Chapter 65 Syncope and Paroxysmal Disorders Other than Epilepsy
Paroxysmal disorders, including epilepsy and syncope, represent one of the most common neurological problems in the pediatric population. Although the clinical manifestations of paroxysmal disorders are highly heterogeneous, recent advances in molecular genetic analysis have highlighted striking similarities in their molecular pathophysiology [Crompton and Berkovic, 2009]. Paroxysmal disorders other than epilepsy are described in this chapter (Box 65-1). Breath-holding spells, migraine, pseudoseizures, and sleep-related paroxysmal disorders are reviewed elsewhere in this book.
Box 65-1 Syncope and Other Nonepileptic Paroxysmal Disorders




































Syncope
Syncope is defined as the temporary loss of consciousness and postural tone resulting from transient and diffuse cerebral hypoperfusion, followed by spontaneous recovery with no neurological sequelae [Feit, 1996]. In the young patient, syncope often results from a fall in systolic pressure below 70 mmHg or a mean arterial pressure of 30–40 mmHg [Kaufmann, 2004]. The event is typically preceded by a prodrome lasting several seconds to 1–2 minutes, which has distinctive premonitory features such as nausea, epigastric discomfort, blurred or tunnel vision, muffled hearing, dizziness, lightheadedness, diaphoresis, hyperventilation, palpitations, pallor, cold and clammy skin, or weakness [Sapin, 2004; Strieper, 2005]. These symptoms may occur in any combination or be variably present in any given patient from one episode to the next. If the prodrome is of sufficient duration, patients may learn to recognize their symptoms and lie down to relieve the symptoms and prevent syncope [McLeod, 2003]. Although syncope is commonly a benign self-limiting event, rarely it may be the first warning sign of a serious underlying cardiac or noncardiac disease.
Epidemiology
Syncope is a common clinical problem affecting an estimated 15–25 percent of children and adolescents prior to adulthood. It is not often brought to medical attention, thus accounting for only about 1 percent of pediatric emergency room visits [Vlahos et al., 2007; Longin et al., 2008]. Its true incidence therefore remains unknown. In the 26-year surveillance of the Framingham study, syncope occurred in 3 percent of men and 3–5 percent of women [Savage et al., 1985]. In the Rochester Epidemiologic Project, Driscoll et al. reported on older children and adolescents studied over two 5-year periods [Driscoll et al., 1997]. In the first 5-year period (1950–1954), the incidence of syncope cases requiring medical attention equaled 71.9 per 100,000 population; the second period of study (1987–1991) reported an incidence of 125.8 cases per 100,000 population, with this incidence peaking among adolescents, in particular females aged 15–19 years [Batra and Balaji, 2005]. According to Sheldon et al., the most common age for a child’s first vasovagal syncopal episode is approximately 13 [Sheldon et al., 2006]. The recurrence rate of syncope ranges from 33 to 51 percent when patients are followed for up to 5 years [Kapoor et al., 1983].
Etiology
Syncope may result from cardiovascular or neurological causes (cardiovascular-mediated or neurally mediated syncope). Each of these categories accounts for about 50 percent of adult syncope but, in children, cardiovascular-mediated syncope is less frequent than it is in adults [Kapoor, 2000].
Cardiovascular-Mediated Syncope
Cardiovascular-mediated syncope has a higher mortality and a higher incidence of sudden death than neurally mediated syncope [Kapoor et al., 1983]. It is therefore imperative to distinguish syncope from seizures, and to triage patients with syncope due to malignant cardiac causes for urgent and appropriate investigations and management [Crompton and Berkovic, 2009]. Risk factors that suggest structural or conduction heart defects are given in Box 65-2. If any of these risk factors is present, an urgent referral should be made for pediatric cardiology evaluation. Cardiovascular causes of syncope in children are given in Box 65-3.



















Clinical Features
A diagnosis of syncope rests mainly on clinical grounds [Lerman-Sagie et al., 1994]. The patient’s history, physical examination, and electrocardiography (EKG) have a combined diagnostic yield of about 50 percent [Kaufmann, 2004]. A prodromal phase of presyncope consists of lightheadedness, blurred vision, epigastric discomfort, nausea, pallor, or diaphoresis [Manolis et al., 1990; Feit, 1996; McLeod, 2003]. When present, these clinical features help to differentiate syncope from epilepsy. A detailed history usually reveals contributory environmental factors before the loss of consciousness and postural tone. These environmental factors include upright posture, prolonged standing, change in posture (orthostasis), crowding, heat, fatigue, hunger, or a concurrent illness [Sutton, 1996]. Emotional or stress factors, such as venipuncture, public speaking, “fight-or-flight” situations, pain, and fear, are also commonly identified [Driscoll et al., 1997]. The loss of consciousness is usually brief, lasting from a few seconds to 1–2 minutes, followed by rapid spontaneous recovery without neurological deficits. During the ictus the patient may have tonic posturing or a brief clonic seizure, rarely associated with incontinence. Rarely, myoclonic jerks mimicking an epileptic seizure may occur during syncope [Crompton and Berkovic, 2009]. The postictal period may be accompanied by persistent nausea, pallor, diaphoresis, and a generally “washed-out” appearance. Complete recovery usually evolves in less than an hour [McLeod, 2003].
Distinguishing between neurocardiogenic syncope and seizure is the most common clinical dilemma and a frequent source of diagnostic error. Often, interobserver agreement is poor when a retrospective diagnosis is made on information obtained after a single syncopal episode. In distinguishing patients with syncope due to cardiac causes, a history or physical signs of cardiac disease can be 95 percent sensitive for a cardiac etiology [Crompton and Berkovic, 2009]. These features are, however, nonspecific, as neurocardiogenic syncope might occur in patients with heart disease [Crompton and Berkovic, 2009]. Patients with pseudosyncope and pseudoseizures typically use the events consciously or unconsciously to avoid an unpleasant emotional situation [Wieling and Shen, 2008]. Most of these patients are young females. A very high number of events, episodes without injury, and florid symptomatology are typical. Furthermore, in these patients, recovery after a syncopal event is often prolonged (10–30 minutes), despite a supine posture. In true syncope, consciousness returns within 1 minute of lying down, and unconsciousness for more than 5 minutes is rare [Wieling and Shen, 2008].
The evaluation of syncope may therefore result in unnecessary and costly investigations [Landau and Nelson, 1996]. A complete evaluation would include neuroimaging studies, electroencephalography (EEG), EKG, echocardiography, Holter monitoring, selected metabolic testing, and, in certain cases, intracardiac electrophysiologic studies or implantable continuous loop recordings. Despite extensive investigations, more than 40 percent of patients with recurrent syncope do not have a specific diagnosis [Kapoor, 1991; Grubb et al., 1992].
Pathophysiology
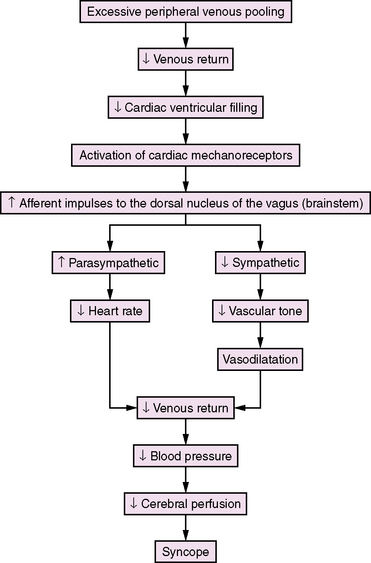
Lewis first introduced the term “vasovagal syncope” in 1932 to indicate that the blood vessels (vaso) and the heart (vagal activity) were involved in the syncopal event [Lewis, 1932]. Hence, the classical clinical signs of vasovagal syncope are marked bradycardia and hypotension. There is no consensus concerning the mechanisms underlying the vasovagal reaction, but several theories have been proposed [Abboud, 1993; Kosinski et al., 1995; Grubb and Kosinski, 1996]. The ventricular theory (Figure 65-1) proposes that, among predisposed individuals who experience recurrent syncope, excess peripheral venous pooling on prolonged standing results in diminished venous return. Decreased cardiac ventricular filling activates mechanoreceptors located mainly in the inferoposterior wall of the left ventricle, which send afferent impulses via C-fibers to the dorsal nucleus of the vagus [van Lieshout et al., 1991]. Arterial baroreceptors and carotid sinus afferent activation may also contribute to the complex pathophysiology of syncope [Kinsella and Tuckey, 2001]. These inhibitory cardiac and arterial receptors mediate increased parasympathetic activity, and inhibit sympathetic activity that results in bradycardia, vasodilatation, and hypotension (Bezold–Jarisch reflex) [Kinsella and Tuckey, 2001; Shen and Gersh, 1993]. The normal response during upright posture is an increased heart rate and diastolic pressure, and an unchanged or slightly decreased systolic pressure [Rea and Thames, 1993]. In individuals susceptible to recurrent syncope, a “paradoxical” reflex bradycardia and peripheral vascular dilatation occur [Grubb and Kosinski, 1996]. The previous emphasis on parasympathetic (vagal) output is shifting to sympathetic withdrawal as the main mechanism responsible for the bradycardia or asystole (cardioinhibitory response) and hypotension (vasodepressor response) that accompany neurocardiogenic syncope [Hannon and Knilans, 1993]. Although parasympathetic-mediated bradycardia remains a contributory factor in syncope, the responsible phenomena are vasodilatation and hypotension [Kosinski et al., 1995]. Current hypotheses propose that the primary efferent event is systemic vasodilatation, and that this vasodepressor element is mediated by a profound, centrally mediated sympathetic withdrawal [Kosinski et al., 1995]. The persistence of neurocardiogenic syncope in subjects who had cardiac transplants and, therefore, technically denervated hearts, as well as the findings of increased epinephrine levels during upright position and syncope, are strong evidence supporting a sympathetic withdrawal mechanism in syncope [Fitzpatrick et al., 1993; Njemanze, 1993; Sra et al., 1994]. Altered cerebral autoregulation may also be contributory in neurocardiogenic syncope [Rodriguez-Nunez et al., 1997]. Transcranial Doppler studies have demonstrated cerebral vasoconstriction during tilt-table-induced syncope [Grubb et al., 1991]. These observations may stimulate future research and modify current insight about neurocardiogenic syncope.
Diagnostic Evaluation
The history, clinical examination, and EKG have a combined diagnostic yield of 50 percent [Kaufmann, 2004]. The patient history is the cornerstone on which the diagnosis of syncope is made. Important historical details to take from patients are given in Box 65-4. On clinical examination, blood pressure and heart rate should be taken in the supine and upright positions, noting any orthostatic hypotension with or without an increase in heart rate. Special attention should be paid to detecting cardiac anomalies. An EKG should be obtained on all patients who present with syncope, especially if it is recurrent or occurs with exercise. All patients with recurrent syncope, family history of syncope or sudden unexplained death should be referred to cardiology for further evaluation. This may include echocardiography and a Holter or event monitor. Rarely, cardiac catheterization with right ventricular endomyocardial biopsy may be necessary before a patient can resume activities [Strieper, 2005].











Tilt-Table Testing
Until the mid-1980s, the diagnosis of neurocardiogenic syncope was made primarily by a careful and detailed patient and family history, the physical examination, and EKG. Tilt-table testing as a potential diagnostic tool for neurocardiogenic syncope was introduced only in 1986 after the ground-breaking report by Kenny et al. [Kenny et al., 1986]. A number of reports have since emerged, attesting to the utility of the test in reproducing syncopal episodes in patients who are predisposed to neurocardiogenic hypotension and bradycardia.
Despite criticism that this provocative test suffers from “naïve rationale,” lacks patient selection standards, and needs controlled treatment and outcome studies [Landau and Nelson, 1996], the tilt-table test continues to enjoy popularity as a noninvasive and physiologically appropriate neurophysiologic test for the diagnosis of neurocardiogenic syncope [Kapoor, 1999; Dijane et al., 1996].
The test is done by positioning the patient head upright at an angle of 60–80 degrees for 15–60 minutes on a tilt table with a supporting footboard. A tilt-table test result is positive when the symptoms of syncope or presyncope are reproduced [Sra et al., 1991]. If the result is negative, isoproterenol is administered intravenously and the dose increased until the heart rate goes up by at least 20 percent. To standardize the test, the duration of head-up tilting has been increased to 45 minutes or 2 standard deviations (SD) of the mean time required to reproduce syncope [Sneddon and Camm, 1993]. A comprehensive analysis of available tilt-table data suggests that administration of isoproterenol has no added benefit, and 60 degrees for 45–60 minutes is recommended [Kapoor et al., 1994].
Experience with tilt-table test among pediatric populations is limited. Of 35 adolescents who had recurrent presyncope or syncope, 26 had positive tilt-table test results at 60 degrees for 30–60 minutes and isoproterenol infusion from 1 to 5 μg/minute [Thilenius et al., 1991]. Among 54 pediatric patients who had recurrent syncope, tilt-table testing at 80 degrees for 30 minutes was superior to studies, such as chest radiograph, EKG, echocardiogram, EEG, or neuroimaging, in arriving at a diagnosis of neurocardiogenic syncope [Strieper et al., 1994]. Tilt-table studies in 20 children with unexplained syncope and 10 controls, using 60 degrees for 25 minutes and isoproterenol infusion of 0.02–0.08 μg/kg/minute, were positive in 75 percent of patients and in 10 percent of controls, with a sensitivity of 75 percent and a specificity of 90 percent [Alehan et al., 1996]. The tilt-table test has been used in children as young as 3 years of age [Grubb et al., 1992].
In addition to heart rate and blood pressure monitoring during tilt-table testing, EKG recording may be used to differentiate anoxic from epileptic seizures [Grubb et al., 1992]. More controlled studies and standardization of degree and duration of tilting are necessary to validate the tilt-table test as a safe, practical, and useful diagnostic tool for neurocardiogenic syncope in children [Benditt and Lurie, 1996; Mansourati and Blanc, 1996; Moya et al., 1996a, b; Victor, 1996].
Treatment
The objective of treatment for neurocardiogenic syncope is to prevent recurrent syncope, which leads to impaired quality of life, psychological distress, and substantial morbidity. Once a diagnosis of neurocardiogenic syncope is confirmed, treatment requires counseling of the patient (when appropriate) and his or her parents. The benign nature of these events should be explained to allay concerns about epilepsy or sudden death. Neurocardiogenic syncope almost always resolves within months to 3–5 years after onset [Strieper, 2005]. Most patients presenting after a single uncomplicated syncopal event require simple reassurance, education about the disease, and advice on recognizing prodromal symptoms and how to avoid provocative situations – in particular, prolonged standing, sudden postural changes, dehydration, and irregular meal times. If a prodromal phase is consistently present, the patient may by taught to recline or sit to avoid injury from a fall. Supplemental fluids and electrolytes may be beneficial; up to 1500–2500 mL per day are recommended for adolescents. Patients should be instructed to increase dietary salt, either as salt tablets or liberal use of salt with meals (Box 65-5).













If, despite these conservative measures, the syncopal episodes become refractory, pharmacologic therapy may be tried. Favorable but not consistent response to treatment has been reported with β-adrenergic receptor antagonists, α-adrenergic receptor agonists, anticholinergic agents, theophylline, serotonin reuptake inhibitors, and mineralocorticosteroids [Milstein et al., 1990; Scott et al., 1995; Raviele et al., 1996; Boehm et al., 1997; Sra et al., 1997] (Box 65-6). Beta blockers, fludrocortisone and midodrine, and an α-adrenergic agonist, are often prescribed in children but none of these agents has shown a consistent therapeutic benefit in clinical trials [Kaufmann and Freeman, 2004; Freeman, 2008]. Low-dose midodrine is promising and is currently recommended as first-line therapy for vasovagal syncope in children by some authorities [Stewart, 2006]. When using fludrocortisone, combine with increased salt intake for an optimal effect. Subjects who require isoproterenol to induce syncope during tilt-table testing or who experience tachycardia before syncope may respond better to beta-blocker therapy [Sra et al., 1992; Leor et al., 1994; Wieling and Shen, 2008].
Box 65-6 Medications for Treatment of Neurocardiogenic Syncope*
















* Pediatric doses are given for those medications used in children for which dosages have been recommended.
(Data from Grubb and Kosinski, 1996; Lazarus and Mauro, 1996; Raviele et al., 1996; Sra et al., 1997.)
Prognosis
The prognosis for recovery is excellent in neurocardiogenic syncope. Most patients show spontaneous resolution of their syncope and presyncope within the first year after onset; 5–10 percent will however, continue to show symptoms over an extended period of time, often up to 5 years [Strieper, 2005].
Convulsive Syncope
A brief tonic or, rarely, a clonic seizure may accompany syncope. In a study of blood donors, 0.05 percent suffered a convulsion associated with syncope (convulsive syncope) [Lin et al., 1982]. Among 216 children who had a positive tilt-table test, 25 (11.6 percent) had seizures during the test [Fernandez Sanmartin et al., 2003]. Most convulsions consisted of tonic spasms (65 percent), characterized by eye rolling, nuchal rigidity, arms flexed at the elbow, and fists clenched, followed usually by prompt recovery [Lin et al., 1982]. Other types of convulsions were myoclonic (23 percent), clonic (6 percent), and tonic-clonic (6 percent). No difference was found in the severity of bradycardia or hypotension among those who had syncope associated with convulsion and those who did not [Lin et al., 1982]. In subjects who had cardiac asystole induced by ocular compression, only those who remained asystolic for more than 14 seconds experienced convulsive phenomena [Lin et al., 1982]. Convulsive syncope results from cerebral ischemia and is not indicative of an epileptic predisposition. The EEG reveals diffuse slowing, followed by loss of electrocerebral activity; epileptiform activity is absent [Fernandez Sanmartin et al., 2003; Stephenson, 1990d]. Rarely, an epileptic seizure is triggered by syncope. In such cases, the EEG, in contrast to what is seen in convulsive syncope, reveals epileptiform activity [Stephenson, 1990g].
Reflex Syncope
Syncope that is triggered by specific factors or events is known as reflex or situational syncope. The most common of these among infants and children is breath-holding spells. These are discussed in greater detail in Chapter 64. A related but distinctive type of syncope (known by various names in the past, including pallid breath-holding spells or pallid infantile syncope) is frequently confused with breath-holding spells. A simple and more appropriate designation for this type of paroxysm is reflex syncope.
In reflex syncope, the antecedent event is minor trauma (usually to the head) before loss of postural tone and consciousness, without any audible inspiratory stridor or expiratory cry. Reflex syncopal episodes are easily confused with typical breath-holding spells, but evidence is lacking that these result from transient cerebral hypoxia because of breath holding. The rapid or immediate onset of syncope after minor trauma or other unexpected painful stimuli differentiates reflex syncope from breath-holding spells. In breath-holding spells, several seconds may elapse before loss of consciousness. There is general agreement that reflex syncopal attacks do not represent epileptic seizures, but the mechanism responsible is less clear. Some refer to these events as reflex anoxic seizures [Stephenson, 1978]. According to Stephenson, cardiac arrest is inducible in individuals susceptible to reflex syncope by the ocular compression test. This test is performed by applying pressure over the closed eyelids for 10 seconds during cardiac and EEG monitoring [Stephenson, 1980]. After 7–15 seconds of asystole, a typical paroxysm is reproduced, thus confirming the diagnosis of reflex syncope. Reproduction of reflex syncope by ocular compression suggests that these children have an exaggerated oculocardiac reflex [Stephenson, 1990f]. Despite assurances about the safety and diagnostic usefulness of the ocular compression test to reproduce this type of syncope [Gastaut, 1974; Stephenson, 1980, 1990f], its acceptance as a provocative test has been limited.
Situational Syncope
Other triggering factors associated with syncope include cough, deglutition (cold liquids), defecation, diving, micturition, sneezing, trumpet playing, weight lifting, and Valsalva maneuver [Hannon and Knilans, 1993]. These events are more common in adults than in children and are referred to as situational syncope. A common denominator in situational syncope is the fact that most of the triggering factors are accompanied by a Valsalva-like maneuver. Hair-grooming syncope is an uncommon type of situational syncope among adolescent females; it is often followed by brief seizure activity [Lewis and Howell, 1986; Igarashi et al., 1988; Lewis and Frank, 1993]. The convulsive syncope is almost invariably preceded by a prodrome of presyncope symptoms of nausea, lightheadedness, diaphoresis, and visual blurring. The reaction is thought to be a variant of neurocardiogenic syncope that is triggered by hair pulling or scalp stimulation, which activates the trigeminal nerve [Kosinski and Grubb, 2005].
Suffocation or Strangulation Syncope
Meadow’s syndrome (Munchausen’s by proxy) is a rarely suspected cause of syncope. A caretaker induces loss of consciousness by obstructing the infant’s airway using a pillow or by pressing the infant’s face against the caretaker’s trunk [Stephenson, 1990e]. In other cases, compression of the neck by strangulation results in cerebral hypoxia and, after repeated attempts, brain damage. A solicitous and omnipresent caretaker should raise suspicion of foul play [Folks, 1995]. The morbid events have been documented in some cases during video/EEG monitoring, but the incriminating evidence may not be admissible in court.
