Representative values for surfactant from adult mammalian species. The primary surface active components are the saturated phosphatidylcholine and the hydrophobic proteins SP-B and SP-C.
Surfactant Proteins
Four proteins that are relatively specific for surfactant are recovered with bronchoalveolar lavage fluid. These proteins have been widely referred to as Surfactant Protein-A (SP-A), SP-B, SP-C, and SP-D. The human genes encoding these proteins are SFTPA, SFTPB, SFTPC, and SFTPD, respectively. Insights into the function of these proteins have come primarily from gene-targeted mice and from in vitro assays of surface properties. The four proteins associate with surfactant lipids to different degrees: SP-A and SP-D are primarily water-soluble collectins (collagen-lectin proteins), whereas SP-B and SP-C are remarkably hydrophobic small peptides that are exclusively associated with surfactant phospholipids (11).
Surfactant Protein-A
SP-A monomer is a water-soluble 24-kDa protein that is heavily glycosylated to yield a protein of about 36 kDa. SP-A monomers contain a collagenous domain that assembles into a collagen-like triple helix to form a trimer; six trimeric subunits associate to form a multimeric protein with a molecular mass of about 650 kDa (12)(Figure 8-2). SP-A associates primarily with saturated phosphatidylcholine and is associated with tubular myelin, a unique square lattice membrane structure that may be involved in host defense. Surfactant from mice that completely lack SP-A does not form tubular myelin, but the mice have normal lung function (13). The absence of SP-A in mice also does not disrupt the secretion, clearance, or catabolism of surfactant lipids.
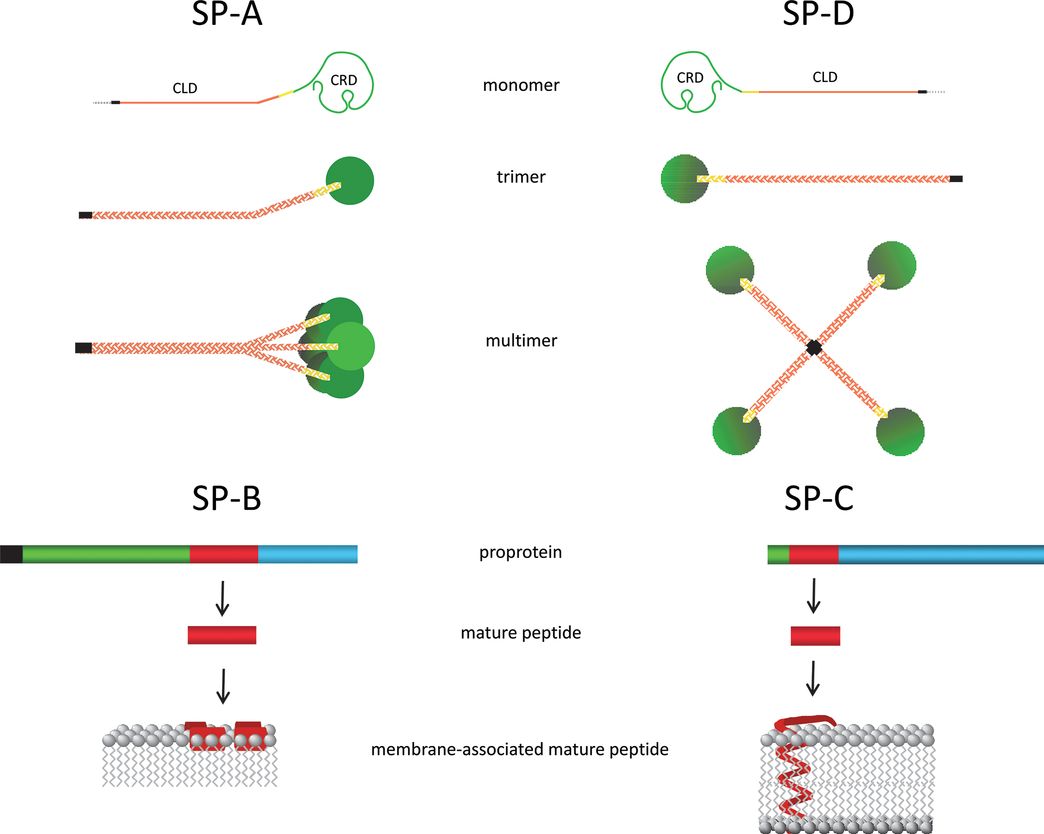
Surfactant Protein Structures SP-A and SP-D are collectins that each contain a collagen-like domain (CLD) and a carbohydrate recognition domain (CRD). The CLD facilitates formation of an SP-A octadecamer and an SP-D dodecamer. The dashed line represents the signal peptide, the black line is the NH2-terminal domain, and the yellow line is the neck region. SP-B and SP-C are synthesized as proproteins that are proteolytically processed to very hydrophobic mature peptides that associate with surfactant membranes: Mature SP-B forms amphipathic helices that associate with the surface of the membrane, whereas mature SP-C forms a metastable α helix that spans the membrane bilayer. Black represents the signal peptide, green is the NH2-terminal propeptide domain, red is the mature peptide, and blue is the COOH-terminal peptide domain.
The major function of SP-A is as an innate host defense protein and regulator of inflammation in the lung (14). SP-A binds to gram-positive and gram-negative bacteria, viruses, and fungi primarily through its carbohydrate recognition domain (CRD). SP-A facilitates phagocytosis of pathogens by macrophages and may directly kill some microbes via membrane permeabilization. SP-A can inhibit inflammation induced by lipopolysaccharide (LPS), peptidoglycan, or zymosan by binding to cell surface receptors (e.g. CD14 and TLR2) for these pathogen-derived molecules. Binding of SP-A to SIRPα also inhibits pro-inflammatory signaling. SP-A can also evoke a context-dependent pro-inflammatory response by binding to rough LPS (via the CRD) and CD14 (via the neck region) or to calreticulin/CD91 on the surface of macrophages via the collagen-like domain. SP-A levels are low in surfactant from preterm lungs and increase as the type II cell numbers increase and mature. The fetal lung increases expression of SP-A in response to corticosteroid or chorioamnionitis in animal models (15).
SP-D
SP-D is a collectin that shares structure and innate host defense functions with SP-A (12). The 43 kDa monomer also forms trimers through its collagen-like domain. Twelve monomers associate to form a multimeric protein composed of four trimeric subunits. SP-D expression in the lung is restricted primarily to type II epithelial and Clara cells, but it is widely expressed by other epithelial cells in the body. SP-D is present in lower amounts in bronchoalveolar lavage than SP-A and is primarily recovered in the water-soluble fraction. As for SP-A, SP-D binds to pathogen-associated molecular patterns (PAMPs) through its CRD; facilitates uptake of bacteria, viruses, and fungal pathogens by macrophages; and may directly kill some pathogens by membrane permeabilization (11). Although SP-D does not appear to interact with intracellular surfactant synthesis, storage and secretion pathways, it does bind to phosphatidylinositol. Absence of SP-D in mouse models is associated with altered surfactant metabolism and a greatly increased alveolar surfactant pool size. SP-D deficient mice also have progressive emphysema related to increased oxidant stress associated with altered macrophage function (16). Addition of SP-D to surfactant used to treat preterm sheep decreases ventilator-mediated inflammation (17). SP-D expression is increased by antenatal corticosteroids and by fetal exposure to inflammation in animal models. Exposure to both corticosteroids and inflammation further increases expression (15).
