Special Neurologic Tests and Procedures
Neuro-otologic tests and procedures are used in a variety of clinical scenarios ranging from evaluation of a dizzy patient to diagnosing brain death. Some of these procedures have become less common with the advent of advanced neuroimaging and electrophysiologic testing; nonetheless, their simplicity and effectiveness give them continued relevance, especially in practice environments with limited resources.1
Caloric Testing
Historical Perspective
Brown-Séquard2 first described the effects of introducing cold water into the ear canal in the mid-19th century. The clinical importance of the phenomenon was first realized in 1906 by Bárány,3 who developed a caloric procedure involving an ice water stimulus. He postulated, correctly, that caloric stimulation of the auditory canal induces the formation of convection currents within the semicircular canals of the vestibular labyrinth.
Standardization of the procedure awaited introduction of the Fitzgerald-Hallpike technique in 1942. This technique used both warm and cool water stimuli under rigidly specified conditions, which permitted quantification of normal and abnormal caloric responses. Today, most formal caloric testing of conscious patients is based on variations of the original Fitzgerald-Hallpike procedure.4
The value of caloric testing in the assessment of comatose patients was emphasized by the work of Klingon5 and Bender and associates.6 Electronystagmography (ENG) can provide a graphic record of reflex eye movements and permit precise determination of the intensity of the caloric response.
Physiology and Functional Anatomy
Impulses of the primary neuron travel via Scarpa’s ganglion and cranial nerve VIII to the brainstem and synapse with secondary vestibular neurons in the superior and medial vestibular nuclei of the upper medulla and lower pons (Fig. 61-1). Although the connections between the vestibular and oculomotor nuclei in the brainstem are quite complex, two main pathways exist. The direct projection runs from the vestibular complex to the nuclei of cranial nerves III and VI via the medial longitudinal fasciculus (MLF) and involves only three neurons: (1) primary vestibular, (2) secondary vestibular, and (3) oculomotor. The indirect projection between these nuclei occurs over multisynaptic circuits in the tegmental reticular formation. Another brainstem structure that contributes to the vestibulo-ocular interaction is the paramedian pontine reticular formation (PPRF), a poorly characterized group of pontine neurons that coordinate both voluntary and involuntary lateral gaze. The PPRF receives multiple inputs. Including projections from the vestibular system and the contralateral frontal cortex, and sends output to oculomotor neurons through both direct and indirect pathways. Excitatory impulses originating in the lateral canal finally travel via the oculomotor and abducens nerves to the ipsilateral medial rectus and contralateral lateral rectus muscles.7
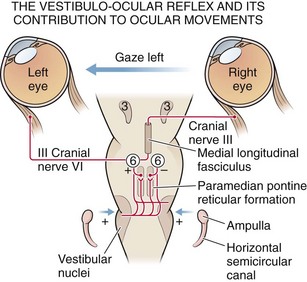
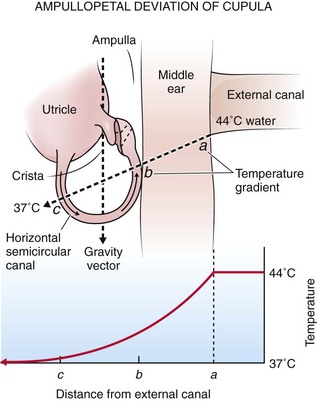
Rotation of the head generates flow of endolymphatic fluid within the semicircular canals. The firing rate of the primary vestibular neuron is dependent on the direction of flow. For example, in the lateral canal, flow toward the ampulla (ampullopetal) increases the firing rate, whereas flow away from the ampulla (ampullofugal) decreases the firing rate. Increased firing on one side results in conjugate deviation of the eyes toward the opposite side, whereas decreased firing causes deviation to the same side. This principle forms the physiologic basis of caloric testing. When the lateral canal is in the vertical position (patient placed supine) and ice water is infused into the ear, the endolymph nearest the canal cools and sinks, which results in ampullofugal flow (Fig. 61-2). As the firing rate decreases, the eyes deviate conjugately toward the side of irrigation. When warm water is used in the same position or when the canal is inverted 180 degrees, the opposite occurs.
Procedure
Variations of the caloric technique may be useful in certain situations. If there is no response to bilateral ice water caloric testing or in cases in which only one ear can be tested, perform warm water caloric testing. Keep the water temperature below 50°C. The response elicited will be the opposite of that obtained with ice water. In patients who fail to respond to ice water caloric testing alone, additional stimulation may be provided by combining irrigation with repeated head turning away from the irrigated side (doll’s-eye maneuver). This should not be performed in trauma patients unless cervical injury has definitely been ruled out. This combination of techniques may produce eye movements in patients who do not respond to caloric testing alone.8 Eviatar and Goodhill9 described a technique for caloric testing in patients with tympanic perforations that involves placing a small latex finger cot in the ear canal to prevent water from entering the middle ear.
Interpretation
Analyze initial eye position and spontaneous movements before irrigation. A description of eye movement abnormalities in comatose patients is beyond the scope of this chapter and is only briefly summarized here. The eyes of comatose patients with intact oculomotor pathways are usually directed straight ahead or are slightly divergent. Unilateral destructive lesions of the cerebral hemisphere can cause conjugate deviation of the eyes toward the side of the lesion, whereas irritative foci, as might be seen with status epilepticus, can cause conjugate deviation away from the affected side. Deviations of this type can usually be overcome by caloric stimulation, although combined irrigation and head turning may be required in the first hours after the insult. Lesions in or near the PPRF in the brainstem cause conjugate deviation away from the side of the lesion. This finding cannot usually be overcome by caloric stimulation. Conjugate downward deviation can be seen with structural lesions of the brainstem or in the deeper phases of metabolic coma. Dysconjugate gaze might indicate damage at the level of the oculomotor nuclei or below or might reflect disruption of the ocular muscles themselves.10 Dysconjugate gaze may also be seen with drug-induced coma in the presence of a structurally intact brainstem. In the very late stages of brainstem dysfunction, the eyes usually return to the central position. Spontaneous roving movements of the eyes, either conjugate or dysconjugate, may be seen with supratentorial insults, but these, too, disappear with brainstem involvement.11 Ocular “bobbing” is an intermittent, spontaneous downward jerking of the eyes that may occur with massive pontine lesions. Ocular “dipping” is a more prolonged, downward conjugate deviation of the eyes that has been reported in cases of severe anoxic encephalopathy (e.g., carbon monoxide poisoning).11 The pathophysiologic basis of these eye movements is poorly understood.12
Second Phase of Interpretation
After irrigation, ocular movements should be observed for any response to the stimulus. Again, typically there is a latency of response of 10 to 40 seconds. Reactions to ice water irrigation may be divided into four categories: (1) caloric nystagmus, (2) conjugate deviation, (3) dysconjugate deviation, and (4) absent responses (Fig. 61-3). The first reaction, caloric nystagmus with the fast component beating away from the side of ice water irrigation, is seen in normal, alert individuals, in cases of psychogenic unresponsiveness, and in those who have very mild organic disturbances in consciousness. The intensity of nystagmus is highly variable in conscious subjects and depends on the degree of visual fixation and the level of mental alertness. The response is present in more than 90% of children by 6 months of age and declines in magnitude only after the seventh decade of life.13

With acute supratentorial lesions, the development of dysconjugate caloric responses is a significant sign that may indicate compression of the brainstem and impending herniation. Caloric responses of this type are less common with metabolic and drug-induced coma; when present in metabolic coma, their significance is less ominous. Reversible internuclear ophthalmoplegia has been reported in patients with hepatic coma and may occur during toxic responses to phenytoin, barbiturates, or amitriptyline. Forced downward deviation of the eyes, either conjugate or dysconjugate, may be seen in sedative-hypnotic drug–induced coma when unilateral caloric testing is performed.14
Palsies of the oculomotor nerves are another cause of dysconjugate reactions, although most should be apparent before irrigation. Causes include diabetic neuropathy, increased intracranial pressure, and Wernicke’s encephalopathy. Finally, Plum and Posner10 reported that unusual and poorly characterized caloric responses may be obtained when testing comatose patients with long-standing severe brain injury.