Drug-Induced Syncope
When episodes begin with a slow onset and gradual recovery, consider a toxic or metabolic cause, such as hypoglycemia, alcohol, or drugs (illicit or prescribed) [Olshansky, 2005]. Among the medications that can cause syncope are those that induce ventricular tachycardia or cause hypotension. Cardiovascular medications (vasodilators and antiarrhythmics), psychotropics, diuretics, and glucose-controlling medications are the most common drugs associated with syncope [Lazarus and Mauro, 1996]. Illicit drugs may also cause syncope (particularly alcohol, but also cocaine and marijuana). Alcohol and illicit drugs can cause syncope by several mechanisms, including exacerbation of a supraventricular or ventricular tachyarrhythmia [Strieper, 2005]. Drug-induced syncope is common among the elderly but rare among children. However, illicit drug use is increasing significantly in the younger age group. Toxicology screens often provide clues to this diagnosis.
Psychogenic Syncope
One of the most important causes of syncope in pediatric patients, in particular adolescents, is psychogenic syncope. Several features help distinguish psychogenic syncope from neurocardiogenic syncope [Benbadis and Chichkova, 2006; Thijs et al., 2009]:
Paroxysmal Dyskinesias
Paroxysmal dyskinesias are episodic attacks of involuntary hyperkinetic (dystonic, choreoathetoid, or ballistic) movements with preserved consciousness. In 1940, Mount and Reback first described a familial paroxysmal movement disorder manifested by involuntary writhing and posturing of the trunk and extremities, later labeled as paroxysmal dystonic choreoathetosis [Mount and Reback, 1940]. Kertesz introduced the term paroxysmal kinesigenic choreoathetosis to differentiate cases in which the episodes were brief and precipitated by movement, instead of by prolonged immobility or intake of specific beverages (coffee, tea, cola drinks, alcohol), as in the cases of Mount and Reback [Kertesz, 1967]. These hyperkinetic movements or dyskinesias may manifest as dystonia (abnormal distorted posturing), chorea (arrhythmic, bizarre, jerky, dancing movement), athetosis (distal slow, sinuous limb movement), or ballism (violent flailing limb movement). They may occur individually or in various combinations. The most widely used classification system for the paroxysmal dyskinesias is the one proposed by Demirkiran and Jankovic in1995 [Demirkiran and Jankovic, 1995]. The paroxysmal dyskinesias are classified into four types: paroxysmal kinesigenic dyskinesia (PKD), paroxysmal nonkinesigenic dyskinesia (PNKD), paroxysmal exercise-induced dyskinesia (PED), and paroxysmal hypnogenic dyskinesia (PHD). Each category is further subdivided into idiopathic (familial and sporadic) and, rarely, symptomatic [Blakeley and Jankovic, 2002] secondary to stroke, trauma, multiple sclerosis, central nervous system infections, and other known causes, with resultant abnormality on brain magnetic resonance imaging. The majority of cases are primary or idiopathic, and of the PKD or PNKD type. PHD, though included in this classification scheme due to semiologic similarity, has been proven to consist of nocturnal seizures secondary to autosomal-dominant nocturnal frontal lobe epilepsy (ADNFLE) [Luders, 1996; Phillips et al., 2001].
The etiology, anatomy, physiology, and pathogenesis of the paroxysmal dyskinesias are not yet completely understood. Evidence supports the striatum as the primary area of brain involvement, based on current clinical, radiological, and experimental data. Symptomatic cases have been shown to have structural pathology of the striatum; abnormalities of the striatum have been found on MR spectroscopy, positron emission tomography (PET), and single photon emission computerized tomography (SPECT); and in in vivo experimental studies, using the dtsz mutant hamster animal model of paroxysmal dyskinesia, striatal increase of extracellular dopamine during dystonic episodes has been shown [Ko et al., 2001; Sanger, 2003; Hamann and Richter, 2004]. However, in another animal study using the lethargic mouse model (produced by calcium channel mutation), abnormal cerebellar output was shown to be responsible for the paroxysmal dyskinesia [Devanagondi et al., 2007]. Excess cytochrome oxidase activity in the red nucleus disappeared after surgical removal of the cerebellum. A similar conclusion was reached in the tottering mouse model [Campbell et al., 1999].
The etiology of most of these paroxysmal dyskinesias, as with many periodic syndromes, is thought to be secondary to an underlying channelopathy. They share common clinical characteristics with epilepsy, migraine headaches, periodic paralysis, episodic ataxia with myokymia, including episodic attacks with normal interictal examination, common precipitating mechanisms such as stress, fatigue, diet, alcohol, and caffeine, and treatment response to similar medications. Both hypokalemic and hyperkalemic periodic paralysis and episodic ataxia with myokymia are due to mutations in ion-channel genes. Carbamazepine is effective in certain epilepsies, as in PKD, and acetazolamide is useful in periodic paralysis, myotonia, episodic ataxia, and some paroxysmal dyskinesias [Bhatia et al., 2000; Ptacek and Fu, 2001; Celesia, 2001; Margari et al., 2005; Cannon, 2001]. Genetic mutations in the alpha subunit of calcium-sensitive potassium channel on chromosome 10q22 cause epilepsy and PNKD [Du et al., 2005]. Mutations in voltage-gated neuronal K+ channel Kv7 (KCNQ) produces dyskinesia in the dtsz mutant hamster model of paroxysmal nonkinesigenic dyskinesia. Retigabine and flupirtine, which result in opening of the above type of K+ channel, effectively treated the paroxysmal dystonia, proving the role of Kv7 channel mutation in the causation of PNKD [Richter et al., 2006]. Interestingly, however, many of the familial PNKDs are caused by the mutation of MR-1 (myofibrillogenesis regulator 1) gene on chromosome 2. The MR-1 gene is not involved with an ion channel; rather, it is predicted to be involved in the stress response pathway [Lee et al., 2004; Rainier et al., 2004; Chen et al., 2005a; Hempelmann et al., 2006; Bruno et al., 2007]. Some symptomatic forms of PKDs are caused by the X-linked monocarboxylate transporter 8 (MCT 8) gene mutation responsible for transport of T3 (tri-iodothyronin) across the neuronal membrane [Dumitrescu et al., 2004; Brockmann et al., 2005; Fuchs et al., 2009].
The paroxysmal dyskinesias are also described in Chapter 68.
Paroxysmal Kinesigenic Dyskinesia
This is the most common form of all the paroxysmal dyskinesias, although the exact prevalence is unknown. The attacks are brief (usually lasting less than 1 minute) and characterized by dystonic posturing, choreoathetosis, or ballistic movements, either singly or in combination; they affect one or both sides concomitantly [Lance, 1977; Bruno et al., 2004]. The patient remains conscious throughout the episode, without any postictal impairment. In the largest series of 121 cases of paroxysmal kinesigenic dyskinesia, 51 were male and 44 female [Bruno et al., 2004]. Ninety-five cases were familial, and the majority of these were consistent with autosomal-dominant inheritance. Males also outnumbered females in sporadic cases. The mean age at onset was 11.7 ± 3.4 years in the familial cases, and similar for the sporadic ones. The age range, however, varied from 1 to 20 years. The dyskinesias are always triggered by sudden movement; however, an intention to move or startle may also bring them on. Anxiety or stress may also play an important role, causing confusion, at times, with a psychogenic movement disorder with consequent delay in the diagnosis. Spells occur more frequently following physical exertion or during sickness. An aura is present in more than 80 percent of cases. These auras consist of a feeling of tightness, numbness, tingling, or paresthesia in the involved extremities [Lance, 1977; Bruno et al., 2004]. The ictus consists of dystonic posturing in up to two-thirds of all cases. Choreoathetosis or ballistic movements occur in some, and a combination of movements occurs in one-third. In about one-third of cases, the attacks may be focal or unilateral, one-third may have bilateral involvement, and the remainder has either unilateral or alternating bilateral involvement. In focal cases, the movement usually starts with the limb in action. The attacks are typically short, with more than 90 percent lasting less than 30 seconds, and usually never more than a minute. The attacks occur daily in more than 80 percent of cases, and the frequency may reach up to 100 attacks in a day [Lance, 1977; Houser et al., 1999; Bruno et al., 2004].
Associated neurological disorders, like infantile convulsions, are common (11–19 percent) in children with PKD; almost half of their family members had infantile seizure [Margari et al., 2000, 2002; Bhatia, 2001; Bruno, et al., 2004]. This association brought up a homogenous autosomal-dominant syndrome described as infantile convulsions and choreoathetosis (ICCA), with benign seizures in infancy, followed by later-onset paroxysmal kinesigenic dyskinesia [Rochette et al., 2008]. In familial forms of PKD, febrile seizures are seen in 9 percent of the affected individuals and in 30 percent of other family members. Migraine with and without aura occurs in about one-third of all PKD cases and in two-thirds of their family members. Other neurological abnormalities include writer’s cramp, essential tremor, and myoclonus [Bruno et al., 2004; Margari et al., 2005].
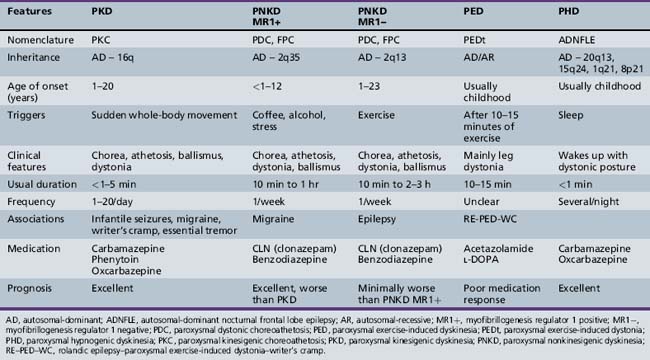
Differential diagnoses depend on age of onset of the symptoms. These include other forms of paroxysmal dyskinesias, dopa-responsive dystonia, complex motor tics, complex motor stereotypies, seizure, pseudoseizure, psychogenic movement disorder, malingering, shuddering attacks, and Sydenham’s chorea. Complex motor stereotypies in younger children are sometimes difficult to distinguish from PKD or PNKD. Excitement precipitating the complex movement pattern and involvement of predominantly the upper limbs or upper part of the body in a child with autistic behaviors or cognitive impairment is suggestive of a complex motor stereotypy rather than PKD. Dopa-responsive dystonia can be distinguished clinically by the predominant involvement of the lower limbs and a diurnal variation, with worsening in the evening. Chorea in Sydenham’s chorea may start unilaterally and intermittently on attempted activity, but the preceding history of sore throat, laboratory evidence of streptococcal infection, and self-limiting course of the disorder help in making the diagnosis. The distinguishing features of various types of paroxysmal dyskinesias are shown in Table 65-1. Increased awareness of PKD among clinicians is needed, as the diagnosis is delayed in most cases. The mean time from first seeking medical attention to confirmation of diagnosis is approximately 5 years [Bruno et al., 2004]. Proposed diagnostic criteria for PKD are shown in Box 65-7 [Bruno et al., 2004].






Despite the existence of several PKD gene loci found through linkage studies, no PKD gene has been identified so far. A form of autosomal-dominant PKD with episodic ataxia and spasticity has been mapped at chromosome 1p [Auburger et al., 1996]. ICCA, or ICCA-like syndromes, has been mapped to the pericentromeric region of chromosome 16 (16p12–q12). This involves the PKC1 or EKD1 gene locus [Tomita et al., 1999; Swoboda et al., 2000; Bennett et al., 2000; Rochette et al., 2008]. Another locus (EKD 2) has been identified on the same chromosome in an Indian family with PKD [Valente et al., 2000].
Neurophysiology and neuroimaging studies are normal in familial and sporadic idiopathic cases, except for occasional slowing noted on the EEG [Buruma and Roos, 1986]. No epileptiform activity is usually seen, even during an episode. Nevertheless, reports of epilepsy among members of the same family raise the question about the possible association between these disorders [Demirkiran and Jankovic, 1995; Margari et al., 2000, 2002; Rochette et al., 2008]. Lombroso demonstrated ictal discharges emanating from the supplementary sensorimotor cortex and ipsilateral caudate nucleus by invasive long-term monitoring in a female with paroxysmal kinesigenic choreoathetosis [Lombroso, 1995]. Thus, some authors believe that these paroxysmal movement disorders are an expression of subcortical seizures involving cortico-striato-thalamo-cortical circuits [Tan et al., 1998]. The pathology in this circuit underlies the pathophysiology of most movement disorders in general. In primary (idiopathic or genetic) movement disorders, there is no detectable pathology on MRI of the brain, whereas in secondary or symptomatic cases there is involvement of basal ganglia or thalamus, with or without cortical involvement. Electromyography shows doublet or triplet discharges. Somatosensory-evoked potential (SSEP) shows reduction in amplitude of cortical sensory response. Motor-evoked potential, however, shows an increased motor cortical excitability with diminished stimulation threshold, high amplitude, and motor facilitation. Spinal and brainstem reflexes, like H reflex and blink reflex, suggest lack of inhibition. This phenomenon has led to the alternate hypothesis of spinal or brainstem modulation of central motor activity, producing paroxysmal dyskinesias [Lee et al., 1999].
Once diagnosed, there are only a few neurological disorders with a more satisfying treatment response, with an exquisite sensitivity to antiepileptic medications [Wein et al., 1996]. The attacks can be prevented, even with subtherapeutic levels of phenytoin or carbamazepine. Other medications found to be effective, but to a lesser degree, are barbiturates, benzodiazepines, valproate, lamotrigine, levetiracetam, scopolamine, l-DOPA, belladonna, chlordiazepoxide, and diphenhydramine [Lance, 1977; Kinast et al., 1980; Bruno et al., 2004]. Most children tried with other medication were eventually switched back to either phenytoin or carbamazepine. In a recent report, oxcarbazepine worked very well in four children with PKD [Chillag and Deroos, 2009]. This marked sensitivity to antiepileptic medications has been used to differentiate paroxysmal kinesigenic dyskinesia from the nonkinesigenic type. Whether treated or not, the attack frequency generally decreases with increasing age. More than one-quarter of patients enjoy complete remission by age 20 years, and a further quarter have marked reduction in attack frequency. Females in the idiopathic category have the best prognosis [Bruno et al., 2004].
Paroxysmal Nonkinesigenic Dyskinesia
PNKD is characterized by episodes of dystonia and/or choreoathetosis involving the face, trunk, and extremities, often associated with dysarthria and dysphasia, lasting from minutes to several hours and occurring up to several times a week [Pryles et al., 1952; Lance, 1977; Tibbles and Barnes, 1980; Kinast et al., 1980; Bressman et al., 1988; Demirkiran and Jankovic, 1995]. Historically, in 1940, Mount and Reback reported the first family with PNKD, labeling them as cases of “familial paroxysmal choreoathetosis” [Mount and Reback, 1940]. Various eponyms have been used to describe this condition but, due to overlapping semiologic features, the term paroxysmal nonkinesigenic dyskinesia is preferred. Proposed diagnostic criteria for PNKD are indicated in Box 65-8.





Mutations of the myofibrillogenesis regulator 1 (MR-1) gene (also known as the PNKD1 gene) form a distinct homogenous subset of PNKD [Bruno et al., 2007]. Onset is usually in infancy or early childhood, with a mean of 4 years, but late onset has been reported. This is in contradistinction to the MR-1 gene-negative group, which has a later age of onset with a mean of 12 ± 10.8 years. The attacks begin spontaneously at rest or after intake of caffeine or alcohol. Alcohol and caffeine sensitivity is noted in almost 100 percent of MR-1 gene-positive cases, which is distinctly uncommon in the other group. Stress is another common precipitating factor that occurs in more than 80 percent of the MR-1 gene-positive group. Other precipitating factors include fatigue, hunger, chocolate, and excitement. Exercise has been noted to bring on the majority of the attacks in MR-1 gene-negative cases, implicating more heterogeneity in these patients and causing confusion with some cases of paroxysmal exercise-induced dyskinesia. An aura, consisting of focal limb stiffening, twitching, numbness, a generalized “funny feeling,” or lightheadedness, has been noted [Williams and Stevens, 1963; Bruno et al., 2007]. About 12 percent present only with dystonia, and the rest with a combination of dystonia and chorea in the MR-1 gene-positive cases, whereas the other group may have dystonia, chorea, a combination of both, and ballism as part of the clinical manifestations. Almost half may have speech involvement. Typical attack duration is 10 minutes to 1 hour, but may last up to 12 hours. Attack frequency is highly variable, but 86 percent have at least one attack every week.
Clonazepam and diazepam are the most effective preventive and abortive agents in 97 percent of cases studied by Bruno et al. [Bruno et al., 2007]. Sleep benefit is also noted in the majority of cases [Byrne et al., 1991; Bruno et al., 2007]. The MR-1-negative group, however, did not have sustained benefit from clonazepam in at least half of the cases treated. Other antiepileptic medications, including valproate, carbamazepine, topiramate, and gabapentin, proved partially beneficial for MR-1-negative patients, but MR-1-positive patients did not respond well. l-DOPA, acetazolamide, and haloperidol did not help either group.
Prognosis is not as good as in PKD; however, almost two-thirds of the mutation-positive group improved with age, with only a minority (18 percent) becoming worse. The prognosis in the mutation-negative group is a little worse than in the mutation-positive group. Migraine is coexistent in 47 percent of cases in the MR-1-positive cases, whereas seizures are reported in 23 percent of the MR-1-negative cases [Kinast et al., 1980; Przuntek and Monninger, 1983; Bruno et al., 2007].
PNKD follows an autosomal-dominant transmission pattern, with almost equal sex distribution. The majority of familial PNKD are caused by the mutation of the MR-1 gene on chromosome 2q35 [Fouad et al., 1996; Fink et al., 1996; Jarman et al., 1997; Raskind et al., 1998]. Recently, linkage studies in a Canadian family with European descent identified a second locus at the 2q31 region [Spacey et al., 2006]. The MR-1 gene is not involved in the ion channel, contrary to previous postulations; rather, it works in the stress response pathway. Although the exact function of MR-1 is unknown, its gene product is homologous to the hydroxyacylglutathione hydrolase (HAGH) of the glyoxalase system. HAGH catalyzes the final step in conversion of methylglyoxal (a byproduct of glycolysis, also found in coffee and alcoholic beverages) to lactic acid and reduced glutathione. This relationship explains the exquisite sensitivity to coffee and alcohol in attack precipitation; emotional stress may also act in a similar manner [Lee et al., 2004; Rainier et al., 2004; Chen et al. 2005a; Hempelmann et al., 2006; Bruno et al., 2007]. Mutations in voltage-gated neuronal K+ channel Kv7 (KCNQ) has been shown to produce dyskinesia in dtsz mutant hamster model of PNKD [Richter et al., 2006]. In some patients with coexistent generalized epilepsy and PNKD, a mutation in the alpha subunit of the calcium-sensitive potassium channel (BK channel) located on chromosome 10q22 has been discovered [Du et al., 2005].
Paroxysmal Exercise-Induced Dyskinesia
This interesting autosomal-dominant disorder is not as common or well understood as the others. No specific genetic defect has yet been found. Children or young adults with otherwise normal health develop dystonic posturing, with or without pain, after a prolonged period of exercise, usually more than 10–15 minutes. Fasting and stress can bring on the attacks. The attack may last 10–30 minutes and usually involves the limb/s being used in exercise. Association with absence or complex partial seizures has been described [Plant et al., 1984; Munchau et al., 2000]. In a Sardinian family with rolandic epilepsy, paroxysmal exertional dyskinesia, and writer’s cramp, and showing an autosomal-recessive pattern of inheritance, the disorder has been has been mapped to 16p12–11.2, which overlaps the ICCA locus [Guerrini et al., 1999]. Linkage of one family pedigree with typical PED was not established to the PNKD or ICCA loci [Munchau et al., 2000]. PED cases may represent a variant of either PKD or PNKD where there is precipitation by exercise. Cases of dopa-responsive dystonias may pose a great challenge in differential diagnosis when the lower limbs are primarily involved; however, the excellent response to l-DOPA in dopa-responsive dystonias, along with gene testing, should clarify that doubt. The responses to medication in PED are modest at best; drugs used have included acetazolamide, l-DOPA, and trihexyphenidyl.
