An infant boy was born at 36 weeks gestation after a pregnancy complicated by idiopathic polyhydramnios, a prenatal evaluation showing normal chromosome karyotype, and a fetal echocardiogram demonstrating a moderately dilated right ventricle with normal function, mild right ventricular hypertrophy, and a mildly dilated right atrium, pulmonary valve annulus, and main pulmonary artery. The infant had respiratory failure at delivery with resuscitation complicated by difficulty in intubation and ventilation. MRI showed a small fistulous connection between the carina and air distended esophagus (A–B, arrows) that was confirmed at autopsy (C, arrow). The carina was continuous with the right and left main stem bronchi distally (A, arrowheads), but there was tracheal agenesis proximally with a fluid-filled structure in the midneck at the level of the glottis (D, arrow). Tracheal agenesis was confirmed at autopsy showing absence of the trachea below the larynx (E, left arrow), with the lower portion of the larynx terminating in a cystic blind-ended sac (E, right arrow; laryngotracheal cross sections shown proximal to distal from top to bottom). The cystic sac was lined by respiratory type epithelium transitioning to squamous epithelial metaplasia (F, arrow), consistent with trachea agenesis with a blind-ended sac just below the larynx. The lungs were developed (G, br = bronchiole; bv = blood vessels) because the fistulous connection between the carina and esophagus (A–C, arrows) allowed amniotic fluid into the lung. Histologic features of aspiration pneumonia including intraluminal mucus with inflammatory cells (H, arrow) and numerous squames (I, arrow), as well as features of pulmonary hypertension, were present at autopsy, resulting in respiratory failure as the immediate cause of death at 3 days of age. As is common with bronchoesophageal fistulas and tracheal agenesis, multiple other congenital anomalies were identified by radiography and at autopsy, including multilevel anomalies of the thoracic vertebrae, absence of the right superior vena cava with persistence of the left superior vena cava, Meckel’s diverticulum, and a bifid thumb. This constellation of anomalies is consistent with VATER/VACTERL association. Original magnifications: 4x (F-G), 20x (H), 100x (I).
Congenital Bronchogenic Cysts
Congenital bronchogenic cysts, also known as foregut duplications, are commonly found in the mediastinum and are caused by abnormal budding of the ventral foregut during the embryonic stage of lung development (Figure 6-2). Bronchogenic cysts may also be found in the peripheral lung, most commonly in the lower lobes, and arise from abnormal branching of the tracheobronchial tree at a later time (11,21,23–27). Approximately two-thirds are located in the mediastinum and one-third in the parenchyma. Although bronchogenic cysts are rare, with an incidence of 1 per 68,000 (26), they are the most common primary cysts of the mediastinum (21,28).
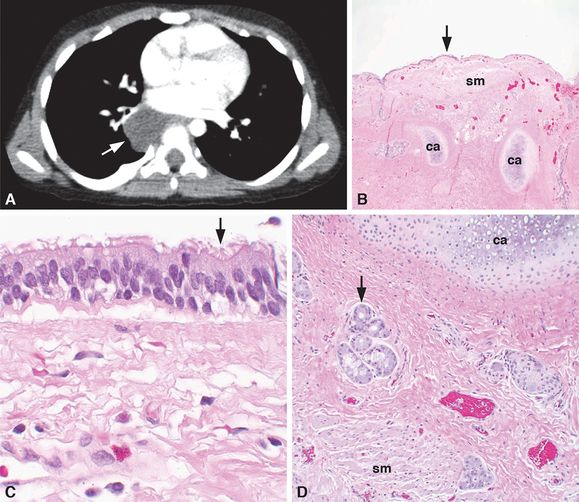
A 3-year-old boy had a right posterior mediastinal, hypodense, fluid-filled, 3-cm mass just inferior and posterior to the right hilum by CT imaging (A, arrow). No evidence of extension into the adjacent neural foramen was identified, and the mass did not enhance with intravenous contrast nor was there an aberrant vessel to the mass. The radiographic imaging favored the diagnosis of a foregut duplication cyst. At the time of surgery, the lesion was confirmed to be in the posterior mediastinum and to lack a connection to any surrounding structures. The lesion was resected with gross examination, revealing a unilocular, fluid-filled cyst. Histologic examination demonstrated that the cyst was lined by a pseudostratified, ciliated, columnar, respiratory epithelium (B–C, arrows) and that the cyst wall (B and D) contained cartilage (ca), smooth muscle (sm), and mucus glands (D, arrow), confirming the diagnosis of a bronchogenic cyst. Original magnifications: 4x (B), 20x (D), 100x (C).
Bronchogenic cysts found in the mediastinum can arise at any point along the tracheobronchial tree, commonly in paratracheal, carinal, hilar, or paraesophageal locations with the carinal location being the most frequent (21,26). Typically, they do not communicate with the conducting airway, but may be attached to the trachea or bronchus by a strand of tissue. Bronchogenic cysts found in the lungs as peripheral cysts, usually in the medial third of the lung, may or may not communicate with the tracheobronchial tree (21). Bronchogenic cysts are unilocular, typically solitary, thin-walled cystic cavities that are filled with fluid or mucus. They are lined with bronchial epithelium composed of pseudostratified, ciliated, columnar, or cuboidal epithelial cells, and their walls contain cartilage, smooth muscle, and mucus glands (Figure 6-2B–D). On chest X-ray, they appear as round or oval masses with smooth walls and may either be fluid-filled, air-filled, or have distinct air-fluid levels, unless infected (Figure 6-2A) (28). Most bronchogenic cysts are asymptomatic, but communication between an intrapulmonary cyst and the tracheobronchial tree may result in rapid expansion of the cyst, causing respiratory distress and cyanosis in the neonate, or if obstructed, atelectasis of the distal lung may occur. Mediastinal cysts may cause compression of the trachea, bronchi, and/or esophagus and can present as wheezing, stridor, dyspnea, and/or dysphagia. Intrapulmonary cysts may present as recurrent infection in older children and more rarely with hemoptysis or pneumothorax. Treatment is surgical excision (28).
Conducting Airway Abnormalities
Tracheal Stenosis
Congenital tracheal stenosis is a rare malformation in which the trachea is narrowed, due either to intrinsic abnormalities in cartilage formation or to external compression by abnormal vessel formation or vascular rings (21,29). Narrowing of the trachea by compression results in local obstruction to the passage of air, while cartilage deformities may cause obstruction of the airway on both inspiration and expiration. The major underlying causes of intrinsic tracheal stenosis are diffuse or focal abnormalities in cartilaginous ring formation, either due to posterior fusion of the C-shaped rings or formation of a complete cartilaginous sleeve (16). Several types of intrinsic stenosis have been described: (1) segmental stenosis with local narrowing of the trachea; (2) generalized or complete stenosis of the trachea, which is associated with other anomalies excluding heart and lung disease; and (3) tracheal stenosis with marked heart and lung disease (30,31). Segmental stenosis, which can occur anywhere in the tracheobronchial tree, is seen in 50% of cases, while generalized or complete stenosis is found in 30% of cases (32). Interestingly, the latter malformation has been reported in children with craniosynostosis, including Apert (OMIM #101200) and Pfeiffer (OMIM #101600) syndromes (33–42). These syndromes involve abnormal fusion of skeletal or osseous structures and are associated with autosomal dominant, heterozygous mutations in the fibroblast growth factor receptor, FGFR2 (OMIM *176943) (35,37,38,42). Craniosynostosis, cleft palate, and tracheal stenosis, with thickened, cartilaginous tracheal sleeves and atelectasis of the distal lung, have also been observed in transgenic mice wherein Fgfr2 has been mutated or partially deleted (43–45). These malformations represent mesenchymal defects in which the cells do not respond normally to FGF signaling, affecting both chondrogenesis and osteogenesis.
Recently, tracheal abnormalities consistent with intrinsic tracheal stenosis have been reported in young children with cystic fibrosis (CF; OMIM #219700), as well as in genetic animal models of CF caused by mutations or deletion of the CFTR gene (OMIM *602421). Examination of the conducting airway in neonatal CF pigs demonstrated luminal narrowing of both the trachea and main stem bronchi, as well as the presence of irregularly shaped cartilage rings, hypoplastic submucosal glands, and abnormal airway smooth muscle (46). Likewise, analysis of previously published morphometric data and chest CT scans from children with CF (less than 2 weeks old) showed alterations in the circular shape of the trachea, as well as a reduction in the size of the lumen (46). Subsequent functional studies demonstrated air trapping and airflow obstruction in young children with CF (47), as well as in the neonatal CF pig (48). Previous studies in Cftr knockout and/or mutated mice also revealed disrupted or incomplete tracheal rings with tracheal stenosis in the upper trachea, as well as altered breathing patterns in both newborn and adult mutant mice (49,50).
As for TEF and tracheal agenesis, patients with congenital tracheal stenosis have additional malformations, including other airway and lung abnormalities, esophageal and diaphragmatic abnormalities, as well as cardiovascular, skeletal, GU, and GI tract anomalies (16,31). Infants with tracheal stenosis present with respiratory distress, stridor, cyanosis, cough, and difficulty feeding. Older patients may present with recurrent pneumonia. Diagnosis is confirmed by bronchoscopy and radiologic imaging.
Extrinsic tracheal stenosis is caused by external compression of the trachea, usually associated with abnormally situated blood vessels, termed vascular rings (20,31). These can include a double aortic arch, a right aortic arch with a left ligamentum arteriosum, an aberrant (retroesophageal) right subclavian artery, a right aortic arch with aberrant left subclavian artery, an anomalous left innominate or carotid artery, or a pulmonary artery sling (retrotracheal), which is found in 20% of tracheal stenosis cases (31,32). In the case of pulmonary artery sling, the left pulmonary artery originates from the right pulmonary artery, encircling and compressing the right main stem bronchus and distal trachea.
Tracheomalacia
Congenital tracheomalacia occurs when there is an absence or abnormality of the cartilaginous rings with hypotonia of the membranous or muscular posterior wall (the trachealis muscle), which causes the trachea to collapse on expiration, obstructing the airway. It is the most common congenital abnormality of the trachea with an estimated incidence of 1 per 1,445 infants (51). Tracheomalacia can also be caused by external compression from cardiovascular structures, tumors, lymph nodes, or other masses. Primary tracheomalacia is caused by congenital immaturity of the tracheal cartilage and may be associated with other lung/foregut defects, such as TEF, EA, and bronchopulmonary dysplasia (51). Tracheomalacia is often seen in connective tissue disorders, which result in the formation of abnormal cartilaginous structures, as well as in many other genetic syndromes (51).