Absent caloric response is the fourth category of reactions to ice water stimuli. As a general rule, the VOR is preserved more than other brainstem reflexes; however, the oculocephalic, or doll’s eye, response may persist in the absence of caloric responses. Loss of caloric responses in comatose patients with structural lesions is usually a sign of brainstem damage. With supratentorial lesions, progressive loss of caloric responses may be seen in the final stages of transtentorial herniation. The VOR may also be transiently absent or decreased on the side opposite massive supratentorial damage during the first hours after injury.15 Absent caloric responses may occur with any subtentorial lesion that affects the vestibular reflex pathways, including pontine hemorrhage, basilar artery occlusion, cerebellar hemorrhage, or infarction with encroachment on the brainstem, and with any expanding mass lesion within the posterior fossa. Caloric responses may disappear in patients with deep coma resulting from subarachnoid hemorrhage, perhaps because of pressure on the brainstem.
The VOR is usually retained until the late stages of metabolic coma. Nevertheless, caloric responses may be transiently absent in certain types of drug-induced coma, with eventual complete recovery of the patient. The VOR seems to be particularly sensitive to the effects of sedative-hypnotic drugs, antidepressants (e.g., amitriptyline, doxepin), and anticonvulsants (e.g., phenytoin, carbamazepine).16,17 As one would expect, neuromuscular blocking agents (e.g., succinylcholine) will abolish caloric-induced ocular movements.
Finally, the caloric response may be absent for reasons other than the neurologic causes responsible for the coma. Inadequate irrigation because of excessive cerumen or poor technique and unilateral or bilateral dysfunction of the peripheral vestibular apparatus must be considered. Bilateral loss of the caloric response (areflexia vestibularis) is uncommon in conscious patients, constituting 1.7% and 0.2% of the ENG clinical population in two large series of patients.18,19
The VOR has prognostic as well as diagnostic significance in comatose patients. In a study of 100 patients who were comatose as a result of head trauma, absence of caloric responses 1 to 3 days after injury was associated with extremely high mortality.20 Testing in the immediate posttraumatic period may yield inconsistent responses and is of considerably less prognostic value. Levy and coworkers21 studied 500 cases of nontraumatic, non–drug-induced coma in a large multicenter study. Absence of the VOR correlated with less than a 5% chance of achieving functional recovery within 1 year when tested within 6 to 24 hours of the onset of coma. In one study of comatose patients, the combination of absent VOR and absent pupillary light reflex at 24 hours was associated with 100% mortality.22 Complete loss of caloric responses is part of the criteria for the diagnosis of brain death and correlates with irreversible cessation of cerebral function at least as well as an isoelectric electroencephalogram (EEG) does.23 Excessive reliance on a single clinical sign must be avoided during consideration of brain death and in decisions regarding neurologic prognosis, and therapy should be based on complete evaluation of all the evidence available (see the later section “Brain Death Testing”).
DIX-Hallpike Test For The Diagnosis of Positional Vertigo
Vertigo occurring only and repeatedly with change in position is probably benign paroxysmal positional vertigo (BPPV); the head-hanging positioning maneuver (Dix-Hallpike test, sometimes referred to as the Nylen-Báràny maneuver) is useful in confirming clinical suspicion of BPPV because the abnormal nystagmus provoked is characteristic of the disorder.24,25 In BPPV, it is thought that calcium crystal material displaced from the vestibule floats within the endolymph of the posterior semicircular canal. Head movement induces bidirectional forces in the fluid acting on the cupula that trigger the attack of BPPV.26–29 The posterior semicircular canal is most commonly affected,25,28 but at times the horizontal canal is thought to be involved, thereby leading to variants of typical BPPV.30–32
Background
BPPV is a common mechanical disorder of the inner ear in which vertigo is precipitated by certain head movements; nystagmus and autonomic symptoms such as nausea and vomiting commonly accompany the vertigo. Although patients may have quite dramatic findings with severe autonomic symptoms, the actual episodes of vertigo are extremely brief and typically last less than 1 minute. Syndromes of positional vertigo and provocative maneuvers have been described by clinicians for more than 100 years; the description of BPPV has been attributed to Adler, Báràny, Nylèn, Bruns, Borries, Dix, and Hallpike.33 Dix and Hallpike most fully defined the disorder, and the provocative technique that they described is superior; thus it most accurately should bear the eponym Dix-Hallpike positioning test or the Dix-Hallpike test.25–33 Rarely, paroxysmal positional vertigo from a central cause has also been described in patients with small cerebellar hemorrhages.34
Indications and Contraindications
The Dix-Hallpike test may be a useful diagnostic test in confirming BPPV by provoking a specific type of nystagmus and in selecting patients suitable for positional therapy (discussed later). The maneuver should not be performed on patients with severe cervical spine disease, unstable spinal injury, high-grade carotid stenosis, or unstable heart disease.25 Patient discomfort and physical infirmity are relative contraindications; some elderly patients or those with ongoing nausea or vertigo may not tolerate the changes in body position necessary for performance of the procedure. If nystagmus is present at rest without any provocation or if associated neurologic signs or symptoms exist, the diagnosis of BPPV is probably excluded and the maneuver is not clinically indicated.