Dopa-Responsive Dystonia
Only a few disorders in the practice of pediatric neurology provide the opportunity to reverse a disabling condition in the dramatic manner afforded by dopa-responsive dystonia (DRD). The condition was first reported by Segawa in 1971, with an autosomal-dominant mode of inheritance [Segawa et al., 1986]. The estimated prevalence in England and Japan is 0.5 per million [Nygaard, 1993]. The onset is between 1 and 9 years of age (average 5 years). Females outnumber males by 3–4:1. Symptoms usually start with unilateral leg or foot dystonia, precipitated by actions such as walking or running, and improvement with rest. The most characteristic feature is the diurnal variation of the dystonia, which worsens in the evening and improves markedly in the morning. Sleep acts as the most important relieving factor. Classically, the dystonia starts with one foot, spreading to affect the whole of the ipsilateral lower limb. It then spreads to the ipsilateral upper limb, contralateral lower limb, and, finally, the contralateral upper limb, following the pattern of the letter N. The last area involved is the craniocervical region. Involvement spreading from one lower limb to the other and, finally, involving upper limbs and craniocervical region may occur. It may take 5–6 years from the onset for the dystonia to be generalized [Segawa et al., 1986; Trender-Gerhard et al., 2009]. The periodicity, action induction, fatigability, asymmetry, and diurnal variation may not be very prominent with advancing disease pathology. This is an important point to consider in the history. Dystonic posturing of the big toe (striatal toe) may mimic an upgoing plantar (Babinski) response [Iivanainen and Kaakkola, 1993]. Features of parkinsonism, such as rigidity, bradykinesia, postural instability, action or postural tremor of the arms, and, rarely, a resting tremor, may eventually develop in some cases [Nygaard et al., 1994]. The presence of rigidity and dystonia in the lower limbs may mimic a spastic diplegic form of cerebral palsy, resulting in misdiagnosis. If in doubt, a trial of l-DOPA usually results in dramatic improvement. It is worth noting that the improvement is expected, even if this condition is diagnosed very late. There is a need for a high index of suspicion on the part of the clinician to think about this disease in the clinical context and consider a trial of l-DOPA [Nygaard et al., 1994; Segawa, 2000; Segawa et al., 2003]. Less frequently, DRD may be inherited in an autosomal-recessive fashion with much younger age of onset, but retaining all the other characteristics of the above description. Dystonia as the predominant clinical entity represents the “pure form” of DRD. In a subset of children, often referred to as having DRD plus syndrome, seizures, developmental delay, other dyskinesias, tremor, chorea, and oculogyric phenomena may occur [Schiller et al., 2004; Clot et al., 2009].
Neuroimaging studies are typically normal. PET studies have been normal in most cases [Snow et al., 1993]. Pathologically, there is no cell degeneration in the nigrostriatal dopaminergic system. There is inadequate dopamine synthesis in the striatum secondary to dysfunction of the enzyme tyrosine hydroxylase, either as a primary deficiency or secondary to a lack of the co-factor tetrahydrobiopterin (BH4). There is reduction of the dopamine metabolite homovanillic acid (HVA) in the cerebrospinal fluid. BH4 acts as essential co-factor for tyrosine, phenylalanine, and tryptophan hydroxylases. Defect in the biosynthetic pathway of BH4 causes a reduction of dopamine and serotonin (5-hydroxytryptamine) metabolites, HVA, and 5-hydroxy indole acetic acid (5-HIAA), respectively [Nygaard, 1995]. Neuropathologic data in one case demonstrated marked reduction of dopamine levels in the substantia nigra and in the striatum [Rajput et al., 1994].
Autosomal-dominant DRD is caused by mutations in the GCH1 gene on chromosome 14 (14q22.1–q22.2), which encodes the enzyme guanosine triphosphate cyclohydrolase 1 (GTPCH-1) in the BH4 biosynthetic pathway [Furukawa et al., 1995; Tanaka et al., 1995; Segawa, 2000; Segawa et al., 2003; Clot et al., 2009]. Autosomal-recessive DRD may be secondary to tyrosine hydroxylase (TH) gene mutation. Tyrosine hydroxylase deficiency is associated with a broader phenotype than just a dopa-responsive dystonia and gait disorder, and includes a more severe infantile parkinsonism or progressive infantile encephalopathy phenotype. DRD plus disorders may be due to sepiapterin reductase gene mutation or other defects in the BH4 biosynthesis pathway.
The response to treatment with l-DOPA in typical cases of DRD is dramatic. Fatigability is usually alleviated within a week, and dystonia within 6 weeks of optimum dose. A combination of l-DOPA and dopa decarboxylase inhibitor should be used. Preparations with a higher ratio of dopa decarboxylase inhibitor, e.g., carbidopa-l-DOPA 25–100 mg, are generally preferred to the 10–100 mg preparation. Young adolescents, more particularly females, have much higher dopa decarboxylase activity, thus requiring a higher dose of carbidopa. A dose of 5 mg/kg/day of carbidopa-l-DOPA is effective in most cases. The medication is generally well tolerated and the effect sustained, even after prolonged use of 30 years [Segawa et al., 1986]. The reason for the lack of any significant side effect is the paucity of dopamine synthesis in this condition with preserved structure of the nigrostriatal system with no cell damage or death. The use of l-DOPA is, therefore, the equivalent of supplementing dopamine in the deficient state.
Episodic Ataxias
The episodic ataxias (EA) are a group of autosomal-dominant disorders characterized by recurrent episodes of cerebellar ataxia, vertigo, dysarthria, and nystagmus, starting in childhood and lasting for minutes or hours, with otherwise normal brain functions. Although patients are asymptomatic between attacks, neurologic examination may reveal ocular abnormalities, including downbeating or gaze-evoked nystagmus, abnormal optokinetic nystagmus, hypermetric saccades, saccadic pursuit, or myokymia [Van Dyke et al., 1975; Jen et al., 2004]. Two main forms are recognized: episodic ataxia type 1 (EA1) and type 2 (EA2). The number of identified EA phenotypes and genotypes is, however, expanding, and is now up to seven (EA7). These disorders share some common features: namely, all are channelopathies, with autosomal-dominant inheritance pattern, and are responsive to acetazolamide. Some show spontaneous improvement with advancing age [Rajakulendran et al., 2007; Strupp et al., 2007; Jen et al., 2007]. The differential diagnoses include episodic neurological disorders, such as epilepsy, paroxysmal dyskinesia, and migraine. To make things more complicated, these disorders may frequently coexist in the same patient. Some metabolic disorders, like Hartnup’s disease, organic aciduria, and mitochondrial cytopathies, may present with intermittent or episodic ataxia.
Episodic Ataxia Type 1
Historically, EA1 was first described by Van Dyke et al. in 1975 in 11 members spanning three generations of a family that had periodic ataxia and continuous muscle movement [Van Dyke et al., 1975]. Children with EA1 have brief attacks of truncal and limb ataxia, coarse tremor, and titubation lasting seconds to minutes, starting in the first or second decade. The attacks may be associated with nausea, vomiting, dysarthria, or nystagmus. They may have visual symptoms of oscillopsia and visual blurring. The frequency is highly variable, varying from multiple times a day to only a few times a year. Physical exertion, emotional stress, startles, and sudden change in posture may bring on the attacks. Brain MRI scan is normal. Rest and sleep may abort an acute attack. The attack frequency decreases with age. One diagnostic clinical feature is the nearly constant presence of continuous muscle activity, either in a rippling pattern of myokymia, or as neuromyotonia. Myokymia may involve eyelids, facial muscles, or the muscles acting on the fingers. This feature, a manifestation of peripheral nerve hyperexcitability, is present almost all the time, irrespective of the ataxic spell. If it is clinically absent, electromyography invariably shows the changes of myokymia, with or without grouped discharges of doublet, triplet, and multiplets [Hanson et al., 1977]. In some cases, mild ischemia in the muscle tested may bring on the myokymic discharges [Lubbers et al., 1995]. Seizures are seen more commonly in these patients [Brunt and van Weerden, 1990]. Seizures are ten times more common in patients with EA1 compared to the general population [Zuberi et al., 1999].
Early genetic linkage studies mapped the EA1 locus to chromosome 12p13, and studies later confirmed a mutation of the KCNA1 gene encoding the voltage-gated potassium channel subunit Kvα1.1. This leads to a reduction in potassium permeability that results in prolongation of the action potential and failure to repolarize, thus producing repetitive myokymic discharges [Benatar, 2000]. The pathogenesis of cerebellar ataxia may be related to hyperactivity of the gamma-aminobutyric acid (GABA)ergic basket cell inhibition over cerebellar Purkinje cells [Zhang et al., 1999]. The other pathogenesis is spreading acidification in cerebellar cortex [Chen et al., 2005b]. KCNA1 gene mutation in the knockout mouse model has been shown to have epilepsy, thus leading to the postulation that KCNA1 gene mutation may be a susceptibility factor for epilepsy in humans [Smart et al., 1998].
Acetazolamide, a carbonic anhydrase inhibitor, is effective in reducing the frequency of ataxic episodes in some patients. The starting dose is 125–250 mg daily and can be slowly increased to a maximum of 500 mg twice daily. Mechanism of action is postulated to be through increasing pH in the vicinity of the ion channel, causing hyperpolarization of the cell membrane and thus reducing neuronal hyperexcitability. Common side effects include tingling, numbness, altered taste, and some impairment of concentration and memory. One bothersome side effect is renal stones, which can be prevented with proper hydration and drinking citrus juice or potassium chloride [Griggs et al., 1978; Lubbers et al., 1995]. Some patients not responding to acetazolamide may respond to antiepileptic medications, such as carbamazepine, phenytoin, valproic acid, or phenobarbital [Eunson et al., 2000; Klein et al., 2004].
Episodic Ataxia Type 2
EA2 is the most common of all episodic ataxias. The ataxic symptoms start in early childhood. Though ataxic symptoms are the same as in EA1, several features distinguish this condition. The duration of each attack is more prolonged, is measured in hours to days, and is more commonly associated with nausea, vomiting, and vertigo. The attack frequency varies from daily to once a year. These attacks are provoked by exertion, stress, intercurrent illness, or alcohol, and never by sudden movement (common in EA1). More than half have migraine headache during the attack; familial hemiplegic migraine is an allelic disorder coexisting with EA2 at times. Myokymia is not a feature. Interictally, more than 90 percent of patients usually manifest downbeating nystagmus. Weakness during, or preceding, the attack is well described in EA2 secondary to a myasthenic phenomenon due to impaired neuromuscular transmission. Patients with EA2 may develop progressive cerebellar ataxia with time [Baloh et al., 1997; Jankovic and Demirkiran, 2002; Jen et al., 2004], and brain MRI scan often demonstrates anterosuperior vermian cerebellar atrophy [Vighetto et al., 1988]. Decreased phosphate, reduced creatine, increased pH, and high lactate in the cerebellum are the hallmarks on MR spectroscopy [Bain et al., 1992; Harno et al., 2005]. Intermittent rhythmic delta activity, sometimes associated with low-amplitude spikes resulting in irregular spike-and-wave patterns, has been reported in EEG studies [Van Bogaert and Szliwowski, 1996]. Vestibular migraine is the closest differential diagnosis, where vertigo is the predominant symptom and ataxia is typically of a vestibular type (rather than cerebellar type). The associated nystagmus is also of a vestibular type and not downbeating, as is classical for EA2. Peripheral vestibular deficits can be seen in up to 20 percent of cases of vestibular migraine [Jen et al., 2004; Brandt and Strupp, 2006].
The responsible gene has been mapped to the short arm of chromosome 19 [von Brederlow et al., 1995]. Localization of the gene responsible for familial hemiplegic migraine also mapped to chromosome 19p, suggesting that these paroxysmal disorders were allelic [Joutel et al., 1993]. Subsequent studies showed that both EA2 and familial hemiplegic migraine are both associated with loss-of-function mutations in the gene CACNAIA, which encodes the alpha1A subunit of voltage-gated neuronal calcium channels located on chromosome 19p13 [Ophoff et al., 1996; Ducros et al., 2001]. This gene encodes the Cav2.1 subunit of the P/Q-type calcium channel, which acts as the voltage sensor and ion-conducting pore [Shapiro et al., 2001].
Approximately 50–75 percent of all patients with EA2 are responsive to treatment with acetazolamide, regarding both the frequency and the severity of the attacks. Favorable response has been sustained for up to 5 years [Griggs et al., 1978]. The mechanism of action is by alteration of intracellular pH. The attacks are precipitated by high intracellular pH values, precipitated by exercise and stress through hyperventilation and consequent alkalosis [Bain et al., 1992]. Acetazolamide lowers the intracellular pH, which in turn reduces potassium conductance and thereby restores excitability and resting activity of the neurons [Shapiro et al., 2001]. Starting dose is usually 250 mg a day, increasing gradually to a maximum of 1000 mg per day. Sulthiame, another carbonic anhydrase inhibitor, has also been used successfully. It causes fewer side effects, and is most effective in dosages between 50 and 300 mg per day [Brunt and van Weerden, 1990]. Acetazolamide is, however, the choice of treatment. For those who are allergic, intolerant, or unresponsive to acetazolamide, an alternative treatment to consider is 4-aminopyridine (4AP), a potassium channel blocker. At a dose of 5 mg three times a day, 4AP has been shown to be beneficial [Strupp et al., 2004]. Similarly, 3,4 diamino pyridine (DAP) has been shown to improve the downbeating nystagmus often observed with EA2 [Strupp et al., 2003]. The loss-of-function mutations in EA2 lead to the reduction of calcium-dependent neurotransmitter GABA release in Purkinje cells. Aminopyridines increase the release of GABA in the cerebellar Purkinje cells. Increase in excitability of Purkinje cell and the prolongation of the action potential duration by blocking potassium channels have been proven in animal studies [Shapiro et al., 2001; Etzion and Grossman, 2001].
Other Types of Episodic Ataxias
Other types of episodic ataxias are much rarer. EA3 was described in a large Canadian family with episodic vertigo, tinnitus, and ataxia, linked to chromosome 1q42. These patients are normal in between attacks [Steckley et al., 2001; Cader et al., 2005]. EA4 is described in two kindreds from North Carolina with late-onset episodic vertigo and ataxia with persistent interictal nystagmus not responsive to acetazolamide. No gene locus has been detected yet [Farmer and Mustian, 1963; Small et al., 1996]. The EA cases with mutation in the CACNB4 gene, encoding the beta4 subunit of the P/Q-type voltage-gated calcium channel, have been designated as EA5 [Escayg et al., 2000]. EA6 was first observed in a child with episodic attacks of hemiplegia and migraine in a setting of fever and epilepsy. A mutation in the SLC1A3 gene encoding glial glutamate transporter, EAAT1, has been found [Jen et al., 2005]. A family with EA, triggered by exertion and excitement lasting hours to days, and associated with weakness, vertigo, and slurred speech, mapped to chromosome 19q13, has been designated as EA7 [Kerber et al., 2007].
Childhood Periodic Syndromes
The childhood periodic syndromes include a diverse group of disorders, which have at their core periodic or paroxysmal occurrences with a return to normal baseline functioning and symptom-free interval between attacks. They are believed to be precursors to migraine and are classified as such by the International Classification of Headache Disorders, Second Edition [Headache Classification Committee, 2004]. The term childhood periodic syndromes was first used by Wylie and Schlesinger in 1933 [Wyllie and Schlesinger, 1933] and has gained increasing acceptance in the past decade [Winner, 2005; Cuvellier and Lepine, 2010]. Sometimes referred to as “migraine equivalents,” it includes benign paroxysmal vertigo, benign paroxysmal torticollis, and cyclic vomiting syndrome, which are discussed below.
Benign Paroxysmal Vertigo
Benign paroxysmal vertigo (BPV) in childhood is a paroxysmal, nonepileptic event first described by Basser in 1964 [Basser, 1964]. The syndrome is characterized by vertigo of sudden onset lasting seconds to minutes, an inability to maintain posture, stance, or gait without support, and no change in sensorium. The child is usually pale and scared. Nystagmus, vomiting, and diffuse sweating may occur but are uncommon [Fenichel, 1967; Koenigsberger et al., 1968; Dunn and Snyder, 1976; Eeg-Olofsson et al., 1982; Drigo et al., 2001]. The paroxysms tend to be stereotypic and the frequency is variable, ranging from several times a week to once a year. Recovery is rapid and complete. Specific trigger factors are uncommon. BPV is easily overlooked due to its benign, paroxysmal, and transient nature, coupled with the difficulty that a young child has in describing vertiginous sensations. Onset is usually under 4 years of age [Eeg-Olofsson et al., 1982; Drigo et al., 2001], but later age of onset has been described [Abu-Arafeh and Russell, 1995b; Mira et al., 1984b; Mierzwinski et al., 2007]. The episodes gradually abate over months to years. Prevalence data are poor; one population study estimated the prevalence rate at 2.6 percent [Abu-Arafeh, Russell, 1995b], although the population characteristics were not typical for BPV. The neurological examination between spells is normal.
Etiology is unknown but thought to involve the central or peripheral vestibular system. This is supported, in part, by the presence of nystagmus during the acute attack [Eeg-Olofsson et al., 1982]. Normal hearing and caloric test would suggest that central vestibular pathways are more likely to be affected than the peripheral part of the vestibular nerve or inner ear [Mira et al., 1984a; Finkelhor and Harker, 1987]; earlier studies, however, reported abnormal caloric and rotational tests [Basser, 1964; Koenigsberger et al., 1968; Dunn and Snyder, 1976]. A relationship between BPV and migraine has been suggested [Fenichel, 1967; Koehler, 1980; Mira et al., 1984a; Lanzi et al., 1994; Abu-Arafeh and Russell, 1995b], and BPV is often considered a precursor to migraine headaches later on in life. There is a greater prevalence of migraine in BPV patients (24 versus 10.6 percent), and of BPV in migraine sufferers (8.8 versus 2.6 percent), than controls [Abu-Arafeh and Russell, 1995b]. Basilar artery migraine may present with similar symptoms [Golden and French, 1975; Lempert et al., 2009], suggesting a possible vascular basis for the symptoms [Basser, 1964; Fenichel, 1967; Perez Plasencia et al., 1998].
Benign paroxysmal torticollis (BPT) appears to be related to BPV and sometimes precedes it. First described by Snyder in 1969 [Snyder, 1969], the spells tend to occur at a slightly younger age (2–8 months) and are paroxysmal, but may last minutes to days. Spells begin with a sudden onset of torticollis with tilting of the head to one side and rotation of the chin to the opposite side. This may occasionally be accompanied by torsion or dystonia of the trunk or pelvis [Chutorian, 1974]. The frequency and duration of the episodes decline as the child gets older. A number of children will subsequently go on to develop BPV [Dunn and Snyder, 1976; Eeg-Olofsson et al., 1982; Lindskog et al., 1999]. Linkage to the CACNA1A gene has been described in four children [Giffin et al., 2002], but for the most part, BPT is also considered a migraine equivalent. This is discussed in more detail in the next section.
The diagnosis of BPV is based on a characteristic history and normal neurological events, but certain differential diagnoses should be considered. The most common differential diagnosis is epilepsy, especially temporal lobe epilepsy, but the brief nature of the spells, their occurrence only in the awake state, and lack of any change in sensorium should all help to differentiate BPV from seizure. Other differential diagnostic considerations include posterior fossa tumors (these usually have other neurological signs and symptoms) and acute vestibular neuronitis (which tends to occur more commonly in adults, is invariably associated with nystagmus, and lasts days to weeks rather than seconds). Ménière’s disease is rare in childhood and is usually associated with tinnitus and hearing loss. BPV is differentiated by the postural nature of this syndrome, which precipitates vertigo and nystagmus. The typical age of onset of BPV would make a functional disorder unlikely, although this should be considered in older children [Mierzwinski et al., 2007].
Benign Paroxysmal Torticollis of Infancy
Involuntary twisting of the neck (wryneck or torticollis), with abnormal head positioning, followed by subsequent spontaneous resolution, is characteristic of BPT of infancy. Usual age of onset is the first 6 months of life. The condition may involve either side of the neck, and the side may alternate between spells. In addition to the torticollis, the affected child may develop a pelvic tilt (tortipelvis) or retrocollis [Chutorian, 1974; Rosman et al., 2009]. The episodes may last from 10 minutes to 30 days (average about 5 days), and may recur every 7 days to 5 months (mean 37 days). The abnormal neck posturing may persist during sleep, which goes against the dystonic theory as to the possible underlying mechanism of BPT [Snyder, 1969; Drigo et al., 2000; Rosman et al., 2009]. An attack may be anticipated by the onset of irritability, distress, or vomiting [Chaves-Carballo, 1996]. There may be associated vertigo, ataxia, pallor, apathy, and gaze abnormalities during an acute attack. Some children are found to have motor developmental delay. The episodes spontaneously remit in most children by 2–3 years of age without treatment. There are few cases of familial occurrence [Gilbert, 1977; Lipson and Robertson, 1978]. Since the original description of BPT by Snyder more than 40 years ago [Snyder, 1969], there have been a further 23 reports of 113 cases described in the medical literature that conform to the features as outlined above [Sanner and Bergstrom, 1979; Hanukoglu et al., 1984; Bratt and Menelaus, 1992; Cataltepe and Barron, 1993; Cohen et al., 1993; Rosman et al., 2009].
The pathophysiology of BPT is subject to speculation. The observation, in some cases, of eye rolling or deviation suggests labyrinthine involvement. Abnormal oculovestibular function was found in 9 of 12 cases [Snyder, 1969], but not reproduced by future studies. Others believe that BPT is a forerunner of migraine, and it is thus considered as migraine equivalent. Evidence supporting this hypothesis includes:
Another theory involves an ion-channel disorder, since two patients in a kindred with familial hemiplegic migraine linked to the CACNA1A gene initially presented with BPT [Giffin et al., 2002].
Treatment with diphenhydramine, meclizine, and chlorpromazine has not been successful [Snyder, 1969; Rosman et al., 2009]. The prognosis, however, is excellent, with complete disappearance of torticollis in almost 100 percent of cases by 3 years of age. Motor developmental delay, found in some cases, also improves with time [Dunn and Snyder, 1976; Drigo et al., 2000; Rosman et al., 2009].
Benign paroxysmal torticollis is also described in Chapter 68.
