As a measure of superoxide production, cyanide-resistant O2 consumption was measured in lung homogenates from rat pups. Rat pups that had been exposed to 60% O2 for 14 days (light gray) had double the superoxide production of those exposed to air for 14 days (black), despite both sets of homogenates being equilibrated with 21% O2, indicative of phagocyte-generated superoxide. To emulate superimposed hyperoxia, homogenates from rat pups that had been exposed to 60% O2 for 14 days were equilibrated with 100% O2 (dark gray), resulting in a several-fold increase in superoxide production.
We have begun to question a uniformly toxic role for O2-derived ROS in the development of BPD, based on observations in a rat model of BPD. Rat pups exposed to 60% O2 for 14 days that have received interventions to limit lung inflammation actually have acceleration alveolar formation and increased estimated alveolar numbers (22,24,25). This would suggest, at least in this model, that it is the inflammatory cells rather than ROS that are important for the development of the lung injury and associated vascular remodeling. The stimulatory effect of 60% O2 on alveologenesis is likely to be mediated by lipid hydroperoxides, which have been shown to regulate normal alveologenesis in the newborn rat. Rat pups have immature alveolar development at birth, but do have mature antioxidant enzyme development, so this does not completely mimic the human situation with ELBW infants. However, the possibility that ROS are less uniformly toxic than previously thought may help to explain the disappointing results with antioxidant interventions, such as recombinant superoxide dismutase and N-acetylcysteine in human trials.
Ventilator-Induced Lung Injury
Ventilator-induced lung injury has also been recognized as a possible cause of, or contributor to, BPD since the time that BPD was first described. There is a plethora of literature to support the induction of lung injury by excessive tidal volumes, but evidence to support lung injury at clinically applicable tidal volumes is more limited. Attempts to minimize volutrauma by the use of high-frequency ventilation (26) and permissive hypercapnia (27) have shown no benefit. The sustained use of high-frequency ventilation would not seem to be an appropriate clinical option, given the requirement for cyclic lung expansion during fetal breathing movements to sustain lung growth. “Therapeutic” hypercapnia has been extremely effective in animal models of BPD (25) and chronic neonatal PHT (28), but this approach is unlikely to be destined for clinical use given that passive hypercapnia is associated with adverse neurological outcomes (27) and, based on animal data, an increased risk of retinopathy of prematurity. Recent data in adult animals suggests that benefits of therapeutic hypercapnia may be replicated by induction of mild–moderate metabolic (nonhypercapnic) acidosis (29). While requiring confirmation of efficacy in neonatal animal models, “permissive” or “therapeutic” acidosis could represent a potentially more translatable strategy than hypercapnia.
A large before-and-after quality improvement initiative, including delivery room extubation and the decreased use of conventional ventilation in favor of CPAP, actually showed an increase in BPD during the quality improvement cycle (30). However, when assessed by meta-analysis in isolation, there seem to be small, but significant, benefits from avoiding endotracheal intubation (31) and applying nasal CPAP in the delivery room after surfactant therapy in ELBW infants (32). Unfortunately, more than half of these infants will go on to require subsequent intubation and mechanical ventilation. The use of noninvasive modes of intermittent positive pressure ventilation has shown no reduction in the rate of BPD when compared to nasal CPAP alone (33). However, the use of volume-targeted ventilation rather than pressure-limited ventilation has been associated with a reduced combined outcome of death or BPD (34). Induction of pro-inflammatory cytokine release occurs rapidly and at relatively low distending pressures. Any adverse effects of ventilation are likely to be associated with the release of pro-inflammatory cytokines and a resultant phagocyte influx, which would be additive to that caused by hyperoxia and any concurrent sepsis (5).
Hyperbilirubinemia
Given the known antioxidant properties of bilirubin, it might seem that some degree of jaundice in the early days of life may have evolved as a protective mechanism to assist in adapting to the acute oxidative stress induced by birth, particularly by preterm birth. However, the opposite may in fact be true. In a study comparing aggressive with conservative phototherapy, there were significant beneficial effects of aggressive phototherapy on the development of BPD and the combined outcome of BPD and death (35).
Sepsis
There is very strong evidence to support a role for both early and late bacterial sepsis with the development of BPD (36). This is most likely attributable to the inflammatory response induced by sepsis.