Procedure
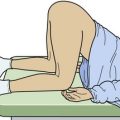
The Dix-Hallpike maneuver is illustrated in Figure 61-4. Place the patient initially in the seated position on the stretcher with the head turned 45 degrees to one side. With the patient instructed to keep the eyes open and focused on the examiner, quickly lay the patient down flat with the head hanging over the edge of the bed and observe the eyes for induced nystagmus. Repeat the entire maneuver with the head turned 45 degrees toward the opposite side.25
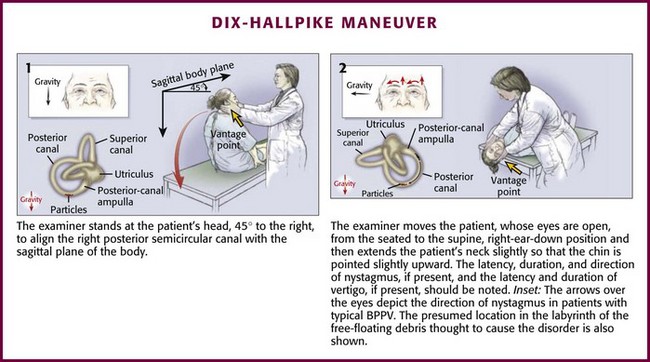
Interpretation
The Dix-Hallpike head-hanging positioning maneuver produces vertigo in patients with positional vertigo that is most commonly characterized as BPPV. The stereotypic positive response in BPPV is provoked vertigo developing after a brief delay (1 to 10 seconds), lasting less than a minute, and with direction-fixed rotary nystagmus. Nausea or other systemic symptoms are often present.24,25 The eye movements are mixed torsional and vertical nystagmus of both eyes with the upper pole of the eye beating toward the dependent ear and the vertical nystagmus beating toward the forehead. The side with the ear in the downward position during the Dix-Hallpike test that elicits greater nystagmus usually identifies the affected ear. After the patient is returned to the sitting position, the nystagmus may again be transiently observed in a reverse direction. If the positional nystagmus is atypical or if the maneuver fails to elicit nystagmus in a patient with ongoing symptoms of vertigo, another diagnostic possibility should be considered.25,35
Canalith-Repositioning Maneuvers
If the clinical evaluation of a patient with vertigo is consistent with the diagnosis of BPPV and the Dix-Hallpike test is supportive of the diagnosis and lateralizes to one ear, the patient may be a candidate for attempted canalith-repositioning maneuvers. These techniques have largely been described in the otologic and neurologic literature and have recently been described in the ED setting. Success rates of 44% to 100% are reported.25 One small ED-based study concluded that the Epley maneuver was more efficacious than a placebo maneuver.36
Background
With theory suggesting that stray material in the posterior semicircular canal causes the symptoms of BPPV, maneuvers were designed that involved sequential head movements to reposition the debris to the vestibule.25,29,37,38 Manipulation of head position theoretically allows the debris (“canaliths”) to sequentially fall from the problematic location in the semicircular canal to the vestibule of the labyrinth, where they presumably adhere. The currently recommended maneuver was introduced by Epley.29 Other maneuvers are described but require repeated trials or are more difficult to perform and perhaps more uncomfortable for the patient.25,37,39 The Semont maneuver is also described because it may be performed at the bedside. In theory, the Semont maneuver would be effective primarily in patients in whom the displaced canaliths were adhering to the cupola of the posterior semicircular canal (Schuknecht cupolith theory), although it might be effective as well when the debris are free floating in the long arm of the semicircular canal; the Epley maneuver would be effective only in patients with debris floating in the long arm of the posterior semicircular canal (canalith theory).40 In one small trial comparing the efficacy of either the Semont or the Epley maneuver in a carefully selected outpatient otolaryngology population, the maneuvers were found to be of roughly the same effectiveness (90%) in relieving or improving symptoms of BPPV.40
Procedure
The Epley procedure is illustrated in Figure 61-5. Briefly, as in the Dix-Hallpike maneuver, place the patient initially in the seated position on the stretcher with the head turned 45 degrees toward the affected side. Lay the patient down flat with the head hanging over the edge of the bed. After 20 seconds or after the symptoms subside, rotate the patient’s head so that it faces the opposite shoulder while maintaining the head-hanging orientation. Roll the patient further onto the side and rotate the head further into a face-down position. Again, after 20 seconds or after any symptoms subside, return the patient to a seated position.26,35,39 Some authors suggest keeping the patient’s head in each position long enough for any provoked symptoms of nystagmus or vertigo to resolve.25 Others suggest a period of 4 minutes after the head is placed in the hanging position and again after rotation of the head (“modified Epley maneuver”).40 The maneuver may be repeated a few times until some improvement in symptoms occurs, although this is not typically described in the literature.40 After a successful procedure, advise the patient to remain in a head-upright position for 24 hours.
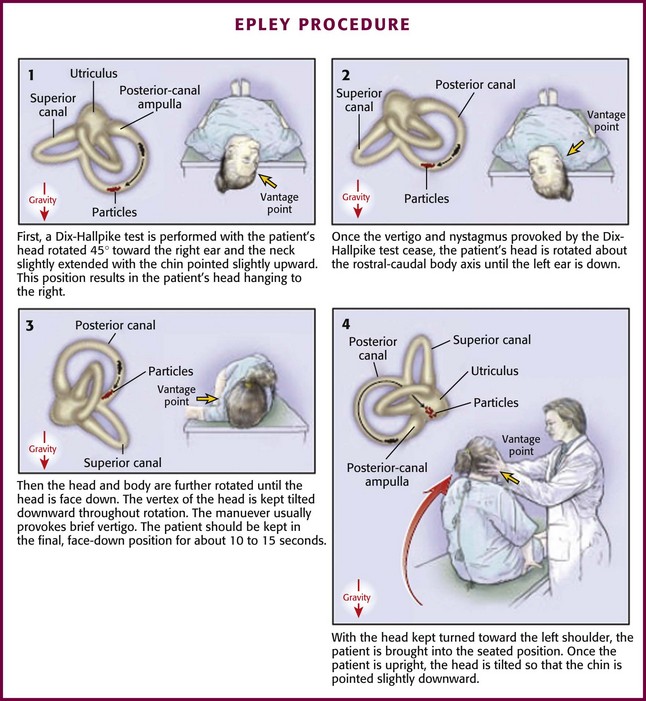
The Semont maneuver involves larger and more abrupt body movements. Identify the affected ear by the Dix-Hallpike maneuver. With the patient seated on the side of an examination table or bed, turn the patient’s head toward the unaffected side. Quickly lay the patient down into a side-lying position (Fig. 61-6) and keep the patient there until the symptoms subside. Move the patient abruptly through the sitting position to the opposite side-lying position and keep the patient there until the symptoms subside. Return the patient to the upright position.39,40 Advise the patient to remain in a head-upright position for 24 hours after a successful procedure.
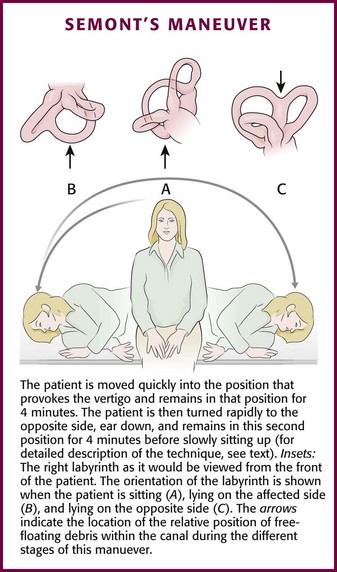
Figure 61-6 Semont’s maneuver.
Tests to Distinguish Central From Peripheral Lesions in Patients with AVS
Unfortunately, a certain percentage of patients with AVS (as many as 25% in some series) harbor a more sinister central pathology such as a brainstem or cerebellar stroke.41 Moreover, unlike BPPV, which tends to be very specific in its findings, it can be very difficult to distinguish a peripheral from a central process in patients with AVS. Traditional teaching has emphasized a careful neurologic examination in an attempt to find other cranial nerve, cerebellar, or long-tract (descending motor or sensory) findings to corroborate the presence of a central lesion. However, in some patients with stroke involving the posterior circulation, the vertigo is isolated and routine neurologic examination will reveal none of these findings. Magnetic resonance imaging (MRI) may help unravel such cases, but it is not standard to routinely perform MRI in patients with AVS simply because of the rare incidence of occult stroke. Stroke mimicking AVS is rare in young healthy patients, but a stroke causing AVS symptoms may be considered in the elderly or in patients with hypertension, diabetes, previous stroke, atherosclerotic vascular disease, or other vasculopathies.
