Chapter 115 Surgery for Pediatric Vitreoretinal Disorders
General aspects
Development of the child’s eye and surgical consequences
The development of spatial vision and binocular fusion (sensitive period) starts in the human infant at the age of 3–4 months.1 A reduction in the visual acuity of one or both eyes with morphologic intactness is called amblyopia. The child’s age at exposure to an amblyopia-inducing condition is the most important determinant for its development. Children up to 8 years can develop an amblyopia; development at later age is rare. The therapy of choice for all forms of amblyopia is an appropriate occlusion therapy which sometimes must be continued until the child is 12 or 13 years old in order to avoid a recurrence.
Untreated anisometropia resulting from vitreoretinal operations induces amblyopia, especially in small children (Table 115.1). Whereas a refractive error averaging −2.75 diopters (D) is induced by a cerclage in the adult,3 the anisomyopia resulting from a cerclage may be greater in pediatric eyes. Myopia occurs as a result of the axial elongation induced by the encircling band which has a more pronounced effect on refraction due to the very short axial length of the infant eye. Furthermore, buckling surgery with a cerclage induces forward displacement of the lens, which causes severe myopia in the infant lens with its relatively high refractivity.4 The refractive change is generally less than 6 D and amblyopia is rarely manifest, since patients still have sharp near vision.5 These patients can generally be fitted with spectacles. High axial myopia of −11 to −15 D following buckling surgery in infants with retinopathy of prematurity (ROP) is more difficult to treat. Therefore, once successful reattachment of the retina has been achieved, the cerclage can be severed thus reducing the refractive error to values around −5 D.4
| Stimulus deprivation | Stimulus deprivation and suppression | |
|---|---|---|
| Bilateral deprivation: amblyopia due to media opacity of the same severity, e.g., bilateral congenital cataract or bilateral vitreous hemorrhage | 1. | All predominantly unilateral stimulus deprivations or bilaterally differing stimulus deprivations |
| 2. | Relative amblyopia: congenital or early manifestation of defects in the foveal region | |
| 3. | Uncorrected anisometropia because of blurred image and aniseikonia (e.g., anisomyopia due to cerclage anisohyperopia in silicone oil tamponade, cycloplegic eye drops) | |
| 4. | Acquired medium opacity | |
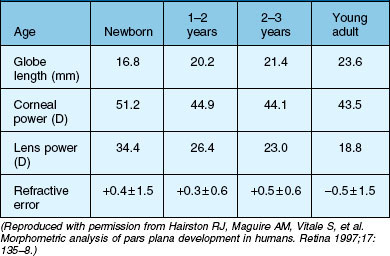
The most obvious and important anatomic consideration in pediatric retinal surgery is the relatively smaller size of the child’s globe and orbit compared with that of the adult (Table 115.2). At birth, the average axial eye length is 17.5 mm; which is 70% of the axial length of an adult eye. In the first year, the eyeball length increases by nearly 3.5 mm. From age 3–14, the eye grows very slowly; an average of 0.1 mm/year.6 The average horizontal and vertical corneal diameters are 9.8 ± 0.33/10.4 ± 0.35 mm (newborn boys) and 10.1 ± 0.33/10.7 ± 0.29 mm (newborn girls). By age 7, the cornea has an average diameter of 11.7 mm.7 In the newborn, the anterior segment already comprises 80% of its final area, while the surface of the posterior segment comprises only 20% of an adult eye; in the first 6 months of life, the posterior segment surface increases by 50%. The lens of the newborn is spherical and is relatively thicker than the adult lens. As the lens flattens, the power of the lens is reduced by approximately 8 D; in the first 2 years, it is reduced by approximately 11.5 D and it reaches adult values between 7 and 10 years.8 Since the cornea flattens, its power is also reduced: in the first 6 weeks of life, the power is reduced from 51 D to 44 D.9
A vision-dependent feedback mechanism is necessary for the development of emmetropia.10 It is assumed that various forms of stimulus deprivation induces a myopic shift during development.11 Dense hemorrhages persisting in the neonatal vitreous for 4 weeks or longer appear to cause axial myopia and severe amblyopia. Surgical intervention before this time should be considered to avert deprivation amblyopia and to retard axial myopia.12
The ciliary body of the eye is divided into the pars plicata ciliaris and the pars plana ciliaris. The pars plicata of the mature newborn is nearly adult in size, whilst the pars plana is relatively small (Table 115.3).13,14 The pars plana of mature newborns measures 1.6–2.0 mm.6
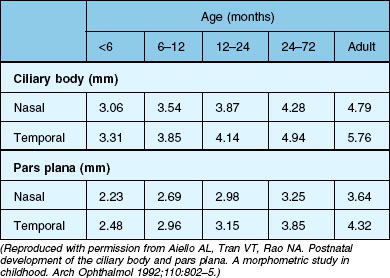
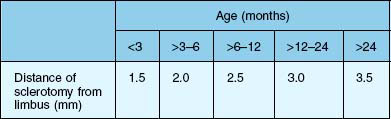
The anterior–posterior extension of the pars plana begins in the postnatal phase (Fig. 115.1). In a 6-month-old infant, the pars plana is >3.0 mm6,14 this means that in infants younger than 6 months, the pars plana width is <3.0 mm. However, the extension of the entire ciliary body is quite variable in individual eyes, its total extension in the age group 24–72 months ranges from 3.75 mm to 5.5 mm. A total of 75% of the final length of the ciliary body at adulthood is reached by 24 months.14 A positive correlation between the anteroposterior diameter of the eye and the distance from the sclerocorneal limbus to the ora serrata was demonstrated in the four meridians.13
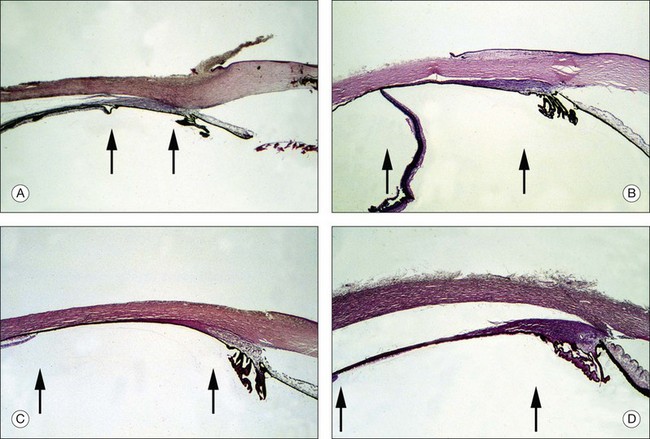
The results of the morphometric investigations are of great importance for the choice of the vitreoretinal–surgical entrance. The anterior–posterior expansion of the ciliary body corresponds approximately to the distance from the corneoscleral limbus measured externally (in millimeters). For an accurate external determination of the location of the ora serrata, 0.3–0.4 mm must be added to the measured distance starting from the corneoscleral limbus for the values of the ciliary body extension.14 Accordingly, in infants, one must select a pars plicata entrance for a vitrectomy. Only then is it ensured that the retina is not artificially damaged. The disadvantage of this entrance is the direct proximity of the lens and therefore manipulations and lens-saving operations are often not possible. Because of the relatively larger size of the lens in relation to the anterior segment in children compared with adults, special care is required if the eye is operated on without removing the lens. For a standard three-port vitrectomy, the width of the pars plana should be at least 3 mm,6 so that at the earliest a pars plan a approach can be selected for a 1-year-old child. The measurements mentioned here are based exclusively on histological slides, i.e., after appropriate fixation of the material. The real dimensions are therefore larger.
Scleral thickness is clearly age-dependent. During the first half-year of life the average scleral thickness is approximately 0.4 mm, by age two, the thickness has doubled. After that age, the scleral thickness increases very slowly and becomes stiffer with age.15
The vitreous body is avascular, transparent, and gel-like in consistency, and comprises 80% of the eyeball volume. The vitreous body is attached to the surrounding structures; a firm attachment exists within the area of the vitreous base and Wieger’s ligament forms the firm attachment between the soft lens and anterior vitreous. One differentiates between the anterior and posterior vitreous body. The posterior vitreous cortex (posterior vitreous hyaloid) is 100–110 µm thick and consists of closely packed collagen fibrils. In pediatric eyes the posterior vitreous cortex lies adjacent to the retinal surface. This structure is known as the vitreoretinal border region, the vitreoretinal junction, or the vitreoretinal interface. The vitreoretinal interface consists of the internal limiting membrane (ILM) and the posterior hyaloid. In the newborn, the vitreous body appears homogeneous with a fine radial stripe pattern. It is interspersed with so-called transvitreal channels. With increasing age vitreous strands develop, which in the adult eye form typical concentrically arranged diaphragm-like condensations.16 The secondary vitreous in the term infant without ocular disease consists of a very dense collagenous gel. Some authors believe that in term infants, there is no hyaluronic acid present until 4 years of age. There are, however, many diseases that are associated with a lack of densely formed vitreous (e.g., myopia, retinitis pigmentosa, ROP, familial exudative vitreoretinopathy (FEVR), Goldmann–Favre disease, and congenital retinoschisis). In pediatric eyes, the attachment between the vitreous cortex and retina is very firm. Formed vitreous and increased scarring seem to be correlated. It is believed that children develop proliferative vitreoretinopathy (PVR) more frequently than adults.
Surgical considerations and techniques
Cornea and keratoprosthesis
The placement of a temporary keratoprosthesis (e.g. Eckhardt keratoprosthesis (DORC) or Landers keratoprosthesis (Ocular instruments) allows a clear view for vitreoretinal surgery, which is then followed by a corneal transplant. Several retrospective studies have documented acceptable outcomes with combined surgery in adults, but only a few cases with children have been well documented.17–19 The best outcome was of a 7-year-old boy whose operation included usage of a keratoprosthesis followed by lentectomy, scleral buckling, vitrectomy, C3F8-fluid/gas-exchange, and penetrating keratoplasty. At 10 months postoperatively, the best corrected visual acuity was 20/30.17 In summary, 10 of 17 corneal grafts in children remained clear in the follow-up period, but in 16 eyes, visual acuity did not exceed finger counting. Phthisis occurred in about 40%.17–19
In general, the prognosis of keratoplasty in children is poorer than in adults. There is a greater risk of an immune reaction with development of neovascularization. Perforating keratoplasties in children following trauma have a poorer general prognosis, especially in cases of aphakia and after injuries involving the posterior segment of the eye as opposed to phakic eyes and after isolated injuries to the anterior segment. In a meta analysis,20 55–100% of transplants were not opaque 1 year after keratoplasty in children.
The corneal sutures in children can be removed much sooner than in adults. Removal should be performed within the time intervals specified in Table 115.4, in order to enable immediate provision of contact lenses, especially in aphakic eyes.21
Table 115.4 Recommendation for removal of corneal suture by age
| Age | Removal of corneal suture |
|---|---|
| 1–6 months | 4–6 weeks |
| 6–12 months | 6–8 weeks |
| 12–24 months | 8–12 weeks |
| 24–48 months | 12–16 weeks |
| 5–15 years | 4–6 months |
(Reproduced with permission from Reidy JJ. Penetrating keratoplasty in infancy and early childhood. Curr Opin Ophthalmol 2001;12:258–61.)
Lens management
In some cases, especially in eyes with a high risk of developing proliferative vitreoretinopathy, such as after severe open globe injury and in uveitis, an intraocular lens (IOL) should not be implanted. Opacity of the remaining capsule fragments with optically disadvantageous postcataract membranes is inevitable. In addition, remnants of the lens capsule and the zonular fibers lead to development of synechiae with distortion of the pupil which is associated with a reduction in the visibility of the peripheral retina. Surgical treatment of retinal detachment is impeded. For this reason, complete removal of the entire lens, including the complete capsule should be aimed for in eyes with a high risk of developing PVR. In addition, a current study shows a significantly increased endophthalmitis risk if an intraocular lens implanted placed in eye immediately after open globe injury.22
The risk of a severe intraocular fibrin reaction is one of the further drawbacks of intraocular lens implantation in children. Even after operation of a nontraumatic cataract in a pediatric eye, a significantly less pronounced fibrin reaction is observed after pars plana lentectomy than with a limbal approach, as necessary for implantation of an intraocular lens.23,24 In darkly pigmented eyes, an even more intense fibrin reaction is observed. Further disadvantages of primary intraocular lens implantation are the imprecise biometry and unclear prognosis concerning eye growth.
Posterior-segment surgical techniques
The smaller size of infant eyes results in differences in surgical landmarks, relative instrument size, and fluid dynamics of vitreous surgery (Fig. 115.2) in addition to increased vitreous adhesion. We will address these specific issues below.