Ureaplasma
There is a clear association between the development of BPD and pulmonary colonization with Ureaplasma species. What has been a bone of contention for many years has been whether this is a causal association. This issue has been recently comprehensively reviewed (37). There is a general consensus that Ureaplasma from the lower genital tract can ascend during pregnancy to cause chorioamnionitis and result in preterm delivery. Injection of Ureaplasma into the amniotic fluid of early-gestation sheep does result in a mild inflammatory response in the fetal sheep lung. There were some transient architectural changes in lung structure. Similar studies in nonhuman primates showed a greater degree of inflammation and significant changes in pulmonary function. There are, therefore, species differences in respect to sensitivity to Ureaplasma. With respect to reports from studies with human infants the available literature is confusing and contradictory and provides no clear answers as to any causal role for Ureaplasma in the development of BPD. To date, attempts to prevent BPD by treatment with macrolide antibiotics have been disappointing.
Packed Red Blood Cell Transfusion
There is a reported association between packed red blood cell transfusions and the development of BPD (38). Consistent with such a finding has been a single report that there is a decrease in the incidence of BPD with erythropoietin administration, prescribed for treatment of anemia of prematurity (39). However, there remain significant concerns about the association between erythropoietin administration and increased severity of retinopathy of prematurity (40), as well as there being paradoxical animal data suggesting an increased risk of ventilator-induced lung injury in premature lambs treated with erythropoietin. These latter observations argue for caution in the use of erythropoietin for prevention of BPD until both safety and efficacy have been confirmed.
Patent Ductus Arteriosus and Its Treatments
This issue has been the subject of a recent excellent review (41). In brief, the presence of a persistent left-to-right shunt across a PDA impairs lung mechanics due to an increased rate of hydrostatic fluid filtration into lung tissue, with a resultant prolongation of the need for mechanical ventilation. Use of pharmacological agents to induce an early closure of the PDA does seem to reduce the incidence of both intraventricular and pulmonary hemorrhage, but there is no conclusive evidence to support a role for persistent PDA in the development of BPD. There is, however, convincing evidence that surgical ligation of a PDA is an independent risk factor for the development of BPD. Diuretics have been commonly used in an attempt to improve lung mechanics. Parenteral administration of loop diuretics, such as furosemide, do have a short-term beneficial effect on lung compliance and O2 requirement, but evidence for long-term benefit is lacking. Similarly, evidence for long-term benefits of thiazide diuretics or spironolactone are also lacking.
Interventions Aimed at Reducing the Incidence/Severity of BPD
Therapies Targeting Lung Growth
As stated earlier, a cardinal feature of BPD in the current era is impaired alveologenesis, angiogenesis, and lung growth. Impaired pulmonary angiogenesis has been suggested to represent a primary event (42). Postnatal treatment with a stimulator of growth related genes, vitamin A, has been shown to reduce the incidence of BPD (43). Despite this, many NICUs do not use vitamin A prophylaxis. This may relate to whether the size of the benefit is seen to justify a prolonged course of intramuscular injections. As recently reviewed (44), there are also uncertainties about dosing, safety, and efficacy of high-dose vitamin A in the current population of ELBW infants. Widespread acceptance of vitamin A prophylaxis likely depends on the results of current trials assessing an intravenous delivery approach, with ongoing assessments of efficacy and safety.
Alveologenesis begins at approximately 35 weeks of gestation in the human. It is a process in which secondary crests grow into large precursor saccules to subdivide them into smaller alveoli and increase gas-exchange surface area. All appropriately grown ELBW infants are delivered prior to the onset of alveologenesis. Growth factor regulation of alveologenesis, at least in rodents, requires a number of individual growth factors such as VEGF, platelet-derived growth factor (PDGF)-AA and-BB, hepatocyte growth factor (HGF), FGF-7 (also known as KGF), and insulin-like growth factor (IGF)-I (45). That interventions targeted to each of these individual growth factors can arrest/impair alveologenesis (45) suggests that they act in concert to coordinate the process of secondary crest formation during rodent alveologenesis, which is dysregulated during lung injury, leading to impaired alveologenesis. A number of observations suggest that the same pathways may be dysregulated in human infants developing BPD. Serum IGF-I concentrations are reduced in infants developing BPD, as are levels of VEGF in their bronchoalveolar lavage and urine. In premature human neonates, a high concentration of FGF-7 in their airways is associated with a reduced risk for BPD. Reduced concentrations of HGF in tracheal aspirates are also associated with the development of BPD.
