[level-membership-for-pathology-category]22
Systemic metabolic diseases
HYPOGLYCEMIA
MACROSCOPIC APPEARANCES
 ETIOLOGY OF HYPOGLYCEMIC BRAIN INJURY
ETIOLOGY OF HYPOGLYCEMIC BRAIN INJURY
 The principal source of energy in the CNS is glucose. Its uptake by nerve cells is independent of insulin. Neuronal stores of glucose and glycogen are relatively small and the requirement practically continuous. During periods of fasting, adults and, to a lesser extent, children are able to obtain some energy from metabolism of ketones, but a blood glucose level of less than 1.5 mmol/L (25–30 mg/100 mL) leads to brain damage within 1–2 hours.
The principal source of energy in the CNS is glucose. Its uptake by nerve cells is independent of insulin. Neuronal stores of glucose and glycogen are relatively small and the requirement practically continuous. During periods of fasting, adults and, to a lesser extent, children are able to obtain some energy from metabolism of ketones, but a blood glucose level of less than 1.5 mmol/L (25–30 mg/100 mL) leads to brain damage within 1–2 hours.



 HYPOGLYCEMIA
HYPOGLYCEMIA




MICROSCOPIC APPEARANCES
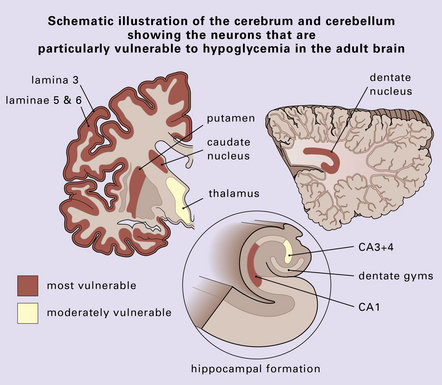
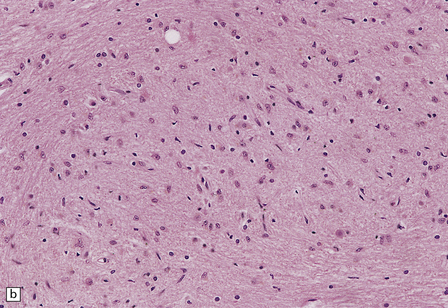
The lesions are similar to those of acute hypoxia–ischemia (see Chapter 8), but not identical. In general, the pattern of injury is that of selective degeneration of neurons rather than frank infarction. Affected neurons are shrunken with hypereosinophilic cytoplasm. The nuclei are initially pyknotic, but later become more eosinophilic and appear to blend in with the cytoplasm (nuclear dropout) (Fig. 22.1). As in hypoxia, the subiculum and CA1 field of the hippocampus are particularly vulnerable, while CA3 and 4 are less, and CA2 is least vulnerable. In the cerebral neocortex, the large neurons in laminae 3, 5, and 6 are most likely to be involved. The caudate nucleus and putamen, particularly the small neurons, are highly vulnerable to hypoglycemia. In infants, the dentate nucleus may also be affected (Fig. 22.1). In contrast to hypoxic–ischemic brain injury, Purkinje cells are usually spared. The regions of the brain particularly vulnerable to hypoglycemia are shown in Figure 22.2.
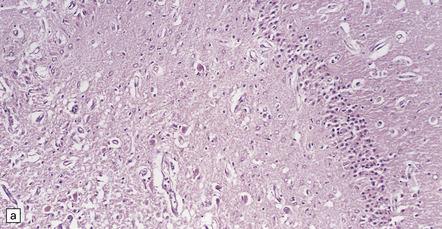
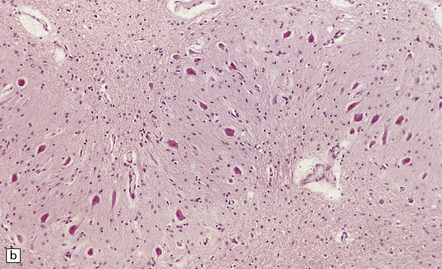
22.1 Acute hypoglycemic injury. (a) Severe hypoglycemic hippocampal injury due to insulin overdose. Most of the nuclei in the dentate fascia are shrunken and pyknotic. Many of the large neurons in the end folium (CA4) show nuclear dropout. (b) Hypereosinophilic neurons in the dentate nucleus of an infant with acute fatal hypoglycemia due to nesidioblastosis. In adults with hypoglycemia, the dentate nucleus is, like the Purkinje cell layer, usually spared.
Infants dying from hypoglycemia may show widespread neuronal degeneration throughout the brain.
FINDINGS IN LONG-TERM SURVIVORS OF SEVERE HYPOGLYCEMIA
MACROSCOPIC APPEARANCES
The cerebral cortex may appear thinned and the hippocampi shrunken and discolored (Fig. 22.3). The white matter is reduced in bulk and the ventricles are dilated. There may be marked atrophy of the caudate nucleus and putamen (Fig. 22.3).
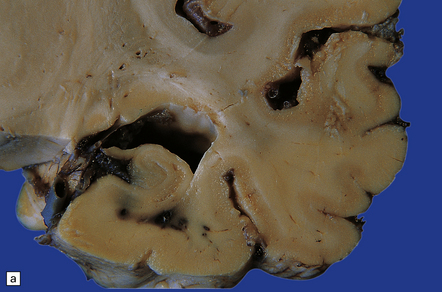
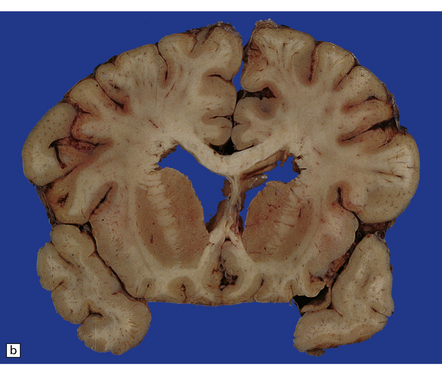
22.3 Chronic changes after hypoglycemic injury. (a) Macroscopic findings in long-term survivor of severe hypoglycemia. Granular and atrophic neocortex in a survivor of an insulin overdose. The hippocampus and tail of the caudate nucleus are also severely shrunken. (b) Macroscopic features several weeks after an insulin overdose. The cerebral cortex is congested and focally thinned, particularly in the orbital frontal region and in the depths of sulci over the cerebral convexities. The corpus striatum is congested and there is irregular shrinkage of the caudate nucleus so that the medial surface is uneven.
MICROSCOPIC APPEARANCES
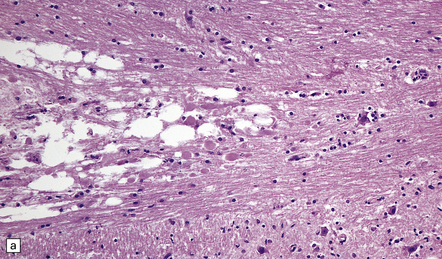
The cerebral cortex shows laminar neuronal loss and gliosis associated with capillary proliferation (Fig. 22.4). There is often dense subpial gliosis. The hippocampal pyramidal cell layer and subiculum are replaced by a loose meshwork of glial tissue (Fig. 22.5). The white matter is usually rarefied and gliotic. The caudate nucleus and putamen are diffusely gliotic (Fig. 22.4). The globus pallidus is relatively spared. Moderate neuronal loss and gliosis may be evident in the thalamus. As in acute hypoglycemia, the cerebellar cortex, including the Purkinje cells, is relatively spared (Fig. 22.4).
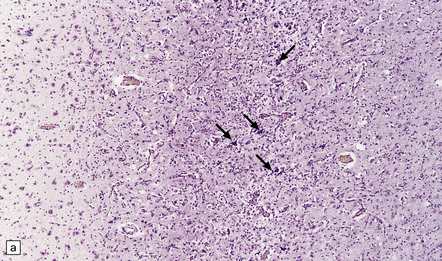
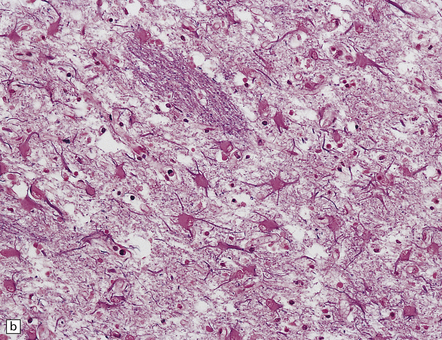
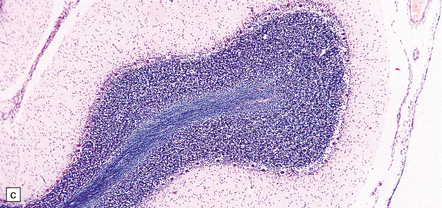
22.4 Chronic effects of severe hypoglycemia. (a) The cerebral cortex shows laminar neuronal loss, gliosis, and capillary proliferation. Several remaining neurons are mineralized (arrows). (b) The putamen is diffusely gliotic. (c) There is relative sparing of the cerebellar cortex, including the Purkinje cells.
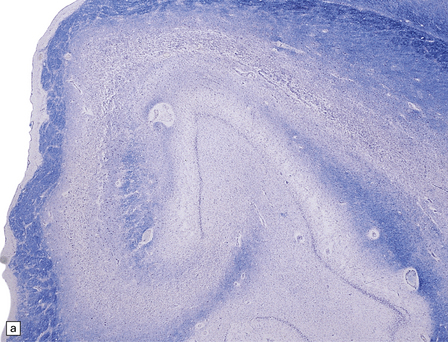
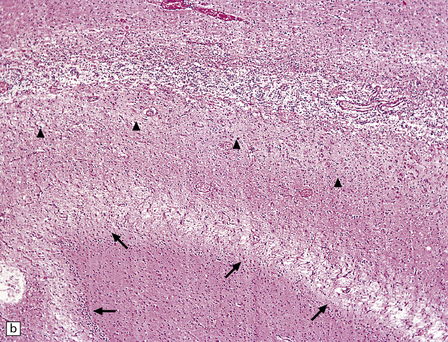
22.5 Microscopic features 6 weeks after an insulin overdose. (a) The hippocampal pyramidal cell layer and subiculum are replaced by a loose meshwork of glial tissue and capillaries. (b) Higher magnification view of the pyramidal cell layer (arrowheads) and the dentate fascia (arrows), both of which are severely gliotic.
DISTURBANCES OF BODY TEMPERATURE
HYPERTHERMIA
 ETIOLOGY OF HYPERTHERMIA
ETIOLOGY OF HYPERTHERMIA This is most commonly due to strenuous physical exertion under hot and humid conditions, but may also be a consequence of an infection or drug reaction. Predisposing conditions include diabetes mellitus, alcoholism, intoxication with certain drugs (particularly 3,4–methylenedioxymethamphetamine (Ecstasy), and other amphetamines) and disorders in which there is impaired sweating (e.g. anhidrotic ectodermal dysplasia).
This is most commonly due to strenuous physical exertion under hot and humid conditions, but may also be a consequence of an infection or drug reaction. Predisposing conditions include diabetes mellitus, alcoholism, intoxication with certain drugs (particularly 3,4–methylenedioxymethamphetamine (Ecstasy), and other amphetamines) and disorders in which there is impaired sweating (e.g. anhidrotic ectodermal dysplasia).



