Chapter 11 Stiffness syndromes
Muscle stiffness may be the presenting symptom in many disorders of the motor nervous system and muscles (Table 11.1) (Rowland, 1985; Thompson, 1993, 1994; Brown and Marsden, 1999). Spasticity is the most classic, and the others must be distinguished from it. In this chapter, therefore, spasticity will be considered first. The stiff-person syndrome and its variants is perhaps the major entity confronting the movement disorder specialist, and it will be considered next. Stiffness can arise not only from dysfunction of the central nervous system but also from disorders where there is continuous muscle activity where the pathology lies in the muscle, nerve, or anterior horn cell. These conditions will be considered last.
Table 11.1 Causes of muscle stiffness, cramps, spasms, rigidity, or contracture
| Cerebral – brainstem |
| Spinal cord |
| Peripheral nerve |
| Muscle |
| Unknown origin |
| Contracture |
Stiffness is assessed by the amount of force needed to get a movement. Tone, the more general clinical term, can be defined as the resistance to passive stretch of a joint. Normal tone is very low, and it is difficult to appreciate a decrease in tone, but some authorities say that they can detect a hypotonia in cerebellar dysfunction. Increased tone, hypertonia, is characteristic of several different states and can come from three theoretical mechanisms: (1) altered mechanical properties of the muscle or joint; (2) background co-contraction of muscles acting on the joint; and (3) increase in reflex response to the stretch opposing the movement (Hallett, 1999, 2000). A clear example of altered mechanical properties is contracture. Increased background contraction is often seen with difficulty in relaxation, such as commonly characterizes Parkinson’s disease. There are many different reflexes that can occur in response to stretch and these can help differentiate different states of hypertonia. In differentiating spasticity and rigidity, in simple terms, spasticity shows exaggerated short latency reflexes and rigidity shows exaggerated long latency reflexes.
Spasticity
Spasticity is a form of hypertonia with a number of characteristic features (Benecke et al., 2002; Sheean, 2002; Sanger et al., 2003). The increased resistance to stretch is velocity sensitive; there is more resistance the faster the joint is moved. There may be a clasped-knife phenomenon where the resistance increases and then suddenly gives way. There are also several other “positive” features such as increased tendon jerks, clonus, increased flexor reflexes, spontaneous flexor spasms, and abnormal postures (spastic dystonia). Importantly, there are also “negative” features including weakness (in a “pyramidal distribution”), fatigue, loss of coordination, and a decrease of some cutaneous reflexes. It can be noted that the increased stiffness can be valuable to a patient with significant weakness, as it might, for example, allow the patient to stand.
Loosely, neurologists usually say that spasticity arises from a lesion in the pyramidal tract or that it is from damage to the “upper motor neuron.” The first seems false and the second is vague. Supraspinal control of movement is complicated and consists of many tracts. Briefly, those fibers that go through the pyramid arise from the cortex and go to the spinal cord and can be called also the corticospinal tract (Wiesendanger, 1984; Davidoff, 1990). Approximately 30% of those fibers arise from the primary motor cortex; there are also significant contributions from premotor cortex and sensory cortex. The fibers largely cross in the pyramid, but some remain uncrossed. Some terminate as monosynaptic projections onto alpha-motoneurons, and others terminate on interneurons including those in the dorsal horn. Other cortical neurons project to basal ganglia, cerebellum, and brainstem; these structures can also originate spinal projections. Particularly important is the reticular formation that originates several tracts with different functions (Nathan et al., 1996). The dorsal reticulospinal tract may have particular relevance for spasticity and is normally inhibitory onto the spinal cord (Habaguchi et al., 2002; Takakusaki et al., 2001, 2003). Thinking about the cortical innervation of the reticular formation, it is possible to speak of a cortico-reticulo-spinal tract. Lesions of the primary motor area alone and lesions of the pyramid alone do not cause spasticity (Sherman et al., 2000). It appears that premotor damage is necessary and likely involvement of cortico-reticulo-spinal pathways. Dysfunction of the dorsal reticulospinal tract will disinhibit the spinal cord and may give rise to the hyperexcitability characteristic of spasticity. The term “corticofugal syndrome” has been suggested to indicate that “spasticity” has important negative as well as positive features and that the lesions involve descending tracts other than the corticospinal tracts, but it is not commonly accepted (Thilmann, 1993).
Clinical features of spasticity that help with the diagnosis, in addition to the velocity-dependent increased tone, include brisk tendon reflexes, the Babinski sign, Hoffman reflex (indicating brisk finger flexor reflexes), and loss of cutaneous abdominal reflexes. The negative features will often be seen as well, with weakness in the lower extremities of flexors more than extensors and in the upper extremities of extensors more than flexors. In the clinical neurophysiology laboratory, there will be increased H reflexes, identified with an increase of the maximum amplitude H reflex compared to the M wave (muscle response to direct supramaximal stimulation of the nerve), called the H/M ratio (Hallett, 1999, 2000; Benecke et al., 2002). There is also a diminished decrease of the H reflex with vibration of the body part. Characteristics of the tonic stretch reflex can also be assessed for threshold and gain to stretches of varying velocity. In spasticity, there is some controversy, but both lowered velocity threshold and an increased gain have been found (Powers et al., 1988; Katz and Rymer, 1989; Thilmann et al., 1991; Ibrahim et al., 1993; Musampa et al., 2007). It is important to recognize that there are both reflex and nonreflex contributions to spastic hypertonia (Chung et al., 2008).
There are many methods to treat spasticity, but this must be done carefully as correction of the positive features may not be all that helpful, and, as noted before, may even be detrimental. For many patients, the much more important aspects of their corticofugal syndrome are the negative features such as the weakness and these cannot be dealt with easily. Increased tone can be improved with a variety of oral agents including benzodiazepines, baclofen, and tizanidine (Krach, 2001; Abbruzzese, 2002; Ronan and Gold, 2007; Kamen et al., 2008). Baclofen can be given intrathecally by pump, and this can be much more efficacious, likely because of the ability to increase the dose at the target tissue without side effects (Ivanhoe et al., 2001; Albright et al., 2003; Dykstra et al., 2007; Rietman and Geertzen, 2007; Saval and Chiodo, 2010). Tolperisone has been evaluated in patients after stroke (Stamenova et al., 2005) and might be another consideration (Quasthoff et al., 2008).
Direct blockade of muscle contraction with agents such as phenol has been used for some time and the introduction of botulinum toxin for this purpose has been welcomed with enthusiasm (Boyd and Hays, 2001; Moore, 2002; Barnes, 2003; Mancini et al., 2005; Mohammadi et al., 2009). An evidence-based review gave botulinum toxin its highest recommendation for treatment in both adults and children (Simpson et al., 2008). For post-stroke spasticity, there is very good evidence for reduction of spasticity. Evidence is developing for an increase in functional ability (Elia et al., 2009; McCrory et al., 2009; Foley et al., 2010; Fridman et al., 2010; Sun et al., 2010), but often this is rather limited (Kaji et al., 2010). Children with cerebral palsy can be much improved (Baird and Vargus-Adams, 2009; Kanellopoulos et al., 2009; Lukban et al., 2009; Coutinho Dos Santos et al., 2011), but are a vulnerable population and need to be treated with care (Albavera-Hernandez et al., 2009). Several evidence-based reviews document the utility of botulinum toxin for this indication (Delgado et al., 2010; Hoare et al., 2010). It is interesting to note that the effect of botulinum toxin is mediated not only on the alpha motoneuron neuromuscular junction but also via the gamma motoneuron effect on the muscle spindle (Trompetto et al., 2008). Surgical methods such as rhizotomy can also be used in some cases for symptomatic relief of severe spasticity (Lazorthes et al., 2002).
Stiff-person (stiff-man) syndrome
The stiff-person syndrome (originally called the stiff-man syndrome) (Table 11.2) consists of progressive fluctuating muscular rigidity (Moersch and Woltman, 1956; Blum and Jankovic, 1991; Thompson, 1993, 1994; Stayer and Meinck, 1998; Brown and Marsden, 1999; Levy et al., 1999; Thompson, 2001; Meinck and Thompson, 2002). Typically, the rigidity affects axial muscles of the back, abdomen, hips, and shoulders, causing excessive lordosis with prominent contraction of paraspinal muscles, a “board-like” abdomen, and stiffness of the legs on walking (Fig. 11.1) (Video 11.1). Superimposed upon this continuous stiffness are spasms provoked by excitement, anxiety, voluntary movement, sudden noise, or peripheral stimuli. These spasms can be intensely painful and forceful such as to fracture bones or dislocate joints. Voluntary movement can provoke similar spasms that sometimes may cause falls “like a wooden man.” 
Table 11.2 Criteria for the diagnosis of the stiff-person syndrome
| Clinical |
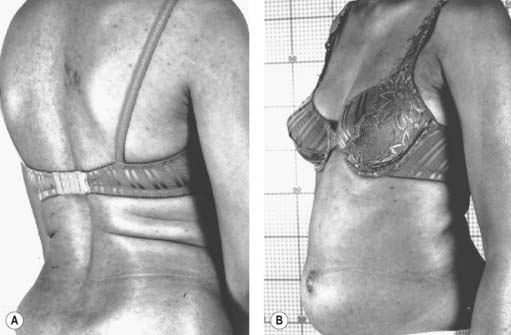
Figure 11.1 Patient with stiff-person syndrome. Note the marked lumbar lordosis (A) and the prominent abdomen (B).