If several growth factors act in concert to regulate alveolar development, it seems unlikely that treatment with any one growth factor will restore the impaired alveologenesis induced by neonatal lung injury. Recombinant human FGF-7 (rhFGF-7) protected newborn rats from hyperoxia-induced lethality, but not from hyperoxia-induced inhibition of postnatal alveologenesis (46,47). However, in contrast, rhHGF did partially improve hyperoxia-induced impairment of alveologenesis in neonatal mice (48), and both adenovirus mediated VEGF gene therapy (49) and treatment with rhVEGF (50) preserved alveolar development in hyperoxia-exposed newborn rats. It is difficult to understand why therapy with a single growth factor, such as rhHGF or rhVEGF, can restore impaired alveologenesis if physiological alveologenesis requires the coordinated expression of several growth factors acting independently. Perhaps their protective effects are mediated by their anti-inflammatory properties, rather than their effect on DNA synthesis. Any consideration of the chronic use of such agents as rhVEGF in human infants must be tempered by observations in transgenic mice. Chronic overexpression of VEGF in the lungs of transgenic mice resulted in increased infant mortality and caused pulmonary hemorrhage, hemosiderosis, alveolar remodeling, and inflammation.
Rather than supplementing positive effectors of lung growth, a more profitable approach may be to use antagonists of growth inhibitors, which are upregulated during lung injury. The obvious candidate is TGFβ1, which may be upregulated by a peroxynitrite-mediated effect on myofibroblasts. Bronchoalveolar lavage fluid of human infants who go on to develop BPD has been reported to have both reduced latent TGFβ content and increased TGFβ1 content (51). Increased TGFβ immunoreactivity is observed in lung tissue at autopsy of infants with BPD (52). Overexpression of TGFβ1 in mutant mice (53) or neonatal rats overexpressing TGFβ due to treatment with an engineered adenovirus (54) have BPD-like lung pathology. Increased TGFß expression has been shown to be induced by hyperoxia in a mouse model of BPD, and we have observed this also to occur in a rat model of BPD. Inhibition of TGFβ signaling has been shown to protect against hyperoxia-mediated inhibition of alveologenesis in rodents (55). The peroxisome proliferator-activated receptor gamma (PPARγ) agonist, rosiglitazone, blocks the increase in TGFβ signaling induced in neonatal rat lung by hyperoxia and protects against hyperoxia-mediated inhibition of alveologenesis. Protection is also seen using the PPARγ regulator, curcumin. One way in which TGFβ may act is through a downstream stimulation of CTGF expression, which reversibly stimulates an inflammatory response and an inhibition of alveologenesis.
Optimizing nutrition in ELBW infants through the standardized use of feeding guidelines has a number of advantages, which may impact the risk of BPD (56). Earlier achievement of full enteral feeds with discontinuation of parenteral nutrition lessens risks of central-line-related sepsis. Earlier achievement of birth weight reflects better nutrition, and undernutrition has been demonstrated in animal models to impair lung growth and enhance lung injury.
Caffeine
A significant reduction in BPD was observed in a large randomized controlled trial (RCT) in which infants were treated with caffeine for apnea of prematurity or to facilitate extubation (57). Concerns were raised about the generalizability of this finding based on the maturity of the population being studied and the fact that the finding was a secondary, rather than a primary, outcome (58). Subsequent large retrospective analyses support a preventive effect of early caffeine therapy against the development of BPD but, concerningly, identified increased risks of either death (59) or necrotizing enterocolitis (60) in the caffeine-treated population. The mechanism by which caffeine exerts any beneficial effect on lung injury is unclear. A study in hyperoxic neonatal rats suggested that its protective effects were due to a reduction in pulmonary inflammation, but another study in hyperoxic newborn mice reported that caffeine therapy is associated with increased inflammation, lung cell apoptosis, and impairment of alveolar development.
Steroids and Nonsteroidal Anti-Inflammatory Drugs (NSAIDs)
Antenatal steroids are given to reduce the risk of RDS, but have no impact on the incidence of BPD, even when given as multiple courses. Postnatal dexamethasone was shown, many years ago, to improve pulmonary function and facilitate extubation in patients with BPD (61), and its use was widely adopted. This widespread use was curtailed due to its association with adverse neurologic outcomes (62). The pendulum has swung back to a limited extent with the realization that in severe cases of BPD benefits of therapy may outweigh the associated risks. Both the American Academy of Pediatrics and the Canadian Paediatric Society have made position statements supporting the use of a short course of low-dose dexamethasone in selected infants at very high risk of BPD after one to two weeks of age. The effects of dexamethasone on the lung appear to vary, depending on the duration of exposure and timing in relation to developmental stage. Dexamethasone reverses established chronic PHT in adult rats, yet prolonged treatment of neonatal rats with dexamethasone, during a period (days 3–14) when alveolarization occurs, caused permanent lung hypoplasia and augmented the severity of hypoxia-induced PHT when pups reached maturity.
Alternate strategies, such as the systemic use of a less-potent steroid, hydrocortisone, or inhaled steroids to reduce systemic side effects are as yet of no proven benefit. Results from ongoing RCTs are awaited.