MACROSCOPIC AND MICROSCOPIC APPEARANCES
The brain often appears normal or only mildly edematous. Some patients develop a bleeding diathesis, which may be associated with parenchymal or meningeal hemorrhages. Parenchymal petechial hemorrhages may be found adjacent to the third and fourth ventricles. Cerebellar Purkinje cell loss has been reported. Other described abnormalities are similar to those of hypoxic–ischemic damage (see Chapter 8) and probably result from a combination of cardiovascular collapse and an increased metabolic rate (Fig. 22.6). In patients with malignant hyperthermia, skeletal muscle may show central cores.
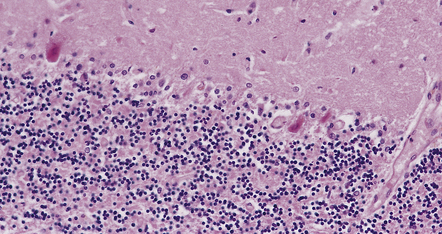
22.6 Hyperthermia. Cerebellar cortex from a patient who collapsed and later died after spending several hours in a steam tent. The systemic necropsy findings were in keeping with heat stroke. Examination of the brain revealed changes indistinguishable from those of acute hypoxic–ischemic neuronal injury, as illustrated by these Purkinje cells with shrunken, pyknotic nuclei, and hypereosinophilic cytoplasm.
DISORDERS OF SERUM ELECTROLYTES
HYPONATREMIA
 ETIOLOGY OF HYPONATREMIA
ETIOLOGY OF HYPONATREMIA





 HYPONATREMIA
HYPONATREMIA Acute and severe hyponatremia causes neurological syndromes necessitating rapid correction with hypertonic saline. Hyponatremic encephalopathy and hepatic encephalopathy (see below) may coexist in patients with cirrhosis. The clinical picture is common to any metabolic encephalopathy and is characterized by a confusional syndrome that may evolve into coma.
Acute and severe hyponatremia causes neurological syndromes necessitating rapid correction with hypertonic saline. Hyponatremic encephalopathy and hepatic encephalopathy (see below) may coexist in patients with cirrhosis. The clinical picture is common to any metabolic encephalopathy and is characterized by a confusional syndrome that may evolve into coma.
 Patients with hyponatremia develop lethargy, headache, and nausea and vomiting.
Patients with hyponatremia develop lethargy, headache, and nausea and vomiting.
 If severe, hyponatremia may cause seizures and coma, leading to death.
If severe, hyponatremia may cause seizures and coma, leading to death.
HYPERNATREMIA
 ETIOLOGY OF HYPERNATREMIA
ETIOLOGY OF HYPERNATREMIA




 HYPERNATREMIA
HYPERNATREMIA

OSMOTIC DEMYELINATION SYNDROME
 ETIOLOGY OF ODS
ETIOLOGY OF ODS Most predisposing conditions are associated with hyponatremia (i.e. alcoholic liver damage, extensive skin burns, inappropriate secretion of antidiuretic hormone, psychogenic polydipsia, hyperemesis gravidarum).
Most predisposing conditions are associated with hyponatremia (i.e. alcoholic liver damage, extensive skin burns, inappropriate secretion of antidiuretic hormone, psychogenic polydipsia, hyperemesis gravidarum).
 ODS occasionally develops in patients with a normal serum sodium concentration.
ODS occasionally develops in patients with a normal serum sodium concentration.
 The incidence of ODS is relatively high in liver transplant patients.
The incidence of ODS is relatively high in liver transplant patients.

 ODS has occurred after infusion of hypertonic saline into normonatremic patients.
ODS has occurred after infusion of hypertonic saline into normonatremic patients.
 ODS occasionally complicates chronic malnourishment or debilitation.
ODS occasionally complicates chronic malnourishment or debilitation.
 ODS
ODS