Photos courtesy of Dr M. Dalakas.
The syndrome usually begins in the fourth and fifth decades and affects men and women equally. The onset of the illness usually is gradual with increasing painful tightness, stiffness, clumsiness of the trunk and legs, and limitation of range of motion (Fig. 11.2). On examination there is continuous muscular contraction of the paraspinal and abdominal muscles, but there are no other neurologic signs, other than brisk reflexes. The illness is slowly progressive with stiffness spreading from the trunk to hip and then shoulder muscles, but the face and distal limbs usually are spared. Sphincter function is normal. While the onset of the disorder seems typically spontaneous, one case has been reported where the onset appears to have been triggered by West Nile fever (Hassin-Baer et al., 2004). There has also been a report of a father and daughter, each with anti-GAD-positive stiff-person syndrome (Burns et al., 2003).

Figure 11.2 Patient with stiff-person syndrome. Note the limitation in bending forward.
Photo courtesy of Dr M. Dalakas.
Electromyography shows continuous normal motor unit activity, despite attempted relaxation, with no signs of denervation and normal peripheral motor and sensory nerve conduction velocity. Other physiologic abnormalities include exaggerated, non-habituating exteroceptive or cutaneomuscular reflexes, brainstem myoclonus, and an exaggerated startle reflex (“jerking stiff-man”) (Leigh et al., 1980; Meinck et al., 1983; Matsumoto et al., 1994; Meinck et al., 1995; Stayer and Meinck, 1998; Brown and Marsden, 1999).
Insulin-dependent diabetes mellitus occurs in up to two-thirds of patients. Diabetic ketoacidosis is the commonest cause of death in such patients. Other autoimmune endocrine diseases include thyroid disease, pernicious anemia, and vitiligo (Solimena et al., 1990). Epilepsy occurs in 10% of cases, although this is debatable.
A central, perhaps spinal cord origin for the spasms, rigidity, and continuous motor unit activity is suggested by their disappearance after peripheral nerve block, sleep, and general anesthesia. Other neurophysiologic tests suggest that spinal motoneuron excitability is normal and that the condition is due to defective input of inhibitory pathways onto motoneurons. To test inhibitory spinal circuits in patients, Floeter et al. (1998) used H-reflexes to test reciprocal inhibition in the forearm and thigh, vibration-induced inhibition of flexor carpi radialis and soleus H-reflexes, recurrent inhibition, and nonreciprocal (1b) inhibition of soleus H-reflexes. Vibration-induced inhibition of H-reflexes was diminished in eight of nine patients tested, but the presynaptic period of reciprocal inhibition was normal in most patients. Both circuits are presumed to involve presynaptic inhibition and GABAergic interneurons. Presumed glycinergic circuits, including the first period of reciprocal inhibition and nonreciprocal (1b) inhibition, showed occasional abnormalities. Recurrent inhibition was normal in all five patients tested. It appears that some, but not all, populations of GABAergic neurons are affected. The involvement of presumptive glycinergic circuits in some patients could point to impairment of non-GABAergic neurons, unrecognized involvement of GABAergic neurons in these inhibitory circuits, or, more likely, alterations of supraspinal systems that exert descending control over spinal circuits. Studies of cortical excitability with transcranial magnetic stimulation show decreased intracortical inhibition likely due to loss of GABA function at this level (Sandbrink et al., 2000). Hyperexcitability of the brainstem has been demonstrated by increased R2 recovery in the blink reflex recovery curve (Molloy et al., 2002). Direct measurement of GABA with magnetic resonance spectroscopy shows a deficiency in the sensorimotor cortex (Levy et al., 2005). There is also a cortical loss of flumazenil binding, a measure of postsynaptic benzodiazepine receptors associated with GABA receptors (Perani et al., 2007; Galldiks et al., 2008).
The significance of the association of insulin-dependent diabetes mellitus with the stiff-person syndrome has been emphasized by the discovery of antibodies directed against glutamic acid decarboxylase (GAD), the enzyme responsible for the synthesis of GABA, in both blood and cerebrospinal fluid in 60% or more of patients (Solimena et al., 1990; Walikonis and Lennon, 1998). The great majority of these patients also have antibodies directed against pancreatic islet cells, as well as gastric parietal cells and the thyroid. The anti-GAD antibodies are the same as those found in insulin-dependent diabetes mellitus, but it appears that the antibodies do have subtle differences (Lohmann et al., 2000). Currently the best test is the radioimmunoassay for GAD65 (Walikonis and Lennon, 1998). The luciferase immunoprecipitation technology (LIPS) seems also to be very good for this purpose (Burbelo et al., 2008). Present evidence raises the possibility that the anti-GAD antibodies may destroy GABAergic inhibitory mechanisms in the spinal cord, but, alternatively, they may be secondary to some other process leading to an appropriate immunologic response (Raju et al., 2005; Raju and Hampe, 2008). Anti-GAD antibodies have been demonstrated to block GABAergic neurotransmission in rat cerebellar slices (Ishida et al., 1999; Mitoma et al., 2000). One study reported that motor cortex excitability correlated with antibody levels suggesting an etiologic role (Koerner et al., 2004). In a study of 18 patients with stiff-person syndrome and serum antibodies, all had high titers as well in the cerebrospinal fluid (CSF) and 11 of 13 patients had an increased anti-GAD(65)-specific IgG index (Dalakas et al., 2001b). In the same study, the mean level of GABA in the CSF was found to be lower in patients than in controls. On the other hand, the levels of antibodies do not correlate with the severity of the disorder (Rakocevic et al., 2004).
The role of anti-GAD antibodies continues to be somewhat confusing, in part because they are not specific to stiff-person syndrome. In one study of 61 neurologic patients with high antibody titers, 22 (36%) had stiff-person syndrome and 17 (28%) had cerebellar ataxia (Saiz et al., 2008). Intrathecal synthesis of antibody gives more assurance that the antibody might be relevant (Jarius et al., 2010).
Another antibody found in a large number of patients is that directed against GABA(A) receptor-associated protein (GABARAP) (Raju et al., 2006; Dalakas, 2008, 2009). This has been identified in up to 65% of patients. Such antibodies would inhibit GABA(A) receptor expression on GABAergic neurons and certainly would be pathophysiologically relevant.
Stiff-person syndrome can also be seen in association with anti-amphiphysin I antibodies in patients with breast cancer (Saiz et al., 1999; Wessig et al., 2003). Anti-amphiphysin antibodies can block GABAergic neurotransmission (Geis et al., 2010). A report of 11 patients with anti-amphiphysin antibodies found that all were women, and 10 had breast cancer (Murinson and Guarnaccia, 2008). Average age was 60, and cervical involvement was particularly prominent. Marked improvement was seen in three of five patients with tumor excision and chemotherapy. Other neurologic disorders such as sensory neuropathy, cerebellar ataxia, and opsoclonus may be present as well, and the syndrome can occur with other tumor types (Antoine et al., 1999). Another antibody in some cases is directed against 17-beta-hydroxysteroid dehydrogenase type 4 (Dinkel et al., 2002).
Oligoclonal bands have been reported in the CSF in a number of cases (Meinck and Ricker, 1987; Williams et al., 1988; Meinck et al., 1994), and white matter lesions have been seen on brain MRI. However, so far no consistent pathology has been demonstrated in the few cases that have come to autopsy.
The treatment of this condition relies upon a combination of benzodiazepines and baclofen in high dosage. These drugs may decrease the superimposed severe spasms, but are not entirely effective in controlling the background sustained continuous muscle hyperactivity. Sodium valproate and tizanidine have also been reported to be beneficial (Meinck and Conrad, 1986; Stayer and Meinck, 1998). Intrathecal baclofen has been used (Silbert et al., 1995; Stayer et al., 1997; Stayer and Meinck, 1998). Patients have been reported to respond to steroid therapy (Blum and Jankovic, 1991), intravenous human immunoglobulin (IVIg) infusions (Khanlou and Eiger, 1999; Souza-Lima et al., 2000), and plasmapheresis (Vicari et al., 1989; Blum and Jankovic, 1991; Brashear and Phillips, 1991; Hayashi et al., 1999), but others have gained no benefit from plasmapheresis (Harding et al., 1989). A double-blind, placebo-controlled study documented the value of IVIg (Dalakas et al., 2001a). Another study has shown improvement in quality of life with IVIg (Gerschlager and Brown, 2002). Rituximab has been reported to be useful in a few patients (Bacorro and Tehrani, 2010; Dupond et al., 2010; Katoh et al., 2010).
Progressive encephalomyelitis with rigidity, sometimes known as spinal interneuronitis, may present with similar clinical features to the stiff-person syndrome. However, such patients go on to develop a relentless and progressive course, with the emergence of cranial nerve dysfunction producing bulbar symptoms and disorders of eye movement, along with cognitive impairment and long tract signs (Whiteley et al., 1976; Howell et al., 1979; Brown and Marsden, 1999; Gouider-Khouja et al., 2002). The condition may be isolated, or may occur in the setting of neoplasia associated with the pathologic changes of paraneoplastic encephalomyelitis (Roobol et al., 1987; Bateman et al., 1990).