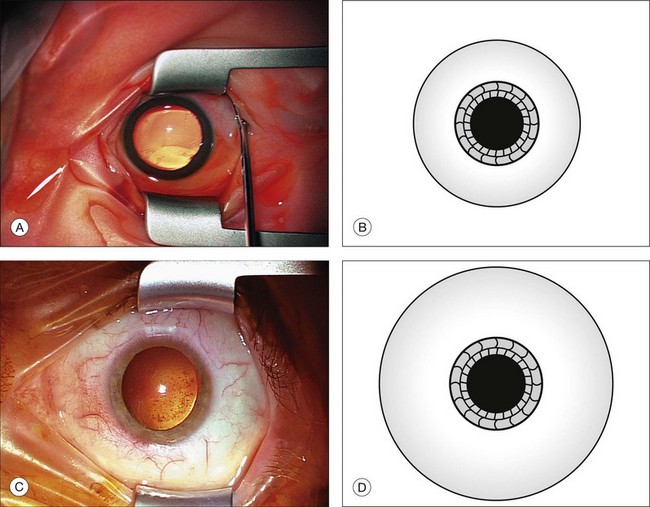
Fig. 115.2 Comparison of a globe from a neonate (A,B) with that of an adult (C,D) shows different proportions.
Buckling surgery (see also Chapter 100, Techniques of scleral buckling): after a peritomy, traction sutures (silk 5/0) are placed beneath the insertion of two to four rectus muscles. Then the retinal holes are located, marked, and a transscleral cryoretinopexy is performed under ophthalmoscopic control. Depending on location of the hole, appropriate radial or circumferential scleral buckles are placed and fixed, if necessary with an additional segmental silicone-rubber sponge. For a cerclage, one suture per quadrant is placed, and the ends are secured with a sleeve. Due to spatial considerations, the sleeve is preferably placed in the lower temporal quadrant. The perfusion of the central retinal artery must be checked. Rarely, a paracentesis is necessary. The scleral thickness is thinner than in adult eyes, therefore thinner (6/0) suture (polyester fiber, polyamide fiber) should be used for partial-thickness scleral sutures in babies. We also recommend the use of flat spatula needles (0.28 mm diameter).
A child’s eyes are more prone to vigorous reproliferation than those of adult. This can be attributed to biochemistry of this age group which support cell growth more actively, or to a longer delay between the time of detachment and the establishing of diagnosis and therapy. For primary detachments, we use a sculpted 3 × 5 mm sponge with an encircling 2.5 mm solid silicone band. It is often assumed that the children’s orbits cannot accommodate a large sponge, but this type of exoplant is well tolerated. Drainage of subretinal fluid is controversial. Postoperative complications of scleral buckling in children range from refractive amblyopia to alteration of eye growth. To avoid these complications, the band is cut in all children younger than 3 years, approximately 3 months after the scleral buckling operation, once a stable reattachment has been achieved.4 We choose to cut rather than to remove the element because we believe that continued support is given to the retina by the encapsulated exoplant. To reduce amblyopia development in the postoperative period, we prescribe 1% atropine drops for 5 days; if both eyes have good visual potential, we prescribe the drops for both eyes. In addition to amblyopia therapy, a refractive error needs to be treated with adequate prescriptions.
Usually a three-port vitrectomy is performed in children. The infusion is preferably placed temporally. The sclerotomies are placed 3.5 mm behind the limbus in infants older than 2 years. In children younger than 2 years, the sclerotomies are performed within the range of the pars plicata and/or pars plana, in accordance with Table 115.5. In the infant eye, especially in babies, the scleral incisions are made posterior to the limbus with incisions through the pars plicata. Displacement of the conjunctiva for adequate coverage of the sclerotomies necessary for transconjunctival sutureless surgery is not possible.25 Therefore, conjunctival dissection is thus preferred before the sclerotomy incisions are made. Because of the very thin sclera in babies and infants, we recommend using trocars beginning at age two. In preschool children, we recommend placing the trocars perpendicular through the sclera. In young eyes, oblique placement of the trocars is associated with an increased risk of accidental dislocation, since the trocars have only a short path through the pediatric sclera compared to the longer path through an adult sclera. A young patient age is a risk factor for intraoperative sclerotomy leakage: in a study of 322 eyes, 61% of patients aged <40 years needed intraoperative suture placement for leaking sclerotomies at the end of 23G transscleral vitrectomy.26 Due to the decreased scleral stiffness and the increased elasticity of young sclera, suturing of sclerotomies in children is necessary to ensure a leakproof closure. For the closure of the conjunctiva, absorbable Vicryl 10/0 can be used.
A complete range of small incision instruments which permit a complete vitrectomy through small (23G and 25G) incisions has been developed. Performing surgical maneuvers with smaller instruments allows the surgeon to work more precisely and increases the potential for minimizing surgical trauma. We prefer the use of 23G instruments, but case selection remains important. In case of very dense cellular membranes, the 23G or 25G cutter cannot adequately address this tissue and additional instrumentation may be necessary. In more complicated cases, such as traction retinal detachments, eyes with extensive fibrovascular proliferation, complex retinal detachments requiring scleral buckles for proliferative vitreoretinopathy, and giant retinal tears, surgical maneuvers can be performed with small gauge instruments, but are often more easily performed using 20G instrumentation.27 For cases requiring enhanced panoramic illumination, it is possible to use the 20G shielded bullet light pipe instead of the 23G illumination light pipe. The sclerotomies are made with a 23-MVR blade. In babies, we use a 23G disposable Tornambe infusion cannula (DORC 1272). Larger sclerotomies may allow for better manipulation of the 23G or 25G cutter within the sclerotomy, avoiding unintentional bending of the instrument and potential damage to the lens or retina.25
Vitrectomies are always performed with wide-angle observation systems. In addition, wide-field vitrectomy contact lenses are specifically designed to facilitate pediatric vitreoretinal surgery.28
The wide-angle observation systems for vitreous surgery (binocular indirect ophthalmomicroscope, vitreopanfundoscope, and stereoscopic diagonal inverter have many advantages: (1) wide angle of view; (2) large depth of field; (3) stereopsis; (4) good visualization in hazy media, even with narrow pupil; (5) upright, tru-to-side image; (6) magnification with the microscope zoom; (7) beam splitter for the assistant; (8) good visualization in gas-filled or silicone-filled eyes; (9) free mobility of the eye for peripheral surgery. With these systems, surgery is possible from the posterior pole to the vitreous base and the anterior loop traction, regardless of whether the eye is phakic, pseudophakic, or aphakic. The vitreous body can be removed with a 23G high-speed cutter with a cutting rate of 5000 revolutions/minute and a suction power of 400 mmHg. With the cutter near the peripapillary retina, suction should be applied to separate the posterior hyaloid from the retina in slow circular movements. Special attention is necessary to create a posterior vitreous detachment; “tenting of the retina” is a sign of very strong vitreoretinal adherence and poses a high risk for development of iatrogenic retinal holes. A compromise is a judicious core vitrectomy, leaving as thin a layer of cortical vitreous on the retina as possible. If it is not possible to remove vitreous in the periphery, a thorough shaving is necessary. This can be performed by a small gauge high-speed cutter; if 20G instruments are used, special 20G shaver-cutters are available. Creation of a posterior vitreous detachment with suction alone to lift the vitreous off of the optic nerve is more difficult with small gauge vitrectomy probes. The smaller port opening decreases the ability to engage and hold the vitreous. Creating a posterior vitreous detachment in a child with adherent posterior hyaloid remains substantially easier with 20G instruments because of larger port sizes and greater flow rates.27 For small-gauge vitrectomies, higher infusion rate in the range of 35–50 mmHg are necessary to ensure adequate intraocular fluid dynamics. Due to the lower systolic blood pressure of babies and infants, iatrogenic occlusion of the central retinal artery can be induced, and the surgeon has to observe the optic nerve.29 Retinal breaks should be avoided because they are often difficult to treat. Posterior drainage retinotomies are associated with profound postoperative proliferation. Iatrogenic breaks are best avoided by abandoning peeling in favor of bimanual dissection with forceps and scissors. If a retinotomy is unavoidable, it must be performed as close as possible to the ora serrata and following diathermy coagulation. Retinopexy of retinotomy edges is done with argon laser coagulation. If the extension of the retinotomy is larger than 2 clock-hours, a silicone oil tamponade is used. Diagnostic vitrectomy can be used to evaluate conditions such as inflammatory conditions, and amyloidosis. The cytopathologic analysis of ocular fluids obtained for diagnostic purposes has proven to be effective for establishing and confirming diagnosis.
It is hoped that pharmacologic agents will become available to assist in separating the youthful posterior cortical vitreous from the retina. The goals of enzymatic vitrectomy (dispase, plasmin, tissue plasminogen activator, or chondroitinase) are either to disinsert the posterior hyaloid from the retinal surface in an atraumatic, precise, cleavage plane or to try to disinsert the peripheral vitreous from the neurosensory retina. Enzymatic (collagenase or hyaluronidase) manipulation of the central vitreous in terms of liquefaction has been evaluated. At present microplasmin is used in clinical studies.30–32 This is certainly only the beginning of this type of vitreal surgery, whether adjuvant or alternative.
Silicone oil and gas tamponade
The physical properties of silicone oil include a combination of specific gravity, refractive index, and surface tension. Heavy silicone oil may be of interest for inferior traumatic retinal detachments. Use of 1000 cs or 5000 cs silicone oil can be considered in the management of pediatric complex retinal detachments associated with multiple etiologies.33 The choice of viscosity offers an optimal balance between easy injection and a longtime tamponade. Indications and the long-term results of pars plana vitrectomy in children are comparable with those in the adult. Retinal reattachment and preserved visual acuity can be achieved in the majority of eyes using silicone oil retinal tamponade. In complicated retinal detachments, especially after injuries, a radical approach with primary silicone oil injection and later silicone oil removal has proved useful. In children, with their long life expectancy, timely removal of silicone oil vital for maintaining the function of the eye.
Biostaining of the ILM and intraocular membranes is a technique that is still evolving, and despite good results in adults, it should be used in children with utmost care. We recommend Trypan blue and Brilliant blue as “safe” dyes for staining, assuming they have nontoxic or at least minimal toxic effects.34 Visualization of the stained posterior hyaloid greatly facilitates complete removal and reduces surgical time. Control staining following membrane removal is possible, preferable by the use of triamcinolone to check if all membranes have been successfully removed.
Indications for surgery
Injuries
(See also Chapter 98, Pathophysiology of ocular trauma, and Chapter 100, Techniques of scleral buckling.)
Direct injury
Open globe injury
Some 29–47% of pediatric open globe injuries involve both the anterior and posterior segments of the eye.35–37 Whereas the primary surgery includes external reconstruction of the globe, internal reconstruction can be performed simultaneously with the primary wound closure or, in most cases, in a second step.
The timing of secondary comprehensive anatomical internal reconstruction is crucial. Various authors have recommended a secondary vitreoretinal operation for internal reconstruction in the period between the 7th and 10th post-traumatic day in adults38–40; others recommend 3–10 days after trauma.39,41 The general rule is to delay comprehensive anatomical reconstruction of the seriously injured eye for approximately 1 week, when the risk of intraoperative hemorrhage is dramatically reduced and a separation of the posterior vitreous begins to develop.38,39 Kuhn et al. recommend early vitrectomy within 3 days to prevent development of vitreoretinal proliferation.39
The value of a prophylactic encircling band in severe globe-opening injuries is controversial. There are no prospective studies documenting the advantage in children. In adults, the frequency of retinal detachment is less on the whole after applying an encircling band. An encircling buckle procedure is recommended in the presence of a corneoscleral or scleral wound >5 mm with ciliary body involvement with marked vitreous loss and vitreous hemorrhage.42–44 Despite optimal follow-up, the functional results after combined pediatric anterior and posterior segment injuries are limited: about 12%33 to 69%37 of the children attain a visual acuity of 0.1 or better after primary wound closure and secondary surgery for reconstruction of the anterior and posterior segments.
