,223). Bourgeois (161) showed that, on average, coadministration of primidone with either phenytoin or carbamazepine, or both, resulted in 50% reduction in the trough primidone level at steady state compared to that expected with monotherapy. PEMA levels increased by 17% and phenobarbital levels by 60% under those polytherapeutic conditions, and the trough ratio of phenobarbital to primidone increased by as much as three-fold. Application of the data obtained in Bourgeois’s study predicts that with primidone monotherapy a steady-state phenobarbital level of 16.5 μg/mL can be expected to be associated with an average primidone level of 10 μg/mL, but that the coadministration of phenytoin or carbamazepine, or both, increases the average phenobarbital level to a level of 26.5 μg/mL, while the average primidone level falls to 5 μg/mL. If the total daily dose were increased by approximately 100% to maintain the serum PRM level at 10 μg/mL, the phenobarbital level at steady state could be expected to rise to as much as 58.3 μg/mL. Thus, polytherapy with these agents can be expected to lower the therapeutic index of primidone significantly because of the unfavorable ratio of primidone to phenobarbital in the brain compared to the experimentally ideal 1:1 ratio.
When polytherapy is considered necessary, problems of enzymatic induction are avoided by combining primidone with noninducing agents such as valproate, gabapentin, lamotrigine, topiramate, tiagabine, or benzodiazepines (224). However, combination with valproate produces the unfavorable kinetic problems encountered when phenobarbital and valproate are combined (225). Combination with benzodiazepines may produce intolerable sedation. Coadministration of primidone and valproate tends to produce lesser degrees of phenobarbital accumulation than are observed with phenobarbital and valproate coadministration (as stated previously). This may be because valproate inhibits not only phenobarbital clearance but also the conversion of primidone to phenobarbital (225). Thus, if it is judged necessary to coadminister valproate with phenobarbital, it might be better to administer the latter as primidone. Acetazolamide may diminish the gastrointestinal absorption of primidone, and carbamazepine may in some cases inhibit biotransformation of primidone to phenobarbital (221).
Toxicity
Animal studies demonstrate that primidone has less toxicity to the nervous system and other organs than phenobarbital (5,200). Nonetheless, toxic effects, particularly those of the CNS, are common. They are reported at some point in their therapy by one-half to two-thirds of patients who are treated with this drug (149,226–228). Primidone therapy is unsuccessful and is discontinued in 10% to 30% of patients. Discontinuation is much more commonly the result of intolerable side effects than to lack of therapeutic efficacy (149,227). Side effects of primidone therapy closely resemble those seen with phenobarbital. It has been difficult to distinguish the contributions of primidone compared to phenobarbital in the elicitation of these problems. The time of onset of side effects and the development of tolerance assist in making this determination.
Sensations variously characterized as sleepiness, light-headedness, dizziness, weakness, or intoxication are very common, if not universal, at the initiation of primidone therapy (217,229). They develop within a few hours of ingestion of primidone, but there is considerable individual variability in susceptibility to these dose-related effects, which range from mild to severe and incapacitating affectation (230,231). Cross-tolerance occurs in patients who are in the process of changing from phenobarbital to primidone therapy, but those changing from phenytoin or carbamazepine do not experience such cross-tolerance (229). These very early effects are due to primidone, because they occur before any significant accumulation of phenobarbital and usually wane within a few days, despite the fact that that is the time at which phenobarbital levels begin to rise (232,233). The severe effects can be avoided by administration of a small “test dose” at the onset of therapy to determine whether the individual patient is highly susceptible. These effects can be minimized in all patients by careful attention to the speed and amplitude of the acceleration of the dosing schedule to achieve full doses (217,231).
There is a second family of more persistent sedative side effects that develop during the chronic phase of primidone therapy. These chronic side effects are quite similar, if not identical, to those reported by some patients treated with phenobarbital. Patients and families report abnormalities of energy level, attentiveness, behavior, and learning. The frequency with which they are reported, and their amelioration in at least some cases when primidone is discontinued, suggest that the medication may be in part or entirely responsible. Rodin and coworkers (234) compared the effects of primidone and carbamazepine as adjuncts to phenytoin therapy and found that primidone produced more significant impairment of performance in a cognitive-perceptual test battery than carbamazepine, especially with regard to concentration and fine motor performance (235). The fact that these types of difficulties are quite similar, if not identical, to the problems reported during phenobarbital therapy, and the observation that they are most troublesome during the chronic phase of therapy, when the phenobarbital-to-primidone ratio is highest, suggest that phenobarbital is the likely culprit. It is unlikely that PEMA, which contributes little to the anticonvulsant potency of primidone, plays any significant role in CNS or other forms of primidone-related toxicity (211).
Virtually all of the other phenobarbital-related side effects can occur with primidone therapy, including the various connective tissue problems such as contracture formation, joint problems, and Peyronie’s disease. One study has suggested that in children, primidone is less likely to produce any more significant side effects than phenytoin or phenobarbital (119). Transient nausea, vomiting, dizziness, and drowsiness may occur with initiation of primidone therapy, a peculiar combination of side effects that are seldom seen in patients who start on phenobarbital (149).
Overdosage
Little is known about the relationship between peak primidone concentrations and short-term toxic effects. Acute primidone overdosage (primidone levels exceeding 80–100 μg/mL in tolerant patients) may produce varying degrees of CNS depression ranging from somnolence or lethargy to deep coma, flaccidity, and loss of deep tendon reflexes (233,236,237). The degree of these abnormalities tends to correlate with primidone levels rather than phenobarbital or PEMA levels in the acute interval, although the ensuing rise in PB levels produces additional CNS depression (233,238).
Massive overdosage may result in hypotension and acute renal failure in association with crystalluria, especially when serum primidone levels exceed 200 μg/mL (239). Fatal cases of primidone ingestion have been reported. However, with appropriate management (gastric lavage, administration of activated charcoal, forced diuresis, and supportive measures) patients have recovered without permanent sequelae from ingestion of as much as 22 grams of primidone (231). Crystalluria (largely crystallized PEMA), after administration of large amounts of primidone, was first observed in rats (200) and is an almost constant feature of serious PRM overdosage in humans (240). It has not been shown to result in chronic renal failure even after massive overdosage with acute renal dysfunction (237). Massive overdosage can be effectively treated with hemoperfusion (239).
Clinical Use
Early uncontrolled, retrospective studies demonstrated that primidone could be used with considerable success as an adjuvant drug in the management of generalized tonic–clonic seizures, localization-related seizures, and myoclonus epilepsies (226,241–245); however, some more recent controlled studies have shown less favorable results. These data, combined with the availability of a wider range of major anticonvulsants and decreased enthusiasm for polytherapy, have reduced the popularity of this agent. Early comparison studies failed to demonstrate that primidone had some unique value in the treatment of any particular type of epilepsy or population of patients, compared to phenobarbital or phenytoin, drugs that were less expensive and in some ways easier to use (214,246,247). Therefore, it appears that the benefits of primidone therapy and the potential toxicity do not support the use of this agent as first-line therapy for any seizure type or syndrome.
One possible exception is the treatment of epilepsy occurring in patients with long QT syndrome, because primidone may have some value in the treatment of the cardiac dysrhythmia (248). However, most patients who have seizures in association with the long QT syndrome have them only in association with cardiac decompensation, which may be effectively treated with other medications. On the other hand, primidone continues to be useful as a third-line agent for treatment of occasional patients affected with almost any form of epilepsy for which phenobarbital is indicated, including age groups ranging from the neonate (249) to the adult. The exception to this principle is the fact that primidone has little value in the management of status epilepticus, because there is no commercially available parenteral formulation. Primidone may be effective in the prophylaxis of febrile seizures (179). Sapin and coworkers (250) found that primidone exerted antiepileptic effects in neonates that were independent of the effects of phenobarbital.