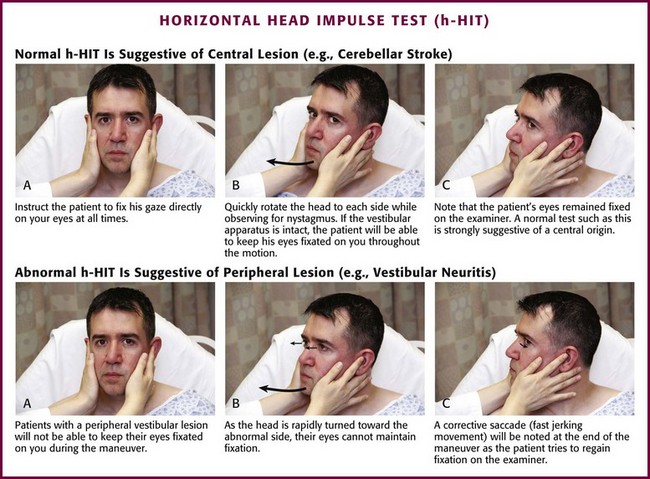
In 1988, Halmagyi and Curthoys42 described the horizontal head impulse test (h-HIT), a test of the VOR, as a bedside evaluation to detect peripheral vestibular disease. Subsequent studies have revealed that although a normal finding on the h-HIT is able to reliably identify patients with a central cause of vertigo, it is not as reliable in ruling out central pathology; a positive (abnormal) h-HIT may also occur in patients with lateral pontine and cerebellar stroke syndromes.43
The ability of the h-HIT to exclude stroke may be enhanced by the addition of two bedside maneuvers, an examination of nystagmus and a test of skew. The nystagmus that results from a peripheral lesion is unidirectional; its fast or corrective phase beats away from the affected side when the head is rapidly rotated toward that side. Nystagmus that changes its direction (the fast beating phase) with horizontal gaze to either side is highly predictive of a central lesion, as is spontaneous vertical and multidirectional nystagmus.44 Skew deviation refers to a misalignment of the eyes in the vertical plane; it is usually associated with a central lesion. Although skew deviation can be quite subtle, it can be unmasked by alternately covering each eye in rapid succession while the patient fixes his gaze on the examiner.45
Using the h-HIT, Newman-Toker and colleagues43 were able to distinguish central vertigo from peripheral vertigo in most cases. However, the test is far from perfect; in one study of 43 patients with AVS, 3 patients with stroke (9%) had a positive result on the h-HIT. In a more recent study, when an examination for bidirectional nystagmus and test of skew were added, diagnostic accuracy improved.46 In 108 patients with chronic risk factors for cerebrovascular disease and a new onset of AVS, the presence of either a normal h-HIT result, direction-changing nystagmus on eccentric gaze, or skew deviation was 100% sensitive and 96% specific for stroke. In this series, 25 peripheral and 76 central lesions were diagnosed by using a combination of MRI and clinical follow-up. Consistent with previous studies, the h-HIT erroneously suggested a peripheral lesion in a small minority (two) of cases. Interestingly, initial diffusion-weighted MRI was falsely negative in 12% of patients in whom a central lesion was ultimately diagnosed.
Procedure
Have the patient seated in a comfortable position. Ask the patient to gaze directly on the examiner’s eyes at all times. Quickly rotate the patient’s head to each side while carefully observing the eyes for nystagmus. A negative and positive h-HIT maneuver is simulated in Figure 61-7. Next, note any spontaneous vertical nystagmus or nystagmus that is bidirectional with horizontal gaze to both sides. To test for skew, alternately cover each eye in rapid succession while the patient looks directly ahead—note any vertical misalignment that occurs.
Brain Death Testing
A textbook of emergency medicine from 1988 once stated that because emergency medicine is a life support–oriented specialty, determination of brain death was outside the practice of the emergency clinician.47 The date of publication reflects practice at that time; neuroimaging was not often performed while patients were in the ED, and delays for inpatient beds were less common. With increased use of neuroimaging in the ED and general trends toward increased ED length of stay, the occasion may arise for emergency clinicians to be involved in the assessment of brain death. The duty to identify potential organ and tissue donors is another consideration in identifying patients with irreversible loss of brain function. Local practices, policies, regulations, and laws differ, and these remarks are general; the clinician is urged to be familiar with local practices and administrative policies regarding the issue of brain death. Brain death determination has been considered in court cases in the United States, and court rulings have upheld the medical practice of determination of brain death.48
For purposes of this discussion, brain death is defined as irreversible loss of functioning of the cerebral hemispheres and brainstem consistent with definitions in the literature.49–52 The cause of the brain injury should be known or identified because some toxicologic syndromes, notably barbiturate toxicity, exactly duplicate the clinical syndrome of brain death. This section focuses on clinical procedures commonly used to delineate brain death. Radiologic, electrophysiologic, and other tests that are sometimes used are not covered in detail. It should also be noted that in most cases a determination of brain death is not necessary for withdrawal of life-supporting measures. If a cause of coma or severe neurologic injury is identified and a poor prognosis is shared with the family, the option of withholding or withdrawing life support is often exercised. This is entirely within the realm of the patient, family, and clinician, and formal determination of brain death need not occur in these circumstances.
Background
With the advancement of intensive care techniques, patients were identified with continuing spontaneous cardiovascular function while on ventilatory support but without evidence of CNS activity. Observation of these patients revealed that cardiovascular function would eventually fail, although rare prolonged survival has been reported.53,54 Criteria to reliably identify these patients have been sought for multiple reasons, including to help in family counseling, improve resource allocation, and identify potential organ donors. Part of the impetus for a reliable set of criteria for brain death was legal; a legal pronouncement of death allows discontinuation of advanced life support in cases in which disagreement about continued life support measures may exist.
Death had long been defined as cessation of cardiac function; deeply comatose patients with persistent cardiac activity but without demonstrable brain function challenged the traditional definition of death. The concept of brain death has been entangled in continuing ethical, legal, and policy discussions roughly since the advent of the clinical application of mechanical ventilation in the 1960s.49,55,56
Indications and Contraindications
Evaluation for brain death implies that severe CNS dysfunction has been identified, that the cause of the CNS dysfunction is known, and that reversible causes of profound coma have been confidently excluded.50,52 Formal assessment of brain death is made in preparation for pronouncement of death to allow organ harvest or in uncommon cases of disparate family or caregiver convictions regarding prognosis of the patient. Complex medical issues that may confound the assessment should be considered and ruled out, including severe electrolyte disturbances, hypothermia (defined as a core temperature < 32°C), hypotension, drug intoxication or poisoning, and pharmacologic neuromuscular blockade.52 Neuroimaging studies should be carefully reviewed.
Procedure
Establishment of Coma and Cortical Assessment
By definition, a patient under evaluation for brain death will be in a coma without spontaneous respirations. Certain examination techniques are used to establish loss of function of the cerebral cortex and brainstem; clinical neurologic examination remains the standard for determination of brain death.52 It typically involves assessment for cortical function and brainstem reflexes, including respiratory drive.
