Surgery
Prenatal Consultation and Fetal Interventions
Sacrococcygeal teratoma: Fetuses with evidence of hydrops have been treated with trials of radiofrequency ablation of feeding vessels or fetal resection of the teratoma. The benefit of these treatment modalities is unknown. 12345
Congenital Diaphragmatic Hernia (CDH)
4. What are three causes of respiratory distress in a baby born with a CDH?



5. What is the common clinical presentation of a baby with a CDH?
 KEY POINTS: MOST COMMON CHARACTERISTICS OF A NEWBORN INFANT WITH CDH
KEY POINTS: MOST COMMON CHARACTERISTICS OF A NEWBORN INFANT WITH CDHMany studies have looked at lung-to-head ratio (LHR, the ratio of contralateral lung diameter to head circumference measured during 24-28 weeks’ gestation), liver position, and mediastinal shift as tools to predict mortality. Although reports have been conflicting, LHR is increasingly used to predict mortality in infants with left-sided CDH. Recent studies of infants with left-sided CDH have shown that an LHR value of less than 0.85 carries a very poor prognosis and is predictive of mortality 95% of the time. An LHR greater than 1.4, however, is virtually always associated with survival. 67
The most useful tool is a chest x-ray, which will usually demonstrate air-filled intestinal loops in the chest (once the baby has had time to swallow air); the diaphragmatic contour on the affected side is obliterated, and the mediastinum is often shifted to the opposite side ( Fig. 19-1). In babies with the less common right-sided CDH, the findings may be more confusing, with opacification of the right lower chest from the herniated liver; in these cases, ultrasonography will provide clarification.
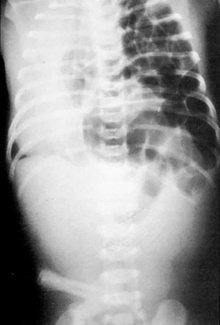
Figure 19-1 Left-sided diaphragmatic hernia with air-filled loops of intestine on the left side of the chest and deviation of the mediastinum to the right.
Endotracheal intubation with mechanical ventilation, supplemental oxygen, and orogastric decompression are used immediately in the presence of respiratory distress. Positive pressure ventilation through a face mask is not recommended because gas will enter the gastrointestinal tract and further compress the lungs. Exogenous surfactant, high-frequency ventilation, and inhaled nitric oxide are occasionally used but have no proven benefit. Barotrauma to the lungs caused by aggressive ventilation should be avoided. The level of PCO2 may be allowed to rise to 50 to 60 mmHg (permissive hypercapnia) as long as the arterial pH remains greater than 7.25. The arterial PaO2 should be kept between 50 and 80 mmHg but not above 100 mm Hg. 8910
ECMO, the use of a modified heart–lung machine to provide cardiorespiratory support independent of the lungs, may be used before or after corrective surgery if the baby does not respond to the ventilatory therapy described previously. Supporting an infant on ECMO and delaying surgery allow time for pulmonary hypertension to improve while avoiding lung damage caused by barotrauma and excessive oxygen concentrations from the ventilator. The availability of ECMO may be associated with an increased chance of survival among infants with CDH. 11
CDH was once thought to be a surgical emergency, but now repair is deferred intentionally to allow for normal physiologic changes to occur in the postnatal circulation. Current recommendations are for resuscitation followed by a period of stabilization until the neonate’s clinical condition improves. If the baby requires ECMO preoperatively, surgical repair is usually delayed until the ECMO settings have been lowered and the patient is considered ready to come off ECMO, but before decannulation. 1213
11. What is the current survival rate for infants with CDH? Which factors are most responsible for the recent improvements?
Several institutions are now reporting survival rates of 80% to 90% (compared with historical survival rates of 50% to 60%) for infants with left-sided CDH and approximately 55% for right-sided CDH. Most of the improvement is believed to be attributable to referral to high-volume tertiary care centers for management of these babies, as well as minimization of iatrogenic pulmonary injury through the avoidance of high ventilatory settings. However, many single institution–based reports are confounded by case selection bias, which fails to consider those CDH patients who do not reach referral centers. This is referred to as the “hidden mortality” of CDH. 14151617
ECMO
ECMO support can provide heart–lung bypass (venoarterial support) or simply lung bypass (venovenous support). For infants with signs of hemodynamic instability such as in sepsis, heart failure, or CDH, venoarterial support is most commonly used. A cannula is placed into the right atrium via the right internal jugular vein for venous return, and a second cannula is placed into the aortic arch by way of the right common carotid artery for arterial delivery. In cases of isolated respiratory failure such as in meconium aspiration, venovenous support can be used. A double-lumen cannula is placed into the right internal jugular vein, and the tip of the cannula lies in the right atrium. Blood is removed from the right atrium, gas exchange occurs in the ECMO circuit ( Fig. 19-2), and the blood is returned to the right atrium.