Cyclic Vomiting Syndrome
Cyclic vomiting syndrome (CVS) is a chronic, disabling disorder characterized by “recurrent, discrete, self-limited episodes of vomiting and is defined by symptom-based criteria and the absence of positive laboratory, radiographic, and endoscopic testing” [Li et al., 2008]. First described in the British literature by Gee in 1882 [Gee, 1882], the prevalence has been estimated at 1.9 percent in community-based studies [Abu-Arafeh and Russell, 1995a]. In an Irish study, the incidence was 3.15 per 100,000 children per year, with a median age of onset of symptoms of 4 years (range 0–14 years) [Fitzpatrick et al., 2008]. Onset is typically in early childhood; however, onset in infancy and adults also occurs [Prakash and Clouse, 1999; Fleisher et al., 2005]. Although the majority of cases abate by adolescence, about one-third of individuals continue to experience vomiting during their teenage years [Fleisher and Matar, 1993; Dignan et al., 2001; Fitzpatrick et al., 2007]. It is not known what percentage of persons continue with vomiting into adult life. A female predominance has been reported in CVS [Prakash et al., 2001; Li and Misiewicz, 2003]; however, in the population-based Irish study by Fitzpatrick et al., females accounted for only 49 percent of the cohort [Fitzpatrick et al., 2008].
In a consensus paper from the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition [Li et al., 2008], an operational definition of CVS was developed that stipulates inclusion of all of the following criteria for diagnosis:






The term cyclic vomiting syndrome plus (CVS+) has been used to describe a subset of children with CVS who have an underlying neuromuscular or neurological disorder [Boles et al., 2003]. In this group of children, the median age of onset of cyclic vomiting episodes was 4 years – 3 years younger than the children with CVS and no underlying neurological or neuromuscular disorders, suggesting a more severe phenotype [Boles et al., 2006]. Population studies, however, have suggested a similar median age of onset of cyclic vomiting episodes (see above). Maternal inheritance in many cases, mitochondrial DNA mutations in some, and biochemical markers of disturbed electron transport chain/energy metabolism (lactic acidosis, energy-depleted patterns on urine organic acid testing) initially raised the suggestion that CVS+ syndrome was a mitochondrial cytopathy [Boles and Williams, 1999; Wang et al., 2004]. Further analysis of this cohort, and comparisons to CVS children without associated neuromuscular or neurological features, concluded that mitochondrial DNA sequence-related mitochondrial dysfunction is a risk factor for disease development in CVS in general, and is not specific to the CVS+ group [Boles et al., 2005, 2006].
The etiology and pathophysiology of CVS is unknown. Since CVS shares certain characteristics with migraine headaches, it is sometimes referred to as a migraine variant. These characteristics include similar triggers and prodromal symptoms, recurrent, discrete episodes, vasomotor/autonomic changes, and responsiveness to antimigraine medication in CVS. It is one of the childhood periodic syndromes that is a common precursor to subsequent migraine. Migraine headaches have been reported in 11–38 percent of children with CVS [Fleisher and Matar, 1993; Symon and Russell, 1995]. The prevalence rate of migraine in children with CVS is twice that of the general childhood population (21 vs, 10.6 percent) [Abu-Arafeh and Russell, 1995a]. In approximately 22–27 percent of children with CVS, the condition transforms into more typical migraine headaches [Fleisher and Matar, 1993; Dignan et al., 2001; Stickler, 2005]. Mitochondrial dysfunction is felt to play a role in selected cases (see below). Autonomic dysregulation has been shown in a number of studies in children with CVS [Rashed et al., 1999; Chelimsky and Chelimsky, 2007], and sympathetic hyperresponsiveness has been postulated as a potential mechanism contributing to the CVS. The episodic nature and natural history of CVS have led some to postulate that CVS may represent an ion-channel disorder, despite the fact that it is not inherited in a simple mendelian fashion [Ptacek, 1999]. The corticotrophin-releasing factor (CRF) hypothesis proposes that CVS is precipitated by stimuli or factors associated with CRF release, and that the resultant endocrine, autonomic, and visceral changes are reminiscent of CRF activation in the paraventricular nucleus of the hypothalamus and dorsal vagal complex in the brain [Tache, 1999]. Central CRF has been shown to delay gastric emptying and stasis in animal studies [Tache, 1999].
Clinically, the attacks are explosive in onset, with recurrent severe bouts of emesis, retching, and nausea lasting hours to days. The vomiting is often bilious [Li et al., 2008], with a peak median intensity of six emeses per hour [Li, 2001]. Only half of the children with CVS have a “predictable” periodicity, typically every 2–4 weeks [Li, 2001]. Other symptoms include abdominal pain, diarrhea, anorexia, lethargy, pallor, headache, photophobia, and phonophobia [Li, 2001; Prakash et al., 2001; Li and Misiewicz, 2003; Fitzpatrick et al., 2008]. Low-grade fever may be present [Fleisher, 1995; Li, 2001]. The spells often occur in the early hours of the morning or upon waking, and they are frequently triggered by physical or psychological stress or excitement [Fleisher and Matar, 1993; Li et al., 2008]. Onset may be abrupt but a prodrome is reported in 22–38 percent of cases [Prakash et al., 2001; Fitzpatrick et al., 2008]. The paroxysms often end abruptly, much like the onset. For any particular child, the episodes tend to be stereotypical with respect to time of onset, duration, and symptoms. Intravenous hydration is often required [Prakash et al., 2001; Li and Misiewicz, 2003]. There is frequently a very long delay from disease onset to diagnosis [Prakash et al., 2001; Fitzpatrick et al., 2008]. Between episodes, children are completely well.
A family history of migraine in a first- and second-degree relative is common (35–56 percent) [Fleisher and Matar, 1993; Symon and Russell, 1995; Pfau et al., 1996; Stickler, 2005]; a figure of 20 percent was found in one population-based study [Fitzpatrick et al., 2008], although a family history of CVS is uncommon. There appears to be a high prevalence of anxiety and mood symptoms in children with CVS, as well as their parents [Forbes et al., 1999; Tarbell and Li, 2008].
Diagnosis relies on a careful history and exclusion of other serious underlying causes of vomiting. Vomiting in CVS tends to occur with a much higher intensity but lower frequency than other chronic vomiting disorders. There are no specific laboratory or neurophysiologic markers that lead to a diagnosis of CVS. Testing to exclude all of the possible differential diagnoses would subject many children to unnecessary and costly radiographic and endoscopic procedures [Li et al., 2008]. Expert consensus recommends that electrolytes, glucose, and upper gastrointestinal radiographs should be obtained in order to exclude malrotation in all children; in refractory cases, a renal ultrasound should also be performed to exclude transient hydronephrosis [Li et al., 2008]. These studies should preferably be carried out during an acute crisis, and blood work should be done prior to intravenous hydration. Abnormal neurological findings on examination, a history that suggests a possible underlying metabolic disorder (such as an association of the spells with fasting, illness, or certain food groups), or hypoglycemia on presentation warrant further diagnostic evaluation; this should include neuroimaging and testing for inborn errors of metabolism (fatty acid oxidation disorders, urea cycle defects, mitochondrial cytopathies, or amino/organic acidurias). The North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition task force for CVS also recommends a neurometabolic work-up for children under the age of 2 with suspected CVS [Li et al., 2008]. Significant abdominal pain and tenderness, together with bilious vomiting and progressive worsening of symptoms, require further gastroenterology evaluation.
Differential diagnosis is extensive and includes both gastrointestinal and nongastrointestinal disorders. The most common gastrointestinal differential diagnosis is from viral gastroenteritis. Children with CVS are usually substantially sicker, requiring intravenous fluids for dehydration [Li and Misiewicz, 2003]. Nongastrointestinal disorders include neurological, metabolic, renal, and endocrine etiologies.
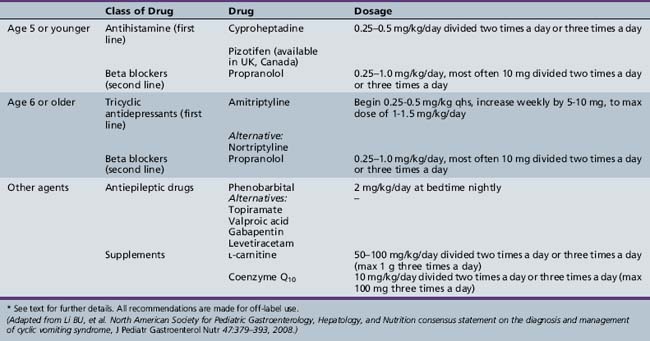
Treatment includes prevention, prophylaxis, and acute management of vomiting episodes. Prevention depends on the identification of triggers and avoidance of precipitants, if possible. If anticipatory anxiety or stress is a factor, lifestyle changes, behavioral intervention, biofeedback, and counseling may be effective. Prophylactic medication should be considered in children with frequent cycles of emesis, frequent school absences, and/or severe, debilitating spells. A wide variety of medications have been used for prophylactic treatment of CVS; however, good evidence-based data to support any therapy are lacking [Li et al., 2008]. Table 65-2 is modified from consensus recommendations from a recent expert task force review on CVS [Li et al., 2008]. A recent study showed the effectiveness of valproic acid in a small group of children with refractory CVS, in whom “organic causes” were excluded [Hikita et al., 2009]. There are no evidence-based guidelines to help direct treatment and there is a very high placebo effect [Li and Misiewicz, 2003]. A sequential trial of medications, over an appropriate length of time and vomiting cycles, should be tried. Acute management involves rapid recognition and identification of a spell. If the child experiences a prodrome, efforts should focus on abortive measures to prevent a full-blown attack. Nonsteroidal analgesia (e.g., Ibuprofen) or off-label use of triptans, if appropriate, together with an antiemetic, especially sublingual or oral disintegrating tablets, should be tried [Li et al., 2008]. In the case of an established attack, intravenous fluids containing glucose and intravenous antiemetic medication (5HT3 antagonists) should be administered [Li et al., 2008]. Ketosis should be avoided. Proton pump inhibitors or H2 receptor antagonists should be considered for children who have prolonged or frequent episodes of vomiting or epigastric discomfort [Forbes et al., 1999; Forbes and Fairbrother, 2008; Li et al., 2008]. The child should be placed in a quiet, dark environment. Sedation may be of some benefit if there is a lot of anxiety or distress. Pain medication may be necessary for severe abdominal pain or headache.
Sandifer’s Syndrome
Named for the British neurologist, Paul Sandifer [Sutcliffe, 1969], the full syndrome comprises tonic neck extension, deviation of the head to one side (“spastic torticollis”), and dystonic posturing of the trunk [Kinsbourne, 1964; Mandel et al., 1989]. Torticollis in the absence of truncal dystonia may occur [Murphy and Gellis, 1977], and must be differentiated from other positional torticollis in which there is typically contracture or tightness of the sternocleidomastoid muscles. Apnea, tonic body stiffening, and writhing movements of the limbs may be seen in the neonatal or early infantile period [Werlin et al., 1980; Mandel et al., 1989]. Initially described in association with hiatus hernia [Kinsbourne, 1964; Sutcliffe, 1969; Gellis and Feingold, 1971; Murphy and Gellis, 1977], Sandifer’s syndrome more commonly occurs in association with gastroesophageal reflux disease (GERD) [Bray et al., 1977; Werlin et al., 1980; Shepherd et al., 1987; Gorrotxategi et al., 1995]. The abnormal posturing is felt possibly to represent the body’s response to discomfort associated with reflux [Werlin et al., 1980; Deskin, 1995].
The incidence is unknown, although the syndrome is said to occur in anywhere from 1 to 8 percent of cases of GERD [Shepherd et al., 1987; Lehwald et al., 2007]. There appears to be no correlation between the degree of reflux or esophagitis and the severity of symptoms [Mandel et al., 1989]. Although Sandifer’s syndrome is commonly seen in clinical practice, there are scant reports in the medical literature. In one series of children aged 2 months to 5 years monitored by video EEG for paroxysmal nonepileptic events, approximately 15 percent were found to be due to GERD. Onset is usually in infancy or early childhood, but older-onset cases have been described [Kinsbourne, 1964; Mandel et al., 1989; Lehwald et al., 2007]. The diagnosis can be difficult in children with underlying neurological or metabolic disorders, as the dystonic movements or neck/head posturing is often attributed to their primary underlying disorder [Mandel et al., 1989; Gorrotxategi et al., 1995; Lehwald et al., 2007].
Sandifer’s syndrome is most commonly associated with a normal neurological examination, especially in infants. The history should provide clues to the diagnosis, given the intermittent nature of the torticollis, and the relationship to feeding or the immediate postprandial period. Reflux or vomiting of feeds is not always present. Ways to differentiate Sandifer’s syndrome from other causes of torticollis (muscular, cervical, and vertebral abnormalities, or posterior fossa tumors) include the intermittent nature of the torticollis, lack of muscle contracture of the sternocleidomastoid, and typical relationship to feeding. Other differential diagnostic considerations include extensor spasms/seizures [Kabakus and Kurt, 2006; Lehwald et al., 2007] or primary movement disorders [Mandel et al., 1989].
Spasmus Nutans
Spasmus nutans is a benign, self-limiting condition manifest by an intermittent triad of asymmetric nystagmus, head nodding, and torticollis in infancy. It was first described in the medical literature by Raudnitz in 1897 [Weissman et al., 1987]. Differentiation of spasmus nutans from other similar but more serious disorders has been hampered by the fact that patients often do not demonstrate all of the typical diagnostic features [Gottlob et al., 1990]. Furthermore, clinical detection of asymmetric nystagmus at such a young age may be challenging. The nystagmus, typically, is of low amplitude (3 degrees), high frequency (up to 15 Hz), and horizontal, but may rarely be vertical, oblique, jerky, or pendular. Monocular or dissociated nystagmus may occur in spasmus nutans [Farmer and Hoyt, 1984]. The head nodding possibly corresponds to a compensatory oculovestibular reflex [Gottlob et al., 1992]. The mechanism of head tilt is unclear, possibly related to asymmetry in the type of nystagmus, with partial compensated correction in a specific head posture. The pathophysiology, mechanisms, and substrate involved in spasmus nutans are undetermined. Initial speculation that “spasmus nutans results from environmental deprivation of sunlight” has not been substantiated. Certain demographic populations (African-American and Hispanic ancestry) and low socioeconomic conditions may, however, represent risk factors for the development of spasmus nutans [Wizov et al., 2002].
Numerous cases of spasmus nutans have been associated with chiasmatic lesions [Kelly, 1970; Antony et al., 1980; Farmer and Hoyt, 1984; Albright et al., 1984], diencephalic syndrome, porencephalic cysts [Gottlob et al., 1990], opsoclonus-myoclonus syndrome [Allarakhia and Trobe, 1995], empty sella syndrome [Gottlob et al., 1990], ependymoma [Gottlob et al., 1990], and retinal disorders [Lambert and Newman, 1993; Gottlob et al., 1995a; Kiblinger et al., 2007]. Spasmus nutans should no longer be regarded as a benign entity when first seen until serious ocular, intracranial, or systemic abnormalities are excluded by brain MRI scan and electroretinography [Norton and Cogan, 1954; Hoefnagel and Biery, 1968; Kiblinger et al., 2007].
To differentiate congenital nystagmus from spasmus nutans, eye and head movements were recorded in 23 patients with spasmus nutans, 10 patients with spasmus nutans-like disease (associated with central nervous system lesions), and 25 patients with congenital nystagmus. The mean onset of nystagmus was 8 months and head nodding 15 months in the spasmus nutans group. The following findings helped differentiate spasmus nutans from infantile nystagmus: ocular oscillations were of later onset; head nodding was more frequent, of larger amplitude, and clinically easier to detect; and nystagmus was asymmetric and intermittent in spasmus nutans compared with infantile nystagmus. Opticokinetic nystagmus was usually present in spasmus nutans and absent in most patients with infantile nystagmus. Head tilt was not found to be helpful in the differentiation. The ocular movement recordings were not useful for differentiating idiopathic spasmus nutans from spasmus nutans associated with neurologic abnormalities; neuroimaging studies are required for this [Gottlob et al., 1990]. A normal electroretinogram may help to rule out retinal pathology, thereby substantiating a diagnosis of spasmus nutans [Smith et al., 2000]. Other differential diagnoses include bobble-head doll syndrome, which mimics head nodding but without head tilt or nystagmus, and is secondary to third ventricular tumors or colloid cysts. Long-term follow-up studies are important to substantiate a diagnosis of spasmus nutans. Most patients eventually attain good visual acuity. Subclinical nystagmus persists until at least 5–12 years of age [Gottlob et al., 1990, 1995b).
Paroxysmal Tonic Upgaze of Childhood
First described in 1988 as a benign paroxysmal extraocular movement disorder [Ouvrier and Billson, 1988], paroxysmal tonic upgaze of childhood (PTUC) involves transient episodes of sustained tonic upward deviation of the eyes, with or without ataxia. Although it was initially thought to be a benign condition, neurological and developmental abnormalities occur in approximately half of the cases. These include chronic ataxia, persistent mild ocular movement abnormalities (nystagmus, strabismus, and saccadic abnormalities), learning disabilities, pervasive developmental disorder, and cognitive deficits [Hayman et al., 1998; Ouvrier and Billson, 2005]. Most cases are sporadic; however, familial cases have been reported, suggesting an autosomal-dominant mode of inheritance [Campistol et al., 1993; Guerrini et al., 1998; Roubertie et al., 2008]. Etiology remains uncertain, although neurotransmitter depletion affecting the pathways controlling supranuclear vertical eye movements has been postulated [Ouvrier and Billson, 1988]. Others have suggested that it may be the result of an age-dependent immature corticomesencephalic control of vertical eye movement [Hayman et al., 1998]. Abnormal GABA transmission was suggested in one case with coexisting absence epilepsy in which valproic acid seemed to be associated with the development or “unmasking” of PTUC [Luat et al., 2007]. In some instances, brain MRI scan has demonstrated structural abnormalities in the mesencephalic region, including a pinealoma [Spalice et al., 2000] and a vein of Galen malformation [Hayman et al., 1998]. A single case report showed an association between PTUC and a partial tetrasomy of chromosome 15 in a 3-month-old child [Joseph et al., 2005], and mutations in the CACNA1A gene have been shown in at least one family pedigree, in which PTUC was an early clinical manifestation in some family members [Roubertie et al., 2008].
Abnormalities of ocular movement typically occur in the first few years of life, often the first year, but may occur as early as the first few weeks of life [Ahn et al., 1989; Mets, 1990; Hayman et al., 1998]. The eye movement abnormalities include brief, conjugate, upward deviation of the eyes lasting seconds to minutes, compensatory neck flexion, and incomplete downward saccades on attempted downgaze [Ouvrier and Billson, 1988; Campistol et al., 1993]. They tend to exacerbate with fatigue and are relieved by sleep. Horizontal eye movements are normal. The episodes tend to dissipate gradually and resolve over years [Hayman et al., 1998; Verrotti et al., 2001]. There is no consistent precipitating factor with regard to onset; however, a relationship to febrile illness and immunization has been raised in some cases [Hayman et al., 1998; Spalice et al., 2000; Verrotti et al., 2001].
Ataxia is a frequent accompaniment of the paroxysmal events and appears to be predominantly truncal in nature. It persists in a significant number of affected cases as a chronic, permanent disability [Ouvrier and Billson, 2005].
In the majority of children, laboratory, neuroimaging, and neurophysiological studies are normal. Metabolic evaluation, including spinal fluid analysis, has been normal, although the specific nature of the work-up has not been well defined [Ouvrier and Billson, 1988; Campistol et al., 1993; Spalice et al., 2000; Lispi and Vigevano, 2001; Verrotti et al., 2001]. In all cases, video EEG has been normal, with captured spells confirming their nonepileptic nature. Neuroimaging studies are typically normal, but a number of different abnormalities have been described, including a pinealoma [Spalice et al., 2000], vein of Galen malformation [Hayman et al., 1998], periventricular leukomalacia [Sugie et al., 1995], and delayed myelination (Luat et al., 2007], suggesting that neuroimaging studies should be a consideration in the evaluation of these children.
The paroxysmal episodes are generally brief and self-limiting, making medication unnecessary. Response to l-DOPA has been reported in some cases [Ouvrier and Billson, 1988; Campistol et al., 1993], but this is not consistent [Hayman et al., 1998; Ouvrier and Billson, 2005]. Adrenocorticotropic hormone (ACTH), acetazolamide, and anticonvulsant medications appear to be ineffective [Ouvrier and Billson, 2005].
Benign Myoclonus of Infancy
Benign myoclonus of infancy (BMI) was first described in 1976 by Fejerman [Fejerman, 1976]. Infants aged 1–12 months develop sudden onset of flexor or extensor spasms while awake; these usually occur in clusters. The paroxysmal activity resembles infantile spasms, without EEG correlate of hypsarrhythmia and the associated neurologic abnormalities seen in West’s syndrome. Development is normal. The clinical features have been best summarized by Caraballo et al. in 2009; their report includes 102 infants with long-term follow-up [Caraballo et al., 2009]. The paroxysmal motor phenomena may involve brief myoclonus or tonic contractions mimicking infantile spasm, shuddering, atonic or negative myoclonus, or a combination of the above. Involved body parts include neck, trunk, upper limbs, head (cephalic myoclonus), or eyes (blinking) [Lombroso and Fejerman, 1977; Maydell et al., 2001; Caraballo et al., 2009]. The myoclonic activity increases for a few weeks or months after onset, then stabilizes and starts decreasing after about 3 months, before disappearing spontaneously by 2 years of age [Lombroso and Fejerman, 1977]. There is no obvious gender predilection, and familial occurrence has only rarely been reported [Galletti et al., 1989]. The etiology is unknown but prognosis is excellent, with no significant morbidity, even after 40 years of follow-up. No specific treatment is needed; however, it is very important to avoid over-investigation in these cases [Caraballo et al., 2009].