Various direct comparisons of primidone to phenobarbital, phenytoin, or carbamazepine treatment in patients of various ages have shown these drugs to have similar efficacy for oral treatment of focal, secondarily generalized, and generalized tonic–clonic seizures (149,214,234,247,251). However, most of these studies do not control for phenobarbital levels. Reinterpretation of one of the best of these studies (214) has suggested that a subgroup of patients may enjoy better control at any given phenobarbital level if that level is achieved as the result of primidone rather than phenobarbital oral therapy (252). One crossover comparison showed that primidone was superior to orally administered phenobarbital in the management of generalized tonic–clonic seizures (253). The VA Epilepsy Cooperative study comparison of primidone to phenobarbital, phenytoin, and carbamazepine showed the highest rate of failures in treatment of focal or secondarily generalized seizures occurred with primidone therapy, failure that usually was due to drug discontinuation or poor adherence early in the course of treatment because of intolerable side effects (150).
Two potential benefits of the choice of primidone rather than phenobarbital are (a) the possibility of achieving seizure control in a given subject at lower serum PB levels than are required with the administration of phenobarbital itself, and (b) the possibility that in certain settings primidone is a “better” drug than phenobarbital. Others have argued that most or all of the clinically significant antiepileptic effect of primidone is simply due to its conversion into phenobarbital (254,255). This is particularly likely to be true when primidone is administered too infrequently during the day to achieve the most favorable serum ratio of primidone to phenobarbital. In most patients, this requires administration every 6 hours, which may be inconvenient. Because of unfavorable effects in the primidone-to-phenobarbital ratio, there are reasons to believe that there is no advantage to employing primidone in combination with phenytoin, acetazolamide, or carbamazepine. Unfortunately, most patients who now receive primidone are patients with intractable epilepsy on polytherapy with complicated dosing schedules.
When administered to adults as monotherapy, primidone can be administered twice daily because the half-time of elimination for primidone typically is 10 to 15 hours. However, the shorter elimination half-time that is typical of most children and most patients on polytherapy mandates administration of this drug at 6-hour intervals if advantage is to be taken of the presumed antiepileptic potency of primidone. Administration to newborns is little studied, but in the event that administration of primidone were judged important, once-daily dosing is theoretically possible. So great is the variation of elimination half-times observed in the newborn (23) that individualization of dosage for newborns would be prudent, as would careful follow-up to ensure that toxic accumulation of metabolites does not occur.
Treatment Guidelines
If there are no coadministered drugs that interfere with metabolism or distribution, an initial daily primidone dose of approximately 20 mg/kg/day will result, after 2 to 3 weeks, in a steady-state phenobarbital level ranging from 10 to 30 μg/mL. Neonates typically require maintenance doses of 15 to 25 mg/kg/day, infants 10 to 25 mg/kg/day, and children 10 to 20 mg/kg/day (23). The total daily dose should be divided into three or four times of administration, as noted previously, given the short half-time of elimination of primidone; the peak serum concentration typically is achieved 3 to 5 hours after each dose. Initiation of primidone monotherapy in patients who are not already receiving barbiturates should in many cases start with a low single daily dose at bedtime (eg, one-fourth of a 250-mg tablet for patients who weigh more than 20 kg) with subsequent upward titration at 3-day intervals (161). Not every patient will require or tolerate steady-state dosage at 20 mg/kg/day. In some cases, relatively large initial doses may be tried to more rapidly achieve serum levels that are effective in controlling seizures. For example, it has been shown that the administration to neonates of 25 mg/kg/day, divided into three doses, produces a serum primidone level of approximately 10 mg/dL by day 3, a level that is sufficient to control the seizures of many neonates independent of the associated phenobarbital level (250). Monitoring primidone or PEMA levels confers little therapeutic advantage in most clinical situations.
Changing patients from chronic oral phenobarbital therapy to primidone is usually easily accomplished by administering four to five times as much primidone each day as the discontinued total daily phenobarbital dose. In patients who are not on phenobarbital, rapid initiation of primidone therapy can be achieved by first loading with phenobarbital (256). Rapid escalation of oral phenobarbital loading can be achieved using phenobarbital doses of 3 mg/kg/day on the first day of treatment, 3.5 mg/kg/day on the second day, 4 mg/kg/day on the third day, and 5 mg/kg/day on the fourth day. This approach results in a linear accumulation of phenobarbital in the serum to approximately 20 μg/mL on day 4 without significant sedation. The phenobarbital can be discontinued on the following day and replaced with a full maintenance primidone dose of between 12.5 and 20 mg/kg/day (161).
Immediate initiation of primidone at an amount expected to produce serum concentration of 20 to 30 μg/mL can be undertaken by IV loading of 20 mg/kg of phenobarbital (producing a serum concentration of 20 μg/mL) and initiation of 20 mg/kg/day of primidone divided into three or four doses. Any patient loaded with enough phenobarbital to achieve serum phenobarbital concentrations of at least 20 μg/mL within 3 days after initiation will experience significant degrees of sedation (256).
Discontinuation
In general, the considerations that arise with regard to drug discontinuation are similar to those previously noted for phenobarbital. One small study suggested that withdrawal-related seizures may be more likely with primidone than with phenobarbital (257). Theodore and coworkers (183) found no relationship between the peak dose or rate of withdrawal of primidone or phenobarbital and the occurrence of seizures. In their study, seizures were most likely to occur as the phenobarbital level fell between 15 and 20 μg/mL.
ACKNOWLEDGMENTS
The authors would like to gratefully acknowledge Dr. Robert Rust for his extensive contribution to an earlier version of this chapter.
REFERENCES
1. Butler TC. Metabolic oxidation of phenobarbital to p-hydroxyphenobarbital. Science. 1954;120:494.
2. Bush MT. Sedatives and hypnotics: absorption, fate and excretion. In: Rot WS, Hofmann FG, eds. Physiological Pharmacology. New York, NY: Academic Press; 1963:185–218.
3. Nishihara K, Uchino K, Saitoh Y, et al. Estimation of plasma unbound phenobarbital concentration by using mixed saliva. Epilepsia. 1979;20(1):37–45.
4. Wade A. Barbiturates. Pharmaceutical Handbook. London, UK: The Pharmaceutical Press; 1980.
5. Bourgeois BF, Dodson WE, Ferrendelli JA. Primidone, phenobarbital, and PEMA: I. seizure protection, neurotoxicity, and therapeutic index of individual compounds in mice. Neurology. 1983;33(3):283–290.
6. Bourgeois BF, Dodson WE, Ferrendelli JA. Primidone, phenobarbital, and PEMA: II. seizure protection, neurotoxicity, and therapeutic index of varying combinations in mice. Neurology. 1983;33(3):291–295.
7. Morrell F, Bradley W, Ptashne M. Effect of drugs on discharge characteristics of chronic epileptogenic lesions. Neurology. 1959;9(7):492–498.
8. Nicoll RA. Pentobarbital: action on frog motoneurons. Brain Res. 1975;96(1):119–123.
9. Macdonald RL, Barker JL. Different actions of anticonvulsant and anesthetic barbiturates resolved by use of cultured mammalian neurons. Science. 1978;200:775–777.
10. Ransom BR, Barker JL. Pentobarbital modulates transmitter effects of mouse spinal neurones grown in tissue culture. Nature. 1975;254:703–705.
11. Ransom BR, Barker JL. Pentobarbital selectively enhances GABA-mediated post-synaptic inhibition in tissue cultured mouse spinal neurons. Brain Res. 1976;114(3):530–535.
12. Schulz DW, MacDonald RL. Barbiturate enhancement of GABA-mediated inhibition and activation of chloride ion conductance: correlation with anticonvulsant and anesthetic actions. Brain Res. 1981;209(1):177–188.