The condition may start at any age in adults (Kraemer and Berlit, 2008). Initial symptoms may be those of pain, dysesthesia or sensory loss in the limbs, or weakness, stiffness, clumsiness, and rigidity. Extensor trunk spasm and/or brainstem myoclonus may be striking (Video 11.2). The tendon reflexes often are absent and the plantar responses are extensor. Nystagmus, opsoclonus, ophthalmoplegia, deafness, dysarthria, and dysphagia can occur. The illness usually leads to death within about 3 years. As in the stiff-person syndrome, there is continuous motor unit activity with particular involvement of trunk muscles, which disappears after a peripheral nerve or spinal nerve root block, or general anesthesia. EMG exploration may reveal evidence of denervation of muscles. A few have reticular reflex myoclonus. Progressive myoclonus with rigidity and myoclonus has been called PERM. Some of these patients may also exhibit anti-GAD (Burn et al., 1991), or antineuronal (anti-Ri) antibodies (Casado et al., 1994). The CSF may contain a lymphocytic pleocytosis, elevated protein and immunoglobulin levels, and oligoclonal IgG bands. MRI may show brainstem atrophy and altered signal in the brainstem and spinal cord. 
Pathologic examination has shown widespread encephalomyelitis with perivascular lymphocytic cuffing and infiltration, associated with neuronal loss throughout the brainstem and spinal cord, mainly involving interneurons. The relation of progressive encephalomyelitis with rigidity to the classic stiff-person syndrome, particularly in those with anti-GAD antibodies, remains to be established (Brown and Marsden, 1999). Antibodies to the glycine receptor have also been described (Hutchinson et al., 2008; Mas et al., 2010). A model of the disease in mice can be produced by glutamic acid decarboxylase-specific CD4(+) T cells, and this suggests that cellular immunity may play an important role (Burton et al., 2010).
As with the stiff-person syndrome, treatment is with high doses of diazepam and baclofen. One case, with evidence of myelitis on spinal cord biopsy, improved on steroids (McCombe et al., 1989). Rituximab has also been used (Saidha et al., 2008).
Spinal alpha rigidity is a related condition resulting from isolation of spinal motor neurons from inhibitory interneuronal control (Gelfan and Tarlov, 1959). Examples have been described with trauma, cord vascular disease, cord tumors, and syringomyelia, as well as with myelitis. Most of these lesions have involved the cervical cord. Characteristically, there are stimulus-induced spasms, rigidity and abnormal limb postures involving rigid adduction, extension, and internal rotation of the affected body parts. These postures are produced by continuous motor activity which is not influenced by voluntary effort. In addition, there is wasting, weakness and loss of tendon reflexes in the arms, with long tract signs in their legs.
A variant of the stiff-person syndrome has been recognized, namely the “stiff leg” syndrome (Brown et al., 1997; Barker et al., 1998; Brown and Marsden, 1999; Fiol et al., 2001; Gurol et al., 2001; Bartsch et al., 2003). In contrast to the classic stiff-person, where continuous motor unit activity affects the back and thighs, those with the stiff-leg syndrome present with stiffness and painful spasms of one or both legs, which are rigid and dystonic. EMG findings are characteristic with continuous motor unit activity at rest, spasms of repetitive grouped discharges, and abnormal cutaneomuscular reflexes. Anti-GAD antibodies are present in about 15% of cases. Whether this is a partial syndrome or a separate disorder is debated. The prognosis is generally relatively benign with absence of other neurologic symptoms and signs for up to 16 years, but other cases can progress to the syndrome of progressive encephalomyelitis with rigidity (Gouider-Khouja et al., 2002).
Syndromes of continuous muscle activity
Continuous muscle fiber activity or neuromyotonia
A variety of disorders of peripheral nerve origin may produce continuous muscle activity causing stiffness and cramps (Table 11.3). This condition has been described as “continuous muscle fiber activity or Isaacs syndrome” (Isaacs, 1961), “neuromyotonia” (Mertens and Zschocke, 1965), “myokymia with impaired muscle relaxation” (Gardner-Medwin and Walton, 1969), or “pseudomyotonia and myokymia” (Hughes and Matthews, 1969). This variable terminology reflects the overlap between the clinical and electromyographic use of terms such as myokymia (a wave-like rippling of muscle or motor unit discharges in doublets or triplets) and neuromyotonia (delayed muscle relaxation or high-frequency EMG discharges).
| Clinical |
The characteristic and fairly stereotyped clinical picture of this condition is the gradual onset of muscle stiffness at rest, with continuous twitching (fasciculation) or rippling (myokymia) of muscles, with cramps following voluntary contractions due to delay in muscle relaxation (pseudomyotonia). Pain is rare, but muscle aching is common. The distribution of muscle contraction often is predominantly distal (in contrast to the axial involvement in stiff-person syndrome), producing a pseudo-tetany picture. However, proximal and cranial muscles can be affected. Involvement can be focal, as exemplified by one patient who presented with finger flexion that resembled focal hand dystonia (Jamora et al., 2006). The symptoms of muscle contraction, often accompanied by profuse sweating, persist during sleep, and following peripheral nerve or spinal nerve root block, or general anesthesia. The continuous muscle fiber activity is abolished by peripheral neuromuscular blockade with curare, indicating its origin at the neuromuscular junction.
The condition can be inherited (Ashizawa et al., 1983; Auger et al., 1984), or sporadic (Isaacs, 1961; Gardner-Medwin and Walton, 1969), may occur as a result of a paraneoplastic process (Walsh, 1976; Lahrmann et al., 2001), or thymoma with acetylcholine receptor antibodies with or without myasthenia gravis (Halbach et al., 1987; Garcia-Merino et al., 1991), or be associated with many types of inherited (Vasilescu et al., 1984b; Hahn et al., 1991), inflammatory (Valenstein et al., 1978; Vasilescu et al., 1984a), or metabolic (Wallis et al., 1970; Vasilescu and Florescu, 1982) peripheral neuropathies. A case has been described in the setting of lupus (Taylor, 2005). A search for a remote neoplasm is required.
An autoimmune basis for neuromyotonia has been demonstrated in those with no obvious precipitating cause, on the basis of antibodies to specific nerve membrane voltage-gated potassium ion channels (VGKC) (Sinha et al., 1991; Shillito et al., 1995; Nagado et al., 1999; Hart, 2000; Hayat et al., 2000; Arimura et al., 2002; Vernino and Lennon, 2002; Lang and Vincent, 2003; Newsom-Davis et al., 2003). Indeed, it seems that the autoimmune production of antibodies to voltage-gated potassium channels is the common thread for all the etiologies (Rueff et al., 2008).
Electrophysiologically, the hallmark of the syndrome is the presence of continuous motor unit activity that persists during sleep and usually following peripheral nerve block. This continuous muscle activity may originate in proximal nerve segments (when distal nerve block suppresses activity) or distal segments (when nerve block has no effect). The motor unit activity often is increased by hyperventilation or ischemia. Fasciculations and grouped high-frequency discharges are evident on electromyography, with repetitive bursts of motor units of normal appearance (myokymia) (Fig. 11.3) and also bizarre high-frequency (150–300 Hz) discharges (neuromyotonia) (Fig. 11.4). Prolonged high frequency after discharges following nerve stimulation, voluntary contraction, or muscle percussion are characteristic. Evidence of muscle denervation and reinnervation may be found, and measurement of peripheral nerve motor and sensory conduction may confirm the presence of peripheral neuropathy.
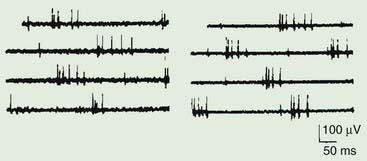
Figure 11.3 Two examples of myokymic discharges as might be seen in patients with neuromyotonia.
From Kimura J. Electrodiagnosis in Diseases of Nerve and Muscle: Principles and Practice. Philadelphia: F.A. Davis; 1984, with permission.
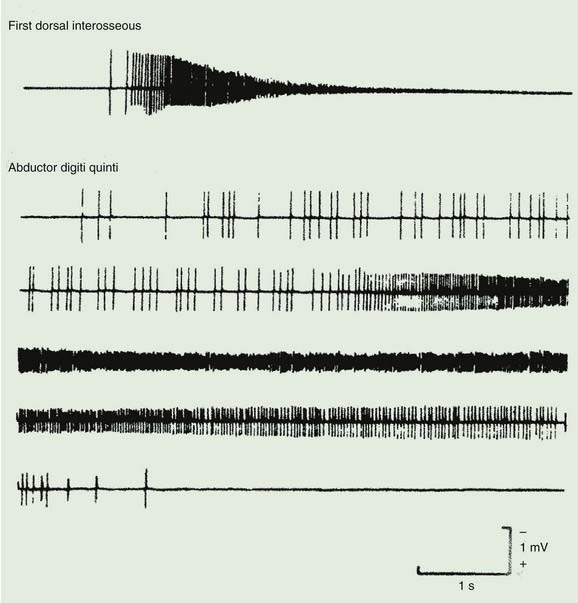
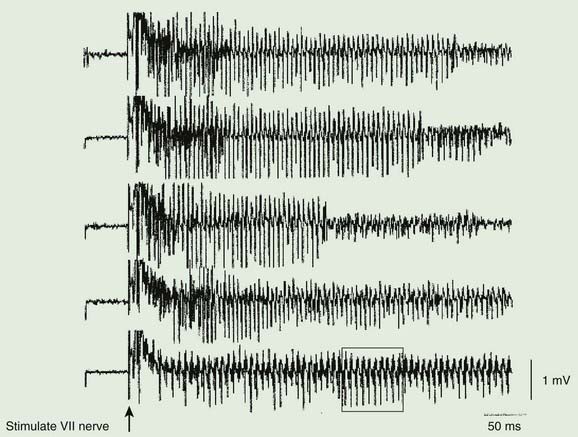
Figure 11.4 Two examples of neuromyotonic discharges as might be seen in patients with neuromyotonia.