The condition should be differentiated from stimulus-sensitive or action myoclonus as seen in posthypoxic encephalopathies, which are severe and disabling, often resulting in the patient falling to the ground on attempted standing or walking. Periodic myoclonus may result from subacute sclerosing panencephalitis, but the myoclonus is a bit slower, as these are reticular in origin. Other differential diagnoses include epileptic cortical myoclonus, secondary to herpes simplex encephalitis, for example; progressive degenerative disorders of gray or white matter, including gangliosidoses; leukodystrophies; and progressive myoclonic epilepsy of early childhood, such as Unverricht–Lundborg and Lafora body diseases [Dravet et al., 1986] or myoclonic epilepsy and ragged red fibers (MERRF). Other protracted but eventually self-limiting disorders, such as hereditary essential myoclonus, benign essential myoclonus, and shuddering attacks, should be considered and excluded. The condition should also be differentiated from benign neonatal sleep myoclonus, which represents an exaggeration of normal physiologic phenomena during sleep [Noone et al., 1995]. EEG studies fail to reveal any epileptiform activity during the paroxysmal episodes of myoclonus; however, more prolonged video EEG monitoring may be required in certain situations to establish the nonepileptic nature of the myoclonus [Bleasel and Kotagal, 1995; Pachatz et al., 1999].
Benign myoclonus of infancy is also described in Chapter 68.
Hereditary Hyperekplexia
An exaggerated startle response may be a component of epilepsy (startle epilepsy) or of a nonepileptiform paroxysmal disorder: namely, hyperekplexia, or startle disease. The term hyperekplexia is derived from Greek, and means excessive jerking or jumping (startle). Kirstein and Silfverskiold first described this entity in 1958 [Kirstein and Silfverskiold, 1958]. It involves a strikingly excessive response to startle elicited by unexpected sudden visual, auditory, or somatosensory stimuli that fail to produce a startle response in most normal individuals. In the normal individual, the startle response is a basic alerting reaction, with stereotyped features consisting of eye blinking, facial grimacing, flexion of the head, elevation of the shoulders, and flexion of the elbows, trunk, and knees. This involuntary reflex appears during infancy at the same time as the Moro reflex [Andermann and Andermann, 1988]. The abnormal or pathologic response consists of an exaggerated startle response associated with generalized muscle stiffness, attaining a fetal position and loss of postural control, causing the subject to fall “en statue” without loss of consciousness [Aicardi, 1992]. The generalized stiffness may compromise breathing in the neonatal and early infantile period, sometimes with fatality; frequent falls are common at a later age which may result in injuries, including head trauma [Kirstein and Silfverskiold, 1958; Andermann et al., 1980; Andermann and Andermann, 1988; Aicardi, 1992; Praveen et al., 2001].
Hyperekplexia presents in its major form during the neonatal period as stiff-baby syndrome (“stiff-man syndrome in the newborn”) [Klein et al., 1972; Lingam et al., 1981]. The onset of stiffness becomes evident a few hours after birth. Shoulder girdle muscles are particularly stiff. There may be difficulty in swallowing and frequent choking. Apnea may result from hyperekplexia and may cause death [Kurczynski, 1983; Nigro and Lim, 1992]. The hypertonia usually disappears during sleep, although repetitive and violent movements of the extremities may be seen during the hypnagogic stage, which may lift the child off the bed [Andermann and Andermann, 1988; Aicardi, 1992]. The neonatal form improves spontaneously during the first year of life, although later on there may be absence of crawling and delay in walking. Hip dislocation, as well as umbilical, inguinal, and diaphragmatic hernias, may result from increased intra-abdominal pressure due to generalized muscle stiffness [Aicardi, 1992; Gordon, 1993]. A clinically useful maneuver is to tap the bridge of the nose or the glabella (glabellar tap). This maneuver will elicit an exaggerated, nonhabituating startle response in affected individuals [Shahar et al., 1991; Nigro and Lim, 1992]. Similar results may be obtained by blowing air directly on the face of neonates and infants with hyperekplexia [Shahar and Raviv, 2004].
Neurophysiologic studies demonstrate that hyperekplexia is not simply an exaggerated normal startle response [Hallett et al., 1986]. Electromyogram latencies are shorter than normal. EEG studies are mostly normal; however, they may reveal an initial myogenic spike, maximally in the frontocentral region, followed by slow waves and desynchronization of background activity corresponding to the phase of apnea, bradycardia, and cyanosis [Andermann and Andermann, 1988; Tohier et al., 1991; Praveen et al., 2001]. Auditory and somatosensory-evoked potentials may be exaggerated or normal [Hallett et al., 1986; Andermann and Andermann, 1988]. Rostrocaudal recruitment of cranial nerve-innervated muscles supports a brainstem reticular origin for the abnormal startle response in hyperekplexia [Brown, 2002].
The condition is familial and transmitted as an autosomal-dominant trait. Linkage analyses initially demonstrate genetic homogeneity in typical cases to the long arm of chromosome 5 [Ryan et al., 1992; Shiang et al., 1993]. Point mutations or deletions in the alpha subunit of the inhibitory glycine receptor (GLRA1) gene, located on chromosome 5q33–35, were subsequently found [Shiang et al., 1995; Tsai et al., 2004]. Some sporadic cases of hyperekplexia may show abnormalities in the beta subunit of the glycine receptor (GLRβ), located on chromosome 4q32.1 [Hejazi et al., 2001; Rees et al., 2002]. Rare mutations of the genes coding for receptor clustering proteins gephyrin and collybistin have also been described as causing hyperekplexia [Rees et al., 2003; Harvey et al., 2004]. New research suggests that mutation in the genes encoding presynaptic glycine transporter GlyT2 is also a cause of human hyperekplexia [Eulenburg et al., 2006]. Glycine receptors are found primarily in the brainstem and spinal cord [Rajendra et al., 1997]. The main inhibitory neurotransmitter receptor is the GABA type A receptor. Affected individuals usually reveal a favorable response to treatment with clonazepam, an agonist of GABA type A receptors [Shiang et al., 1995].
The differential diagnoses in the neonatal period are congenital stiff-person syndrome, Schwartz–Jampel syndrome, startle epilepsy, myoclonic seizures, neonatal tetany, and phenothiazine toxicity [Andermann and Andermann, 1988; Praveen et al., 2001]. The relationship of hyperekplexia to other nonepileptic startle disorders, such as jumping (jumping Frenchmen of Maine), latah (ticklishness associated with echopraxia and coprolalia in Malaysia), and myriachit (to act foolishly, as reported from Siberia, Asia, and Africa), as discussed over a century ago by Gilles de la Tourette [1884; 1899] in the context of “tic convulsif,” remains conjectural.
Treatment is most effective with clonazepam (0.1–0.2 mg/kg/day) but, in some cases, symptoms may not be totally suppressed [Aicardi, 1992; Tijssen et al., 1997]. Valproic acid is recommended in cases of late onset [Dooley and Andermann, 1989]. Vigabatrin did not reduce startle activity among four patients with hyperekplexia [Tijssen et al., 1997]. Phenobarbital, phenytoin, and diazepam have not proved to be effective [Giacoia and Ryan, 1994]. The prognosis is variable. Neonatal hyperekplexia may result in apnea, neonatal encephalopathy, cerebral palsy, and unexpected death [Chaves-Carballo et al., 1999]. Early identification and treatment improve the outcome in most children. A simple maneuver, like forced flexion of the head and legs towards the trunk, is known to be life-saving when prolonged stiffness impedes breathing [Vigevano et al., 1989]. Hypertonia and motor delay improve with increasing age, and muscle tone becomes normal by the age of 3 years. In some families, the exaggerated startle response ameliorates or disappears spontaneously by 2 years of age, although hyperekplexia may persist or reoccur in adult life, resulting in falls [Shahar et al., 1991].
In the minor form of hyperekplexia, there is only an abnormal startle response without generalized stiffness or tonic spasms. The minor form is not associated with neurologic or catastrophic sequelae, and in this group no mutations have been detected in the genes encoding the glycine receptor [Tijssen et al., 1997, 2002].
Shuddering Attacks
Shuddering or shivering attacks are uncommon paroxysmal events rarely reported and poorly understood. A retrospective series of paroxysmal nonepileptic events in 666 children found 7 percent of all events to be due to shuddering attacks [Bye et al., 2000]. These may start as early as 4–6 months of age, and rarely occur after the age of 3. They are precipitated or aggravated by excitement, fear, anger, frustration, or embarrassment [Vanasse et al., 1976; Holmes and Russman, 1986]. The episodes last usually for a few seconds, and are characterized by rapid shivering or stiffening of the body with abnormal posturing, with adduction of the knees and arms, flexion of the head, elbows, trunk, and knees, and flexion or extension of the neck. There is no alteration of consciousness with the abnormal movements. The attacks are very frequent, occurring multiple times a day, and in excess of 100 per day in some cases. The pathophysiology of shuddering attacks is unknown. In one series, in 5 of 6 cases, one of the parents had an essential tremor. These children also had some postural tremor on examination [Vanasse et al., 1976]. Electromyography studies have revealed the frequency of these shuddering attacks to be similar to that of essential tremor [Kanazawa, 2000]. Head tremor may evolve from shuddering attacks [DiMario, 2000]. These characteristics suggested that shuddering attacks may represent the expression of an “essential tremor in the immature brain.” Later studies, however, failed to find an increased frequency of family history of essential tremor in children with shuddering attacks [Kanazawa, 2000]. The other theory is that shuddering attacks are a variant of benign myoclonus of early infancy [Fejerman, 1997]. The differential diagnoses include generalized seizures or infantile spasm. Ictal EEG is normal [Holmes and Russman, 1986; Kanazawa, 2000]. No symptomatic cases of shuddering attacks have been reported so far. Treatment with antiepileptic mediations has not been effective. Propranolol has been effective in eliminating shuddering attacks [Barron and Younkin, 1992]. Monosodium glutamate has been implicated in some cases, and its avoidance or elimination from the diet has been effective [Reif-Lehrer and Stemmermann, 1975]. The prognosis is favorable; most children improve spontaneously before 10 years of age [Vanasse et al., 1976; Kanazawa, 2000].
Stereotypies, Self-Stimulation, and Masturbation
Repetitive stereotyped movements are very common among children with autism, mental retardation, and sensory deprivation; however, they are also observed in nonautistic children with normal cognition. These movements involve rhythmic motor behaviors, self-stimulating movements, and gratification phenomena, an extreme example of which is masturbatory behavior. Examples of self-stimulating movements or gratification phenomena include body rocking, head banging (jactatio capitis), and head rolling [Sallustro and Atwell, 1978]. Rocking motions of the trunk observed among mentally and visually impaired children may represent a form of vestibular stimulation akin to the maternal rocking that effectively comforts a crying, tired, or sleepy infant. The characteristic, purposeless, writhing, repetitive hand movements seen among autistic children and in Rett’s syndrome may also be grouped into the category of self-stimulating behavior. Children who have a photoconvulsive response may learn to induce a seizure by repetitive hand movements in front of their eyes as a form of photic stimulation. Broadly, all the above movements fall in the category of stereotypies. More specifically, stereotypies are abnormal involuntary, repetitive, rhythmic, seemingly purposeless, suppressible, distractible, and predictable in regard to pattern, amplitude, and location of the movement. In the majority of cases, emotional excitement precedes the stereotypies. These may take the form of hand flapping, hand waving or rotation, finger wiggling, wing-beating movements of the arm, head/shoulder/body gyration, and ritualistic complex movement pattern, with or without vocalization. Stereotypies may be motor or vocal (associated with sound production). Each type may be simple or complex, depending on the complexity of the movement pattern or content of the vocalization [Harris et al., 2008; Goldman et al., 2009]. Among the vocal stereotypies, common ones involve grunting sounds, hissing, whistling, acting out a movie character, sound of engine, imaginary video game. Very often, complex motor stereotypies may be combined with vocal stereotypies.
Stereotypy and posturing during masturbation are also described in Chapter 68.
Tics, paroxysmal dyskinesias, and complex partial seizures are the major differential diagnoses to be considered. Tics are differentiated from stereotypies by the brevity of the phenomena, tics being much briefer; the majority of the tics affect craniofacial muscles, whereas upper limbs are predominantly affected in stereotypies. Emotional excitement is the prime precipitant for stereotypies, whereas stress/anxiety or relaxation after stress is a major factor in tic production. Distractibility is also more suggestive of stereotypies. Family history is more common with tics and less common in stereotypies. Association with autism, mental retardation, and sensory deprivation also characterizes stereotypies [Singer, 2009]. The pathophysiology of stereotypies is unclear. As in other involuntary movements, the cortico-striato-pallido-thalamic pathway is thought to be involved. Improvement of stereotyped behavior in primates with stereotypies by high-frequency deep-brain stimulation of anterior subthalamic nucleus gives credence to the above hypothesis [Baup et al., 2008]. Medications have not been successful in treating stereotypies. Applied behavioral therapy may be of some help in autistic children [Miller et al., 2006]. Habit reversal was beneficial in reducing motor stereotypies in nonautistic children [Miller et al., 2006]. Prognosis depends on the primary underlying diagnosis. Even in primary stereotypies without underlying autism, mental retardation, or sensory deprivation, stereotypies may persist in 94 percent of cases [Harris et al., 2008].
More difficult to recognize is masturbatory behavior, particularly when this occurs in young children and infants [Fleisher and Morrison, 1990; Nechay et al., 2004]. The repetitive movements usually involve the lower trunk and may be accompanied by pelvic thrusting or contractions of the gluteal muscles. Thighs may adduct and rub against each other. Use of midline seat belts in a car seat or while on a high chair may stimulate the genitalia and provoke this masturbatory behavior. The physical effort may be prolonged until the inciting stimulus is removed or the child is distracted. Other manifestations may include diaphoresis, hyperpnea, flushing, and grunting [Fleisher and Morrison, 1990]. The paroxysms may terminate in fatigue, exhaustion, or sleep. During a typical episode, parents may give a history of partial responsiveness, as it may require a lot of effort to distract the infant or child from a pleasurable act. Differential diagnosis is from an epileptic seizure, paroxysmal dyskinesia, or a dystonic disorder. The prominent hyperventilation and grunting sounds may unnecessarily lead to investigations for asthma. Detailed observation of an episode or review of a videotaped event can be especially helpful in clarifying the event and making the diagnosis, particularly in infants younger than 1 year of age [Casteels et al., 2004]. Any explanation of the benign nature of the paroxysms should take into account unusually sensitive or unbelieving parents, more so in baby girls [Leung and Robson, 1993]. Reassurance that the episodes are benign and self-limiting should help to avoid unjustified concerns and unnecessary investigations [Mink and Neil, 1995].
Hyperventilation Syndrome in Childhood
The hyperventilation syndrome (HVS) may best be defined as a syndrome characterized by “a variety of somatic symptoms induced by physiologically inappropriate hyperventilation and usually reproduced in whole or in part by voluntary hyperventilation” [Lewis and Howell, 1986]. In addition to somatic symptoms, psychological symptoms are also common. The term was first used in by Kerr et al. in 1937 [Kerr et al., 1938]. Hyperventilation as a paroxysmal event may also be a primary manifestation of certain neurological conditions, metabolic disorders, or genetic syndromes but differs from HVS in which, by definition, there is no underlying organic disease.
The incidence of HVS in the general (adult) population is about 5–11 percent; however, in a selected population of patients being evaluated for dizziness, it accounted for 24 percent of the cases [Evans, 1995]. It is more common in females than males [Enzer and Walker, 1967; Joorabchi, 1977; Perkin and Joseph, 1986]. In two small series, age at onset ranged from 5 to 18 years, with just over half occurring in the 13–16-year-old age group [Enzer and Walker, 1967; Herman et al., 1981]. Symptoms may last from several minutes to hours [Enzer and Walker, 1967].
The clinical presentations of HVS are protean [Joorabchi, 1977; Perkin and Joseph, 1986; Hanna et al., 1986; Evans, 1995] and often do not involve the nervous system. Common non-neurological manifestations include symptoms referable to the heart (palpitations, shortness of breath, chest pain), abdomen (abdominal distention, abdominal pain, flatulence, belching, diarrhea), and lungs (shortness of breath, feeling of suffocation, or inability to draw in an adequate breath). Neurological symptoms may involve the autonomic, peripheral, or central nervous system. Autonomic manifestations include palpitations, tachycardia, sweating, and nausea from excessive sympathetic activity. Peripheral nervous system manifestations include numbness, tingling, generalized weakness or muscle stiffness, carpopedal spasm, or generalized tetany. The parasthesias may be asymmetric or even unilateral [Lewis, 1953; Tavel, 1964; Perkin and Joseph, 1986; Evans, 1995] and may involve the distal extremities, face, or trunk. Dizziness, vertigo, ataxia, tinnitus, headache, syncope, tremulousness, and visual disturbances are common central nervous symptoms. Visual symptoms include blurring of vision, loss of vision, and flashing lights. Syncope may result in secondary seizure. Psychological manifestations include nervousness, disorientation, “out of body” sensation, fear, and anxiety.
The pathophysiology underlying HVS includes reduction in arterial PCO2 with resultant respiratory alkalosis, causing a left shift in the oxygen dissociation curve and increased binding of oxygen to hemoglobin. This results in decreased oxygen delivery to tissue. Hypocapnea also leads to cerebral vasoconstriction, with resultant diminished cerebral blood flow. Alkalosis also causes a reduction of plasma calcium concentration. Hypophosphatemia has also been implicated. Finally, hyperventilation may be triggered by beta-adrenergic stimulation from anxiety or stress [Herman et al., 1981; Evans, 1995]. In a double-blind, placebo-controlled trial, hypocapnea was not necessary to induce symptoms and appeared to be an epiphenomenon or consequence of the attack, suggesting that other mechanisms may be the cause for the symptoms [Hornsveld et al., 1996].
The diagnosis of HVS is often unrecognized or misdiagnosed, as the patients generally do not complain of hyperventilation, the diagnosis is not considered, or the signs and symptoms from the hyperventilation may be atypical [Joorabchi, 1977]. Psychological factors, including stress, anxiety, and panic attacks, commonly underlie HVS [Enzer and Walker, 1967; Herman et al., 1981]. The diagnosis can often be made at the bedside by having the patient breathe rapidly or draw in exaggerated deep breaths, with reproduction of the symptoms. This should not be performed in persons with cardiac or cerebrovascular disease, or in persons with hypercoagulable states or sickle cell disease. The validity of the hyperventilation provocation test has, however, been brought into question [Hornsveld et al., 1996]. EEG is typically normal, although hyperventilation may induce an absence seizure (3 Hertz spike and wave) in a person with absence epilepsy and whose presentation and symptoms, especially transient alteration in consciousness, may be confused with HVS. Other causes should be excluded by appropriate laboratory tests when indicated. Pain, fever, sepsis, and certain drugs (such as caffeine or salicylate toxicity) can result in HVS. Topiramate, an antiepileptic medication, may also cause central hyperventilation due to its inhibition of carbonic anhydrase [Laskey et al., 2000; Philippi et al., 2002]. Neurological causes include brainstem strokes or tumors, malignant hyperthermia, and encephalitis. A number of genetic syndromes cite primary hyperventilation as a clinical manifestation. These include Joubert’s syndrome [Joubert et al., 1969; Boltshauser and Isler, 1977]; Pitt–Hopkins syndrome, caused by mutations of the TCF4 gene on chromosome 18q21 [Pitt and Hopkins, 1978; Giurgea et al., 2008]; Rett’s syndrome, caused by a mutation of the MECP2 gene [Southall et al., 1988; Murakami et al., 1998; Kerr and Julu, 1999]; and Leigh’s syndrome, due to SURF1 gene mutations [Pronicka et al., 2001]. There is also a single case report of two brothers with novel duplication in the ARX gene and intermittent hyperventilation [Demos et al., 2009].
Treatment is predominantly supportive and includes reassurance, education, and respiratory control procedures. Nonmedical interventions include breathing exercises, holding one’s breath or breathing more slowly, biofeedback, counseling, and occasionally breathing into a paper bag. Medications may occasionally be necessary if there is a significant component of stress, anxiety, or depression. Beta blockers may also be of use in selective cases; however, there is no good evidence-based medicine to support any of these approaches. Signs and symptoms of hyperventilation continued to occur in adulthood in 40 percent in one series [Herman et al., 1981].
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Abboud F.M. Neurocardiogenic syncope. N Engl J Med. 1993;328:1117-1120.
Abu-Arafeh I., Russell G. Cyclical vomiting syndrome in children: a population-based study. J Pediatr Gastroenterol Nutr. 1995;21:454-458.
Abu-Arafeh I., Russell G. Paroxysmal vertigo as a migraine equivalent in children: a population-based study. Cephalalgia. 1995;15:22-25.
