[level-membership-for-pathology-category]33
Thalamic and pallidal degenerations, neuroaxonal dystrophy, and autonomic failure
THALAMIC DEGENERATIONS
Thalamic degeneration is a feature of several multisystem neurodegenerative disorders (Table 33.1), but can also rarely occur in a ‘pure’ form.
Table 33.1
Diseases in which thalamic degeneration may be prominent
Multiple system atrophy (glial cytoplasmic inclusions)
Spinocerebellar degenerations
Wernicke’s encephalopathy
Huntington’s disease
Creutzfeldt–Jakob disease
Menkes syndrome
Membranous lipodystrophy
Neuroaxonal dystrophy
Fatal familial insomnia
PURE THALAMIC ATROPHY
 PURE THALAMIC ATROPHY
PURE THALAMIC ATROPHY


 DIFFERENTIAL DIAGNOSIS OF PURE THALAMIC ATROPHY
DIFFERENTIAL DIAGNOSIS OF PURE THALAMIC ATROPHY
 The conditions listed in Table 33.1 should be excluded. These include fatal familial insomnia, which has a primary thalamic pattern of involvement and is a form of prion disease (see Chapter 32).
The conditions listed in Table 33.1 should be excluded. These include fatal familial insomnia, which has a primary thalamic pattern of involvement and is a form of prion disease (see Chapter 32).
 Neuronal loss from the thalamus has been described in patients that have undergone leukotomy.
Neuronal loss from the thalamus has been described in patients that have undergone leukotomy.
PALLIDAL DEGENERATIONS
The grouping together of pallidal degenerations is based on the morphologic finding of degeneration centered on the globus pallidus, either alone or in combination with degeneration of the subthalamic nucleus or substantia nigra. These disorders have been subdivided according to the regions of the brain showing pathologic changes (Table 33.2). Clinically, pallidal degenerations are associated with a variety of movement disorders, with or without dementia. Some are familial and others sporadic.
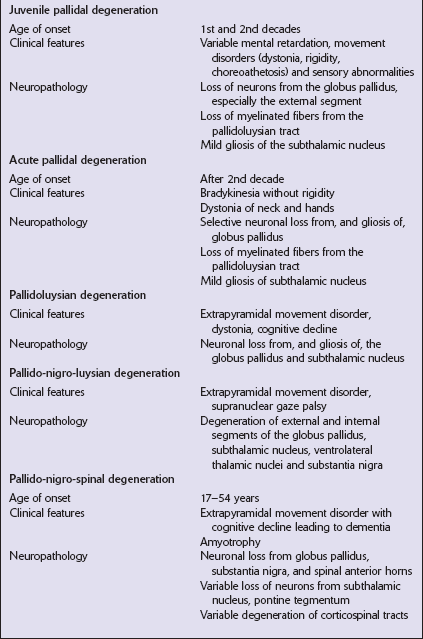
It is difficult to evaluate the nosologic status of many of the cases described in the literature as their relationship to disorders that can now be better defined by molecular genetic and immunohistochemical techniques is uncertain. In particular, cases of dentatorubropallidoluysian atrophy (DRPLA) (see Chapter 29), spinocerebellar atrophies (see Chapter 29), multiple system atrophy (see Chapter 28), and diseases characterized by the ubiquitinated inclusions seen in ALS (see Chapter 27) are probably included in most of the historic series. Once these entities are excluded, a group of pallidal degenerations remains that can be classified on a purely descriptive basis, although it is not clear to what extent they represent distinct diseases.
 DIFFERENTIAL DIAGNOSIS OF PALLIDAL DEGENERATIONS
DIFFERENTIAL DIAGNOSIS OF PALLIDAL DEGENERATIONS

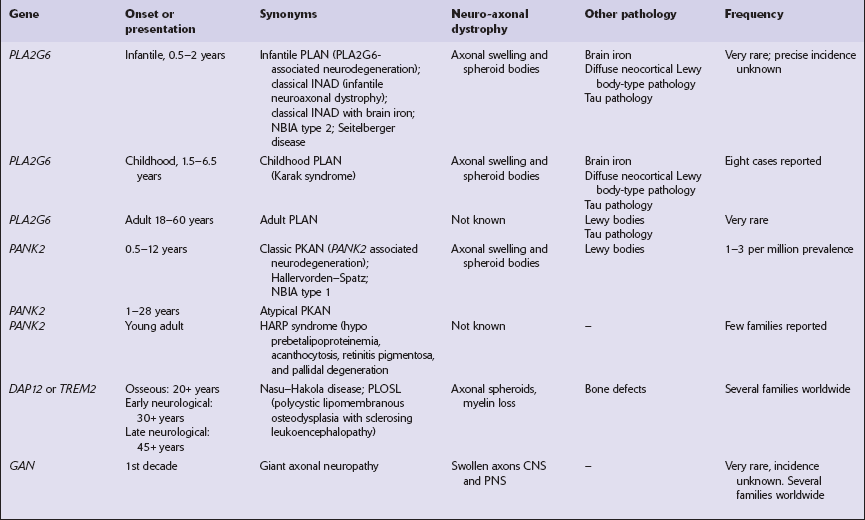
NEUROAXONAL DYSTROPHY
Several conditions, grouped as neuroaxonal dystrophies, are characterized pathologically by the presence of axonal swellings, which are thought to develop as a result of neuronal dysfunction that produces distal axonal degeneration. In most cases, the nature of the neuronal dysfunction and the pathogenesis of the axonal swelling are poorly understood. Neuroaxonal dystrophy occurs in three contexts (Tables 33.3, 33.4)).