In addition, children may develop more extensive postoperative inflammation, scarring, and severe proliferative vitreoretinopathy than adults, which may also affect the anatomical and functional outcomes.45 The results after pediatric vitreoretinal surgery depend greatly on the extent and severity of the primary damage (Fig. 115.3). A wound size exceeding 10 mm, a lens lesion, and a patient age under 4 years are factors with unfavorable prognoses. Further negative influential factors include: severe intravitreal bleeding, poor initial vision, and gunshot injuries. Retinal detachment always correlates with significantly poorer results. Preoperative macular attachment is a crucial prognostic factor for attaining a postoperative vision better than 0.1.46,47
Closed globe injury
Oradialysis
A common finding in closed injuries is an oradialysis. An oradialysis is located most often inferotemporally, which may only become symptomatic years after the trauma. In the absence of cataract, minimal vitreous haemorrhage or haze, treatment consists of a circumferential segmental buckle. The edge of the segmental buckle should exceed the edge of the dialysis by twice.48 Anatomical reattachment was achieved in 67–88% of cases, whereas the rate of PVR was low.49,50
Traumatic macular hole
Most traumatic macular holes result from closed-globe contusion injuries. Furthermore, the formation of a macular hole secondary to a penetrating injury has been documented in an 8-year-old boy.51
Traumatic macular holes may result from blunt trauma when it causes separation of the vitreous from the retina, contusion necrosis, or subfoveal hemorrhage. The cause of visual deterioration is necrosis resulting from contrecoup forces or tearing of the retina by a sudden separation of the posterior vitreous face.52 But others found that the posterior vitreous was detached from the macula in only 5% of eyes with traumatic macular holes.53 They supported the theory that rupture of the macula caused by retinal stretching occurs as a result of either ocular deformity in the equatorial region or from the strong force of impact on the macula.53 Small, full-thickness traumatic macular holes with a size of 0.1–0.2 disc diameter (DD), can resolve spontaneously in 64–100% of pediatric cases, 3–4 months following trauma, and this may be associated with good visual recovery, in most cases even 20/20.54–56 In contrast to spontaneous closure of small macular holes (100–200 µm) closure of large holes (400–600 µm) has been rarely described (Fig. 115.4).57
In preschool children, the timing of surgery should be based on the fact that traumatic macular holes may be amblyogenic. We therefore recommend vitreous surgery for the treatment of traumatic macular hole after 4 weeks. In elderly children, a period of observation of 3–4 months before vitreoretinal surgery is recommended.55
Common features of patients with spontaneous closure of traumatic macular hole are young age (<25 years), small size of the macular hole (0.1–0.2 DD), no posterior vitreous detachment, and no epiretinal membrane.55
Vitreous surgery for traumatic macular holes can lead to anatomic success in 92–100% and visual acuity improvement in 92–100% of pediatric patients.51,55,58 Recent studies recommend surgical procedures including removal of the posterior hyaloid, the epiretinal membrane, and internal limiting membrane and expanding gas/air mixture tamponade.55,58,59 Studies with adjunctive treatments have been published and describe the successful use of platelet concentrate,60 and plasmin (0.4 IU of autologous plasmin) for enzyme-assisted pars plana vitrectomy.58 In young patients, poor compliance with face-down positioning may be a factor in surgical failure. It is difficult for a child to maintain a face-down position postoperatively, therefore the use of silicone oil is advocated in children who need long-term intraocular tamponade after vitreoretinal surgery. However, additional surgery for silicone oil removal is necessary.
Vitreous hemorrhage
Differential diagnosis for intraocular hemorrhages in infants include trauma, X-chromosomal retinoschisis, Terson syndrome, shaken-baby syndrome, and Toxocara infection. Complications of infantile vitreous hemorrhage include epiretinal membrane formation, pigmentary retinopathy, strabismus, high anisometropic myopia, and occlusion amblyopia.10 These serious complications occur as early as 5 weeks after the onset of the vitreous hemorrhage. Vitrectomy is an acceptable early therapy for infantile vitreous hemorrhage, and we recommend that vitrectomy be considered as early as 3–4 weeks after the onset of a dense, infantile, vitreous hemorrhage in an attempt to avert serious complications of the hemorrhage.12,61 Furthermore, early removal of severe vitreous blood clots and severe vitreous hemorrhaging is recommended for injured eyes. The tractional retinal detachment arises as a result of contracting epiretinal membranes. Vitrectomy reduces the incidence of these tractional retinal detachments in open-globe injuries.38,62 The timing of vitrectomy for the removal of vitreous hemorrhage remains crucial. If retinal and/or choroidal involvement are not present, serial ultrasonography is necessary to detect complications. In individual cases, vitreous hemorrhage may absorb spontaneously, but because of the danger of amblyopia, vitrectomy may nevertheless be indicated.
Indirect injury
Terson syndrome
Terson syndrome is characterized by intraocular hemorrhage secondary to subarachnoidal or subdural hemorrhage. Vitreous hemorrhage in patients surviving subarachnoid hemorrhage appears to be more common than previously thought, underscoring the need for routine funduscopic screening. In the only published prospective case series of nonabuse instances of intracranial hemorrhage in children,63 2% had intraocular hemorrhages. The authors estimate the incidence of Terson syndrome in children with intracranial hemorrhage to be less than 8%. There is a low incidence of spontaneous intracranial hemorrhage in infants.
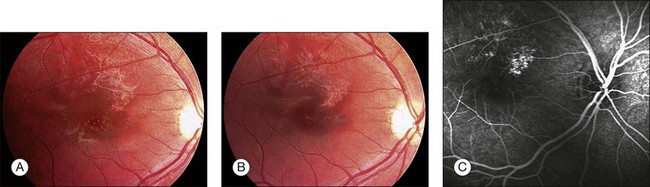
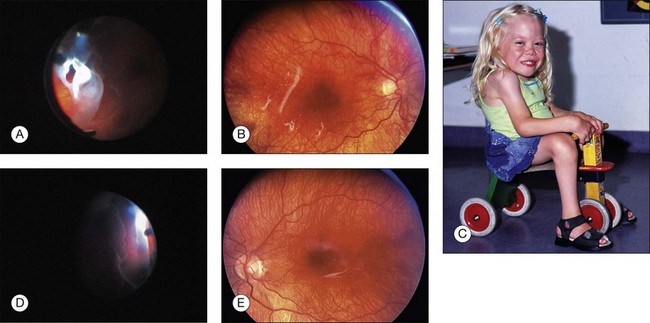
Three case reports of Terson syndrome in babies, from 5 weeks to 7 months old, due to a ruptured angiodysplasia or an aneurysma have been published.64–66 The hemorrhages can occur within the sensory retina, in the subretinal space, and in the vitreous cavity. In approximately one-third of these eyes, a premacular accumulation of blood termed a “hemorrhagic macular cyst (HMC)” may be found.67 Based on the blood’s relation to the ILM, two types of HMC can be distinguished in Terson syndrome. A HMC is preretinal if the blood is located anterior to all layers of the retina, and submembranous if the blood has accumulated beneath the ILM.67 The anterior cyst wall may initially be formed by the posterior hyaloid face and later by proliferation. This newly formed membrane may present even if the blood has cleared from the cyst. Hemorrhagic macular cysts are more often submembranous than preretinal. At the vitreous base and in the equatorial zone, attachment plaques are present between the ILM and the Müller cells. Because attachment plaques are missing in the posterior zone, where the ILM is much thicker, a retinal hemorrhage is capable of detaching the ILM from the retina in that area. The presence of attachment plaques is considered in relation to centripetal vitreous traction, which is absent in the area of the posterior precortical vitreous pocket (PPVP). The posterior wall of the PPVP therefore probably coincides exactly with the thick part of the ILM in the posterior zone.68 Most cases of vitreous and retinal hemorrhages in Terson syndrome do not require surgery because of spontaneous resorption. However, young individuals with a healthy vitreous gel may have delayed hemorrhage clearance, but babies and preschool-age infants can develop deep amblyopia within a few weeks. The formation of epiretinal membranes in severe cases is associated with the development of PVR. To prevent this complication, some authors advocate close follow-up and early, extensive treatment in Terson syndrome.69 Complete cyst removal should be performed, i.e., including contents and anterior wall, to prevent blood-related complications and to allow early rehabilitation of macular function (Fig. 115.5).67
If a pars plana vitrectomy is performed, the possibility of a hydraulically dissected ILM has to be considered. Postoperatively, perimacular retinal folds or fibrotic lines may occur.70 Indications for vitrectomy therefore include: dense vitreous hemorrhage, bilateral hemorrhage, premacular HMC and/or dense vitreous hemorrhage if spontaneous resorption has failed after 4 weeks, development of esotropia, and/or PVR. Whereas the prognosis of vitrectomy for Terson syndrome is excellent in adults, it remains relatively poor in visually immature children. Amblyopia, despite early intervention, and permanent brain damage are potentially responsible for this limited visual outcome. We recommend surgical intervention, management of amblyopia, correction of refractive errors, and intensive orthoptic treatment.
Shaken-baby syndrome
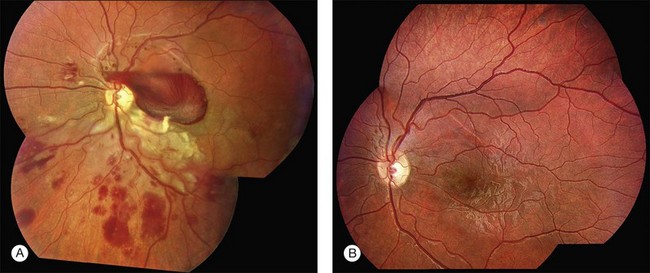
In shaken-baby syndrome, retinal hemorrhages are the most common fundus finding, and dome-shaped hemorrhages are the most distinctive form of hemorrhage (Fig. 115.6). The dome-shaped hemorrhages are most often seen in the macular area, but can be found in any area of the retina on histopathologic examination. They can be caused by separation of the ILM or a more vision-threatening deep retinoschisis. Recent studies have shown, hand-held spectral domain optical coherence tomography is helpful in the evaluation of patients with shaken-baby syndrome to detect vitreoretinal abnormalities, e.g., multilayered retinoschisis, disinsertion of the internal limiting membrane and foveal detachment in these eyes.71,72 In some cases, the blood breaks through the ILM and/or hyaloid face and causes a vitreous hemorrhage. This can occur immediately after injury or a few days after the cerebrovascular accident from rupture of the ILM over a dome-shaped hemorrhage.
In addition to extensive retinal and preretinal hemorrhages, bilateral symmetric white ring-shaped retinal folds are seen encircling the macula outside the vascular arcades. These retinal folds may be a hallmark of shaking injuries in child-abuse victims.73 Short-term follow-up is necessary with large dome-shaped hemorrhages because they can induce severe amblyopia by themselves or can break into the vitreous and linger for months in these visually immature patients. We recommend vitrectomy for large premacular hemorrhage and/or after absence of vitreous hemorrhage clearing after 4 weeks. If esotropia develops, vitrectomy should be performed immediately. During vitrectomy, the hyaloid is peeled, and the membrane covering the macula is completely removed, i.e., the ILM is ruptured: following careful incision of the anterior wall of the HMC with a bent microvitreoretinal blade in a selected macular quadrant, the flap is grasped by end-gripping forceps, and the membrane is slowly torn in a motion respecting the visible edge of the HMC. In most eyes, this is a nearly circular motion concentric to the fovea. Vitreous surgery for HMC leads to excellent anatomic success,70 and it offers immediate visual rehabilitation.
Diseases of the pediatric retina
Myopia
Myopia is one of the important predisposing factors of nontraumatic rhegmatogenous retinal detachment in childhood.74 In east Asia, about 40% of children younger than 18 years who underwent surgery for retinal detachment suffered from high myopia (>−6 diopters).75 due to the significant differences in the refractive error prevalences as a function of ethnicity, the rate of myopic non-Asian children, whom develop a retinal detachment in the first two decades of life, is described to be 11–28%.76 According to the results of the scleral buckling versus primary vitrectomy in rhegmatogenous retinal detachment study (SPR-study),77 in pediatric almost phakic eyes, primary treatment should be performed by a scleral buckling procedure. Primary vitrectomy is reserved for posterior tears, severe PVR, or for cases with obscured ocular media. The value of an additional encircling buckle is controversial, we recommend it for eyes with PVR.
In a study78 of highly myopic children <10 years of age, the authors found retinal holes in 4%; lattice degeneration in 20%, and white-without-pressure in 11% of the eyes. The authors recommend performing laser photocoagulation of retinal holes and prophylactic laser photocoagulation of lattice degenerations because children are less likely to report symptoms, thus delaying diagnosis and treatment of retinal detachment.78 Prophylactic laser photocoagulation remains a controversial issue; to date the database on prophylactic treatment in children is lacking.