Ahn J.C., Hoyt W.F., Hoyt C.S. Tonic upgaze in infancy. A report of three cases. Arch Ophthalmol. 1989;107:57-58.
Aicardi J. Paroxysmal disorders other than epilepsy. In: Aicadi J., editor. Diseases of the nervous system in childhood. London: MacKeith Press, 1992.
Albright A.L., Sclabassi R.J., Slamovits T.L., et al. Spasmus nutans associated with optic gliomas in infants. J Pediatr. 1984;105:778-780.
Alehan D., Celiker A., Ozme S. Head-up tilt test: a highly sensitive, specific test for children with unexplained syncope. Pediatr Cardiol. 1996;17:86-90.
Allarakhia I.N., Trobe J.D. Opsoclonus-myoclonus presenting with features of spasmus nutans. J Child Neurol. 1995;10:67-68.
Andermann F., Andermann E. Startle disorders of man: hyperekplexia, jumping and startle epilepsy. Brain Dev. 1988;10:213-222.
Andermann F., Keene D.L., Andermann E., et al. Startle disease or hyperekplexia: further delineation of the syndrome. Brain. 1980;103:985-997.
Antony J.H., Ouvrier R.A., Wise G. Spasmus nutans: a mistaken identity. Arch Neurol. 1980;37:373-375.
Auburger G., Ratzlaff T., Lunkes A., et al. A gene for autosomal dominant paroxysmal choreoathetosis/spasticity (CSE) maps to the vicinity of a potassium channel gene cluster on chromosome 1p, probably within 2 cm between D1S443 and D1S197. Genomics. 1996;31:90-94.
Bain P.G., O’Brien M.D., Keevi S.F., et al. Familial periodic cerebellar ataxia: a problem of cerebellar intracellular pH homeostasis. Ann Neurol. 1992;31:147-154.
Baloh R.W., Yue Q., Furman J.M., et al. Familial episodic ataxia: clinical heterogeneity in four families linked to chromosome 19p. Ann Neurology. 1997;41:8-16.
Barron T.F., Younkin D.P. Propranolol therapy for shuddering attacks. Neurology. 1992;42:258-259.
Basser L.S. Benign paroxysmal vertigo of childhood. (A variety of vestibular neuronitis). Brain. 1964;87:141-152.
Batra A.S., Balaji S. Management of syncope in pediatric patients. Curr Treat Options in Cardiovasc Med. 2005;7:391-398.
Baup N., Grabli D., Karachi C., et al. High-frequency stimulation of the anterior subthalamic nucleus reduces stereotyped behaviors in primates. J Neurosci. 2008;28:8785-8788.
Benatar M. Neurological potassium channelopathies. QJM. 2000;93:787-797.
Benbadis S.R., Chichkova R. Psychogenic pseudosyncope: an underestimated and provable diagnosis. Epilepsy Behav. 2006;9:106-110.
Benditt D.G., Lurie K.G., Adler S.W., et al. Pathophysiology of vasovagal syncope. In: Blanc J.J., Benditt D., Sutton R., editors. Neurally mediated syncope: Pathophysiology, investigations, and treatment. Armonk, NY: Futura, 1996.
Bennett L.B., Roach E.S., Bowcock A.M. A locus for paroxysmal kinesigenic dyskinesia maps to human chromosome 16. Neurology. 2000;54:125-130.
Bhatia K.P. Familial (idiopathic) paroxysmal dyskinesias: an update. Sem Neurol. 2001;21:69-74.
Bhatia K.P., Griggs R.C., Ptacek L.J. Episodic movement disorders as channelopathies. Mov Disord. 2000;15:429-433.
Blakeley J., Jankovic J. Secondary causes of paroxysmal dyskinesia. Adv Neurol. 2002;89:401-420.
Bleasel A., Kotagal P. Paroxysmal nonepileptic disorders in children and adolescents. Sem Neurol. 1995;15:203-217.
Boehm K.E., Kip K.T., Grubb B.P., et al. Neurocardiogenic syncope: response to hormonal therapy. Pediatrics. 1997;99:623-625.
Boles R.G., Adams K., Ito M., et al. Maternal inheritance in cyclic vomiting syndrome with neuromuscular disease. Am J Med Genet. 2003;120A:474-482.
Boles R.G., Adams K., Li B.U. Maternal inheritance in cyclic vomiting syndrome. Am J Med Genet. 2005;133A:71-77.
Boles R.G., Powers A.L., Adams K. Cyclic vomiting syndrome plus. J Child Neurol. 2006;21:182-188.
Boles R.G., Williams J.C. Mitochondrial disease and cyclic vomiting syndrome. Dig Dis Sci. 1999;44:103S-107S.
Boltshauser E., Isler W. Joubert syndrome: episodic hyperpnea, abnormal eye movements, retardation and ataxia, associated with dysplasia of the cerebellar vermis. Neuropadiatrie. 1977;8:57-66.
Brandt T., Strupp M. Migraine and vertigo: Classification, clinical features and special treatment considerations. Headache Currens. 2006;3:12-19.
Bratt H.D., Menelaus M.B. Benign paroxysmal torticollis of infancy. J Bone Joint Surg Br. 1992;74:449-451.
Bray P.F., Herbst J.J., Johnson D.G., et al. Childhood gastroesophageal reflux. Neurologic and psychiatric syndromes mimicked. JAMA. 1977;237:1342-1345.
Bressman S.B., Fahn S., Burke R.E. Paroxysmal non-kinesigenic dystonia. Adv Neurol. 1988;50:403-413.
Brockmann K., Dumitrescu A.M., Best T.T., et al. X-linked paroxysmal dyskinesia and severe global retardation caused by defective MCT8 gene. J Neurol. 2005;252:663-666.
Brown P. Neurophysiology of the startle syndrome and hyperekplexia. Adv Neurol. 2002;89:153-159.
Bruno M.K., Hallett M., Gwinn-Hardy K., et al. Clinical evaluation of idiopathic paroxysmal kinesigenic dyskinesia: new diagnostic criteria. Neurology. 2004;63:2280-2287.
Bruno M.K., Lee H.Y., Auburger G.W., et al. Genotype-phenotype correlation of paroxysmal nonkinesigenic dyskinesia. Neurology. 2007;68:1782-1789.
Brunt E.R., van Weerden T.W. Familial paroxysmal kinesigenic ataxia and continuous myokymia. Brain. 1990;113:1361-1382.
Buruma O.J.S., Roos R.A.C.. Paroxysmal choreoathetosis. Vinken P.J., Bruyn G.W., Klawans H.L., editors. Handbook of Clinical Neurology. Extrapyramidal disorders. vol 49. Amsterdam: Elsevier; 1986. (Revised Series 5)
Bye A.M., Kok D.J., Ferenschild F.T., et al. Paroxysmal non-epileptic events in children: a retrospective study over a period of 10 years. J Paediatr Child Health. 2000;36:244-248.
Byrne E., White O., Cook M. Familial dystonic choreoathetosis with myokymia; a sleep responsive disorder. J Neurol Neurosurg Psychiatry. 1991;54:1090-1092.
Cader M.Z., Steckley J.L., Dyment D.A., et al. A genome-wide screen and linkage mapping for a large pedigree with episodic ataxia. Neurology. 2005;65:156-158.
Campbell D.B., North J.B., Hess E.J. Tottering mouse motor dysfunction is abolished on the Purkinje cell degeneration (pcd) mutant background. Exp Neurol. 1999;160:268-278.
Campistol J., Prats J.M., Garaizar C. Benign paroxysmal tonic upgaze of childhood with ataxia. A neuro-ophthalmological syndrome of familial origin? Devel Med Child Neurol. 1993;35:436-439.
Cannon S.C. Voltage-gated ion channelopathies of the nervous system. Clin Neurosci. 2001;1:104-117.
Caraballo R.H., Capovilla G., Vigevano F., et al. The spectrum of benign myoclonus of early infancy: Clinical and neurophysiologic features in 102 patients. Epilepsia. 2009;50:1176-1183.
Casteels K., Wouters C., Van Geet C., et al. Video reveals self-stimulation in infancy. Acta Paediatr. 2004;93:844-846.
Cataltepe S.U., Barron T.F. Benign paroxysmal torticollis presenting as “seizures” in infancy. Clin Pediatr. 1993;32:564-565.
Celesia G.G. Disorders of membrane channels or channelopathies. Clin Neurophysiol. 2001;112:2-18.
Chaves-Carballo E. Paroxysmal torticollis. Sem Pediatr Neurol. 1996;3:255-256.
Chaves-Carballo E., Dabbagh O., Essa M., et al. Neurological complications of hyperekplexia in infancy. Ann Neurol. 1999;46:466.
Chelimsky T.C., Chelimsky G.G. Autonomic abnormalities in cyclic vomiting syndrome. J Pediatr Gastroenterol Nutr. 2007;44:326-330.
Chen D.H., Matsushita M., Rainier S., et al. Presence of alanine-to-valine substitutions in myofibrillogenesis regulator 1 in paroxysmal nonkinesigenic dyskinesia: confirmation in 2 kindreds. Arch Neurol. 2005;62:597-600.
Chen G., Gao W., Reinert K.C., et al. Involvement of Kv1 potassium channels in spreading acidification and depression in the cerebellar cortex. J Neurophysiol. 2005;94:1287-1298.
Chillag K.L., Deroos S.T. Oxcarbazepine use in paroxysmal kinesigenic dyskinesia: report on four patients. Pediatr Neurol. 2009;40:295-297.
Chutorian A.M. Benign paroxysmal torticollis, tortipelvis and retrocillis in infancy. Neurology. 1974;24:366-367.
Clot F., Grabl D., Cazeneuve C., et al. Exhaustive analysis of BH4 and dopamine biosynthesis genes in patients with Dopa-responsive dystonia. Brain. 2009;132:1753-1763.
Cohen H.A., Nussinovitch M., Ashkenasi A., et al. Benign paroxysmal torticollis in infancy. Pediatr Neurol. 1993;9:488-490.
Crompton D.E., Berkovic S.F. The borderland of epilepsy: clinical and molecular features of phenomena that mimic epileptic seizures. Lancet Neurol. 2009;8:370-381.
Cuvellier J.-C., Lepine A. Childhood periodic syndromes. Pediatrc Neurolo. 2010;42:1-11.
Demirkiran M., Jankovic J. Paroxysmal dyskinesias: clinical features and classification. Ann Neurol. 1995;38:571-579.
Demos M.K., Fullston T., Partington M.W., et al. Clinical study of two brothers with a novel 33 bp duplication in the ARX gene. Am J Med Genet. 2009;149A:1482-1486.
Deonna T., Martin D. Benign paroxysmal torticollis in infancy. Arch Dis Child. 1981;56:956-959.
Deskin R.W. Sandifer syndrome: a cause of torticollis in infancy. Int J Pediatr Otorhinolaryngol. 1995;32:183-185.
Devanagondi R., Egami K., LeDoux M.S., et al. Neuroanatomical substrates for paroxysmal dyskinesia in lethargic mice. Neurobiol Dis. 2007;27:249-257.
Dignan F., Symon D.N., AbuArafeh I., et al. The prognosis of cyclical vomiting syndrome. Arch Dis Child. 2001;84:55-57.
Dijane P., Deharo J.-P., Macaluso G. Use of tilt table testing in clinical practice: Its role in the evaluation of syncope and dizziness. In: Blanc J.J., Benditt D., Sutton R., editors. Neurally mediated syncope: Pathophysiology, investigations, and treatment. Armonk, NY: Futura, 1996.
DiMario F.J.Jr. Childhood head tremor. J Child Neurol. 2000;15:22-25.
Dooley J.M., Andermann F. Startle disease or hyperekplexia: adolescent onset and response to valproate. Pediatr Neurol. 1989;5:126-127.
Dravet C., Giraud N., Bureau M., et al. Benign myoclonus of early infancy or benign non-epileptic infantile spasms. Neuropediatr. 1986;17:33-38.
Drigo P., Carli G., Laverda A.M. Benign paroxysmal torticollis of infancy. Brain Dev. 2000;22:169-172.
Drigo P., Carli G., Laverda A.M. Benign paroxysmal vertigo of childhood. Brain Dev. 2001;23:38-41.
Driscoll D.J., Jacobsen S.J., Porter C.J., et al. Syncope in children and adolescents. J Am Coll Card. 1997;29:1039-1045.
Ducros A., Denier C., Joutel A., et al. The clinical spectrum of familial hemiplegic migraine associated with mutations in a neuronal calcium channel. N Engl J Med. 2001;345:17-24.
Dumitrescu A.M., Liao X.H., Best T.B., et al. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet. 2004;74:168-175.
Dunn D.W., Snyder C.H. Benign paroxysmal vertigo of childhood. Am J Dis Child. 1976;130:1099-1100.
Du W., Bautista J.F., Yang H., et al. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet. 2005;37:733-738.
Eeg-Olofsson O., Odkvist L., Lindskog U., et al. Benign paroxysmal vertigo in childhood. Acta Otolaryngol. 1982;93:283-289.
Enzer N.B., Walker P.A. Hyperventilation syndrome in childhood. A review of 44 cases. J Pediatr. 1967;70:521-532.
Escayg A., De Waard M., Lee D.D., et al. Coding and noncoding variation of the human calcium-channel beta4-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am J Hum Genet. 2000;66:1531-1539.
Etzion Y., Grossman Y. Highly 4-aminopyridine sensitive delayed rectifier current modulates the excitability of guinea pig cerebellar Purkinje cells. Exp Brain Res. 2001;139:419-425.
Eulenburg V., Becker K., Gomeza J., et al. Mutations within the human GLYT2 (SLC6A5) gene associated with hyperekplexia. Biochem Biophys Res Commun. 2006;348:400-405.
Eunson L.H., Rea R., Zuberi S.M., et al. Clinical, genetic, and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability. Ann Neurol. 2000;48:647-656.
Evans R.W. Neurologic aspects of hyperventilation syndrome. Sem Neurol. 1995;15:115-125.
Eviatar L. Benign paroxysmal torticollis. Pediatr Neurol. 1994;11:72.
Farmer J., Hoyt C.S. Monocular nystagmus in infancy and early childhood. Am J Ophthalmol. 1984;98:504-509.
Farmer T.W., Mustian V.M. Vestibulocerebellar ataxia. A newly defined hereditary syndrome with periodic manifestations. Arch Neurol. 1963;8:471-480.
Feit L.R. Syncope in the pediatric patient: diagnosis, pathophysiology, and treatment. Adv Pediatr. 1996;43:469-494.
Fejerman N. Mioclonias benignas de la infancia temprana. Co-municacion preliminary. Actas IV Jornadas Rioplatenses de Neurologia Infantil (Montevideo-Uruguay). 1976.
Fejerman N. Non-epileptic neurologic paroxysmal disorders and episodic symptoms in infants. In: Pedley T.A., editor. Epilepsy: A Comprehensive Textbook. Philadelphia: Lippincott-Raven, 1997.
Fenichel G.M. Migraine as a cause of benign paroxysmal vertigo of childhood. J Pediatr. 1967;71:114-115.
Fernandez Sanmartin M., Rodriguez Nunez A., Martinon-Torres F., et al. Convulsive syncope: characteristics and reproducibility using the tilt test. An Pediatr (Barc). 2003;59:441-447.
Finkelhor B.K., Harker L.A. Benign paroxysmal vertigo of childhood. Laryngoscope. 1987;97:1161-1163.
Fink J.K., Rainer S., Wilkowski J., et al. Paroxysmal dystonic choreoathetosis: tight linkage to chromosome 2q. Am J Hum Genet. 1996;59:140-145.
Fitzpatrick A.P., Banner N., Cheng A., et al. Vasovagal reactions may occur after orthotopic heart transplantation. J Am Coll Cardiol. 1993;21:1132-1137.
Fitzpatrick E., Bourke B., Drumm B., et al. Outcome for children with cyclical vomiting syndrome. Arch Dis Child. 2007;92:1001-1004.
Fitzpatrick E., Bourke B., Drumm B., et al. The incidence of cyclic vomiting syndrome in children: population-based study. Am J Gastroenterol. 2008;103:991-995.
Fleisher D.R. The cyclic vomiting syndrome described. J Pediatr Gastroenterol Nutr. 1995;21(Suppl 1):S1-S5.
Fleisher D.R., Gornowicz B., Adams K., et al. Cyclic Vomiting Syndrome in 41 adults: the illness, the patients, and problems of management. BMC Med. 2005;3:20.
Fleisher D.R., Matar M. The cyclic vomiting syndrome: a report of 71 cases and literature review. J Pediatr Gastroenterol Nutr. 1993;17:361-369.
Fleisher D.R., Morrison A. Masturbation mimicking abdominal pain or seizures in young girls. J Pediatr. 1990;116:810-814.
Folks D.G. Munchausen’s syndrome and other factitious disorders. Neurol Clin. 1995;13:267-281.
Forbes D., Fairbrother S. Cyclic nausea and vomiting in childhood. Aust Fam Physician. 2008;37:33-36.
Forbes D., Withers G., Silburn S., et al. Psychological and social characteristics and precipitants of vomiting in children with cyclic vomiting syndrome. Dig Dis Sci. 1999;44(Suppl):19S-22S.
Fouad G.T., Servidei S., Durcan S., et al. A gene for familial paroxysmal dyskinesia (FPD1) maps to chromosome 2q. Am J Hum Genet. 1996;59:135-139.
Freeman R. A treatment for neurally mediated syncope? (Don’t) hold your breath. Ann Neurol. 2008;63:265-267.
Fuchs O., Pfarr N., Pohlenz J., et al. Elevated serum triiodothyronine and intellectual and motor disability with paroxysmal dyskinesia caused by a monocarboxylate transporter 8 gene mutation. Dev Med Child Neurol. 2009;51:240-244.
Furukawa Y., Mizuno Y., Nishi K., et al. A clue to the pathogenesis of dopa-responsive dystonia. Ann Neurol. 1995;37:139-140.
Galletti F., Brinciotti M., Emanuelli O. Familial occurrence of benign myoclonus of early infancy. Epilepsia. 1989;30:579-581.
Gastaut H.. Syncope: Generalized anoxic cerebral seizures. Vinken P.J., Bruyn G.W., editors. Handbook of Clinical Neurology. The Epilepsies. vol 15. Amsterdam: North-Holland; 1974.
Gee S. On Fitful or Recurrent Vomiting. Saint Bartholomew’s Hospital Reports. 1882;18:1-6.
Gellis S.S., Feingold M. Syndrome of hiatus hernia with torsion spasms and abnormal posturing (Sandifer’s syndrome). Am J Dis Child. 1971;121:53-54.
Giacoia G.P., Ryan S.G. Hyperekplexia associated with apnea and sudden infant death syndrome. Arch Pediatr Adolesc Med. 1994;148:540-543.
Giffin N.J., Benton S., Goadsby P.J. Benign paroxysmal torticollis of infancy: four new cases and linkage to CACNA1A mutation. Dev Med Child Neurol. 2002;44:490-493.
Gilbert G.J. Familial spasmodic torticollis. Neurology. 1977;27:11-13.
Gilles de la Tourette G. Jumping. latah, myriachit. Arch Neurol. 1884;8:68.
Giurgea I., Missirian C., Cacciagli P., et al. TCF4 deletions in Pitt-Hopkins Syndrome. Hum Mutat. 2008;29:E242-E251.
Glees de la Tuorette G. La maladie des tics convulsifs. La Semaine Medicale. 1899;19:153.
Golden G.S., French J.H. Basilar artery migraine in young children. Pediatr. 1975;56:722-726.
Goldman S., Wang C., Salgado M.W., et al. Motor stereotypies in children with autism and other developmental disorders. Dev Med Child Neurol. 2009;51:30-38.
Gordon N. Startle disease or hyperekplexia. Dev Med Child Neurol. 1993;35:1015-1018.
Gorrotxategi P., Reguilon M.J., Arana J., et al. Gastroesophageal reflux in association with the Sandifer syndrome. Eur J Pediatr Surg. 1995;5:203-205.
Gottlob I., Wizov S.S., Reinecke R.D. Quantitative eye and head movement recordings of retinal disease mimicking spasmus nutans. Am J Ophthal. 1995;119:374-376.
Gottlob I., Wizov S.S., Reinecke R.D. Spasmus nutans. A long-term follow-up. Invest Ophthalmol Vis Sci. 1995;36:2768-2771.
Gottlob I., Zubcov A.A., Wizov S.S., et al. Head nodding is compensatory in spasmus nutans. Ophthalmol. 1992;99:1024-1031.
Gottlob I., Zubcov A., Catalano R.A., et al. Signs distinguishing spasmus nutans (with and without central nervous system lesions) from infantile nystagmus. Ophthalmol. 1990;97:1166-1175.
Griggs R.C., Moxley R.T.3rd, Lafrance R.A., et al. Hereditary paroxysmal ataxia: response to acetazolamide. Neurology. 1978;28:1259-1264.
Grubb B.P., Gerard G., Roush K., et al. Cerebral vasoconstriction during head-upright tilt-induced vasovagal syncope. A paradoxic and unexpected response. Circulation. 1991;84:1157-1164.
Grubb B.P., Kosinski D. Current trends in etiology, diagnosis, and management of neurocardiogenic syncope. Curr Opin Cardiol. 1996;11:32-41.