One of the mechanisms by which dexamethasone may have exerted its beneficial effects on the immature lung is through its anti-inflammatory properties. This may be mediated by its highly selective inhibition of the cyclooxygenase-2 (COX-2) isoform. In a neonatal rat model of BPD, exposure to 60% O2 resulted in an increased lung tissue content of COX-2, but not COX-1. Two recent studies in rodent models of BPD have confirmed that selective COX-2 inhibitors are effective anti-inflammatory agents in hyperoxia-induced lung injury in both rat (24) and mouse (63) models of BPD, suggesting that these agents are worthy of further exploration.
Bronchodilators
Bronchodilators do have a place in the management of bronchospasm observed in some patients with BPD. They improve dynamic compliance by reducing airway resistance. Their effects are transient, and there is no evidence to date to support any long-term benefits.
Inflammation
There is a widely held view, with which we agree, that BPD is a disease caused by pulmonary inflammatory cell influx and inflammatory cell-mediated tissue injury (5). Both O2 therapy and ventilation-induced lung injury induce an inflammatory response, whereas the protection against BPD associated with caffeine and some growth factor therapies may be mediated by their anti-inflammatory effects. Whether the prenatal inflammation of chorioamnionitis is a significant contributor to the development of BPD has been an issue of recent vigorous debate (64,65), which appears, as yet, to be unresolved.
Much of our understanding of the role of inflammation in neonatal lung injury comes from animal models of BPD. Depletion of neutrophils in hyperoxia-mediated lung injury in neonatal rodents protects against inhibition of alveologenesis (22, 66). As a start to our discussion of potential targets for intervention, we will use the sequence of events that regulates the BPD-like injury observed in neonatal rats exposed to 60% oxygen for fourteen days (Figure 11-2). The likely initiating event is injury to the alveolar epithelium with the release of proinflammatory cytokines. A critical proinflammatory cytokine in this model is IL-1 because competitive inhibition of its receptor prevents the lung injury (67). IL-1 is an upstream regulator of the neutrophil chemokine, CINC-1, and blocking CINC-1 binding with a CXCR2 antagonist also prevents lung injury (22). Our current studies have shown that neutrophil elastase causes elastin fragmentation, with elastin fragments being the chemotactic agents responsible for the macrophage influx, as previously described by others in a model of adult emphysema. The macrophage influx is essential for the generation of PHT in this model (68) and in another neonatal rat model of BPD-like lung injury induced by systemic bleomycin (28). Macrophage-derived NO combines with superoxide to form peroxynitrite (69), which acts on arachidonic acid to generate F2-isoprostanes, which bind to thromboxane A2 receptors to stimulate endothelin-1 generation (70). Peroxynitrite has been shown to play specific roles in the pathogenesis of experimental chronic neonatal PHT, causing pulmonary vasoconstriction (71), vascular remodeling (69,72), and right ventricular dysfunction (73). Treatment with a peroxynitrite decomposition catalyst prevents the PHT, parenchymal thickening, and impairment of alveolarization observed in the 60% O2 exposure model (69). This is likely to be due to normalization of the expression of critical growth factors, normalization of impaired physiological apoptosis, and as our recent observations suggest, prevention of nitration and inactivation of critical growth factor receptors.
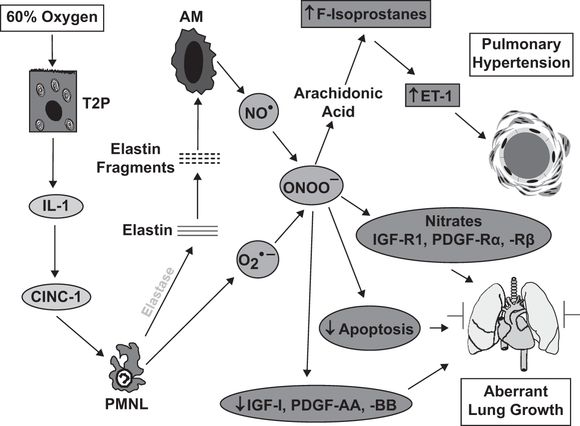
Schematic illustrating the sequence of events leading to lung injury in the neonatal rat exposed to 60% O2 for fourteen days from birth. Abbreviations: T2P = type II pneumocyte; IL-1 = interleukin-1; CINC-1 = cytokine-induced neutrophil chemoattractant-1; PMNL = polymorphonuclear leukocytes/neutrophils; AM = alveolar macrophages; NO● = nitric oxide; O2●– = superoxide; ONOO– = peroxynitrite; ET-1 = endothelin-1; IGF-R = insulin-like growth factor receptor; PDGF-R = platelet-derived growth factor receptor.