While holding the patient’s eyes open, give loud verbal commands such as “Look up!” and assess for voluntary eye movements, particularly important for patients with the locked-in syndrome. Additionally, deliver a strong painful stimulus by forcefully pressing on the brow, sternum, or nail bed. Should any cerebral or brainstem function be discovered, the patient is not by definition brain-dead even if severe brain injury is present. Some institutions require evaluation by two clinicians with particular specialty training or repeated examinations several hours apart. The need for a second brain death determination has recently been questioned.57,58 The clinician must be familiar with local practices and policies, which may also require an ancillary EEG, nuclear angiography, or other techniques. Brain death in the pediatric population is more complex, with varying recommendations for repeated examinations and ancillary tests; such discussion is outside the scope of this chapter but is summarized elsewhere.52,59
Brainstem Reflex Testing
Pupillary Response: The pupils in brain-dead patients are unreactive and midposition to dilated. Shine a bright light into the pupil and observe for a reaction; none will be seen in a brain-dead patient. Should any reactivity be noted, the patient is not brain-dead.
Auditory Reflex: Deliver a loud handclap into each ear. Observe for eye blink or other reaction. Any reaction establishes that some brainstem function remains and excludes brain death.
Caloric Testing: Perform cold water irrigation of the external auditory canals with large volumes (≥100 mL) to elicit any eye movements through the VOR (described in detail earlier in this chapter). In a brain-dead patient, there will be no movement of the eyes in response to irrigation. Any eye movement excludes brain death.
Corneal Reflex: Stimulate the cornea with a cotton wisp or applicator. Observe for any eye closure, which indicates that the cranial nerve V to VII reflex arc remains intact and excludes the diagnosis of brain death.
Cough Reflex: Stimulate the trachea or main stem bronchi by deep suctioning and observe for coughing. A cough excludes brain death.
Apneic Oxygenation Test: CNS control of respiratory drive resides in the medulla. Establishing apnea is necessary to confirm medullary failure. Mechanical hyperventilation may artificially depress respiratory effort. It is hypercapnia, not hypoxia, that triggers respiratory effort. Simply disconnecting the ventilator to allow the development of hypercapnia for apnea testing may lead to hypoxia.60 A variety of techniques have been described that allow adequate hypercapnia for medullary stimulation to develop but ensure that oxygenation is adequate during the test. The most commonly described technique is to disconnect the patient from the ventilator and deliver oxygen at 10 to 15 L/min through a catheter inserted into the trachea and then observe for respiratory effort for 8 minutes. Any observed excursion of the abdomen or chest sufficient to produce a tidal volume suggests that brain death is not present. If no respiratory excursions are observed, arterial blood gas analysis is obtained and the patient is reconnected to the ventilator pending results. An arterial partial carbon dioxide pressure (Pco2) of 60 mm Hg or higher and the absence of respiratory excursions are the criteria for a positive apnea test.51 Others suggest that a possibly safer technique is to simply set the ventilator rate to zero while allowing continuous oxygen flow to continue and maintaining any necessary continuous positive pressure through the ventilator.61
Declaration of Death
If the criteria for brain death are satisfied, the family and all clinicians involved in patient care should be informed to allow further management decisions. At some institutions the patient is declared dead at the time that the criteria are met, and further care is assumed by the transplant services if that is the anticipated course. Per institutional protocol, confirmation of brain death by two clinicians may be required. Families are generally given the option of being present at the bedside while mechanical ventilation is discontinued, although some advise against this policy because spontaneous reflex movements such as the Lazarus sign (discussed below) may occur and disturb the family.8 For patients 18 years of age or younger, particularly infants, repeated examinations and confirmatory tests are generally recommended.52,56,59 A model for direct family conversation in this sensitive interaction has been described and includes a sample script and procedure.62
Complications
A variety of movements have been observed in brain-dead patients; at times these spinal level–mediated reflexes may be dramatic and erroneously lead observers to believe that the brain-dead patient is demonstrating voluntary movements or brain-mediated reflex movements. Finger jerks and facial myokymia (spontaneous, fine fascicular muscular contractions) have been reported to be spontaneously present in brain-dead patients. Decerebrate-like extensor posturing, the Lazarus sign (flexion of the arms at the elbows, shoulder adduction, lifting the arms, dystonic posturing of the hands with crossing of the hands), undulating toe flexion, triple-flexion response in the lower extremities, and flexion of the trunk (giving the appearance of sitting up) are among the signs described during apnea testing and after the termination of ventilation or triggered by tactile stimuli.11,63–67
MG Testing
Myasthenia gravis (MG) is the most common disease of neuromuscular transmission, but the incidence is just 1 in 20,000 in the general population. Patients with MG may be grouped into two major categories. The first group shows weakness in the proximal muscles that increases with activity and improves with rest. The other group has ocular complaints of diplopia or ptosis. Patients in the ocular weakness group may or may not have generalized symptoms as well. Fatigue is the hallmark of the disease; the symptoms typically wax and wane. Patients often see a number of clinicians before a correct diagnosis is made.68,69
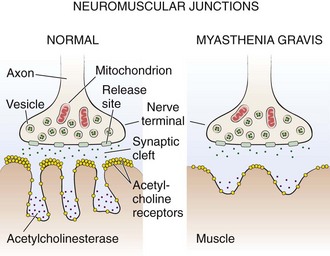
MG results from immune-mediated destruction of postsynaptic acetylcholine (ACh) receptors, with variable failure of neuromuscular transmission.68,69 ACh is the transmitter at the neuromuscular junction. When the nerve terminal is stimulated, ACh is released in a quantity far in excess of that needed for effective activation of the ACh receptor. ACh diffuses across the synaptic cleft to transiently interact with the ACh receptor, and an electrical potential is generated at the myoneural end plate. If of sufficient magnitude, the end plate potential initiates an action potential that is propagated along the muscle membrane, and muscle fiber contraction follows. The ACh is rapidly hydrolyzed by acetylcholinesterase in the synaptic cleft. Figure 61-8 summarizes neurotransmitter action at the neuromuscular junction. Of the millions of receptors at each myoneural junction, only a fraction must depolarize to stimulate muscle fiber contraction. Any factor that decreases the interaction of ACh with ACh receptors decreases the probability of an action potential being generated and may lead to failure of neuromuscular transmission with resulting weakness. Acetylcholinesterase inhibitors (anticholinesterases) have been the mainstay of therapy for MG for years, but they have been supplemented by immunosuppressive regimens and thymectomy. Failure of neuromuscular transmission may also occur with excessive inhibition of acetylcholinesterase; persistence of ACh in the synaptic cleft leads to continuous depolarization of the receptor.
A battery of tests can be used for the assessment of a patient with suspected MG, but only a few are available to the emergency clinician.70,71 ACh receptor antibody assay is positive in more than 80% of patients with MG but has a turnaround time of several days.72,73 Repetitive nerve stimulation and single-fiber electromyography tests are available in the electrophysiology laboratory.74 Several pharmacologic tests have been used to aid in the diagnosis of suspected MG, including parenteral administration of edrophonium chloride (Tensilon), neostigmine, or curare. Administration of edrophonium chloride for the diagnosis of MG (Tensilon test) is described in detail because of the drug’s rapid onset, short duration of action, and widespread acceptance for this diagnostic challenge; the emergency clinician may find occasion to use this test. The ice pack test is discussed because of favorable reports of its utility and noninvasive nature.71,75,76 Other noninvasive tests such as the “sleep test” (improvement in ptosis after 30 minutes of rest) and the “peek sign” (failure of the patient to maintain gentle eye closure) have been described.71 Both systemic and regional administration of curare has also been reported to aid in the diagnosis of MG. A technique is described for administration of curare into an ischemic arm; this modification is known as the regional curare test.74 The use of curare in this setting is outside the realm of practice of the emergency clinician.