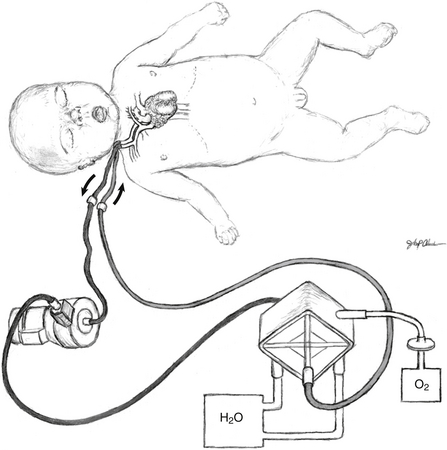
The selection of neonates as potential ECMO candidates remains controversial and varies according to the institution. Relative contraindications that must be considered are the presence of an irreversible cardiopulmonary disorder, coexisting anomalies incompatible with life (e.g., trisomy 13 or 18), uncorrectable bleeding diathesis, and existing intracranial hemorrhage (above grade II). Infants who are younger than 35 weeks’ gestation are at a high risk of developing intracranial hemorrhage with systemic heparinization.
Vascular Anomalies
Vascular anomalies represent a spectrum of conditions that result from focal aberrations of blood vessel development. According to the International Society for the Study of Vascular Anomalies (ISSVA), vascular anomalies are classified as hemangiomas (proliferating endothelial tumors) and congenital vascular malformations ( Table 19-1). Hemangiomas are proliferative lesions that typically undergo periods of rapid growth and involution after birth. Hemangiomas can be distinguished from congenital vascular malformations by immunoreactivity for the glucose-1 transporter (GLUT-1). Congenital vascular malformations have been defined as lesions that are present at birth that do not further proliferate postnatally, although more recent data suggest that remodeling and growth can occur in some settings. Congenital vascular malformations can be subclassified further according to hemodynamic characteristics. Fast-flow lesions include arteriovenous fistulas and malformations, and slow-flow lesions include venous, lymphatic, and mixed malformations. 181920
TABLE 19-1
MAJOR DIFFERENCES BETWEEN HEMANGIOMAS AND VASCULAR MALFORMATIONS
| INFANTILE HEMANGIOMAS | VASCULAR MALFORMATIONS | |
| Clinical | Variably visible at birth Subsequent rapid growth Slow, spontaneous involution |
Usually visible at birth (AVMs may be quiescent) Growth proportionate to the skin’s growth (or slow progression); present lifelong |
| Sex ratio (female : male) | 3 : 1 to 5 : 1 and 9 : 1 in severe cases | 1 : 1 |
| Pathology | Proliferating stage: hyperplasia of endothelial cells and SMC-actin cells Multilaminated basement membrane Higher mast cell content in involution |
Flat endothelium Thin basement membrane Often irregularly attenuated walls (VM, LM) |
| Radiology | Fast-flow lesion on Doppler sonography Tumoral mass with flow voids on MRI Lobular tumor on arteriogram |
Slow flow (CM, LM, VM) or fast flow (AVM) on Doppler sonography MR: Hypersignal on T2 when slow flow (LM, VM); flow voids on T1 and T2 when fast flow (AVM) Arteriography of AVM demonstrates AV shunting |
| Bone changes | Rarely mass effect with distortion but no invasion | Slow-flow VM: distortion of bones, thinning, underdevelopment Slow-flow CM: hypertrophy Slow-flow LM: distortion, hypertrophy, and invasion High-flow AVM: destruction, rarely extensive lytic lesions Combined malformations (e.g., slow-flow [CVLM = Klippel–Trénaunay–Weber syndrome] or fast-flow [CAVM = Parkes Weber syndrome]): overgrowth of limb bones, gigantism |
| Immunohistochemistry on tissue samples | Proliferating hemangioma: high expression of PCNA, type IV collagenase, VEGF, urokinase, and bFGF Involuting hemangioma: high TIMP-1, high bFGF (at all growth stages) Express GLUT-1, merosin, FcγRII and Lewis Y antigen |
Lack expression of PCNA, type IV collagenase, urokinase, VEGF, and bFGF Lack expression of GLUT-1, merosin, FcγRII, and Lewis Y antigen One familial (rare) form of VM linked to a mutated gene on 9p (VMCM1) |
| Hematology | No coagulopathy (Kasabach–Merritt syndrome is a complication of other vascular tumors of infancy, e.g., kaposiform hemangioendothelioma and tufted angioma, with an LM component) | Slow-flow VM or LM or LVM may have an associated LIC with risk of bleeding (DIC) |
Treatment of hemangiomas is selective, with intervention reserved for lesions that threaten vital functions, such as vision or respiration, or cause deformity or pain ( Box 19-1). Current first-line medical therapy for common hemangiomas of infancy has shifted in recent years from corticosteroids to beta blockers. The molecular mechanisms of response are still not fully defined, but both agents appear to induce or accelerate involution.
BOX 19-1 INDICATIONS FOR CONSIDERING EARLY HEMANGIOMA TREATMENT
 Life-threatening or function-threatening hemangiomas, including those causing impairment of vision, respiratory compromise, or congestive heart failure
Life-threatening or function-threatening hemangiomas, including those causing impairment of vision, respiratory compromise, or congestive heart failure