Can any of these observations suggest novel approaches to therapy in BPD? An obvious target is IL-1. Transgenic mice overexpressing IL-1β develop a lung injury similar to BPD, with lack of alveolar septation and impaired vascular development of the lung, which may be mediated through effects on the retinoic acid pathway. They also have inflammation mediated by the increased expression of neutrophil and macrophage chemokines (74). Antagonism of IL-1β binding to its receptor protects against neonatal animal models of lung injury induced by hyperoxia (67) but not that induced by excessive ventilation (75). IL-1β is increased in the lungs and circulations of infants developing BPD, so it does appear to be a rational target for clinical intervention studies.
One of the most interesting, and promising, observations from other animal models relates to gastrin-releasing peptide (GRP), which is homologous to amphibian bombesin. GRP appears to be a potent pro-inflammatory agent. Studies in neonatal mice demonstrated that exogenous GRP impaired alveolarization. Of great clinical relevance was the observation in the 125-day gestation baboon model of BPD that GRP was elevated, and that an intervention with an antibombesin antibody was protective against impaired alveolarization (76). Elevated urine bombesin-like peptide levels confer a tenfold increased risk of developing BPD in human infants born at or less than 28 weeks gestation (77).
While targeting IL-1β or bombesin-like peptides in ELBW human infants do seem like reasonable approaches to reducing BPD, it must be remembered that the ELBW population that would be the target for such interventions is already immunodeficient and at high risk of septic episodes, so suppressing their inflammatory response to reduce the risk of BPD could concurrently increase their risk of sepsis.
Inhibition of Neutrophil Elastase
The concept that proteases, such as neutrophil elastase, are critical for the development of chronic neonatal lung injury has been around for many years. An antiprotease intervention with α1-antitrypsin protected neonatal rats from O2-induced chronic neonatal lung injury (78). Unfortunately, a subsequent trial in human infants failed to show benefit, likely due to rapid degradation of intratracheally delivered α1-antitrypsin. Interest in inhibition of neutrophil elastase as a means of protecting against neonatal lung injury has been reactivated by two recent publications, in which increased levels of the specific neutrophil elastase inhibitor elafin protected against ventilator-induced lung injury (79,80). The authors attributed the observed protective effects to a preservation of the intact elastin scaffold necessary for alveologenesis. There is ample evidence to support this contention, in that elastin-knockout mice or rats chronically exposed to an inhibitor of lysyl oxidase and elastin cross-linking have impaired alveologenesis. Our recent studies (Figure 11-2) suggest the alternative or supplemental explanation that inhibition of neutrophil elastase limits elastin fragmentation and macrophage recruitment, thus limiting peroxynitrite formation.
Therapies for BPD-Associated PHT
The mainstays of therapy for BPD-associated PHT remain the same as those for BPD, including supplemental O2 when necessary, diuretics, adequate nutrition, prevention of infection, and correction of comorbidities that may contribute to lung injury. Unfortunately, therapies specific to pulmonary arterial hypertension employed with success in older children and adults are unsupported by good quality clinical data in patients with BPD and are likely of doubtful value in this context. Ideally, long-term treatment with pulmonary vasodilators should only be instituted following comprehensive evaluation of cardiopulmonary hemodynamics by cardiac catheterization, which allows for accurate determination of PHT severity, evaluation of acute vasodilator responsiveness, and definitive exclusion of major collateral vessels, pulmonary vein stenosis, and left heart disease as contributing factors (81).
Agents Targeting Peroxynitrite
Peroxynitrite is a major reactive nitrogen species formed by the reaction of superoxide with NO. Peroxynitrite can directly injure lung tissue by nitration of proteins and lipids or, following degradation, by generation of the hydroxyl radical (Figure 11–3). NO can act as either a pro-oxidant or an antioxidant, depending on the relative concentration of superoxide. When superoxide is in excess, NO is a pro-oxidant through peroxynitrite formation, whereas when NO is in excess, it is an antioxidant by limiting lipid peroxidation (Figure 11–4). This antioxidant property of NO is not widely appreciated by clinicians but may account for a number of its biological and pharmacological effects. Our data, using a peroxynitrite decomposition catalyst in neonatal rats, suggests a critical role for peroxynitrite in the 60% O2-induced lung injury (69) and in pulmonary vascular injury secondary to chronic hypoxia exposure (72). The peroxynitrite decomposition catalyst results in peroxynitrite decomposing to nitrate, rather than the toxic hydroxyl radical generated by spontaneous decomposition, and reverses the impaired lung growth and physiological apoptosis observed in this model. Protection has been observed in other models in which heavy metal–containing synthetic antioxidants that also react with peroxynitrite have been used.