13. French-Mullen JM, Barker JL, Rogawski MA. Calcium current block by (-)-pentobarbital, phenobarbital, and CHEB but not (+)-pentobarbital in acutely isolated hippocampal CA1 neurons: comparison with effects on GABA-activated Cl– current. J Neurosci. 1993;13(8):3211–3221.
14. Sawada S, Yamamoto C. Blocking action of pentobarbital on receptors for excitatory amino acids in the guinea pig hippocampus. Brain Res. 1985;59(2):226–231.
15. Newberry NR, Nicoll RA. Comparison of the action of baclofen with gamma-aminobutyric acid on rat hippocampal pyramidal cells. J Physiol. 1984;360:161–185.
16. Pritchett DB, Sontheimer H, Shivers BD, et al. Importance of a novel GABAA receptor subunit for benzodiazepine pharmacology. Nature. 1989;338:582–585.
17. MacDonald R, Twyman R. Kinetic properties and regulation of GABAA receptor channels. Ion Channels. 1992;3:15–343.
18. Graves NM, Holmes GB, Kriel RL, et al. Relative bioavailability of rectally administered phenobarbital sodium parenteral solution. DICP. 1989;23(7–8):565–568.
19. Svensmark O, Buchthal F. Accumulation of phenobarbital in man. Epilepsia. 1963;4:199–206.
20. Svensmark O, Buchthal F. Dosage of phenytoin and phenobarbital in children. Dan Med Bull. 1963;10:234–235.
21. Fouts JR, Hart LG. Hepatic drug metabolism during the perinatal period. Ann N Y Acad Sci. 1965;123:245–251.
22. Langslet A, Meberg A, Bredesen JE, et al. Plasma concentrations of diazepam and N-desmethyldiazepam in newborn infants after intravenous, intramuscular, rectal and oral administration. Acta Paediatr Scand. 1978;67(6):699–704.
23. Nau H, Rating D, Häuser I, et al. Placental transfer and pharmacokinetics of primidone and its metabolites phenobarbital, PEMA and hydroxyphenobarbital in neonates and infants of epileptic mothers. Eur J Clin Pharmacol. 1980;18(1):31–42.
24. Kaneko S, Suzuki K, Sato T, et al. The problems of antiepileptic medication in the neonatal period: is breastfeeding advisable? In: Janz D, Dam M, Richens A, et al, eds. Epilepsy, Pregnancy, and the Child. New York, NY: Raven Press; 1982:343–348.
25. Boréus LO, Jalling B, Kållberg N. Phenobarbital metabolism in adults and in newborn infants. Acta Paediatr Scand. 1978;67(2):193–200.
26. Boréus LO, Jalling B, Wallin A. Plasma concentrations of phenobarbital in mother and child after combined prenatal and postnatal administration for prophylaxis of hyperbilirubinemia. J Pediatr. 1978;93(4):695–698.
27. Bossi L, Battino D, Caccamo ML, et al. Pharmacokinetics and clinical effects of antiepileptic drugs in newborns of chronically treated epileptic mothers. In: Janz D, Dam M, Richens A, et al, eds. Epilepsy, Pregnancy, and the Child. New York, NY: Raven Press; 1982:373–381.
28. Rating D, Nau H, Kuhnz W, et al. [Antiepileptic drugs in the newborn. Clinical and pharmacological data]. Monatsschr Kinderheilkd. 1983;131(1):6–12.
29. Boreus LO, Jalling B, Kallberg N. Clinical pharmacology of phenobarbital in the neonatal period. In: Morselli PL, Garattini S, Sereni F, eds. Basic and Therapeutic Aspects of Perinatal Pharmacology. New York, NY: Raven Press; 1975:331–340.
30. Dodson WE. Antiepileptic drug utilization in pediatric patients. Epilepsia. 1984;25:S132–S139.
31. Heinze E, Kampffmeyer HG. Biological half-life of phenobarbital in human babies. Klin Wochenschr. 1971;49(20):1146–1147.
32. Ehrnebo M, Agurell S, Jalling B, et al. Age differences in drug binding by plasma proteins: studies on human foetuses, neonates and adults. Eur J Clin Pharmacol. 1971;3(4):189.
33. Painter MJ, Pippenger C, MacDonald H, et al. Phenobarbital and diphenylhydantoin levels in neonates with seizures. J Pediatr. 1978;92(2):315–319.
34. Lockman LA, Kriel R, Zaske D, et al. Phenobarbital dosage for control of neonatal seizures. Neurology. 1979;29(11):1445–1449.
35. Pitlick W, Painter M, Pippenger C. Phenobarbital pharmacokinetics in neonates. Clin Pharmacol Ther. 1978;23(3):346–350.
36. Strandjord RE, Johannessen SI. Serum levels of phenobarbitone in healthy subjects and patients with epilepsy. In: Gardner-Thorpe C, Janz D, Meinardi H, et al, eds. Antiepileptic Drug Monitoring. Tunbridge Wells (Kent), UK: Pitman Medical; 1977:89–103.
37. Brachet-Liermain A, Goutieres F, Aicardi J. Absorption of phenobarbital after the intramuscular administration of single doses in infants. J Pediatr. 1975;87(4):624–626.
38. Jalling B. Plasma and cerebrospinal fluid concentrations of phenobarbital in infants given single doses. Dev Med Child Neurol. 1974;11:781–793.
39. Butler TC, Waddell WJ. Metabolic conversion of primidone (mysoline) to phenobarbital. Proc Soc Exp Biol Med. 1956;93(3):544–546.
40. Tang BK, Kalow W, Grey AA. Metabolic fate of phenobarbital in man. N-glucoside formation. Drug Metab Dispos. 1979;7(5):315–318.
41. Kwan P, Brodie MJ. Phenobarbital for the treatment of epilepsy in the 21st century: a critical review. Epilepsia. 2004;45(9):1141–1149.
42. Whyte MP, Dekaban AS. Metabolic fate of phenobarbital. A quantitative study of p-hydroxyphenobarbital elimination in man. Drug Metab Dispos. 1977;5(1):63–70.
43. Wilensky AJ, Friel PN, Levy RH, et al. Kinetics of phenobarbital in normal subjects and epileptic patients. Eur J Clin Pharmacol. 1982;23(1):87–92.
44. Conney AH. Pharmacological implications of microsomal enzyme induction. Pharmacol Rev. 1967;19(3):317–366.
45. Butler TC, Mahaffee C, Waddell WJ. Phenobarbital: studies of elimination, accumulation, tolerance, and dosage schedules. J Pharmacol Exp Ther. 1954;111(4):425–435.
46. Lous P. Blood serum and cerebrospinal fluid levels and renal clearance of phenemal in treated epileptics. Acta Pharmacol Toxicol. 1954;10:166–177.
47. Lous P. Plasma levels and urinary excretion of three barbituric acids after oral administration to man. Acta Pharmacol Toxicol. 1954;10:147–165.
48. Morselli PL, Rizzo M, Garattini S. Interaction between phenobarbital and diphenylhydantoin in animals and in epileptic patients. Ann N Y Acad Sci. 1971;179:88–107.
49. Patel IH, Levy RH, Cutler RE. Phenobarbital—valproic acid interaction. Clin Pharmacol Ther. 1980;27(4):515–521.
50. Domek NS, Barlow CF, Roth LJ. An ontogenetic study of phenobarbital-C-14 in cat brain. J Pharmacol Exp Ther. 1960;130:285–293.
51. Melchior JC, Svensmark O, Trolle D. Placental transfer of phenobarbitone in epileptic women, and elimination in newborns. Lancet. 1967;11:860–861.
52. Painter MJ, Pippenger CE, MacDonald H, et al. Phenobarbital and phenytoin blood levels in neonates. Pediatrics. 1977;92:315–319.