SP-B
SP-B is a hydrophobic peptide of 79 amino acids that is cleaved from a precursor protein of approximately 40 kDa prior to association with surfactant lipids in type II epithelial cells (18). Surfactant lipids and proteins (SP-B and SP-C) are stored as concentric bilayer membranes (lamellae) in specialized secretory organelles (lamellar bodies) prior to secretion into the alveolar airspaces. The ability of SP-B to both lyse and fuse bilayer membranes is critical for organization of surfactant membranes within lamellar bodies and for transition of newly secreted membranes to a surface active film at the alveolar air–liquid interface. SP-B is absolutely essential for lung function as knockout mice have normal lung structure at birth, but cannot initiate air breathing because of a lack of functional surfactant (19). In the absence of SP-B, type II cells fail to form lamellar bodies or process SP-C. Thus, SP-B is required for both the synthesis and assembly of surfactant in type II cells as well as for its function in the alveolar compartment. SP-B comprises only about 2% of surfactant, and when this amount is decreased in adult mice, the animals develop lung injury and respiratory failure (20). Production of large amounts of appropriately folded active synthetic SP-B peptide has not been achieved, but shorter peptide mimetics have been developed for use in synthetic surfactant preparations with some success. As with the other surfactant components, SP-B expression and amounts increase with advancing gestational age and increase with antenatal corticosteroid or fetal exposure to inflammation (15).
SP-C
SP-C is the other hydrophobic protein in surfactant that comprises up to 4% of surfactant. Organic solvent extracts of surfactant recover both SP-B and SP-C with lipids. Like SP-B, SP-C is synthesized as a precursor protein that is processed to an extremely hydrophobic 35 amino acid (4 kDa) protein that associates with lipids in lamellar bodies (18). The mRNA for SP-C appears in the epithelium of the developing airways from early gestation. With advancing lung maturation, SP-C expression is restricted to type II epithelial cells. The amino acid sequence and cellular localization of SP-C are remarkably similar across mammalian species consistent with an important, but poorly understood, function. Although mice that lack SP-C have normal lung structure and surfactant function at birth (21), these animals develop strain-dependent, progressive interstitial lung disease (ILD) and emphysema as they age. Although not critical for survival, SP-C contributes to surface film stability by interacting with SP-B and lipids, thus contributing to the unique surface properties of surfactant. SP-C expression increases in the fetal lung as type II cell numbers and maturation increase.
Metabolism of Surfactant
Synthesis and Secretion
Type II cells and macrophages are the cell types responsible for the major pathways involved in surfactant metabolism (Figure 8-3). The synthesis and secretion of surfactant is a complex sequence of biochemical synthetic events that results in the release by exocytosis of lamellar bodies to the alveolus (18). The basic pathways for the synthesis of phospholipids are common to all mammalian cells. Specific enzymes use glucose, phosphate, and fatty acids as substrates for phospholipid synthesis. Surfactant lipids are synthesized primarily in the endoplasmic reticulum (ER) and transported to lamellar bodies, the storage organelle for surfactant. An important component of the pathway involved in the intraorganelle transfer of newly synthesized surfactant lipids is ABCA3, a phospholipid import protein located in the limiting membrane of the lamellar body. The orientation of ABCA3 in the membrane and the absence of typical lamellar bodies in ABCA3 null mice strongly suggest that surfactant phospholipids are transported directly from the ER to the lamellar body, via as yet unidentified lipid transfer proteins located in the cytosol of type II cells. Candidate phosphatidylcholine transfer proteins and other potential lipid importers have been identified in the lamellar body proteome, but the involvement of these proteins in surfactant biosynthesis has yet to be evaluated (22). In the adult lung, surfactant secretion is presumed to be regulated by the microenvironment of the alveolus. Secretion can be stimulated by beta-agonists, purinergic agonists, or hyperventilation. In the fetus, surfactant is released into fetal lung fluid in increasing amounts as gestation advances. Surfactant is secreted into fetal lung fluid and is carried out of the lung with fetal breathing and subsequently swallowed or diluted in amniotic fluid. Thus, amniotic fluid can be used to evaluate the development of the surfactant system.
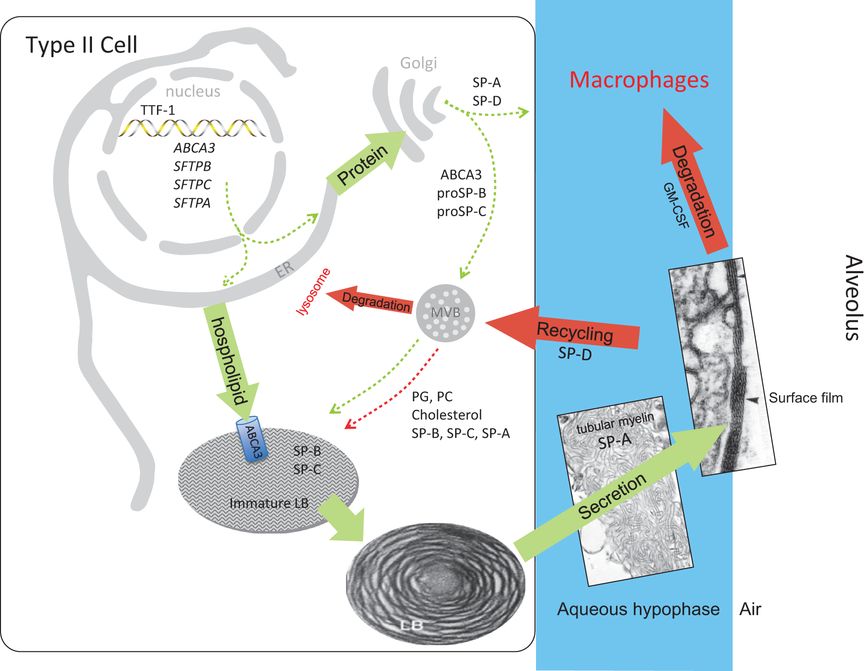
Surfactant metabolism biosynthesis of surfactant likely involves distinct pathways for surfactant proteins and lipids (green arrows). Newly synthesized SP-B and SP-C are trafficked from the ER to lamellar bodies (LB) via the Golgi and multivesicular body (MVB), whereas SP-A and SP-D are constitutively secreted by the type II epithelial cells (dashed green arrows). In contrast, surfactant phospholipids are likely directly transported from the ER to specified lipid importers (ABCA3) in the lamellar body-limiting membrane (solid green arrow). Surfactant proteins and lipids are assembled into bilayer membranes that are secreted into the alveolar airspace, where they form a surface film at the air-liquid interface. Cyclical expansion and compression of the bioactive film results in incorporation (green arrow) and loss (red arrows) of lipids/proteins from the multilayered surface film. Surfactant components removed from the film are degraded in alveolar macrophages or are taken up by the type II epithelial cell for recycling (dashed red arrow) or degradation in the lysosome (solid red arrow). The MVB plays a key role in the integration of surfactant synthesis, recycling, and degradation pathways. Green arrows indicate biosynthetic pathways; red arrows indicate degradation and recycling pathways.
Alveolar Cycle of Surfactant
After exocytosis of lamellar bodies by type II cells, surfactant proceeds through a series of form transitions that define its metabolic and functional roles. The exocytosed lamellar bodies “unravel” into the elegant “tubular myelin” structure with SP-A at the corners of the lattice (Figure 8-3). Tubular myelin requires at least SP-A, SP-B, and the phospholipids for its unique structure. Tubular myelin and other loose, less-structured surfactant lipoprotein membrane arrays are the pool in the fluid hypophase that supplies surfactant for the surface film within the alveolus and small airways. Compression of this film squeezes out unsaturated lipids and some protein components of surfactant with concentration of saturated phosphatidylcholine in the surface film. New surfactant enters the surface film from tubular myelin and the loose lipid arrays, and “used” surfactant leaves as small vesicles, which then are cleared from the airspaces. The major differences in composition between the surface-active surfactant structures and the small vesicular forms are that the small forms contain SP-D but little SP-A, SP-B, or SP-C (23). The small vesicles of used surfactant are much less surface active and seem to be the major catabolic form of the lipids that are taken up by type II cells and by alveolar macrophages for recycling or catabolism.