Table 33.3
Classification of neuroaxonal dystrophic processes
Physiologic neuroaxonal dystrophy: normal brain aging
Gracile and cuneate nuclei
Globus pallidus
Substantia nigra
Spinal anterior horns
Primary neuroaxonal dystrophy: diseases in which the main pathology is neuroaxonal dystrophy
Neuroaxonal dystrophies with PLA2G6 mutations (phospholipase A2 group VI)
Infantile neuroaxonal dystrophy (INAD)
Late infantile neuroaxonal dystrophy
Juvenile neuroaxonal dystrophy
Adult neuroaxonal dystrophy
Childhood onset phospholipase A2 group 6-associated neurodegeneration (PLAN; atypical neuroaxonal dystrophy)
Schindler disease (PLA2G6 mutation and co-occurrence of α-N-acetylgalactosaminidase (α-NAGA) deficiency)
Neuroaxonal leukodystrophy
Hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS)
Familial pigmentary orthochromatic leukodystrophy (POLD)
Pantothenate kinase-associated neurodegeneration (PKAN; formerly Hallervorden–Spatz disease)
Classical PKAN
Atypical PKAN
Hypoprebetalipoproteinemia, acanthocytosis, retinitis pigmentosa, and pallidal degeneration (HARP syndrome)
Nasu–Hakola disease
Giant axonal neuropathy (caused by mutation of GAN, the gigaxonin gene)
Secondary neuroaxonal dystrophy: accentuation of neuroaxonal dystrophy in other disease processes
Neurodegenerative disease
Metabolic disease
Infective disease
MICROSCOPIC APPEARANCES
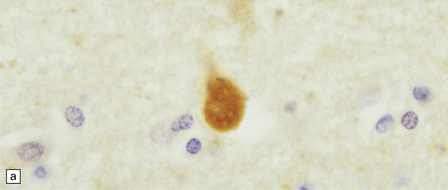
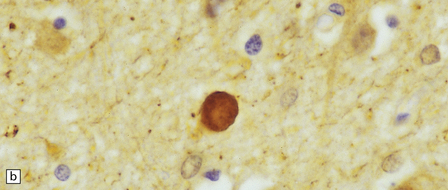
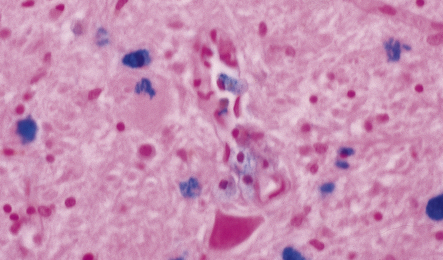
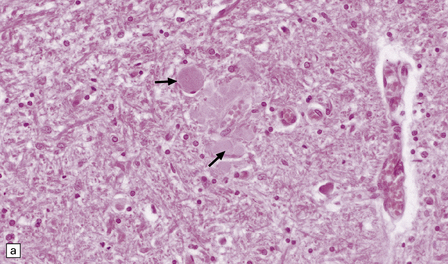
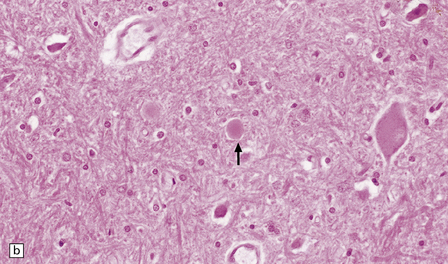
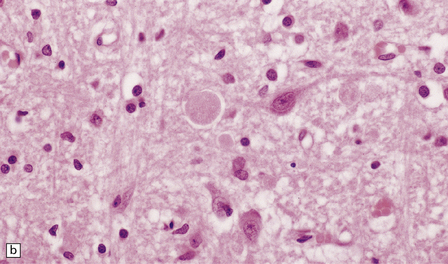
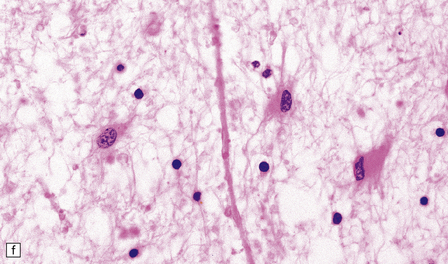
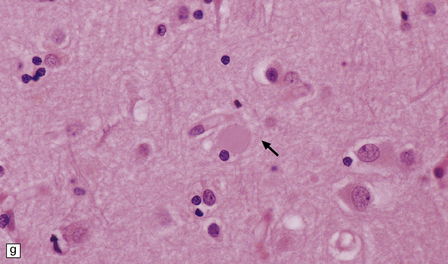
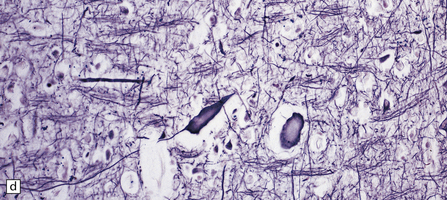
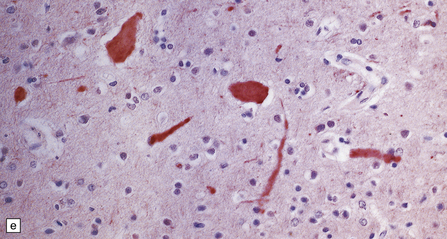
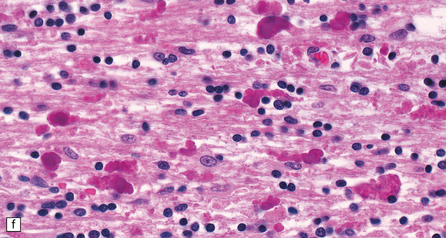
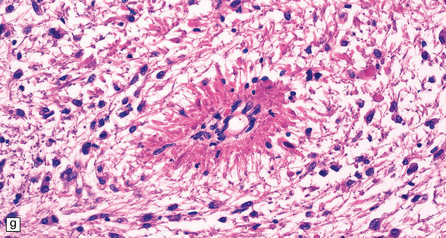
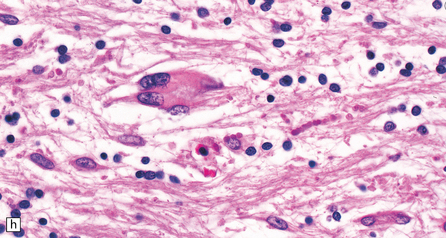
Dystrophic axonal swellings can be identified in sections stained with hematoxylin and eosin as rounded or elongated eosinophilic structures varying from 20 μm to 120 μm in diameter (Fig. 33.1a,b). Some dystrophic axons contain an intensely stained eosinophilic core surrounded by a paler zone (Fig. 33.1c). The swollen axons can be demonstrated by silver impregnation techniques (Fig. 33.1d). In toluidine-blue-stained resin sections, the swellings are seen to contain granular material (Fig. 33.1e). Electron microscopy shows this to consist of mitochondria, electron-dense lysosome-related bodies, tubulomembranous structures, and amorphous matrix material (Fig. 33.1f). Generally, relatively few neurofilaments are present and those that are may be displaced towards the periphery of the axon. Immunoreactivities for neurofilament protein and ubiquitin are largely confined to axonal swellings smaller than 30 μm in diameter (Fig. 33.2). Iron-containing and lipofuscin-like pigment may accumulate in axonal spheroids (Fig. 33.3), leading to the descriptive term pigment-spheroidal dystrophy.