From AAEM. Glossary of terms in electromyography. Muscle Nerve 1987;10(8S):G1–G60, with permission.
Carbamazepine and phenytoin may be successful in abolishing most of the symptoms of neuromyotonia and continuous muscle fiber activity. When the disorder is autoimmune, plasmapheresis may be effective (Hayat et al., 2000; Nakatsuji et al., 2000) as may IVIg (Alessi et al., 2000). Neuromyotonia without malignancy or peripheral neuropathy may prove to be relatively benign (Isaacs and Heffron, 1974; Wilton and Gardner-Medwin, 1990). Focal neuromyotonia can be treated with injection of botulinum toxin (Hobson et al., 2009).
Myokymia is also a feature of episodic ataxia type 1 (EA-1). In this disorder, the myokymia is prominent around the eyes or lips or in the fingers. See Chapters 21 and 22 for more details.
Morvan syndrome (Morvan’s fibrillary chorea) is a disorder related to neuromyotonia (Kleopa et al., 2006). It is characterized by generalized myokymia, burning pain, cramping, weakness, pruritus, hyperhidrosis, weight loss, sleeplessness, and hallucinations (Madrid et al., 1996; Bajaj and Shrestha, 2007). Some cases are of unknown cause, but others are associated with mercury intoxication, chrysotherapy, thymoma, and other remote neoplasms. In one case with thymoma, muscle histopathology disclosed chronic denervation and myopathic changes and in-vitro electrophysiology demonstrated both presynaptic and postsynaptic defects in neuromuscular transmission (Lee et al., 1998). Probably most cases are associated with increased antibodies to voltage-gated potassium channels (Liguori et al., 2001; Kleopa et al., 2006). Other serum antibodies, such as those to acetylcholine receptors, titin, and N-type calcium channels, can also be detected. Plasmapheresis (Liguori et al., 2001), thymectomy, and long-term immunosuppression may induce a dramatic resolution of symptoms.
Rippling muscle disease is an autosomal dominant inherited disorder of skeletal muscle in which patients present with muscle cramps, pain, stiffness especially on exercise, and exhibit a characteristic lateral rolling movement of muscle after contraction and balling of muscle on percussion (Stephan et al., 1994; Vorgerd et al., 1999; Torbergsen, 2002). In one family with 11 affected members, muscle stiffness and myalgia were the most prominent symptoms (So et al., 2001). Muscle rippling was present in only 6 of affected family members, whereas persistent muscle contraction to muscle percussion was present in all affected adults. Phenotypes clearly vary in the same family (Jacobi et al., 2010). Curiously, EMG shows normal recruitment and motor unit potentials. One hypothesis is that the abnormal muscle contractions are evoked by “silent” action potentials traveling in the muscle’s tubular system (Lamb, 2005). In a genetic study of five families, a genome-wide linkage analysis identified a locus on chromosome 3p25 with missense mutations in CAV3 (encoding caveolin 3) (Betz et al., 2001). Other cases with mutations in the same gene have been found (Vorgerd et al., 2001; Lorenzoni et al., 2007; Traverso et al., 2008). Mutations in CAV3 have also been described in limb-girdle muscular dystrophy type 1C (LGMD1C) and other neuromuscular syndromes, again demonstrating that different phenotypes can come from mutations in the same gene (Sotgia et al., 2003; Woodman et al., 2004; Aboumousa et al., 2008; Gazzerro et al., 2010). This disorder can be associated with myasthenia gravis and might be improved with immunosuppression (Muller-Felber et al., 1999; Schoser et al., 2009). Indeed, it appears that rippling muscle disease can be immune-mediated without a mutation in the CAV3 gene (Schulte-Mattler et al., 2005).
Schwartz–Jampel syndrome (chondrodystrophic myotonia) is a rare familial condition, usually occurring with an autosomal recessive pattern of inheritance (Schwartz and Jampel, 1962; Taylor et al., 1972; Fowler et al., 1974; Fontaine et al., 1996), but occasionally as an autosomal dominant trait (Pascuzzi et al., 1990; Spaans, 1991). The typical picture is of continuous muscle fiber activity and muscle stiffness, especially of the face, with an abnormal facial appearance (blepharophimosis, a small jaw and a puckered chin, and low-set ears), skeletal abnormalities (spondylo-epiphyseal dysplasia), and short stature (Naharajakumar et al., 2006). Percussion myotonia may be present. The muscle stiffness is due to semi-continuous motor unit discharges, with high frequency and after-discharges of muscle fibers (Fig. 11.5). These abnormalities persist after ischemia and curare (Spaans et al., 1990). An abnormality of muscle fiber sodium channels has been demonstrated (Lehmann-Horn et al., 1990). Procainamide abolishes such abnormal muscle activity (Lehmann-Horn et al., 1990). In a number of patients, a mutation in the gene for perlecan, the major proteoglycan of basement membranes, has been identified (Nicole et al., 2000; Arikawa-Hirasawa et al., 2002; Stum et al., 2006). Botulinum toxin can be used to improve the blepharospasm in this condition (Vargel et al., 2006; Aburahma et al., 2009).
Satoyoshi syndrome (Satoyoshi, 1978) begins in childhood or adolescence with severe intermittent painful muscle spasms and twitches, twisting the body into abnormal postures. The spasms last up to a few minutes and occur frequently, up to hundreds per day. They are precipitated by movement or stimulation of the affected area. At the same time, complete alopecia and intestinal malabsorption with diarrhea develops. There also is metaphyseal dysplasia with growth retardation. It may also affect adults (Ikeda et al., 1998). One case had manifestations only unilaterally (Uddin et al., 2002). The etiology is likely to be a systemic autoimmune disorder (Heger et al., 2006; Matsuura et al., 2007). Treatment with steroids can be effective (Wisuthsarewong et al., 2001; Endo et al., 2003), as can IVIg (Arita et al., 1996) and methotrexate (Heger et al., 2006).
AAEM. Glossary of terms in electromyography. Muscle Nerve. 1987;10(8S):G1-G60.
Abbruzzese G. The medical management of spasticity. Eur J Neurol. 2002;9(Suppl 1)):30-34. discussion 53–61
Aboumousa A., Hoogendijk J., Charlton R., et al. Caveolinopathy – new mutations and additional symptoms. Neuromuscul Disord. 2008;18(7):572-578.
Aburahma S.K., Al-Khateeb T., Alrefai A., Amarin Z. Botulinum toxin A injections for the treatment of Schwartz-Jampel syndrome: a case series. J Child Neurol. 2009;24(1):5-8.
Albavera-Hernandez C., Rodriguez J.M., Idrovo A.J. Safety of botulinum toxin type A among children with spasticity secondary to cerebral palsy: a systematic review of randomized clinical trials. Clin Rehabil. 2009;23(5):394-407.
Albright A.L., Gilmartin R., Swift D., et al. Long-term intrathecal baclofen therapy for severe spasticity of cerebral origin. J Neurosurg. 2003;98(2):291-295.
Alessi G., De Reuck J., De Bleecker J., Vancayzeele S. Successful immunoglobulin treatment in a patient with neuromyotonia. Clin Neurol Neurosurg. 2000;102(3):173-175.
Antoine J.C., Absi L., Honnorat J., et al. Antiamphiphysin antibodies are associated with various paraneoplastic neurological syndromes and tumors. Arch Neurol. 1999;56(2):172-177.
Arikawa-Hirasawa E., Le A.H., Nishino I., et al. Structural and functional mutations of the perlecan gene cause Schwartz-Jampel syndrome, with myotonic myopathy and chondrodysplasia. Am J Hum Genet. 2002;70(5):1368-1375.
Arimura K., Sonoda Y., Watanabe O., et al. Isaacs’ syndrome as a potassium channelopathy of the nerve. Muscle Nerve. 2002;Suppl 11:S55-S58.
Arita J., Hamano S., Nara T., Maekawa K. Intravenous gammaglobulin therapy of Satoyoshi syndrome. Brain Dev. 1996;18(5):409-411.
Ashizawa T., Butler I.J., Harati Y., Roongta S.M. A dominantly inherited syndrome with continuous motor neuron discharges. Ann Neurol. 1983;13(3):285-290.
Auger R.G., Daube J.R., Gomez M.R., Lambert E.H. Hereditary form of sustained muscle activity of peripheral nerve origin causing generalized myokymia and muscle stiffness. Ann Neurol. 1984;15(1):13-21.
Bacorro E.A., Tehrani R. Stiff-person syndrome: persistent elevation of glutamic acid decarboxylase antibodies despite successful treatment with rituximab. J Clin Rheumatol. 2010;16(5):237-239.
Baird M.W., Vargus-Adams J. Outcome measures used in studies of botulinum toxin in childhood cerebral palsy: a systematic review. J Child Neurol. 2009;25:793-794.
Bajaj B.K., Shrestha S. An interesting case report of Morvan’s syndrome from the Indian subcontinent. Neurol India. 2007;55(1):67-69.
Barker R.A., Revesz T., Thom M., et al. Review of 23 patients affected by the stiff man syndrome: clinical subdivision into stiff trunk (man) syndrome, stiff limb syndrome, and progressive encephalomyelitis with rigidity. J Neurol Neurosurg Psychiatry. 1998;65(5):633-640.