53. Painter MJ, Pippenger C, Wasterlain C, et al. Phenobarbital and phenytoin in neonatal seizures: metabolism and tissue distribution. Neurology. 1981;31(9):1107–1112.
54. Heimann G, Gladtke E. Pharmacokinetics of phenobarbital in childhood. Eur J Clin Pharmacol. 1977;12:305–310.
55. Jalling B. Plasma concentrations of phenobarbital in the treatment of seizures in newborns. Acta Paediatr Scand. 1975;64(3):514–524.
56. Dodson WE, Prensky AL, Devivo DC, et al. Management of seizure disorders: selected aspects. Part I. J Pediatr. 1976;89(4):527–540.
57. Baumel I, Defeo JJ, Lal H. Effects of acute hypoxia on brain-sensitivity and metabolism of barbiturates in mice. Psychopharmacologia. 1970;17:1983–1987.
58. Dauber IM, Krauss AN, Symchych PS, et al. Renal failure following perinatal anoxia. J Pediatr. 1976;88(5):851–855.
59. Gal P, Toback J, Erkan NV, et al. The influence of asphyxia on phenobarbital dosing requirements in neonates. Dev Pharmacol Ther. 1984;7(3):145–152.
60. Morselli PL. Antiepileptics. In: Morselli PL, ed. Drug Disposition During Development. New York, NY: Spectrum; 1977:311–360.
61. Neuvonen PJ, Elonen E. Effect of activated charcoal on absorption and elimination of phenobarbitone, carbamazepine and phenylbutazone in man. Eur J Clin Pharmacol. 1980;17(1):51–57.
62. Viswanathan CT, Booker HE, Welling PG. Bioavailability of oral and intramuscular phenobarbital. J Clin Pharmacol. 1978;18(2–3):100–105.
63. Yaffe SJ. Developmental factors influencing interactions of drugs. Ann N Y Acad Sci. 1976;281:90–97.
64. Heimann G, Gladtke E. Pharmacokinetics of phenobarbital in childhood. Eur J Clin Pharmacol. 1977;12(4):305–310.
65. Garrettson LK, Dayton PG. Disappearance of phenobarbital and diphenylhydantoin from serum of children. Clin Pharmacol Ther. 1970;11(5):674–679.
66. Dodson WE. Special pharmacokinetic considerations in children. Epilepsia. 1987;28(Suppl 1):S56–S70.
67. Waddell WJ, Butler TC. The distribution and excretion of phenobarbital. J Clin Invest. 1957;36(8):1217–1226.
68. Kapetanovi IM, Kupferberg HJ. Inhibition of microsomal phenobarbital metabolism by valproic acid. Biochem Pharmacol. 1981;30(11):1361–1363.
69. Kapetanovi IM, Kupferberg HJ, Porter RJ, et al. Mechanism of valproate-phenobarbital interaction in epileptic patients. Clin Pharmacol Ther. 1981;29(4):480–486.
70. Giotti A, Maynert EW. The renal clearance of barbital and the mechanism of its reabsorption. J Pharmacol Exp Ther. 1951;101(3):296–309.
71. Myschetzky A, Lassen NA. Forced diuresis in treatment of acute barbiturate poisoning. In: Matthew H, ed. Acute Barbiturate Poisoning. Amsterdam: Excerpta Medica; 1971:223–232.
72. Davis M, Simmons C, Harrison NG, et al. Paracetamol overdose in man: relationship between pattern of urinary metabolites and severity of liver damage. QJ Med. 1976;45:181–191.
73. Vesell ES, Page JG. Genetic control of the phenobarbital-induced shortening of plasma antipyrine half-lives in man. J Clin Invest. 1969;48(12):2202–2209.
74. Cucinell SA, Conney AH, Sansur M, et al. Drug interactions in man. i. lowering effect of phenobarbital on plasma levels of bishydroxycoumarin (dicumarol) and diphenylhydantoin (dilantin). Clin Pharmacol Ther. 1965;6:420–429.
75. Christiansen J, Dam M. Influence of phenobarbital and diphenylhydantoin on plasma carbamazepine levels in patients with epilepsy. Acta Neurol Scand. 1973;49(4):543–546.
76. Lander CM, Eadie MJ, Tyrer JH. Factors influencing plasma carbamazepine concentrations. Clin Exp Neurol. 1977;14:184–193.
77. Spina E, Martines C, Fazio A, et al. Effect of phenobarbital on the pharmacokinetics of carbamazepine-10,11-epoxide, an active metabolite of carbamazepine. Ther Drug Monit. 1991;13(2):109–112.
78. Krasinski K, Kusmiesz H, Nelson JD. Pharmacologic interactions among chloramphenicol, phenytoin and phenobarbital. Pediatr Infect Dis. 1982;1(4):232–235.
79. Lader M. Drug interactions and the major tranquilizers. In: Grahame-Smith DG, ed. Drug Interactions. Baltimore: University Park Press; 1977:159–170.
80. Loga S, Curry S, Lader M. Interactions of orphenadrine and phenobarbitone with chlorpromazine: plasma concentrations and effects in man. Br J Clin Pharmacol. 1975;2(3):197–208.
81. Somogyi A, Gugler R. Drug interactions with cimetidine. Clin Pharmacokinet. 1982;7(1):23–41.
82. Carstensen H, Jacobsen N, Dieperink H. Interaction between cyclosporin A and phenobarbitone. Br J Clin Pharmacol. 1986;21(5):550–551.
83. Neuvonen PJ, Penttilä O, Lehtovaara R, et al. Effect of antiepileptic drugs on the elimination of various tetracycline derivatives. Eur J Clin Pharmacol. 1975;9(2–3):147–154.
84. Ioannides C, Parke D. Mechanism of induction of hepatic microsomal drug metabolizing enzymes by a series of barbiturates. J Pharm Pharmacol. 1975;27(10):739–746.
85. Fonné R, Meyer UA. Mechanisms of phenobarbital-type induction of cytochrome P-450 isozymes. Pharmacology Therapeu. 1987;33(1):19–22.
86. Treiman DM, Pledger GW, Degiorgio C, et al. Increasing plasma concentration tolerability study of flunarizine in comedicated epileptic patients. Epilepsia. 1993;34(5):944–953.
87. Beurey J, Weber M, Vignaud JM. [Treatment of tinea microsporum. Metabolic interference between phenobarbital and griseofulvin]. Annales de dermatologie et de vénéréologie. 1982;109(6–7):567–570.
88. Linnoila M, Viukari M, Vaisanen K, et al. Effect of anticonvulsants on plasma haloperidol and thioridazine levels. Am J Psychiatry. 1980;137(7):819–821.
89. Stambaugh JE, Hemphill DM, Wainer IW, et al. A potentially toxic drug interaction between pethidine (meperidine) and phenobarbitone. Lancet (London, England). 1977;1(8008):398–399.
90. Liu SJ, Wang RI. Case report of barbiturate-induced enhancement of methadone metabolism and withdrawal syndrome. Am J Psychiatry. 1984;141(10):1287–1288.
91. Rambeck B. Pharmacological interactions of mesuximide with phenobarbital and phenytoin in hospitalized epileptic patients. Epilepsia. 1979;20(2):147–156.
92. Braithwaite RA, Flanagan RJ, Richens A. Steady-state plasma nortriptyline concentrations in epileptic patients. Br J Clin Pharmacol. 1975;2(5):469–471.
93. Jonkman JH, Upton RA. Pharmacokinetic drug interactions with theophylline. Clin Pharmacokinet. 1984;9(4):309–334.
94. May T, Rambeck B. Serum concentrations of valproic acid: influence of dose and comedication. Ther Drug Monit. 1985;7(4):387–390.
95. Perucca E, Gatti G, Frigo GM, et al. Disposition of sodium valproate in epileptic patients. Br J Clin Pharmacol. 1978;5(6):495–499.