MACROSCOPIC APPEARANCES
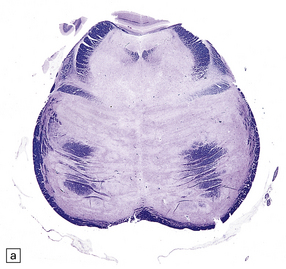
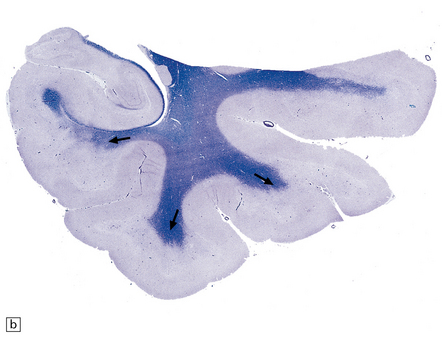
Typically, the basis pontis includes a fusiform region of gray discoloration, which is abnormally soft and appears granular on sectioning (Fig. 22.7). The extent of the lesion is variable. Its cross-sectional area is usually greatest in the upper part of the pons, where only a narrow rim of subpial tissue may be spared (Fig. 22.8). It may involve the middle cerebral peduncles, but rarely extends rostrocaudally beyond the confines of the pons and lower midbrain. The lesion may be asymmetric, being largely or completely confined to one side of the pons.
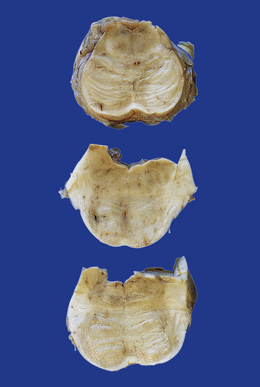
22.7 ODS. Transverse sections through the brain stem in CPM showing gray discoloration and slight granularity of much of the basis pontis.
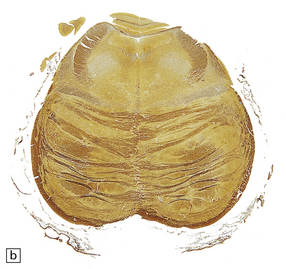
22.8 Sections of the pons in CPM. (a) There is extensive loss of myelin. Note the sparing of myelin in a narrow rim of subpial tissue and in small ‘islands’ within the base of the pons. (b) In an adjacent section, axons are seen to be well preserved.
The reported frequency of extrapontine lesions varies, but careful examination will reveal lesions in other parts of the CNS such as the cerebellum (Fig. 22.9), lateral geniculate body, capsula externa or extrema, subcortical cerebral white matter (Fig. 22.10), basal ganglia, thalamus, or internal capsule in 25–50% of cases. In up to 25% of patients the lesions may be exclusively extrapontine.
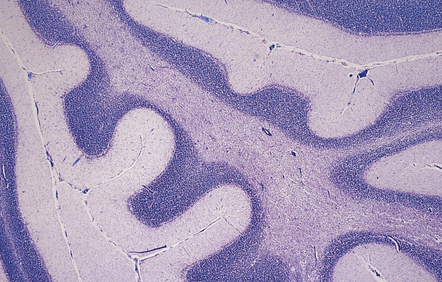
22.9 ODS: extrapontine demyelination. This section shows extrapontine demyelination in the white matter in the cerebellum. (Luxol fast blue/cresyl violet)
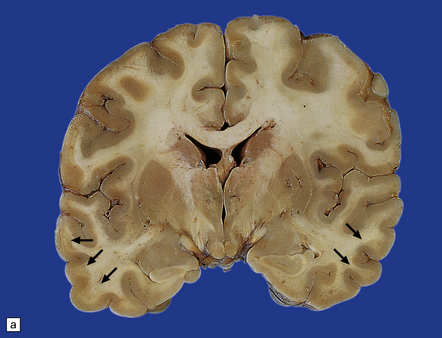
22.10 Subcortical demyelination in ODS. (a) Gray discoloration of the demyelinated subcortical white matter (arrows) in a case of extrapontine myelinolysis associated with ODS. (b) Solochrome cyanin staining confirms the presence of subcortical demyelination involving white matter in the crests of gyri (arrows).
MICROSCOPIC APPEARANCES
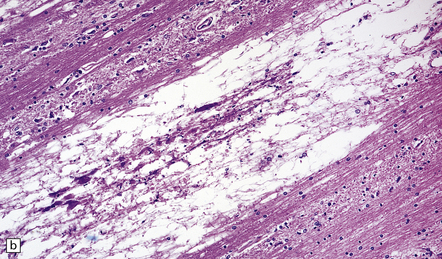
The microscopic appearances of CPM are of active demyelination (Fig. 22.11). The lesions contain reactive astrocytes and large numbers of foamy lipid-laden macrophages (Fig. 22.12), but only very scanty lymphocytes. Silver impregnation may reveal some axonal fragmentation, but most neuronal somata and axons are intact. Within the lesions, cranial nerves or central ‘islands’ of transverse pontine fibers or corticospinal tracts may be preserved (see Fig. 22.8).
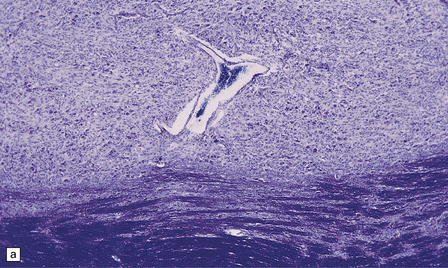
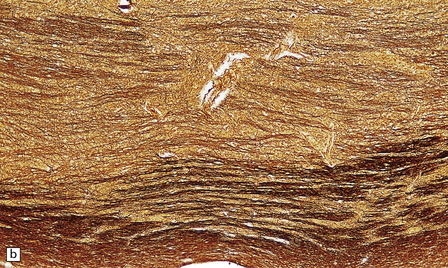
22.11 Preservation of neuronal somata and axons in ODS. (a) Section of pons stained to show myelin. (b) Section of pons adjacent to (a) stained for axons. There is good preservation of neuronal somata and axons within the zone of demyelination.
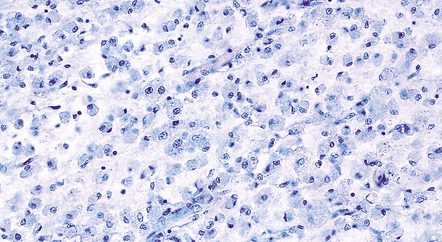
22.12 Foamy macrophages in ODS. Higher magnification of the zone of demyelination in ODS reveals large numbers of foamy macrophages.
 DIFFERENTIAL DIAGNOSIS OF ODS
DIFFERENTIAL DIAGNOSIS OF ODS

CALCIUM DISTURBANCES AND FAHR’S DISEASE
Mild cerebral calcification, particularly of the basal ganglia, is a frequent incidental finding, especially in the elderly. More extensive calcification can occur in numerous medical conditions, including a variety of infections (e.g. intrauterine cytomegalovirus infection or toxoplasmosis), metabolic and endocrine (especially parathyroid) disorders, and genetic syndromes (e.g. Cockayne syndrome, see Chapter 5). Fahr’s disease refers to familial idiopathic calcification of the basal ganglia, also known as striopallidodentate calcinosis. The more general term Fahr syndrome is sometimes used to encompass both Fahr’s disease and basal ganglia calcification secondary to other disorders.
 FAHR’S DISEASE, AND ETIOLOGY OF BASAL GANGLIA CALCIFICATION
FAHR’S DISEASE, AND ETIOLOGY OF BASAL GANGLIA CALCIFICATION
 Fahr’s disease may be inherited as an autosomal dominant or recessive disorder.
Fahr’s disease may be inherited as an autosomal dominant or recessive disorder.
 Linkage to chromosome 14q was demonstrated in one family with dominantly inherited Fahr’s disease.
Linkage to chromosome 14q was demonstrated in one family with dominantly inherited Fahr’s disease.

 A sporadic dementing disease, termed diffuse neurofibrillary, tangles with calcification (DNTC), largely confined to Japanese patients, is characterized by mineralization of the brain in a pattern identical to that of Fahr’s disease, but also neuronal loss, gliosis, and neurofibrillary tangles in the hippocampus, cerebral neocortex, basal ganglia, thalamus, subthalamus, substantia nigra, locus ceruleus, and occasionally in other brain stem structures and the dentate nucleus. Lewy bodies and neurites are present in the amygdala, hippocampus, and usually also the substantia nigra and dorsal motor nucleus of the vagus, and astrocytes immunopositive for α-synuclein occur in the hippocampus and temporal neocortex. A high proportion of patients also have TDP-43-positive neuronal and glial cytoplasmic inclusions, predominantly in the temporal lobe (see also Chapter 28).
A sporadic dementing disease, termed diffuse neurofibrillary, tangles with calcification (DNTC), largely confined to Japanese patients, is characterized by mineralization of the brain in a pattern identical to that of Fahr’s disease, but also neuronal loss, gliosis, and neurofibrillary tangles in the hippocampus, cerebral neocortex, basal ganglia, thalamus, subthalamus, substantia nigra, locus ceruleus, and occasionally in other brain stem structures and the dentate nucleus. Lewy bodies and neurites are present in the amygdala, hippocampus, and usually also the substantia nigra and dorsal motor nucleus of the vagus, and astrocytes immunopositive for α-synuclein occur in the hippocampus and temporal neocortex. A high proportion of patients also have TDP-43-positive neuronal and glial cytoplasmic inclusions, predominantly in the temporal lobe (see also Chapter 28).

 FAHR’S DISEASE
FAHR’S DISEASEAge at onset of clinical symptoms in Fahr’s disease is 30–60 years.