Barnes M. Botulinum toxin – mechanisms of action and clinical use in spasticity. J Rehabil Med. 2003;41 Suppl:56-59.
Bartsch T., Herzog J., Baron R., Deuschl G. The stiff limb syndrome – a new case and a literature review. J Neurol. 2003;250(4):488-490.
Bateman D.E., Weller R.O., Kennedy P. Stiffman syndrome: a rare paraneoplastic disorder? J Neurol Neurosurg Psychiatry. 1990;53(8):695-696.
Benecke R., Classen J., Dressler D. Tone and its disorders. In: Brown W.F., Bolton C.F., Aminoff M.J., editors. Neuromuscular Function and Disease. Philadelphia: W.B. Saunders Company; 2002:1781-1801.
Betz R.C., Schoser B.G., Kasper D., et al. Mutations in CAV3 cause mechanical hyperirritability of skeletal muscle in rippling muscle disease. Nat Genet. 2001;28(3):218-219.
Blum P., Jankovic J. Stiff-person syndrome: an autoimmune disease. Mov Disord. 1991;6(1):12-20.
Boyd R.N., Hays R.M. Current evidence for the use of botulinum toxin type A in the management of children with cerebral palsy: a systematic review. Eur J Neurol. 2001;8(Suppl 5):1-20.
Brashear H.R., Phillips L.H.2nd. Autoantibodies to GABAergic neurons and response to plasmapheresis in stiff-man syndrome. Neurology. 1991;41(10):1588-1592.
Brown P., Marsden C.D. The stiff man and stiff man plus syndromes. J Neurol. 1999;246(8):648-652.
Brown P., Rothwell J.C., Marsden C.D. The stiff leg syndrome. J Neurol Neurosurg Psychiatry. 1997;62(1):31-37.
Burbelo P.D., Groot S., Dalakas M.C., Iadarola M.J. High definition profiling of autoantibodies to glutamic acid decarboxylases GAD65/GAD67 in stiff-person syndrome. Biochem Biophys Res Commun. 2008;366(1):1-7.
Burn D.J., Ball J., Lees A.J., et al. A case of progressive encephalomyelitis with rigidity and positive antiglutamic acid decarboxylase antibodies [corrected]. J Neurol Neurosurg Psychiatry. 1991;54(5):449-451.
Burns T.M., Jones H.R., Phillips L.H.2nd, et al. Clinically disparate stiff-person syndrome with GAD65 autoantibody in a father and daughter. Neurology. 2003;61(9):1291-1293.
Burton A.R., Baquet Z., Eisenbarth G.S., et al. Central nervous system destruction mediated by glutamic acid decarboxylase-specific CD4(+) T cells. J Immunol. 2010;184(9):4863-4870.
Casado J.L., Gil-Peralta A., Graus F., et al. Anti-Ri antibodies associated with opsoclonus and progressive encephalomyelitis with rigidity. Neurology. 1994;44(8):1521-1522.
Chung S.G., van Rey E., Bai Z., et al. Separate quantification of reflex and nonreflex components of spastic hypertonia in chronic hemiparesis. Arch Phys Med Rehabil. 2008;89(4):700-710.
Coutinho Dos Santos L.H., Bufara Rodrigues D.C., Simões de Assis T.R., Bruck I. Effective results with botulinum toxin in cerebral palsy. Pediatr Neurol. 2011;44(5):357-363.
Dalakas M.C. Advances in the pathogenesis and treatment of patients with stiff person syndrome. Curr Neurol Neurosci Rep. 2008;8(1):48-55.
Dalakas M.C. Stiff person syndrome: advances in pathogenesis and therapeutic interventions. Curr Treat Options Neurol. 2009;11(2):102-110.
Dalakas M.C., Fujii M., Li M., et al. High-dose intravenous immune globulin for stiff-person syndrome. N Engl J Med. 2001;345(26):1870-1876.
Dalakas M.C., Li M., Fujii M., Jacobowitz D.M. Stiff person syndrome: quantification, specificity, and intrathecal synthesis of GAD65 antibodies. Neurology. 2001;57(5):780-784.
Davidoff R.A. The pyramidal tract. Neurology. 1990;40(2):332-339.
Delgado M.R., Hirtz D., Aisen M., et al. Practice parameter: pharmacologic treatment of spasticity in children and adolescents with cerebral palsy (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2010;74(4):336-343.
Dinkel K., Rickert M., Moller G., et al. Stiff-man syndrome: identification of 17 beta-hydroxysteroid dehydrogenase type 4 as a novel 80-kDa antineuronal antigen. J Neuroimmunol. 2002;130(1–2):184-193.
Dupond J.L., Essalmi L., Gil H., et al. Rituximab treatment of stiff-person syndrome in a patient with thymoma, diabetes mellitus and autoimmune thyroiditis. J Clin Neurosci. 2010;17(3):389-391.
Dykstra D., Stuckey M., DesLauriers L., et al. Intrathecal baclofen in the treatment of spasticity. Acta Neurochir Suppl. 2007;97(Pt 1):163-171.
Elia A.E., Filippini G., Calandrella D., Albanese A. Botulinum neurotoxins for post-stroke spasticity in adults: a systematic review. Mov Disord. 2009;24(6):801-812.
Endo K., Yamamoto T., Nakamura K., et al. Improvement of Satoyoshi syndrome with tacrolimus and corticosteroids. Neurology. 2003;60(12):2014-2015.
Fiol M., Cammarota A., Rivero A., et al. Focal stiff-person syndrome. Neurologia. 2001;16(2):89-91.
Floeter M.K., Valls-Sole J., Toro C., et al. Physiologic studies of spinal inhibitory circuits in patients with stiff-person syndrome. Neurology. 1998;51(1):85-93.
Foley N., Murie-Fernandez M., Speechley M., et al. Does the treatment of spastic equinovarus deformity following stroke with botulinum toxin increase gait velocity? A systematic review and meta-analysis. Eur J Neurol. 2010 May 13. Epub ahead of print
Fontaine B., Nicole S., Topaloglu H., et al. Recessive Schwartz-Jampel syndrome (SJS): confirmation of linkage to chromosome 1p, evidence of genetic homogeneity and reduction of the SJS locus to a 3-cM interval. Hum Genet. 1996;98(3):380-385.
Fowler W.M.Jr., Layzer R.B., Taylor R.G., et al. The Schwartz-Jampel syndrome. Its clinical, physiological and histological expressions. J Neurol Sci. 1974;22(1):127-146.
Fridman E.A., Crespo M., Gomez Arguello S., et al. Kinematic improvement following botulinum Toxin-A injection in upper-limb spasticity due to stroke. J Neurol Neurosurg Psychiatry. 2010;81(4):423-427.
Galldiks N., Thiel A., Haense C., et al. (11)C-Flumazenil positron emission tomography demonstrates reduction of both global and local cerebral benzodiazepine receptor binding in a patient with stiff person syndrome. J Neurol. 2008;255:1361-1364.
Garcia-Merino A., Cabello A., Mora J.S., Liano H. Continuous muscle fiber activity, peripheral neuropathy, and thymoma. Ann Neurol. 1991;29(2):215-218.
Gardner-Medwin D., Walton J.N. Myokymia with impaired muscular relaxation. Lancet. 1969;1(7586):127-130.
Gazzerro E., Sotgia F., Bruno C., et al. Caveolinopathies: from the biology of caveolin-3 to human diseases. Eur J Hum Genet. 2010;18(2):137-145.
Geis C., Weishaupt A., Hallerman S., et al. Stiff person syndrome-associated auto-antibodies to amphiphysin mediate reduced GABAergic inhibition. Brain. 2010;133(11):3166-3180.
Gelfan S., Tarlov I.M. Interneurons and rigidity of spinal origin. J Physiol (London). 1959;146:594-617.
Gerschlager W., Brown P. Effect of treatment with intravenous immunoglobulin on quality of life in patients with stiff-person syndrome. Mov Disord. 2002;17(3):590-593.
Gouider-Khouja N., Mekaouar A., Larnaout A., et al. Progressive encephalomyelitis with rigidity presenting as a stiff-person syndrome. Parkinsonism Relat Disord. 2002;8(4):285-288.
Gurol M.E., Ertas M., Hanagasi H.A., et al. Stiff leg syndrome: case report. Mov Disord. 2001;16(6):1189-1193.
Habaguchi T., Takakusaki K., Saitoh K., et al. Medullary reticulospinal tract mediating the generalized motor inhibition in cats: II. Functional organization within the medullary reticular formation with respect to postsynaptic inhibition of forelimb and hindlimb motoneurons. Neuroscience. 2002;113(1):65-77.
Hahn A.F., Parkes A.W., Bolton C.F., Stewart S.A. Neuromyotonia in hereditary motor neuropathy. J Neurol Neurosurg Psychiatry. 1991;54(3):230-235.
Halbach M., Homberg V., Freund H.J. Neuromuscular, autonomic and central cholinergic hyperactivity associated with thymoma and acetylcholine receptor-binding antibody. J Neurol. 1987;234(6):433-436.
Hallett M. Electrophysiologic evaluation of movement disorders. In: Aminoff M.J., editor. Electrodiagnosis in Clinical Neurology. fourth ed. New York: Churchill Livingstone; 1999:365-380.