Subfoveal membranes
Choroidal neovascularization membranes (CNVMs) in children are rare. The cause of the CNVM is presumed to be an ocular histoplasmosis syndrome, idiopathic, optic nerve coloboma and drusen, ocular toxoplasmosis, Toxocara canis, rubella retinopathy, serpiginous choroidopathy, trauma, degenerative myopia, and Best disease. The treatment of CNVM is a controversial topic and the natural history of idiopathic CNVM is not necessarily associated with a profound loss of vision.79 Photodynamic therapy is impractical in babies and infants, and surgical excision might have visually threatening risks. Worsening of visual acuity, protracted neurosensory detachment with the development of cystoid macular edema, or subfoveal hemorrhage may be factors that make treatment of CNVM by intravitreal anti-VEGF a favorable option. Various authors reported children (aged 19 months to 15 years) with CNVM that regressed with documented improvement in visual acuity following treatment with intravitreal anti-VEGF.80–83 Some authors used bevacizumab in a dose of 1.25 mg/0.05 mL,80,82,83 others used ranibizumab, in a dose of 0.25 mg/0.025 mL to 0.5 mg/0.05 mL81,83 or 0.3 mg/0.09 mL of Pegaptanib.83 Most patients required 2–5 injections of the anti-VEGF agent. No ocular or systemic adverse events were observed in any of our treated patients.
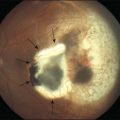
Coats disease
Coats disease is characterized by exudative retinitis and telangiectasias of the retina.84 Usually (95%) only one eye is affected, and the etiology is unknown.85
The main goal of treatment of Coats disease should be to eradicate the telangiectasias by laser photocoagulation or cryocoagulation. If telangiectasias are associated with retinal detachment, cryotherapy alone can be performed to destroy vascular abnormalities. The preferred method of cryotherapy is a double freeze-thaw technique applied directly to the telangiectasias. But excessive cryotherapy can induce an increase in the extent of retinal detachment. Therefore, diffuse telangiectasias involve all quadrants, it is advisable to initially treat two quadrants only and the other quadrants 4 weeks later. A second treatment of the same area should be withheld for 3 months, because resolution of exudation is very slow process.86
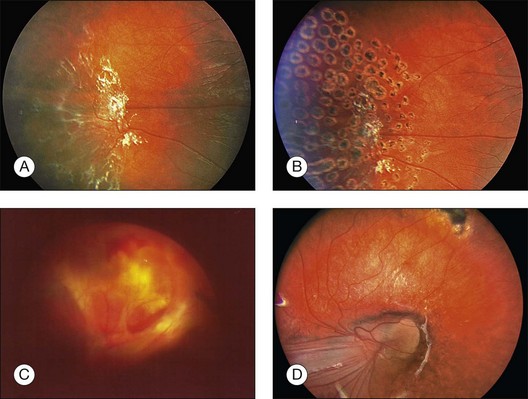
A classification of Coats disease has been proposed by Jerry and Carol Shields.85 Stage 1 is characterized by telangiectasia only and can be managed by either periodic observation or laser photocoagulation. In stage 1 disease, there is a high probability that the eye can be salvaged, and the visual prognosis is usually favorable. Stage 2 is characterized by telangiectasia and exudation, and is generally best managed with cryotherapy or laser photocoagulation. If the exudation is limited to one quadrant or located nasally, a reasonably good visual outcome can be anticipated. In stage 2A, the fovea is not involved by exudation, and the visual prognosis is generally good. Eyes with stage 2B shows foveal exudation. A dense yellow-gray nodule, a macular fibrosis, centered within the foveal exudation is usually associated with a worse visual outcome.87 Stage 3A disease (subtotal retinal detachment) is generally managed by laser photocoagulation or more effectively in most cases by cryotherapy. Even if the retinal detachment involves the fovea, it will resolve when the peripheral telangiectasias are eradicated (Fig. 115.7). Stage 3B disease (total retinal detachment) can be managed with cryotherapy if the detachment is shallow, but pars plana vitrectomy may be required if the retina is immediately posterior to the lens. Stage 4 disease (total retinal detachment with glaucoma) is often best managed by enucleation to relieve severe ocular pain. In some instances, attempts to control the glaucoma medically may be undertaken. Stage 5 disease is defined as a blind, nonpainful eye with a total retinal detachment, often with cataract and phthisis These patients require no aggressive treatment.85
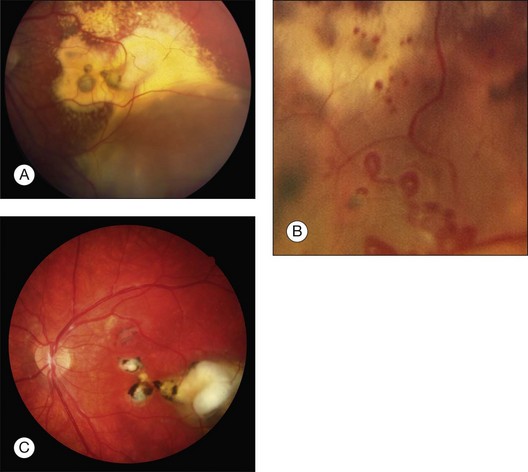
Recent studies have shown that Coats disease is associated with an increased intraocular VEGF level, and therefore intravitreal anti-VEGF injection may be a valuable adjunctive treatment for pediatric Coats disease. Since sole therapy with intravitreal bevacizumab may not be effective, combining it with laser photocoagulation or cryotherapy may be a viable option for managing Coats disease, reducing leakage, and preventing neovascular complications.82,88 The optimal time period between injection and coagulation treatment is unclear. The employed dosage for bevacizumab was 1.25 mg/0.05 mL (14–17 years old)88–90 to 2.5 mg/0.1 mL (6 months to 12 years old ).91 All patients received one to three injections and no side-effects were observed.
It is paramount that ophthalmologists maintain a high clinical suspicion for the possibility of retinoblastoma before performing an injection that could spread tumor cells to the subconjunctival space and orbit.85,92 Unless the retina and the abnormal telangiectatic vessels found in Coats disease can be examined by indirect ophthalmoscopy, no injection should be performed, even with the absence of calcification on B-scan. Furthermore, a diffuse infiltrating retinoblastoma and advanced Coats disease can be indistinguishable from each other on magnetic resonance imaging. Therefore, enucleation must be performed in patients with poor visual potential and when it is impossible to rule out the possibility of retinoblastoma.93
Hereditary vitreoretinopathies
Stickler syndrome
Hereditary vitreoretinopathies are potentially blinding disorders characterized by an abnormal-appearing vitreous gel with associated retinal changes. Stickler et al.94,95 described an autosomal dominant disorder with characteristic ophthalmological and systemic abnormalities, orofacial features, deafness, and arthritis. Abnormalities of vitreous gel architecture are a pathognomonic feature, usually associated with high myopia, which is congenital and nonprogressive, and cataract (Fig. 115.8). Nonocular features show great variation in expression. Stickler syndrome type 1 (STL1) arises due to mutations in COL2A1 gene and shows membranous vitreous phenotype, while type 2 (STL2) disease arises from mutations in COL11A1 and affected patients have a fibrillar appearance of the vitreous.96 Only patients with STL1 or STL2 show ocular changes, whereas STL3 patients do not suffer eye problems. The majority of patients have STL1. Retinal features of Stickler syndrome are radial perivascular pigmented retinal degenerations, lattice degeneration, anterior giant retinal tears, and posterior retinal tears. The first retinal detachment (RD) is observed between the ages of 10 and 30 years; 8% of affected children have RD between 0–9 years, and 26% between the ages of 10 and 19 years.95,97
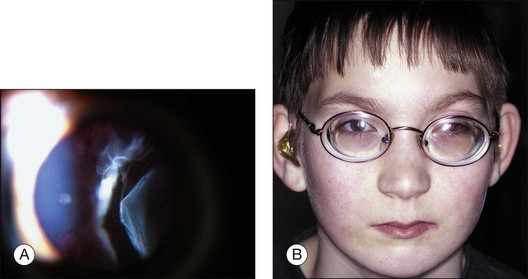
With a high rate of bilateral spontaneous retinal detachment, “unaffected” family members without retinal detachment or other associated symptoms of Stickler syndrome should be approached and offered molecular genetic analysis to rule out the presence and type of Stickler syndrome, and should be examined regularly.98
Positive effects of prophylactic intervention is have been described.99,100 In a recent systematic review,98 two principal retrospective cohort studies with control groups in populations with STL1 were analyzed. One study evaluated 360° cryotherapy (n = 204) on the post-oral retina and reported a statistically significant lower rate in the treatment group compared with the untreated STL1 group (n = 22). Leiba et al.100 performed a subgroup analysis based on age and reported that 0/6 eyes treated prophylactically by laser coagulation (circumferential treatment for eyes with extensive contiguous retinal lesions where lesions were present in at least three quadrants; focal laser treatment for eyes with small localized lesions of lattice degeneration or isolated breaks) in children aged ≤13 years detached compared with 1/4 eyes treated prophylactically in children aged ≥13 years. In the control group, which did not receive any prophylaxis, the retina detached in 46% eyes of children aged ≤13 years, but detached in 60% in adolescents and adults aged ≥13 years in the follow-up of 1–15 years. However, both studies were subject to a high risk of bias and the optimal age for prophylaxis is uncertain.98
Despite complicated surgery and often multiple procedures, Stickler patients undergoing primary vitrectomy for rhegmatogenous detachment shows good anatomical outcomes as well as useful functional visual results in comparison to outcomes of buckling. Vitrectomy may be considered as the operation of choice in the Stickler group.97 Almost three-quarters of re-detachments occur within the first 4 postoperative months. Complications are predominantly re-detachment and subsequently multiple surgeries. As such, the use of primary vitrectomy to improve the primary success rate is also likely to reduce the overall complication rate.
Kniest dysplasia
Kniest dysplasia is an autosomal dominant disorder characterized by kyphoscoliosis, severe short trunked dwarfism, cleft palate, flat face, hearing defects, and joint contractures.101,102 Patients with Kniest dysplasia have severe congenital myopia and vitreoretinal degeneration. The vitreous cavity contains fibrous, clouded, membranous structures floating in the retrolental space (Fig. 115.9). Other eyes have cortical and posterior subcapsular opacity of the lens, and also veil-like vitreous opacities in the periphery. The common retinal changes include perivascular lattice degeneration and white-without-pressure of various degrees. Bullous retinal detachment with Kniest dysplasia can be treated successfully, especially by vitreous surgery with silicone oil tamponade.103
Familial exudative vitreoretinopathy
Familial exudative vitreoretinopathy (FEVR) is a bilateral disorder of aberrant peripheral retinal development, including peripheral avascular retina, abnormal vascularization with retinal neovascularization, subretinal exudation, formation of an abnormal vitreoretinal interface, and retinal detachment (Fig. 115.10). The phenotype of FEVR resembles a form fruste of ROP, occurring in a larger and often term infant. Aberrant vasculogenesis and subsequent abnormal angiogenesis are thought to cause the vitreoretinal abnormalities in FEVR.104 FEVR patients have a mutation in the Norrie disease gene. This results in dysregulation of the Wnt-receptor: ß-catenin pathway and has been associated with increased levels of vascular endothelial growth factor (VEGF). These can potentially explain the lifelong chronic nature of FEVR, which is characterized by exacerbations in exudation secondary to upregulated vascular activity. The clinical classification is based on five stages.105
Systemic associations are absent. The management of FEVR is similar to that for ROP: in the early stages, peripheral coagulation by laser or cryocoagulation of the avascular retinal periphery is used to reduce subretinal and intraretinal exudation. Anti-VEGF therapy may provide a valuable adjunctive treatment for FEVR. In one study,106 three children (6–14 years), with persistent vascular activity and increasing exudation, despite aggressive laser photocoagulation and cryocoagulation, were successfully treated by intravitreal injection of pegaptanib sodium (Macugen; 0.3 mg/90 µL). All patients showed a reduction of exudation, but two children developed vitreous hemorrhage due to tractional retinal detachment and needed pars plana vitrectomy. After injection of bevacizumab (Avastin; 1.25 mg/0.05 mL) in an adult patient, rapid regression and accelerated fibrosis of neovascular tissues was observed.107 In traction retinal detachment vitreoretinal surgery is indicated to release vitreous traction by either scleral buckling or vitrectomy. The vitreoretinal adhesions are so strong in the peripheral avascular area that iatrogenic retinal breaks occur easily. A bimanual technique with vitreous scissors and forceps is preferred to dissect the vitreous membranes from the retinal surface. Dissection of the vitreous in the peripheral avascular area is very difficult, and if this procedure cannot successfully be performed, the prognosis is poor.105,108 If an eye is symptomatic in the first 3 years of life, the prognosis is poor. Despite laser photocoagulation of the peripheral avascular retina, most of these eyes develop tractional retinal detachment. Vitreoretinal surgery can preserve some degree of vision in FEVR patients but amblyopia, reproliferation, and vitreous hemorrhage may limit long-term visual improvement.109 The ultimate goal of prophylactic laser treatment in FEVR is the prevention of retinal detachment or secondary macular exudate.