MACROSCOPIC AND MICROSCOPIC APPEARANCES
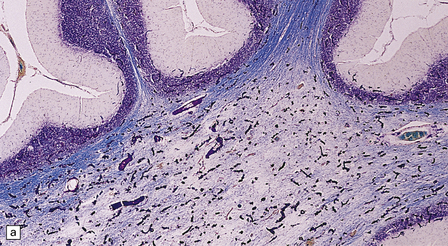
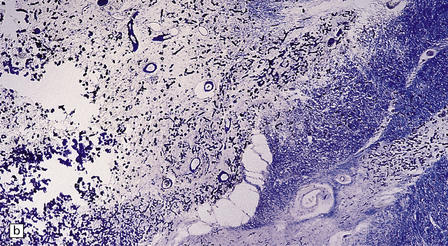
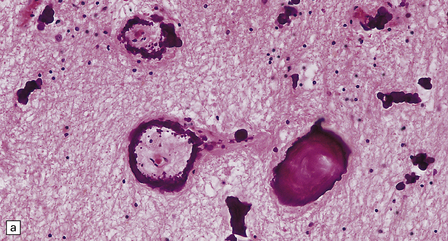
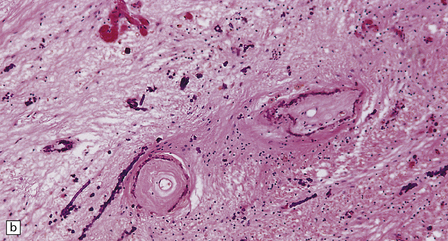
Vascular calcification and scanty parenchymal mineral deposits are a common incidental finding in the pallidum and, to a lesser extent, in the hippocampus and dentate nucleus, particularly in old age. The mineralization in Fahr’s disease is, however, much more extensive and can involve the cerebral sulci, basal ganglia (particularly the globus pallidus), dentate nucleus, subthalamus, red nucleus, and other regions (Fig. 22.13). In addition to calcium, the concretions contain iron, magnesium, aluminum, and glycoproteins. The calcification of the media and adventitia of blood vessels may be associated with intimal fibrosis and narrowing, in some cases completely occluding the lumen (Fig. 22.14). The extent of associated neuronal degeneration and gliosis is variable and may be partly related to the degree of ischemia.
HYPERCALCEMIC ENCEPHALOPATHY
 HYPERCALCEMIC ENCEPHALOPATHY
HYPERCALCEMIC ENCEPHALOPATHY The most common causes are primary hyperparathyroidism and malignancies (particularly in association with widespread involvement of bone).
The most common causes are primary hyperparathyroidism and malignancies (particularly in association with widespread involvement of bone).
 The clinical presentation is typically with confusion, seizures, visual abnormalities and headaches.
The clinical presentation is typically with confusion, seizures, visual abnormalities and headaches.

 The neuropathological findings in fatal cases include microvascular necrosis, perivascular exudates, microhemorrhages and edema (see Chapter 10 for further description of the microscopic changes in accelerated hypertension).
The neuropathological findings in fatal cases include microvascular necrosis, perivascular exudates, microhemorrhages and edema (see Chapter 10 for further description of the microscopic changes in accelerated hypertension).
LIVER DISEASE
ACQUIRED HEPATIC ENCEPHALOPATHY
 ETIOLOGY AND PATHOGENESIS OF ACQUIRED HEPATIC ENCEPHALOPATHY
ETIOLOGY AND PATHOGENESIS OF ACQUIRED HEPATIC ENCEPHALOPATHY




 ACQUIRED HEPATIC ENCEPHALOPATHY
ACQUIRED HEPATIC ENCEPHALOPATHY






MICROSCOPIC APPEARANCES
 Deep layers of the cerebral cortex.
Deep layers of the cerebral cortex.
 Caudate nucleus and putamen (Fig. 22.15).
Caudate nucleus and putamen (Fig. 22.15).
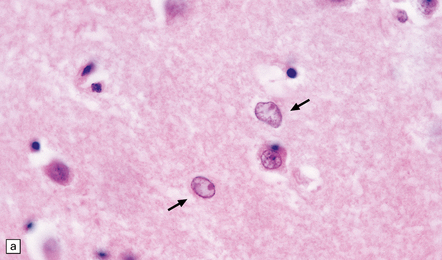
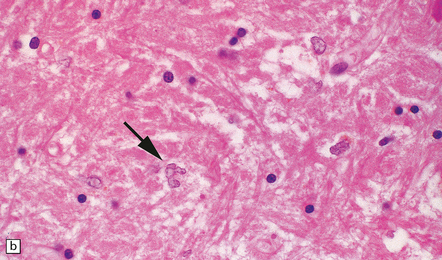
22.15 Alzheimer type II astrocytes. (a) Alzheimer type II astrocytes (arrows) in the putamen tend to have an enlarged, oval, vesicular nucleus and very scanty cytoplasm. (b) Alzheimer type II astrocytes in the pallidum tend to have an irregularly lobulated nucleus (arrow).
 Pallidum (where they are often particularly prominent) (Fig. 22.15).
Pallidum (where they are often particularly prominent) (Fig. 22.15).
 Thalamus, subthalamus, and hypothalamus.
Thalamus, subthalamus, and hypothalamus.


Chronic or recurrent hepatic encephalopathy may also cause:
 Patchy pseudolaminar necrosis and microcavitation in the depths of sulci at the junction of cerebral cortex and white matter (Fig. 22.16). The necrosis may extend into the cerebral white matter and can also involve the cerebellar white matter in severe cases.
Patchy pseudolaminar necrosis and microcavitation in the depths of sulci at the junction of cerebral cortex and white matter (Fig. 22.16). The necrosis may extend into the cerebral white matter and can also involve the cerebellar white matter in severe cases.
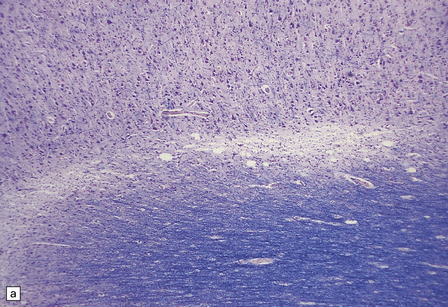
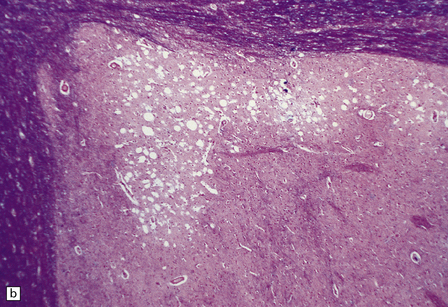
22.16 Chronic hepatic encephalopathy. (a) Microcavitation at the junction of cerebral cortex and white matter. (b) Neuronal loss and microcavitation in the dorsal pole of the putamen.
 Neuronal loss, gliosis and, in severe cases, microcavitation, which are most prominent in the dorsal pole of the putamen (Fig. 22.16).
Neuronal loss, gliosis and, in severe cases, microcavitation, which are most prominent in the dorsal pole of the putamen (Fig. 22.16).
 In chronic hepatic myelopathy, there is degeneration of corticospinal tract fibers.
In chronic hepatic myelopathy, there is degeneration of corticospinal tract fibers.
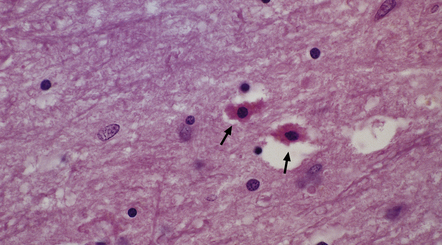
Opalski cells are altered astrocytes with a small central nucleus and abundant, finely granular, deeply eosinophilic cytoplasm. More usually associated with Wilson’s disease (see below), they may also be seen in non-Wilsonian chronic hepatic encephalopathy (Fig. 22.17).
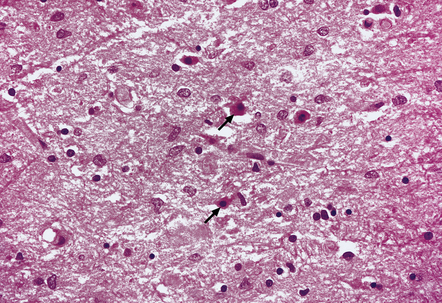
22.17 Opalski cells in the subthalamus in hepatic encephalopathy due to portacaval shunting. These cells have a small hyperchromatic nucleus and abundant granular deeply eosinophilic cytoplasm (arrows). Note also the presence of numerous Alzheimer type II astrocytes with irregularly lobulated nuclei.
HEPATOLENTICULAR DEGENERATION (WILSON’S DISEASE)
 ETIOLOGY OF HEPATOLENTICULAR DEGENERATION (WILSON’S DISEASE)
ETIOLOGY OF HEPATOLENTICULAR DEGENERATION (WILSON’S DISEASE)
 This autosomal recessive disorder is caused by mutations in a copper-transporting ATPase gene (ATP7B) encoded on chromosome 13q14.3.
This autosomal recessive disorder is caused by mutations in a copper-transporting ATPase gene (ATP7B) encoded on chromosome 13q14.3.