 Large facial hemangiomas, particularly those with a large dermal component
Large facial hemangiomas, particularly those with a large dermal component

 Pedunculated hemangiomas that are virtually certain to leave fibrofatty residua
Pedunculated hemangiomas that are virtually certain to leave fibrofatty residua
Treatment of congenital vascular malformations is highly dependent on the type of lesion and its location. Some lymphatic malformations, such as unilocular macrocystic malformations of the neck, may be amenable to surgical excision; other macrocystic lesions can often be treated successfully by sclerotherapy with doxycycline or other agents. Arteriovenous and venous malformations are generally treated using interventional radiologic techniques, such as transarterial embolization or sclerotherapy. Others, such as Klippel–Trénaunay–Weber syndrome, are in general treated conservatively and supportively. Current trials examining the role of oral therapy for diffuse, extensive, refractory, and recurrent lymphatic or mixed lesions with agents such as sirolimus [ http://clinicaltrials.gov/ct2/show/NCT00975819] and sildenafil are currently under way. 2122
17. How are severe cases of cervical vascular anomalies with tracheal compression treated at the time of delivery?
The ex utero intrapartum treatment (EXIT) procedure is available at selected centers for fetuses with evidence of airway compression in utero. A standard cesarean section is performed, and the baby is partially delivered but remains attached by its umbilical cord to the placenta. While the infant is maintained on placental circulation, an airway can be established, the mass resected, or extracorporeal life support can be initiated. Studies have shown that the EXIT procedure can be performed with minimal maternal morbidity and effective rescue of threatened infants. 23
Congenital Lung Abnormalities
18. What are the various types of congenital lung malformations in newborn infants?
 Pulmonary sequestration: This malformation of the lung usually receives its blood supply from anomalous systemic vessels; they may be intralobar (i.e., incorporated within the normal lung) or extralobar (i.e., separate from the normal lung) and do not communicate with the bronchial tree.
Pulmonary sequestration: This malformation of the lung usually receives its blood supply from anomalous systemic vessels; they may be intralobar (i.e., incorporated within the normal lung) or extralobar (i.e., separate from the normal lung) and do not communicate with the bronchial tree.

 Congenital lobar emphysema: This represents overinflation of a lobe or segment of the lung usually caused by cartilaginous deficiency of the bronchial tree, leading to distal air trapping. It may also result from trauma caused by mechanical ventilation ( Fig. 19-3). 24
Congenital lobar emphysema: This represents overinflation of a lobe or segment of the lung usually caused by cartilaginous deficiency of the bronchial tree, leading to distal air trapping. It may also result from trauma caused by mechanical ventilation ( Fig. 19-3). 24
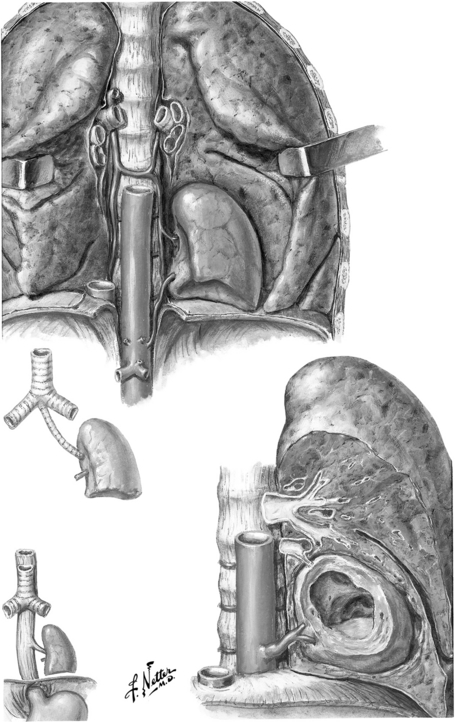
The treatment is almost always surgical excision, although the timing of surgery remains controversial. Increasingly, thoracoscopic resection is safe and feasible in infancy. CPAMs may resolve after a course of prenatal steroids with bethamethasone given during the second trimester. Some evidence suggests that CPAMs may develop into pleuropulmonary blastoma if left untreated. Asymptomatic congenital lobar emphysema may be observed, and many cases will regress over time. 25
Esophageal Atresia and Tracheoesophageal Fistula
22. Describe the five possible configurations of esophageal atresia and tracheoesophageal fistulas. Which is the most common?
 Type A: isolated esophageal atresia (rare)
Type A: isolated esophageal atresia (rare)
 Type B: esophageal atresia with a proximal fistula (rare)
Type B: esophageal atresia with a proximal fistula (rare)
 Type C: The upper esophagus ends blindly with a fistulous connection between the distal esophagus and the trachea (by far the most common type, accounting for approximately 85% of cases)
Type C: The upper esophagus ends blindly with a fistulous connection between the distal esophagus and the trachea (by far the most common type, accounting for approximately 85% of cases)


 KEY POINTS: PATTERNS OF ESOPHAGEAL ATRESIA AND TRACHEOESOPHAGEAL FISTULA
KEY POINTS: PATTERNS OF ESOPHAGEAL ATRESIA AND TRACHEOESOPHAGEAL FISTULA
1. Isolated esophageal atresia (5% to 10% of cases)
2. Esophageal atresia with a tracheal fistula to the upper esophageal segment (rare)
3. Esophageal atresia with a tracheal fistula to the lower esophageal segment (most common, in 85% of cases)
4. Esophageal atresia with tracheal fistulas to both esophageal segments (rare)
5. Isolated tracheoesophageal (“H” type) fistula (5% to 10% of cases)







24. How do babies born with esophageal atresia usually present? How can the diagnosis be established?
If the baby is stable and the gap between esophageal segments is short, operative division of the fistula and a primary esophageal anastomosis is performed. When the infant is extremely premature or sick or has a long esophageal gap (as frequently occurs in isolated esophageal atresia without a fistula), the repair is done in stages. Division of any fistula and placement of a feeding gastrostomy are the initial procedures. Numerous classification systems have been developed to predict the outcome of infants with tracheoesophageal fistulas, such as the Waterson and Spitz criteria. Generally, infants weighing less than 1.5 kg and those with cardiac abnormalities carry a poor prognosis. Infants with one risk factor generally have good outcomes; those with both factors have a poor prognosis. 26
Congenital Obstruction of the Intestinal Tract
28. What are some clinical findings indicating that a newborn infant may have an obstruction of the intestinal tract?
 Polyhydramnios: The fetus swallows large quantities of amniotic fluid, which is absorbed in the upper intestinal tract in the latter stages of pregnancy; an obstruction in the proximal intestine will cause this fluid to back up and accumulate in excessive quantities.
Polyhydramnios: The fetus swallows large quantities of amniotic fluid, which is absorbed in the upper intestinal tract in the latter stages of pregnancy; an obstruction in the proximal intestine will cause this fluid to back up and accumulate in excessive quantities.