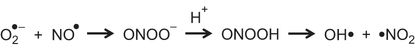
Superoxide (O2●–) reacts with nitric oxide (NO●) to form peroxynitrite (ONOO–). This becomes protonated to form peroxynitrous acid (ONOOH), which breaks down to form hydroxyl (OH•) and nitrogen dioxide (•NO2).
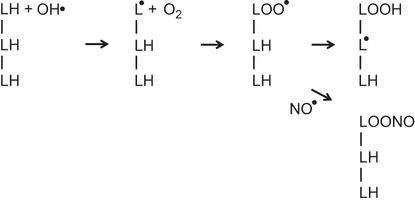
The illustration represents three adjacent fatty acids (LH) in a membrane. Exposure to a hydroxyl radical (OH•) results in the formation of a lipid radical (L●). A reaction with O2 leads to the formation of a lipid peroxyl radical (LOO●). This abstracts a hydrogen from an adjacent fatty acid to form a lipid hydroperoxide (LOOH) and a new lipid radical (L●). The process can then repeat itself sequentially generating lipid hydroperoxides and lipid radicals on adjacent fatty acids in a chain reaction. Nitric oxide (NO●) can terminate this chain reaction by the formation of alkyl peroxynitrite (LOONO) from the lipid peroxyl radical (LOO●).
Whether peroxynitrite plays as significant a role in human BPD is unknown. Plasma 3-nitrotyrosine content, a marker for peroxynitrite-mediated reactions, is increased in the first month of life in human infants developing BPD (82), consistent with sustained peroxynitrite-mediated reactions contributing to the development of the lung pathology. However, were peroxynitrite a critical mediator in human BPD, one might have expected a beneficial effect of early treatment with inhaled NO, which could potentially minimize peroxynitrite formation, but no such effect has been observed (83), despite ample experimental data from animal studies to suggest that such an effect would be observed. Possible explanations for the lack of the expected effects of NO therapy in human infants with BPD are discussed later.
Moreover, interventions targeting peroxynitrite, such as the use of peroxynitrite decomposition catalysts, involve the use of heavy metal–containing compounds, which are potentially toxic to the immature liver, based on observations in premature baboons delivered at gestational age comparable to ELBW human infants. This would make the testing of the current generation of such compounds in ELBW infants unlikely to meet the required safety standards.
Nitric Oxide
Nitric oxide (NO) is a readily diffusible and highly reactive free radical gas, first identified as the “endothelium-derived relaxing factor” in 1987. NO-mediated vasorelaxation is critical to the rapid decrease in pulmonary vascular resistance following birth and maintenance of normally low pulmonary vascular tone. NO is also critical to alveolar development, in which angiogenesis, mediated by NO downstream of VEGF tyrosine kinase receptor activation plays a critical role. Attenuated vascular NO signaling also contributes to the pathogenesis of acute and chronic PHT in early life. Properties of NO on the lung that are protective of experimental injury also include anti-inflammatory, antioxidant, anti-(smooth muscle) proliferative, and cytoprotective effects. Augmentation of NO signaling has therefore been shown to reverse sustained vasoconstriction, inhibit smooth muscle proliferation, and stimulate angiogenesis and alveolarization in various experimental lung injuries (84). NO signals in the vasculature by binding to soluble guanylate cyclase, thus increasing production of cyclic guanosine monophosphate (cGMP). NO also signals via oxidation to nitrite, or by direct nitrosylation of cysteine thiols to produce S-nitrosothiols, both of which mediate the extrapulmonary effects of inhaled NO. However, NO also contributes to disease states via generation of reactive nitrogen species, which causes oxidation and nitration.
Inhaled NO was rapidly adopted into clinical practice in the early 1990s as a short-acting pulmonary-selective vasodilator that remains in common use in neonatal intensive care. Failure of the postnatal transition leading to hypoxemic respiratory failure, known as persistent PHT of the newborn (PPHN), remains the only approved indication for this therapy. An appreciation for a critical role for attenuated lung NO signaling in BPD-like injury derived from animal studies has led to off-label use of inhaled NO, in an effort to prevent BPD. However, despite promising results in pilot studies, large trials have shown no benefit of early prolonged exposure to iNO as preventive therapy for BPD, on either short- or long-term outcomes (83).