96. MacDonald MG, Robinson DS. Clinical observations of possible barbiturate interference with anticoagulation. JAMA. 1968;204:95–100.
97. Rettie AE, Rettenmeier AW, Howald WN, et al. Cytochrome P-450-catalyzed formation of delta-four VPA, a toxic metabolite of valproic acid. Science. 1987;235:890–893.
98. Kesterson JW, Granneman GR, Machinist JM. The hepatotoxicity of valproic acid and its metabolites in rats. I. toxicologic, biochemical and histopathologic studies. Hepatology. 1984;4(6):1143–1152.
99. Kochen W, Schneider A, Ritz A. Abnormal metabolism of valproic acid in fatal hepatic failure. Eur J Pediatr. 1983;141(1):30–35.
100. Prickett KS, Baillie TA. Metabolism of unsaturated derivatives of valproic acid in rat liver microsomes and destruction of cytochrome P-450. Drug Metab Dispos. 1986;14(2):221–229.
101. Segura-Bruna N, Rodriguez-Campello A, Puente V, et al. Valproate-induced hyperammonemic encephalopathy. Acta Neurol Scand. 2006;114(1):1–7.
102. Booker HE, Tormey A, Toussaint J. Concurrent administration of phenobarbital and diphenylhydantoin: lack of an interference effect. Neurology. 1971;21(4):383–385.
103. Browne TR, Szabo GK, Evans J, et al. Phenobarbital does not alter phenytoin steady-state serum concentration or pharmacokinetics. Neurology. 1988;38(4):639–642.
104. Perucca E, Zaccara G. Interactions between antiepileptic drugs, and between antiepileptic drugs and other drugs. Epileptic Disord. 2014;16(4):409–432.
105. Perucca E, Cloyd J, Critchley D, et al. Rufinamide: clinical pharmacokinetics and concentration-response relationships in patients with epilepsy. Epilepsia. 2008;49(7):1123–1141.
106. Contin M, Albani F, Riva R, et al. Lacosamide therapeutic monitoring in patients with epilepsy: effect of concomitant antiepileptic drugs. Ther Drug Monit. 2013;35(6):849–852.
107. Brooks SM, Werk EE, Ackerman SJ, et al. Adverse effects of phenobarbital on corticosteroid metabolism in patients with bronchial asthma. N Engl J Med. 1972;286(21):1125–1128.
108. Burstein S, Klaiber EL. Phenobarbital-induced increase in 6-beta-hydroxycortisol excretion: clue to its significance in human urine. J Clin Endocrinol Metab. 1965;25:293–296.
109. Janz D, Schmidt D. Letter: anti-epileptic drugs and failure of oral contraceptives. Lancet (London, England). 1974;1(7866):1113.
110. Levin W, Kuntzman R, Conney AH. Stimulatory effect of phenobarbital on the metabolism of the oral contraceptive 17 alpha-ethynylestradiol-3-methyl ether (mestranol) by rat liver microsomes. Pharmacology. 1979;19(5):249–255.
111. Kleinman PD, Griner PF. Studies of the epidemiology of anticoagulant-drug interactions. Arch Intern Med. 1970;126(3):522–523.
112. Bourgeois BF. Pharmacologic interactions between valproate and other drugs. Am J Med. 1988;84(1A):29–33.
113. Vining EP. Use of barbiturates and benzodiazepines in treatment of epilepsy. Neurol Clin. 1986;4(3):617–632.
114. Buchthal F, Svensmark O, Simonsen H. Relation of EEG and seizures to phenobarbital in serum. Arch Neurol. 1968;19(6):567–572.
115. Hutt SJ, Jackson PM, Belsham A, et al. Perceptual-motor behaviour in relation to blood phenobarbitone level: a preliminary report. Dev Med Child Neurol. 1968;10(5):626–632.
116. Mattson RH, Cramer JA, Collins JF; et al. Comparison between carbamazepine and valproate for complex partial and secondarily generalized tonic–clonic seizures. Epilepsia. 1991;32:S18.
117. Addy DP. Phenobarbitone and febrile convulsions. Arch Dis Child. 1990;65(9):921.
118. Sylvester CE, Marchlewski A, Manaligod JM. Primidone or phenobarbital use complicating disruptive behavior disorders. Clin Pediatr. 1994;33:252–253.
119. Herranz JL, Armijo JA, Arteaga R. Clinical side effects of phenobarbital, primidone, phenytoin, carbamazepine, and valproate during monotherapy in children. Epilepsia. 1988;29(6):794–804.
120. Camfield CS, Chaplin S, Doyle AB, et al. Side effects of phenobarbital in toddlers; behavioral and cognitive aspects. J Pediatr. 1979;95(3):361–365.
121. Mitchell WG, Chavez JM. Carbamazepine versus phenobarbital for partial onset seizures in children. Epilepsia. 1987;28(1):56–60.
122. Young RS, Alger PM, Bauer L, et al. A randomized, double-blind, crossover study of phenobarbital and mephobarbital. J Child Neurol. 1986;1(4):361–363.
123. Hayes SG. Barbiturate anticonvulsants in refractory affective disorders. Ann Clin Psychiatry. 1993;5(1):35–44.
124. Glauser TA. Behavioral and psychiatric adverse events associated with antiepileptic drugs commonly used in pediatric patients. J Child Neurol. 2004;19(Suppl 1):S25–S38.
125. Lennox WG. Brain injury, drugs and environment as causes of mental decay in epilepsy. Am J Psychiatry. 1942;99:174–180.
126. Hillesmaa VK, Teramo K, Granstrom ML, et al. Fetal head growth retardation associated with maternal antiepileptic drugs. Lancet. 1981;1:165–167.
127. Somerfeld-Ziskind E, Ziskind E. Effect of phenobarbital on the mentality of epileptic patients. Arch Neurol Psychol. 1940;43:70–79.
128. Wapner I, Thurston DL, Holowach J. Phenobarbital. Its effect on learning in epileptic children. JAMA. 1962;182:937.
129. Stores G. Behavioural effects of anti-epileptic drugs. Dev Med Child Neurol. 1975;17(5):647–658.
130. Schain RJ, Ward JW, Guthrie D. Carbamazepine as an anticonvulsant in children. Neurology. 1977;27(5):476–480.
131. Vining EP, Mellitis ED, Dorsen MM, et al. Psychologic and behavioral effects of antiepileptic drugs in children: a double-blind comparison between phenobarbital and valproic acid. Pediatrics. 1987;80(2):165–174.
132. Calandre EP, Dominguez-Granados R, Gomez-Rubio M, et al. Cognitive effects of long-term treatment with phenobarbital and valproic acid in school children. Acta Neurol Scand. 1990;81(6):504–506.
133. Hirtz DG, Sulzbacher SI, Ellenberg JH, et al. Phenobarbital for febrile seizures—effects on intelligence and on seizure recurrence. N Engl J Med. 1990;322:364–369.
134. Farwell JR, Lee YJ, Hirtz DG, et al. Phenobarbital for febrile seizures—effects on intelligence and on seizure recurrence. N Engl J Med. 1990;322(6):364–369.
135. Hollister LE. Nervous system reactions to drugs. Ann N Y Acad Sci. 1965;123:342–353.
136. Sulzbacher S, Farwell JR, Temkin N, et al. Late cognitive effects of early treatment with phenobarbital. Clin Pediatr. 1999;38:387–394.
137. Tomson T, Battino D, Bonizzoni E, et al. Dose-dependent risk of malformations with antiepileptic drugs: an analysis of data from the EURAP epilepsy and pregnancy registry. The Lancet. Neurology. 2011;10(7):609–617.
138. Tomson T, Battino D. Teratogenic effects of antiepileptic drugs. Lancet Neurol. 2012;11(9):803–813.
139. Harden CL, Meador KJ, Pennell PB, et al. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): teratogenesis and perinatal outcomes: report of the Quality Standards Subcommittee and Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and American Epilepsy Society. Neurology. 2009;73(2):133–141.