Edrophonium (Tensilon) Test
Edrophonium chloride (Tensilon) is an acetylcholinesterase inhibitor that has been used for the diagnosis of MG since the 1960s. The short duration of action of edrophonium that made it unsatisfactory as a therapeutic agent for MG makes it useful as a diagnostic agent. The drug’s onset of action is rapid, and the duration of maximal effect is short, usually less than 2 minutes. Any effect resolves within 5 to 10 minutes.77
Edrophonium administration has also been recommended in the past to monitor acetylcholinesterase inhibitor therapy.78,79 However, edrophonium is not sufficiently reliable for titrating the effect of anticholinesterase medication.80 Use of the Tensilon test in a patient with known MG is controversial and should be done only in consultation with or in the presence of the treating neurologist, if at all.
Earlier types of crises described included “myasthenic” crisis from insufficient drug administration, “cholinergic” crisis from overdosage of a cholinesterase inhibitor, and the “brittle” patient with rapidly changing drug requirements,81,82 but terminology in this setting remains controversial.83 Even the clinical existence of the different types of crises has been debated.80 Several authorities advocate withdrawing all cholinesterase inhibitors in patients with worsening symptoms because most patients show increased responsiveness to cholinesterase inhibitors after several days without taking the drug.80,83 Others advocate a trial of edrophonium chloride at reduced dosage (1 to 2 mg) only after ventilator support.81,84
Indications and Contraindications
The bedside Tensilon test is indicated for the diagnosis of patients with suspected MG when there is a clinical need to make this diagnosis immediately. If other bedside tests such as the ice pack test (discussed later) are conclusive or if eyelid fatigue on prolonged upgaze can be demonstrated, there is no need to administer edrophonium.85 If the tempo of the clinical scenario is such that consultation or admission and performing specialized electromyelograms or serologic testing are feasible, deferring the Tensilon test in the ED may be preferred in recognition of the fact that serologic and neurophysiologic testing will not be completed for several days.
Administration of edrophonium is commonly performed in office settings by neuro-ophthalmologists, and occasionally complications of bradycardia occur that at times may be symptomatic with hypotension and loss of consciousness.86,87 One study reported the complication rate in office settings to be 0.16%.86 A history of asthma or cardiac dysrhythmias is a relative contraindication to the administration of cholinesterase inhibitors. Complications reported in the literature from edrophonium testing largely date from the era when it was used in cardiology to differentiate supraventricular rhythms.85 Reported complications include disturbances in cardiac rhythm and even death; several of these patients were taking digoxin or β-blockers.88–91 One report noted transient asystole after the administration of edrophonium in a patient with suspected MG; the patient was critically ill and had been receiving intravenous labetalol.85 Hence, caution is advised in administering edrophonium to patients receiving β-blocking agents, digoxin, or other drugs with atrioventricular-blocking properties.
Administration of edrophonium to a patient with MG being treated with cholinesterase inhibitors is controversial. Many investigators consider myasthenic crisis to be a contraindication to edrophonium administration, although in series of patients with myasthenic crisis, its use continues to be reported.81,84
Equipment
The following material is needed for testing an adult. Intravenous access should be secured with a saline lock. Ten milligrams of edrophonium chloride should be drawn up in a tuberculin syringe. Edrophonium chloride is supplied in 1- and 10-mL vials at a concentration of 10 mg/mL. A second syringe of normal saline should be available to administer as placebo, although some clinicians have recommended nicotine, calcium chloride, or atropine for this purpose.79 Atropine and other cardiovascular drugs and resuscitative equipment should be readily available. Cardiac monitoring is generally recommended. Photographic recording equipment is desirable to objectively document any improvement in motor function.
Procedure
Again, ptosis is an easy sign to test and is generally used if present (Fig. 61-9). The principles involved in assessing the effect of edrophonium on ptosis may be extended to testing other muscles. Ask the patient to look upward for several moments to fatigue the levator muscles. Note the degree of ptosis and document it by measurements or photographs. After a moment’s rest, ask the patient to look straight ahead. Inject 0.2 mL of the test substance in one syringe (2 mg of edrophonium or saline). If no response is seen within 1 minute, inject further increments of 2 mg up to a maximum of 10 mg. Note any increase in strength as reflected by an increase in size of the palpebral fissure. Repeat the procedure with the other test substance. Any improvement should occur within 30 seconds and disappear in 5 minutes.
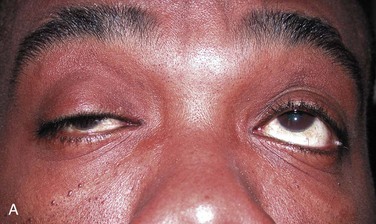
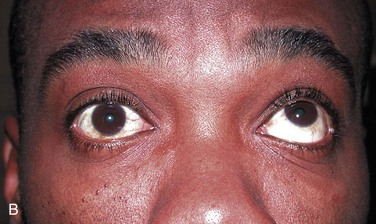
Interpretation
The key to the procedure is its interpretation. If a clearly paretic muscle has been identified, objective signs of improvement in the strength of that muscle within a moment of administration of edrophonium and fading of that improvement over the next 5 minutes are the criteria for a positive test result. Up to 90% of patients with MG have a positive test result under ideal circumstances.78,79 False-negative results do occur consistently, however.
Reproducible and unequivocal reversal of weakness in a specific muscle is extremely specific for MG.92,93 False-positive test results have been reported in patients with Eaton-Lambert syndrome and rarely in patients with intracranial lesions.94–96 Other rare reports of positive test results involve patients with amyotrophic lateral sclerosis. A “perverse” reaction in which a paretic extraocular muscle, weak from other causes, becomes even weaker with edrophonium administration has been noted rarely.97
Ice Pack Test
It has been observed clinically that myasthenic patients have exacerbations of weakness with environmental heat and improvement in strength with cold temperatures. A simple bedside test uses these observations to evaluate ptosis.71,75,76 Ice placed in a surgical glove or wrapped in a towel is placed lightly over the eyelid of the patient. Cooling of the eyelid below 29°C is accomplished within 2 minutes. Ptosis has been noted to improve in 80% or more of patients tested and may be more sensitive than the edrophonium test in detecting ocular MG. Although the reported number of patients evaluated by this method continues to be small, the test is included here because of its potential application in the ED, its lack of side effects, and its noninvasive nature.
Indications
Unilateral or bilateral ptosis of uncertain etiology in which MG is a diagnostic possibility is the sole indication for this test. Use of the ice pack test for evaluating diplopia possibly related to MG has also been described, but this role has yet to be established and its utility is unclear.98,99
Procedure
Ice and a surgical glove or towel are the only material required. A camera to record any response is optional. Measure or photograph the degree of the patient’s ptosis. Ask the patient to look upward, which often provokes the ptosis. If bilateral ptosis is present, use the more affected eye for evaluation. Cool the eyelid by lightly holding the wrapped ice to the patient’s eyelid for 2 minutes or until patient discomfort limits application. Compare the width of the palpebral fissure with the pretest width (Fig. 61-10; see p. 1258).