 ATP7B shares over 80% amino acid identity with ATP7A, the copper-transporting ATPase that is defective in Menkes’ disease (see Chapter 6), and is probably involved in the import of copper into the cell.
ATP7B shares over 80% amino acid identity with ATP7A, the copper-transporting ATPase that is defective in Menkes’ disease (see Chapter 6), and is probably involved in the import of copper into the cell.
 HEPATOLENTICULAR DEGENERATION
HEPATOLENTICULAR DEGENERATION





MACROSCOPIC AND MICROSCOPIC APPEARANCES
The putamen and caudate nucleus appear brown and shrunken, particularly the middle third of the putamen (Fig. 22.18). The putamen may be centrally cavitated. Histology of these nuclei reveals neuronal loss, scattered lipid- and pigment-laden macrophages, and many fibrillary astrocytes (Fig. 22.19). Alzheimer type II astrocytes (see acquired hepatic encephalopathy above) are abundant. The globus pallidus, subthalamic nucleus, thalamus, and brain stem are often involved, but less severely.
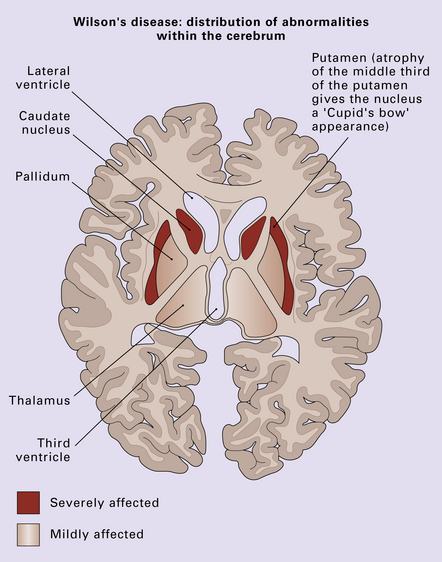
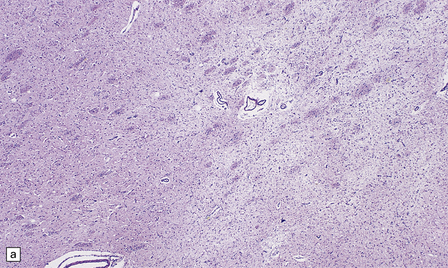
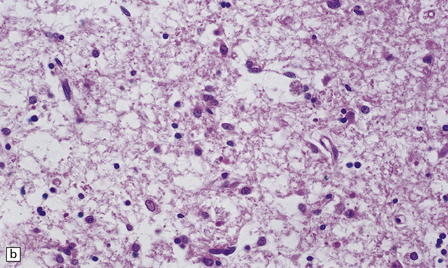
22.19 Histology of the putamen in Wilson’s disease. (a,b) show neuronal loss, pigment-laden macrophages, and gliosis of the putamen in a case of Wilson’s disease.
A distinctive but not entirely specific feature (see acquired hepatic encephalopathy, above) is the presence of Opalski cells, particularly in the globus pallidus (Fig. 22.20). Expression of glial antigens has been noted in some studies. Cells showing varied degenerative nuclear and cytoplasmic changes are often found in the basal ganglia, thalamus, and zona reticulata of the substantia nigra. Other abnormalities that may be present include foci of spongy degeneration in the cerebral cortex and white matter.
OTHER, RARE CAUSES OF BRAIN IRON ACCUMULATION
These include the primary neuroaxonal dystrophies (see Chapter 33) and neuroferritinopathy caused by mutation of the ferritin light chain gene, in which accumulation of iron and ferritin, formation of neuroaxonal spheroids and neuronal degeneration are most pronounced in the posterior putamen and cerebellar cortex.
REYE SYNDROME
MACROSCOPIC APPEARANCES
 ETIOLOGY OF REYE SYNDROME
ETIOLOGY OF REYE SYNDROME


 REYE SYNDROME
REYE SYNDROME




 DIFFERENTIAL DIAGNOSIS OF REYE SYNDROME
DIFFERENTIAL DIAGNOSIS OF REYE SYNDROME

PORPHYRIA
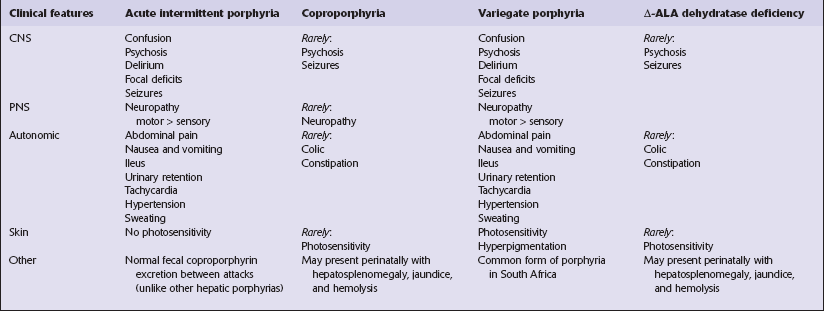
The porphyrias are inherited defects of metabolism characterized by overproduction and excretion of porphyrins or their precursors (Table 22.1):
Table 22.1
Enzyme defects and genetic loci in hepatic porphyrias (all are autosomal dominant disorders)
| Subtype | Enzyme defect | Locus |
| Acute intermittent | Phorphobilinogen deaminase | 11q24 |
| Coproporphyria | Coproporphyrinogen III oxidase | 3q12 |
| Variegate porphyria | Protoporphyrinogen oxidase | 1q22 |
| Δ-aminolevulinic acid Dehydratase deficiency | 9q34 |
 In the erythropoietic porphyrias, porphyrins accumulate in normoblasts and red blood cells.
In the erythropoietic porphyrias, porphyrins accumulate in normoblasts and red blood cells.
 In the hepatic porphyrias, porphyrins or their precursors are overproduced by the liver.
In the hepatic porphyrias, porphyrins or their precursors are overproduced by the liver.
Only the hepatic porphyrias produce neurologic disease.
The symptoms and signs of hepatic porphyrias are summarized in Table 22.2. They rarely begin before puberty. Factors that may precipitate acute disease include:
 Certain drugs (e.g. barbiturates, sulfonamides, griseofulvin, meprobamate, phenytoin, succinimides, steroids).
Certain drugs (e.g. barbiturates, sulfonamides, griseofulvin, meprobamate, phenytoin, succinimides, steroids).


 Menstruation (some women experience attacks just before menstruation).
Menstruation (some women experience attacks just before menstruation).
MACROSCOPIC AND MICROSCOPIC APPEARANCES
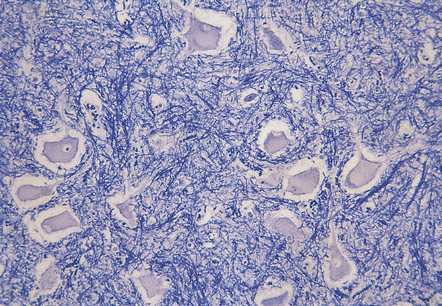
The autonomic, somatomotor, and sensory disturbances are due to an axonal neuropathy. Motor neurons in the spinal cord and brain stem nuclei may appear chromatolytic (Fig. 22.21). Distal degeneration of posterior column fibers has been reported in some cases.
GASTROINTESTINAL DISORDERS
 ETIOLOGY OF CELIAC DISEASE
ETIOLOGY OF CELIAC DISEASE




 CELIAC DISEASE
CELIAC DISEASE This usually presents with malabsorption and diarrhea, but gastrointestinal symptoms may be mild or absent.
This usually presents with malabsorption and diarrhea, but gastrointestinal symptoms may be mild or absent.