In infants, the airway cartilage is normally soft, so that all infants have some degree of collapse on expiration, that is, when external pressure on the trachea is greater than the internal pressure. In the normal trachea, the cartilage-to-soft tissue ratio is 4.5:1, a ratio that remains constant throughout childhood. In tracheomalacia, there is a reduction in this ratio, in some instances as low as 2:1 (20,51). In secondary, or acquired tracheomalacia, normal cartilage undergoes degeneration, which may be caused by prolonged positive pressure ventilation or by infection or inflammation. Tracheomalacia may also be a frequent complication of surgical repair of EA/TEF. Symptoms include wheeze, cough, stridor, dyspnea, tachypnea, cyanosis, and recurrent infection. Most affected infants improve by 6–12 months of age as the structural integrity of the trachea is gradually restored by further cartilage development and growth of the trachea (16).
Bronchial Atresia, Bronchial Stenosis, and Bronchomalacia
Congenital bronchial atresia is a rare anomaly and is often identified as an incidental finding on a chest X-ray in asymptomatic older children or adults, appearing as a hyperinflated or hyperlucent area that may compress adjacent tissue and cause a mediastinal shift (Figure 6-3). The most commonly affected lobe is the left upper lobe, but bronchial atresia of the right upper and lower lobes has also been reported. The segmental bronchus is the most common site of atresia, but subsegmental and lobar bronchi can also be affected along with multiple segments (21,29). The lung distal to the obstruction may be hypoplastic, often with regions of microcystic maldevelopment (9), emphysema, or hyperinflation. Air may enter the affected lobe via collateral airways, causing mild overinflation or air trapping. Mucus may accumulate in the distal bronchial segments, causing a mucus plug or a mucus-filled cyst. Often there is a loss of bronchi and vessels in the affected lobe, as well as an absence of segmentation and interlobular septa. It has been suggested that bronchial atresia is a secondary process rather than a primary developmental failure, and that these lesions arise after bronchial formation is complete (weeks 5 to 17) (9).
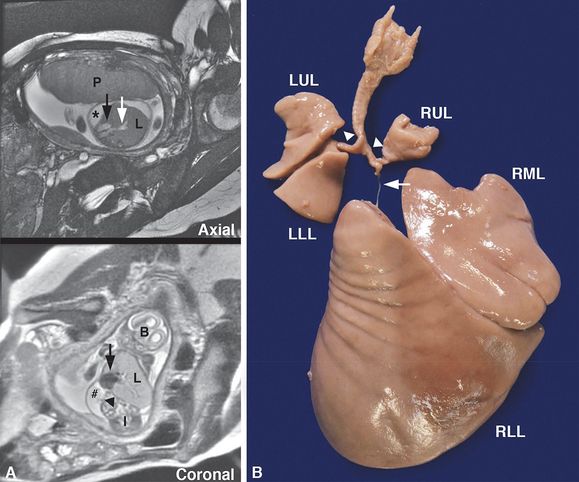
Intrauterine fetal death occurred at 25 weeks gestation after a pregnancy complicated by hydrops fetalis and a fetal MRI showing right bronchial atresia and a large right lung (A, L = lung) with dilated intrapulmonary airways (A, white arrow) associated with a mediastinal shift to the left, displacing the heart (A, black arrows) to abut the left chest wall. The diaphragm was inverted (A, arrowhead), and pleural (A, *) and peritoneal (A, #) fluid was noted. The placenta (A, P), brain (A, B) and intestines (A, I) are designated in the fetal MRI for orientation. Autopsy confirmed atresia of the right middle and lower lobe bronchi (B, arrow) with hypertrophy of the right middle (B, RML) and lower lobes (B, RLL) and hypoplasia of the right upper lobe (B, RUL) and left upper (B, LUL) and lower lobes (B, LLL) with patent bronchi (B, arrowheads).
Like tracheal stenosis, congenital bronchial stenosis, or narrowing of the bronchi, may be intrinsic or due to external compression (25). Intrinsic bronchial stenosis is rare and is usually associated with anomalous cartilage segmentation. Extrinsic bronchial stenosis due to compression is often associated with congenital heart disease. Compression occurs when the pulmonary arteries enlarge in response to pulmonary hypertension, compressing the left upper lobe bronchus. An enlarged left atrium, a bronchogenic cyst, or a teratoma may also compress the left main bronchus.
Congenital bronchomalacia, or dynamic narrowing of the bronchi, is caused by congenital abnormalities or absence of the bronchial cartilage, which lead to collapse, or bronchiectasis, of the affected airway during respiration. Bronchomalacia is a relatively common abnormality of the lower airways and is often associated with tracheomalacia. It may also be caused by extrinsic compression, or it may be secondary to infection or to lung/heart-lung transplant. Bronchomalacia is often associated with other anomalies, such as various skeletal dysplasias or diffuse congenital cartilage deficiency (29).
Pulmonary Parenchymal Malformations
Pulmonary Agenesis and Aplasia
Pulmonary agenesis and aplasia represent two forms of arrested lung development with complete absence of lung tissue (52,53). In pulmonary agenesis the absence of lung parenchyma is accompanied by complete absence of the bronchi and vasculature, whereas in pulmonary aplasia a blind-ended rudimentary bronchus is present. Pulmonary agenesis and aplasia differ from pulmonary hypoplasia wherein bronchi, vasculature and distal lung parenchyma are present but incompletely or defectively developed resulting in an overall decrease in lung size.
Pulmonary agenesis is a very rare condition estimated to affect 1 of 15,000 births (52,53). Bilateral pulmonary agenesis is extremely unusual and incompatible with life. Unilateral pulmonary agenesis is more common with the prognosis being variable and best predicted by the severity of the associated anomalies and the presence of genetic abnormalities. Pulmonary agenesis is suspected to result from a disruption of normal lung bud development that begins during the sixth week of gestation. Although the pathogenesis remains unknown, teratogenic insults and defective dorsal aortic arch blood flow in the fourth week of gestation have been proposed as contributing factors (54). The hypothesis that abnormalities in embryonic aortic arch development contribute to the pathogenesis of pulmonary agenesis is based on frequent association of unilateral pulmonary agenesis with ipsilateral malformations of derivatives of the first and second branchial arches and/or radial ray defects (54). Review of 72 cases of pulmonary agenesis associated with other malformation revealed that 82% of cases had malformations of the first and second arch derivatives and/or radial ray defects. Moreover, in all cases, the face and radial ray malformations were ipsilateral to the unilateral pulmonary agenesis, and bilateral facial and/or radial ray anomalies appeared to be indicative of bilateral pulmonary agenesis. Branchial arch and/or radial ray malformations were occasionally on the side of the less-involved lung in bilateral pulmonary agenesis cases, however, providing evidence that additional etiologic factors are operative in the pathogenesis.
Pulmonary agenesis is frequently associated with other malformations, including cardiovascular, GI, skeletal, GU, limb, and facial anomalies (53,54). Pulmonary agenesis is also a component in several syndromes, including Goldenhar syndrome, Vici syndrome (OMIM #242840), VACTERL syndrome, trisomy 21, and DiGeorge syndrome with microdeletions in 22q11.2 (53). In cases of unilateral pulmonary agenesis, multiple other anomalies are detected in ~75% of cases (53). Pulmonary aplasia can also be associated with aberrant vascular development resulting in a pulmonary artery sling that can result in tracheobronchial compression leading to severe respiratory symptoms (55). The pulmonary arteries are formed by joining of the vascular buds derived from the sixth branchial arches (central pulmonary arteries) to the lung buds derived from the postbranchial vessels (peripheral pulmonary arteries). In the case of right lung aplasia with left pulmonary artery sling, the right vascular bud is believed to connect to the left lung bud because no right lung bud exists. This process implies that in right pulmonary aplasia, the right sixth branchial arch develops initially despite the absence of the right lung bud and persists long enough to connect to the left lung bud (55). Surgical repositioning of the aberrant pulmonary artery can be successful in alleviating respiratory systems (55).
Isolated unilateral pulmonary agenesis that occurs in ~25% of patients has a much improved prognosis compared with those cases associated with other structural abnormalities (53). Compensatory growth of the unaffected lung occurs in individuals with isolated unilateral pulmonary atresia who can live without limitations. Despite concerns related to diminished lung capacity and susceptibility to recurrent pulmonary infections, isolated unilateral pulmonary atresia has been diagnosed incidentally in adults, providing further evidence that unilateral pulmonary agenesis can be associated with adequate respiratory function (56,57).
Pulmonary Dysplasia
These are a group of fatal lung disorders that include congenital acinar dysplasia (also known as acinar dysgenesis or acinar aplasia), congenital alveolar dysplasia, and alveolar capillary dysplasia with or without misalignment of the pulmonary veins (58–63). Infants with these disorders have persistent pulmonary hypertension and unexplained, severe respiratory distress requiring ventilation and/or ECMO, but deteriorate quickly when support is withdrawn.
These disorders represent a rare form of diffuse interstitial lung disease characterized by uniform developmental impairment of the distal pulmonary airspaces, or acini, resulting in severe pulmonary hypoplasia. Although there is considerable overlap between the clinical and histological features of these disorders, acinar dysplasia is the most severe phenotype, exhibiting an almost complete lack of mature alveoli, with little to no development of the pulmonary acini distal to the bronchioles. The lung lobules are composed of bronchioles lined by ciliated columnar epithelia surrounded by smooth muscle fibers, which terminate directly at the pleura and interlobular septa. Although acinar dysplasia is thought to represent arrest in the early pseudoglandular stage of lung development (8–16 weeks), the bronchiolar epithelium is well differentiated and representative of the term lung (64). In some cases, a few immature canalicular or saccular structures may be found, but this is rare (59).
In comparison, congenital alveolar dysplasia represents arrest of acinar development in the late canalicular or early saccular stage of lung development (17–24 weeks) with the formation of primitive, somewhat simplified canalicular and/or saccular structures (59,62). The interstitial regions are very wide and composed of loose, primitive mesenchyme with little collagen. Although the capillary bed is extensive, only a few of the capillaries are adjacent to the epithelial surface of the distal airspaces. Thus, development of the alveolar–capillary membrane is impaired, and gas exchange is severely compromised.