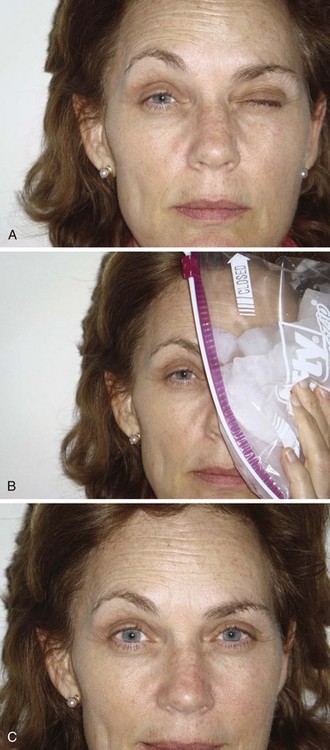
Interpretation
A clear improvement in ptosis in the cooled eye is the criterion for a positive test. The effect should be reproducible. In small clinical studies, the ice pack test was found to be at least as sensitive as administration of edrophonium in improving ptosis in patients with ocular MG. False-negative results do occur, probably at about the same frequency as with Tensilon testing. One individual has been reported to have a negative ice pack test result with a positive Tensilon test result. Negative or equivocal Tensilon tests have been reported in other individuals who had clearly positive ice pack test results. Normal individuals showed no change in width of the palpebral fissure after cold exposure. False-positive results are rare.75,76,100
Summary
The bedside Tensilon test has a long history of utility in diagnosing MG but it has largely been replaced by acetylcholinesterase receptor assay and electrodiagnostic studies in the ambulatory setting.72,74,85,93 On occasion, when rapid diagnosis is desired or MG is suspected in the presence of a normal ACh receptor titer, a carefully performed Tensilon test is still clinically valuable. Use of the Tensilon test in the setting of myasthenic crisis is controversial and is discouraged.
It is often the case with neuromuscular diseases, given the broad diagnostic possibilities, that the emergency clinician is unable to establish a confident diagnosis at a single patient encounter.101 Appropriate consultation and referral are necessary.
References
1. Booth, CM, Boone, RH, Tomlinson, G, et al. Is this patient dead, vegetative, or severely neurologically impaired? Assessing outcome for comatose survivors of cardiac arrest. JAMA. 2004;291:870.
2. Brown-Séquard, C. Course of Lectures on the Physiology and Pathology of the Central Nervous System. Philadelphia: Collins; 1860.
3. Bárány, R. Untersuchungen über den vom Vestibularapparat des Ohres reflektorisch ausgelosten rhythmischen Nystagmus und seine Begleiterscheinungen Mschr. Ohrenheilk. 1906;40:193.
4. Fitzgerald, G, Hallpike, CS. Studies in human vestibular function: I. Observations of the directional preponderance (“nystagmus bereitschaft”) of caloric nystagmus resulting from cerebral lesions. Brain. 1942;65:115.
5. Klingon, GH. Caloric stimulation in localization of brain stem lesions in a comatose patient. Arch Neurol Psychiatry. 1952;68:233.
6. Bender, MB, Bergman, PS, Nathanson, M. Ocular movements on passive head turning and caloric stimulation in comatose patients. Neurol Assoc. 1955;80:184.
7. Baloh, RW, Honrubia, V, Vestibular function: an overview. Clinical Neurophysiology of the Vestibular System, Oxford, Oxford University Press, 2001;Vol 63:3.
8. Fischer, CM. The neurological exam of the comatose patient. Acta Neurol Scand. 1960;45:43.
9. Eviatar, A, Goodhill, V. A dry calorization method for vestibular function studies. Laryngoscope. 1968;78:1746.
10. Plum, F, Posner, JB. The Diagnosis of Stupor and Coma. Philadelphia: Davis; 1980.
11. Ropper, AH. Unusual spontaneous movements in brain-dead patients. Neurology. 1984;34:1089.
12. Rosenberg, ML. Spontaneous vertical eye movements in coma. Ann Neurol. 1986;20:625.
13. Bruner, A, Norris, TW. Age-related changes in caloric nystagmus. Acta Otolaryngol Suppl. 1971;282:1.
14. Simon, RP. Forced downward ocular deviation. Occurrence during oculovestibular testing in sedative drug–induced coma. Arch Neurol. 1978;35:456.
15. Posner, JB, Plum, F. Diagnostic significance of the vestibulo-ocular response. J Neurol Neurosurg Psychiatry. 1975;38:727.
16. Spector, RH, Davidoff, RA, Schwartzman, RJ. Phenytoin-induced ophthalmoplegia. Neurology. 1976;26:1031.
17. Spector, RH, Schnapper, R. Amitriptyline-induced ophthalmoplegia. Neurology. 1981;31:1188.
18. Simmons, FB. Patients with bilateral loss of caloric response. Ann Otol Rhinol Laryngol. 1973;82:175.
19. Steensen, SH, Toxman, J, Zilstorff, K. Bilateral loss of caloric response in conscious patients (areflexia vestibularis). Clin Otolaryngol. 1980;5:373.
20. Poulsen, J, Zilstorff, K. Prognostic value of the caloric-vestibular test in the unconscious patient with cranial trauma. Acta Neurol Scand. 1972;48:282.
21. Levy, DE, Bates, D, Caronna, JJ, et al. Prognosis in nontraumatic coma. Ann Intern Med. 1981;94:293.
22. Mueller-Jensen, A, Neunzig, HP, Emskotter, T. Outcome prediction in comatose patients: significance of reflex eye movement analysis. J Neurol Neurosurg Psychiatry. 1987;50:389.
23. Hicks, RG, Torda, TA. The vestibulo-ocular (caloric) reflex in the diagnosis of cerebral death. Anaesth Intensive Care. 1979;7:169.
24. Drachman, DA. A 69-year-old man with chronic dizziness. JAMA. 1998;280:2111.
25. Furman, JM, Cass, SP. Benign paroxysmal positional vertigo. N Engl J Med. 1999;341:1590.
26. Brandt, T, Steddin, S, Daroff, RB. Therapy for benign paroxysmal positioning vertigo, revisited. Neurology. 1994;44:796.
27. Brandt, T, Steddin, S. Current view of the mechanism of benign paroxysmal positioning vertigo: cupulolithiasis or canalolithiasis? J Vestib Res. 1993;3:373.
28. Parnes, LS, McClure, JA. Posterior semicircular canal occlusion in the normal hearing ear. Otolaryngol Head Neck Surg. 1991;104:52.
29. Epley, JM. The canalith repositioning procedure: for treatment of benign paroxysmal positional vertigo. Otolaryngol Head Neck Surg. 1992;107:399.
30. McClure, JA. Horizontal canal BPV. J Otolaryngol. 1985;14:30.
31. De la Meilleure, G, Dehaene, I, Depondt, M, et al. Benign paroxysmal positional vertigo of the horizontal canal. J Neurol Neurosurg Psychiatry. 1996;60:68.
32. von Brevern, M, Clarke, AH, Lempert, T. Continuous vertigo and spontaneous nystagmus due to canalolithiasis of the horizontal canal. Neurology. 2001;56:684.
33. Lanska, DJ, Remler, B. Benign paroxysmal positioning vertigo: classic descriptions, origins of the provocative positioning technique, and conceptual developments. Neurology. 1997;48:1167.