Hallett M. Electrodiagnosis in movement disorders. In: Levin K.H., Lüders H.O., editors. Comprehensive Clinical Neurophysiology. Philadelphia: W.B. Saunders Company; 2000:281-294.
Harding A.E., Thompson P.D., Kocen R.S., et al. Plasma exchange and immunosuppression in the stiff man syndrome. Lancet. 1989;2(8668):915.
Hart I.K. Acquired neuromyotonia: a new autoantibody-mediated neuronal potassium channelopathy. Am J Med Sci. 2000;319(4):209-216.
Hassin-Baer S., Kirson E.D., Shulman L., et al. Stiff-person syndrome following West Nile fever. Arch Neurol. 2004;61(6):938-941.
Hayashi A., Nakamagoe K., Ohkoshi N., et al. Double filtration plasma exchange and immunoadsorption therapy in a case of stiff-man syndrome with negative anti-GAD antibody. J Med. 1999;30(5–6):321-327.
Hayat G.R., Kulkantrakorn K., Campbell W.W., Giuliani M.J. Neuromyotonia: autoimmune pathogenesis and response to immune modulating therapy. J Neurol Sci. 2000;181(1–2):38-43.
Heger S., Kuester R.M., Volk R., et al. Satoyoshi syndrome: a rare multisystemic disorder requiring systemic and symptomatic treatment. Brain Dev. 2006;28(5):300-304.
Hoare Hoare, B.J., Wallen, M.A., Imms, C., et al., 2010. Botulinum toxin A as an adjunct to treatment in the management of the upper limb in children with spastic cerebral palsy (UPDATE). Cochrane Database Syst Rev (1), CD003469.
Hobson D.E., Kerr P., Hobson S. Successful use of botulinum toxin for post-irradiation unilateral jaw neuromyotonia. Parkinsonism Relat Disord. 2009;15(8):617-618.
Howell D.A., Lees A.J., Toghill P.J. Spinal internuncial neurones in progressive encephalomyelitis with rigidity. J Neurol Neurosurg Psychiatry. 1979;42(9):773-785.
Hughes R.C., Matthews W.B. Pseudo-myotonia and myokymia. J Neurol Neurosurg Psychiatry. 1969;32(1):11-14.
Hutchinson M., Waters P., McHugh J., et al. Progressive encephalomyelitis, rigidity, and myoclonus: a novel glycine receptor antibody. Neurology. 2008;71(16):1291-1292.
Ibrahim I.K., Berger W., Trippel M., Dietz V. Stretch-induced electromyographic activity and torque in spastic elbow muscles. Differential modulation of reflex activity in passive and active motor tasks. Brain. 1993;116(Pt 4):971-989.
Ikeda K., Satoyoshi E., Kinoshita M., et al. Satoyoshi’s syndrome in an adult: a review of the literature of adult onset cases. Intern Med. 1998;37(9):784-787.
Isaacs H. A syndrome of continuous muscle-fibre activity. J Neurol Neurosurg Psychiatry. 1961;24:319-325.
Isaacs H., Heffron J.J. The syndrome of “continuous muscle-fibre activity” cured: further studies. J Neurol Neurosurg Psychiatry. 1974;37(11):1231-1235.
Ishida K., Mitoma H., Song S.Y., et al. Selective suppression of cerebellar GABAergic transmission by an autoantibody to glutamic acid decarboxylase. Ann Neurol. 1999;46(2):263-267.
Ivanhoe C.B., Tilton A.H., Francisco G.E. Intrathecal baclofen therapy for spastic hypertonia. Phys Med Rehabil Clin N Am. 2001;12(4):923-938. viii–ix
Jacobi C., Ruscheweyh R., Vorgerd M., et al. Rippling muscle disease: variable phenotype in a family with five afflicted members. Muscle Nerve. 2010;41(1):128-132.
Jamora R.D., Umapathi T., Tan L.C. Finger flexion resembling focal dystonia in Isaacs’ syndrome. Parkinsonism Relat Disord. 2006;12(1):61-63.
Jarius S., Stich O., Speck J., et al. Qualitative and quantitative evidence of anti-glutamic acid decarboxylase-specific intrathecal antibody synthesis in patients with stiff person syndrome. J Neuroimmunol. 2010;229(1–2):219-224.
Kaji R., Osako Y., Suyama K., et al. Botulinum toxin type A in post-stroke lower limb spasticity: a multicenter, double-blind, placebo-controlled trial. J Neurol. 2010;257(8):1330-1337.
Kamen L., Henney H.R.3rd, Runyan J.D. A practical overview of tizanidine use for spasticity secondary to multiple sclerosis, stroke, and spinal cord injury. Curr Med Res Opin. 2008;24(2):425-439.
Kanellopoulos A.D., Mavrogenis A.F., Mitsiokapa E.A., et al. Long lasting benefits following the combination of static night upper extremity splinting with botulinum toxin A injections in cerebral palsy children. Eur J Phys Rehabil Med. 2009;45(4):501-506.
Katoh N., Matsuda M., Ishii W., et al. Successful treatment with rituximab in a patient with stiff-person syndrome complicated by dysthyroid ophthalmopathy. Intern Med. 2010;49(3):237-241.
Katz R.T., Rymer W.Z. Spastic hypertonia: mechanisms and measurement. Arch Phys Med Rehabil. 1989;70(2):144-155.
Khanlou H., Eiger G. Long-term remission of refractory stiff-man syndrome after treatment with intravenous immunoglobulin. Mayo Clin Proc. 1999;74(12):1231-1232.
Kimura J. Electrodiagnosis in Diseases of Nerve and Muscle: Principles and Practice. Philadelphia: F.A. Davis; 1984.
Kleopa K.A., Elman L.B., Lang B., et al. Neuromyotonia and limbic encephalitis sera target mature Shaker-type K+ channels: subunit specificity correlates with clinical manifestations. Brain. 2006;129(Pt 6):1570-1584.
Koerner C., Wieland B., Richter W., Meinck H.M. Stiff-person syndromes: motor cortex hyperexcitability correlates with anti-GAD autoimmunity. Neurology. 2004;62(8):1357-1362.
Krach L.E. Pharmacotherapy of spasticity: oral medications and intrathecal baclofen. J Child Neurol. 2001;16(1):31-36.
Kraemer M., Berlit P. Progressive encephalomyelitis with rigidity and myoclonus in an 81-year-old patient. Clin Neurol Neurosurg. 2008;110(3):279-281.
Lahrmann H., Albrecht G., Drlicek M., et al. Acquired neuromyotonia and peripheral neuropathy in a patient with Hodgkin’s disease. Muscle Nerve. 2001;24(6):834-838.
Lamb G.D. Rippling muscle disease may be caused by “silent” action potentials in the tubular system of skeletal muscle fibers. Muscle Nerve. 2005;31(5):652-658.
Lang B., Vincent A. Autoantibodies to ion channels at the neuromuscular junction. Autoimmun Rev. 2003;2(2):94-100.
Lazorthes Y., Sol J.C., Sallerin B., Verdie J.C. The surgical management of spasticity. Eur J Neurol. 2002;9(Suppl 1)):35-41. discussion 53–61
Lee E.K., Maselli R.A., Ellis W.G., Agius M.A. Morvan’s fibrillary chorea: a paraneoplastic manifestation of thymoma. J Neurol Neurosurg Psychiatry. 1998;65(6):857-862.
Lehmann-Horn F., Iaizzo P.A., Franke C., et al. Schwartz-Jampel syndrome: II. Na+ channel defect causes myotonia. Muscle Nerve. 1990;13(6):528-535.
Leigh P.N., Rothwell J.C., Traub M., Marsden C.D. A patient with reflex myoclonus and muscle rigidity: “jerking stiff-man syndrome. J Neurol Neurosurg Psychiatry. 1980;43(12):1125-1131.
Levy L.M., Dalakas M.C., Floeter M.K. The stiff-person syndrome: an autoimmune disorder affecting neurotransmission of gamma-aminobutyric acid. Ann Intern Med. 1999;131(7):522-530.
Levy L.M., Levy-Reis I., Fujii M., Dalakas M.C. Brain gamma-aminobutyric acid changes in stiff-person syndrome. Arch Neurol. 2005;62(6):970-974.
Liguori R., Vincent A., Clover L., et al. Morvan’s syndrome: peripheral and central nervous system and cardiac involvement with antibodies to voltage-gated potassium channels. Brain. 2001;124(Pt 12):2417-2426.
Lohmann T., Hawa M., Leslie R.D., et al. Immune reactivity to glutamic acid decarboxylase 65 in stiffman syndrome and type 1 diabetes mellitus [see comments]. Lancet. 2000;356(9223):31-35.
Lorenzoni P.J., Scola R.H., Vieira N., et al. A novel missense mutation in the caveolin-3 gene in rippling muscle disease. Muscle Nerve. 2007;36(2):258-260.
Lukban M.B., Rosales R.L., Dressler D. Effectiveness of botulinum toxin A for upper and lower limb spasticity in children with cerebral palsy: a summary of evidence. J Neural Transm. 2009;116(3):319-331.
Madrid A., Gil-Peralta A., Gil-Neciga E., et al. Morvan’s fibrillary chorea: remission after plasmapheresis. J Neurol. 1996;243(4):350-353.
Mancini F., Sandrini G., Moglia A., et al. A randomised, double-blind, dose-ranging study to evaluate efficacy and safety of three doses of botulinum toxin type A (Botox) for the treatment of spastic foot. Neurol Sci. 2005;26(1):26-31.