140. Isbell H, Fraser HF. Addiction to analgesics and barbiturates. Pharmacol Rev. 1959;2:355–397.
141. Berman LB, Jeghers HJ, Schreiner GE, et al. Hemodialysis, an effective therapy for acute barbiturate poisoning. JAMA. 1956;161:820–827.
142. Christiansen C, Rodbro P, Lund M. Incidence of anticonvulsant osteomalacia and effect of vitamin D: controlled therapeutic trial. Br Med J. 1973;4(5894):695–701.
143. Richens A, Rowe DJ. Disturbance of calcium metabolism by anticonvulsant drugs. Br Med J. 1970;4(5727):73–76.
144. Hahn TJ, Birge SJ, Scharp CR, et al. Phenobarbital-induced alterations in vitamin D metabolism. J Clin Invest. 1972;51(4):741–748.
145. Hunter J. Effects of enzyme induction on vitamin D3 metabolism in man. In: Richens A, Woodford FP, eds. Anticonvulsant Drugs and Enzyme Induction. New York, NY: Elsevier; 1976:77–84.
146. Lazzari AA, Dussault PM, Thakore-James M, et al. Prevention of bone loss and vertebral fractures in patients with chronic epilepsy—antiepileptic drug and osteoporosis prevention trial. Epilepsia. 2013;54(11):1997–2004.
147. Mockli G, Crowley M, Stern R, et al. Massive hepatic necrosis in a child after administration of phenobarbital. Am J Gasteroenterol. 1989;84:820–822.
148. Morkunas AR, Miller MB. Anticonvulsant hypersensitivity syndrome. Crit Care Clin. 1997;13(4):727–739.
149. Mattson RH, Cramer JA, Collins JF, et al. Comparison of carbamazepine, phenobarbital, phenytoin, and primidone in partial and secondarily generalized tonic–clonic seizures. N Engl J Med. 1985;313(3):145–151.
150. Smith DB, Mattson RH, Cramer JA, et al. Results of a nationwide Veterans Administration Cooperative Study comparing the efficacy and toxicity of carbamazepine, phenobarbital, phenytoin, and primidone. Epilepsia. 1987;28(Suppl 3):S50–S58.
151. Feely M, O’Callagan M, Duggan G, et al. Phenobarbitone in previously untreated epilepsy. J Neurol Neurosurg Psychiatry. 1980;43:364–368.
152. Schmidt D, Richter K. Alternative single anticonvulsant drug therapy for refractory epilepsy. Ann Neurol. 1986;19(1):85–87.
153. Forsythe WI, Sills MA. One drug for childhood grand mal: medical audit for three-year remissions. Dev Med Child Neurol. 1984;26(6):742–748.
154. Schmidt D. Single drug therapy for intractable epilepsy. J Neurol. 1983;229(4):221–226.
155. Schmidt D. Two antiepileptic drugs for intractable epilepsy with complex-partial seizures. J Neurol Neurosurg Psychiatry. 1982;45(12):1119–1124.
156. Cornaggia CM, Canevini MP, Giuccioli D, et al. Carbamazepine, phenytoin and phenobarbital in drug-resistant partial epilepsies. Ital J Neurol Sci. 1986;7(1):113–117.
157. Callaghan N, O’Dwyer R, Keating J. Unnecessary polypharmacy in patients with frequent seizures. Acta Neurol Scand. 1984;69(1):15–19.
158. Painter MJ. How to use phenobarbital. In: Morselli PL, Pippinger CE, Penry JK, eds. Antiepileptic Drug Therapy in Pediatrics. New York, NY: Raven Press; 1983:245–252.
159. Painter MJ. Phenobarbital clinical use. In: Levy RH, Dreifuss FE, Mattson RH, et al, eds. Antiepileptic Drugs. 3rd ed. New York, NY: Raven Press; 1989:329–340.
160. Elwes RD, Johnson AL, Shorvon SD, et al. The prognosis for seizure control in newly diagnosed epilepsy. N Engl J Med. 1984;311(15):944–947.
161. Bourgeois BFD. Primidone. In: Wyllie E, ed. The Treatment of Epilepsy: Principles and Practices. Philadelphia, PA: Lea & Febiger; 1993:909–913.
162. Tokola RA, Neuvonen PJ. Pharmacokinetics of antiepileptic drugs. Acta Neurol Scand Suppl. 1983;97:17–27.
163. Alix D, Berthou F, Riche C, et al. [Plasma concentrations of phenobarbital and dosage according to age]. Dev Pharmacol Ther. 1984;7(Suppl 1):164–170.
164. Rossi LN, Nino LM, Principi N. Correlation between age and plasma level/dosage ratio for phenobarbital in infants and children. Acta Paediatr Scand. 1979;68(3):431–434.
165. Mizrahi EM, Kellaway P. Characterization and classification of neonatal seizures. Neurology. 1987;37(12):1837–1844.
166. Painter MJ, Gaus LM. Neonatal seizures: diagnosis and treatment. J Child Neurol. 1991;6(2):101–108.
167. Fischer J, Lockman L, Zoske D, et al. Phenobarbital maintenance dose requirements in treating neonatal seizures. Neurology. 1982;31:1042–1044.
168. Donn SM, Grasela TH, Goldstein GW. Safety of a higher loading dose of phenobarbital in the term newborn. Pediatrics. 1985;75(6):1061–1064.
169. Painter MJ, Scher MS, Stein AD, et al. Phenobarbital compared with phenytoin for the treatment of neonatal seizures. N Engl J Med. 1999;341(7):485–489.
170. Painter MJ, Gaus LM. Phenobarbital. In: Wyllie E, eds. The Treatment of Epilepsy: Principles and Practices. Philadelphia, PA: Lea & Febiger; 1993:900–908.
171. Gal P, Boer HR, Toback J, et al. Phenobarbital dosing in neonates and asphyxia. Neurology. 1982;32(7):788–789.
172. Gilman JT, Gal P, Duchowny MS, et al. Rapid sequential phenobarbital treatment of neonatal seizures. Pediatrics. 1989;83(5):674–678.
173. Connell J, Oozeer R, de Vries L, et al. Clinical and EEG response to anticonvulsants in neonatal seizures. Arch Dis Child. 1989;64:459–464.
174. Svenningsen NW, Blennow G, Lindroth M, et al. Brain-orientated intensive care treatment in severe neonatal asphyxia. Effects of phenobarbitone protection. Arch Dis Child. 1982;57(3):176–183.
175. Grasela TH, Donn SM. Neonatal population pharmacokinetics of phenobarbital derived from routine clinical data. Dev Pharmacol Ther. 1985;8(6):374–383.
176. Glauser TA, Shinnar S, Gloss D, et al. Evidence-based guideline: treatment of convulsive status epilepticus in children and adults: report of the guideline committee of the American Epilepsy Society. Epilepsy. 2016;16(1):48–61.
177. Siemes H. [New aspects in prevention of febrile convulsions]. Klinische Pädiatrie. 1992;204(2):67–71.
178. Faero O, Kastrup KW, Lykkegaard Nielsen E, et al. Successful prophylaxis of febrile convulsions with phenobarbital. Epilepsia. 1972;13(2):279–285.
179. Minagawa K, Miura H. Phenobarbital, primidone and sodium valproate in the prophylaxis of febrile convulsions. Brain Deve. 1981;3(4):385–393.
180. McKinlay I, Newton R. Intention to treat febrile convulsions with rectal diazepam, valproate or phenobarbitone. Dev Med Child Neurol. 1989;31(5):617–625.
181. Schmidt D. [Pharmacotherapy of epilepsy—current problems and controversies]. Fortschr Neurol Psychiatr. 1983;51(11):363–386.