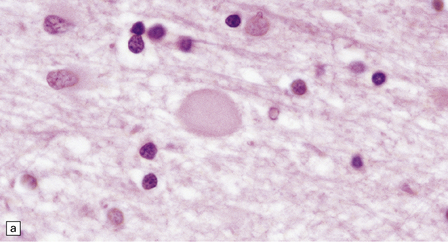
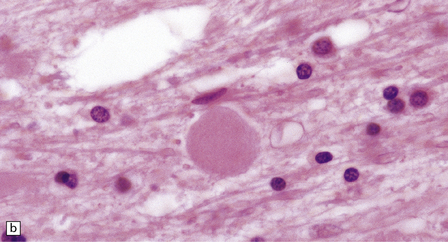
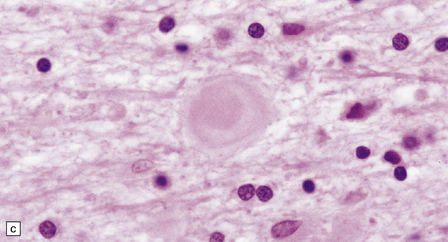
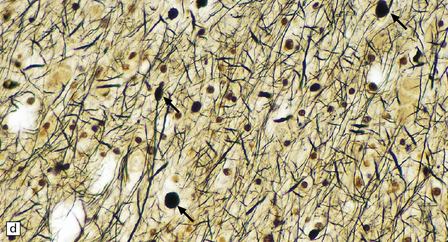
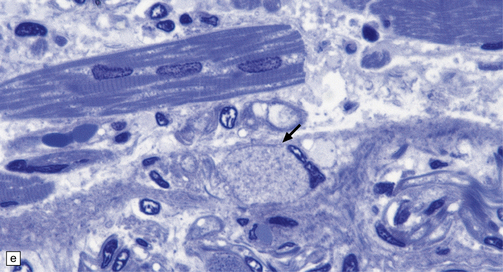
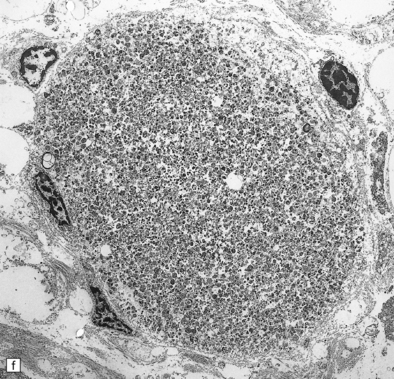
33.1 Microscopic features of neuroaxonal dystrophy.
(a) Axonal spheroids appear as pale eosinophilic rounded structures. (b) Many have a granular appearance in sections stained with hematoxylin and eosin. (c) Some spheroids show slight central pallor. Others, particularly the large spheroids, may contain a densely eosinophilic central region and a paler surrounding zone, as shown here. (d) The axonal swellings can be demonstrated by silver impregnation (arrows). (e) In resin sections, spheroids (arrow) appear pale and granular. (f) Electron microscopy shows that the spheroids contain abundant electron dense granules, mostly membrane-bound, and derived from lysosomes and degenerating mitochondria.
PHYSIOLOGIC NEUROAXONAL DYSTROPHY
 Gracile and cuneate nuclei in the medulla (Fig. 33.4a).
Gracile and cuneate nuclei in the medulla (Fig. 33.4a).
33.4 Physiologic neuroaxonal dystrophy.
(a) Pale eosinophilic axonal swellings (arrows) in the gracile nucleus of an elderly person. (b) In the substantia nigra, axonal swellings (arrow) are typically more eosinophilic.


PRIMARY NEUROAXONAL DYSTROPHY
 Diseases with PLA2G6 mutations: infantile, late infantile, juvenile, and adult neuroaxonal dystrophy.
Diseases with PLA2G6 mutations: infantile, late infantile, juvenile, and adult neuroaxonal dystrophy.




NEUROAXONAL DYSTROPHY WITH PLA2G6 Mutations
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Axonal spheroids and reactive astrocytic gliosis are widely distributed in the central and peripheral nervous system (Fig. 33.5). Degeneration of the corticospinal and spinobulbar tracts is usually prominent. There is often a moderate to severe diffuse cortical Lewy body pathology as well as tau pathology with neurofibrillary tangles and neuropil threads.
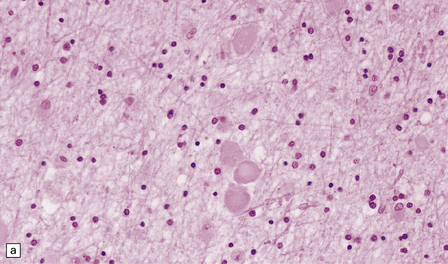
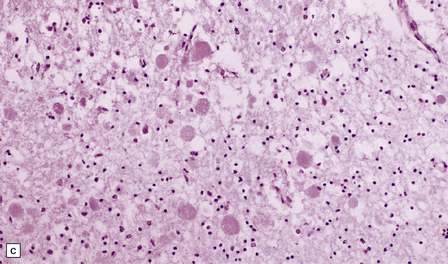
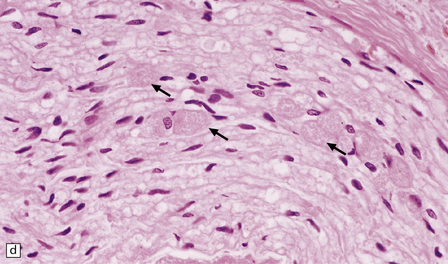
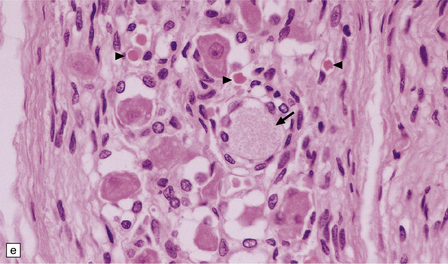
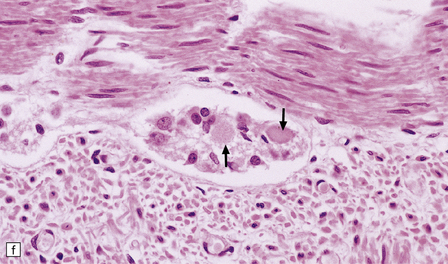
33.5 Infantile neuroaxonal dystrophy.
(a) Axonal spheroids are widely distributed in both white and gray matter in the CNS, as shown here in the hemispheric white matter. (b) Axonal spheroids in the basal ganglia. (c) Axonal spheroids are especially prominent in long tracts in the spinal cord. (d) Dystrophic axonal swellings (arrows) composed of granular eosinophilic material are also seen in peripheral nerves. (e) Dystrophic axonal swellings (arrowheads) and a degenerating neuron (arrow) in sympathetic ganglia. (f) Dystrophic axonal swellings (arrows) in the myenteric plexus of the gut. In the CNS, the formation of spheroids is followed by loss of myelin, neuronal degeneration, and astrocytic gliosis.