Mas N., Saiz A., Leite M.I., et al. Antiglycine-receptor encephalomyelitis with rigidity. J Neurol Neurosurg Psychiatry. 2010. [epub ahead of print]
Matsumoto J.Y., Caviness J.N., McEvoy K.M. The acoustic startle reflex in stiff-man syndrome. Neurology. 1994;44:1952-1955.
Matsuura E., Matsuyama W., Sameshima T., Arimura K. Satoyoshi syndrome has antibody against brain and gastrointestinal tissue. Muscle Nerve. 2007;36(3):400-403.
McCombe P.A., Chalk J.B., Searle J.W., et al. Progressive encephalomyelitis with rigidity: a case report with magnetic resonance imaging findings. J Neurol Neurosurg Psychiatry. 1989;52(12):1429-1431.
McCrory P., Turner-Stokes L., Baguley I.J., et al. Botulinum toxin A for treatment of upper limb spasticity following stroke: a multi-centre randomized placebo-controlled study of the effects on quality of life and other person-centred outcomes. J Rehabil Med. 2009;41(7):536-544.
Meinck H.M., Benecke R., Kuster S., Conrad B. Cutaneomuscular (flexor) reflex organization in normal man and in patients with motor disorders. Adv Neurol. 1983;39:787-796.
Meinck H.M., Conrad B. Neuropharmacological investigations in the stiff-man syndrome. J Neurol. 1986;233(6):340-347.
Meinck H.M., Ricker K. Long-standing “stiff-man” syndrome: a particular form of disseminated inflammatory CNS disease? J Neurol Neurosurg Psychiatry. 1987;50(11):1556-1557.
Meinck H.M., Ricker K., Hulser P.J., et al. Stiff man syndrome: clinical and laboratory findings in eight patients. J Neurol. 1994;241(3):157-166.
Meinck H.M., Ricker K., Hulser P.J., Solimena M. Stiff man syndrome: neurophysiological findings in eight patients. J Neurol. 1995;242(3):134-142.
Meinck H.M., Thompson P.D. Stiff man syndrome and related conditions. Mov Disord. 2002;17(5):853-866.
Mertens H.-G., Zschocke S. Neuromyotonie. Klin Wochenschr. 1965;43:917-925.
Mitoma H., Song S., Ishida K., et al. Presynaptic impairment of cerebellar inhibitory synapses by an autoantibody to glutamate decarboxylase. J Neurol Sci. 2000;175(1):40-44.
Moersch F.P., Woltman H.W. Progressive fluctuating muscular rigidity and spasm (“stiff-man” syndrome): report of a case and some observations in 13 other cases. Mayo Clinic Proceedings. 1956;31:421-427.
Mohammadi B., Balouch S.A., Dengler R., Kollewe K. Long-term treatment of spasticity with botulinum toxin type A: an analysis of 1221 treatments in 137 patients. Neurol Res. 2009;32(3):309-313.
Molloy F.M., Dalakas M.C., Floeter M.K. Increased brainstem excitability in stiff-person syndrome. Neurology. 2002;59(3):449-451.
Moore A.P. Botulinum toxin A (BoNT-A) for spasticity in adults. What is the evidence? Eur J Neurol. 2002;9(Suppl 1)):42-47. discussion 53–61
Muller-Felber W., Ansevin C.F., Ricker K., et al. Immunosuppressive treatment of rippling muscles in patients with myasthenia gravis. Neuromuscul Disord. 1999;9(8):604-607.
Murinson B.B., Guarnaccia J.B. Stiff-person syndrome with amphiphysin antibodies: distinctive features of a rare disease. Neurology. 2008;71(24):1955-1958.
Musampa N.K., Mathieu P.A., Levin M.F. Relationship between stretch reflex thresholds and voluntary arm muscle activation in patients with spasticity. Exp Brain Res. 2007;181(4):579-593.
Nagado T., Arimura K., Sonoda Y., et al. Potassium current suppression in patients with peripheral nerve hyperexcitability. Brain. 1999;122(Pt 11):2057-2066.
Naharajakumar, Subbulakshmi C., John E., et al. Crying or smiling? The Schwartz-Jampel syndrome. J Pediatr. 2006;148(5):702.
Nakatsuji Y., Kaido M., Sugai F., et al. Isaacs’ syndrome successfully treated by immunoadsorption plasmapheresis. Acta Neurol Scand. 2000;102(4):271-273.
Nathan P.W., Smith M., Deacon P. Vestibulospinal, reticulospinal and descending propriospinal nerve fibres in man. Brain. 1996;119(Pt 6):1809-1833.
Newsom-Davis J., Buckley C., Clover L., et al. Autoimmune disorders of neuronal potassium channels. Ann NY Acad Sci. 2003;998:202-210.
Nicole S., Davoine C.S., Topaloglu H., et al. Perlecan, the major proteoglycan of basement membranes, is altered in patients with Schwartz-Jampel syndrome (chondrodystrophic myotonia). Nat Genet. 2000;26(4):480-483.
Pascuzzi R.M., Gratianne R., Azzarelli B., Kincaid J.C. Schwartz-Jampel syndrome with dominant inheritance. Muscle Nerve. 1990;13(12):1152-1163.
Perani D., Garibotto V., Moresco R.M., et al. PET evidence of central GABAergic changes in stiff-person syndrome. Mov Disord. 2007;22(7):1030-1033.
Powers R.K., Marder-Meyer J., Rymer W.Z. Quantitative relations between hypertonia and stretch reflex threshold in spastic hemiparesis. Ann Neurol. 1988;23(2):115-124.
Quasthoff S., Mockel C., Zieglgansberger W., Schreibmayer W. Tolperisone: a typical representative of a class of centrally acting muscle relaxants with less sedative side effects. CNS Neurosci Ther. 2008;14(2):107-119.
Raju R., Foote J., Banga J.P., Hall T.R., et al. Analysis of GAD65 autoantibodies in stiff-person syndrome patients. J Immunol. 2005;175(11):7755-7762.
Raju R., Hampe C.S. Immunobiology of stiff-person syndrome. Int Rev Immunol. 2008;27(1–2):79-92.
Raju R., Rakocevic G., Chen Z., et al. Autoimmunity to GABAA-receptor-associated protein in stiff-person syndrome. Brain. 2006;129(Pt 12):3270-3276.
Rakocevic G., Raju R., Dalakas M.C. Anti-glutamic acid decarboxylase antibodies in the serum and cerebrospinal fluid of patients with stiff-person syndrome: correlation with clinical severity. Arch Neurol. 2004;61(6):902-904.
Rietman J.S., Geertzen J.H. Efficacy of intrathecal baclofen delivery in the management of severe spasticity in upper motor neuron syndrome. Acta Neurochir Suppl. 2007;97(Pt 1):205-211.
Ronan S., Gold J.T. Nonoperative management of spasticity in children. Childs Nerv Syst. 2007;23(9):943-956.
Roobol T.H., Kazzaz B.A., Vecht C.J. Segmental rigidity and spinal myoclonus as a paraneoplastic syndrome. J Neurol Neurosurg Psychiatry. 1987;50(5):628-631.
Rowland L.P. Cramps, spasms and muscle stiffness. Rev Neurol. 1985;141(4):261-273.
Rueff L., Graber J.J., Bernbaum M., Kuzniecky R.I. Voltage-gated potassium channel antibody-mediated syndromes: a spectrum of clinical manifestations. Rev Neurol Dis. 2008;5(2):65-72.
Saidha S., Elamin M., Mullins G., et al. Treatment of progressive encephalomyelitis with rigidity and myoclonic jerks with rituximab: a case report. Eur J Neurol. 2008;15(5):e33.
Saiz A., Blanco Y., Sabater L., et al. Spectrum of neurological syndromes associated with glutamic acid decarboxylase antibodies: diagnostic clues for this association. Brain. 2008;131(Pt 10):2553-2563.
Saiz A., Dalmau J., Butler M.H., et al. Anti-amphiphysin I antibodies in patients with paraneoplastic neurological disorders associated with small cell lung carcinoma. J Neurol Neurosurg Psychiatry. 1999;66(2):214-217.
Sandbrink F., Syed N.A., Fujii M.D., et al. Motor cortex excitability in stiff-person syndrome. Brain. 2000;123(Pt 11):2231-2239.
Sanger T.D., Delgado M.R., Gaebler-Spira D., et al. Classification and definition of disorders causing hypertonia in childhood. Pediatrics. 2003;111(1):e89-97.
Satoyoshi E. A syndrome of progressive muscle spasm, alopecia, and diarrhea. Neurology. 1978;28(5):458-471.
Saval A., Chiodo A.E. Intrathecal baclofen for spasticity management: a comparative analysis of spasticity of spinal vs cortical origin. J Spinal Cord Med. 2010;33(1):16-21.
Schoser B., Jacob S., Hilton-Jones D., et al. Immune-mediated rippling muscle disease with myasthenia gravis: a report of seven patients with long-term follow-up in two. Neuromuscul Disord. 2009;19(3):223-228.
Schulte-Mattler W.J., Kley R.A., Rothenfusser-Korber E., et al. Immune-mediated rippling muscle disease. Neurology. 2005;64(2):364-367.
Schwartz O., Jampel R.S. Congenital blepharophimosis associated with a unique generalized myopathy. Arch Ophthalmol. 1962;68:52-57.