182. Herranz JL, Armijo JA, Arteaga R. Effectiveness and toxicity of phenobarbital, primidone, and sodium valproate in the prevention of febrile convulsions, controlled by plasma levels. Epilepsia. 1984;25(1):89–95.
183. Theodore WH, Porter RJ, Raubertas RF. Seizures during barbiturate withdrawal: relation to blood level. Ann Neurol. 1987;22(5):644–647.
184. Offringa M, Newton R. Prophylactic drug management for febrile seizures in children (review). Evid-Based Child Health. 2013;8(4):1376–1485.
185. Butler TC, Waddell WJ. N-methylated derivatives of barbituric acid, hydantoin and oxazolidinedione used in the treatment of epilepsy. Neurology. 1958;8(Suppl 1):106–112.
186. Hooper WD, Kunze HE, Eadie MJ. Pharmacokinetics and bioavailability of methylphenobarbital in man. Ther Drug Monit. 1981;3(1):39–44.
187. Hooper WD, Qing MS. The influence of age and gender on the stereoselective metabolism and pharmacokinetics of mephobarbital in humans. Clin Pharmacol Ther. 1990;48(6):633–640.
188. Lim WH, Hooper WD. Stereoselective metabolism and pharmacokinetics of racemic methylphenobarbital in humans. Drug Metab Dispos. 1989;17(2):212–217.
189. Eadie MJ, Bochner F, Hooper WD, et al. Preliminary observations on the pharmacokinetics of methylphenobarbitone. Clin Exp Neurol. 1978;15:131–144.
190. Kunze HE, Hooper WD, Eadie MJ. High performance liquid chromatographic assay of methylphenobarbital metabolites in urine. Ther Drug Monit. 1981;3(1):45–49.
191. Craig CR, Shideman FE. Metabolism and anticonvulsant properties of mephobarbital and phenobarbital in rats. J Pharmacol Exp Ther. 1971;176(1):35–42.
192. Lowenstein DH, Aminoff MJ, Simon RP. Barbiturate anesthesia in the treatment of status epilepticus: clinical experience with 14 patients. Neurology. 1988;38(3):395–400.
193. Delgado-Escueta AV, Wasterlain C, Treiman DM, et al. Current concepts in neurology: management of status epilepticus. N Engl J Med. 1982;306(22):1337–1340.
194. Simon RP. Management of status epilepticus. In: Pedley TA, Meldrum BS, eds. Recent Advances in Epilepsy. London, UK: Churchill Livingstone; 1985.
195. Fraser HF, Wikler A, Essig CF, et al. Degree of physical dependence induced by secobarbital or pentobarbital. JAMA. 1958;166:126–129.
196. Essig CF. Clinical and experimental aspects of barbiturate withdrawal convulsions. Epilepsia. 1967;8(1):21–30.
197. Narahashi T, Moore JW, Poston RN. Anesthetic blocking of nerve membrane conductances by internal and external applications. J Neurobiol. 1969;1(1):3–22.
198. Proctor WR, Weakly JN. A comparison of the presynaptic and post-synaptic actions of pentobarbitone and phenobarbitone in the neuromuscular junction of the frog. J Physiol. 1976;258(1):257–268.
199. Handley R, Stewart AS. Mysoline; a new drug in the treatment of epilepsy. Lancet (London, England). 1952;1(6711):742–744.
200. Bogue JY, Carrington HC. The evaluation of mysoline—a new anticonvulsant drug. Br J Pharmacol. 1953;8:230–235.
201. Frey HH, Hahn I. Untersuchungen uber die bedeutung des durch biotransformation gebildeten phenobarbital fur die antikonvulsive wirkung von primidon. Arch Int Pharmacodyn Ther. 1960;128:281–290.
202. Baumel IP, Gallagher BB, DiMicco J, et al. Metabolism and anticonvulsant properties of primidone in the rat. J Pharmacol Exp Ther. 1973;186(2):305–314.
203. Leal KW, Rapport RL, Wilensky AJ, et al. Single-dose pharmacokinetics and anticonvulsant efficacy of primidone in mice. Ann Neurol. 1979;5(5):470–474.
204. Frey HH, Löscher W. [Is primidone more efficient than phenobarbital? An attempt at a pharmacological evaluation (author’s transl)]. Nervenarzt. 1980;51(6):359–362.
205. Schottelius DD, Fincham RW. Clinical application of serum primidone levels. In: Pip-Penger CE, Penry JK, Kutt H, eds. Antiepileptic Drugs: Quantitative Analysis and Interpretation. New York, NY: Raven Press; 1978:273–282.
206. Kauffman RE, Habersang R, Lansky L. Kinetics of primidone metabolism and excretion in children. Clin Pharmacol Ther. 1977;22(2):200–205.
207. Gallagher BB, Baumel IP, Mattson RH. Metabolic disposition of primidone and its metabolites in epileptic subjects after single and repeated administration. Neurology. 1972;22(11):1186–1192.
208. Gallagher BB, Baumel IP. Primidone. Absorption, distribution and excretion. In: Woodbury DM, Penry JK, Schmidt RP, eds. Antiepileptic Drugs. New York, NY: Raven Press; 1972:357–359.
209. Wyllie E, Pippenger CE, Rothner AD. Increased seizure frequency with generic primidone. JAMA. 1987;258(9):1216–1217.
210. Cloyd JC, Miller KW, Leppik IE. Primidone kinetics: effects of concurrent drugs and duration of therapy. Clin Pharmacol Ther. 1981;29(3):402–407.
211. Baumel IP, Gallagher BB, Mattson RH. Phenylethylmalonamide (PEMA). An important metabolite of primidone. Arch Neurol. 1972;27(1):34–41.
212. Pisani F, Richens A. Pharmacokinetics of phenylethylmalonamide (PEMA) after oral and intravenous administration. Clin Pharmacol. 1983;8:272–276.
213. Zavadil P, Gallagher BB. Metabolism and excretion of 14C-primidone in epileptic patients. In: Janz D, ed. Epileptology. Stuttgart: Georg Thieme; 1976:129–139.
214. Olesen OV, Dam M. The metabolic conversion of primidone (mysoline) to phenobarbitone in patients under long-term treatment. An attempt to estimate the independent effect of primidone. Acta Neurol Scand. 1967;43(3):348–356.
215. Battino D, Avanzini G, Bossi L, et al. Plasma levels of primidone and its metabolite phenobarbital: effect of age and associated therapy. Ther Drug Monit. 1983;5(1):73–79.
216. Armijo JA, Herranz JL, Arteaga R, et al. Poor correlation between single-dose data and steady-state kinetics for phenobarbitone, primidone, carbamazepine and sodium valproate in children during monotherapy. possible reasons for the lack of correlation. Clin Pharmacokinet. 1986;11(4):323–335.
217. Booker HE, Hosokowa K, Burdette RD, et al. A clinical study of serum primidone levels. Epilepsia. 1970;11(4):395–402.
218. Bourgeois BFD. Primidone. In: Resor SR, Kutt H, eds. The Medical Treatment of Epilepsy. New York, NY: Marcel Dekker; 1992:371–378.
219. Sutton G, Kupferberg HJ. Isoniazid as an inhibitor of primidone metabolism. Neurology. 1975;25(12):1179–1181.
220. Bourgeois BF, Dodson WE, Ferrendelli JA. Isoniazid and other drugs. Pediatrics. 1982;70(5):824–825.
221. Bourgeois BF, Dodson WE, Ferrendelli JA. Interactions between primidone, carbamazepine, and nicotinamide. Neurology. 1982;32(10):1122–1126.
222. Fincham RW, Schottelius DD, Sahs AL. The influence of diphenylhydantoin on primidone metabolism. Arch Neurol. 1974;30(3):259–262.