Phosphodiesterase Inhibitors
Neonatal rats exposed to 95% O2 develop pulmonary hypertension and have arrested alveologenesis. Treatment with Sildenafil, a phosphodiesterase-5 (PDE-5) inhibitor, which, like NO, acts by increasing cGMP signaling, attenuates both the PHT and the impairment of alveologenesis (85). These results have been verified by other groups. Effects of Sildenafil were attributed to stimulated capillary formation with vasculogenesis driving alveologenesis. This is consistent with the protective effect of the angiogenic agent, adrenomedullin, in the same model. The enhanced vasculogenesis may be mediated by an upregulation of HIF-1/2α and its downstream mediator VEGF. Despite the promising animal studies, a recent pilot study of sildenafil for prevention of BPD was disappointing (86). Inhibition of PDE-4, which increases cyclic adenosine monophosphate (cAMP) signaling, has been reported both to have, and not to have, protective effects against hyperoxia-induced lung injury.
Novel NO-Based Therapies
Assuming that the biological rationale for NO-based therapy in the prevention of neonatal lung and pulmonary vascular injury is sound, there are several possible explanations for the disappointing results of human studies. First, that the beneficial effects of exogenous NO are counterbalanced by adverse ones (e.g., leading to enhanced, rather than diminished, nitration) and/or that inhalation of NO gas is a suboptimal means of providing NO to tissues in which endogenous production is deficient. Circulating and tissue-bound S-nitrosothiols contribute importantly to NO-cGMP signaling (87) and cause reversible posttranslational regulation of protein function in a manner akin to phosphorylation. Inhaled NO has been shown inferior as a means of improving tissue NO function in experimental animals, when compared to S-nitrosothiol-based (inhaled ethyl nitrite) therapy (88). In pilot human studies, inhaled ethyl nitrite improved oxygenation and hemodynamics in term infants with PPHN (89); however, no studies have been carried out to date in preterm infants. Another potential means to boost the potential benefits of exogenous NO may be as combination therapy with other agents known to improve lung growth and decrease lung injury, such as vitamin A.
Nitrite was until recently considered a physiologically inert by-product of NO oxidation. It is now apparent that circulating nitrite is recycled in tissues to form NO, thereby acting as a stable endocrine pool for “NO-like” bioactivity that is complementary to endogenous nitric oxide synthase (NOS) (90). Systemic or inhaled inorganic nitrite possesses many theoretical advantages over other forms of NO-based therapy in that tachyphylaxis does not occur with chronic dosing, effects are of relatively rapid onset and last many hours, and (sodium) nitrite is inexpensive and stable. Protective effects of sodium nitrite on adult experimental models of ischemic injury and chronic PHT have been reported (90), but no studies in neonates have been reported to date.
Novel Approaches to Improve Endogenous NO Function
Endogenous NO production by endothelial NOS (eNOS) requires an adequate supply of substrate, L-arginine, and arginine precursors, including L-citrulline. In the absence of sufficient substrate, “uncoupling” of eNOS results in a shift from NO to superoxide production, leading to oxidative and nitrative stress. Upregulation of arginases are an important cause of substrate deficiency directly contributing to inflammation and lung injury, which is preventable by hypercapnic acidosis (91) or by arginase-specific inhibitors (92). Supplementation of L-citrulline has also been shown to inhibit arginase and to prevent hyperoxia-induced lung injury in neonatal rats. Tetrahydrobiopterin (BH4) is an important cofactor for eNOS to remain in a coupled state. Newborn mice haploinsufficient for GTP cyclohydrolase I, a rate-limiting enzyme in BH4 synthesis, spontaneously develop chronic PHT (93). eNOS function may be restored, at least in vitro, by treatment with L-sepiapterin, which serves as a substrate for BH4 synthesis.
Rho-Kinase Inhibition
Activation of the small GTPase, RhoA, and its effector protein, Rho-kinase (ROCK), on stimulation by G-protein-coupled receptor ligands, including endothelin-1, thromboxane A2 and 8-isoprostane, is strongly implicated as a key pathway regulating changes in pulmonary vascular tone and smooth muscle phenotype. Therapies that enhance NO-cGMP signaling also suppress RhoA/ROCK activation (94), and beneficial effects of “statins” on pulmonary vascular disease are believed to act through attenuation of this pathway (95). Numerous animal studies and pilot reports using single doses or brief infusions of Fasudil, a ROCK-selective kinase inhibitor, in human adults and children (96) with chronic PHT have confirmed an efficacy that is equal or superior to existing pulmonary vasodilators. In neonatal rats with BPD-like lung injury, ROCK inhibitors prevent and reverse pulmonary vasoconstriction, vascular remodeling, and inhibited pulmonary angiogenesis and alveolarization (97). ROCK inhibitors are also effective when given by inhalation. No studies employing ROCK inhibitors have been conducted to date in human neonates.