NEUROAXONAL LEUKODYSTROPHY
Macroscopic and microscopic appearances
There is cerebral atrophy with ventricular dilatation, and softening and gray discoloration of the white matter. Histology reveals severe loss of myelin from the hemispheric white matter, with astrocytic gliosis and numerous axonal swellings (Fig. 33.6a–f). Smaller numbers of dystrophic axonal swellings are present in gray matter areas, especially the cerebral cortex (Fig. 33.6g). There is distal tract degeneration in the spinal cord, particularly in the corticospinal tracts and posterior columns.
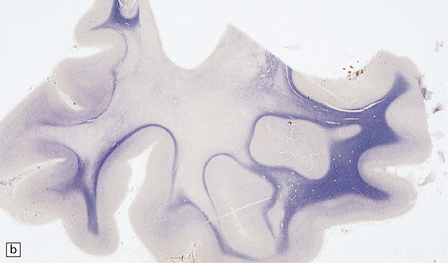
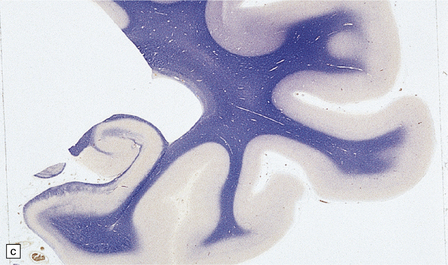
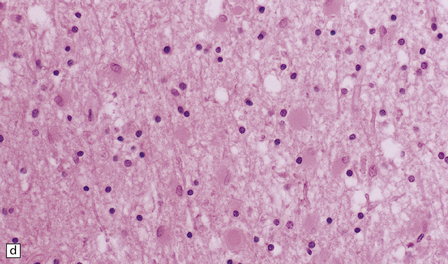
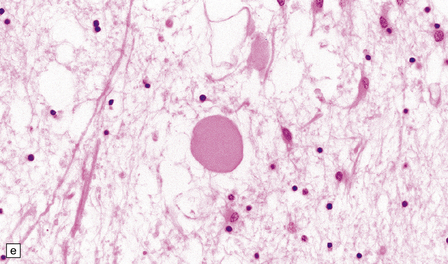
33.6 Neuroaxonal leukodystrophy.
(a) There is severe loss of fibers from the cerebral hemispheres, producing pallor in sections stained with H&E. (b) The severe loss of fibers from the cerebral hemispheres also produces pallor in sections stained for myelin. The subcortical fibers are spared. (c) In a personally studied case, the temporal lobes were also relatively spared, as shown here. (d) Mildly affected regions show axonal spheroids with relative preservation of fiber density. (e) In severely affected regions, there are scattered spheroids amidst few preserved axons. (f) Astrocytic gliosis of white matter is prominent in severely affected white matter. (g) Small numbers of spheroids (arrow) are seen in the cerebral cortex.
PANTOTHENATE KINASE-ASSOCIATED NEURODEGENERATION (FORMERLY HALLERVORDEN–SPATZ DISEASE)
MACROSCOPIC AND MICROSCOPIC APPEARANCES
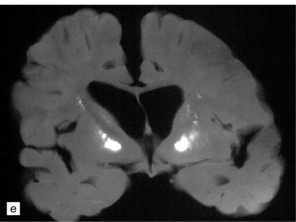
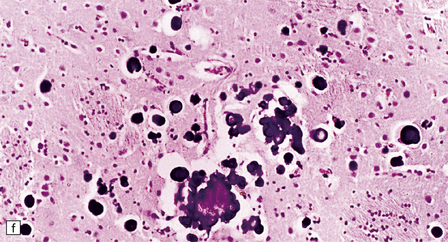
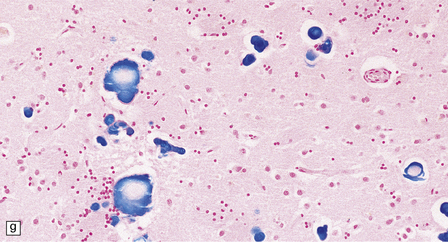
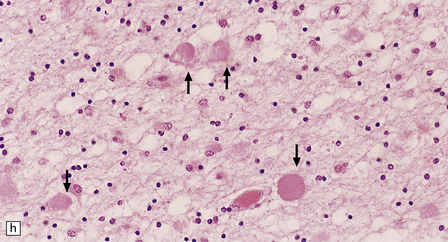
The globus pallidus and pars reticularis of the substantia nigra appear shrunken, and rust-brown in color (Fig. 33.7a). Histology shows neuronal loss, astrocytic gliosis, and accumulation of iron-containing pigment in these regions, which also contain large numbers of axonal swellings (Fig. 33.7b–d). Spheroids may also be seen in the cerebral cortex and the brain stem nuclei (Fig. 33.7e,f). Some patients have associated Lewy body pathology, some have associated neurofibrillary tangles. Pigment can be found in the renal tubular epithelium.
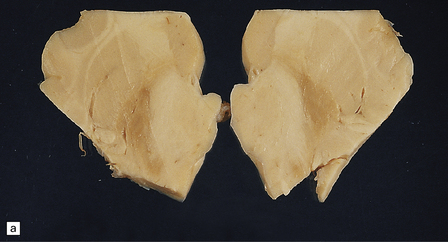
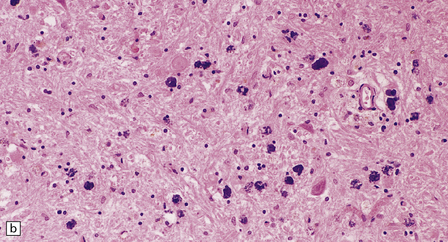
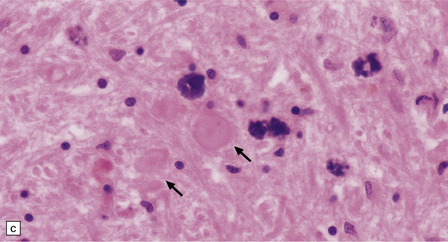
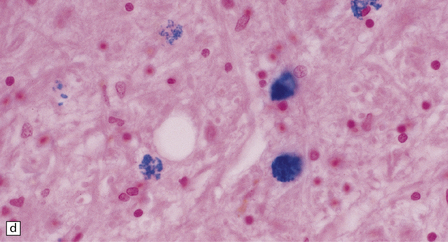
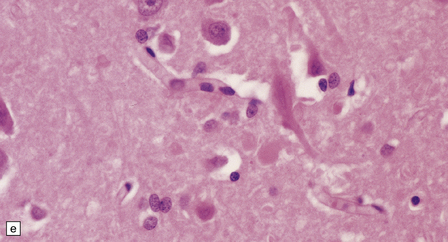
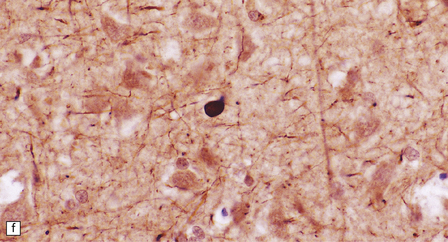
33.7 Pantothenate kinase-associated neurodegeneration.
(a) Macroscopically, the globus pallidus is rust-brown and atrophic. (b) The globus pallidus shows neuronal loss and gliosis and a conspicuous accumulation of pigment, seen as darkly stained material. (c) Closer examination reveals rounded eosinophilic axonal spheroids (arrows). (d) Pigment is present in surviving neurons, within spheroids, and in astrocytes. Some of the pigment, mostly around blood vessels, appears to be extracellular. The pigment contains lipofuscin, neuromelanin, and iron, which appears blue in this section by Perls’ method. (e) Small numbers of spheroids can be seen in the cerebral cortex. (f) The spheroids in the cerebral cortex can be highlighted by silver impregnation, or immunohistochemistry for neurofilamant protein, as shown here.