Alveolar macrophages are the sentinel immune cells of the lung. These cells are in the airspaces directly in contact with the alveolar hypophase and surfactant. Fetal monocytes seed the developing lung and undergo granulocyte-macrophage colony-stimulating factor (GM-CSF) mediated differentiation to alveolar macrophages shortly after birth (24). Once differentiated, alveolar macrophages have a relatively long life span under normal conditions. Important functions of alveolar macrophages include immune surveillance, phagocytosis of invading microorganisms, antigen presentation, interactions with adaptive immune cells, and surfactant homeostasis. Fetuses have very few alveolar macrophages. In mice, primitive macrophages can be detected in the lung interstitium from early gestation, while in other species including nonhuman primates and sheep, few macrophages are found in the fetal lung (25). Fetal exposure to inflammation and lung injury can mature lung monocytes into macrophages and stimulate their migration into the fetal alveolar spaces (26).
The number and maturity of type II epithelial cells increase in the fetal human after about 20 weeks gestation. Immature type II cells with large intracellular lakes of glycogen mature with the disappearance of glycogen and the appearance of more and larger lamellar bodies. Just preceding and following birth, lamellar bodies are secreted to yield an alveolar pool that is primarily lamellar bodies and tubular myelin (27). This surfactant then begins to function with aeration of the lung. As the newborn goes through neonatal transition, the percentage of surface-active forms falls, and the small vesicular forms increase. At equilibrium approximately 50% of the surfactant in the airspaces is in a surface-active form, and 50% is in the inactive vesicular form. Conversion from surface active to inactive surfactant forms occurs more rapidly in the preterm, presumably because there is a lower pool size and less-effective surfactant with lower amounts of the surfactant proteins. Pulmonary edema can further accelerate the process, with the net result being a depletion of the surface-active fraction of surfactant despite normal surfactant pool sizes.
Surfactant Turnover
The Adult Lung
In the adult human, the amount of surfactant in the airspaces recoverable by bronchoalveolar lavage is about 5 mg/kg body weight (28). There are no reliable measurements of turnover of the surfactant components in the normal adult human. In animal models (primarily the rabbit) the airspace pool size of surfactant is about 16.5 mg/kg, and the lamellar body pool size is about 24 mg/kg (Figure 8-4). The kinetics of secretion were measured with radiolabeled precursors of surfactant components. Clearance kinetics were measured using radiolabeled surfactant components given into the airspaces (29–31). The lag from synthesis to peak accumulation of surfactant lipids in the airspace is about 15 hours. Once secreted, the half-life of the subsequent linear loss from airspace is about 10 hours. The curves have been modeled to demonstrate that the surfactant lipids SP-B and SP-C from the airspace appear in lamellar bodies for resecretion with an efficiency of about 25% (31). The residual surfactant lost from the airspaces is catabolized by alveolar macrophages. Some surfactant moves from the alveoli into small airways, but large amounts of surfactant are not lost up the airways. This elaborate steady-state control of surfactant pool sizes and therefore function in the adult lung can be disrupted by injury to type II cells and by a block in surfactant catabolism by alveolar macrophages from a lack of GM-CSF signaling resulting in alveolar proteinosis (32).
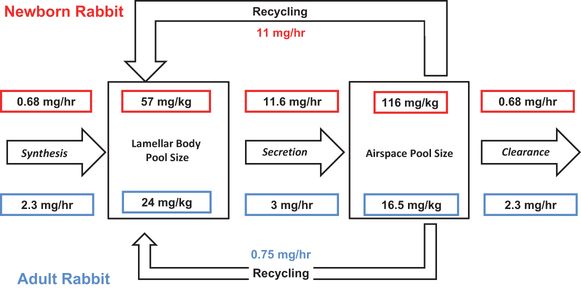
Estimates of pool sizes and flux rates of surfactant in adult rabbits (blue) and in 3-day-old newborn rabbits (red). All values were measured as saturated phosphatidylcholine and have been converted to mg/kg total surfactant (1 μ mol saturated phosphatidylcholine is equivalent to about 1.5 mg total surfactant). The 3-day rabbits weighed about 65 g. The values are from modeling studies using radiolabeled precursors of saturated phosphatidylcholine by Jacobs and colleagues(29–31).
The Newborn Lung
Surfactant pool sizes and turnover times are quite different in preterm and term newborns (Figure 8-4). Following the observation of Avery and Mead that saline extracts of the lungs of infants with RDS had high minimum surface tensions (2), decreased alveolar and tissue surfactant pools were demonstrated in preterm animals. Increasing surfactant pool sizes correlated with improving compliances, although other factors such as structural maturation also influence lung function (33). Premature infants who died with RDS without mechanical ventilation in the 1960s had surfactant pool sizes that were less than about 5 mg/kg of body weight. Preterm lambs with RDS can be managed with respiratory support if their surfactant pool sizes exceed about 4 mg/kg (34). Of note, the quantity of surfactant recovered from the airspaces of infants with RDS is about the same as the amount of surfactant found in the alveoli of healthy adult animals or humans (28). Nevertheless, much less surfactant is recovered from preterms than healthy term animals that have surfactant pool sizes of about 100 mg/kg of body weight (35). The large amount of surfactant in amniotic fluid in the human at term indirectly indicates that the term human fetal lung also has large pool sizes. The fetal lung at term has more surfactant in lamellar bodies and the fetal lung fluid than at any time during life as a biological adaptation to ensure neonatal transition to air breathing. Surfactant function is concentration dependent, and the high amounts of surfactant in the fetal lung fluid facilitate film formation and the establishment of surface forces that promote fluid clearance. The high surfactant pool sizes present at term birth progressively decrease to normal values for the adult animal by about 7 days in the rabbit. There is no information for the time of transition of surfactant pool sizes in the human.
Preterm infants that develop RDS often have a “honeymoon” period of relatively normal lung function that can last for several hours prior to progressive respiratory failure. Their small surfactant pool sizes are sufficient for initial transition to air breathing, but that surfactant has decreased function relative to surfactant from mature infants and is more sensitive to inactivation. Part of the problem may be that following preterm birth the surfactant stores in the type II cells are depleted, limiting the potential to quickly increase alveolar surfactant pool sizes. The increase in the pool size of alveolar surfactant after preterm birth has been measured in ventilated preterm monkeys recovering from RDS (36). The surfactant pool size increased toward the 100 mg/kg value measured in term monkeys within 3 to 4 days. Similarly, the concentration of saturated phosphatidylcholine in airway aspirates from infants with RDS increased over a 4- to 5-day period to become comparable to values for normal or surfactant-treated infants (37). This slow increase in pool size is consistent with a clinical course of RDS of about 5 days without surfactant treatment.
The only surfactant pool that can be sampled from the newborn human is that which can be recovered by a tracheal aspirate procedure, which severely limits the analyses that are possible. In term newborn rabbits turnover studies using radiolabeled precursors demonstrated that synthesis and clearance was about 10-fold less in the newborns than the adults (29) (Figure 8-4), but the high lamellar body and airspace pools were maintained by a recycling rate for saturated phosphatidylcholine of >90%. The phospholipids were recycled as intact molecules without degradation and resynthesis.
Metabolic measurements have been made in preterm infants with the material recovered in tracheal aspirates using stable isotopes given by intravascular injection to measure endogenous synthesis and secretion (38,39). Following the intravascular administration of labeled glucose or palmitic acid precursors, rapid incorporation into surfactant phosphatidylcholine is followed by long time delays for the movement of surfactant components from the ER to lamellar bodies for secretion. In infants with RDS, glucose-labeled phosphatidylcholine was detected in the airway samples after about 20 hours, and peak enrichment of the stable isotope in the airspaces occurred at about 70 hrs (38). Therefore, delays between synthesis and secretion and the interval to peak airway accumulation of endogenously synthesized surfactant lipid is very long in the preterm human.