29. If congenital obstruction is suspected on the basis of the scenarios just mentioned, what should be done next?
 KEY POINTS: SIGNS OF CONGENITAL OBSTRUCTION OF THE INTESTINAL TRACT IN NEONATES
KEY POINTS: SIGNS OF CONGENITAL OBSTRUCTION OF THE INTESTINAL TRACT IN NEONATES30. Which imaging study should be performed first if congenital intestinal obstruction is suspected?
Normal rotation consists of a 270-degree turning of the midgut on the axis of the superior mesenteric artery, resulting in the duodenojejunal junction being fixed in the left upper quadrant and the cecum attached in the right lower quadrant. In malrotation this process does not occur or is incomplete, resulting in a narrow base mesentery that puts the child at risk for development of a volvulus and obstruction ( Fig. 19-4).
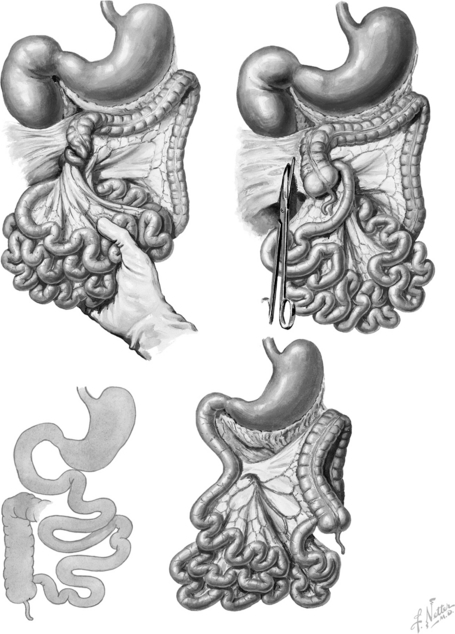
Figure 19-4 Congenital intestinal abnormalities, including malrotation of the colon with volvulus of the midgut. (Netter illustration from www.netterimages.com. © Elsevier Inc. All rights reserved.)
The twisting of the mesentery leads to vascular compromise and intestinal ischemia. Gangrene of the entire small intestine may occur within as short a period as several hours from the onset of symptoms.
37. How can the etiology of jejunal and ileal atresia be differentiated from that of duodenal atresia?
The leading theory is that duodenal atresia results from a failure of recanalization in the eighth through tenth week of fetal development; there is no similar solid phase of development of the jejunum or ileum. Jejunal and ileal atresia are believed to result from an intrauterine vascular accident that produces infarction. Because there are no bacteria within the intestine at this time, gangrene and bacterial peritonitis do not develop and the involved segment atrophies, resulting in an atresia.
42. What are the dangers associated with attempted contrast enema treatment of simple meconium ileus?
Two thirds of the time the transition zone is in the rectosigmoid region, but the zone of aganglionosis may involve the entire colon or even extend into the small intestine (i.e., total colonic Hirschsprung disease). Although Hirschsprung disease affects boys four times as often as girls, long-segment disease affects boys and girls equally.
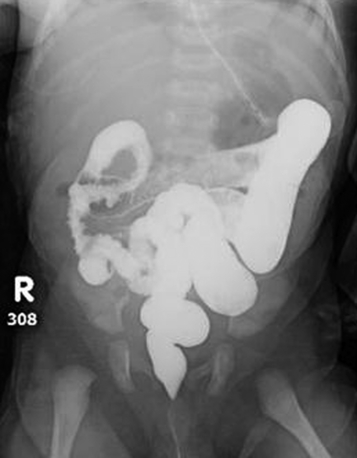
The gold standard is a rectal biopsy that typically demonstrates an absence of ganglion cells and hypertrophy of parasympathetic nerve fibers, which stain intensively for acetylcholinesterase. This biopsy can be done at the bedside in the neonate. A contrast enema is suggestive of Hirschsprung disease if it shows a change in the caliber of the colon at the transition zone ( Fig. 19-5). It is important that the study be delayed if the baby has had an enema or a digital rectal examination or even if a rectal thermometer was inserted because any rectal manipulation may temporarily obliterate the radiographic appearance of the transition zone. The contrast enema may identify a transitional zone, which may be useful for operative planning.
Anorectal Malformations
Inspection and urinalysis allow the clinician to determine the anatomy in most cases. A perineal fistula always means the lesion is low and a colostomy is not necessary. If such a fistula cannot be detected initially, there should always be a 16- to 24-hour waiting period to allow increased luminal pressure to force meconium through a possible fistula so that it becomes visible on examination. If there is meconium in the urine, an internal fistula to the urinary tract is confirmed. If there is no visible fistula, a cross-table lateral film with the baby in the prone position can be used to measure the most distal aspect of the rectum relative to the perineal skin. The work-up should also include a search for other possible components of the VACTERL association (see answer to Question 23).
55. When is a colostomy not necessary as the initial operative procedure? Is surgery always necessary?
Necrotizing Enterocolitis (NEC)
 Damage to the intestinal mucosa, which may result from ischemia caused by perinatal hypoxia, low-flow states (e.g., premature infants with patent ductus arteriosus), or reperfusion injury
Damage to the intestinal mucosa, which may result from ischemia caused by perinatal hypoxia, low-flow states (e.g., premature infants with patent ductus arteriosus), or reperfusion injury