 PANTOTHENATE KINASE-ASSOCIATED NEURODEGENERATION
PANTOTHENATE KINASE-ASSOCIATED NEURODEGENERATION
 The pantothenate kinase gene (PANK2) is defective in this disease.
The pantothenate kinase gene (PANK2) is defective in this disease.
 Patients affected before 15 years of age:
Patients affected before 15 years of age:
• develop a gait disorder with rigidity of the legs, and dystonia
• show progressive disease that leads to motor slowing, dysarthria, cognitive decline, and, frequently, choreoathetosis
 Patients affected in later life:
Patients affected in later life:
 Computerized tomographic (CT) scans show high-signal lesions in the globus pallidus on both sides.
Computerized tomographic (CT) scans show high-signal lesions in the globus pallidus on both sides.


NASU–HAKOLA DISEASE
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Adipocytes in subcutaneous tissue, bone marrow, and elsewhere show membranocystic (lipomembranous) change (Fig. 33.8). The brain typically weighs of the order of 1000 g but can be reduced to less than 700 g. Atrophy is marked frontally. There is considerable loss of myelinated fibers from the hemispheric white matter, which contains many axonal spheroids. Severe neuronal loss with mineralization affects the basal ganglia, and in some cases, the thalamus (Fig. 33.8f). Peripheral nerves may also be affected.
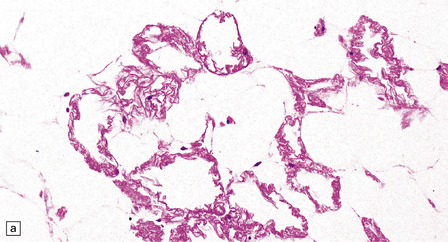
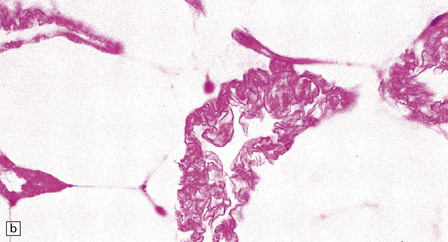
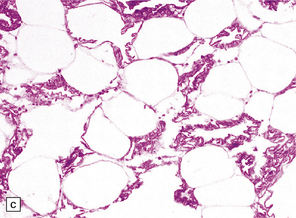
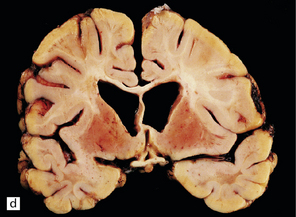
33.8 Nasu–Hakola disease.
(a) Histology reveals membranocystic lesions in adipose tissue, including that of bone marrow. The lesions are characterized by marked thickening and wrinkling of the plasma membrane of adipocytes. (b) At a higher magnification, the complex infolding of the adipocyte membrane can be better appreciated. (c) This is well demonstrated on periodic acid–Schiff (PAS) staining. Note the complex infolding of the adipocyte membrane. (d) The brain shows atrophy with ventricular dilatation. There is softening and loss of hemispheric white matter. The basal ganglia appear yellow-brown. (e) X-ray image of the brain slice showing prominent mineralization in the basal ganglia, particularly of the globus pallidus. (f) Sections of the basal ganglia show atrophy, neuronal loss, astrocytic gliosis, microcysts, and severe mineralization. (g) The mineral deposits contain iron, which appears blue in sections stained by Perls’ method. (h) The hemispheric white matter appears rarefied due to loss of myelinated fibers, and contains many axonal spheroids (arrows). Foci of mineralization may also be found. (i) There is relative sparing of subcortical fibers, stained blue in this preparation. (Courtesy of Dr David Hilton, Derriford Hospital, Plymouth.)
 NASU–HAKOLA DISEASE
NASU–HAKOLA DISEASE




GIANT AXONAL NEUROPATHY
MACROSCOPIC AND MICROSCOPIC APPEARANCES
The peripheral nerves contain swollen axons with closely packed neurofilaments. There is gray discoloration and atrophy of the deep white matter in the cerebrum (Fig. 33.9a), the cerebellum, and the long tracts in the spinal cord. The brain and spinal cord contain many dystrophic axonal swellings, particularly in the corticospinal tracts, middle and inferior cerebellar peduncles, and posterior columns of the spinal cord (Fig. 33.9b). Smaller numbers of axonal swellings are present in the cerebral cortex (Fig. 33.9c–e) and basal ganglia. Numerous Rosenthal fibers are present in the white matter (Fig. 33.9f) and may cluster around blood vessels in a pattern resembling that of Alexander’s disease (Fig. 33.9g). Occasional large multinucleated astrocytes may be seen (Fig. 33.9h). Abnormal accumulations of intermediate filaments have been noted in Schwann cells, fibroblasts, melanocytes, endothelial, and Langerhans cells in some cases. Electron microscopy has shown longitudinal grooving of the kinky hairs.
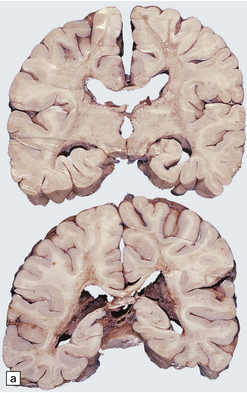
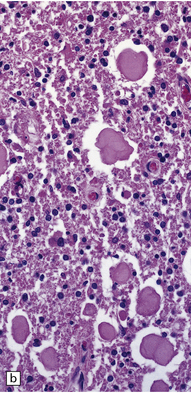
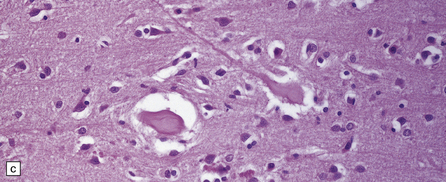
33.9 Giant axonal neuropathy.
(a) Shrinkage and gelatinous gray discoloration of the white matter in longstanding giant axonal neuropathy. The periventricular and parasagittal frontal white matter is most severely affected. (b) Numerous dystrophic axonal swellings in the posterior columns of the cervical spinal cord. (c) Scattered axonal swellings in the cerebral cortex in sections stained with H&E. (d) Scattered axonal swellings in the cerebral cortex in sections stained by Bielschowsky’s silver impregnation (toned). (e) Scattered axonal swellings in the cerebral cortex in sections stained with antibody to neurofilament protein. (f) The white matter contains large numbers of deeply eosinophilic Rosenthal fibers. (g) The clustering of Rosenthal fibers around blood vessels may simulate that of Alexander’s disease. (h) Large multinucleated astrocyte in the centrum semiovale in giant axonal neuropathy.