Marfan syndrome
Retinal detachment is the most serious eye complication, occurring in 10–25% of patients with Marfan syndrome. A total of 75% of retinal detachments occur before patients are 20 years old.110 Retinal detachment in Marfan syndrome is bilateral in about one-third of cases. Most Marfan syndrome patients with retinal detachment have a dislocated lens or have undergone previous lens surgery.111 These patients should, therefore be checked especially carefully.
Retinal detachment surgery can be complicated by insufficient pupil dilation, possible ectopia lentis, and multiple retinal breaks.112 The edge of the clear or cataractous subluxated lens hampers evaluation of the peripheral fundus.
Vitreous surgery should be considered in the following situations: failed scleral buckling; proliferative vitreoretinopathy; a posteriorly dislocated lens; a subluxated or cataractous lens not allowing an adequate evaluation of the fundus periphery, and giant retinal tears.113 Due to the thin sclera, the use of trochars should be avoided.
The results of surgery vary depending on the nature of the retinal tear and the presence of PVR. Generally, the results are comparable with those patients without Marfan syndrome and successful reattachment of the retina is achieved in 86%113 to 100%.112 But poorer visual outcome is likely to be encountered when there is concomitant lens dislocation or a history of intraocular surgery.112 The increased incidence of high myopia and peripheral lattice degeneration predisposes these eyes to retinal breaks. The high bilateral rate of retinal detachment may justify prophylactic laser treatment of lattice degenerations the fellow eye of these patients.111,112
Congenital X-linked retinoschisis
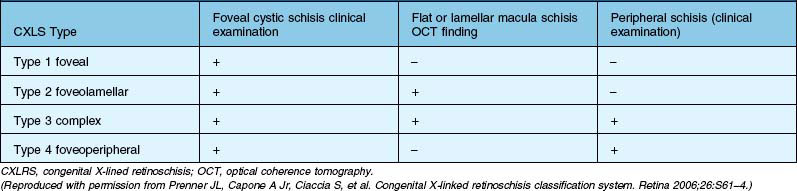
Congenital X-linked retinoschisis (CX-LRS) is an inherited retinal disease characterized by abnormal Müller’s cell pillars that allow schisis cavities to form. In 98–100% of the cases, these cavities in the foveal area, and the peripheral retinoschisis, usually inferotemporal, is present in about 50% of cases and is associated with severe vision-threatening complications, such as vitreous hemorrhage and retinal detachment. Visual function is often stable from childhood until age 40s, when deterioration occurs. The most severe disease form, which manifests as bullous, peripheral retinoschisis cavities, occurs primarily in patients younger than 10 years. The age of onset follows a bimodal distribution with one group of patients presenting in infancy with strabismus and nystagmus and a second group with poor vision and reading difficulties presenting at school age.114 There are a few case reports on children suffering from retinoschisis in the first year of life. In a series of five children who presented in the first 18 months of life, highly elevated bullous retinoschisis involving the macula was observed. In 9 of 10 eyes of these patients, the bullous retinoschisis cavity flattened, leaving only pigment demarcation lines within an observation time of 1–6.5 years.115 Surgical intervention for congenital retinoschisis is recommended under certain clinical conditions: rapid progression of the peripheral schisis cavity involving or threatening the macula, obscuration of the macula by the overhanging inner wall of a schisis cavity, dense nonclearing vitreous hemorrhage with a large schisis cavity hemorrhage, schisis combined with rhegmatogenous retinal detachment, and schisis combined with tractional retinal detachment, and macular pucker.31 Prenner et al.116 developed a new classification system for CX-LRS based on an improved understanding of the underlying pathologic lesion, and identified better surgical approach (Table 115.6).
Recent studies advocate vitrectomy for a bullous schisis cavity with or without hemorrhage in children with successful surgically induced posterior vitreous detachment.31,117,118 García-Arumí et al.117 performed posterior hyaloid dissection after injection of 0.05 mL of diluted preservative-free triamcinolone acetonide in an 8-month old child. Trese32 describe the adjuvant use of autologous plasmin for hyaloid separation. In pediatric eyes is it very difficult to mechanically achieve a complete posterior vitreous detachment. In most cases, the only way to relieve the vitreous traction is to remove the inner wall of the retinoschisis, since there is no effective way to remove the posterior cortical vitreous in children.119,120 Because the area of retinoschisis corresponds to an absolute scotoma on the visual field, removing the inner wall of retinoschisis has no effect on the patient’s visual field,119 but it results in loss of retinal ganglion cells and interneurons, eliminating any chance for future schisis cavity adhesion. Furthermore, eyes that undergo inner-wall retinotomy are prone to proliferation along the anterior leaflet of the schisis. Trese recommends the application of autologous plasmin, since this method eliminates the need for inner-wall retinectomy in up to 82% of eyes.32 They only performed inner-wall retinectomy on eyes with type 4 CX-LRS. Incision into the inner wall of the schisis cavity should be performed 2–3 mm anterior to the posterior border of the schisis cavity, some authors describe fluid drainage by use of a 42G cannula.117 Photocoagulation should applied to the border of the attached retina and the schisis cavity to prevent the progression of retinal splitting. Gas or silicone can be used for postoperative tamponade. Silicone oil is particularly recommended for younger children.
Authors described postoperative retinal reattachment in the pediatric eyes, but the functional results, however, are limited, the visual acuity improved in only 53% of cases, best reported postoperative visual acuity was 20/50 attained by a 6-year-old child.117,120 In a current series of cases, the main indication for unilateral vitrectomy in a 7 year old and bilaterally in a 17-year-old was foveal retinoschisis, which was associated with peripheral retinoschisis or peripheral inner holes. The vitreous surgery consisted of core vitrectomy, surgically induced posterior vitreous detachment, removal of the internal limiting membrane, and a 30% sulfur hexafluoride gas tamponade. The follow-up periods ranged from 6 months to 12 years. Restoration of the foveal depression with flattening of the schisis cavity was achieved in the first surgery in all three eyes. The visual acuity improved in all three cases. The anatomical result was normal foveal configuration with small cystoids spaces in the retina layer, but negative b-waves persisted in the affected eyes even 3 years after clinical resolution of the retinoschisis due to the inability of the intraretinal nerve fibers to reconnect despite repositioning of the retinoschisis.118
Rhegmatogenous retinal detachment in retinoschisis occurs if outer- and inner-wall holes develop in an area of peripheral schisis develop. The surgical principle is to coagulate outer-wall breaks and perform scleral buckling, if the location and size of the outer-wall breaks allow for support along the element. The advantage of scleral buckling is that it is less invasive than vitrectomy and provides support to the vitreous base. If scleral buckling fails, vitrectomy is recommended because it allows to improved treatment of the outer-wall break with laser after internal drainage and flattening the retina.31 Further indication for vitrectomy is tractional retinal detachment. In such cases, a combination of primary scleral buckling and vitrectomy is recommended. The surgical approach is scleral buckling for retinal detachment, particularly in the absence PVR and, the outer wall holes are located anterior to the equator, and vitrectomy for vitreous hemorrhage or PVR. PVR is the major reason for reoperation. However, multiple operations and the use of advanced vitreoretinal techniques to manage PVR-related complications are necessary for ultimate success in certain cases.121
Knobloch syndrome
Knobloch syndrome is a recessive syndrome of hereditary vitreoretinal degeneration with retinal detachment, high myopia, cataract, telecanthus, hypertelorism, and a high-arched palate.122 There is also a defect of the anterior midline scalp with involvement of the frontal bone. The presence of a congenital midline scalp defect should alert the clinician to possible underlying central nervous system and/or ocular pathology and should lead to consideration of further diagnostic evaluations and prophylactic measures. It is reported that fellow eyes should be treated with prophylactic retinal cryotherapy, but new reports are not available.123,124
Incontinentia pigmenti
Incontinentia pigmenti is a rare, X-linked, dominant disorder in which affected female infants develop characteristic abnormalities of the skin, central nervous system, hair, teeth, and eyes. Ocular abnormalities occur in about 35% of patients125 and consist of nystagmus, strabismus, microphthalmos, ptosis, blue sclera, pigmentation of the conjunctiva, corneal changes, cataract, optic atrophy, vitreous haemorrhage, myopia, and in 11.5% of cases, fibroblastic retinal detachment secondary to ischemic vasculopathy similar in appearance to retinopathy of prematurity. Fluorescein angiography indicates profound peripheral retinal ischemia, and abnormal arteriovenous connections in these eyes. The affected eyes develop neovascularizations, followed by preretinal fibrotic tissue at the temporal equator, and in a late stage, an extensive fibrovascular tissue pulls the retina anteriorly behind the lens. It seems likely that most retinal detachments occur within the first year of life, and all retinal detachments occur within the first 3 years of life. Most of the ocular abnormalities are unilateral, in those with bilateral involvement; one eye is usually less affected. Patients with NEMO mutation may have an increased risk of retinal pathology.126 For affected babies retinal screening should be performed under anesthesia as soon as possible after birth, and fluorescein angiography should be performed, when areas of avascular peripheral retina are noted by indirect ophthalmoscopy. Therapeutic intervention with indirect laser photocoagulation to the peripheral ischemic retina should be performed as soon as the diagnosis is made, some authors describe successfully resolution of the vasculopathy after early laser photocoagulation.127 Today, the intravitreal injection of anti-VEGF could be a further adjunctive treatment option. Five days after of intravitreal injection of 1.25 mg/0.05 mL bevacizumab in an 11-month-old boy, extensive neovascularization transformed into sclerotic ghost vessels, and vitrectomy was facilitated.128
Norrie disease
Norrie disease is a bilateral X-linked recessive syndrome of ocular dysgenesis with progressive auditory and mental impairment. Norrie disease is believed to result from mutations in the 28-kb Norrie disease protein (NDP) gene, located on chromosome Xp11.3. The disease is characterized by a retinal falciform fold, retinal detachment, repeated vitreous hemorrhage, retrolental membrane formation, and finally clinical progression from these early retinal and vitreous changes to the more typical retrolental vascularized detached retina. Most surgical interventions have been disappointing.129 Only one successful vitrectomy of a 15-month-old child has been described.130 Chow et al. reported the first case of prophylactic laser treatment in a patient who was diagnosed with Norrie disease by genetic testing with amniocentesis at week 23 of gestation; 1 day postpartum laser photocoagulation was applied to the avascular retina bilaterally. Complete regression of extraretinal fibrovascular proliferation was observed 1 month after laser treatment. No retinal detachment had occurred at 24 months. Teller visual acuity at 23 months of life was 20/100 in both eyes. The patient’s vision and developmental milestones were age appropriate.131
Malformations
Anomalies of the papilla
Morning-glory disc anomaly is a descriptive classification for a characteristic lesion of the optic nerve that is often associated with serous retinal detachment. Systemic associations are present in some patients. A wide range of posterior segment abnormalities can be observed. Mild optic disc dysplasia or pit formation has no functional consequences and is underdiagnosed. More severe colobomas or related abnormalities, such as the morning-glory anomaly, often lead to poor visual acuity. Molecular biology allows detection of the mutations in the PAX2 gene in approximately 50% of cases. The observation of an optic disc coloboma or related abnormality must stimulate nephrologic investigations to check for renal hypoplasia, a potentially life-threatening disease.131a Nonrhegmatogenous retinal detachment occurs in one-third of cases. Detachment may be limited to the peripapillary retina or be extensive. Retinal detachment is usually seen after the first year of life. The benefit of vitreous surgery with drainage of subretinal fluid, gas tamponade, or silicone oil, and peripapillary photocoagulation has been established.