The slow secretion and alveolar accumulation of surfactant are balanced in the term and preterm lung by slow catabolism and clearance. Trace amounts of radiolabeled surfactant phospholipid mixed with treatment doses of surfactant and given into the airspaces to infants with RDS had half-life values of several days (39). Surfactant phosphatidylcholine labeled with intravascular glucose had a half-life after peak secretion of about 80 hours for infants with RDS on conventional or high-frequency oscillation (40). Fractional synthetic rates also were similar at 4.5% per day. The half-life of surfactant phosphatidylcholine was 62 hours in term infants, but infants with pneumonia had much shorter half-life for surfactant phosphatidylcholine of about 30 hours.
Less is known about the metabolism of the surfactant proteins in the preterm lung. In animal models, SP-A, SP-B, and SP-C seem to have alveolar clearance kinetics that are similar to saturated phosphatidylcholine. These proteins also seem to be recycled to some degree from the airspace back into lamellar bodies for resecretion with surfactant (35). In the ventilated preterm human, SP-B was labeled using leucine and its secretion and clearance from airway samples measured (41). The estimate of catabolic rate was about one alveolar pool equivalent per day with a half-life of about 16 hours. The relationships between the metabolism of surfactant proteins and lipids remain to be studied in the human and during development. The presumed function of the recycling pathways is to reassemble the components to regenerate biophysically active surfactant.
Surfactant Physiology in the Lung
The gas exchange surfaces of alveoli are complex polygonal shapes that are interdependent in that their structures are determined by the shapes and elasticity of neighboring alveoli and airways. The forces acting on the pulmonary microstructure are chest wall elasticity, lung tissue elasticity, and surface tensions of the air-fluid interfaces in the small airways and alveoli. At static equilibrium, a surfactant film will reduce surface tension from the value of 72 mN/m for water to about 23 mN/m. Surface area compressions of about 25% will decrease surface tension to close to 0. In the normal lung, the surface area changes of the alveolar surface with tidal breathing are not large. Nevertheless, surface forces balance the inflation of the lung across the approximately 500 million alveoli and their connecting small airways in all lung regions. As noted earlier, the problem for the term fetus transitioning to air breathing is to move fluid from the airspaces while establishing alveolar expansion. Surfactant is critical for this process together with active Na + clearance. The infant makes high negative-pressure breaths that move fluid down the airway tree. The surface film is quickly formed on the expanding air–fluid interface to retain air as a functional residual capacity. The fluid moves into the lung interstitium to be cleared from the lung over hours. Surfactant also facilitates both clearance and the maintenance of patency of the small airways.
The effects of surfactant on the preterm surfactant-deficient lung are demonstrated by pressure-volume relationships during quasi-static inflation and deflation. The preterm surfactant-deficient lung does not begin to inflate until pressures exceed 20 cm H2O (42) (Figure 8-5A). Multiple airways connect to distal saccules/alveoli with different radii. The pressure needed to open a lung unit (airway plus distal structures) is related to the radius of curvature and surface tension of the meniscus of fluid in the airspace leading to each lung unit. The units with larger radii and lower surface tensions will “pop” open first because, with partial expansion, the radius increases and the forces needed to finish opening the unit decrease. The movement of an air–fluid interface with high surface tensions in the airways causes very high sheer forces that can disrupt the airway epithelium (43). With surfactant treatment, the fluid menisci in the airways have lower surface tensions that decrease the opening pressure from about 25 to 15 cm H2O in this example. The subsequent inflation is more uniform as more units open at lower pressures, resulting in less epithelial injury and less overdistention of the open units.
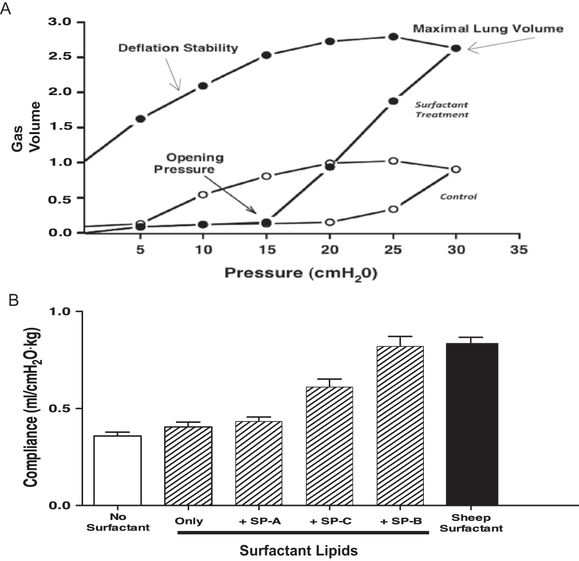
(A) Surfactant treatment of the preterm rabbit lung greatly increases lung gas volumes by the combined effects of decreasing opening pressure, increasing maximal lung volume, and increasing deflation stability. Lung gas volumes have been normalized to 1 for the maximal volume at 30 cmH2O pressure for the control lung. Data derived from (42). (B) Natural sheep surfactant was fractionated into a lipid-only fraction and each of the sheep surfactant proteins. Surfactants were reconstructed using lipids only and lipids plus each of the surfactant proteins. Preterm rabbits were treated with the surfactants and ventilated with similar tidal volumes, and compliances were measured. Sheep surfactant greatly increased compliance relative to the control value in untreated rabbits.
A particularly important effect of surfactant on the surfactant-deficient lung is the increase in maximal volume at maximal pressure. In this example, maximal volume at 35 cm H2O is increased about 2.5-fold with surfactant treatment. Pressures above 35 cm H2O in control lungs result in lung rupture with little further volume accumulation. The opening pressures of many distal lung units in the surfactant-deficient lung exceed 35 cm H2O, and an attempt to inflate the lung to full volume with higher pressure will rupture the preterm lung. This volume increase with surfactant treatment improves gas exchange because it is primarily distal lung gas volume. Another important effect of surfactant is lung stabilization on deflation. The surfactant-deficient lung collapses at low transpulmonary pressures, whereas the surfactant-treated lung retains about 40% of the gas volume on deflation to 5 cm H2O, which is the static equivalent of functional residual capacity.
Surfactant lipid composition and the surfactant proteins SP-B and SP-C strikingly change the behavior of surfactant in the airspaces. For example, natural sheep surfactant can be fractionated into its lipid components and SP-A, S-B, and SP-C for reconstruction experiments to demonstrate physiologic responses of each component in the ventilated preterm rabbit lung (44). In the example in Figure 8-5B, treatment of the surfactant-deficient preterm lung with the surfactant lipids does not improve lung compliance relative to untreated rabbits. Addition of SP-A has minimal effects, while addition of only SP-C improves the pressure volume relationships substantially. However, SP-B plus the surfactant lipids is equivalent to the natural surfactant. The deficit in function of a surfactant with only SP-C can be overcome by adding positive end expiratory pressure during ventilation of the preterm rabbits (45). The physiologic effects of surfactant on the lung are striking and can be demonstrated by giving surfactant to the surfactant deficient lung or removing surfactant from the normal lung. The changes in lung mechanics are accompanied by large changes in gas exchange and lung injury with ventilation of the surfactant-deficient lung.
Surfactant Inactivation
Surfactant function is easily disrupted by multiple factors that frequently occur in the preterm lung (Table 8-2). Once the preterm lung has released surfactant stores at delivery, the surfactant will be depleted with time by the normal conversion of the pool from the tubular myelin and loose surfactant arrays into the catabolic liposomes unless there is active replacement. The generation of new secretion capacity from lamellar bodies from de novo synthesis and recycling is a slow process, and efficient recycling depends on normal lamellar body and alveolar pool sizes. The preterm with just enough surfactant at birth to transition to air breathing may become functionally deficient over hours. Further, the surfactant of the preterm lung has lower amounts of saturated to total phosphatidylcholine and lower amounts of the surfactant proteins (46). The multiple proteins, lipids, and other factors that can interfere with film formation listed in Table 8-2 will be more inhibitory for surfactant from the immature lung than mature surfactant that contains more saturated phosphatidylcholine, more SP-B and SP-C, and particularly more SP-A. Finally, surfactant lipids can function as a thromboplastin and promote clotting of plasma that leaks into the airspaces. The surfactant deficient preterm lung is easily injured by spontaneous or mechanical ventilation with increased permeability of the epithelium, resulting in proteinaceous pulmonary edema (46). Plasma components will clot to form hyaline membranes that can trap surfactant, removing it from the functional pool. Multiple substances interfere with film formation by competing with surfactant for the air–water interface. These inactivation phenomena are concentration dependent, as high concentrations of normal surfactant can form stable surface films in the presence of plasma or inhibitors, while surface film formation by low surfactant concentrations in the hypophase are easily disrupted (47). Thus, the preterm lung is at substantial disadvantages because the immature surfactant with functional deficits is likely to be present in small amounts. The term lung also can have surfactant inactivation by meconium and the inflammatory products from pneumonia.