Sheean G. The pathophysiology of spasticity. Eur J Neurol. 2002;9(Suppl 1)):3-9. discussion 53–61
Sherman S.J., Koshland G.F., Laguna J.F. Hyper-reflexia without spasticity after unilateral infarct of the medullary pyramid. J Neurol Sci. 2000;175(2):145-155.
Shillito P., Molenaar P.C., Vincent A., et al. Acquired neuromyotonia: evidence for autoantibodies directed against K+ channels of peripheral nerves. Ann Neurol. 1995;38(5):714-722.
Silbert P.L., Matsumoto J.Y., McManis P.G., et al. Intrathecal baclofen therapy in stiff-man syndrome: a double-blind, placebo-controlled trial. Neurology. 1995;45(10):1893-1897.
Simpson D.M., Gracies J.M., Graham H.K., et al. Assessment: Botulinum neurotoxin for the treatment of spasticity (an evidence-based review): report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2008;70(19):1691-1698.
Sinha S., Newsom-Davis J., Mills K., et al. Autoimmune aetiology for acquired neuromyotonia (Isaacs’ syndrome). Lancet. 1991;338(8759):75-77.
So Y.T., Zu L., Barraza C., et al. Rippling muscle disease: evidence for phenotypic and genetic heterogeneity. Muscle Nerve. 2001;24(3):340-344.
Solimena M., Folli F., Aparisi R., et al. Autoantibodies to GABA-ergic neurons and pancreatic beta cells in stiff- man syndrome. N Engl J Med. 1990;322(22):1555-1560.
Sotgia F., Woodman S.E., Bonuccelli G., et al. Phenotypic behavior of caveolin-3 (R26Q), a mutant associated with hyperCKemia, distal myopathy, and rippling muscle disease. Am J Physiol Cell Physiol. 2003;285:C1150-C1160.
Souza-Lima C.F., Ferraz H.B., Braz C.A., et al. Marked improvement in a stiff-limb patient treated with intravenous immunoglobulin. Mov Disord. 2000;15(2):358-359.
Spaans F. Schwartz-Jampel syndrome with dominant inheritance. Muscle Nerve. 1991;14(11):1142-1144.
Spaans F., Theunissen P., Reekers A.D., et al. Schwartz-Jampel syndrome: I. Clinical, electromyographic, and histologic studies. Muscle Nerve. 1990;13(6):516-527.
Stamenova P., Koytchev R., Kuhn K., et al. A randomized, double-blind, placebo-controlled study of the efficacy and safety of tolperisone in spasticity following cerebral stroke. Eur J Neurol. 2005;12(6):453-461.
Stayer C., Meinck H.M. Stiff-man syndrome: an overview. Neurologia. 1998;13(2):83-88.
Stayer C., Tronnier V., Dressnandt J., et al. Intrathecal baclofen therapy for stiff-man syndrome and progressive encephalomyelopathy with rigidity and myoclonus. Neurology. 1997;49(6):1591-1597.
Stephan D.A., Buist N.R., Chittenden A.B., et al. A rippling muscle disease gene is localized to 1q41: evidence for multiple genes. Neurology. 1994;44(10):1915-1920.
Stum M., Davoine C.S., Vicart S., et al. Spectrum of HSPG2 (Perlecan) mutations in patients with Schwartz-Jampel syndrome. Hum Mutat. 2006;27(11):1082-1091.
Sun S.F., Hsu C.W., Sun H.P., et al. Combined botulinum toxin type a with modified constraint-induced movement therapy for chronic stroke patients with upper extremity spasticity: a randomized controlled study. Neurorehabil Neural Repair. 2010;24(1):34-41.
Takakusaki K., Kohyama J., Matsuyama K. Medullary reticulospinal tract mediating a generalized motor inhibition in cats: III. Functional organization of spinal interneurons in the lower lumbar segments. Neuroscience. 2003;121(3):731-746.
Takakusaki K., Kohyama J., Matsuyama K., Mori S. Medullary reticulospinal tract mediating the generalized motor inhibition in cats: parallel inhibitory mechanisms acting on motoneurons and on interneuronal transmission in reflex pathways. Neuroscience. 2001;103(2):511-527.
Taylor P.W. Isaacs’ syndrome (autoimmune neuromyotonia) in a patient with systemic lupus erythematosus. J Rheumatol. 2005;32(4):757-758.
Taylor R.G., Layzer R.B., Davis H.S., Fowler W.M.Jr. Continuous muscle fiber activity in the Schwartz-Jampel syndrome. Electroencephalogr Clin Neurophysiol. 1972;33(5):497-509.
Thilmann A.F. Spasticity: History, definitions, and usage of the term. In: Thilmann A.F., Burke D.J., Rymer W.Z., editors. Spasticity: Mechanisms and Management. Berlin: Springer-Verlag; 1993:1-5.
Thilmann A.F., Fellows S.J., Garms E. The mechanism of spastic muscle hypertonus. Variation in reflex gain over the time course of spasticity. Brain. 1991;114(Pt 1A):233-244.
Thompson P.D. Stiff muscles. J Neurol Neurosurg Psychiatry. 1993;56(2):121-124.
Thompson P.D. Stiff people. In: Marsden C.D., Fahn S., editors. Movement Disorders 3. Oxford: Butterworth-Heinemann; 1994:373-405.
Thompson P.D. The stiff-man syndrome and related disorders. Parkinsonism Relat Disord. 2001;8(2):147-153.
Torbergsen T. Rippling muscle disease: a review. Muscle Nerve. 2002;Suppl 11:S103-S107.
Traverso M., Bruno C., Broccolini A., et al. Truncation of caveolin-3 causes autosomal-recessive rippling muscle disease. J Neurol Neurosurg Psychiatry. 2008;79(6):735-737.
Trompetto C., Bove M., Avanzino L., et al. Intrafusal effects of botulinum toxin in post-stroke upper limb spasticity. Eur J Neurol. 2008;15(4):367-370.
Uddin A.B., Walters A.S., Ali A., Brannan T. A unilateral presentation of “Satoyoshi syndrome. Parkinsonism Relat Disord. 2002;8(3):211-213.
Valenstein E., Watson R.T., Parker J.L. Myokymia, muscle hypertrophy and percussion “myotonia” in chronic recurrent polyneuropathy. Neurology. 1978;28(11):1130-1134.
Vargel I., Canter H.I., Topaloglu H., Erk Y. Results of botulinum toxin: an application to blepharospasm Schwartz-Jampel syndrome. J Craniofac Surg. 2006;17(4):656-660.
Vasilescu C., Alexianu M., Dan A. Muscle hypertrophy and a syndrome of continuous motor unit activity in prednisone-responsive Guillain-Barre polyneuropathy. J Neurol. 1984;231(5):276-279.
Vasilescu C., Alexianu M., Dan A. Neuronal type of Charcot-Marie-Tooth disease with a syndrome of continuous motor unit activity. J Neurol Sci. 1984;63(1):11-25.
Vasilescu C., Florescu A. Peripheral neuropathy with a syndrome of continuous motor unit activity. J Neurol. 1982;226(4):275-282.
Vernino S., Lennon V.A. Ion channel and striational antibodies define a continuum of autoimmune neuromuscular hyperexcitability. Muscle Nerve. 2002;26(5):702-707.
Vicari A.M., Folli F., Pozza G., et al. Plasmapheresis in the treatment of stiff-man syndrome. N Engl J Med. 1989;320(22):1499.
Vorgerd M., Bolz H., Patzold T., et al. Phenotypic variability in rippling muscle disease. Neurology. 1999;52(7):1453-1459.
Vorgerd M., Ricker K., Ziemssen F., et al. A sporadic case of rippling muscle disease caused by a de novo caveolin-3 mutation. Neurology. 2001;57(12):2273-2277.
Walikonis J.E., Lennon V.A. Radioimmunoassay for glutamic acid decarboxylase (GAD65) autoantibodies as a diagnostic aid for stiff-man syndrome and a correlate of susceptibility to type 1 diabetes mellitus. Mayo Clin Proc. 1998;73(12):1161-1166.
Wallis W.E., Van Poznak A., Plum F. Generalized muscular stiffness, fasciculations, and myokymia of peripheral nerve origin. Arch Neurol. 1970;22(5):430-439.
Walsh J.C. Neuromyotonia: an unusual presentation of intrathoracic malignancy. J Neurol Neurosurg Psychiatry. 1976;39(11):1086-1091.
Wessig C., Klein R., Schneider M.F., et al. Neuropathology and binding studies in anti-amphiphysin-associated stiff-person syndrome. Neurology. 2003;61(2):195-198.
Whiteley A.M., Swash M., Urich H. Progressive encephalomyelitis with rigidity. Brain. 1976;99(1):27-42.
Wiesendanger M. Pyramidal tract function and the clinical “pyramidal syndrome. Hum Neurobiol. 1984;2(4):227-234.
Williams A.C., Nutt J.G., Hare T. Autoimmunity in stiff man syndrome. Lancet. 1988;2(8604):222.
Wilton A., Gardner-Medwin D. 21-year follow-up of myokymia with impaired muscle relaxation. Lancet. 1990;336(8723):1138-1139.
Wisuthsarewong W., Likitmaskul S., Manonukul J. Satoyoshi syndrome. Pediatr Dermatol. 2001;18(5):406-410.
Woodman S.E., Sotgia F., Galbiati F., et al. Caveolinopathies: mutations in caveolin-3 cause four distinct autosomal dominant muscle diseases. Neurology. 2004;62(4):538-543.