Alveolar capillary dysplasia is similar to congenital alveolar dysplasia but exhibits reduced alveolar capillary formation, as well as misalignment of the pulmonary veins (ACD/MPV; Figure 6-4). In general, the capillaries are located in the interior of thickened alveolar septa instead of in close proximity to the alveolar epithelia (58,60,63). Although the largest pulmonary veins may be located in the interlobular septa, the smaller pulmonary veins are displaced, or “misaligned,” in that they are located adjacent to the pulmonary arteries in the peribronchiolar connective tissue (60). Both the pulmonary veins and the lymphatics are thin walled and dilated, while there is increased medial hypertrophy and hyperplasia of the pulmonary arteries and muscularization of the smaller, peripheral arterioles. One-third of the patients have lymphangiectasis. In addition, there is significant underdevelopment of the pulmonary lobules with reduction and simplification of the distal acinar structures with some alveolar type II cell hyperplasia (63). Recently, three-dimensional reconstruction of lung tissue from patients with ACD/MPV demonstrated that there is a right-to-left vascular shunt linking the systemic and pulmonary circulation, which bypasses the alveolar capillary bed and causes respiratory insufficiency and persistent hypertension (65,66). Patients with ACD/MPV often have additional organ abnormalities, including GI, GU, musculoskeletal, and cardiovascular malformations, as well as disruption of the normal right–left symmetry of intrathoracic or intraabdominal organs (58,67–69). Haploinsufficiency of FOXF1 (OMIM *601089), a transcription factor important for vascular and alveolar development, causes ACD/MPV. At least 60 distinct mutations and/or genomic deletions in FOXF1 have been identified in patients with this disorder (70). Most are sporadic, autosomal dominant mutations, although several have been inherited as autosomal recessive disorders with maternal inheritance, consistent with paternal imprinting (71). Inactivation of Foxf1 in murine endothelial cells inhibited VEGF signaling and decreased expression of endothelial genes critical for vascular development (72).
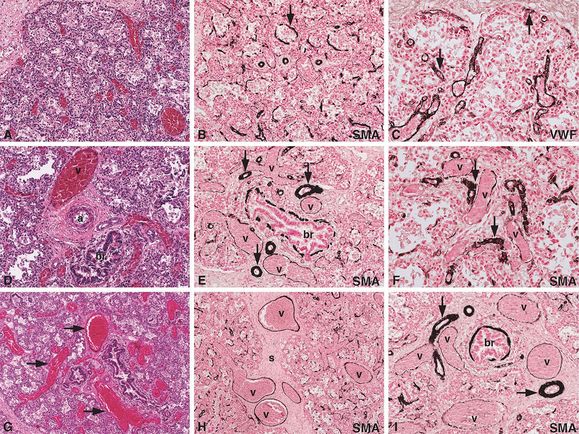
An infant girl born at term developed progressive cyanosis and respiratory distress several hours after birth, requiring mechanical ventilation and eventually extracorporeal membrane oxygenation (ECMO). After several attempts to discontinue the ECMO, a diagnosis of persistent pulmonary hypertension due to an irreversible, pulmonary lesion was made, and ECMO was discontinued. Histologic examination of the lungs at autopsy revealed diffuse alveolar capillary dysplasia (A) with muscularization of the alveolar septa (B, arrow; SMA = alpha smooth muscle actin immunostain) and reduced alveolar capillary formation (C, arrows; VWF = von Willebrand’s factor immunostain for endothelial cells), as well as misalignment of the pulmonary veins and arteries (D, br = bronchiole, v = vein, a = artery). In addition, there was muscular hypertrophy of the pulmonary arteries (E, arrows), muscularization of the small pulmonary arterioles (F, arrows), and dilated venules (F, v = dilated venules). Thin-walled, dilated, and congested venous vessels were found to be traveling indiscriminately through the peripheral lobules (G, arrows), as well as in the perilobular septa (H, s = septum, v = veins). In addition, misalignment of the muscular arteries (I, arrows) was observed, many of which were found accompanying the normally positioned pulmonary veins (I, v = veins; br = bronchiole) in the perilobular septa. Original magnifications: 5x (G, H), 10x (A, B, D, E, I), 20x (C, F).
Congenital Malformations Associated with Lung Growth and Structure
Congenital pulmonary malformations have an estimated incidence of 2–7% (73). The most commonly encountered congenital lung anomalies associated with deficient lung growth and aberrant lung structure include lung agenesis–hypoplasia complex (pulmonary underdevelopment), bronchogenic cysts, congenital pulmonary airway malformations (CPAM), pulmonary sequestrations (PS) and congenital lobar overinflation (CLO). CPAM and PS have a reported incidence of 50% and 33% of prenatally diagnosed lung lesions, respectively (74). It is widely accepted that congenital lung lesions result from perturbations in lung and airway embryogenesis. Although congenital lung malformations are a heterogeneous group of lesions, there is considerable overlap, and frequently the lesions occur together. Based on these observations, airway obstruction during development has been proposed as a unifying pathogenetic mechanism, with the location of the obstruction within the tracheobronchial tree, completeness of the obstruction, and timing of the developmental insult determining the type of lesion and histopathology (9,75).
In the past, congenital pulmonary malformations were noticed within the first weeks to months of life, but currently these lesions often are diagnosed in utero by ultrasonography and magnetic resonance imaging (MRI) due to advances in technology and implementation of routine investigations at weeks 18–20 of gestation (76–78). Prenatal MRI is highly accurate in defining congenital lung anomalies, with one study showing postnatal confirmation of the prenatal MRI diagnosis in 91% (51/56) of lesions, providing evidence that fetal MRI can provide a specific diagnosis to guide prenatal counseling and patient care (78). Prenatal diagnosis also provides an opportunity to sequentially follow these lesions to better understand their pathophysiological mechanisms. Others warn that those who interpret prenatal imaging should not attempt to make a definitive diagnosis because CPAM, PS, bronchial atresia, and CLO have overlapping features. It has thus been emphasized that only a histologic examination can definitively support a diagnosis, and diagnoses based on imaging alone should be avoided to not complicate interpretation of the literature (79).
Most congenital lung malformations have a favorable prognosis, with a mortality rate of <5% for antenatally detected lesions, and symptoms at birth occurring in 17% of cases (76,79). Poor prognostic factors include mediastinal shift, polyhydramnios and hydrops (76). Timely resection remains the treatment of choice for all symptomatic postnatal lesions; however, much controversy remains around the management of asymptomatic congenital lung malformations, which are usually discovered by routine fetal ultrasound examination (79,80). Incomplete knowledge surrounding the natural history of congenital lung malformations compromises the ability to define relative risks and benefits of early resection. Arguments for resection of asymptomatic lesions include infection risk, malignant potential, uncertainty in radiographically distinguishing lesions, prevention of pneumothorax, decreasing radiation exposure associated with surveillance, and greater compensatory lung growth early in life. These risks vary among the specific types of congenital lung malformations (80,81). On the basis of risk of infection and malignancy, current recommendations are for resection of bronchogenic cysts, CPAM, and intralobular PS, while asymptomatic CLO and extralobular PS may be observed (80). A large multicenter registry of congenital lung malformations would enhance understanding of the onset, timing, natural history, and prognosis of congenital lung malformations to improve evidence-based approaches to patient diagnosis and management.
Pulmonary Hypoplasia
Pulmonary hypoplasia is defective or incomplete development of the lung resulting in reduced lung size due to decreased numbers or size of acini (Figure 6-5). Lung weight, lung weight/body weight ratios, radial alveolar counts, and lung volume measurements are used to determine the presence of pulmonary hypoplasia (82). Clinically, fetal lung size is determined by two- and three-dimensional ultrasound and MRI (83–85). Primary pulmonary hypoplasia occurs in the absence of an identifiable cause or association and is believed to result from alterations in transcription factor and/or growth factor signaling (86). Retinoic acid signaling deficiencies in animal models results in severe respiratory phenotypes including lung hypoplasia and agenesis (87,88). Retinoic acid also influences perinatal alveolus formation in rodents, which has led to its clinical use for the prevention of bronchopulmonary dysplasia/chronic lung disease of prematurity (89,90). Identification of NKX2-1 (OMIM *600635) mutations in patients with brain-lung-thyroid syndrome (OMIM #610878) and corresponding phenotypes in Nkx2-1 deficient murine models, established NKX2-1 as a critical gene driving lung development. NKX2-1 encodes the protein thyroid transcription factor-1 (TTF-1). Expression of TTF-1 is restricted to the developing lung, thyroid, and ventral forebrain, corresponding with the respiratory distress syndrome, congenital hypothyroidism, and benign hereditary chorea phenotypes seen in syndromic patients with heterozygous NKX2-1 mutations (91–93). Although the pulmonary histopathology is heterogeneous, there are often clear growth abnormalities with alveolar simplification, lobular remodeling, and cyst formation (94). Related phenotypes in Nkx2-1 deficient mice provide evidence for a causative role for TTF-1 loss in the human syndrome. Heterozygous Nkx2-1 loss in mice results in neurological and thyroid dysfunctions, whereas homozygous deletion results in complete absence of the thyroid, as well as severe brain defects and lung hypoplasia (92,95). The lungs in Nkx2-1 null mice consist of bilateral sac-like structures that originate from a short, common tracheoesophageal tube and are lined by primitive epithelial cells (96).
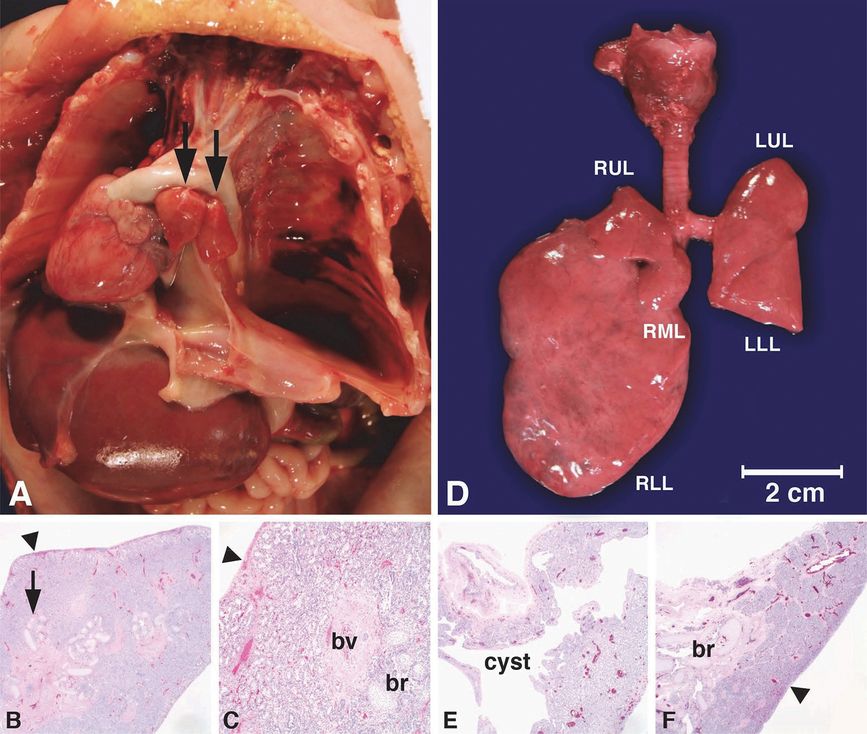
Images from two different patients are shown. Patient 1 (A–C) is an infant born at 31 weeks gestation by Cesarean section after pregnancy complicated by fetal hydrops, ascites, pleural effusions, and an abnormal chromosome karyotype of 46XX with a 13q deletion (13q32.3→13q34) and duplication of ~1.3 Mb from 12q11.1, detected by cytogenetic studies on amniocentesis fluid, microarray studies, and fluorescence in situ hybridization (FISH). Autopsy revealed a hydropic infant with severe bilateral pulmonary hypoplasia (A, arrows) with a combined lung weight = 2.1 grams (normal for 31 weeks gestation = 19.4 ± 6.1 grams), bilateral pleural effusions, ascites, and a small muscular interventricular heart defect. Histologic examination of the lungs revealed development of normal lung structures, including conducting airways (B, arrow and C, br = bronchus) with accompanying arterial vessels (C, bv = blood vessel) and a reduced number of small acini (B–C, arrowheads = pleural surface). Patient 2 (D–F) is a fetus with unilateral, macrocystic, congenital pulmonary airway malformation (CPAM) with a mediastinal shift diagnosed at 22 weeks gestation. Prenatal evaluation also revealed fetal hydrops with congestive heart failure, pleural effusion, ascites, and polyhydramnios. Intermittent variable and late decelerations as well as absent end diastolic blood flow were noted with death in utero occurring at 29 weeks gestation, despite amnioreduction and shunt placement in the right pleural cavity. A torso restricted autopsy revealed ascites, pericardial effusion, and a mediastinal shift with an enlarged right lower lobe (D, RLL) containing a CPAM with multiple large cysts, consistent with CPAM, type 1 (E), and hypoplastic right upper (D, RUL), right middle (D, RML), left upper (D, LUL), and left lower (D, LLL) lobes. Histologic examination of the hypoplastic lobes revealed normally developed conducting airways (F, br = bronchus) with accompanying vascular structures and a reduced number of small acini, resulting in a decreased distance between the hilar bronchus (F, br = bronchus) and the pleural surface (F, arrowhead). Original magnifications: 2x (B, E, F), 10x (C).