Grubb B.P., Orecchio E., Kurczynski T.W. Head-upright tilt table testing in evaluation of recurrent, unexplained syncope. Pediatr Neurol. 1992;8:423-427.
Guerrini R., Belmonte A., Carrozzo R. Paroxysmal tonic upgaze of childhood with ataxia: a benign transient dystonia with autosomal dominant inheritance. Brain Dev. 1998;20:116-118.
Guerrini R., Bonanni P., Nardocci N., et al. Autosomal recessive rolandic epilepsy with paroxysmal exercise-induced dystonia and writer’s cramp: delineation of the syndrome and gene mapping to chromosome 16p12–11.2. Ann Neurol. 1999;45:344-352.
Hallett M., Marsden C.D., Fahn S.. Myoclonus. Vinken P.J., Bruyn, Klawans H.L., et al. Handbook of Clinical Neurology. Extrapyramidal disorders. vol 49. Amsterdam: Elsevier; 1986. (Revised series 5)
Hamann M., Richter A. Striatal increase of extracellular dopamine levels during dystonic episodes in a genetic model of paroxysmal dyskinesia. Neurobiol Dis. 2004;16:78-84.
Hanna D.E., Hodgens J.B., Daniel W.A.Jr. Hyperventilation syndrome. Pediatr Ann. 1986;15:708-712.
Hannon D.W., Knilans T.K. Syncope in children and adolescents. Current Probl Pediatr. 1993;23:358-384.
Hanson P.A., Martinez L.B., Cassidy R. Contractures, continuous muscle discharges, and titubation. Ann Neurol. 1977;1:120-124.
Hanukoglu A., Somekh E., Fried D. Benign paroxysmal torticollis in infancy. Clin Pediatr. 1984;23:272-274.
Harno H., Heikkinen S., Kaunisto M.A., et al. Decreased cerebellar total creatine in episodic ataxia type 2: a 1H MRS study. Neurology. 2005;64:542-544.
Harris K.M., Mahone E.M., Singer H.S. Nonautistic motor stereotypies: clinical features and longitudinal follow-up. Pediatr Neurol. 2008;38:267-272.
Harvey K., Duguid I.C., Alldred M.J., et al. The GDP-GTP exchange factor collybistin: an essential determinant of neuronal gephyrin clustering. J Neuroscience. 2004;24:5816-5826.
Hayman M., Harvey A.S., Hopkins I.J., et al. Paroxysmal tonic upgaze: a reappraisal of outcome. Ann Neurol. 1998;43:514-520.
Headache Classification Committee. The international classification of headache disorders, cranial neuralgia and facial pain, 2nd edn. Cephalgia. 2004;24(Suppl 1):1-160.
Hejazi N.S., Chaves E.C., Boumah C., et al. Linkage of hyperekplexia (HEK) in three Sausi families to chromosome 4q31.3 associated with the glycine receptor beta subunit (glrβ). Neurology. 2001;56(Suppl 3):A132.
Hempelmann A., Kumar S., Muralitharan S., et al. Myofibrillogenesis regulator 1 gene (MR-1) mutation in an Omani family with paroxysmal nonkinesigenic dyskinesia. Neurosci Lett. 2006;402:118-120.
Herman S.P., Stickler G.B., Lucas A.R. Hyperventilation syndrome in children and adolescents: long-term follow-up. Pediatrics. 1981;67:183-187.
Hikita T., Kodama H., Nakamoto N., et al. Effective prophylactic therapy for cyclic vomiting syndrome in children using valproate. Brain Dev. 2009;31:411-413.
Hoefnagel D., Biery B. Spasmus nutans. Dev Med Child Neurol. 1968;10:32-35.
Holmes G.L., Russman B.S. Shuddering attacks. Evaluation using electroencephalographic frequency modulation radiotelemetry and videotape monitoring. Am J Dis Child. 1986;140:72-73.
Hornsveld H.K., Garssen B., Dop M.J., et al. Double-blind placebo-controlled study of the hyperventilation provocation test and the validity of the hyperventilation syndrome. Lancet. 1996;348:154-158.
Houser M.K., Soland V.L., Bhatia K.P., et al. Paroxysmal kinesigenic choreoathetosis: a report of 26 patients. J Neurol. 1999;246:120-126.
Igarashi M., Boehm R.Jr, May W.N., et al. Syncope associated with hair-grooming. Brain Dev. 1988;10:249-251.
Iivanainen M., Kaakkola S. Dopa-responsive dystonia of childhood. Dev Med Child Neurol. 1993;35:362-367.
Jankovic J., Demirkiran M. Classification of paroxysmal dyskinesias and ataxias. Adv Neurol. 2002;89:387-400.
Jarman P.R., Davis M.B., Hodgson S.V., et al. Paroxysmal dystonic choreoathetosis. Genetic linkage studies in a British family. Brain. 1997;120:2125-2130.
Jen J.C., Wan J., Palos T.P., et al. Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology. 2005;65:529-534.
Jen J., Kim G.W., Baloh R.W. Clinical spectrum of episodic ataxia type 2. Neurology. 2004;62:17-22.
Jen J.C., Graves T.D., Hess E., et al. Primary episodic ataxias: Diagnosis, pathogenisis and treatment. Brain. 2007;130:2484-2493.
Joorabchi B. Expressions of the hyperventilation syndrome in childhood: studies in management, including an evaluation of the effectiveness of propranolol. Clin Pediatr. 1977;16:1110-1115.
Joseph K., Avallone J., Difazio M. Paroxysmal tonic upgaze and partial tetrasomy of chromosome 15: a novel genetic association. J Child Neurol. 2005;20:165-168.
Joubert M., Eisenring J.J., Robb J.P., et al. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 1969;19:813-825.
Joutel A., Bousser M.G., Biousse V., et al. A gene for familial hemiplegic migraine maps to chromosome 19. Nat Gen. 1993;5:40-45.
Kabakus N., Kurt A. Sandifer Syndrome: a continuing problem of misdiagnosis. Pediatrics International. 2006;48:622-625.
Kanazawa O. Shuddering attacks – report of four children. Pediatr Neurol. 2000;23:421-424.
Kapoor W.N. Diagnostic evaluation of syncope. Am J Med. 1991;90:91-106.
Kapoor W.N. Syncope. N Engl J Med. 2000;343:1856-1862.
Kapoor W.N. Using a tilt table to evaluate syncope. Am J Med Sci. 1999;317:110-116.
Kapoor W.N., Karpf M., Wieand S., et al. A prospective evaluation and follow-up of patients with syncope. N Engl J Med. 1983;309:197-204.
Kapoor W.N., Smith M.A., Miller N.L. Upright tilt testing in evaluating syncope: a comprehensive literature review. Am J Med. 1994;97:78-88.
Kaufmann H. Evaluation of the patient with syncope. In Robertson D., Biaggioni I., Burnstock G., et al, editors: Primer on the autonomic nervous system, ed 2, San Diego, CA: Elsevier Academic Press, 2004.
Kaufmann H., Freeman R. Pharmacological treatment of reflex syncope. Clin Auton Res. 2004;14(Suppl 1):71-75.
Kelly T.W. Optic glioma presenting as spasmus nutans. Pediatrics. 1970;45:295-296.
Kenny R.A., Ingram A., Bayliss J., et al. Head-up tilt: a useful test for investigating unexplained syncope. Lancet. 1986;1:1352-1355.
Kerber K.A., Jen J.C., Lee H., et al. A new episodic ataxia syndrome with linkage to chromosome 19q13. Arch Neurol. 2007;64:749-752.
Kerr A.M., Julu P.O. Recent insights into hyperventilation from the study of Rett syndrome. Arch Dis Child. 1999;80:384-387.
Kerr W.J., Gliebe P.A., Dalton J.W. Physical Phenomena Associated with Anxiety States: The Hyperventilation Syndrome. Cal West Med. 1938;48:12-16.
Kertesz A. Paroxysmal kinesigenic choreoathetosis. An entity within the paroxysmal choreoathetosis syndrome. Description of 10 cases, including 1 autopsied. Neurology. 1967;17:680-690.
Kiblinger G.D., Wallace B.S., Hines M., et al. Spasmus nutans-like nystagmus is often associated with underlying ocular, intracranial, or systemic abnormalities. J Neuroophthalmol. 2007;27:118-122.
Kinast M., Erenberg G., Rothner A.D. Paroxysmal choreoathetosis: report of five cases and review of the literature. Pediatrics. 1980;65:74-77.
Kinsbourne M. Hiatus Hernia with Contortions of the Neck. Lancet. 1964;1:1058-1061.
Kinsella S.M., Tuckey J.P. Perioperative bradycardia and asystole: relationship to vasovagal syncope and the Bezold-Jarisch reflex. Br J Anaesth. 2001;86:859-868.
Kirstein L., Silfverskiold B.P. A family with emotionally precipitated drop seizures. Acta Psychiatr Neurol Scand. 1958;33:471-476.
Klein A., Boltshauser E., Jen J., et al. Episodic ataxia type 1 with distal weakness: a novel manifestation of a potassium channelopathy. Neuropediatrics. 2004;35:147-149.
Klein R., Haddow J.E., DeLuca C. Familial congenital disorder resembling stiff-man syndrome. Am J Dis Child. 1972;124:730-731.
Ko C.H., Kong C.K., Ngai W.T., et al. Ictal (99m)Tc ECD SPECT in paroxysmal kinesigenic choreoathetosis. Pediatr Neurol. 2001;24:225-227.
Koehler B. Benign paroxysmal vertigo of childhood: a migraine equivalent. Eur J Pediatr. 1980;134:149-151.
Koenigsberger M.R., Chutorian A.M., Gold A.P., et al. Benign paroxysmal vertigo of childhood. Neurology. 1968;18:301-302.
Kosinski D., Grubb B.P., Temesy-Armos P. Pathophysiological aspects of neurocardiogenic syncope: current concepts and new perspectives. Pacing Clin Electrophysiol. 1995;18:716-724.
Kosinski D.J., Grubb B.P. Miscellaneous causes of syncope. In Grubb B.P., Olshanky B., editors: Syncope, mechanisms and management, ed 2, Malden, MA: Blackwell Publishing, 2005.
Kurczynski T.W. Hyperekplexia. Arch Neurol. 1983;40:246-248.
Lambert S.R., Newman N.J. Retinal disease masquerading as spasmus nutans. Neurology. 1993;43:1607-1609.
Lance J.W. Familial paroxysmal dystonic choreoathetosis and its differentiation from related syndromes. Ann Neurol. 1977;2:285-293.
Landau W.M., Nelson D.A. Clinical neuromythology XV. Feinting science: Neurocardiogenic syncope and collateral vasovagal confusion. Neurology. 1996;46:609-618.
Lanzi G., Balottin U., Fazzi E., et al. Benign paroxysmal vertigo of childhood: a long-term follow-up. Cephalalgia. 1994;14:458-460.
Laskey A.L., Korn D.E., Moorjani B.I., et al. Central hyperventilation related to administration of topiramate. Pediatr Neurol. 2000;22:305-308.
Lazarus J.C., Mauro V.F. Syncope: pathophysiology, diagnosis, and pharmacotherapy. Ann Pharmacother. 1996;30:994-1005.
Lee H.Y., Xu Y., Huang Y., et al. The gene for paroxysmal non-kinesigenic dyskinesia encodes an enzyme in a stress response pathway. Hum Mol Genet. 2004;13:3161-3170.
Lee M.S., Kim W.C., Lyoo C.H., et al. Reciprocal inhibition between the forearm muscles in patients with paroxysmal kinesigenic dyskinesia. J Neurolog Sci. 1999;168:57-61.
Lehwald N., Krausch M., Franke C., et al. Sandifer syndrome – a multidisciplinary diagnostic and therapeutic challenge. Eur J Pediatr Surg. 2007;17:203-206.
Lempert T., Neuhauser H., Daroff R.B. Vertigo as a symptom of migraine. Ann N Y Acad Sci. 2009;1164:242-251.
Leor J., Rotstein Z., Vered Z., et al. Absence of tachycardia during tilt test predicts failure of beta-blocker therapy in patients with neurocardiogenic syncope. Am Heart J. 1994;127:1539-1543.
Lerman-Sagie T., Lerman P., Mukamel M., et al. A prospective evaluation of pediatric patients with syncope. Clin Pediatr. 1994;33:67-70.
Leung A.K., Robson W.L. Childhood masturbation. Clin Pediatr. 1993;32:238-241.
Lewis B.I. The hyperventilation syndrome. Ann Int Med. 1953;38:918-927.
Lewis D.W., Frank L.M. Hair-grooming syncope seizures. Pediatrics. 1993;91:836-838.
Lewis R.A., Howell J.B. Definition of the hyperventilation syndrome. Bull Eur Physiopathol Respir. 1986;22:201-205.
Lewis T. A lecture on vasovagal syncope and the carotid sinus mechanism with comments on Gowers’ and Nothnagel’s syndrome. BMJ. 1932;1:873.
Li B.U. Cyclic vomiting syndrome: age-old syndrome and new insights. Sem Pediatr Neurol. 2001;8:13-21.
Li B.U., Lefevre F., Chelimsky G.G., et al. North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition consensus statement on the diagnosis and management of cyclic vomiting syndrome. J Pediatr Gastroenterol Nutr. 2008;47:379-393.
Li B.U., Misiewicz L. Cyclic vomiting syndrome: a brain-gut disorder. Gastroenterol Clin N Am. 2003;32:997-1019.
Lindskog U., Odkvist L., Noaksson L., et al. Benign paroxysmal vertigo in childhood: a long-term follow-up. Headache. 1999;39:33-37.
Lingam S., Wilson J., Hart E.W. Hereditary stiff-baby syndrome. Am J Dis Child. 1981;135:909-911.
Lin J.T., Ziegler D.K., Lai C.W., et al. Convulsive syncope in blood donors. Ann Neurol. 1982;11:525-528.
Lipson E.H., Robertson W.C.Jr. Paroxysmal torticollis of infancy: familial occurrence. Am J Dis Child. 1978;132:422-423.
Lispi M.L., Vigevano F. Benign paroxysmal tonic upgaze of childhood with ataxia. Epileptic Dis. 2001;3:203-206.
Lombroso C.T. Paroxysmal choreoathetosis: an epileptic or non-epileptic disorder? Ital J Neurol Sci. 1995;16:271-277.
Lombroso C.T., Fejerman N. Benign myoclonus of early infancy. Ann Neurol. 1977;1:138-143.
Longin E., Reinhard J., von Buch C., et al. Autonomic function in children and adolescents with neurocardiogenic syncope. Pediatr Cardiol. 2008;29:763-770.
Luat A.F., Asano E., Chugani H.T. Paroxysmal tonic upgaze of childhood with co-existent absence epilepsy. Epileptic Dis. 2007;9:332-336.
Lubbers W.J., Brunt E.R., Scheffer H., et al. Hereditary myokymia and paroxysmal ataxia linked to chromosome 12 is responsive to acetazolamide. J Neurol Neurosurg Psychiatry. 1995;59:400-405.
Luders H.O. Paroxysmal choreoathetosis. Eur Neurol. 1996;36(Suppl 1):20-23.
Mandel H., Tirosh E., Berant M. Sandifer syndrome reconsidered. Acta Paediatr Scand. 1989;78:797-799.
Manolis A.S., Linzer M., Salem D., et al. Syncope: current diagnostic evaluation and management. Ann Int Med. 1990;112:850-863.
Mansourati J., Blanc J.J. Tilt test procedure: Angle, duration, positive criteria. In: Blanc J.J., Benditt D., Sutton R., editors. Neurally mediated syncope: Pathophysiology, investigations, and treatment. Armonk, NY: Futura, 1996.
Margari L., Perniola T., Illiceto G., et al. An uncommon disorder: Familial paroxysmal exercise-induced dyskinesia and benign epilepsy. A clinical and neurophysiological study. Neurol Rev J. 2002;2:8-11.
Margari L., Perniola T., Illiceto G., et al. Familial paroxysmal exercise-induced dyskinesia and benign epilepsy: a clinical and neurophysiological study of an uncommon disorder. Neurolog Sci. 2000;21:165-172.
Margari L., Presicci A., Ventura P., et al. Channelopathy: hypothesis of a common pathophysiologic mechanism in different forms of paroxysmal dyskinesia. Pediatr Neurol. 2005;32:229-235.
Maydell B.V., Berenson F., Rothner A.D., et al. Benign myoclonus of early infancy: an imitator of West’s syndrome. J Child Neurol. 2001;16:109-112.
McLeod K.A. Syncope in childhood. Arch Dis Child. 2003;88:350-353.
Mets M. Tonic upgaze in infancy. Arch Ophthalmol. 1990;108:482-483.
Mierzwinski J., Polak M., Dalke K., et al. Benign paroxysmal vertigo of childhood. Otolaryngol Pol. 2007;61:307-310.
Miller J.M., Singer H.S., Bridges D.D., et al. Behavioral therapy for treatment of stereotypic movements in nonautistic children. J Child Neurol. 2006;21:119-125.
Milstein S., Buetikofer J., Dunnigan A., et al. Usefulness of disopyramide for prevention of upright tilt-induced hypotension-bradycardia. Am J Cardiol. 1990;65:1339-1344.
Mink J.W., Neil J.J. Masturbation mimicking paroxysmal dystonia or dyskinesia in a young girl. Mov Disord. 1995;10:518-520.
Mira E., Piacentino G., Lanzi G., et al. Benign paroxysmal vertigo in childhood. Diagnostic significance of vestibular examination and headache provocation tests. Acta Otolaryngol Suppl. 1984;406:271-274.
Mira E., Piacentino G., Lanzi G, et al. Benign paroxysmal vertigo in childhood: a migraine equivalent. ORL. 1984;46:97-104.
Mount L.A., Reback A. Familial paroxysmal choreoathetosis: Preliminary report on a hitherto undescribed clinical syndrome. Arch Neurol. 1940;44:841.
Moya A., Permanyer-Miralda G., Sagrista J., et al. Is there a role for tilt testing in the evaluation of treatment of vasovagal syncope? In: Blanc J.J., Benditt D., Sutton R., editors. Neurally mediated syncope: Pathophysiology, investigations, and treatment. Armonk, NY: Futura, 1996.
Moya A., Sgrista J., Permanyer-Miralda G., et al. Isoproterenol and tilt test: Protocol, importance, containdications. In: Blanc J.J., Benditt D., Sutton R., editors. Neurally mediated syncope: Pathophysiology, investigations, and treatment. Armonk, NY: Futura, 1996.
Munchau A., Valente E.M., Shahidi G.A., et al. A new family with paroxysmal exercise induced dystonia and migraine: a clinical and genetic study. J Neurol Neurosurg Psych. 2000;68:609-614.
Murakami Y., Yamashita Y., Matsuishi T., et al. Cerebral oxygenation and hemodynamics during hyperventilation and sleep in patients with Rett syndrome. Brain Dev. 1998;20:574-578.
Murphy W.J., Gellis S.S. Torticollis with hiatus hernia in infancy. Sandifer syndrome. Am J Dis Child. 1977;131:564-565.
Nechay A., Ross L.M., Stephenson J.B., et al. Gratification disorder (“infantile masturbation”): a review. Arch Dis Child. 2004;89:225-226.
Nigro M.A., Lim H.C. Hyperekplexia and sudden neonatal death. Pediatr Neurol. 1992;8:221-225.
Njemanze P.C. Isoproterenol induced cerebral hypoperfusion in a heart transplant recipient. Pacing Clin Electrophysiol. 1993;16:491-495.
Noone P.G., King M., Loftus B.G. Benign neonatal sleep myoclonus. Irish Med J. 1995;88:172.
Norton E.W., Cogan D.G. Spasmus nutans; a clinical study of twenty cases followed two years or more since onset. Arch Ophthalmol. 1954;52:442-446.
Nygaard T.G. Dopa-responsive dystonia. Curr Opin Neurol. 1995;8:310-313.
Nygaard T.G. Dopa-responsive dystonia. Delineation of the clinical syndrome and clues to pathogenesis. Adv Neurol. 1993;60:577-585.
Nygaard T.G., Waran S.P., Levine R.A., et al. Dopa-responsive dystonia simulating cerebral palsy. Pediatr Neurol. 1994;11:236-240.
Olshansky B. Syncope: Overview and approach to management. In Grubb B.P., Olshanky B., editors: Syncope, mechanisms and management, ed 2, Malden, MA: Blackwell Publishing, 2005.
Ophoff R.A., Terwindt G.M., Vergouwe M.N., et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543-552.
Ouvrier R.A., Billson F. Benign paroxysmal tonic upgaze of childhood. J Child Neurol. 1988;3:177-180.
Ouvrier R., Billson F. Paroxysmal tonic upgaze of childhood – a review. Brain Dev. 2005;27:185-188.
Pachatz C., Fusco L., Vigevano F. Benign myoclonus of early infancy. Epileptic Dis. 1999;1:57-61.
Perez Plasencia D., Beltran Mateos L.D., del Canizo Alvarez A., et al. Benign Paroxysmal Vertigo in Childhood. Acta Otorrinolaringol Esp. 1998;49:151-155.
Perkin G.D., Joseph R. Neurological manifestations of the hyperventilation syndrome. J Royal Soc Med. 1986;79:448-450.
Pfau B.T., Li B.U., Murray R.D., et al. Differentiating cyclic from chronic vomiting patterns in children: quantitative criteria and diagnostic implications. Pediatrics. 1996;97:364-368.