 Impaired host defense mechanisms, as is the case in premature infants, allowing intestinal bacteria to invade the wall of the intestine (It is believed that the excessive immature inflammatory response associated with abnormal intestinal microbiota is the most likely basis for the pathogenesis of NEC.) ( Fig. 19-6) 27
Impaired host defense mechanisms, as is the case in premature infants, allowing intestinal bacteria to invade the wall of the intestine (It is believed that the excessive immature inflammatory response associated with abnormal intestinal microbiota is the most likely basis for the pathogenesis of NEC.) ( Fig. 19-6) 27
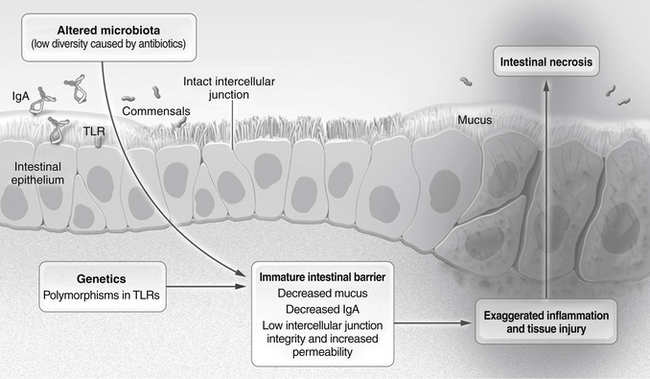
Figure 19-6 Pathophysiology of Necrotizing Enterocolitis. Factors conferring a predisposition to necrotizing enterocolitis include genetic factors and several immature characteristics of the fetal intestine, including altered microbiota, inadequate intestinal barrier function, and an excessive inflammatory response. These factors contribute to the severe necrosis of the small intestine that is characteristic of this disease. TLR denotes toll-like receptor. (From Neu J, Walker WA. Necrotizing enterocolitis. New Engl J Med 2011;364:255–64.)
Although the diagnosis may be strongly suspected by the clinical findings outlined in the previous passages, the presence of bubbly lucencies in the intestinal wall on x-ray, called pneumatosis intestinalis, is pathognomonic. Pneumatosis represents gas in the bowel wall produced by enteric organisms and is seen in 80% of NEC cases ( Fig. 19-7). Other radiographic features may include irregularly dilated air-filled loops of bowel and the visualization of branching lucencies in the liver, which may signify gas in the portal venous system.
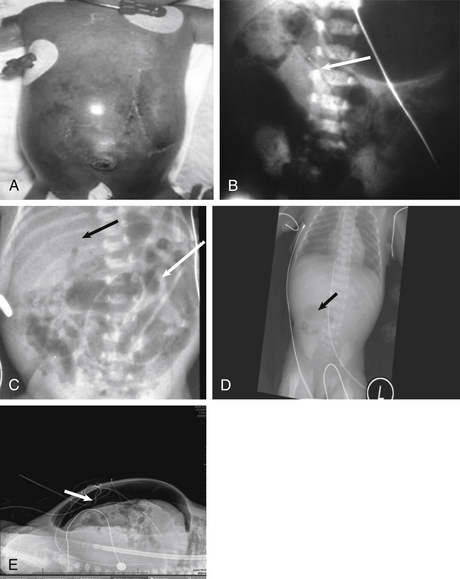
Figure 19-7 Features of Necrotizing Enterocolitis. A, Tense abdominal distension. B, Pneumaosis intestinalis. C, Portal venous gas and thickened bowel wall. D, Featureless, fixed loop of bowel. E, Pneumoperitoneum, or free air.
 KEY POINTS: INDICATIONS FOR SURGERY IN INFANTS WITH NEC
KEY POINTS: INDICATIONS FOR SURGERY IN INFANTS WITH NECTraditionally, a laparotomy is performed with inspection of the entire intestinal tract. Necrotic or perforated segments are resected, and an ostomy is performed. Bedside placement of an abdominal drain has been shown to have similar outcomes with regard to mortality, dependence on total parenteral nutrition, and length of hospital stay compared with laparotomy. Peritoneal drainage can be used as a temporizing procedure followed by subsequent operation. Close observation is required after any operative intervention. 28
Babies with SIP are generally younger (<1 week old) and more premature than NEC babies and often have not been fed. The exact etiology is unknown, but a very localized area of ischemia may be causative. Unlike with NEC, the surrounding bowel is not affected. Treatment consists of a localized resection with either an ostomy or possibly a primary anastomosis. 29
Abdominal Wall Defects: Omphalocele and Gastroschisis
69. What are the different embryologic events that result in the development of an omphalocele and a gastroschisis?
Between the fifth and tenth weeks of embryologic development, the intestine protrudes out of the umbilical ring and into the yolk sac. An omphalocele results when the lateral abdominal folds do not close and the exteriorized viscera remain in the sac ( Fig. 19-8). The etiology of gastroschisis is unknown. ( Table 19-2)
TABLE 19-2
DIFFERENCES BETWEEN GASTROSCHISIS AND OMPHALOCELE
| GASTROSCHISIS | OMPHALOCELE | |
| Incidence | 1 in 10,000 (now increasing) | 1 in 5000 |
| Defect location | Right paraumbilical | Central |
| Covering sac | Absent | Present (unless sac ruptured) |
| Description | Free intestinal loops | Firm mass including bowel, liver, etc. |
| Associated with prematurity | 50% to 60% | 10% to 20% |
| Necrotizing enterocolitis | Common (18%) | Uncommon |
| Common associated anomalies | Gastrointestinal (10% to 25%) | Intestinal atresia |
| Malrotation | Cryptorchidism (31%) | Trisomy syndromes (30%) |
| Cardiac defects (20%) | Beckwith–Wiedemann syndrome | Bladder exstrophy |
| Prognosis | Excellent for small defect | Varies with associated anomalies |
| Mortality | 5% to 10% | Varies with associated anomalies (80% with cardiac defect) |
From Chabra S, Gleason CA. Gastroschisis: embryology, pathogenesis, epidemiology. NeoReviews 2005;6:e493–e499.
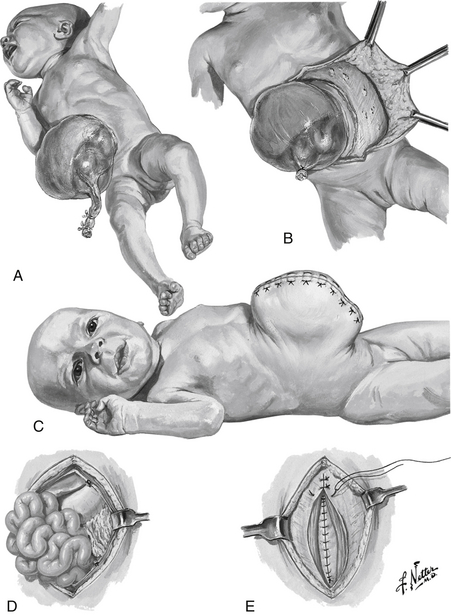
Figure 19-8 Methods of Omphalocele Repair. A, Giant omphalocele. B and C, Mobilization of skin flaps and skin closure, leaving large hernia. D and E, Layered fascial closure. (Netter illustration from www.netterimages.com. © Elsevier Inc. All rights reserved.)
TABLE 19-3
CHARACTERISTICS OF GASTROSCHISIS AND OMPHALOCELE
| OMPHALOCELE | GASTROSCHISIS | |
| Sac | Present | Absent |
| Associated anomalies | Common | Uncommon |
| Location of defect | Umbilicus | Right of umbilicus |
| Maternal age | Average | Younger |
| Mode of delivery | Vaginal/cesarean | Vaginal |
| Surgical management | Not urgent | Urgent |
| Prognostic factors | Condition of bowel | Associated anomalies |
Volume 16, Issue 3 < http://www.sciencedirect.com/science/journal/09575839/16/3> June 2006. p. 192–8 [accessed 27.06.2013].
70. How does the previously described embryology account for the anatomic appearances of omphalocele and gastroschisis?