Secondary pulmonary hypoplasia occurs in association with other abnormalities and accounts for >85% of cases (82,97). A wide variety of associated abnormalities have been described, however, and the most frequent associations are with processes that compromise thoracic space or result in oligohydramnios (82). These abnormalities limit fetal breathing movements and lung distension required for normal lung development. Among the most common associated anomalies are congenital diaphragmatic hernia, obstructive uropathy, and renal anomalies, including renal agenesis, renal dysgenesis, and polycystic kidney disease. Thoracic space occupying lesions including CPAM (Figure 6-5D–F), PS, mediastinal masses, and lymphatic malformations can result in pulmonary hypoplasia. Oligohydramnios due to prolonged premature rupture of membranes has also been associated with compromised lung growth. Potter sequence is seen in cases associated with oligohydramnios of any cause and is characterized by sloping forehead, flattened face and nose, receding chin, large ears, broad spade-like hands, and deformations of the limbs secondary to compression within the uterus due to inadequate amniotic fluid. Finally, pulmonary hypoplasia can be seen in association with chromosomal abnormalities (Figure 6-5A–C), including trisomy 13, 18, and 20, and as a component of multiple syndromes including Scimitar (OMIM 608281), Down, Eagle-Barret, and Pena-Ahokeir syndromes (82). Developmental lung abnormalities, including pulmonary hypoplasia, abnormal pulmonary lobation, and anomalies of laryngeal and tracheal development, are also relatively common in Smith–Lemli–Opitz syndrome (OMIM #270400), an autosomal recessive malformation syndrome caused by mutations in the DHCR7 gene (OMIM *602858), which encodes the enzyme 7-dehydrocholesterol reductase that catalyzes the final step in cholesterol biosynthesis (98, 99). Dhcr7 null mice, a model for the human disease, die within 24 hours of birth with lung saccular hypoplasia characterized by failure to terminally differentiate alveolar sacs, delayed differentiation of type I alveolar epithelial cells, and an immature vascular network (100). Approximately one-quarter of Smith–Lemli–Opitz syndrome patients also have renal anomalies, including renal hypoplasia or agenesis, which may contribute to the lung hypoplasia (98).
Although pulmonary hypoplasia is often diagnosed prenatally or at birth, lung growth disorders can also present as diffuse lung disease in infancy. In review of lung biopsies from children <2 years of age, lung growth abnormalities were the leading diagnosis, accounting for 25% of cases (101). Pulmonary hypoplasia with prenatal conditions associated with deficient lung growth were present in some cases; however, in 85% of the cases, postnatal growth abnormalities occurred in the setting of prematurity, congenital heart disease, and/or a chromosomal abnormality, Trisomy 21 being the most frequent. The deficient lung development was histologically characterized by variable lobar simplification with subpleurally predominant alveolar enlargement.
Lung biopsy is not generally done for a diagnosis of lung growth abnormality, and infants who come to biopsy typically have diffuse lung disease with pulmonary symptoms and morbidity that are disproportionate to the clinical circumstances (101). The diagnosis of lung growth disorders in this setting is often unsuspected clinically and underrecognized histologically. A history of prematurity or congenital heart disease was highly predictive of lung growth abnormality as the primary histologic finding. Congenital heart diseases with right outflow obstruction are a significant risk factor for prenatally acquired pulmonary hypoplasia, and a decrease in alveolar multiplication may also be acquired postnatally as a direct result of the decrease in pulmonary blood flow (102). Perfusion independent mechanisms are also thought to play a role in the pathogenesis of congenital heart disease–associated pulmonary hypoplasia (102). Patient outcomes are related to the associated conditions, with prematurity being an independent clinical predictor of mortality, and congenital heart disease and pulmonary hypertension being associated with trends toward increased mortality (101). Severity of the growth abnormality, as judged histologically by dramatically increased alveolar size and moderate to severe hypertensive changes on the biopsy are also associated with increased mortality. Patients with pulmonary hypoplasia associated with congenital diaphragmatic hernias require more pulmonary support initially as compared to patients with associated omphaloceles or congenital lung malformations (103). In addition to lung volumes, disease specific factors such as pulmonary hypertension in congenital diaphragmatic hernia contributed to pulmonary morbidity and overall outcome. Pulmonary hypoplasia can also rarely present in adults as primary unilateral pulmonary hypoplasia without associated anomalies (104–106). Patients may present with wheezing, have recurrent infections, or be asymptomatic with the diagnosis made as an incidental radiographic finding. Unilateral primary pulmonary hypoplasia is typically accompanied by compensatory hypertrophy of the contralateral lung, which likely accounts for the lack of symptoms, delay of diagnosis into adulthood, and favorable outcome.
Congenital Pulmonary Airway Malformations
Congenital pulmonary airway malformations (CPAM; formerly referred to as congenital cystic adenomatoid malformations) are a heterogeneous group of cystic and noncystic lung lesions resulting from aberrant fetal lung development (Figures 6-5 D-F and 6-6). Although CPAMs are rare, with a variable reported incidence between 1 per 8,300 and 1 per 25,000–35,000 births, CPAMs account for 30–40% of all congenital lung diseases and ~95% of all congenital cystic lung diseases (77,78,107). CPAMs are classified into five major types in the Stocker classification system (types 0–4) based on clinical and pathologic features, including cyst size and histologic resemblance to segments of the normal tracheobronchial tree (82). Challenges in applying this classification scheme clinically include overlap among the CPAM types, atypical forms that do not fit well into a specific category, and importantly, indistinguishable features between type 4 CPAM and the cystic neoplasm, pleuropulmonary blastoma (PPB) (9,108). These challenges have led in some instances to the clinical practice of not dividing CPAM into types or limiting categorization of cystic CPAM to large cyst and small cyst types. Despite the challenges, the Stocker classification system is widely used and has utility in delineating several characteristic developmental lung lesions pathologically as well as radiologically.
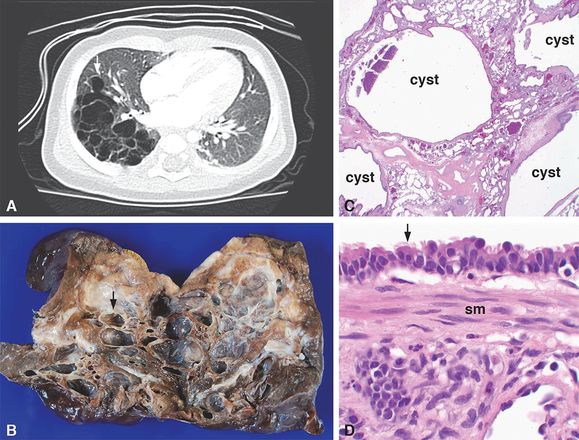
A 4-month-old boy was found to have a right lower lobe multicystic lesion by CT imaging (A, arrow) that lacked an aberrant vascular connection to systemic or pulmonary arteries. A lobectomy was performed with the lesion grossly consisting of multiple cysts ranging in size from 1 mm to 1.5 cm in greatest dimension (B, arrow) separated by intervening parenchyma. Histologic examination revealed multiple cysts surrounded by compressed normal parenchyma (C) and lined by ciliated, pseudostratified, columnar epithelium (D, arrow). Smooth muscle bundles (D, sm = smooth muscle) were present in the cyst walls, characteristic of CPAM, type 1. Original magnifications: 2x (C), 100x (D).
CPAM type 0, also known as acinar dysplasia or agenesis, is composed of bronchus-like structures with muscle, glands, and cartilage plates separated by prominent mesenchymal tissue. Type 0 CPAMs are rare and largely incompatible with life presenting in term or preterm neonates who are cyanotic at birth and survive only a few hours. Cardiovascular abnormalities and dermal hypoplasia are associated findings (82). Type 1 CPAM, the large cyst type, is the most common subtype accounting for nearly 65% of cases (Figures 6-5 D-F and 6-6) (82). CPAM type 1 is characterized by single or multiple large cysts (3–10 cm in diameter) that resemble bronchi and proximal bronchioles, including segments lined by mucinous epithelium that have been suggested as cells of origin for the mucinous adenocarcinomas rarely reported in these lesions. Type 1 CPAMs are clinically distinct from type 0 in that they primarily present in the first week to month of life but can be seen in older children and adults and are associated with a good prognosis (9,82). Occasionally, these lesions are sufficiently large to result in mediastinal shift and pulmonary hypoplasia (Figure 6-5D–F) (9). Type 2 CPAM, the small cyst type, accounts for 10–15% of cases and is composed of smaller cysts (0.5–2.0 cm in diameter), presenting in the first year of life with a poorer outcome due to the frequent association with other anomalies such as bilateral renal agenesis, cardiovascular malformations, diaphragmatic hernia, and pulmonary hypoplasia (82). Back-to-back cysts in type 2 CPAMs resemble distal bronchioles and blend with the adjacent normal parenchyma. Type 3 CPAM occurs infrequently, accounting for 5% of cases, and present exclusively in the first days to month of life with a high mortality rate (82). The lesions present as large, solid, air-containing masses comprised of small cystic spaces (≤0.2 cm) that produce a mediastinal shift, often resulting in hypoplasia of the uninvolved lung. Histologically, the lesions are comprised of immature appearing lung that is devoid of bronchi, consisting of bronchiolar-like structures surrounded by alveolar ducts and saccules. CPAM types 1–3 can be distinguished radiologically, with types 1 and 2 being heterogeneous with multiple discrete different-sized cysts and type 3 CPAM being homogeneous and solid, lacking discernible cystic spaces (11, 78). Type 4 CPAMs present as large thin-walled cystic lesions resembling distal acinar structures that are lined by epithelial cells with an alveolar type 1 and/or type II cell phenotype (109). This variant typically presents in the newborn to 4 years of age range and accounts for 10–15% of cases. The morphology, cellular phenotypes, radiologic findings, and clinical presentation of type 4 CPAMs are indistinguishable from cystic PPB and likely represent the same lesion (9,78,110,111). Erroneous designation of cystic PPB as CPAM needs to be avoided given that cystic PPB is a neoplastic process with potential to progress to an overt sarcoma.