Surfactant Treatment
The treatment responses to surfactant illustrate the effects of surfactant on the developing lung. It must be emphasized that the development of surfactant treatment has transformed the care of preterm infants (48). The reasons that surfactant treatments are so effective go beyond simply acutely improving surface tensions and thus the physiology of the preterm lung. The major reason that surfactant treatments cause persistent clinical responses is that the metabolic characteristics of surfactant phospholipids and proteins in the preterm are favorable (35). Alveolar and tissue pool sizes are small, and the rate of accumulation is slow. Treatment acutely increases both the alveolar and tissue pools because the exogenously administered saturated phosphatidylcholine is taken up by type II cells and processed for resecretion. The surfactants used clinically are not equivalent in composition or function to native surfactant in the mature lung. Furthermore, airway instillation does not achieve an ideal distribution of surfactant. However, within hours following surfactant treatment of preterm animals, the surfactant recovered by alveolar wash has improved function. Therefore, the preterm lung, if uninjured, can rapidly transform surfactants used for treatment with poor function to a better surfactant (47). Also of benefit is the slow catabolic rate of surfactant, with the result being that the surfactant used for treatment remains in the lungs, is recycled, and is not rapidly degraded. The surfactant used for treatment becomes substrate for the endogenous recycling pathways to increase overall surfactant quantities. Treatment doses of surfactant do not feedback-inhibit the endogenous synthesis of saturated phosphatidylcholine or the surfactant proteins (49). No adverse metabolic consequences of surfactant treatment on the endogenous metabolism of surfactant or other lung functions have been identified.
The static mechanics of the preterm lung are strikingly improved by surfactant treatments (Figure 8-5A). The dynamic lung mechanics also are altered by surfactant treatments (35). The time constant for deflation increases, resulting in less-rapid lung emptying. The clinical correlate is that a surfactant treatment can increase the functional residual capacity of infants with RDS by two mechanisms: the improved deflation stability and the longer expiratory time constant. The consistent initial response of infants with RDS to surfactant treatments is a rapid improvement in oxygenation, whereas improvements in PCO2, compliance, and therefore ventilatory support variables tend to change more gradually. The improved oxygenation without changes in ventilation results from the acute increase in lung volumes following surfactant treatments. In experimental animals, these acute physiological responses are accompanied by much more uniform aeration of the preterm lung at the anatomic level, a decreased lung permeability, and less indicators of lung injury with mechanical ventilation (50,51).
Clinical Lung Maturation
RDS and Induced Lung Maturation
At term the total amount of surfactant present in the human airways plus lamellar body pools probably exceeds that in the adult by about 10-fold on a body weight basis. Lamellar bodies first appear within type II cells by 20 to 24 weeks’ gestation in the human fetus, and the amount of saturated phosphatidylcholine in lung tissue progressively increases to term. Lung maturity as defined clinically by the absence of RDS in the human fetus is generally present after 36 weeks of normal gestation, but infants born at 24 weeks can have “mature lungs” based on their ability to exchange oxygen and CO2. Therefore, a 12-week window of “early maturation” is possible for the human, in part because the surfactant synthetic and storage machinery can be induced in the human early in gestation.
The clinical syndrome linked to inadequate amounts of surfactant is RDS, but the identification of surfactant deficiency is problematic clinically. There are no tests to measure surfactant amount in the airspaces, and some preterm infants will have sufficient surfactant, but that surfactant may be inhibited by edema or inflammation associated with pneumonia, for example.
From the epidemiologic perspective, the incidence of RDS increases as gestational age decreases. However, incidence of RDS is not easily defined because of variable definitions and the effect of different clinical care strategies on the diagnosis of RDS. For example, from 1997 to 2002 the NICHD Neonatal Research Network defined RDS as the need for oxygen and some ventilatory support, plus a compatible chest roentgenogram. The Network reported the incidence of RDS for infants less than 1 kg as 63% (52). The definition was changed for 2003 to 2007 to the use of supplemental oxygen for >6 hr and 95% of similar infants than had a diagnosis of RDS (53). In contrast, only about 50% of infants with birth gestations less than 28 weeks that are initially supported with CPAP have sufficient RDS to receive surfactant (54). If the use of surfactant is a surrogate for significant surfactant deficiency, then many very early gestation infants did not have severe RDS. Biologically, this indicates that induced lung maturation is very frequent. This spontaneous early lung maturation in the human fetus is believed to result from stress-induced maturation events that can be maternal, placental, or fetal in origin. Surprisingly, the fetal stress that must accompany fetal growth restriction or preeclampsia, does not consistently induce early lung maturation.
A changing epidemiology of RDS results in part from the more frequent clinical use of antenatal glucocorticoids and changes in obstetric practice that delay preterm delivery, presumably allowing the lung to mature. Numerous clinical trials have documented that maternal corticosteroid treatments decrease the incidence of RDS by about 50%, and those infants with RDS tend to have less severe disease (55). Chronic infection and fetal exposure to inflammation and histologic chorioamnionitis is frequent in pregnancies with preterm labor between 22 and 30 weeks gestation and also is associated with a decreased incidence of RDS (56). In experimental models, fetal pro-inflammatory exposures induce striking increases in surfactant and improvements in postnatal lung function without increasing fetal cortisol levels (57). Therefore, fetal exposure to inflammation may have the short-term benefit of increasing surfactant and decreasing RDS.
Fetal plasma cortisol increases as the fetus approaches term and is associated with the large increase in surfactant at term. Corticotropin-releasing hormone-deficient mice and mice with ablation of the glucocorticoid receptor have inadequate surfactant to survive following term birth (58,59). Therefore, endogenous cortisol is essential for normal lung maturation. The very early gestation human fetal lung also is responsive to corticosteroids. Explants of human lung at 14 to 20 weeks gestational age differentiate in organ culture in the absence of hormonal stimuli, and corticosteroids and thyroid hormones accelerate maturation (60).
The responses of the fetal lung to corticosteroids are multiple and impact many different systems that will influence the clinical outcome (61). Biochemical markers of maturation include glycogen loss from type II cells, increased fatty acid synthesis, increased beta receptors, and increased choline incorporation into surfactant phosphatidylcholine. In vivo, animals demonstrate improved lung function and survival. Corticosteroid treatment also decreases the tendency of the preterm lung to develop pulmonary edema (50). Although the primary effect of corticosteroids on the fetal lung is generally considered to be induction of surfactant synthesis, effects on enzymes in the synthetic pathways for surfactant have not been consistently demonstrated, and surfactant pool sizes do not increase until more than 4 days after maternal glucocorticoid treatments in sheep (61). Corticosteroids induce lung structural maturation by decreasing the amount of mesenchyme and increasing the surface area for gas exchange as is reflected by increased lung volumes within 12 to 24 hrs in fetal sheep (62). In preterm animal models, corticosteroid treatment changes the dose-response curve for surfactant treatments such that less surfactant is needed to achieve larger clinical responses (63). Because of increased lung volume, corticosteroid-treated fetuses also have improved responses to postnatal surfactant (64). There are additive or synergistic effects between the corticosteroid-exposed lungs and surfactant treatments in animal models.