 GIANT AXONAL NEUROPATHY
GIANT AXONAL NEUROPATHY




SECONDARY NEUROAXONAL DYSTROPHY



 Vitamin E deficiency (see Chapter 21), particularly in association with cystic fibrosis, congenital biliary atresia, ataxia with isolated vitamin E deficiency caused by mutations in α-tocopherol transfer protein, and Bassen–Kornzweig syndrome (abetalipoproteinemia).
Vitamin E deficiency (see Chapter 21), particularly in association with cystic fibrosis, congenital biliary atresia, ataxia with isolated vitamin E deficiency caused by mutations in α-tocopherol transfer protein, and Bassen–Kornzweig syndrome (abetalipoproteinemia).
 Niemann–Pick disease type C (see Chapter 23).
Niemann–Pick disease type C (see Chapter 23).
 Some cases of autosomal recessive infantile osteopetrosis (Fig. 33.10).
Some cases of autosomal recessive infantile osteopetrosis (Fig. 33.10).
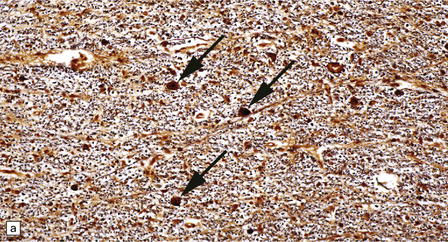
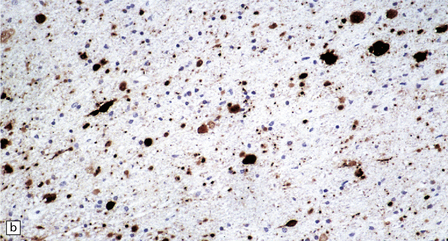
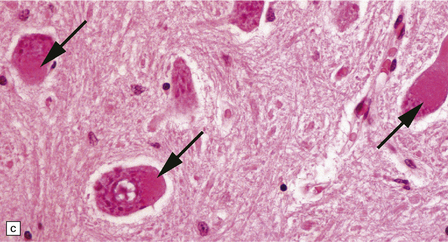
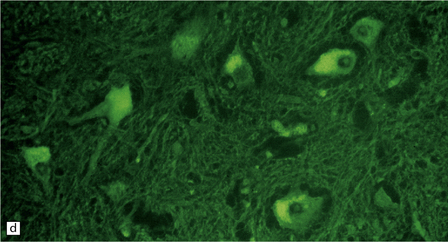
33.10 CNS abnormalities in infantile osteopetrosis with neuroaxonal dystrophy.
(a) The spinal white matter contains numerous dystrophic axonal swellings (arrows) (Palmgren silver impregnation). (b) These are well demonstrated by immunohistochemistry for β-amyloid precursor protein. In addition, many axons that are not obviously swollen show abnormal accumulation of this protein. (c) Neurons show cytoplasmic accumulation of lipofuscin-like, diastase-resistant material that stains strongly with periodic acid-Schiff (arrows). (d) Like lipofuscin, this material autofluoresces under ultraviolet light.

Dystrophic axonal swellings and focal white matter necrosis are also features of methotrexate leukoencephalopathy (see Chapter 25) and the multifocal necrotizing leukoencephalopathy that occurs in several other clinical contexts (see Chapter 22).
CENTRAL AUTONOMIC FAILURE
INTRODUCTION AND CLASSIFICATION
Autonomic failure can be divided into primary and secondary types (Table 33.5). Diabetes mellitus is the commonest cause of autonomic failure. Of the remaining patients, about 50% have a primary autonomic failure and the other 50% autonomic failure secondary to an identifiable cause.
Table 33.5
Classification and causes of autonomic failure
Primary autonomic failure
Multiple system atrophy
Lewy body disease
Progressive autonomic failure due to postganglionic pathology
Dopamine β-hydroxylase deficiency
Bradbury–Eggleston syndrome (idiopathic)
Secondary autonomic failure
Structural lesions of central pathways in corticolimbic, hypothalamic, brain stem or spinal regions
Wernicke’s encephalopathy
Baroreceptor failure
Botulism
Acute autonomic neuropathy
Peripheral neuropathy
diabetic
amyloid
inflammatory
alcoholic
toxic and drug-related
chronic renal failure
paraneoplastic
connective tissue disease
acute intermittent porphyria
familial neuropathy
SHY–DRAGER SYNDROME
 In both multiple system atrophy and Lewy body-associated disease, autonomic dysfunction is related to loss of cells from the intermediolateral column of the spinal cord (Fig. 33.11a–c).
In both multiple system atrophy and Lewy body-associated disease, autonomic dysfunction is related to loss of cells from the intermediolateral column of the spinal cord (Fig. 33.11a–c).