 Rarely, celiac disease results in myelopathy or peripheral neuropathy due to malabsorption and vitamin deficiency, particularly of vitamin E (see Chapter 21). Peripheral neuropathies, of axonal and demyelinating types, have also been reported and may respond to the elimination of dietary gluten.
Rarely, celiac disease results in myelopathy or peripheral neuropathy due to malabsorption and vitamin deficiency, particularly of vitamin E (see Chapter 21). Peripheral neuropathies, of axonal and demyelinating types, have also been reported and may respond to the elimination of dietary gluten.
MACROSCOPIC AND MICROSCOPIC APPEARANCES
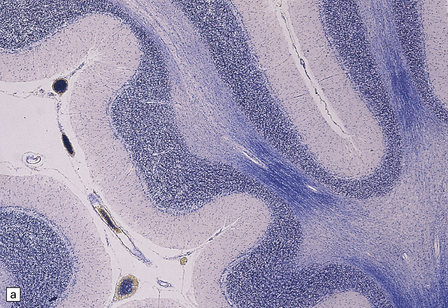
The most consistent neuropathologic abnormality is cerebellar atrophy, with loss of Purkinje cells, Bergmann cell gliosis, and variable loss of granule cells (Fig. 22.22). There may be diffuse neuronal loss and gliosis in the dentate and inferior olivary nuclei (Fig. 22.22). Perivascular lymphocytic infiltrates have been described in the cerebellum in some cases, and probably represent an early phase of the cerebellar lesions in celiac disease. Focal neuronal loss and gliosis are occasionally seen in the basal ganglia, diencephalon, and brain stem nuclei. Little has been published on the neuropathologic findings associated with the cortical and subcortical parieto-occipital calcification that may occur in this disorder.
RENAL DISEASE
Non-specific neuropathologic abnormalities have been described, including cerebral atrophy, gliosis, and foci of perivascular necrosis with accumulation of macrophages. Alzheimer type II astrocytes are often prominent. ODS has been reported (see above). Patients may also develop changes of hypertensive encephalopathy (see Chapter 10). MRI abnormalities can be seen in the basal ganglia, particularly in patients who also have diabetes mellitus; these changes are largely reversible on treatment and their neuropathological substrate is not known.
DIALYSIS ENCEPHALOPATHY
Two distinct CNS disorders have been associated with dialysis for end-stage renal disease:


Dialysis dementia is thought to be due to aluminum toxicity (see Chapter 25). Dialysis dysequilibrium syndrome is more commonly caused by hemodialysis than peritoneal dialysis. It consists of acute headache, nausea, muscle cramps, asterixis, myoclonus, and seizures, and is believed to be due to cerebral water intoxication caused by hypo-osmolality.
MULTIFOCAL NECROTIZING LEUKOENCEPHALOPATHY (MNL)
This is characterized by the development of multiple, usually microscopic, foci of necrosis with calcification, predominantly in the white matter. The basis pontis is often affected and the condition used to be known as focal pontine leukoencephalopathy. The pathogenesis of MNL is not known, but it occurs predominantly in immunosuppressed patients. The most commonly associated diseases are AIDS and leukemia. X-irradiation, amphotericin B, methotrexate, and various other cytotoxic drugs have been implicated in some cases (see Chapter 25).
MACROSCOPIC AND MICROSCOPIC APPEARANCES
The brain usually appears macroscopically normal. Ill-defined foci of chalky white discoloration may be visible in the pons or cerebral white matter (Fig. 22.23). Rarely, the pons is diffusely swollen, simulating a mass lesion.
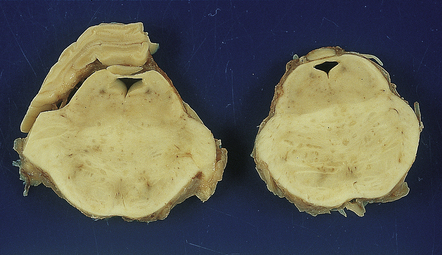
22.23 Multifocal necrotizing leukoencephalopathy. The base of the pons is slightly swollen and includes an ill-defined central zone of chalky white discoloration.
In general, the likelihood of identifying MNL depends on how extensively the brain is examined histologically. Microscopically, the lesions consist of well-demarcated foci of spongy vacuolation and loss of myelin staining, containing swollen fragmented axons (Fig. 22.24), which are often calcified, and scattered macrophages. The lesions are most consistently found in the basis pontis, especially in the transverse pontine fibers. There are also extrapontine foci in some patients.
REFERENCES
Arora, A., Neema, M., Stankiewicz, J., et al. Neuroimaging of toxic and metabolic disorders. Semin Neurol.. 2008;28:495–510.
Sharma, P., Eesa, M., Scott, J.N. Toxic and acquired metabolic encephalopathies: MRI appearance. AJR Am J Roentgenol.. 2009;193:879–886.
Spinazzé, S., Schrijvers, D. Metabolic emergencies. Crit Rev Oncol Hematol.. 2006;58:79–89.
Surtees, R., Leonard, J.V. Acute metabolic encephalopathy: a review of causes, mechanisms and treatment. J Inherit Metab Dis.. 1989;12:42–54.
Weathers, A.L., Lewis, S.L. Rare and unusual … or are they? Less commonly diagnosed encephalopathies associated with systemic disease. Semin Neurol.. 2009;29:136–153.
Alkalay, A.L., Sarnat, H.B., Flores-Sarnat, L., et al. Neurologic aspects of neonatal hypoglycemia. Isr Med Assoc J.. 2005;7:188–192.
Auer, R.N., Siesjo, B.K. Hypoglycaemia: brain neurochemistry and neuropathology. Baillieres Clin Endocrinol Metab.. 1993;7:611–625.
Auer, R.N. Insulin, blood glucose levels, and ischemic brain damage. Neurology.. 1998;51:S39–S43.
Hawdon, J.M. Hypoglycaemia and the neonatal brain. Eur J Pediatr.. 1999;158:S9–S12.
Kang, E.G., Jeon, S.J., Choi, S.S., et al. Diffusion MR imaging of hypoglycemic encephalopathy. AJNR Am J Neuroradiol.. 2010;31:559–564.
Ma, J.H., Kim, Y.J., Yoo, W.J., et al. MR imaging of hypoglycemic encephalopathy: lesion distribution and prognosis prediction by diffusion-weighted imaging. Neuroradiology.. 2009;51:641–649.
Service, F.J. Hypoglycemia. Med Clin North Am.. 1995;79:1–8.
Vannucci, R.C., Vannucci, S.J. Hypoglycemic brain injury. Semin Neonatol.. 2001;6:147–155.
Wass, C.T., Lanier, W.L. Glucose modulation of ischemic brain injury: review and clinical recommendations. Mayo Clin Proc.. 1996;71:801–812.
Disturbances of body temperature
Dietrich, W.D., Bramlett, H.M. Hyperthermia and central nervous system injury. Prog Brain Res.. 2007;162:201–217.
Sarkar, S., Barks, J.D. Systemic complications and hypothermia. Semin Fetal Neonatal Med.. 2010;15:270–275.
Sharma, H.S., Johanson, C.E. Blood-cerebrospinal fluid barrier in hyperthermia. Prog Brain Res.. 2007;162:459–478.
Wang, C.X., Stroink, A., Casto, J.M., et al. Hyperthermia exacerbates ischaemic brain injury. Int J Stroke.. 2009;4:274–284.
Wappler, F. Malignant hyperthermia. Eur J Anaesthesiol.. 2001;18:632–652.
White, M.G., Luca, L.E., Nonner, D., et al. Cellular mechanisms of neuronal damage from hyperthermia. Prog Brain Res.. 2007;162:347–371.
Disorders of serum electrolytes
Achinger, S.G., Moritz, M.L., Ayus, J.C. Dysnatremias: why are patients still dying? South Med J.. 2006;99:353–362.
Agrawal, V., Agarwal, M., Joshi, S.R., et al. Hyponatremia and hypernatremia: disorders of water balance. J Assoc Physicians India.. 2008;56:956–964.
Ayus, J.C., Arieff, A.I. Pathogenesis and prevention of hyponatremic encephalopathy. Endocrinol Metab Clin North Am.. 1993;22:425–446.
Brown, W.D. Osmotic demyelination disorders: central pontine and extrapontine myelinolysis. Curr Opin Neurol.. 2000;13:691–697.
Córdoba, J., García-Martinez, R., Simón-Talero, M. Hyponatremic and hepatic encephalopathies: similarities, differences and coexistence. Metab Brain Dis.. 2010;25:73–80.
Ferreiro, J.A., Robert, M.A., Townsend, J., et al. Neuropathologic findings after liver transplantation. Acta Neuropathol (Berl).. 1992;84:1–14.
Fried, L.F., Palevsky, P.M. Hyponatremia and hypernatremia. Med Clin North Am.. 1997;81:585–609.
Geschwind, D.H., Loginov, M., Stern, J.M. Identification of a locus on chromosome 14q for idiopathic basal ganglia calcification (Fahr Disease). Am J Hum Genet.. 1999;65:764–772.
Gocht, A., Colmant, H.J. Central pontine and extrapontine myelinolysis: a report of 58 cases. Clin Neuropathol.. 1987;6:262–270.
Kleinschmidt-Demasters, B.K., Rojiani, A.M., Filley, C.M. Central and extrapontine myelinolysis: then and now. J Neuropathol Exp Neurol.. 2006;65:1–11.
Kumar, S., Fowler, M., Gonzalez-Toledo, E., et al. Central pontine myelinolysis, an update. Neurol Res.. 2006;28:360–366.
Lampl, C., Yazdi, K. Central pontine myelinolysis. Eur Neurol.. 2002;47:3–10.
Martin, R.J. Central pontine and extrapontine myelinolysis: the osmotic demyelination syndromes. J Neurol Neurosurg Psychiatry. 2004;75:iii22–iii28.
Riggs, J.E. Neurologic manifestations of fluid and electrolyte disturbances. Neurol Clin.. 1989;7:509–523.
Verbalis, J.G., Martinez, A.J., Drutarosky, M.D. Neurological and neuropathological sequelae of correction of chronic hyponatremia. Kidney Int.. 1991;39:1274–1282.
Manyam, B.V. What is and what is not ‘Fah’s disease’. Parkinsonism Relat Disord.. 2005;11:73–80.
Kastrup, O., Maschke, M., Wanke, I., et al. Posterior reversible encephalopathy syndrome due to severe hypercalcemia. J Neurol.. 2002;249:1563–1566.
Lamy, C., Oppenheim, C., Méder, J.F., et al. Neuroimaging in posterior reversible encephalopathy syndrome. J Neuroimaging.. 2004;14:89–96.
Lumachi, F., Brunello, A., Roma, A., et al. Cancer-induced hypercalcemia. Anticancer Res.. 2009;29:1551–1555.
Brewer, G.J. Neurologically presenting Wilson’s disease: epidemiology, pathophysiology and treatment. CNS Drugs.. 2005;19:185–192.
Butterworth, R.F. Altered glial-neuronal crosstalk: cornerstone in the pathogenesis of hepatic encephalopathy. Neurochem Int.. 2010;57:383–388.
Butterworth, R.F. Metal toxicity, liver disease and neurodegeneration. Neurotox Res.. 2010;18:100–105.
Butterworth, R.F. Neuronal cell death in hepatic encephalopathy. Metab Brain Dis.. 2007;22:309–320.
Das, S.K., Ray, K. Wilson’s disease: an update. 1. Nat Clin Pract Neurol. 2006;2:482–493.
Ferrara, J., Jankovic, J. Acquired hepatocerebral degeneration. J Neurol.. 2009;256:320–332.
Forbes, J.R., Cox, D.W. Copper-dependent trafficking of Wilson disease mutant ATP7B proteins. Hum Mol Genet.. 2000;9:1927–1935.
Hoyumpa, A.M., Jr., Desmond, P.V., Avant, G.R., et al. Hepatic encephalopathy. Gastroenterology.. 1979;76:184–195.
Loudianos, G., Gitlin, J.D. Wilson’s disease. Semin Liver Dis.. 2000;20:353–364.
Machado, A., Chien, H.F., Deguti, M.M., et al. Neurological manifestations in Wilson’s disease: Report of 119 cases. Mov Disord.. 2006;21:2192–2196.
Mak, C.M., Lam, C.W. Diagnosis of Wilson’s disease: a comprehensive review. Crit Rev Clin Lab Sci.. 2008;45:263–290.
Meenakshi-Sundaram, S., Mahadevan, A., Taly, A.B., et al. Wilson’s disease: a clinico-neuropathological autopsy study. J Clin Neurosci.. 2008;15:409–417.
Mercer, J.F. The molecular basis of copper-transport diseases. Trends Mol Med.. 2001;7:64–69.
Morita, H., Ikeda, S., Yamamoto, K., et al. Hereditary ceruloplasmin deficiency with hemosiderosis: a clinicopathological study of a Japanese family. Ann Neurol.. 1995;37:646–656.
Pfeiffer, R.F. Wilson’s Disease. Semin Neurol.. 2007;27:123–132.
Sternlieb, I. Wilson’s disease. Clin Liver Dis. 2000;4:229–239. [viii–ix].
Stracciari, A., Mattarozzi, K., D’Alessandro, R., et al. Cognitive functioning in chronic acquired hepatocerebral degeneration. Metab Brain Dis.. 2008;23:155–160.
Strausak, D., Mercer, J.F., Dieter, H.H., et al. Copper in disorders with neurological symptoms: Alzheimer’s, Menkes, and Wilson diseases. Brain Res Bull.. 2001;55:175–185.
Vassiliev, V., Harris, Z.L., Zatta, P. Ceruloplasmin in neurodegenerative diseases. Brain Res Brain Res Rev.. 2005;49:633–640.
Lemberg, A., Fernández, M.A., Coll, C., et al. Reyes’s syndrome, encephalopathy, hyperammonemia and acetyl salicylic acid ingestion in a city hospital of Buenos Aires, Argentina. Curr Drug Saf.. 2009;4:17–21.
Maheady, D.C. Reye’s syndrome: review and update. J Pediatr Health Care.. 1989;3:246–250.
Partin, J.C., Partin, J.S., Schubert, W.K., et al. Brain ultrastructure in Reye’s syndrome. J Neuropathol Exp Neurol.. 1975;34:425–444.
Starko, K.M., Mullick, F.G. Hepatic and cerebral pathology findings in children with fatal salicylate intoxication: further evidence for a causal relation between salicylate and Reye’s syndrome. Lancet.. 1983;1:326–329.
Toovey, S. Influenza-associated central nervous system dysfunction: a literature review. Travel Med Infect Dis.. 2008;6:114–124.
Lai, C.W., Hung, T.P., Lin, W.S. Blindness of cerebral origin in acute intermittent porphyria. Report of a case and postmortem examination. Arch Neurol. 1977;34:310–312.
Pischik, E., Kauppinen, R. Neurological manifestations of acute intermittent porphyria. Cell Mol Biol (Noisy-le-grand). 2009;55:72–83.
Puy, H., Gouya, L., Deybach, J.C. Porphyrias. Lancet.. 2010;375:924–937.
Pitchumoni, C.S., Agarwal, N., Jain, N.K. Systemic complications of acute pancreatitis. Am J Gastroenterol.. 1988;83:597–606.
Zhang, X.P., Tian, H. Pathogenesis of pancreatic encephalopathy in severe acute pancreatitis. Hepatobiliary Pancreat Dis Int.. 2007;6:134–140.
Bhatia, K.P., Brown, P., Gregory, R., et al. Progressive myoclonic ataxia associated with coeliac disease. Brain.. 1995;118:1087–1093.
Farrell, R.J., Kelly, C.P. Celiac sprue. N Engl J Med.. 2002;346:180–188.
Freeman, H.J. Neurological disorders in adult celiac disease. Can J Gastroenterol.. 2008;22:909–911.