Future Directions and Barriers to Progress
Parenchymal Lung Injury in BPD
Sadly, despite numerous initiatives in clinical management, the overall incidence of BPD in ELBW infants has not declined significantly. It may be that there has been some impact on certain subpopulations of affected infants, which are masked within overall data. Use of definitions that allow categorization of mild, moderate, and severely affected infants would seem to be an appropriate first step to overcoming any such hurdle.
There are some approaches to clinical management of ELBW infants for which there is high-quality supportive evidence from the literature:
(i) Applying nasal CPAP after surfactant therapy in the delivery room, and avoiding endotracheal intubation if possible.
(ii) Maintaining O2 saturations in the 90–95% range.
(iii) Using volume-targeted ventilation if mechanical ventilation becomes necessary.
(iv) Using aggressive phototherapy to treat hyperbilirubinemia.
(v) Using pharmacological agents to induce an early closure of the patent ductus arteriosus, in an attempt to avoid surgical ligation.
(vi) Use of short courses of low-dose dexamethasone in selected infants at very high risk of BPD after one to two weeks of age.
Even assuming that these clinical interventions stand the test of time, their impact is likely to be quite small. If major inroads are to be made, it will likely be through pharmacologic interventions based on mechanistic insights derived from animal models. The two current prophylactic pharmacologic interventions, for which there is evidence of efficacy in the literature, are vitamin A and caffeine. Both interventions are currently subject to uncertainties about dosing, safety, and efficacy in the ELBW population. These issues need to be better resolved before we would feel comfortable in recommending their use, but they certainly may have a place once these uncertainties are resolved. Even then, based on currently available data, their impact is likely to be small.
Given the cumulative evidence to support a critical role for inflammatory cell influx in BPD, the development of effective approaches targeting inflammation would seem to offer the most promise. Perhaps the most promising approach, based on data from extremely premature baboons, would be to target bombesin-like peptides. A small number of rodent studies suggest that the use of NSAIDs should be further studied for their clinical potential.
Suppression of the inflammatory response in ELBW infants, as stated earlier, carries a significant risk of being associated with an increased rate of sepsis in this already immunosuppressed population. Dampening the inflammatory response, without completely suppressing it, may be an option after appropriate dosimetry. Alternatively, being able to target a critical downstream mediator of lung injury due to inflammation, without directly suppressing the inflammatory response, is an exciting possibility raised by the recent observations with neutrophil elastase inhibitors, which certainly seem worthy of further exploration.
BPD-Associated PHT
The current lack of large prospective cohort studies documenting the incidence and clinical course of BPD-associated PHT in prematurely-born infants is a critical gap in knowledge that must be addressed. Lung or heart–lung transplantation, the only “curative” option for end-stage disease, is rarely feasible in BPD-associated PHT, which contributes to a paucity of high-quality human tissue available for study and a consequently greater reliance on mechanistic and therapeutic insights from preclinical models. A major determinant of long-term survival in progressive BPD-associated PHT is the ability of the right ventricle to maintain adequate output in the face of increased pressure load, yet this aspect of disease has only recently been considered as a distinct therapeutic target (98). Right ventricular adaptation to increased pressure load evolves from a compensated (hypertrophied) state to a decompensated (dilated) state, in which a progressive decline in contractile function heralds imminent death (99). This evolution appears to proceed more rapidly in infants with chronic PHT than in older children and adults. There is currently no knowledge on the pathogenesis of right heart failure in formerly premature infants and currently no published data from relevant animal models.
There are a number of barriers to progress from our currently poor understanding of the pathogenesis and natural history of chronic PHT in BPD. Diagnosis of PHT and right heart dysfunction is problematic in small infants. Clinical signs are unreliable, and catheterization is often not feasible until well after term corrected gestation, leading to a sole reliance on echocardiography, at least for initial diagnosis. Echocardiography can be challenging, especially for evaluation of the right heart, due to the thin chest wall of premature infants and frequent presence of lung hyperinflation. Echocardiography-derived parameters indicating raised pulmonary arterial pressure, such as tricuspid regurgitant jet velocity, are not measurable in all patients, and when present have been shown to correlate poorly with severity as determined by catheter measurement (100). In addition, there are no agreed-upon definitions for echocardiographic diagnosis of PHT in neonates, and certainly none for evaluation of right heart function. Systematic study of echocardiographic parameters of right ventricular function that are useful in newborns is required, incorporating new methodologies, including tissue Doppler and strain imaging, that have shown potential in children and adults. Finally, the effects of therapies designed to ameliorate injury to the lung and pulmonary vasculature could have unanticipated adverse effects on the right ventricle. Such considerations have important implications for translation of new therapies and for adoption of existing therapies employed in older children and adults.