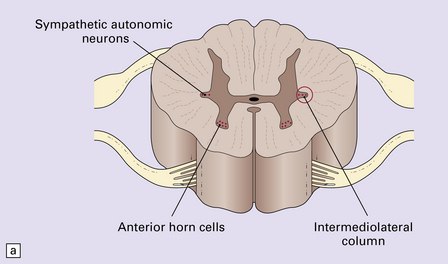
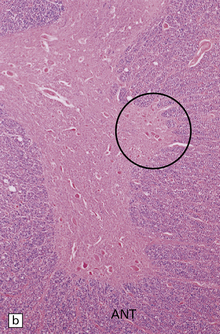
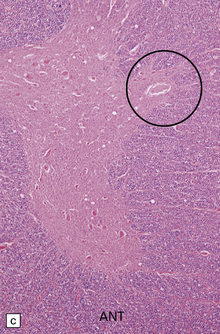
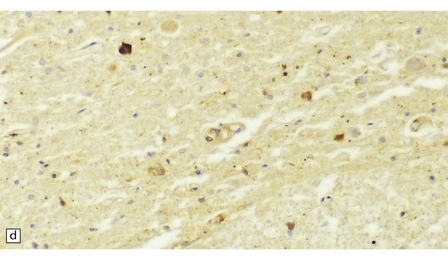
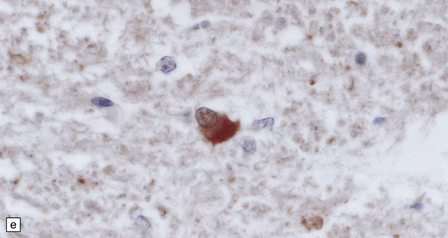
33.11 Spinal autonomic neurons in autonomic failure.
(a) The intermediolateral column contains preganglionic sympathetic neurons. (b) The intermediolateral group of neurons (in ring) is found in the lateral column in the thoracic spinal cord (ANT, anterior). They are preganglionic sympathetic neurons. The number of intermediolateral column neurons is greater in the upper thoracic cord segments than in the lower ones. (c) In central autonomic failure, these intermediolateral column neurons are reduced in number throughout the thoracic cord and there is mild astrocytic gliosis. Because of the variability in the number of intermediolateral column neurons at any one level, counts should be obtained from several levels before a loss of preganglionic sympathetic neurons can be diagnosed. (d) In autonomic failure caused by multiple system atrophy, small numbers of glial cytoplasmic inclusions can be found in the intermediolateral column, as shown in these sections immunostained for ubiquitin. (e) MSA inclusion stained with anti-tau antibody.
 In multiple system atrophy, the typical glial cytoplasmic inclusions are widely distributed (Fig. 33.11d,e), supraspinal autonomic nuclei may be affected, and the characteristic abnormalities are present in the substantia nigra and other regions of the brain (see Chapter 28).
In multiple system atrophy, the typical glial cytoplasmic inclusions are widely distributed (Fig. 33.11d,e), supraspinal autonomic nuclei may be affected, and the characteristic abnormalities are present in the substantia nigra and other regions of the brain (see Chapter 28).
 In Lewy body-associated disease, there are also degenerative changes with Lewy bodies in the substantia nigra and elsewhere in the brain (see Chapter 28), and in the peripheral sympathetic ganglia.
In Lewy body-associated disease, there are also degenerative changes with Lewy bodies in the substantia nigra and elsewhere in the brain (see Chapter 28), and in the peripheral sympathetic ganglia.
PURE PROGRESSIVE AUTONOMIC FAILURE
Pure progressive autonomic failure may be central or peripheral:


 NECROPSY IN AUTONOMIC FAILURE
NECROPSY IN AUTONOMIC FAILURE













REFERENCES
Bianchin, M.M., Capella, H.M., Chaves, D.L., et al. Nasu-Hakola disease (polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy – PLOSL): a dementia associated with bone cystic lesions. From clinical to genetic and molecular aspects. Cell Mol Neurobiol. 2004;24:1–24.
Ching, K.H., Westaway, S.K., Gitschier, J., et al. HARP syndrome is allelic with pantothenate kinase-associated neurodegeneration. Neurology.. 2002;58(11):1673–1674.
Engel, L.A., Jing, Z., O’Brien, D.E., et al. Catalytic function of PLA2G6 is impaired by mutations associated with infantile neuroaxonal dystrophy but not dystonia-parkinsonism. PLoS One.. 2010;5(9):e12897.
Freeman, S.H., Hyman, B.T., Sims, K.B., et al. Adult onset leukodystrophy with neuroaxonal spheroids: clinical, neuroimaging and neuropathologic observations. Brain Pathol.. 2009;19:39–47.
Gordon, N. Infantile neuroaxonal dystrophy (Seitelberger’s disease). Dev Med Child Neurol.. 2002;44(12):849–851.
Gregory, A., Polster, B.J., Hayflick, S.J. Clinical and genetic delineation of neurodegeneration with brain iron accumulation. J Med Genet.. 2009;46(2):73–80.
Gregory, A., Westaway, S.K., Holm, I.E., et al. Neurodegeneration associated with genetic defects in phospholipase A(2). Neurology.. 2008;71(18):1402–1409.
Hayflick, S.J., Westaway, S.K., Levinson, B., et al. Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N Engl J Med.. 2003;348:33–40.
Khateeb, S., Flusser, H., Ofir, R., et al. PLA2G6 mutation underlies infantile neuroaxonal dystrophy. Am J Hum Genet.. 2006;79:942–948.
Kruer, M.C., Hiken, M., Gregory, A., et al. Novel histopathologic findings in molecularly-confirmed pantothenate kinase-associated neurodegeneration. Brain.. 2011;134(Pt 4):947–958.
Kurian, M.A., McNeill, A., Lin, J.P., et al. Childhood disorders of neurodegeneration with brain iron accumulation (NBIA). Dev Med Child Neurol.. 2011;53:394–404.
Matarin, M.M., Singleton, A.B., Houlden, H. PANK2 gene analysis confirms genetic heterogeneity in neurodegeneration with brain iron accumulation (NBIA) but mutations are rare in other types of adult neurodegenerative disease. Neurosci Lett.. 2006;407(2):162–165.
Morgan, N.V., Westaway, S.K., Morton, J.E., et al. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet.. 2006;38:752–754.
Ni, X., Ma, Y., Cheng, H., et al. Cloning and characterization of a novel human pantothenate kinase gene. Int J Biochem Cell Biol.. 2002;34(2):109–115.
Paisan-Ruiz, C., Li, A., Schneider, S.A., et al. Widespread Lewy body and tau accumulation in childhood and adult onset dystonia-parkinsonism cases with PLA2G6 mutations. Neurobiol Aging.. 2010;33(4):814–823.
Sadeh, M. Neurodegeneration associated with genetic defects in phospholipase A2. Neurology.. 2009;73(10):819.
Tofaris, G.K., Revesz, T., Jacques, T., et al. Adult-onset neurodegeneration with brain iron accumulation and cortical alpha-synuclein and tau pathology: a distinct clinicopathological entity. Arch Neurol.. 2007;64(2):280–282.
Yoshino, H., Tomiyama, H., Tachibana, N., et al. Phenotypic spectrum of patients with PLA2G6 mutation and PARK14-linked parkinsonism. Neurology.. 2010;75(15):1356–1361.
Zhou, B., Westaway, S.K., Levinson, B., et al. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet.. 2001;28(4):345–349.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]33
Thalamic and pallidal degenerations, neuroaxonal dystrophy, and autonomic failure
THALAMIC DEGENERATIONS
Thalamic degeneration is a feature of several multisystem neurodegenerative disorders (Table 33.1), but can also rarely occur in a ‘pure’ form.