CPAMs usually present as sporadic, nonhereditary lung abnormalities that are associated with other anomalies in 15–20% of cases, particularly in the cases of type 2 lesions (82,107,110). The pathogenesis of CPAM remains unknown. These lesions are thought to result from abnormal branching morphogenesis during lung development. It has been proposed that airway obstruction is the basis for CPAM (9,75,97,112). CPAMs usually communicate with the normal tracheobronchial tree and receive blood supply from pulmonary vessels, which differentiates CPAM from PS, wherein there is systemic blood supply (11,78). Hypothesized mechanisms for CPAM development include failure of appropriate endoderm–mesoderm signaling, imbalance between increased cell proliferation and decreased programmed apoptotic cell death, altered gene expression, and aberrant growth factor signaling during lung morphogenesis (97,113). Experimental studies including genetic modifications in mouse models identify FGF-7, FGF-9, FGF-10, HOXB-5, and SOX-2 as potential molecular mediators of CPAM (97,113–118). Recent studies suggest that distinct patterns of signaling molecule expression may be helpful in distinguishing CPAM from cystic PPB (119). A genetic basis for CPAM type 0 is supported by a tendency for the lesions to recur in families in up to 40% of cases (three of eight families reported in the literature), suggesting an autosomal recessive inheritance pattern (120).
CPAM can be diagnosed antenatally or present as respiratory difficulty and/or infection after birth (97). Indicators of poor prognosis include large lesions, bilateral lung involvement, and hydrops (Figure 6-5D–F) (11,80). The CPAM volume ratio (CVR) has emerged as a useful prognostic tool to identify fetuses at increased risk of developing hydrops (79,80). CVR measures the volume of the lung lesion divided by the head circumference to normalize for gestational age. Prenatal management includes cyst aspiration, pleuroamniotic shunt, and open fetal lung resection (80). Recent reports suggest that maternal steroid therapy may induce regression of cystic lung lesions and thus represent a novel treatment approach (80). Postnatal treatment of symptomatic patients is surgical resection, which generally consists of lobectomy or segmental resection. Surgical intervention for asymptomatic CPAM remains controversial (11,80). There are multiple reports of adenocarcinoma and adenocarcinoma in situ (formerly bronchioloalveolar carcinoma) arising in CPAM type 1 and PPB occurring in the setting of a CPAM diagnosis (81,110,121). Adenocarcinomas diagnosed in association with CPAM have been shown to have genetic mutations seen in lung cancers occurring outside the setting of CPAM including K-RAS (OMIM *190070) and EGFR (OMIM *131550) point mutations, and echinoderm microtubule-associated protein-like 4 (EML4)-anaplastic lymphoma kinase (ALK, OMIM *105590) rearrangement (122–124). On the basis of risk of malignancy and infection, current recommendations are for resection of asymptomatic as well as symptomatic CPAM (80).
Pleuropulmonary Blastoma
Pleuropulmonary blastoma (PPB) is the most common primary malignant lung neoplasm in childhood (Figure 6-7). PPB presents in three distinct clinicopathologic types that represent tumor progression (125). Type I PPB is characterized by cysts lined by benign-appearing epithelium resting on septa containing undifferentiated mesenchymal cells (Figure 6-7A–G) (108). Overgrowth of the primitive mesenchymal cells results in a combined solid and cystic (type II) or purely solid (type III) PPB with a sarcomatous component that resembles sarcomas occurring in other locations, such as embryonal rhabdomyosarcoma and fibrosarcoma (Figure 6-7H–J). The progression of type I to types II-III PPB is well documented. Importantly, however, not all cystic type I PPBs progress to the more malignant types. Indeed, purely cystic lesions that lack the mesenchymal cell component occur and are subclassified as type 1r or regressed PPBs (108,125). Cystic type I/Ir PPBs are clinically and radiographically indistinguishable from CPAM (126). Thus, these two lesions must be differentiated pathologically by identifying distinguishing features of type I PPB, including the multilocular architecture, presence of primitive mesenchymal cell collections within the septa, a relatively well-defined border with the adjacent normal lung parenchyma, and the predominantly flattened or alveolar type epithelium lining the cysts (108).
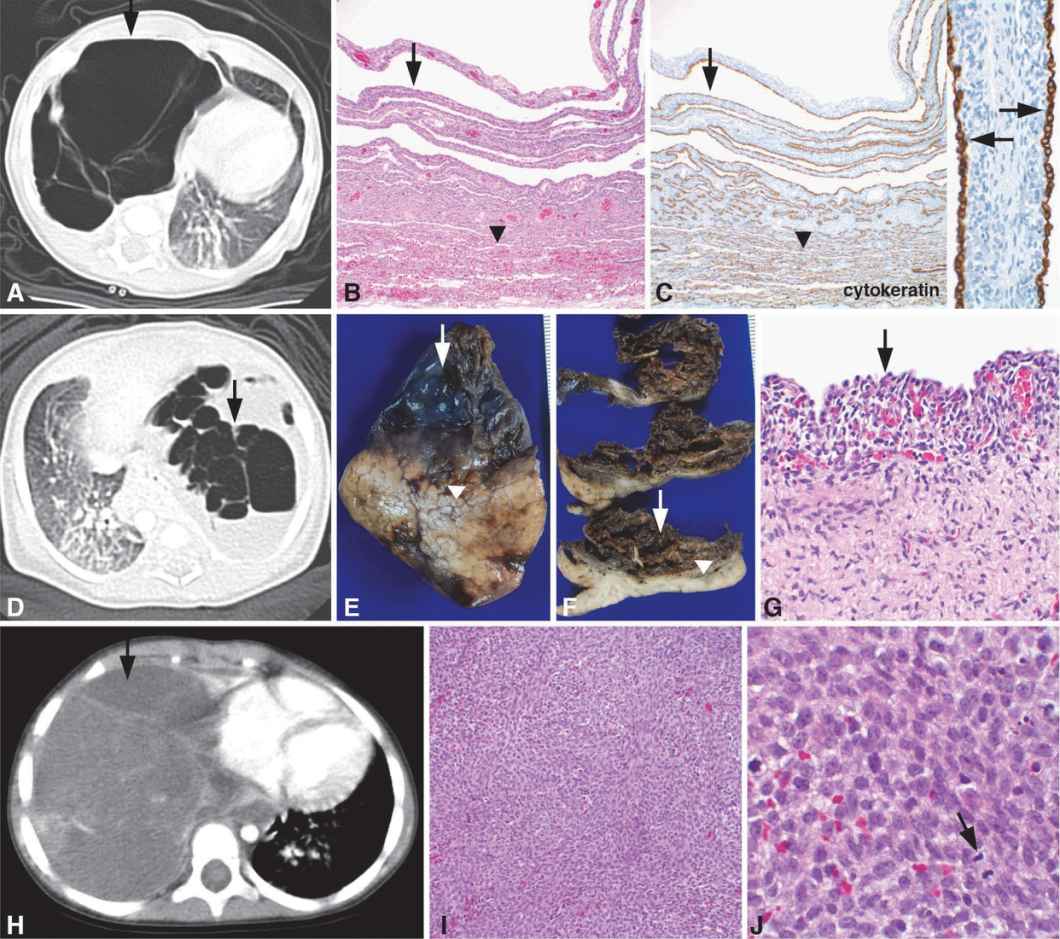
Images from three different patients are shown, demonstrating the spectrum of pleuropulmonary blastoma (PPB). Patient 1 (A–C) is a 2-month-old girl who was found to have a multicystic right lung lesion by CT scan (A, arrow) that was clinically thought to represent a CPAM, highlighting the overlapping clinical and radiographic features between CPAM and PPB. The lobectomy specimen contained a multilocular cystic lesion (B–C, arrows) with the largest cyst measuring 8 cm in maximum diameter. The cyst walls were thin measuring up to 0.2 cm in thickness, and the internal cyst wall was smooth. Some cysts were air-filled, whereas some of the smaller cysts contained fluid. The lesion was fairly well demarcated from the adjacent normal appearing parenchyma (B–C, below arrowheads). The cysts were lined by cuboidal epithelial cells highlighted by positive immunostaining for the general epithelial cell marker, pancytokeratin (C, arrows). The fibromuscular stroma within the cyst walls contained primitive mesenchymal cells, characteristic of type I PPB. Patient 2 (D–G) is a full-term infant boy delivered by scheduled Cesarean section with a prenatal diagnosis of CPAM detected by fetal MRI. A myelomeningocele and Chiari II malformation were also diagnosed prenatally. High-resolution chromosome analysis revealed a 46XY karyotype with no chromosomal abnormalities. A chest CT scan revealed multiple large rounded lucencies in the left lung with mediastinal shift to the right (D, arrow). The lung lesion was resected on day 15 of life with the lung lobe containing a multiloculated cyst (E–F, arrows) with adjacent uninvolved lung parenchyma (E–F, below arrowheads). Histologic examination of the cysts revealed a benign, cuboidal epithelium (G, arrow) lining the cysts with an underlying cambium layer comprised of a condensation of primitive mesenchymal cells directly subjacent to the epithelium (G, underlying arrow), a characteristic feature that distinguishes type I PPB from CPAM. Patient 3 (H–J) is a 3-year-old girl who was diagnosed with a large solid mass occupying the right chest by CT imaging (H, arrow) that was resected and diagnosed as a type III PPB. Local recurrence occurred 4 years later with microscopic examination of the excision specimen revealing a cellular high-grade sarcoma (I) with histologic features identical to the original tumor, including numerous mitotic figures (J, arrow) indicating a high mitotic rate. No cystic or epithelial component was identified, characteristic of type III PPB. Original magnifications: 10x (B, C), 20x (I), 40x (C inset, G), 100x (J).