Congenital pits of the optic nerve vary in size, shape, depth, and location. They often appear as small, hypopigmented, yellow or whitish, oval or round excavated defects, most often within the inferior temporal portion of the optic cup. They are bilateral in 15% of patients, covered or filled with a veil of tissue. Some 40–60% of patients with optic pits develop nonrhegmatogenous serous macular detachments (Fig. 115.11A,B). These fluid-filled cystic maculopathies can develop into lamellar macular holes. There are four etiologic theories: (1) vitreous fluid by way of the pit or a macular hole; (2) leakage from vessels within the pit; (3) fluid from the orbital space surrounding the dura; and (4) cerebrospinal fluid originating from the subarachnoid space.132 A tear in the diaphanous tissue overlying the optic nerve pit may be responsible for the development of serous macular detachment133; this is consistent with findings in similar conditions, such as retinal detachment, in association with chorioretinal coloboma. These tears may be quite subtle, and careful biomicroscopic examination is required to appreciate them. Initially, conservative management is recommended, as 25% of optic disc pit maculopathies resolve spontaneously.132 The value of laser photocoagulation along the temporal disc margin is not very convincing, since even worse retinal detachments or re-detachments were observed.132
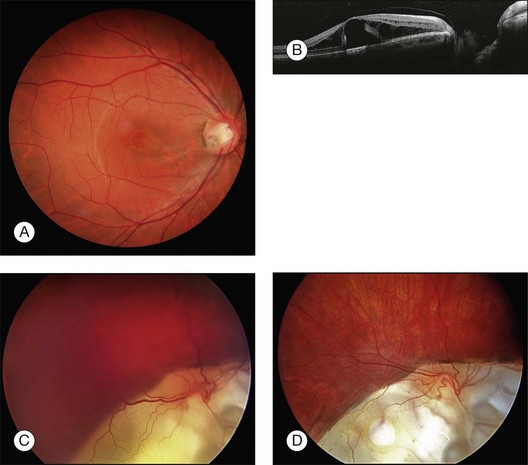
In a recent review of results of vitreoretinal surgery for treatment of an optic pit maculopathy the most widely accepted treatment for both adult patients and children is a pars plana vitrectomy with or without ILM peeling, with or without endolaser photocoagulation and C3F8 endotamponade.132
There are several case reports on successfully performed vitrectomies with and without ILM-peeling in children aged 7–9 years old.134–136 The removal of the posterior hyaloid face from the macula is an essential element of successful treatment, but may be difficult to achieve in pediatric eyes. Georgalas et al.137 reported that surgical PVD seemed impossible in the case of a 5-year-old boy with optic disc pit maculopathy, and therefore the posterior vitreous was peeled off from the macular area within the retinal arcades and air was used as an endotamponade. Some authors135,138 performed laser photocoagulation in the peripapillary area during vitrectomy, but others137 did not recommend laser photocoagulation: first to avoid any adverse effects of the laser treatment in the papillomacular region and, second, because the rationale for inhibition of fluid transportation within the inner retinal layers of the macula through laser photocoagulation was weak.
Jalil et al.139 have suggested an alternative approach with induction of a posterior vitreous detachment and internal limiting membrane peeling. A 42G subretinal cannula was connected to a “backflush” flute handle and introduced into the subretinal space temporal to the fovea within the subretinal fluid cavity. No retinopexy was performed at the site of the subretinal fluid drainage and the retina remained attached with gas tamponade.
Migration of gas or silicone oil into the subretinal space has been reported.140 Furthermore, intracranial migration of silicone oil after vitreoretinal surgery for the treatment of optic disc maculopathy was described.141 Therefore, if possible, a gas tamponade should preferably be used.
Coloboma
Coloboma of the fundus is a congenital defect caused by the faulty closure of the embryonal fissure. It occurs in 0.14% of the general population.142 The literature quotes a 40% occurrence of retinal detachment in these patients, typically in the second decade of life.143 Near the margin of the coloboma, the retina splits into two layers.144 The inner layer continues as an intercalary membrane on to the coloboma, while the outer layer turns back, becomes disorganized, and fuses with the retinal pigment epithelium. The choroid is terminated as a distinct pigmented layer peripheral to this point of reversal. The split in the retinal layer has been identified at the level of inner nuclear layer or outer plexiform layer, or both. The junction where this reversal occurs is a locus minoris resistentiae. The intercalary membrane progressively becomes thinner as it is traced centrally. Breaks can occur at the junction and in the intercalary membrane. Peripheral retinal breaks are also observed. The preferred technique for treatment of retinal detachment associated with choroidal coloboma is vitrectomy with either long-acting gas or oil tamponade. The use of silicone oil has the advantage of being a long-term tamponade of the whole colobomatous border.
Breaks in the intercalary membrane can be identified pre- or intraoperatively, whereas the breaks at the locus minoris resistentiae are not identifiable but can be expected to be located along the coloboma border. Treatment of the pigmented fundus just beyond the coloboma with laser should close these breaks and eliminates the need to close the breaks in the intercalary membrane. The diode laser is preferred to the argon laser because it is less likely to damage the nerve fiber layer. Tansu et al. recommend endolaser barrier of three to four rows along the border of the coloboma. Near the optic disc and papillomacular bundle, we perform a gentle endolaser coagulation as consistent with other authors.146 If adequate chorioretinal adhesion can be achieved around the coloboma, laser of the papillomacular bundle and optic disc may not be necessary (Fig. 115.11C,D).145 After vitrectomy and silicone oil removal, optical coherence tomography showed persistent detachment of the intercalary membrane in most patients. These findings emphasize the importance of sealing the junction between the intercalary membrane and extra colobomatous retina with a laser barrier.146
The placement of an encircling band is done on a purely empirical basis. Several authors observed no difference in the outcome between the buckled and nonbuckled eyes. Peripheral laser photocoagulation along the ora serrata reduces the risk of recurrent retinal detachment after silicone oil removal.143,145
Persistent hyperplastic primary vitreous
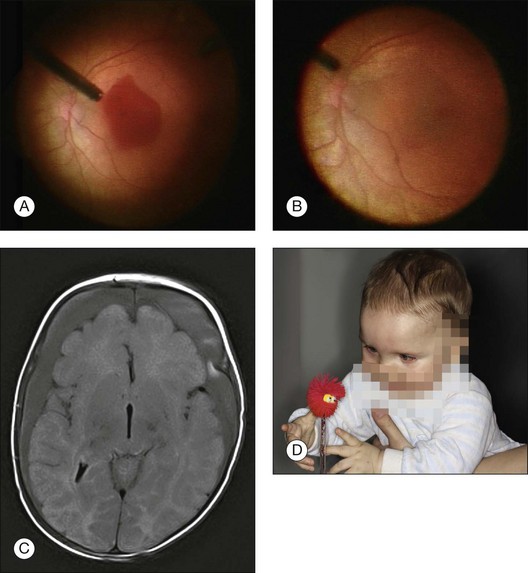
Persistent hyperplastic primary vitreous (PHPV), also known as persistent fetal vasculature, is a congenital anomaly of the eye that results from failure of the embryological, primary vitreous, and the hyaloid vasculature to regress. It typically presents unilaterally, without associated systemic findings in normal full-term infants. In rare cases, bilateral PHPV has also been described.147 Most cases of PHPV are sporadic, but it can be inherited as an autosomal dominant or recessive trait.148 PHPV is subclassified into three types: (1) most often an anterior PHPV (retrolental fibrovascular membrane, elongated ciliary processes, cataract, microphthalmia); (2) posterior PHPV (vitreous membrane and stalk, retinal fold, traction retinal detachment, hypoplastic optic nerve and macula, microphthalmia); (3) a combination of anterior and posterior PHPV (Fig. 115.12). Without surgery, most eyes with PHPV will develop severe glaucoma, retinal detachment, intraocular hemorrhage, and/or phthisis early in life. If left untreated, enucleation may become necessary. To prevent these complications, many authors recommend vitreoretinal surgery without delay if light perception is present, especially in rare cases of bilateral PHPV.149 But in eyes with severe PHPV with an unrecordable visual evoked potential, no light perception, and intense afferent pupillary defect, an operation should not be performed.150
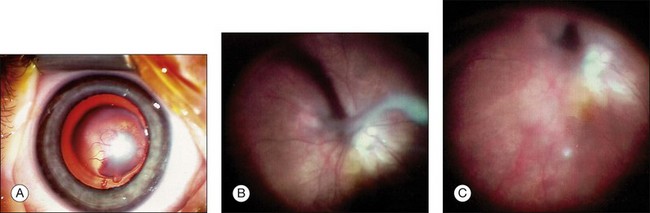
The vitrectomy port location should be varied according to the age of the child, but in these often microphthalmic eyes, the ora serrata may be anteriorly displaced, the pars plana may be absent, or the anterior retina may insert directly onto the pars plicata ciliary body. It is recommended to place the sclerotomies as anteriorly as possible, especially if retinal detachment is determined by preoperative ultrasonography.151 Transillumination is occasionally helpful to locate the ora serrata. To avoid manipulations of the vitreous base or peripheral retina, which may be drawn anteriorly, other authors prefer an approach for vitrectomy through the iris root; incisions are made at the limbus, but this is associated with the danger of severe iris lesions. After lentectomy, diathermy should be used when vessels are visible and the retrolental membrane has to be cut from the ciliary processes using the vitreous cutter. In case of nonaxial lens opacification, lens-sparing vitrectomy should be performed, and Shaikh and Trese152 recommend dividing the stalk immediately on entry. After division of the stalk and hemostasis, with diathermy if necessary, vitrectomy should be carried out with dissection of epiretinal membranes in the posterior pole. The authors presume that the manipulation of the stalk by vitrectomy and diathermy before division result in damage to the lens capsule and resultant cataract formation.
Membrane peeling is necessary in cases of tractional retinal folds or tractional detachment. In the purely anterior form, the cataract should be removed to provide clear media for visual rehabilitation. Eyes with a posterior PHPV have poor visual results due to posterior pole abnormalities. However, despite posterior-segment involvement, surgical treatment of PHPV can result in functional visual outcome. The degree of ocular malformation, however, will ultimately limit the amount of visual improvement. A total of 71% of patients undergoing vitrectomy for a combined form of anterior and posterior PHPV achieve 20/300 or better.151 In another study,150 six of 24 eyes operated for PHPV maintained Snellen visual acuity and approximately 50% of patients undergoing surgery for persistent fetal vasculature may achieve useful vision.153
In a series of patients suffering from the rare condition of bilateral combined anterior and posterior PHPV, Walsh et al.149 found that vitrectomy with or without lensectomy is beneficial: 69% of patients maintained at least light perception vision in at least 1 eye at the last follow-up. Of the 28 operated eyes in 16 patients with follow-up data, only 11% eyes were phthisical at the last follow-up.
After surgery, every child should have a short trial (2 months) of occlusion therapy. This should be terminated if no visual improvement is noted, to avoid undue psychosocial impairment.151 Early age at surgery, prompt optical correction with contact lens, and aggressive occlusion therapy are required for successful visual rehabilitation.
1 Birch EE, Stager DR. The critical period for surgical treatment of dense congenital unilateral cataract. Invest Ophthalmol Vis Sci. 1996;37:1532–1538.
2 Hanse W. Amblyopie. In: Kaufmann H, ed. Strabismus. Stuttgart: Ferdinand Enke; 1995:285–395.
3 Smiddy WE, Loupe DN, Michels RG, et al. Refractive changes after scleral buckling surgery. Arch Ophthalmol. 1989;107:1469–1471.
4 Chow DR, Ferrone PJ, Trese MT. Refractive changes associated with scleral buckling and division in retinopathy of prematurity. Arch Ophthalmol. 1998;116:1446–1448.
5 Lang J, ed. Strabismus, Schielformen, Therapien. Bern: Hans Huber, 2003.
6 Hairston RJ, Maguire AM, Vitale S, et al. Morphometric analysis of pars plana development in humans. Retina. 1997;17:135–138.
7 Möller H. Milestones and normative data. In: Taylor D, ed. Pediatric ophthalmology. Philadelphia: Lippincott, Williams & Wilkins, 1997.
8 Gordon RA, Donzis PB. Refractive development of the human eye. Arch Ophthalmol. 1985;103:785–789.
9 Inagaki Y. The rapid change of corneal curvature in the neonatal period and infancy. Arch Ophthalmol. 1986;104:1026–1027.
10 Miller-Meeks MJ, Bennett SR, Keech RV, et al. Myopia induced by vitreous hemorrhage. Am J Ophthalmol. 1990;109:199–203.
11 Hubel DH, Wiesel TN. The period of susceptibility to the physiological effects of unilateral eye closure in kittens. J Physiol. 1970;206:419–436.
12 Mohney BG. Axial myopia associated with dense vitreous hemorrhage of the neonate. J AAPOS. 2002;6:348–353.
13 Bonomo PP. Pars plana and ora serrata anatomotopographic study of fetal eyes. Acta Ophthalmol (Copenh). 1989;67:145–150.
14 Aiello AL, Tran VT, Rao NA. Postnatal development of the ciliary body and pars plana. A morphometric study in childhood. Arch Ophthalmol. 1992;110:802–805.
15 Friberg TR, Lace JW. A comparison of the elastic properties of human choroid and sclera. Exp Eye Res. 1988;47:429–436.
16 Sebag J. Vitreous pathobiology. In: Tasman W, ed. Duane’s ophthalmology. Philadelphia: Lippincott Williams & Wilkins, 2004.
17 Gallemore RP, Bokosky JE. Penetrating keratoplasty with vitreoretinal surgery using the Eckardt temporary keratoprosthesis: modified technique allowing use of larger corneal grafts. Cornea. 1995;14:33–38.