223. Reynolds EH, Fenton G, Fenwick P, et al. Interaction of phenytoin and primidone. Br Med J. 1975;2(5971):594–595.
224. Windorfer A, Sauer W. Drug interactions during anticonvulsant therapy in childhood: diphenylhydantoin, primidone, phenobarbitone, clonazepam, nitrazepam, carbamazepin and dipropylacetate. Neuropädiatrie. 1977;8(1):29–41.
225. Bruni J. Valproic acid and plasma levels of primidone and derived phenobarbital. Can J Neurol Sci. 1981;8(1):91–92.
226. Smith BH, McNaughton FL. Mysoline, a new anticonvulsant drug; its value in refractory cases of epilepsy. Can Med Assoc J. 1953;68(5):464–467.
227. Sciarra D, Carter S, Vicale CT, et al. Clinical evaluation of primidone (mysoline), a new anticonvulsant drug. JAMA. 1954;154:827–829.
228. Gallagher BB, Baumel IP, Mattson RH, et al. Primidone, diphenylhydantoin and phenobarbital. Aspects of acute and chronic toxicity. Neurology. 1973;23(2):145–149.
229. Leppik IE, Cloyd JC, Miller K. Development of tolerance to the side effects of primidone. Ther Drug Monit. 1984;6(2):189–191.
230. Ajax ET. An unusual case of primidone intoxication. Dis Nerv Syst. 1966;27(10):660–661.
231. Leppik IE, Cloyd JC. Primidone: toxicity. In: Levy RH, Mattson RH, Meldrum BS, eds. Antiepileptic Drugs. 4th ed. New York, NY: Raven Press; 1995:487–490.
232. Goldin S. Toxic effects of primidone. Lancet. 1954;1:102–103.
233. Brillman J, Gallagher BB, Mattson RH. Acute primidone intoxication. Arch Neurol. 1974;30(3):255–258.
234. Rodin EA, Rim CS, Kitano H, et al. A comparison of the effectiveness of primidone versus carbamazepine in epileptic outpatients. J Nerv Ment Dis. 1976;163(1):41–46.
235. Hartlage LC, Stovall K, Kocack B. Behavioral correlates of anticonvulsant blood levels. Epilepsia. 1980;21:185.
236. Dotevall G, Herner B. Treatment of acute primidone poisoning with bemegride and amphenazole. Br Med J. 1957;3:451–452.
237. Matzke GR, Cloyd JC, Sawchuk RJ. Acute phenytoin and primidone intoxication: a pharmacokinetic analysis. J Clin Pharmacol. 1981;21(2):92–99.
238. Lin SL, Chung MY. Acute primidone intoxication: report of a case. J Formosan Med Assoc. 1989;88:1053–1055.
239. van Heijst AN, de Jong W, Seldenrijk R, et al. Coma and crystalluria: a massive primidone intoxication treated with haemoperfusion. J Toxicol Clin Toxicol. 1983;20(4):307–318.
240. Lehmann DF. Primidone crystalluria following overdose. A report of a case and an analysis of the literature. Med Toxicol Adverse Drug Exp. 1987;2(5):383–387.
241. Butter AJ. Mysoline in the treatment of epilepsy. Lancet (London, England). 1953;1(6769):1024.
242. Calnan WL, Borrell YM. Mysoline in the treatment of epilepsy. Lancet. 1953;2:42–43.
243. Jorgenson G. Mysoline in the treatment of epilepsy. Lancet. 1953;2:835.
244. Smith B, Forster FM. Mysoline and milontin, two new medicines for epilepsy. Neurology. 1954;4(2):137–142.
245. Livingston S, Petersen D. Primidone (mysoline) in the treatment of epilepsy; results of treatment of 486 patients and review of the literature. N Engl J Med. 1956;254(7):327–329.
246. Milichap JG, Aymat F. Controlled evaluation of primidone and diphenylhydantoin sodium. Comparative anticonvulsant efficacy and toxicity in children. JAMA. 1968;204(8):738–739.
247. White PT, Plott D, Norton J. Relative anticonvulsant potency of primidone; a double blind comparison. Arch Neurol. 1966;14(1):31–35.
248. DeSilvey DL, Moss AJ. Primidone in the treatment of the long QT syndrome: QT shortening and ventricular arrhythmia suppression. Ann Intern Med. 1980;93(1):53–54.
249. Powell C, Painter MJ, Pippenger CE. Primidone therapy in refractory neonatal seizures. J Pediatr. 1984;105:651–654.
250. Sapin JI, Riviello JJ, Grover WD. Efficacy of primidone for seizure control in neonates and young infants. Pediatr Neurol. 1988;4(5):292–295.
251. Gruber CM, Brock JT, Dyken M. Comparison of the effectiveness of phenobarital, mephobarbital, primidone, diphenylhydantoin, ethotoin, metharbital, and methylphenylethylhydantoin in motor seizures. Clin Pharmacol Ther. 1962;3:23–28.
252. Booker HE. Primidone toxicity. In: Woodbury DM, Penry JK, Schmidt RP, eds. Antiepileptic Drugs. 1st ed. New York, NY: Raven Press; 1972:377–383.
253. Oxley J, Hebdige S, Laidlaw J, et al. A comparative study of phenobarbitone and primidone in the treatment of epilepsy. In: Johannessen SI, Morselli PL, Pippenger CE, et al, eds. Advances in Drug Monitoring. New York, NY: Raven Press; 1980:237–245.
254. Shorvon S. The treatment of epilepsy by drugs. In: Hopkins A, ed. Epilepsy. New York, NY: Demos; 1987:229–282.
255. Eadie MJ. Formation of active metabolites of anticonvulsant drugs. A review of their pharmacokinetic and therapeutic significance. Clin Pharmacokinet. 1991;21(1):27–41.
256. Bourgeois BFD, Luders H, Morris H, et al. Rapid introduction of primidone using phenobarbital loading: acute primidone toxicity avoided. Epilepsia. 1989;30:667.
257. Coulter DL. Withdrawal of barbiturate anticonvulsant drugs: prospective controlled study. Am J Ment Retard. 1988;93(3):320–327.
Related posts:
 Epilepsy Febrile Seizure Plus (GEFS+)Douglas R. Nordli, Jr. and John M. Pellock
Epilepsy Febrile Seizure Plus (GEFS+)Douglas R. Nordli, Jr. and John M. Pellock
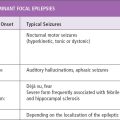 Dominant Focal EpilepsiesSarah Weckhuysen, Mary Kurian, Fabienne Picard, and Stephanie Baulac
Dominant Focal EpilepsiesSarah Weckhuysen, Mary Kurian, Fabienne Picard, and Stephanie Baulac
 in Epilepsy: A Network and Neurodevelopmental PerspectiveRaman Sankar and Edward C. Cooper
in Epilepsy: A Network and Neurodevelopmental PerspectiveRaman Sankar and Edward C. Cooper
 Self-Limited EpilepsiesDouglas R. Nordli, Jr., Colin D. Ferrie, and Chrysostomos P. Panayiotopoulos
Self-Limited EpilepsiesDouglas R. Nordli, Jr., Colin D. Ferrie, and Chrysostomos P. Panayiotopoulos
 and Pharmacologic Consequences of SeizuresShilpa D. Kadam and Michael V. Johnston
and Pharmacologic Consequences of SeizuresShilpa D. Kadam and Michael V. Johnston
 Presurgical Functional MappingAndrew C. Papanicolaou, Roozbeh Rezaie, Shalini Narayana, Marina Kilintari, Asim F. Choudhri, Frederick A. Boop, and James W. Wheless
Presurgical Functional MappingAndrew C. Papanicolaou, Roozbeh Rezaie, Shalini Narayana, Marina Kilintari, Asim F. Choudhri, Frederick A. Boop, and James W. Wheless