Ghezzi, A., Zaffaroni, M. Neurological manifestations of gastrointestinal disorders, with particular reference to the differential diagnosis of multiple sclerosis. Neurol Sci.. 2001:S117–S122.
Lionetti, E., Francavilla, R., Pavone, P., et al. The neurology of coeliac disease in childhood: what is the evidence? A systematic review and meta-analysis. Dev Med Child Neurol.. 2010;52:700–707.
Wills, A.J. The neurology and neuropathology of coeliac disease. Neuropathol Appl Neurobiol.. 2000;26:493–496.
Zois, C.D., Katsanos, K.H., Kosmidou, M., et al. Neurologic manifestations in inflammatory bowel diseases: current knowledge and novel insights. J Crohn’s Colitis.. 2010;4:115–124.
Burks, J.S., Alfrey, A.C., Huddlestone, J., et al. A fatal encephalopathy in chronic haemodialysis patients. Lancet.. 1976;1:764–768.
Heidland, A., Sebekova, K., Klassen, A., et al. Mechanisms of acute uremic encephalopathy: early activation of Fos and Fra-2 gene products in different nuclei/areas of the rat brain. J Ren Nutr.. 2010;20:S44–S50.
Juryñczyk, M., Rozniecki, J., Zaleski, K., et al. Hypoglycemia as a trigger for the syndrome of acute bilateral basal ganglia lesions in uremia. J Neurol Sci.. 2010;297:74–75.
Marsden, S.N., Parkinson, I.S., Ward, M.K., et al. Evidence for aluminium accumulation in renal failure. Proc Eur Dial Transplant Assoc.. 1979;16:588–596.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]22
Systemic metabolic diseases
HYPOGLYCEMIA
MACROSCOPIC APPEARANCES
ETIOLOGY OF HYPOGLYCEMIC BRAIN INJURY
The principal source of energy in the CNS is glucose. Its uptake by nerve cells is independent of insulin. Neuronal stores of glucose and glycogen are relatively small and the requirement practically continuous. During periods of fasting, adults and, to a lesser extent, children are able to obtain some energy from metabolism of ketones, but a blood glucose level of less than 1.5 mmol/L (25–30 mg/100 mL) leads to brain damage within 1–2 hours.
MICROSCOPIC APPEARANCES
The lesions are similar to those of acute hypoxia–ischemia (see Chapter 8), but not identical. In general, the pattern of injury is that of selective degeneration of neurons rather than frank infarction. Affected neurons are shrunken with hypereosinophilic cytoplasm. The nuclei are initially pyknotic, but later become more eosinophilic and appear to blend in with the cytoplasm (nuclear dropout) (Fig. 22.1). As in hypoxia, the subiculum and CA1 field of the hippocampus are particularly vulnerable, while CA3 and 4 are less, and CA2 is least vulnerable. In the cerebral neocortex, the large neurons in laminae 3, 5, and 6 are most likely to be involved. The caudate nucleus and putamen, particularly the small neurons, are highly vulnerable to hypoglycemia. In infants, the dentate nucleus may also be affected (Fig. 22.1). In contrast to hypoxic–ischemic brain injury, Purkinje cells are usually spared. The regions of the brain particularly vulnerable to hypoglycemia are shown in Figure 22.2.
22.1 Acute hypoglycemic injury. (a) Severe hypoglycemic hippocampal injury due to insulin overdose. Most of the nuclei in the dentate fascia are shrunken and pyknotic. Many of the large neurons in the end folium (CA4) show nuclear dropout. (b) Hypereosinophilic neurons in the dentate nucleus of an infant with acute fatal hypoglycemia due to nesidioblastosis. In adults with hypoglycemia, the dentate nucleus is, like the Purkinje cell layer, usually spared.
Infants dying from hypoglycemia may show widespread neuronal degeneration throughout the brain.
FINDINGS IN LONG-TERM SURVIVORS OF SEVERE HYPOGLYCEMIA
MACROSCOPIC APPEARANCES
The cerebral cortex may appear thinned and the hippocampi shrunken and discolored (Fig. 22.3). The white matter is reduced in bulk and the ventricles are dilated. There may be marked atrophy of the caudate nucleus and putamen (Fig. 22.3).
22.3 Chronic changes after hypoglycemic injury. (a) Macroscopic findings in long-term survivor of severe hypoglycemia. Granular and atrophic neocortex in a survivor of an insulin overdose. The hippocampus and tail of the caudate nucleus are also severely shrunken. (b) Macroscopic features several weeks after an insulin overdose. The cerebral cortex is congested and focally thinned, particularly in the orbital frontal region and in the depths of sulci over the cerebral convexities. The corpus striatum is congested and there is irregular shrinkage of the caudate nucleus so that the medial surface is uneven.
MICROSCOPIC APPEARANCES
The cerebral cortex shows laminar neuronal loss and gliosis associated with capillary proliferation (Fig. 22.4). There is often dense subpial gliosis. The hippocampal pyramidal cell layer and subiculum are replaced by a loose meshwork of glial tissue (Fig. 22.5). The white matter is usually rarefied and gliotic. The caudate nucleus and putamen are diffusely gliotic (Fig. 22.4). The globus pallidus is relatively spared. Moderate neuronal loss and gliosis may be evident in the thalamus. As in acute hypoglycemia, the cerebellar cortex, including the Purkinje cells, is relatively spared (Fig. 22.4).
22.4 Chronic effects of severe hypoglycemia. (a) The cerebral cortex shows laminar neuronal loss, gliosis, and capillary proliferation. Several remaining neurons are mineralized (arrows). (b) The putamen is diffusely gliotic. (c) There is relative sparing of the cerebellar cortex, including the Purkinje cells.
22.5 Microscopic features 6 weeks after an insulin overdose. (a) The hippocampal pyramidal cell layer and subiculum are replaced by a loose meshwork of glial tissue and capillaries. (b) Higher magnification view of the pyramidal cell layer (arrowheads) and the dentate fascia (arrows), both of which are severely gliotic.
DISTURBANCES OF BODY TEMPERATURE
HYPERTHERMIA
This is most commonly due to strenuous physical exertion under hot and humid conditions, but may also be a consequence of an infection or drug reaction. Predisposing conditions include diabetes mellitus, alcoholism, intoxication with certain drugs (particularly 3,4–methylenedioxymethamphetamine (Ecstasy), and other amphetamines) and disorders in which there is impaired sweating (e.g. anhidrotic ectodermal dysplasia).
MACROSCOPIC AND MICROSCOPIC APPEARANCES
The brain often appears normal or only mildly edematous. Some patients develop a bleeding diathesis, which may be associated with parenchymal or meningeal hemorrhages. Parenchymal petechial hemorrhages may be found adjacent to the third and fourth ventricles. Cerebellar Purkinje cell loss has been reported. Other described abnormalities are similar to those of hypoxic–ischemic damage (see Chapter 8) and probably result from a combination of cardiovascular collapse and an increased metabolic rate (Fig. 22.6). In patients with malignant hyperthermia, skeletal muscle may show central cores.
22.6 Hyperthermia. Cerebellar cortex from a patient who collapsed and later died after spending several hours in a steam tent. The systemic necropsy findings were in keeping with heat stroke. Examination of the brain revealed changes indistinguishable from those of acute hypoxic–ischemic neuronal injury, as illustrated by these Purkinje cells with shrunken, pyknotic nuclei, and hypereosinophilic cytoplasm.
DISORDERS OF SERUM ELECTROLYTES
HYPONATREMIA
Acute and severe hyponatremia causes neurological syndromes necessitating rapid correction with hypertonic saline. Hyponatremic encephalopathy and hepatic encephalopathy (see below) may coexist in patients with cirrhosis. The clinical picture is common to any metabolic encephalopathy and is characterized by a confusional syndrome that may evolve into coma.
Patients with hyponatremia develop lethargy, headache, and nausea and vomiting.
If severe, hyponatremia may cause seizures and coma, leading to death.
HYPERNATREMIA
OSMOTIC DEMYELINATION SYNDROME
Most predisposing conditions are associated with hyponatremia (i.e. alcoholic liver damage, extensive skin burns, inappropriate secretion of antidiuretic hormone, psychogenic polydipsia, hyperemesis gravidarum).
ODS occasionally develops in patients with a normal serum sodium concentration.
The incidence of ODS is relatively high in liver transplant patients.
ODS has occurred after infusion of hypertonic saline into normonatremic patients.
ODS occasionally complicates chronic malnourishment or debilitation.
MACROSCOPIC APPEARANCES
Typically, the basis pontis includes a fusiform region of gray discoloration, which is abnormally soft and appears granular on sectioning (Fig. 22.7). The extent of the lesion is variable. Its cross-sectional area is usually greatest in the upper part of the pons, where only a narrow rim of subpial tissue may be spared (Fig. 22.8). It may involve the middle cerebral peduncles, but rarely extends rostrocaudally beyond the confines of the pons and lower midbrain. The lesion may be asymmetric, being largely or completely confined to one side of the pons.
22.7 ODS. Transverse sections through the brain stem in CPM showing gray discoloration and slight granularity of much of the basis pontis.
22.8 Sections of the pons in CPM. (a) There is extensive loss of myelin. Note the sparing of myelin in a narrow rim of subpial tissue and in small ‘islands’ within the base of the pons. (b) In an adjacent section, axons are seen to be well preserved.
The reported frequency of extrapontine lesions varies, but careful examination will reveal lesions in other parts of the CNS such as the cerebellum (Fig. 22.9), lateral geniculate body, capsula externa or extrema, subcortical cerebral white matter (Fig. 22.10), basal ganglia, thalamus, or internal capsule in 25–50% of cases. In up to 25% of patients the lesions may be exclusively extrapontine.
22.9 ODS: extrapontine demyelination. This section shows extrapontine demyelination in the white matter in the cerebellum. (Luxol fast blue/cresyl violet)
22.10 Subcortical demyelination in ODS. (a) Gray discoloration of the demyelinated subcortical white matter (arrows) in a case of extrapontine myelinolysis associated with ODS. (b) Solochrome cyanin staining confirms the presence of subcortical demyelination involving white matter in the crests of gyri (arrows).
MICROSCOPIC APPEARANCES
The microscopic appearances of CPM are of active demyelination (Fig. 22.11). The lesions contain reactive astrocytes and large numbers of foamy lipid-laden macrophages (Fig. 22.12), but only very scanty lymphocytes. Silver impregnation may reveal some axonal fragmentation, but most neuronal somata and axons are intact. Within the lesions, cranial nerves or central ‘islands’ of transverse pontine fibers or corticospinal tracts may be preserved (see Fig. 22.8).
22.11 Preservation of neuronal somata and axons in ODS. (a) Section of pons stained to show myelin. (b)
[/not-level-membership-for-pathology-category]