Table 33.1
Diseases in which thalamic degeneration may be prominent
Multiple system atrophy (glial cytoplasmic inclusions)
Spinocerebellar degenerations
Wernicke’s encephalopathy
Huntington’s disease
Creutzfeldt–Jakob disease
Menkes syndrome
Membranous lipodystrophy
Neuroaxonal dystrophy
Fatal familial insomnia
PURE THALAMIC ATROPHY
DIFFERENTIAL DIAGNOSIS OF PURE THALAMIC ATROPHY
The conditions listed in Table 33.1 should be excluded. These include fatal familial insomnia, which has a primary thalamic pattern of involvement and is a form of prion disease (see Chapter 32).
Neuronal loss from the thalamus has been described in patients that have undergone leukotomy.
PALLIDAL DEGENERATIONS
The grouping together of pallidal degenerations is based on the morphologic finding of degeneration centered on the globus pallidus, either alone or in combination with degeneration of the subthalamic nucleus or substantia nigra. These disorders have been subdivided according to the regions of the brain showing pathologic changes (Table 33.2). Clinically, pallidal degenerations are associated with a variety of movement disorders, with or without dementia. Some are familial and others sporadic.
It is difficult to evaluate the nosologic status of many of the cases described in the literature as their relationship to disorders that can now be better defined by molecular genetic and immunohistochemical techniques is uncertain. In particular, cases of dentatorubropallidoluysian atrophy (DRPLA) (see Chapter 29), spinocerebellar atrophies (see Chapter 29), multiple system atrophy (see Chapter 28), and diseases characterized by the ubiquitinated inclusions seen in ALS (see Chapter 27) are probably included in most of the historic series. Once these entities are excluded, a group of pallidal degenerations remains that can be classified on a purely descriptive basis, although it is not clear to what extent they represent distinct diseases.
NEUROAXONAL DYSTROPHY
Several conditions, grouped as neuroaxonal dystrophies, are characterized pathologically by the presence of axonal swellings, which are thought to develop as a result of neuronal dysfunction that produces distal axonal degeneration. In most cases, the nature of the neuronal dysfunction and the pathogenesis of the axonal swelling are poorly understood. Neuroaxonal dystrophy occurs in three contexts (Tables 33.3, 33.4)).
Table 33.3
Classification of neuroaxonal dystrophic processes
Physiologic neuroaxonal dystrophy: normal brain aging
Gracile and cuneate nuclei
Globus pallidus
Substantia nigra
Spinal anterior horns
Primary neuroaxonal dystrophy: diseases in which the main pathology is neuroaxonal dystrophy
Neuroaxonal dystrophies with PLA2G6 mutations (phospholipase A2 group VI)
Infantile neuroaxonal dystrophy (INAD)
Late infantile neuroaxonal dystrophy
Juvenile neuroaxonal dystrophy
Adult neuroaxonal dystrophy
Childhood onset phospholipase A2 group 6-associated neurodegeneration (PLAN; atypical neuroaxonal dystrophy)
Schindler disease (PLA2G6 mutation and co-occurrence of α-N-acetylgalactosaminidase (α-NAGA) deficiency)
Neuroaxonal leukodystrophy
Hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS)
Familial pigmentary orthochromatic leukodystrophy (POLD)
Pantothenate kinase-associated neurodegeneration (PKAN; formerly Hallervorden–Spatz disease)
Classical PKAN
Atypical PKAN
Hypoprebetalipoproteinemia, acanthocytosis, retinitis pigmentosa, and pallidal degeneration (HARP syndrome)
Nasu–Hakola disease
Giant axonal neuropathy (caused by mutation of GAN, the gigaxonin gene)
Secondary neuroaxonal dystrophy: accentuation of neuroaxonal dystrophy in other disease processes
Neurodegenerative disease
Metabolic disease
Infective disease
MICROSCOPIC APPEARANCES
Dystrophic axonal swellings can be identified in sections stained with hematoxylin and eosin as rounded or elongated eosinophilic structures varying from 20 μm to 120 μm in diameter (Fig. 33.1a,b). Some dystrophic axons contain an intensely stained eosinophilic core surrounded by a paler zone (Fig. 33.1c). The swollen axons can be demonstrated by silver impregnation techniques (Fig. 33.1d). In toluidine-blue-stained resin sections, the swellings are seen to contain granular material (Fig. 33.1e). Electron microscopy shows this to consist of mitochondria, electron-dense lysosome-related bodies, tubulomembranous structures, and amorphous matrix material (Fig. 33.1f). Generally, relatively few neurofilaments are present and those that are may be displaced towards the periphery of the axon. Immunoreactivities for neurofilament protein and ubiquitin are largely confined to axonal swellings smaller than 30 μm in diameter (Fig. 33.2). Iron-containing and lipofuscin-like pigment may accumulate in axonal spheroids (Fig. 33.3), leading to the descriptive term pigment-spheroidal dystrophy.
33.1 Microscopic features of neuroaxonal dystrophy.
(a) Axonal spheroids appear as pale eosinophilic rounded structures. (b) Many have a granular appearance in sections stained with hematoxylin and eosin. (c) Some spheroids show slight central pallor. Others, particularly the large spheroids, may contain a densely eosinophilic central region and a paler surrounding zone, as shown here. (d) The axonal swellings can be demonstrated by silver impregnation (arrows). (e) In resin sections, spheroids (arrow) appear pale and granular. (f) Electron microscopy shows that the spheroids contain abundant electron dense granules, mostly membrane-bound, and derived from lysosomes and degenerating mitochondria.
PHYSIOLOGIC NEUROAXONAL DYSTROPHY
Gracile and cuneate nuclei in the medulla (Fig. 33.4a).
33.4 Physiologic neuroaxonal dystrophy.
(a) Pale eosinophilic axonal swellings (arrows) in the gracile nucleus of an elderly person. (b) In the substantia nigra, axonal swellings (arrow) are typically more eosinophilic.
PRIMARY NEUROAXONAL DYSTROPHY
Diseases with PLA2G6 mutations: infantile, late infantile, juvenile, and adult neuroaxonal dystrophy.
NEUROAXONAL DYSTROPHY WITH PLA2G6 Mutations
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Axonal spheroids and reactive astrocytic gliosis are widely distributed in the central and peripheral nervous system (Fig. 33.5). Degeneration of the corticospinal and spinobulbar tracts is usually prominent. There is often a moderate to severe diffuse cortical Lewy body pathology as well as tau pathology with neurofibrillary tangles and neuropil threads.
33.5 Infantile neuroaxonal dystrophy.
(a) Axonal spheroids are widely distributed in both white and gray matter in the CNS, as shown here in the hemispheric white matter. (b) Axonal spheroids in the basal ganglia. (c) Axonal spheroids are especially prominent in long tracts in the spinal cord. (d) Dystrophic axonal swellings (arrows) composed of granular eosinophilic material are also seen in peripheral nerves. (e) Dystrophic axonal swellings (arrowheads) and a degenerating neuron (arrow) in sympathetic ganglia. (f)
[/not-level-membership-for-pathology-category]