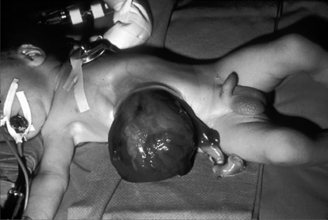

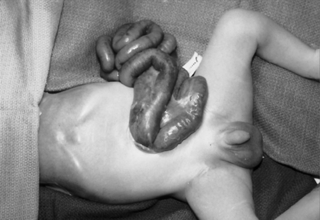
Infants born with abdominal wall defects are prone to three serious problems: hypovolemia, hypothermia, and sepsis. Exposed bowel leads to increased loss of insensible fluid as well as heat loss. Immediate management includes placing the lower half of the infant, including exposed viscera, in a plastic wrapping or moist, sterile gauze; maintaining the infant in a warmer; initiating intravenous access and fluids; and providing nasogastric decompression to minimize bowel distention. Parenteral antibiotics are administered to decrease the risk of sepsis.
Babies with gastroschisis require urgent intervention because the viscera are exposed and vascular compromise may be present. In an infant with omphalocele, surgery is not urgent, and there is time for stabilization and evaluation of potential associated anomalies.
Staged closure involves placing prosthetic material, usually a reinforced Silastic silo, over the viscera and attaching it to the fascia at the edges of the defect ( Fig. 19-11). The silo is manually compressed daily to gradually reduce the viscera and expand the peritoneal cavity. Most infants can be closed within 7 to 10 days with this method. Staged closure decreases the risk of long-term bowel dysfunction and need for reoperation. 30
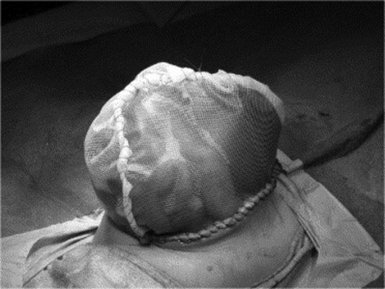
Yes. The omphalocele sac can be painted with an antiseptic, such as silver sulfadiazine or povidone-iodine. The sac will eventually epithelialize and contract, leaving a ventral hernia (which may be quite large) that can be repaired electively if the baby survives. This also allows for shorter duration of mechanical ventilator support and earlier feeds. 31
Abdominal Masses
More than half of all abdominal masses in neonates arise from the urinary tract.
79. List the two most common causes of abdominal masses of urologic origin in the neonate.