18 Garcia-Valenzuela E, Blair NP, Shapiro MJ, et al. Outcome of vitreoretinal surgery and penetrating keratoplasty using temporary keratoprosthesis. Retina. 1999;19:424–429.
19 Roters S, Szurman P, Hermes S, et al. Outcome of combined penetrating keratoplasty with vitreoretinal surgery for management of severe ocular injuries. Retina. 2003;23:48–56.
20 Vanathi M, Panda A, Vengayil S, et al. Pediatric keratoplasty. Surv Ophthalmol. 2009;54:245–271.
21 Reidy JJ. Penetrating keratoplasty in infancy and early childhood. Curr Opin Ophthalmol. 2001;12:258–261.
22 Andreoli CM, Andreoli MT, Kloek CE, et al. Low rate of endophthalmitis in a large series of open globe injuries. Am J Ophthalmol. 2009;147:601–608.
23 Chen TC, Bhatia LS, Walton DS. Complications of pediatric lensectomy in 193 eyes. Ophthalmic Surg Lasers Imaging. 2005;36:6–13.
24 Meier P, Sterker I, Tegetmeyer H, et al. [23-gauge-lentectomy for the treatment of congenital cataract]. Ophthalmologe. 2010;107:241–245.
25 Gonzales CR, Boshra J, Schwartz SD. 25-Gauge pars plicata vitrectomy for stage 4 and 5 retinopathy of prematurity. Retina. 2006;26:S42–S46.
26 Woo SJ, Park KH, Hwang JM, et al. Risk factors associated with sclerotomy leakage and postoperative hypotony after 23-gauge transconjunctival sutureless vitrectomy. Retina. 2009;29:456–463.
27 Thompson JT. Advantages and limitations of small gauge vitrectomy. Surv Ophthalmol. 2011;56:162–172.
28 Peyman GA, Canakis C, Livir-Rallatos C, et al. Small-size pediatric vitrectomy wide-angle contact lens. Am J Ophthalmol. 2003;135:236–237.
29 Recchia FM, Scott IU, Brown GC, et al. Small-gauge pars plana vitrectomy: a report by the American Academy of Ophthalmology. Ophthalmology. 2010;117:1851–1857.
30 Benz MS, Packo KH, Gonzalez V, et al. A placebo-controlled trial of microplasmin intravitreous injection to facilitate posterior vitreous detachment before vitrectomy. Ophthalmology. 2010;117:791–797.
31 Wu WC, Drenser KA, Capone A, et al. Plasmin enzyme-assisted vitreoretinal surgery in congenital X-linked retinoschisis: surgical techniques based on a new classification system. Retina. 2007;27:1079–1085.
32 Trese MT. Enzymatic-assisted vitrectomy. Eye (Lond). 2002;16:365–368.
33 Scott IU, Flynn HW, Jr., Azen SP, et al. Silicone oil in the repair of pediatric complex retinal detachments: a prospective, observational, multicenter study. Ophthalmology. 1999;106:1399–1408.
34 Jackson TL, Vote B, Knight BC, et al. Safety testing of infracyanine green using retinal pigment epithelium and glial cell cultures. Invest Ophthalmol Vis Sci. 2004;45:3697–3703.
35 Gupta A, Rahman I, Leatherbarrow B. Open globe injuries in children: factors predictive of a poor final visual acuity. Eye (Lond). 2009;23:621–625.
36 Jandeck C, Kellner U, Bornfeld N, et al. Open globe injuries in children. Graefe’s Arch Clin Exp Ophthalmol. 2000;238:420–426.
37 Lee CH, Lee L, Kao LY, et al. Prognostic indicators of open globe injuries in children. Am J Emerg Med. 2009;27:530–535.
38 Mittra RA, Mieler WF. Controversies in the management of open-globe injuries involving the posterior segment. Surv Ophthalmol. 1999;44:215–225.
39 Kuhn F, Mester V, Morris R. A proactive treatment approach for eyes with perforating injury. Klin Monbl Augenheilkd. 2004;221:622–628.
40 Kuhn F, ed. Ocular trauma in children and elderly patients. Berlin: Springer; 2008:417–435.
41 Heimann K. Silicone oil in penetrating eye injuries. In: Alfaro DV, ed. Vitreoretinal surgery of the injured eye. Philadelphia: Lippincott-Raven; 1999:211–224.
42 Boiko E, Shishkin M, Dolgih V. Indications for the encircling buckle (EB) procedure in severe open-globe eye injury (OGI). Meeting abstract; 2008. Available at http://www.egms.de/static/de/meetings/rg2008/08rg024.shtml
43 Azad RV, Kumar N, Sharma YR, et al. Role of prophylactic scleral buckling in the management of retained intraocular foreign bodies. Clin Experiment Ophthalmol. 2004;32:58–61.
44 Ahmadieh H, Soheilian M, Sajjadi H, et al. Vitrectomy in ocular trauma. Factors influencing final visual outcome. Retina. 1993;13:107–113.
45 Moisseiev J, Vidne O, Treister G. Vitrectomy and silicone oil injection in pediatric patients. Retina. 1998;18:221–227.
46 Sarrazin L, Averbukh E, Halpert M, et al. Traumatic pediatric retinal detachment: a comparison between open and closed globe injuries. Am J Ophthalmol. 2004;137:1042–1049.
47 Farr AK, Hairston RJ, Humayun MU, et al. Open globe injuries in children: a retrospective analysis. J Pediatr Ophthalmol Strabismus. 2001;38:72–77.
48 Freemann HM. Giant retinal tears. Duane’s clinical ophthalmology. Tasman W, ed. Duane’s clinical ophthalmology. Philadelphia: Lippincott Williams & Wilkins; 1996;Vol. 6(69):1–11.
49 Haring G, Wiechens B. Long-term results after scleral buckling surgery in uncomplicated juvenile retinal detachment without proliferative vitreoretinopathy. Retina. 1998;18:501–505.
50 Wang NK, Chen YP, Yeung L, et al. Traumatic pediatric retinal detachment following open globe injury. Ophthalmologica. 2007;221:255–263.
51 Chen YJ. Vitrectomy and internal limiting membrane peeling of a traumatic macular hole with retinal folds. Case Report Ophthalmol. 2011;2:78–83.
52 Kuhn F, Morris R, Mester V, et al. Internal limiting membrane removal for traumatic macular holes. Ophthalmic Surg Lasers. 2001;32:308–315.
53 Yanagiya N, Akiba J, Takahashi M, et al. Clinical characteristics of traumatic macular holes. Jpn J Ophthalmol. 1996;40:544–547.
54 Lai MM, Joshi MM, Trese MT. Spontaneous resolution of traumatic macular hole-related retinal detachment. Am J Ophthalmol. 2006;141:1148–1151.
55 Mitamura Y, Saito W, Ishida M, et al. Spontaneous closure of traumatic macular hole. Retina. 2001;21:385–389.
56 Kusaka S, Fujikado T, Ikeda T, et al. Spontaneous disappearance of traumatic macular holes in young patients. Am J Ophthalmol. 1997;123:837–839.
57 Yeshurun I, Guerrero-Naranjo JL, Quiroz-Mercado H. Spontaneous closure of a large traumatic macular hole in a young patient. Am J Ophthalmol. 2002;134:602–603.
58 Wu WC, Drenser KA, Trese MT, et al. Pediatric traumatic macular hole: results of autologous plasmin enzyme-assisted vitrectomy. Am J Ophthalmol. 2007;144:668–672.
59 Johnson RN, McDonald HR, Lewis H, et al. Traumatic macular hole: observations, pathogenesis, and results of vitrectomy surgery. Ophthalmology. 2001;108:853–857.
60 García-Arumí J, Corcostegui B, Cavero L, et al. The role of vitreoretinal surgery in the treatment of posttraumatic macular hole. Retina. 1997;17:372–377.
61 Ferrone PJ, de Juan E, Jr. Vitreous hemorrhage in infants. Arch Ophthalmol. 1994;112:1185–1189.
62 Cleary PE, Ryan SJ. Vitrectomy in penetrating eye injury. Results of a controlled trial of vitrectomy in an experimental posterior penetrating eye injury in the rhesus monkey. Arch Ophthalmol. 1981;99:287–292.
63 Schloff S, Mullaney PB, Armstrong DC, et al. Retinal findings in children with intracranial hemorrhage. Ophthalmology. 2002;109:1472–1476.
64 Reddy AR, Clarke M, Long VW. Unilateral retinal hemorrhages with subarachnoid hemorrhage in a 5-week-old infant: is this nonaccidental injury? Eur J Ophthalmol. 2010;20:799–801.
65 McLellan NJ, Prasad R, Punt J. Spontaneous subhyaloid and retinal haemorrhages in an infant. Arch Dis Child. 1986;61:1130–1132.
66 Bhardwaj G, Jacobs MB, Moran KT, et al. Terson syndrome with ipsilateral severe hemorrhagic retinopathy in a 7-month-old child. J AAPOS. 2010;14:441–443.
67 Morris R, Kuhn F, Witherspoon CD. Hemorrhagic macular cysts. Ophthalmology. 1994;101:1.
68 de Vries-Knoppert W. Vitreous findings in a patient with Terson’s syndrome. Doc Ophthalmol. 1995;90:75–80.
69 Velikay M, Datlinger P, Stolba U, et al. Retinal detachment with severe proliferative vitreoretinopathy in Terson syndrome. Ophthalmology. 1994;101:35–37.
70 Meier P, Schmitz F, Wiedemann P. Vitrectomy for pre-macular hemorrhagic cyst in children and young adults. Graefes Arch Clin Exp Ophthalmol. 2005;243:824–828.
71 Koozekanani DD, Weinberg DV, Dubis AM, et al. Hemorrhagic retinoschisis in shaken baby syndrome imaged with spectral domain optical coherence tomography. Ophthalmic Surg Lasers Imaging. 2010. [Epub ahead of print]
72 Muni RH, Kohly RP, Sohn EH, et al. Hand-held spectral domain optical coherence tomography finding in shaken-baby syndrome. Retina. 2010;30:S45–S50.
73 Gaynon MW, Koh K, Marmor MF, et al. Retinal folds in the shaken baby syndrome. Am J Ophthalmol. 1988;106:423–425.
74 Fivgas GD, Capone A, Jr. Pediatric rhegmatogenous retinal detachment. Retina. 2001;21:101–106.
75 Wang NK, Tsai CH, Chen YP, et al. Pediatric rhegmatogenous retinal detachment in East Asians. Ophthalmology. 2005;112:1890–1895.
76 Lee RW, Mayer EJ, Markham RH. The aetiology of paediatric rhegmatogenous retinal detachment: 15 years experience. Eye. 2008;22:636–640.
77 Feltgen N, Weiss C, Wolf S, et al. Scleral buckling versus primary vitrectomy in rhegmatogenous retinal detachment study (SPR Study): recruitment list evaluation. Study report no. 2. Graefes Arch Clin Exp Ophthalmol. 2007;245:803–809.
78 Bansal AS, Hubbard GB, 3rd. Peripheral retinal findings in highly myopic children < or =10 years of age. Retina. 2010;30:S15–S19.
79 Ho AC, Yannuzzi LA, Pisicano K, et al. The natural history of idiopathic subfoveal choroidal neovascularization. Ophthalmology. 1995;102:782–789.
80 Cakir M, Cekic O, Yilmaz OF. Intravitreal bevacizumab for idiopathic choroidal neovascularization. J AAPOS. 2009;13:296–298.
81 Goodwin P, Shields CL, Ramasubramanian A, et al. Ranibizumab for coloboma-related choroidal neovascular membrane in a child. J AAPOS. 2009;13:616–617.
82 Cackett P, Wong D, Cheung CM. Combined intravitreal bevacizumab and argon laser treatment for Coats’ disease. Acta Ophthalmol. 2010;88:e48–e49.
83 Kohly RP, Muni RH, Kertes PJ, et al. Management of pediatric choroidal neovascular membranes with intravitreal anti-VEGF agents: a retrospective consecutive case series. Can J Ophthalmol. 2011;46:46–50.
84 Coats G. Forms of retinal disease with massive exudation. R Lond Ophthalm Hosp Rep. 1908;17:440–525.
85 Shields JA, Shields CL. Review: coats disease: the 2001 LuEsther T. Mertz lecture. Retina. 2002;22:80–91.
86 Shields JA, Shields CL, Honavar SG, et al. Classification and management of Coats disease: the 2000 Proctor Lecture. Am J Ophthalmol. 2001;131:572–583.