Antenatal corticosteroid treatment is now the standard of practice for pregnancies at risk of preterm delivery (55). This therapy is effective and safe, although there is not long-term follow-up for infants born before 28 weeks’ gestation. Repetitive courses of antenatal glucocorticoids have been given at 7- to 10-day intervals, a practice based on the suggestion that the fetal benefit was lost after this interval (61). Maternal glucocorticoid treatments at 7-day intervals in sheep cause fetal growth restriction but augment lung maturation. Randomized trials in women at risk for preterm delivery demonstrate modest benefit, but there is some concern about longer-term outcomes, especially for infants exposed to 4 or more antenatal courses of antenatal corticosteroids (65). There is presently insufficient information for a strong recommendation about the use of repeated corticosteroid treatments.
The only other lung maturation strategy that has been extensively evaluated clinically is the combination of corticosteroids and thyrotropin-releasing hormone (TRH). Thyroid axis hormones induce lung maturation and can act synergistically with corticosteroids in vitro. Thyroid hormones do not cross the human placenta efficiently, but the tripeptide TRH crosses to the fetal circulation and increases fetal thyroid hormone levels. Unfortunately, when evaluated in large randomized, controlled trials, TRH demonstrated no benefit, and possible risks were identified (66).
[level-membership-for-neonatal-and-perinatal-medicine-category]
Tests for Fetal Lung Maturation
Tests for fetal lung maturation depend on amniotic fluid composition reflecting the status of surfactant in the fetal lung. The lung secretes fetal lung fluid at a rate of about 4mL/kg/hr. The flow of fluid out of the lung is episodic and balanced between swallowing and loss of fetal lung fluid to the amniotic cavity. Tests of lung maturation depend on the flow of fetal lung fluid containing surfactant into the amniotic fluid being sufficient to change amniotic fluid composition in a timely manner relative to the state of fetal lung maturation. Tests of lung maturation were developed to identify fetuses with early lung maturation that thus could be delivered without the risk of developing RDS. The lecithin–sphingomyelin (L-S) ratio introduced by Gluck and associates in 1971 remains the standard against which other tests are compared (4). The results are expressed as the ratio of a lecithin (phosphatidylcholine) fraction, that is cold acetone precipitated to enrich for saturated phosphatidylcholine, to sphingomyelin. Sphingomyelin is a membrane lipid and is a nonspecific component of amniotic fluid not related to lung maturation. The sphingomyelin content of amniotic fluid decreases from about 32 weeks gestational age to term, whereas the lecithin content, a large part of which is from the fetal lung, increases.
Each of the surfactant lipids and SPs has a unique developmental profile as surfactant composition changes with development (5). Phosphatidylglycerol normally appears in amniotic fluid at about 35 weeks gestation and indicates lung maturation. Phosphatidylglycerol is present in appreciable amounts only in lung tissue and surfactant. Therefore, phosphatidylglycerol can be measured in amniotic fluid contaminated with blood or meconium. Numerous other tests for lung maturity have been developed. The TDx-FLM assay of the ratio of amniotic fluid surfactant to albumin seems to be equivalent to the L-S ratio for the prediction of RDS. A currently popular test is a measurement of lamellar body numbers in amniotic fluid (67). Tests of fetal lung maturity are less frequently used in clinical practice than in the past because virtually all women at risk of preterm delivery are treated with antenatal corticosteroids, and the clinical decision to deliver a preterm seldom includes considerations of fetal lung maturation because surfactant treatments are now routine.
Genetic Causes of Surfactant Abnormalities
Four single gene disorders affecting surfactant production are currently known to cause newborn lung disease, as well as lung disease in older children and adults. Specific features of these disorders are summarized in Table 8-3 and discussed in detail in the following sections.
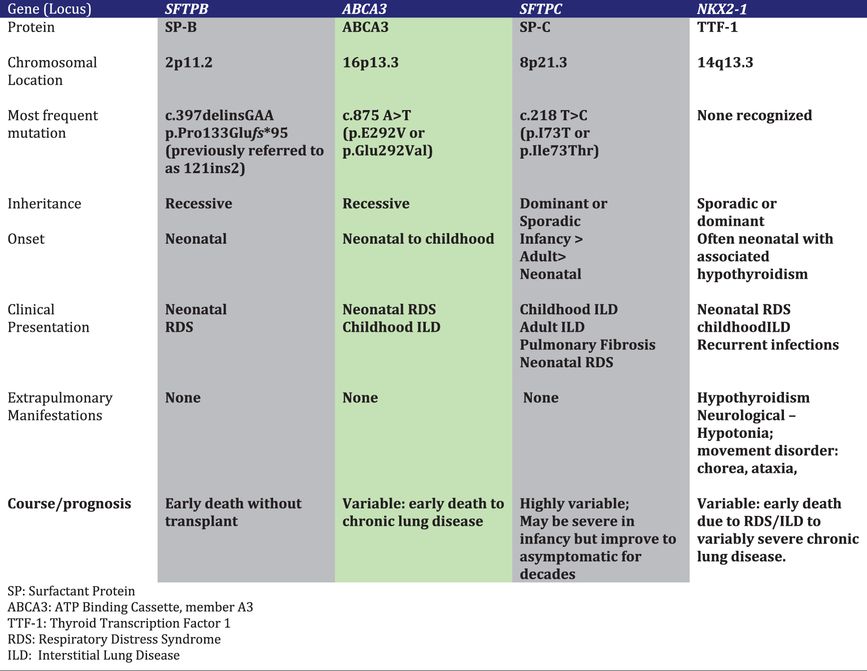 Table 8-3.
Table 8-3.Surfactant Protein-B (SP-B) Deficiency
Loss of function mutations on both alleles of the gene encoding SP-B (SFTPB) cause severe neonatal lung disease in humans and lethal neonatal respiratory failure in genetically engineered mice (68–70). SP-B deficient human newborns are usually born at full-term gestation, but have the clinical and radiographic features of RDS in prematurely born infants. Unlike premature infants with RDS who respond favorably to supportive care and surfactant treatments, SP-B deficient infants have progressive disease that is usually fatal within the first 3 months of life. Rarely affected children may survive for longer periods due to mutations that allow for some SP-B production (71,72). Experiments with genetically engineered mice in which the SP-B gene was able to be turned off indicated that an SP-B level of 20% to 30% of control values was needed to maintain normal lung function (73).
Over 40 different SFTPB mutations have been reported. The most commonly observed is a frameshift mutation precluding any SP-B production that has accounted for approximately two-thirds of the mutant alleles identified to date (70,74). The relative prevalence of this mutation reflects a common ancestral background or “founder” effect of Northern European origin (75). Population-based studies of its frequency have yielded different results depending on the population studied, with carrier rates between 1 in ~600 to 1 in 1000 (76). Combining these data with the relative contribution of this single mutation to disease yields a predicted incidence of SP-B deficiency of less than 1 in one million live births.
The lack of mature SP-B, which is critical for proper surfactant function, is the primary cause of lung disease. Additional changes in surfactant metabolism including markedly reduced amounts of phosphatidylcholine and phosphatidylglycerol in lung fluid, contribute to the severity of the lung disease (74,77). The processing of proSP-C is also impaired and leads to the accumulation and secretion of a partially processed intermediate with retained amino-terminal proSP-C epitopes, with mature SP-C production also likely reduced (78). Thus multiple abnormalities result in secretion of surfactant that is ineffective in lowering surface tension. The impaired processing of SP-C and reductions in surfactant lipids are likely due to disrupted lamellar body formation, the intracellular storage organelle in type II cells for surfactant lipids and proteins, and the cellular compartment where the final processing steps of proSP-C and proSP-B occur. Collectively these observations support an intracellular role for SP-B (or proSP-B) function along with its role in facilitating surface tension reduction at the alveolar air–liquid interface.