80. How do the location and other physical characteristics of the common abdominal masses in newborn infants provide clues for their identification?
Physical examination may significantly narrow the diagnostic possibilities, even if it does not provide any absolute answer ( Table 19-4). The following are of particular note:
TABLE 19-4
COMMON ABDOMINAL MASSES IN NEONATES
| MASS LOCATION | TYPE | CHARACTERISTICS |
| Lateral mass | Multicystic kidney or hydronephrosis | Smooth, moderate mobility, transilluminates |
| Renal tumor (Wilms or mesoblastic nephroma) | Smooth, minimal mobility, does not transilluminate | |
| Neuroblastoma | Irregular contour, minimally mobile, frequently crosses the midline, does not transilluminate | |
| Midabdominal mass | Mesenteric cyst | Smooth, mobile, transilluminates |
| Gastrointestinal duplication cyst | Smooth, mobile, does not transilluminate; may be associated with obstruction | |
| Ovarian cyst | Smooth, mobile, transilluminates | |
| Upper abdominal mass | Hepatic tumors | Hard, immobile, does not transilluminate |
| Choledochal cyst | Smooth, immobile, does not transilluminate; may be associated with jaundice | |
| Lower abdominal mass | Hydrometrocolpos | Smooth, immobile, does not transilluminate; may be associated with imperforate hymen |
| Bladder | Smooth, fixed; associated with lower urinary obstruction | |
| Urachal cyst | Smooth, fixed to abdominal wall, extends to umbilicus | |
| Sacrococcygeal teratoma | Hard, fixed, does not transilluminate; often associated with external sacral component |
 Large masses may fill the entire abdomen, making it impossible to determine the site of origin on examination.
Large masses may fill the entire abdomen, making it impossible to determine the site of origin on examination.
 Hard, nodular masses are usually malignant tumors.
Hard, nodular masses are usually malignant tumors.
 A highly mobile mass is usually a mesenteric cyst, a duplication, or an ovarian cyst.
A highly mobile mass is usually a mesenteric cyst, a duplication, or an ovarian cyst.
81. What is the recommended treatment for a newborn girl with an ovarian cyst that has been detected on antenatal ultrasound?
A plain abdominal radiograph might reveal a mass effect or bowel obstruction; can help localize the mass; and can sometimes provide useful information about the mass itself, such as the presence of calcifications or stool. An abdominal ultrasound is useful in the majority of cases because it can show whether the mass is cystic or solid, can reveal the effect on adjacent anatomic structures, and often can identify the exact anatomic location of the mass. Further information can be provided with abdominal computed tomography, magnetic resonance imaging, or urologic imaging.



Hydronephrosis secondary to ureteropelvic junction obstruction or posterior urethral valves.
85. A newborn infant has a large mass below the spine arising from the presacral region, compressing the rectum and anus anteriorly. What is the most likely diagnosis?
Sacrococcygeal teratomas are the most common congenital tumor ( Fig. 19-12). They can appear alarming because of their large size and compressive effects, but 90% are benign and can be completely resected. The Altman classification system is used to describe the morphology of the tumors relative to their location.
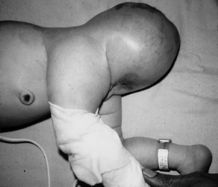
 Altman type I: entirely external, sometimes attached to the body only by a narrow stalk
Altman type I: entirely external, sometimes attached to the body only by a narrow stalk
 Altman type II: mostly outside the body with some intrapelvic extension
Altman type II: mostly outside the body with some intrapelvic extension
 Altman type III: mostly inside the body with some intraabdominal extension
Altman type III: mostly inside the body with some intraabdominal extension
 Altman type IV: entirely internal, also known as a presacral teratoma
Altman type IV: entirely internal, also known as a presacral teratoma
Hernias and Cryptorchidism
86. What are the embryologic causes of an indirect inguinal hernia? Why are they more common in premature infants?
The ovary is the most likely structure.
No. The vast majority of umbilical hernias will close spontaneously by 4 to 5 years of age. The risk of incarceration in the interim is extremely small, and recurrences for early repair are likely.
Urologic Conditions
The incidence of hypospadias is approximately 1 in 300 live male births. 32
Epispadias, abnormal gait, anteriorly displaced anus, and vesicoureteral reflux are often associated with bladder exstrophy. 3334
Bladder exstrophy is caused by a persistence of the cloacal membrane after the fourth gestational week and a lack of medial migration of the lateral mesoderm. 35
Renal agenesis is the most common malformation.
97. What is the approximate percentage of children with spina bifida who have abnormal bladder innervation?
Approximately 90% of children with spina bifida have abnormal bladder innervation.
Circumcision
99. Is the American Academy of Pediatrics in favor of or against routine circumcision in newborn males?
The Academy has recently revised its guidelines on circumcision, now favoring the procedure, though stopping short of recommending it for all male infants. The recent change has been prompted by growing evidence that favors several health benefits including prevention of urinary tract infections, human immunodeficiency virus (HIV), and penile cancer. Circumcision also prevents transmission of certain sexually transmitted infections, including HPV and herpes.


 Lowers the incidence of urinary infections
Lowers the incidence of urinary infections
 Prevents balanoposthitis (i.e., infection of the glans and foreskin)
Prevents balanoposthitis (i.e., infection of the glans and foreskin)

 Decreases the risk of contracting HIV, human papilloma virus (HPV), and herpesvirus
Decreases the risk of contracting HIV, human papilloma virus (HPV), and herpesvirus