PPB was initially recognized as a distinct entity in 1988 (127). Establishment of an International PPB Registry shortly thereafter led to the discovery that PPB was a sentinel tumor of a distinct hereditary syndrome (OMIM #601200) (128). This tumor predisposition syndrome is associated with a broad range of tumors with the most common tumors including PPB, cystic nephroma, ovarian Sertoli-Leydig sex cord-stromal cell tumors, and thyroid multinodular goiter (128–131). In 2009, loss-of-function heterozygous germ line DICER1 mutations (OMIM *606214) were identified as a genetic cause for this familial syndrome with up to 66% of PPB patients having a mutation (125,132). DICER1 is required for the generation of mature miRNAs, which are noncoding small RNAs that play a critical role in regulating fundamental processes, including development, cell growth, cell survival, and oncogenesis (133,134). Most carriers of the DICER1 mutation are unaffected, indicating that tumor risk is modest, and additional events may be required for tumor initiation (131). DICER1 protein expression was specifically lost in the epithelial component of some type I-II PPBs, suggesting that loss of DICER1 function in the lung epithelium may predispose to PPB initiation (111,132). Indeed, genetic targeting of Dicer1 loss to the developing lung epithelium in mice resulted in a cystic PPB phenotype (111). Moreover, studies in mouse models of the disease demonstrated that precise timing of Dicer1 loss during lung development is critical in determining phenotypic outcomes (111). DICER1 loss in the developing lung epithelium was sufficient for initiation of cystic PPB but did not result in tumor progression to sarcoma, providing evidence that additional events may be required for PPB progression. Genetic analysis of human PPBs and ovarian Sertoli-Leydig cell tumors identified frequent heterozygous DICER1 germ line loss-of-function mutations accompanied by somatic mutations in the second DICER1 allele (135–137). The somatic DICER1 mutations occurred within the functional ribonuclease RNase IIIb domain of the protein, which is required for generation of mature miRNAs. Loss of the tumor suppressor, p53, was also frequently detected in PPB (136,137). These studies highlight the potential role of RNase IIIb domain DICER1 mutations and p53 inactivation in PPB pathogenesis.
PPB is diagnosed in children <6 years of age and can arise during fetal development. The clinicobiologic progression from type I to type II and III PPB is reflected in the median age at diagnosis of 8, 35, and 41 months, respectively (125). PPB can also be diagnosed in utero with prenatal diagnosis occurring as early as 23 weeks gestation (125). The vast majority of cases (94%) present in the first 6 years of life, with rare cases presenting in older children and even adults (82). Presenting symptoms include respiratory distress, nonproductive cough, fever, chest pain, and pneumothorax. PPB type is the strongest predictor of outcome (125). Type I/Ir PPB is associated with a 91% 5-year overall survival, with deaths in this group being due to tumor progression to types II or III. Overall survival rates for type II and type III PPB are 71% and 53%, respectively. Surveillance of DICER1 mutation carriers may allow early PPB detection in the cystic type I stage resulting in improved outcomes.
Pulmonary Sequestration
Pulmonary sequestrations (PSs) are discrete masses of nonfunctioning bronchopulmonary tissue that lack communication with the normal bronchial tree and receive blood supply from one or more anomalous systemic arteries (Figures 6-8 and 6-9). PSs are classified into extralobular and intralobular types. Extralobular sequestrations (ELSs) are anatomically separate from the normal lung with a distinct pleural covering and venous drainage to systemic veins. In contrast, intralobar sequestrations (ILSs) are contiguous with the normal lung with a common visceral pleura and venous drainage into the pulmonary veins (Figures 6-8 and 6-9). ILSs are more common that ELSs, accounting for 75–80% of PSs (11,80,138). In addition to the pathologic differences, ELSs and ILSs also have differing clinical features (138). ILSs rarely produce symptoms before 2 years of age, typically presenting in childhood or adulthood as isolated anomalies with no gender predilection (73,139). In contrast, ELSs most commonly present before 6 months of life, with most studies reporting a male:female predominance of 3–4:1. Furthermore, 40–60% of infants with ELSs have other associated anomalies (11,73,113,139). Congenital diaphragmatic hernia is the most frequent coexisting anomaly being present in ~16% of cases. Additional congenital lung abnormalities are present in 25% of ELSs, including pulmonary hypoplasia, CPAM, CLO, congenital pulmonary lymphangiectasia, and bronchogenic cysts. Cardiac abnormalities, foregut duplication cysts, chest wall and vertebral deformities, hindgut duplications, and accessory spleen can also be seen.
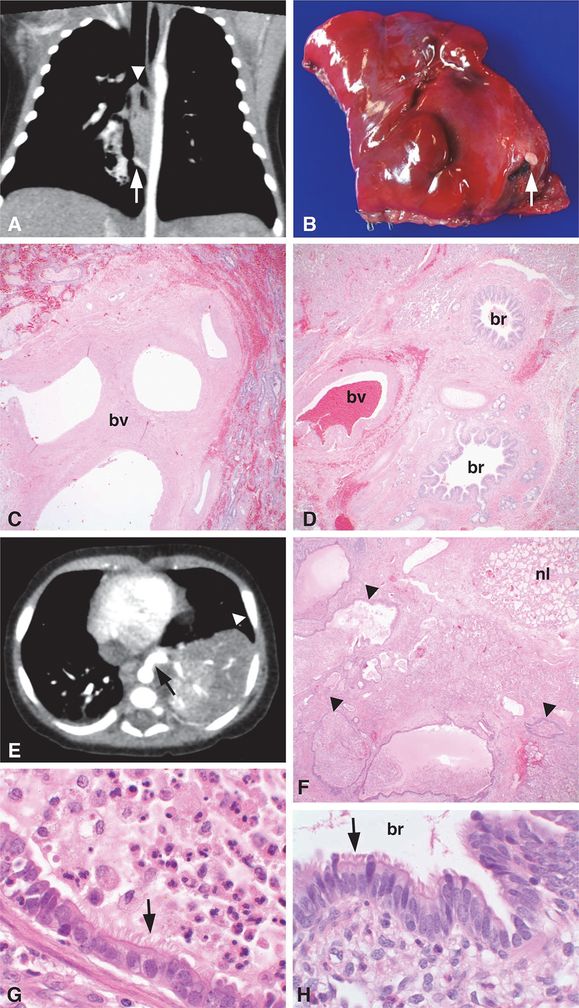
Images from two different patients are shown. Patient 1 (A–D) is a 55-day-old infant with a prenatal diagnosis of CPAM who had a chest CT scan, which showed a 2.6 x 1.5 x 1.5 cm pulmonary opacity in the medial right lower lobe (A, arrowhead) containing air bronchograms and central lucency. A systemic feeding artery to the pulmonary lesion (A, arrow) was present, consistent with PS. The feeding vessel was ~2 mm in diameter, originating from the anterior aspect of the descending thoracic aorta approximately 3 cm cephalad to the diaphragmatic hiatus to the aorta and coursing posterior to the esophagus to end in the pulmonary lesion. A right lower lobe wedge resection was performed. A vessel unpaired with an airway (B, arrow) was present on the opposite side of the specimen as the normal bronchovascular bundle entering the lung wedge. Histologic examination confirmed the diagnosis of ILS with the isolated feeding vessel consisting of a large elastic artery unpaired with an airway structure (C, bv = blood vessel) that was distinct from the paired bronchus and blood vessel entering the opposite side of the lung wedge (D, br = bronchus, bv = blood vessel). Patient 2 (E–H) is a 3-month-old boy who was found to have a left lower lobe lesion (E, arrowhead) with a large systemic feeder vessel originating from the thoracic aorta (E, arrow), consistent with PS. A left lower lobe lobectomy was performed revealing a feeder vessel entering the specimen ~2 cm from the hilar structures. The lobe was comprised of numerous cysts (F, arrowheads) surrounded by a rim of more normal appearing parenchyma (F, nl = normal), consistent with ILS. The cystic structures were lined by ciliated columnar epithelium (G, arrow) resembling the epithelium (H, arrow) lining normal bronchioles (H, br = bronchiole), characteristic of CPAM, type 2. The dilated airways within the lesion contained acute and chronic inflammatory cells including macrophages and neutrophils admixed with amorphous mucinous material (G, top right), indicative of involvement by an inflammatory/infectious process. Original magnifications: 4x (C, D, F), 100x (G, H).
[level-membership-for-neonatal-and-perinatal-medicine-category]
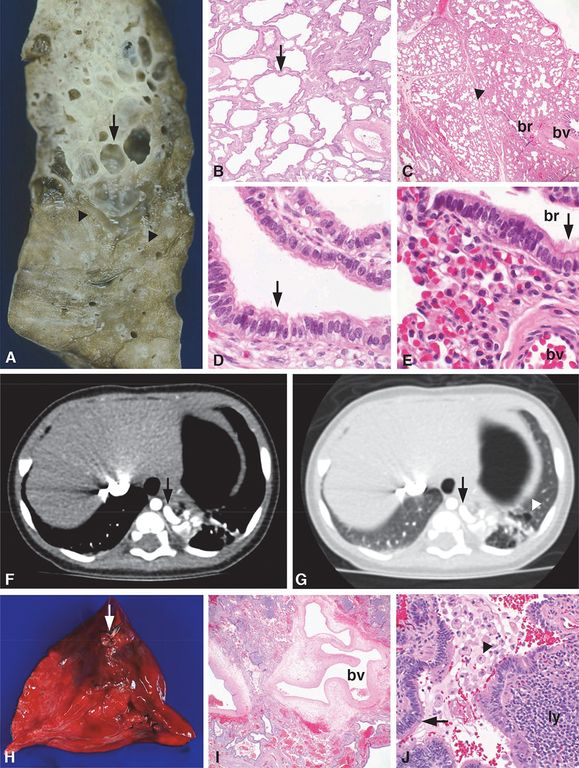
Images from two different patients are shown. Patient 1 (A–E) is a 9-day-old infant who was diagnosed with an ILS and had a left lower lobe resection. A solitary feeder vessel measuring 2 mm diameter entered the lung lobe inferior to the hilar bronchus and blood vessels. Gross examination revealed a distinct region within the lung lobe comprised of numerous cystic structures (A, arrow), some containing intraluminal secretions that contrasted with the adjacent more normal appearing lung parenchyma (A, below arrowheads). Histologic examination revealed irregularly shaped, back-to-back cysts (B, arrow) with a paucity of normal alveoli in the portion of the lung lobe supplied by the feeder artery. This histology was in contrast to the more normal-appearing parenchyma, which was comprised of normally developed, paired bronchioles and blood vessels (C, br = bronchiole, bv = blood vessel) and alveoli (C, arrowhead). The cysts comprising the malformation were lined by cuboidal to columnar, ciliated epithelium (D, arrow) and lacked mucus cells and cartilage in the underlying wall, resembling bronchioles in the normally developed lung (E, arrow, br = bronchiole, bv = blood vessel). Together these gross and histologic features support the diagnosis of a hybrid ILS/CPAM type 2 lesion. Patient 2 (F–J) is a 6-month-old boy who had a left lower lobe lesion with a systemic feeder vessel originating from the thoracic aorta (F–G, arrows) by CT imaging, consistent with PS. The lung windows revealed multiple cystic structures within the left lower lobe lesion (G, arrowhead), consistent with CPAM. Left lower lobe lobectomy was performed, and the diagnosis of a hybrid PS/CPAM lesion was pathologically confirmed. A feeder vessel was identified both grossly (H, arrow) and histologically (I, bv = blood vessel). The lung lesion had features of a CPAM, type 2, including multiple cysts measuring up to 0.8 cm in diameter that were surrounded by chronic inflammation (J, ly = lymphocytes) and lined by bronchiolar type epithelium (J, arrow) with intraluminal foamy macrophages (J, arrowhead) and amorphous eosinophilic material. Original magnifications: 4x (B, C, I), 40x (J), 100x (D, E).