ABCA3 Deficiency
The adenosine triphosphate (ATP) binding cassette family of transporters use energy from the hydrolysis of ATP to move substances across biological membranes, and mutations in their genes often underlie human disease (79). Member A3 of this family (ABCA3) is localized to the limiting membrane of lamellar bodies, and term human newborns with bi-allelic loss-of-function mutations in the ABCA3 gene (ABCA3) develop an RDS-like syndrome (80). Genetically engineered mice unable to produce ABCA3 do not inflate their lungs at birth and die from respiratory failure (81–83). The phospholipid profiles from the lungs of such animals had marked reductions in surfactant phosphatidylcholine and phosphatidylglycerol (83). Analysis of lung fluid from infants who had lung transplantation and were subsequently found to have ABCA3 mutations also demonstrated marked reductions in these same phospholipids compared to infants transplanted for other disorders.(77) Finally, ultrastructural examination of the lung tissue of ABCA3-deficient newborns and animals demonstrates an absence of normally formed lamellar bodies, with numerous small dense bodies with eccentrically placed electron-dense cores giving them a “fried-egg” appearance (80). Collectively these observations support a model where ABCA3 is a key transporter of lipids critical for surfactant function into lamellar bodies.
Over 250 disease-causing mutations scattered throughout the ABCA3 gene (ABCA3) have been recognized along with large deletions spanning one or more exons. Although many infants with ABCA3 mutations have a severe phenotype similar to that of SP-B-deficient infants, others have relatively milder disease and present with findings of diffuse or ILD later in childhood (84,85). Not all have a history of neonatal lung disease, and asymptomatic children with ABCA3 mutations and adult-onset disease have also been recognized (86,87). Thus the pulmonary phenotypic spectrum of ABCA3 deficiency is much broader than that of SP-B deficiency. Genotype appears to be an important determinant of the onset and severity of lung disease, with mutations on both alleles that are predicted to preclude any ABCA3 protein expression and function uniformly associated with neonatal-onset disease and death or need for lung transplantation within the first year of life (85).
The pathophysiology of lung disease due to ABCA3 mutations is incompletely understood. The severe RDS observed in newborns with complete loss-of-function mutations can be attributed to a lack of functional surfactant, but the mechanisms whereby some children do not exhibit signs of surfactant deficiency in the neonatal period yet develop lung disease later in life are unclear. A small number of ABCA3 mutations have been studied in vitro, and missense mutations may cause inappropriate trafficking of the mutant protein, impaired ability to hydrolyze ATP, and/or impaired transport of lipid (88–90). Such mutations may allow for enough surfactant production to prevent neonatal RDS. Alternatively, ABCA3 that is misrouted to the apical surface of the type II cell may allow for secretion of sufficient amounts of surfactant lipids and proteins to prevent neonatal RDS; this hypothesis has not been experimentally tested. The mechanisms leading to ILD later in life are even less well understood. Chronically perturbed surfactant metabolism and stoichiometry resulting from decreased or abnormal ABCA3 function may injure alveolar type II cells, leading to eventual inflammation, tissue injury, and fibrosis. Finally, as ABCA3 expression is developmentally regulated, heterozygosity for an ABCA3 mutation may increase the risk for RDS in prematurely born infants due to the reduced function from one allele (91).
The precise incidence and prevalence of ABCA3 deficiency are unknown. In population-based studies, the carrier rate of the ABCA3 p.Glu292Val mutation, which has accounted for < 10% of identified mutations, has ranged from 1 in 80 (1.3%) to 1 in 125 (76,92). A case-control study of late-preterm infants with and without RDS found a carrier frequency of 3.7% in the European population and 1.5% in the African descent population (91). Databases derived from massive parallel sequencing projects (Exome Variant Server, www.evs.gs.washington.edu/EVS; 1000 genomes, www.1000genomes.org) list multiple ABCA3 coding variants. While many of these are likely not disease-causing, variants known and highly likely to be associated with disease are listed. Thus carrier frequency for any functional ABCA3 mutation may be as high as 1 in 35 individuals, which would translate to a predicted disease incidence as high as 1 in 4,500 births.
SP-C Dysfunction
Mutations in the SP-C gene (SFTPC) result in lung disease that is highly variable in its onset and severity, ranging from severe neonatal RDS to adult-onset ILD and pulmonary fibrosis, with some individuals with SFTPC mutations asymptomatic into their fifth or sixth decade of life (68,93,94). Disease may result from a mutation on just one SFTPC allele, either sporadically due to de novo mutations or inherited in an autosomal dominant pattern. All reported disease-causing SFTPC mutations are predicted to alter the coding sequence of proSP-C, and lung disease due to SFTPC mutations is thought to result from the production of an abnormal protein rather than the lack of production from one allele (haploinsufficiency). One SFTPC mutation that results in the substitution of threonine for isoleucine in codon 73 (p.Ile73Thr) has been identified in multiple unrelated individuals and has accounted for 25%–50% of the reported cases to date (95). The mutation has been associated with both sporadic as well as familial disease and has been found in individuals with different ethnic backgrounds and on different SFTPC haplotypes, indicating that recurrent mutation at this location in this gene may occur. Other mutations are scattered throughout the gene, but many are located in the region encoding the last ~100 amino acids of proSP-C, a domain with homology to a group of proteins associated with familial dementias and malignancy (BRICHOS domain) (96,97).
The pathophysiology of lung disease due to SFTPC mutations is complex and may depend on the specific mutation. Mutations in the BRICHOS domain often result in an unstable proprotein that is misfolded and targeted for degradation in the ER (96,97). Depending on the amount of abnormal protein produced and capacity of the cell to handle it, the misfolded protein may aggregate, and the unfolded protein response (UPR) activated, with subsequent caspase activation, cell death due to apoptosis and inflammation (97). Self-association of normal and mutated proSP-C in the secretory pathway may result in degradation of the protein derived from the normal allele, leading to deficiency of mature SP-C in a dominant negative mechanism. Expression of misfolded proSP-C may also result in cellular stress that can be exacerbated by additional injury. Cells in culture that were stably transfected with a known disease-causing SFTPC mutation did not exhibit toxicity at baseline, but died when subsequently infected with respiratory syncytial virus (98). In addition, transgenic mice expressing a different mutant form of SP-C exposed to bleomycin developed more prominent inflammatory and fibrotic responses than in their wild-type littermates (99).
Mutations located outside the BRICHOS domain may result in proSP-C that is routed to the plasma membrane of alveolar type II cells rather than to lamellar bodies, and then trafficked to early endosomes, and inefficiently or not processed to mature SP-C (100,101). Either the lack of mature SP-C or chronically perturbed alveolar type II cell metabolism may contribute to disease pathophysiology, although precise mechanisms remain to be determined. Depending on the nature and location of the mutation and its particular mechanism for causing disease, different treatment strategies may be necessary. Agents designed to help stabilize misfolded proteins or facilitate transit from the ER to the Golgi may be ineffective for mutations that result in abnormal targeting of proSP-C to the plasma membrane (100).
The incidence and prevalence of lung disease due to SFTPC mutations are unknown. The p.Ile73Thr mutation was not found on > 8000 alleles in samples obtained from a neonatal screening program (76). Only one individual with an SFTPC mutation was identified in a group of adults with sporadic pulmonary fibrosis or ILD, although 25% of familial pulmonary fibrosis kindreds were found to have an SFTPC mutation as the basis for disease in another study (102,103). Multiple SFTPC coding variants are listed in public databases, including some known or likely to cause disease, but the associated pulmonary phenotypes are not available. The majority of published subjects with SFTPC mutations developed lung disease after the neonatal period. Overall SFTPC mutations appear to be a very rare cause of lung disease and an even rarer cause of neonatal RDS.