1Ruano R, Yoshisaki CT, daSilva MM, et al. A randomized controlled trial of fetal endoscopic tracheal occlusion versus postnatal management of severe isolated congenital diaphragmatic hernia. Ultrasound Obstet Gynecol 2012;39:20–7.
2Deprest JA, Gratacos E, Nicolaides K, et al. Changing perspectives on the perinatal management of isolated congenital diaphragmatic hernia in Europe. Clin Perinatol 2009;36:329–47.
3Adzick NS, Thorn EA, Spong CY, et al. A randomized trial of prenatal versus postnatal repair of myelomeningocele. N Engl J Med 2011;364:993–1004.
4Adzick NS. Fetal surgery for myelomeningocele: trials and tribulations. J Pediatr Surg 2012;47:273–81.
5Danzer E, Hubbard AM, Hedrick HL, et al. Diagnosis and characterization of fetal sacrococcygeal teratoma with prenatal MRI. AJR Am J Roentgenol 2006;187:350–6.
6Aspelund G, Fisher JC, Simpson LL. Prenatal lung-head ratio: threshold to predict outcome for congenital diaphragmatic hernia. J Matern Fetal Neonatal Med 2012;25:1011–6.
7Hedrick HL, Danzer E, Merchant A, et al. Liver position and lung-to-head ratio for prediction of extracorporeal membrane oxygenation and survival in isolated left congenital diaphragmatic hernia. Am J Obstet Gynecol 2007;197:422.e1–422.e4.
8Antonoff MB, Hustead VA, Groth SS, et al. Protocolized management of infants with congenital diaphragmatic hernia: effect on survival. J Pediatr Surg 2011;46:39–46.
9Garcia AV, Stolar CJH. Congenital diaphragmatic hernia and protective ventilation strategies in pediatric surgery. Surg Clin North Am 2012;92:659–68.
10Guidry CA, Hranjec T, Rodgers BM, et al. Permissive hypercapnia in the management of congenital diaphragmatic hernia: our institutional experience. J Am Coll Surg 2012;214:640–7.
11Kattan J, Godoy L, Zavala A, et al. Improvement of survival in infants with congenital diaphragmatic hernia in recent years: effect of ECMO availability and associated factors. Pediatr Surg Int 2010;26:671–6.
12Chiu P, Hedrick HL. Postnatal management and long-term outcome for survivors with congenital diaphragmatic hernia. Prenat Diagn 2008;28:592–603.
13Tsao K, Lally KP. Surgical management of the newborn with congenital diaphragmatic hernia. Fetal Diagn Ther 2011;29:46–54.
14Valfre L, Braguglia A, Conforti A, et al. Long-term follow-up in high risk congential diaphragmatic hernia survivors: patching the diaphram affects outcome. J Pediatr Surg 2011;46:52–6.
15Mah VK, Chiu P, Kim PCW. Are we making a real difference? Update on ‘hidden mortality’ in the management of congenital diaphragmatic hernia. Fetal Diagn Ther 2011;29:40–5.
16Logan JW, Cotton CM, Goldberg RN, et al. Mechanical ventilation strategies in the management of congenital diaphragmatic hernia. Semin Pediatr Surg 2007;16:115–25.
17Fisher JC, Jefferson RA, Arkovitz MS, et al. Redefining outcomes in right congenital diaphragmatic hernia. J Pediatr Surg 2008;43:373–9.
18Lang SS, Beslow LA, Bailey RL, et al. Follow-up imaging to detect recurrence of surgically treated pediatric arteriovenous malformations. J Neurosurg Pediatr 2012;9;497–504.
19Litzendorf M, Hoang K, Vaccaro P. Recurrent and extensive vascular malformations in a patient with Bannayan–Riley–Ruvalcaba syndrome. Ann Vasc Surg 2011;25;1138.e15–9.
20Ernemann U, Kramer U, Miller S, et al. Current concepts in the classification, diagnosis and treatment of vascular anomalies. Eur J Radiol 2010;75:2–11.
21Blei F. Congential lymphatic malformations. Ann N Y Acad Sci 2008;1131:185–94.
22Swetman GL, Berk DR, Vasanawala SS, et al. Sildenafil for severe lymphatic malformations. N Engl J Med 2012;366:384–6.
23Olutoye OO, Olutoye OA. EXIT procedure for fetal neck masses. Curr Opin Pediatr 2012;24:386–93.
24Correia-Pinto J, Gonzaga S, Huang Y, et al. Congenital lung lesions—underlying molecular mechanisms. Semin Pediatr Surg 2010;19:171–9.
25Morris LM, Lim F, Livingston JC, et al. High-risk fetal congenital pulmonary airway malformations have a variable response to steroids. J Pediatr Surg 2009;44:60–5.
26Sinha CK, Haider N, Marri RR, et al. Modified prognostic criteria for oesophageal atresia and tracheo-oesophageal fistula. Eur J Pediatr Surg 2007;17:153–7.
27Neu J, Walker WA. Necrotizing enterocolitis. N Engl J Med 2011;364:255–64.
28Moss RL, Dimmitt RA, Barnhart DC, et al. Laparotomy versus peritoneal drainage for necrotizing enterocolitis and perforation. N Engl J Med 2006;354:2224–34.
29Gorson PV, Attridge JT. Understanding clinical literature relevant to spontaneous intestinal perforations. Am J Perinatol 2009;26:309–16.
30Riboh J, Abrajano CT, Garber K, et al. Outcomes of sutureless gastroschisis closure. J Pediatr Surg 2009;44:1947–51.
31Ein SH, Langer JC. Delayed management of giant omphalocele using silver sulfadiazine cream: an 18-year experience. J Pediatr Surg 2012;47:494–500.
32Ardavan A, Stock JA. Long-term follow-up and late complications following treatment of pediatric urologic disorders. Med Clin North Am 2011;95:15–25.
33Stec AA, Baradaran N, Tran C, et al. Colorectal anomalies in patients with classic bladder exstrophy. J Pediatr Surg 2011;46:1790–3.
34Stec AA, Baradaran N, Gearhart JP. Congenital renal anomalies in patients with classic bladder exstrophy. Urology 2012;79:207–9.
35Cheng L, Lopez-Beltran A, Bostwick DG. Congenital disorders and pediatric neoplasms, in bladder pathology. Hoboken, NJ: John Wiley & Sons; 2012.
36Xu X, Patel DA, Dalton VK, et al. Can routine neonatal circumcision help prevent human immunodeficiency virus transmission in the United States? Am J Mens Health 2009;3:79–84.