The etiology of PS has been actively debated with the precise pathogenesis remaining unknown. Proposed etiologies revolve around the themes of vascular traction, vascular insufficiency, acquired pathology following infection, and foregut maldevelopment (73,113). The most widely accepted theory to best explain the spectrum of PS pathology is a congenital malformation wherein a supernumerary lung bud forms ventral to the normal primitive foregut. ILS results if the accessory lung bud develops before formation of the pleura, whereas ELS is the outcome if the accessory lung bud develops after formation of the pleura and the sequestered lung tissue forms its own pleural covering (138). Although the developmental versus acquired nature of ILS was long debated, it now seems clear that most, if not all, of these lesions are truly developmental malformations rather than acquired lesions in the setting of chronic infection (97). The observation that approximately 23% of prenatally detected lung lesions are PS, and the detection of these lesions as early as 16 weeks gestation, provides further evidence that PSs represent a congenital malformation rather than an acquired lesion (11,78). ELSs characteristically have cystic parenchymal maldevelopment with features of small cyst or type 2 CPAM being present in up to 50% of cases (9). The frequent association of PSs with other pulmonary parenchymal abnormalities, including CPAM/PS hybrid lesions (Figures 6-8E–H, and 6-9), supports the concept that multiple congenital lung malformations may have a similar embryologic origin and be part of a malformation sequence rather than representing distinct lesions (9,139). Bronchial atresia has been proposed as the common underlying etiology for congenital bronchopulmonary foregut abnormalities (9,138). This concept is supported by a prospective pathologic review of 47 pulmonary malformations that demonstrated bronchial atresia in all cases of ELS and 82% of ILS (112). Moreover, features of CPAM were present in 91% of both ELS and ILS. Based on these findings, it was proposed that CPAM and PS share the same etiopathogenesis with the spectrum of anatomic manifestations representing aberrant genetic programs and/or other insults that are modified by timing, duration, or completeness of the airway obstruction.
Diagnosis of PS requires a high index of suspicion and visualization of a systemic feeding artery to the lesion. Although newborns with ELS have a broad spectrum of presentations, the majority of patients have feeding difficulties and/or respiratory distress (11,139). Additional manifestations include pneumonia, hemorrhage, hydrops, or congestive heart failure related to the mass effect that can be seen when PS occurs as a hybrid lesion with CPAM or with substantial arteriovenous shunting resulting from the sequestered lobe. ELSs are often discovered on prenatal or neonatal ultrasound or MRI imaging, but may also remain asymptomatic throughout life or be diagnosed after identification of the associated anomalies (81,138). ELSs rarely become infected because the distinct pleural investment prevents contact with inhaled air (138). In contrast to ELSs, most patients with ILSs present in adolescence or early adulthood with recurrent bacterial pneumonia in the affected lower lobe (138). Common clinical symptoms in adult patients include cough, expectoration, hemoptysis, intermittent fever, and chest pain (140). Compared with pediatric patients, adult PS patients have significantly more respiratory infections, including concurrent Aspergillosis, that more often require lobectomy (140). ILSs comprise 93% of adult cases, likely accounting for the high rate of infection that was not seen in any of the adult patients with ELS (140). In ~15% of cases, ILS is an incidental finding on imaging performed for other reasons (138).
PSs are most readily diagnosed by CT (Figures 6-8 and 6-9) or MRI. PSs are characterized by consolidations, masses, or cystic lesions involving the lower lobes, primarily the left posterior basal segment, that are supplied by anomalous arteries arising from the aorta or other systemic artery (80,138). ELSs are almost always airless due to their separate pleural investment, presenting as homogeneous, sharply defined consolidations medial to the lung or in extrathoracic sites including the mediastinum, embedded in the diaphragm, or in the upper abdomen and peritoneum, where ELSs can mimic neuroblastoma or adrenal hemorrhage (11). ILS has three typical radiologic presentations: a solitary mass, a cystic lesion, or a consolidation.
Definitive PS diagnosis relies on identification of the anomalous artery supplying the lesions. The pedicle containing the vascular structure typically does not contain an accompanying airway. If a bronchus is present within the pedicle, a communication with the digestive system should be highly suspected (9). In 80% of cases, ELSs are supplied by a single artery, arising from the thoracic or abdominal aorta with venous drainage into the azygous system or inferior vena cava (73,138). In 15% of cases, ELSs are supplied by small arterial branches or multiple arteries. ILSs receive arterial supply from the descending thoracic aorta in >90% of cases, with the remaining cases being supplied by multiple sources including the subclavian arteries, internal thoracic arteries and arteries feeding the chest wall (73). Contrary to ELS, venous drainage in ILS is to the pulmonary veins in 95% of cases with drainage into the azygous system occurring in a minority of cases (138). The diagnostic differential for PS includes acquired systemic artery supply to the lungs in response to chronic inflammation or pulmonary artery obstruction, congenital systemic arterial supply to an otherwise normal lung such as is seen in Scimitar syndrome (which typically involves the right lower lobe rather than the left lower lobe as seen in PS) and primary metastatic tumor (138,139). ILS must also be differentiated from CPAM and CLO with the key differentiating feature being demonstration of a systemic feeding vessel (78,97).
Surgical resection remains the mainstay of treatment for symptomatic PS, with embolization of the feeding vessel being an additional therapeutic consideration. It is recommended that asymptomatic ILS also be resected based on the risk of future infection, hemoptysis, or malignancy (80,138,140). Carcinoma arising in PS is rare, with eight cases reported in association with ILS and a single report of a BRAF (OMIM *164757) mutant adenocarcinoma arising in ELS (141). ILS-associated malignancies differ pathologically from the mucinous adenocarcinomas and PPBs associated with CPAM, suggesting potential different pathways of carcinogenesis in the two entities. PS-associated malignancies also occur in older patients (>30 years of age) differing from PPBs in CPAM, which occur in children, and the CPAM-related mucinous adenocarcinomas that are typically identified in younger patients (median age of 20 years).
Treatment of asymptomatic ELS is controversial but can generally be observed with serial monitoring because the lesions may remain asymptomatic throughout life (80,138,140). A conservative treatment approach for asymptomatic ELS is also supported by good outcomes of prenatally diagnosed ELS as well as several studies reporting substantial or complete regression of the lesions on sequential scanning during pregnancy (73,74). A significant number of ELSs are diagnosed prenatally, with PS accounting for 33% of all prenatally diagnosed lung lesions (74). A recent postnatal follow-up study of fetal ELS, however, noted that cases diagnosed with complete regression in utero almost invariably had ELS persistence in postnatal CT imaging (142). In 18 ELS cases noted to disappear on sequential scanning through pregnancy, postnatal CT demonstrated persistence in 14 cases with the remaining four infants not undergoing postnatal CT. The apparent disappearance sonographically was explained by PS tissue becoming isoechogenic with adjacent normal lung parenchyma. Even with persistence of the ELC postnatally, however, antenatally diagnosed PS in the absence of hydrops was associated with an excellent prognosis.
Congenital Lobar Overinflation
Congenital lobar overinflation (CLO; also referred to as congenital lobar emphysema, overinflation syndrome and infantile lobar emphysema) is overdistension or hyperplasia of pulmonary segments or lobes resulting from partial or complete bronchial obstruction (Figure 6-10). The term overinflation is used to highlight that this condition is characterized by overdistension of the airways rather than alveolar destruction, as may be implied by the term emphysema. The airway obstruction can be caused by bronchial abnormalities, including bronchial atresia, bronchial stenosis, or most commonly, bronchial cartilaginous dysplasia or congenital bronchial cartilage deficiency resulting in bronchial collapse on expiration (82,143). CLO can also present in association with other conditions that lead to extrinsic or intrinsic bronchial obstruction. Causes of extrinsic bronchial obstruction include vascular anomalies such as pulmonary artery slings and anomalous pulmonary venous return as well as mass lesions such as ELS and bronchogenic cysts. Intrinsic obstructive causes include bronchial mucosal folds, bronchial torsion, or luminal obstruction by aspirated meconium, mucus plug, foreign body, or granulation tissue.
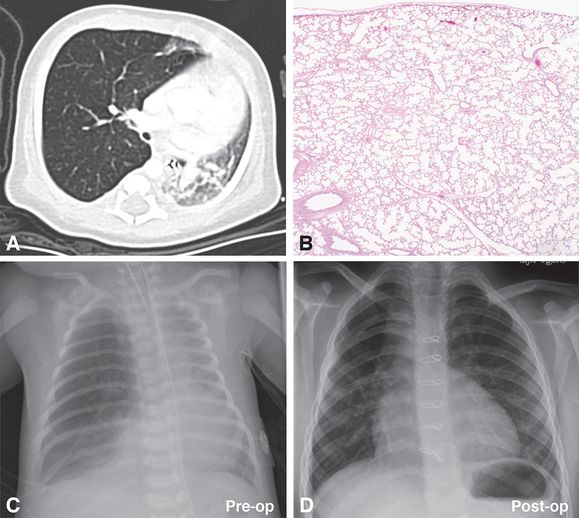
A 14-day-old boy with tetralogy of Fallot had marked hyperinflation of the right upper lobe resulting in leftward deviation of the thymus and anterior junction lines as well as compressive atelectasis of the left lung and right middle and lower lobes by CT scan (A). Cardiac anomalies associated with tetralogy of Fallot and a right-sided aortic arch were also present. The right upper lobe was resected and was without gross lesions. Histologic examination revealed normal lung structure with diffuse overinflation of the alveoli (B). Together the radiographic and histologic features are characteristic of CLO. Resolution of the right lung overinflation and left mediastinal shift present in preoperative chest radiographic images (C, Pre-op) was noted in the postoperative chest radiograph taken at ~7.5 years of age (D, Post-op). Original magnification: 2x (B).