NKX2.1 Haploinsufficiency
The gene NKX2.1 encodes a transcription factor essential for thyroid gland development called thyroid transcription factor 1 (TTF-1). It is also expressed in other tissues, including the central nervous system and lung, where it is necessary for expression of many genes, including those encoding SP-A, SP-B, SP-C, and ABCA3 (104). Chromosomal deletions involving the NKX2.1 locus have been observed in newborns with severe RDS and hypothyroidism, and loss-of-function mutations on one NKX2.1 allele have also been associated with the same phenotype (105,106). NKX2.1 mutations were also recognized as the cause of an autosomal dominantly inherited movement disorder, benign familial chorea. It is now clear that individuals with mutations in or deletions of this gene may have a combination of findings that include pulmonary disease (neonatal RDS, chronic lung disease, and recurrent pulmonary infections), hypothyroidism, and various neurological manifestations (developmental delay, hypotonia, ataxia, dysarthria), a constellation that has been termed “brain–thyroid–lung syndrome” (107). Affected individuals with NKX2.1 mutations may have manifestations in only one organ system, including the lungs. The pulmonary phenotypes are still incompletely described, but range from severe neonatal lung disease with pathology findings of disrupted lung development, to onset of ILD later in childhood (108).
Based on studies of the functional effects of mutations in vitro and the observation that deletions of one copy of NKX2.1 are associated with disease, the primary mechanism whereby NKX2.1 mutations cause disease is haploinsufficiency (105). It is possible that some mutations may result in a gain of function, and specific mutations may have variable effects on different downstream target genes that are important in mediating the phenotype, which might depend on which target genes are most impacted by a given mutation (107). Loss-of-function mutations leading to decreased expression of ABCA3 or SP-B would explain an RDS phenotype, decreased SP-C expression could lead to an ILD phenotype, and decreased expression of pulmonary collectins (SP-A, SP-D) contribute to recurrent infection, given their role in host defense (108).
The incidence and prevalence of lung disease due to NKX2.1 mutations are unknown. Population-based studies of the frequency of such mutations have not yet been carried out. Public databases list variants that are potentially disease causing, but phenotypic correlates are not readily available.
Lung Pathology of Genetic Surfactant Disorders
Lung histopathology findings associated with genetic disorders of surfactant metabolism may be distinctive, but are not specific for the gene involved (Figure 8-6) (109). A common finding in neonates or young infants is an accumulation of granular, eosinophilic material filling distal airspaces, a finding similar to that observed in older subjects with alveolar proteinosis syndromes where surfactant material accumulates in the airspaces due to impaired catabolism. The underlying alveolar architecture is usually preserved in subjects with alveolar proteinosis syndromes, whereas in surfactant production disorders there is often prominent alveolar type II cell hyperplasia, mesenchymal thickening, and variable degrees of fibrosis. In older children the findings of proteinosis are less prominent, and variable accumulations of foamy macrophages within airspaces may be observed, with the lung histopathology interpreted as desquamative interstitial pneumonitis, chronic pneumonitis of infancy, or nonspecific interstitial pneumonia.
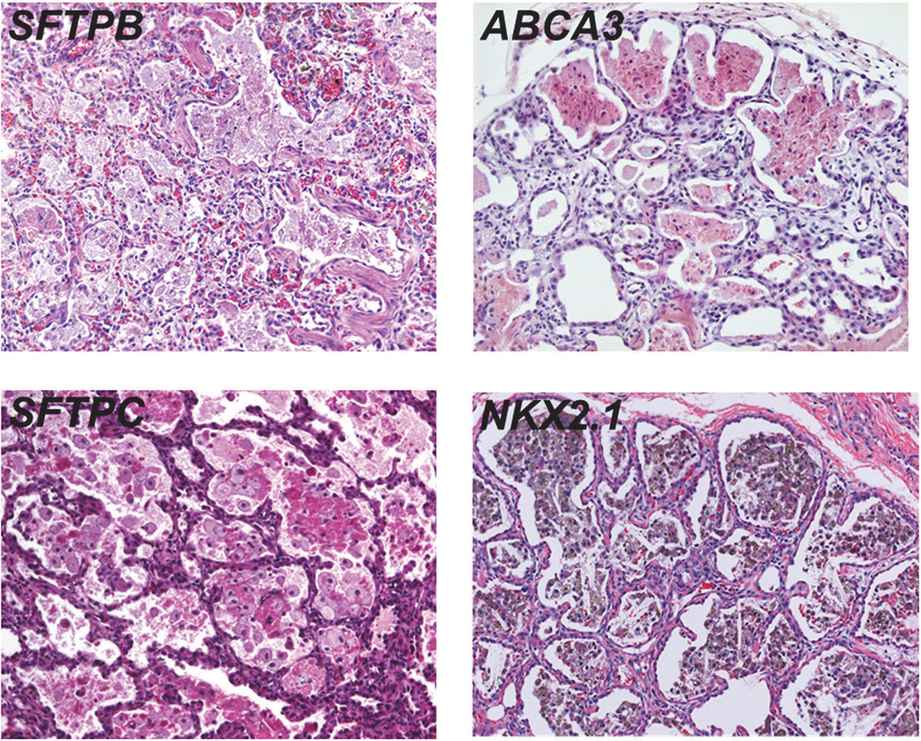
Representative lung histopathology sections (hematoxylin and eosin stain) from infants with genetic surfactant disorders. The specific gene involved is indicated on each panel. Similar findings are observed with each genetic disorder, including extracellular accumulations of proteinaceous material and macrophages, alveolar type II cell hyperplasia, and interstitial thickening.
Electron microscopy studies may provide information on the specific genetic defect. The lamellar bodies in SP-B-deficient infants are disorganized with multiple vesicles within the organelle as opposed to orderly stacked membranes. In ABCA3-deficient infants normal lamellar bodies may not be seen, with small, dense bodies with eccentrically placed electron-dense cores found in the type II cells.
Other Surfactant-Related Genes and Lung Disease
Mutations in other genes involving surfactant function and metabolism have been reported, but have not been associated with lung disease in the perinatal period. Mutations in one of the genes encoding SP-A, SFTPA2, cause familial pulmonary fibrosis and lung cancer, likely from a gain-of-toxic function mechanism (110,111). Mutations in the genes (CSFR2A, CSFR2B) encoding the receptor for GM-CSF lead to impaired alveolar macrophage function and alveolar proteinosis in children and young adults, but have not been reported as causes of perinatal lung disease (112).
Gaps and the Future
Although much has been learned regarding the composition and function of surfactant, virtually nothing is known about the transcriptional networks that integrate molecular pathways involved in prenatal type II cell maturation, surfactant protein and lipid synthesis, and alveolar defense. How these molecular networks “sense” alveolar pool size in the postnatal lung and modulate transcriptional pathways to balance surfactant synthesis with surfactant secretion, recycling, and degradation is completely unknown. Likewise, it is unclear to what extent regulatory networks involved in type II cell maturation are also involved in alveolar repair and reconstitution of surfactant homeostasis following lung injury. These critical knowledge gaps are reflected in the paucity of new therapies for lung immaturity and chronic lung diseases.
Clinically, surfactants isolated from adult animal lungs are widely available and effective. Surfactants containing only synthetic lipids and the surfactant proteins have been tested extensively in animal models and are very effective (17). The lipophilic surfactant proteins B and C are altered to increase stability or used as shorter peptides. These synthetic surfactants will result in more standardized products for clinical use, but clinical responses will probably be similar. A major effort is directed at the development of better ways to deliver surfactant to the preterm lung that can avoid intubation and mechanical ventilation.
Infants who present with a severe and progressive RDS-like syndrome may have a genetic basis for surfactant deficiency. Once infection and other causes of severe respiratory failure are excluded, then tests for abnormalities of SFTPB, SFTPC, ABCA3, or NKx2-1 may be diagnostic. No doubt rarer causes of respiratory failure in the newborn will be discovered. Presently these four diagnoses will not explain the respiratory failure in many infants who have yet to have identified genetic or developmental abnormalities that influence surfactant.
References
[/level-membership-for-neonatal-and-perinatal-medicine-category][not-level-membership-for-neonatal-and-perinatal-medicine-category]
[/not-level-membership-for-neonatal-and-perinatal-medicine-category]