Philippi H., Boor R., Reitter B. Topiramate and metabolic acidosis in infants and toddlers. Epilepsia. 2002;43:744-747.
Phillips H.A., Favre I., Kirkpatrick M., et al. CHRNB2 is the second acetylcholine receptor subunit associated with autosomal dominant nocturnal frontal lobe epilepsy. Am J Hum Genet. 2001;68:225-231.
Pitt D., Hopkins I. A syndrome of mental retardation, wide mouth and intermittent overbreathing. Aust Paediatr J. 1978;14:182-184.
Plant G.T., Williams A.C., Earl C.J., et al. Familial paroxysmal dystonia induced by exercise. J Neurol Neurosurg Psychiatry. 1984;47:275-279.
Prakash C., Clouse R.E. Cyclic vomiting syndrome in adults: clinical features and response to tricyclic antidepressants. Am J Gastroenter. 1999;94:2855-2860.
Prakash C., Staiano A., Rothbaum R.J., et al. Similarities in cyclic vomiting syndrome across age groups. Am J Gastroenter. 2001;96:684-688.
Praveen V., Patole S.K., Whitehall J.S. Hyperekplexia in neonates. Postgrad Med J. 2001;77:570-572.
Pronicka E., Piekutowska-Abramczuk D.H., Popowska E., et al. Compulsory hyperventilation and hypocapnia of patients with Leigh syndrome associated with SURF1 gene mutations as a cause of low serum bicarbonates. J Inher Met Dis. 2001;24:707-714.
Pryles C.V., Livingston S., Ford F.R. Familial paroxysmal choreoathetosis of Mount and Reback; study of a second family in which this condition is found in association with epilepsy. Pediatrics. 1952;9:44-47.
Przuntek H., Monninger P. Therapeutic aspects of kinesiogenic paroxysmal choreoathetosis and familial paroxysmal choreoathetosis of the Mount and Reback type. J Neurol. 1983;230:163-169.
Ptacek L.J. Channelopathies: ion channel disorders of muscle as a paradigm for paroxysmal disorders of the nervous system. Dig Dis Sci. 1999;44(Suppl):94S-96S.
Ptacek L.J., Fu Y.H. Channelopathies: episodic disorders of the nervous system. Epilepsia. 2001;42(Suppl):35-43.
Rainier S., Thomas D., Tokarz D., et al. Myofibrillogenesis regulator 1 gene mutations cause paroxysmal dystonic choreoathetosis. Arch Neurol. 2004;61:1025-1029.
Rajakulendran S, Schorge S, Kullmann DM, et al: Episodic ataxia type 1: a neuronal potassium channelopathy, Neurotherapeutics 4:258–266, 277
Rajendra S., Lynch J.W., Schofield P.R. The glycine receptor. Pharmacol Ther. 1997;73:121-146.
Rajput A.H., Gibb W.R., Zhong X.H., et al. Dopa-responsive dystonia: pathological and biochemical observations in a case. Ann Neurol. 1994;35:396-402.
Rashed H., Abell T.L., Familoni B.O., et al. Autonomic function in cyclic vomiting syndrome and classic migraine. Dig Dis Sci. 1999;44(Suppl):74S-78S.
Raskind W.H., Bolin T., Wolff J., et al. Further localization of a gene for paroxysmal dystonic choreoathetosis to a 5-cm region on chromosome 2q34. Hum Genet. 1998;102:93-97.
Raviele A., Themistoclakis S., Gasparini G. Drug treatment of vasovagal syncope. In: Blanc J.J., Benditt D., Sutton R., editors. Neurally mediated syncope: Pathophysiology, investigations, and treatment. Armonk, NY: Futura, 1996.
Rea R.F., Thames M.D. Neural control mechanisms and vasovagal syncope. J Cardiovasc Electrophysiol. 1993;4:587-595.
Rees M.I., Harvey K., Ward H., et al. Isoform heterogeneity of the human gephyrin gene (GPHN), binding domains to the glycine receptor, and mutation analysis in hyperekplexia. J Biolog Chem. 2003;278:24688-24696.
Rees M.I., Lewis T.M., Kwok J.B., et al. Hyperekplexia associated with compound heterozygote mutations in the beta-subunit of the human inhibitory glycine receptor (GLRB). Hum Mol Genet. 2002;11:853-860.
Reif-Lehrer L., Stemmermann M.G. Monosodium glutamate intolerance in children. N Engl J Med. 1975;293:1204-1205.
Richter A., Sander S.E., Rundfeldt C. Antidystonic effects of Kv7 (KCNQ) channel openers in the dt sz mutant, an animal model of primary paroxysmal dystonia. Br J Pharmacol. 2006;149:747-753.
Rochette J., Roll P., Szepetowski P. Genetics of infantile seizures with paroxysmal dyskinesia: the infantile convulsions and choreoathetosis (ICCA) and ICCA-related syndromes. J Med Genet. 2008;45:773-779.
Rodriguez-Nunez A., Couceiro J., Alonso C., et al. Cerebral oxygenation in children with syncope during head-upright tilt test. Pediatr Cardiol. 1997;18:406-409.
Rosman N.P., Douglass L.M., Sharif U.M., et al. The neurology of benign paroxysmal torticollis of infancy: report of 10 new cases and review of the literature. J Child Neurol. 2009;24:155-160.
Roubertie A., Echenne B., Leydet J., et al. Benign paroxysmal tonic upgaze, benign paroxysmal torticollis, episodic ataxia and CACNA1A mutation in a family. J Neurol. 2008;255:1600-1602.
Ryan S.G., Sherman S.L., Terry J.C., et al. Startle disease, or hyperekplexia: response to clonazepam and assignment of the gene (STHE) to chromosome 5q by linkage analysis. Ann Neurol. 1992;31:663-668.
Sallustro F., Atwell C.W. Body rocking, head banging, and head rolling in normal children. J Pediatr. 1978;93:704-708.
Sanger T.D. Pathophysiology of pediatric movement disorders. J Child Neurol. 2003;18(Suppl 1):S9-S24.
Sanner G., Bergstrom B. Benign paroxysmal torticollis in infancy. Acta Paediatr Scand. 1979;68:219-223.
Sapin S.O. Autonomic syncope in pediatrics: a practice-oriented approach to classification, pathophysiology, diagnosis, and management. Clin Pediatr. 2004;43:17-23.
Savage D.D., Corwin L., McGee D.L., et al. Epidemiologic features of isolated syncope: the Framingham Study. Stroke. 1985;16:626-629.
Schiller A., Wevers R.A., Steenbergen G.C., et al. Long-term course of L-dopa-responsive dystonia caused by tyrosine hydroxylase deficiency. Neurol. 2004;63:1524-1526.
Scott W.A., Pongiglione G., Bromberg B.I., et al. Randomized comparison of atenolol and fludrocortisone acetate in the treatment of pediatric neurally mediated syncope. Am J Cardiol. 1995;76:400-402.
Segawa M. Hereditary progressive dystonia with marked diurnal fluctuation. Brain Dev. 2000;22(Suppl 1):S65-S80.
Segawa M., Nomura Y., Kase M.. Diurnally fluctuating hereditary progressive dystonia. Vinken P.J., Bruyn G.W., Klawans H.L., editors. Handbook of Clinical Neurology, vol 5. Amsterdam: Elsevier, 1986. (Revised Series)
Segawa M., Nomura Y., Nishiyama N. Autosomal dominant guanosine triphosphate cyclohydrolase I deficiency (Segawa disease). Ann Neurol. 2003;54(Suppl 6):S32-S45.
Shahar E., Brand N., Uziel Y., et al. Nose tapping test inducing a generalized flexor spasm: a hallmark of hyperexplexia. Acta Paediatr Scand. 1991;80:1073-1077.
Shahar E., Raviv R. Sporadic major hyperekplexia in neonates and infants: clinical manifestations and outcome. Pediatr Neurol. 2004;31:30-34.
Shapiro M.S., Gomeza J., Hamilton S.E., et al. Identification of subtypes of muscarinic receptors that regulate Ca2+ and K+ channel activity in sympathetic neurons. Life Sci. 2001;68:2481-2487.
Sheldon R.S., Sheldon A.G., Connolly S.J., et al. Age of first faint in patients with vasovagal syncope. J Cardiovasc Electrophysiol. 2006;17:49-54.
Shen W.-K., Gersh B.J. Syncope: mechanisms, approach, and management. In: Low P.A., editor. Clinical autonomic disorders. Boston, MA: Little Brown & Co, 1993.
Shepherd R.W., Wren J., Evans S., et al. Gastroesophageal reflux in children. Clinical profile, course and outcome with active therapy in 126 cases. Clin Pediatr. 1987;26:55-60.
Shiang R., Ryan S.G., Zhu Y.Z., et al. Mutational analysis of familial and sporadic hyperekplexia. Ann Neurol. 1995;38:85-91.
Shiang R., Ryan S.G., Zhu Y.Z., et al. Mutations in the alpha 1 subunit of the inhibitory glycine receptor cause the dominant neurologic disorder, hyperekplexia. Nat Genet. 1993;5:351-358.
Singer H.S. Motor stereotypies. Semin Pediatr Neurol. 2009;16:77-81.
Small K.W., Pollock S.C., Vance J.M., et al. Ocular motility in North Carolina autosomal dominant ataxia. J Neuroophthalmol. 1996;16:91-95.
Smart S.L., Lopantsev V., Zhang C.L., et al. Deletion of the K(V)1.1 potassium channel causes epilepsy in mice. Neuron. 1998;20:809-819.
Smith D.E., Fitzgerald K., Stass-Isern M., et al. Electroretinography is necessary for spasmus nutans diagnosis. Pediatr Neurol. 2000;23(1):33-36.
Sneddon J.F., Camm A.J. Vasovagal syncope: classification, investigation and treatment. Br J Hosp Med. 1993;49:329-334.
Snow B.J., Nygaard T.G., Takahashi H., et al. Positron emission tomographic studies of dopa-responsive dystonia and early-onset idiopathic parkinsonism. Ann Neurol. 1993;34:733-738.
Snyder C.H. Paroxysmal torticollis in infancy. A possible form of labyrinthitis. Am J Dis Child. 1969;117:458-460.
Southall D.P., Kerr A.M., Tirosh E., et al. Hyperventilation in the awake state: potentially treatable component of Rett syndrome. Arch Dis Child. 1988;63:1039-1048.
Spacey S.D., Adams P.J., Lam P.C., et al. Genetic heterogeneity in paroxysmal nonkinesigenic dyskinesia. Neurol. 2006;66:1588-1590.
Spalice A., Parisi P., Iannetti P. Paroxysmal tonic upgaze: physiopathological considerations in three additional cases. J Child Neurol. 2000;15:15-18.
Sra J., Maglio C., Biehl M., et al. Efficacy of midodrine hydrochloride in neurocardiogenic syncope refractory to standard therapy. J Cardiovasc Electrophysiol. 1997;8:42-46.
Sra J.S., Anderson A.J., Sheikh S.H., et al. Unexplained syncope evaluated by electrophysiologic studies and head-up tilt testing. Ann Int Med. 1991;114:1013-1019.
Sra J.S., Murthy V., Natale A., et al. Circulatory and catecholamine changes during head-up tilt testing in neurocardiogenic (vasovagal) syncope. Am J Cardiol. 1994;73:33-37.
Sra J.S., Murthy V.S., Jazayeri M.R., et al. Use of intravenous esmolol to predict efficacy of oral beta-adrenergic blocker therapy in patients with neurocardiogenic syncope. J Am Coll Cardiol. 1992;19:402-408.
Steckley J.L., Ebers G.C., Cader M.Z., et al. An autosomal dominant disorder with episodic ataxia, vertigo, and tinnitus. Neurology. 2001;57:1499-1502.
Stephenson J.B. Size of the Problem. In: Stephenson J.B.P., editor. Fits and faints. Oxford: Blackwell, 1990.
Stephenson J.B. Anoxic seizures or syncope. In: Stephenson J.B.P., editor. Fits and faints. Oxford: Blackwell, 1990.
Stephenson J.B. Specific syncope and anoxic seizure types. In: Stephenson J.B.P., editor. Fits and faints. Oxford: Blackwell, 1990.
Stephenson J.B. Vagocardiac syncope and reflex anoxic seizures. In: Stephenson J.B.P., editor. Fits and faints. Oxford: Blackwell, 1990.
Stephenson J.B. Anoxic-epileptic seizures. In: Stephenson J.B.P., editor. Fits and faints. Oxford: Blackwell, 1990.
Stephenson J.B. Reflex anoxic seizures (‘white breath-holding’): nonepileptic vagal attacks. Arch Dis Child. 1978;53:193-200.
Stephenson J.B. Reflex anoxic seizures and ocular compression. Dev Med Child Neurol. 1980;22:380-386.
Stewart J.M. Midodrine for the treatment of vasovagal syncope (simple faint). J Pediatr. 2006;149:740-742.
Stickler G.B. Relationship between cyclic vomiting syndrome and migraine. Clin Pediatr. 2005;44:505-508.
Strieper M.J. Distinguishing benign syncope from life-threatening cardiac causes of syncope. Semin Pediatr Neurol. 2005;12:32-38.
Strieper M.J., Auld D.O., Hulse J.E., et al. Evaluation of recurrent pediatric syncope: role of tilt table testing. Pediatr. 1994;93:660-662.
Strupp M., Kalla R., Dichgans M., et al. Treatment of episodic ataxia type 2 with the potassium channel blocker 4-aminopyridine. Neurology. 2004;62:1623-1625.
Strupp M., Schuler O., Krafczyk S., et al. Treatment of downbeat nystagmus with 3,4-diaminopyridine: a placebo-controlled study. Neurology. 2003;61:165-170.
Strupp M., Zwergal A., Brandt T. Episodic ataxia type 2. Neurotherapeutics. 2007;4:267-273.
Sugie H., Sugie Y., Ito M., et al. A case of paroxysmal tonic upward gaze associated with psychomotor retardation. Dev Med Child Neurol. 1995;37:362-365.
Sutcliffe J. Torsion spasms and abnormal postures in children with hiatus hernia-Sandifer’s syndrome. Progr Pediatr Radiol. 1969;2:190-197.
Sutton R. Vasovagal syncope: Clinical features, epidemiology, and natural history. In: Blanc J.J., Benditt D., Sutton R., editors. Neurally mediated syncope: Pathophysiology, investigations, and treatment. Armonk, NY: Futura, 1996.
Swoboda K.J., Soong B., McKenna C., et al. Paroxysmal kinesigenic dyskinesia and infantile convulsions: clinical and linkage studies. Neurology. 2000;55:224-230.
Symon D.N., Russell G. The relationship between cyclic vomiting syndrome and abdominal migraine. J Pediatr Gastroenterol Nutr. 1995;21(Suppl 1):S42-S43.
Tache Y. Cyclic vomiting syndrome: the corticotropin-releasing-factor hypothesis. Dig Dis Sci. 1999;44(Suppl):79S-86S.
Tanaka H., Endo K., Tsuji S., et al. The gene for hereditary progressive dystonia with marked diurnal fluctuation maps to chromosome 14q. Ann Neurol. 1995;37:405-408.
Tan L.C., Tan A.K., Tjia H. Paroxysmal kinesigenic choreoathetosis in Singapore and its relationship to epilepsy. Clin Neurol Neurosurg. 1998;100:187-192.
Tarbell S., Li B.U. Psychiatric symptoms in children and adolescents with cyclic vomiting syndrome and their parents. Headache. 2008;48:259-266.
Tavel M.E. Hyperventilation Syndrome with Unilateral Somatic Symptoms. JAMA. 1964;187:301-303.
Thijs R.D., Bloem B.R., van Dijk J.G. Falls, faints, fits and funny turns. J Neurol. 2009;256:155-167.
Thilenius O.G., Quinones J.A., Husayni T.S., et al. Tilt test for diagnosis of unexplained syncope in pediatric patients. Pediatr. 1991;87:334-338.
Tibbles J.A., Barnes S.E. Paroxysmal dystonic choreoathetosis of Mount and Reback. Pediatr. 1980;65:149-151.
Tijssen M.A., Schoemaker H.C., Edelbroek P.J., et al. The effects of clonazepam and vigabatrin in hyperekplexia. J Neurol Sci. 1997;149:63-67.
Tijssen M.A., Vergouwe M.N., van Dijk J.G., et al. Major and minor form of hereditary hyperekplexia. Mov Disord. 2002;17:826-830.
Tohier C., Roze J.C., David A., et al. Hyperexplexia or stiff baby syndrome. Arch Dis Child. 1991;66:460-461.
Tomita H., Nagamitsu S., Wakui K., et al. Paroxysmal kinesigenic choreoathetosis locus maps to chromosome 16p11.2-q12.1. Am J Hum Genet. 1999;65:1688-1697.
Trender-Gerhard I., Sweeney M.G., Schwingenschuh P., et al. Autosomal-dominant GTPCH1-deficient DRD: clinical characteristics and long-term outcome of 34 patients. J Neurol Neurosurg Psychiatry. 2009;80:839-845.
Tsai C.H., Chang F.C., Su Y.C., et al. Two novel mutations of the glycine receptor gene in a Taiwanese hyperekplexia family. Neurology. 2004;63:893-896.
Valente E.M., Spacey S.D., Wali G.M., et al. A second paroxysmal kinesigenic choreoathetosis locus (EKD2) mapping on 16q13-q22.1 indicates a family of genes which give rise to paroxysmal disorders on human chromosome 16. Brain. 2000;123:2040-2045.
Vanasse M., Bedard P., Andermann F. Shuddering attacks in children: an early clinical manifestation of essential tremor. Neurology. 1976;26:1027-1030.
Van Bogaert P., Szliwowski H.B. EEG findings in acetazolamide-responsive hereditary paroxysmal ataxia. Neurophysiol Clin. 1996;26:335-340.
VanDyke D.H., Griggs R.C., Murphy M.J., et al. Hereditary myokymia and periodic ataxia. J Neurolog Sci. 1975;25:109-118.
van Lieshout J.J., Wieling W., Karemaker J.M., et al. The vasovagal response. Clin Sci. 1991;81:575-586.
Verrotti A., Trotta D., Blasetti A., et al. Paroxysmal tonic upgaze of childhood: effect of age-of-onset on prognosis. Acta Paediatr. 2001;90:1343-1345.
Victor J. Tilt test. Environment, material, patient preparation. In: Blanc J.J., Benditt D., Sutton R., editors. Neurally mediated syncope: Pathophysiology, investigations, and treatment. Armonk, NY: Futura, 1996.
Vigevano F., Di Capua M., Dalla Bernardina B. Startle disease: an avoidable cause of sudden infant death. Lancet. 1989;1:216.
Vighetto A., Froment J.C., Trillet M., et al. Magnetic resonance imaging in familial paroxysmal ataxia. Arch Neurol. 1988;45(5):547-549.
Vlahos A.P., Tzoufi M., Katsouras C.S., et al. Provocation of neurocardiogenic syncope during head-up tilt testing in children: comparison between isoproterenol and nitroglycerin. Pediatrics. 2007;119:e419-e425.
von Brederlow B., Hahn A.F., Koopman W.J., et al. Mapping the gene for acetazolamide responsive hereditary paroxysmal cerebellar ataxia to chromosome 19p. Hum Mol Genet. 1995;4:279-284.
Wang Q., Ito M., Adams K., et al. Mitochondrial DNA control region sequence variation in migraine headache and cyclic vomiting syndrome. Am J Med Genet. 2004;131:50-58.
Wein T., Andermann F., Silver K., et al. Exquisite sensitivity of paroxysmal kinesigenic choreoathetosis to carbamazepine. Neurol. 1996;47:1104-1106.
Weissman B.M., Dell’Osso L.F., Abel L.A., et al. Spasmus nutans. A quantitative prospective study. Arch Ophthalmol. 1987;105(4):525-528.
Werlin S.L., D’Souza B.J., Hogan W.J., et al. Sandifer syndrome: an unappreciated clinical entity. Dev Med Child Neurol. 1980;22:374-378.
Wieling W., Shen W.K. Syncope: Approach to management. In Low P.A., Benarroch E.E., editors: Clinical Autonomic Disorders, ed 3, Baltimore, MD: Lippincott, Williams & Wilkins, 2008.
Williams J., Stevens H. Familial paroxysmal chorea-athetosis. Pediatrics. 1963;31:656-659.
Winner P. Childhood periodic syndromes and migraine. Curr Pain Headache Rep. 2005;9:197-2101.
Wizov S.S., Reinecke R.D., Bocarnea M., et al. A comparative demographic and socioeconomic study of spasmus nutans and infantile nystagmus. Am J Ophthalmol. 2002;133:256-262.
Wyllie W.G., Schlesinger B. The periodic group of disorders in childhood. Br J Child Dis. 1933;30:1-21.
Zhang C.L., Messing A., Chiu S.Y. Specific alteration of spontaneous gabaergic inhibition in cerebellar purkinje cells in mice lacking the potassium channel Kv1. 1. J Neuroscience. 1999;19:2852-2864.
Zuberi S.M., Eunson L.H., Spauschus A., et al. A novel mutation in the human voltage-gated potassium channel gene (Kv1.1) associates with episodic ataxia type 1 and sometimes with partial epilepsy. Brain. 1999;122:817-825.