CLO has two patterns that have been hypothesized to be related to the timing of bronchial obstruction during lung development and/or the degree of obstruction (144). The classic pattern, seen in nearly 70% of cases, is characterized by uniform overdistension of normally developed alveolar saccules and alveoli. Radiological imaging in these cases shows a hyperlucent, overdistended lobe compressing the uninvolved lobes and producing a mediastinal shift (Figure 6-10A, C) (143). The remaining 30% of cases have a polyalveolar pattern with little overdistension of complex acini typically seen in hyperplastic lungs (144). This polyalveolar form may present radiographically as a lobe with normal lucency occupying a disproportionate portion of the hemithorax with mediastinal shift. Polyalveolar lobe is generally believed to be a variant of CLO in which there is an increase in alveolar number associated with enlargement of a lobe, rather than overinflation of a lobe with a normal alveolar number (9). It is hypothesized that “pulmonary hyperplasia” results from relatively complete obstruction of a bronchus in early lung development leading to accelerated lung growth in the final trimester similar to the hyperplasia noted in some infants with laryngeal or tracheal atresia (144).
CLO is a rare condition with a prevalence of 1 per 20,000 to 30,000 deliveries (113). CLO presents in the first week of life in ~50% of cases and within the first 6 months in >80% of cases (82). Although most patients present in the neonatal period, CLO can also be detected in utero and occasionally be diagnosed in children and young adults (11,78,145). Age at the time of diagnosis is inversely related to the severity of respiratory distress (143). Presenting symptoms include dyspnea, tachypnea, cyanosis, infection, wheezing, cough, and hoarseness (143,144). Sudden pneumothorax is a rare presentation. Anomalies in other organs can be seen in association with CLO in up to 40% of patients, with 70% of these anomalies occurring within the cardiovascular system (82,143). Less commonly, CLO is associated with renal, GI, musculoskeletal, and cutaneous malformations (113). Treatment is based on the severity of respiratory distress. Traditional treatment is lobectomy to ensure compensatory lung growth; however, recent studies indicate that some patients can be safely managed conservatively (143). Long-term follow-up of 30 patients treated with surgery or conservative management revealed good outcomes with all patients having normal oxygen saturation levels and being free of respiratory symptoms, suggesting that patients can benefit from both surgical and conservative management.
Pulmonary Lymphangiectasia
Pulmonary lymphangiectasis (PL; also referred to as congenital pulmonary lymphangiectasia) is a rare disorder characterized by dilated lymphatic vessels with a normal lymphatic distribution within the subpleural region, in interlobular septa, and around bronchovascular bundles. Diagnosis is based on clinical signs together with radiologic imaging and histologic findings, with open lung biopsy considered the diagnostic gold standard (146). The predominant dilation of nonproliferative lymphatic channels in lymphangiectasis is differentiated from lymphangiomatosis, which is characterized by proliferation of lymphatic channels resulting in an increased number and decreased size of lymphatic spaces with a proliferative spindle cell component. Both conditions preferentially involve lymphatic vessels in a normal lymphangitic distribution, which can lead to challenges in differentiating the two entities clinically and pathologically with limited biopsies.
PL is divided into primary and secondary categories. Primary PL is further subclassified as isolated, generalized, or syndromic based on presentation as an isolated pulmonary lesion, lymphangiectasis involving other tissues in addition to the lung such as bones, viscera and soft tissues, or as a component of a syndrome, respectively (146). Secondary PL occurs in association with obstructive cardiovascular lesions with the most common abnormality being total anomalous pulmonary venous return but also including hypoplastic left heart syndrome, pulmonary vein atresia, congenital mitral stenosis, cor triatriatum, and thoracic duct agenesis (82,146). Cardiovascular anomalies are seen in 60% of PL patients, and renal malformations, generalized lymphangiectasis, and other anomalies are present in another 20% of cases (82,97).
Most PL cases are sporadic with a >2.5:1 male predominance (82,146). The association of PL with numerous syndromes, however, has led to the recommendation that an underlying syndrome should always be considered in PL patients (146). PL has been described in association with chromosomal abnormalities, including Turner, Down, and Phelan McDermid (OMIM #606232) syndromes. PL can also be a component of numerous other syndromes with the RASopathies [Noonan, Cardio-Facio-Cutaneous, and Costello (OMIM #218040) syndromes] being a common association as well as syndromes characterized by lymphedema or generalized lymphatic dysplasia such as Hennekam syndrome. Patients presenting with syndromic PL should be considered for genomic testing. Mutations in genes associated with specific syndromes have been identified in PL patients including PTPN11 (OMIM *176876) and SOS1 (OMIM *182530) signaling molecules within the RAS pathway, and FOXC2 (OMIM *602402) (147–149).
The cause of PL is unknown, and no disease-specific therapies have been developed. Primary congenital PL is believed to be an inherent developmental abnormality of the lymphatic system. Postulated pathogenic mechanisms include a developmental error in which the normal regression of connective tissue elements fails to occur and/or a failure or delay in linkage of isolated lymphatic spaces (97,146). Familial occurrence, although rare, has been described in six affected families suggesting a possible genetic component (146). VEGFR3 (FLT4, OMIM *136352) mutations are described in PL associated with Nonne-Milroy lymphedema syndrome (OMIM #153100), and interestingly, perinatal overexpression of the ligand for this receptor, VEGF-C, was shown to induce PL in a mouse model that phenotypically and histologically resembles the human condition (150). Moreover, VEGFR-3, along with VEGFR-2, was required for the development of lymphangiectasia in the neonatal mice. Together, genetic evaluations of PL patients combined with defining the precise role of the identified molecules in experimental models has great promise for improving our understanding of the molecular mechanisms underlying PL pathogenesis as well as the associated anomalies.
PL is usually a fatal disorder that typically presents in the first hours to days of life as severe respiratory distress resulting from unilateral or bilateral pleural effusions, pulmonary hypoplasia, and surfactant deficiency in combination with prematurity (97,146). The clinical course may be complicated by persistent chylothorax, ventilator dependency, anasarca, arterial hypotension, heart failure, secondary pulmonary hypertension, and progressive respiratory failure. Patients with generalized lymphangiectasis usually have less pulmonary involvement. Neonatal presentation of primary PL limited to the lung was typically considered a uniformly fatal disease, but increased survival has resulted from advances in perinatal care (146). Despite decreased mortality, morbidity may still be high, requiring interdisciplinary long-term follow-up care. PL can also manifest in childhood or even in adult life with a wide spectrum of presentations, including respiratory failure, persistent tachypnea, dyspnea, cough, wheezing, hemoptysis, recurrent airway infections, or as an incidental finding in an asymptomatic patient (146,151). PL can be limited to one or two lobes, which may account for the lack of symptoms and favorable long-term outcomes in some patients (151). Radiographic findings are not specific, but a reticulonodular interstitial pattern with increased interstitial markings or thickening, likely representing dilated pulmonary lymphatics, add to the diagnostic evaluation especially in the absence of biopsy. Treatment of prenatal fetal chylothorax by thoracocentesis, thoracoamniotic shunting, or medical pleurodesis can be performed to prevent severe pulmonary hypoplasia and hydrops. Postnatal treatment of PL is primarily supportive, including mechanical ventilation, surfactant administration, thoracocentesis, ECMO, cardiocirculatory support, and total parenteral nutrition (146). Substitution of electrolytes, coagulation factors, and immunoglobulins may also be required due to chylothorax with persistent, refractory chylothorax requiring prolonged conservative and/or surgical interventions.
Summary and Future Directions
Congenital lung malformations are a heterogeneous group of abnormalities resulting from defective foregut specification, branching morphogenesis and cell proliferation, survival, and differentiation. There is considerable overlap among these anomalies, which commonly occur in combination or as hybrid lesions. Clinical diagnosis is challenging given the overlapping features among the lesions, some of which have distinct natural histories and clinical management. Significant advancements in radiologic imaging and routine investigations in utero have resulted in a shift from postnatal to prenatal fetal diagnoses. Continued focus on developing better imaging modalities capable of differentiating among the distinct malformations is needed to combat the current concern that definitive diagnosis cannot be established by prenatal imaging alone but rather relies on histologic examination. Definite prenatal diagnosis will be beneficial in guiding prenatal counseling and clinical intervention. Incomplete knowledge surrounding the natural history of congenital lung malformations compromises the ability to define relative risks and benefits of early interventions. Large multicenter registries would be beneficial to enhance understanding of the onset, timing, natural history, and prognosis of these rare congenital lung malformations to improve evidence-based approaches to patient diagnosis and management.
Prenatal diagnosis provides an opportunity to follow these congenital malformations sequentially to better understand their pathophysiological mechanisms. Overlapping features among the heterogeneous pulmonary malformations suggest common etiologies, but further studies are needed to define the pathogenesis of both distinct and overlapping phenotypes. Proposed unifying mechanisms include airway obstruction and aberrant, embryonic aortic arch development. Failure of appropriate endoderm–mesoderm signaling, deregulated cell proliferation and programmed cell death, altered gene expression, and aberrant growth factor signaling are additional proposed mechanisms. Defining pathogenic mechanisms underlying congenital lung malformations will provide valuable clinically relevant insights into pulmonary development, as well as define mechanisms of disease pathogenesis for the many syndromic conditions and anomalies in other organ systems commonly seen in conjunction with lung malformations.
Genetic analysis of patients with hereditary lung malformations has led to the identification of associated gene mutations. The products of these genes, such as TTF-1, SOX2, FOXF1, and DICER1, are key molecules that control pulmonary development in genetically modified mouse models. Engineering mice with the corresponding human gene mutations results in lung malformations that phenotypically mimic the human disease, establishing a causative relationship. Continued integration of genomic studies in human patients with basic investigational studies defining molecular pathways that control lung development are needed to identify additional molecular markers and causes for the many lung abnormalities with currently unclear pathogenesis. Defining the molecular basis for lung malformations and disease pathogenesis will provide novel approaches to patient diagnosis, surveillance, and management.
Acknowledgments
We are extremely grateful to Alan Brody, M.D., for providing essential radiology expertise and radiographic images. We also thank Chris Woods for assistance compiling the figures and Ann Maher for clerical support.
References
[/level-membership-for-neonatal-and-perinatal-medicine-category][not-level-membership-for-neonatal-and-perinatal-medicine-category]
[/not-level-membership-for-neonatal-and-perinatal-medicine-category]