87 Jumper JM, Pomerleau D, McDonald HR, et al. Macular fibrosis in Coats disease. Retina. 2010;30:S9–S14.
88 Venkatesh P, Mandal S, Garg S. Management of Coats disease with bevacizumab in 2 patients. Can J Ophthalmol. 2008;43:245–246.
89 Stergiou PK, Symeonidis C, Dimitrakos SA. Coats’ disease: treatment with intravitreal bevacizumab and laser photocoagulation. Acta Ophthalmol. 2009;87:687–688.
90 Kaul S, Uparkar M, Mody K, et al. Intravitreal anti-vascular endothelial growth factor agents as an adjunct in the management of Coats’ disease in children. Indian J Ophthalmol. 2010;58:76–78.
91 Lin CJ, Hwang JF, Chen YT, et al. The effect of intravitreal bevacizumab in the treatment of Coats disease in children. Retina. 2010;30:617–622.
92 Murthy R, Honavar SG, Vemuganti GK, et al. Systemic metastasis following hyphema drainage in an unsuspected retinoblastoma. J Pediatr Ophthalmol Strabismus. 2007;44:120–123.
93 Grabowska A, Calvo JP, Fernandez-Zubillaga A, et al. A magnetic resonance imaging diagnostic dilemma: diffuse infiltrating retinoblastoma versus coats’ disease. J Pediatr Ophthalmol Strabismus. 2010;47:e1–e3.
94 Stickler GB, Belau PG, Farrell FJ, et al. Hereditary Progressive Arthro-Ophthalmopathy. Mayo Clin Proc. 1965;40:433–455.
95 Stickler GB, Hughes W, Houchin P. Clinical features of hereditary progressive arthro-ophthalmopathy (Stickler syndrome): a survey. Genet Med. 2001;3:192–196.
96 Snead MP, Yates JR. Clinical and Molecular genetics of Stickler syndrome. J Med Genet. 1999;36:353–359.
97 Abeysiri P, Bunce C, da Cruz L. Outcomes of surgery for retinal detachment in patients with Stickler syndrome: a comparison of two sequential 20-year cohorts. Graefes Arch Clin Exp Ophthalmol. 2007;245:1633–1638.
98 Carroll C, Papaioannou D, Rees A, et al. The clinical effectiveness and safety of prophylactic retinal interventions to reduce the risk of retinal detachment and subsequent vision loss in adults and children with Stickler syndrome: a systematic review. Health Technol Assess. 2011;15:iii–xiv. 1–62
99 Pollack A, Milstein A, Oliver M, et al. Circumferential argon laser photocoagulation for prevention of retinal detachment. Eye (Lond). 1994;8(Pt 4):419–422.
100 Leiba H, Oliver M, Pollack A. Prophylactic laser photocoagulation in Stickler syndrome. Eye (Lond). 1996;10(Pt 6):701–708.
101 Snead MP. Hereditary vitreopathy. Eye (Lond). 1996;10(Pt 6):653–663.
102 Subramanian S, Gamanagatti S, Sinha A, et al. Kniest syndrome. Indian Pediatr. 2007;44:931–933.
103 Makino I, Watanabe M, Okamoto N, et al. A case of retinal detachment in Kniest dysplasia treated with vitreous surgery. Nippon Ganka Gakkai Zasshi. 1997;101:734–737.
104 Zhao S, Overbeek PA. Elevated TGFbeta signaling inhibits ocular vascular development. Dev Biol. 2001;237:45–53.
105 Pendergast SD, Trese MT. Familial exudative vitreoretinopathy. Results of surgical management. Ophthalmology. 1998;105:1015–1023.
106 Quiram PA, Drenser KA, Lai MM, et al. Treatment of vascularly active familial exudative vitreoretinopathy with pegaptanib sodium (Macugen). Retina. 2008;28:S8–S12.
107 Tagami M, Kusuhara S, Honda S, et al. Rapid regression of retinal hemorrhage and neovascularization in a case of familial exudative vitreoretinopathy treated with intravitreal bevacizumab. Graefes Arch Clin Exp Ophthalmol. 2008;246:1787–1789.
108 Shubert A, Tasman W. Familial exudative vitreoretinopathy: surgical intervention and visual acuity outcomes. Graefes Arch Clin Exp Ophthalmol. 1997;235:490–493.
109 Glazer LC, Maguire A, Blumenkranz MS, et al. Improved surgical treatment of familial exudative vitreoretinopathy in children. Am J Ophthalmol. 1995;120:471–479.
110 Maumenee IH. The eye in the Marfan syndrome. Trans Am Ophthalmol Soc. 1981;79:684–733.
111 Abboud EB. Retinal detachment surgery in Marfan’s syndrome. Retina. 1998;18:405–409.
112 Lee SY, Ang CL. Results of retinal detachment surgery in Marfan syndrome in Asians. Retina. 2003;23:24–29.
113 Sharma T, Gopal L, Shanmugam MP, et al. Retinal detachment in Marfan syndrome: clinical characteristics and surgical outcome. Retina. 2002;22:423–428.
114 Sikkink SK, Biswas S, Parry NR, et al. X-linked retinoschisis: an update. J Med Genet. 2007;44:225–232.
115 George ND, Yates JR, Bradshaw K, et al. Infantile presentation of X linked retinoschisis. Br J Ophthalmol. 1995;79:653–657.
116 Prenner JL, Capone A, Jr., Ciaccia S, et al. Congenital X-linked retinoschisis classification system. Retina. 2006;26:S61–S64.
117 García-Arumí J, Corcóstegui IA, Navarro R, et al. Vitreoretinal surgery without schisis cavity excision for the management of juvenile X linked retinoschisis. Br J Ophthalmol. 2008;92:1558–1560.
118 Ikeda F, Iida T, Kishi S. Resolution of retinoschisis after vitreous surgery in X-linked retinoschisis. Ophthalmology. 2008;115:718–722. e1
119 Ferrone PJ, Trese MT, Lewis H. Vitreoretinal surgery for complications of congenital retinoschisis. Am J Ophthalmol. 1997;123:742–747.
120 Lee JJ, Kim JH, Kim SY, et al. Infantile vitreous hemorrhage as the initial presentation of X-linked juvenile retinoschisis. Korean J Ophthalmol. 2009;23:118–120.
121 Rosenfeld PJ, Flynn HW, Jr., McDonald HR, et al. Outcomes of vitreoretinal surgery in patients with X-linked retinoschisis. Ophthalmic Surg Lasers. 1998;29:190–197.
122 Knobloch WH, Layer JM. Clefting syndromes associated with retinal detachment. Am J Ophthalmol. 1972;73:517–530.
123 Seaver LH, Joffe L, Spark RP, et al. Congenital scalp defects and vitreoretinal degeneration: redefining the Knobloch syndrome. Am J Med Genet. 1993;46:203–208.
124 Sniderman LC, Koenekoop RK, O’Gorman AM, et al. Knobloch syndrome involving midline scalp defect of the frontal region. Am J Med Genet. 2000;90:146–149.
125 Carney RG. Incontinentia pigmenti. A world statistical analysis. Arch Dermatol. 1976;112:535–542.
126 O’Doherty M, Mc Creery K, Green AJ, et al. Incontinentia pigmenti–ophthalmological observation of a series of cases and review of the literature. Br J Ophthalmol. 2011;95:11–16.
127 Sanghi G, Dogra MR, Ray M, et al. Predominant exudative retinopathy in incontinentia pigmenti and clinical course after peripheral laser photocoagulation. Indian J Ophthalmol. 2011;59:255–256.
128 Lin KL, Hirose T, Kroll AJ, et al. Prospects for treatment of pediatric vitreoretinal diseases with vascular endothelial growth factor inhibition. Semin Ophthalmol. 2009;24:70–76.
129 Warden SM, Andreoli CM, Mukai S. The Wnt signaling pathway in familial exudative vitreoretinopathy and Norrie disease. Semin Ophthalmol. 2007;22:211–217.
130 Shima C, Kusaka S, Kondo H, et al. Lens-sparing vitrectomy effective for reattachment of newly developed falciform retinal detachment in a patient with Norrie disease. Arch Ophthalmol. 2009;127:579–580.
131 Chow CC, Kiernan DF, Chau FY, et al. Laser photocoagulation at birth prevents blindness in Norrie’s disease diagnosed using amniocentesis. Ophthalmology. 2010;117:2402–2406.
131a Dureau P, Attie-Bitach T, Salomon R, et al. Renal coloboba syndrome. Ophthalmology. 2001;108:1912–1916.
132 Georgalas I, Ladas I, Georgopoulos G, et al. Optic disc pit: a review. Graefes Arch Clin Exp Ophthalmol. 2011;249:1113–1122.
133 Postel EA, Pulido JS, McNamara JA, et al. The etiology and treatment of macular detachment associated with optic nerve pits and related anomalies. Trans Am Ophthalmol Soc. 1998;96:73–93.
134 Snead MP, James N, Jacobs PM. Vitrectomy, argon laser, and gas tamponade for serous retinal detachment associated with an optic disc pit: a case report. Br J Ophthalmol. 1991;75:381–382.
135 Ishikawa K, Terasaki H, Mori M, et al. Optical coherence tomography before and after vitrectomy with internal limiting membrane removal in a child with optic disc pit maculopathy. Jpn J Ophthalmol. 2005;49:411–413.
136 Hirakata A, Hida T, Wakabayashi T, et al. Unusual posterior hyaloid strand in a young child with optic disc pit maculopathy: intraoperative and histopathological findings. Jpn J Ophthalmol. 2005;49:264–266.
137 Georgalas I, Kouri A, Ladas I, et al. Optic disc pit maculopathy treated with vitrectomy, internal limiting membrane peeling, and air in a 5-year-old boy. Can J Ophthalmol. 2010;45:189–191.
138 Ghosh YK, Banerjee S, Konstantinidis A, et al. Surgical management of optic disc pit associated maculopathy. Eur J Ophthalmol. 2008;18:142–146.
139 Jalil A, Stavrakas P, Dhawahir-Scala FE, et al. Drainage of subretinal fluid in optic disc pit maculopathy using subretinal 42-gauge cannula: a new surgical approach. Graefes Arch Clin Exp Ophthalmol. 2010;248:751–753.
140 Becht C, Senn P, Lange AP. Delayed occurrence of subretinal silicone oil after retinal detachment surgery in an optic disc pit – a case report. Klin Monbl Augenheilkd. 2009;226:357–358.
141 Kuhn F, Kover F, Szabo I, et al. Intracranial migration of silicone oil from an eye with optic pit. Graefes Arch Clin Exp Ophthalmol. 2006;244:1360–1362.
142 Jesberg DO, Schepens CL. Retinal detachment associated with coloboma of the choroid. Arch Ophthalmol. 1961;65:163–173.
143 Gopal L, Badrinath SS, Sharma T, et al. Surgical management of retinal detachments related to coloboma of the choroid. Ophthalmology. 1998;105:804–809.
144 Schubert HD. Structural organization of choroidal colobomas of young and adult patients and mechanism of retinal detachment. Trans Am Ophthalmol Soc. 2005;103:457–472.
145 Teoh SC, Mayer EJ, Haynes RJ, et al. Vitreoretinal surgery for retinal detachment in retinochoroidal colobomata. Eur J Ophthalmol. 2008;18:304–308.
146 Tansu E, Serhad N. Optical coherence tomography after pars plana vitrectomy for retinal detachment related to choroidal coloboma. Retina. 2010;30:1078–1083.
147 Sanghvi DA, Sanghvi CA, Purandare NC. Bilateral persistent hyperplastic primary vitreous. Australas Radiol. 2005;49:72–74.
148 Shastry BS. Persistent hyperplastic primary vitreous: congenital malformation of the eye. Clin Experiment Ophthalmol. 2009;37:884–890.
149 Walsh MK, Drenser KA, Capone A, Jr., et al. Early vitrectomy effective for bilateral combined anterior and posterior persistent fetal vasculature syndrome. Retina. 2010;30:S2–S8.
150 Dass AB, Trese MT. Surgical results of persistent hyperplastic primary vitreous. Ophthalmology. 1999;106:280–284.
151 Mittra RA, Huynh LT, Ruttum MS, et al. Visual outcomes following lensectomy and vitrectomy for combined anterior and posterior persistent hyperplastic primary vitreous. Arch Ophthalmol. 1998;116:1190–1194.
152 Shaikh S, Trese MT. Lens-sparing vitrectomy in predominantly posterior persistent fetal vasculature syndrome in eyes with nonaxial lens opacification. Retina. 2003;23:330–334.
153 Alexandrakis G, Scott IU, Flynn HW, Jr., et al. Visual acuity outcomes with and without surgery in patients with persistent fetal vasculature. Ophthalmology. 2000;107:1068–1072.