34. Johkura, K. Central paroxysmal positional vertigo: isolated dizziness caused by small cerebellar hemorrhage. Stroke. 2007;38:e26–e27. [author reply e28].
35. Fife, TD. Bedside cure for benign positional vertigo. BNI Q. 1994;10:2.
36. Chang, AK, Schoeman, G, Hill, M. A randomized clinical trial to assess the efficacy of the Epley maneuver in the treatment of acute benign positional vertigo. Acad Emerg Med. 2004;11:918.
37. Brandt, T, Daroff, RB. Physical therapy for benign paroxysmal positional vertigo. Arch Otolaryngol. 1980;106:484.
38. Semont, A, Freyss, G, Vitte, E. Curing the BPPV with a liberatory maneuver. Adv Otorhinolaryngol. 1988;42:290.
39. Koelliker, P, Summers, RL, Hawkins, B. Benign paroxysmal positional vertigo: diagnosis and treatment in the emergency department—a review of the literature and discussion of canalith-repositioning maneuvers. Ann Emerg Med. 2001;37:392.
40. Herdman, SJ, Tusa, RJ, Zee, DS, et al. Single treatment approaches to benign paroxysmal positional vertigo. Arch Otolaryngol Head Neck Surg. 1993;119:450.
41. Norrving, B, Magnusson, M, Holtas, S. Isolated acute vertigo in the elderly; vestibular or vascular disease? Acta Neurol Scand. 1995;91:43–48.
42. Halmagyi, GM, Curthoys, IS. A clinical sign of canal paresis. Arch Neurol. 1988;45:737–739.
43. Newman-Toker, DE, Kattah, JC, Alvernia, JE, et al. Normal head impulse test differentiates acute cerebellar strokes from vestibular neuritis. Neurology. 2008;70:2378–2385.
44. Cnyrim, CD, Newman-Toker, D, Karch, C, et al. Bedside differentiation of vestibular neuritis from central “vestibular pseudo-neuritis.”. J Neurol Neurosurg Psychiatry. 2008;79:458–460.
45. Brodsky, MC, Donahue, SP, Vaphiades, M, et al. Skew deviation revisited. Surv Ophthalmol. 2006;51:105–128.
46. Kattah, JC, Talkad, AV, Wang, DZ. HINTS to diagnose stroke in the acute vestibular syndrome three-step bedside oculomotor examination more sensitive than early MRI diffusion-weighted imaging. Stroke. 2009;40:3504–3510.
47. Huff, JS. Coma. In: Rosen P, Barker FJ, Barkin RM, et al, eds. Emergency Medicine: Concepts and Clinical Practice. St. Louis: Mosby; 1988:267.
48. Burkle, CM, Schipper, AM, Wijdicks, EF. Brain death and the courts. Neurology. 2011;76:837–841.
49. A definition of irreversible coma: report of the ad Hoc Committee of the Harvard Medical School to examine the definition of brain death. JAMA. 1968;205:337.
50. Bleck, TP, Smith, MC. Diagnosing brain death and persistent vegetative states. J Crit Illness. 1989;4:60.
51. Wijdicks, EF. Determining brain death in adults. Neurology. 1995;45:1003.
52. Wijdicks, EF. The diagnosis of brain death. N Engl J Med. 2001;344:1215.
53. Cranford, R. Even the dead are not terminally ill anymore. Neurology. 1998;51:1530.
54. Shewmon, DA. Chronic “brain death”: meta-analysis and conceptual consequences. Neurology. 1998;51:1538.
55. Swash, M, Beresford, R. Brain death: still-unresolved issues worldwide. Neurology. 2002;58:9.
56. Wijdicks, EF. Brain death worldwide: accepted fact but no global consensus in diagnostic criteria. Neurology. 2002;58:20.
57. Lustbader, D, O’Hara, D, Wijdicks, EF, et al. Second brain death examination may negatively affect organ donation. Neurology. 2011;76:119–124.
58. Sung, G, Greer, D. The case for simplifying brain death criteria. Neurology. 2011;76:113–114.
59. Nakagawa, TA, Ashwal, S, Mathur, M, et al. Clinical report—guidelines for the determination of brain death in infants and children: an update of the 1987 task force recommendations. Pediatrics. 2011;128:e720–e740.
60. Marks, SJ, Zisfein, J. Apneic oxygenation in apnea tests for brain death. A controlled trial. Arch Neurol. 1990;47:1066.
61. Rockoff, MA, Thompson, JE. The diagnosis of brain death. N Engl J Med. 2001;345:616.
62. Shaner, DM, Orr, RD, Drought, T, et al. Really, most SINCERELY dead. Policy and procedure in the diagnosis of death by neurologic criteria. Neurology. 2004;62:1683.
63. Ivan, LP. Spinal reflexes in cerebral death. Neurology. 1973;23:650.
64. Jordan, JE, Dyess, E, Cliett, J. Unusual spontaneous movements in brain-dead patients. Neurology. 1985;35:1082.
65. Mandel, S, Arenas, A, Scasta, D. Spinal automatism in cerebral death. N Engl J Med. 1982;307:501.
66. Marti-Fabregas, J, Lopez-Navidad, A, Caballero, F, et al. Decerebrate-like posturing with mechanical ventilation in brain death. Neurology. 2000;54:224.
67. Saposnik, G, Bueri, JA, Maurino, J, et al. Spontaneous and reflex movements in brain death. Neurology. 2000;54:221.
68. Drachman, DB. Myasthenia gravis. N Engl J Med. 1994;330:1797.
69. Seybold, ME. The office Tensilon test for ocular myasthenia gravis. Arch Neurol. 1986;43:842.
70. Benatar, M. A systematic review of diagnostic studies in myasthenia gravis. Neuromuscul Disord. 2006;16:459.
71. Scherer, K, Bedlack, RS, Simel, DL. Does this patient have myasthenia gravis? JAMA. 2005;293:1906–1914.
72. Kelly, JJ, Jr., Daube, JR, Lennon, VA, et al. The laboratory diagnosis of mild myasthenia gravis. Ann Neurol. 1982;12:238.
73. Soliven, BC, Lange, DJ, Penn, AS, et al. Seronegative myasthenia gravis. Neurology. 1988;38:514.
74. Patten, BM. Myasthenia gravis: review of diagnosis and management. Muscle Nerve. 1978;1:190.
75. Ertas, M, Arac, N, Kumral, K, et al. Ice test as a simple diagnostic aid for myasthenia gravis. Acta Neurol Scand. 1994;89:227.
76. Sethi, KD, Rivner, MH, Swift, TR. Ice pack test for myasthenia gravis. Neurology. 1987;37:1383.
77. Osserman, KE. Rapid diagnostic test for myasthenia gravis: increased muscle strength, without fasciculations, after intravenous administration of edrophonium (Tensilon) chloride. JAMA. 1952;150:265.
78. Osserman, KE, Genkins, G. Critical reappraisal of the use of edrophonium (Tensilon) chloride tests in myasthenia gravis and significance of clinical classification. Ann N Y Acad Sci. 1966;135:312.
79. Osserman, KE, Genkins, G. Studies in myasthenia gravis: review of a twenty-year experience in over 1200 patients. Mt Sinai J Med. 1971;38:497.
80. Rowland, LP. Controversies about the treatment of myasthenia gravis. J Neurol Neurosurg Psychiatry. 1980;43:644.
81. Gracey, DR, Divertie, MB, Howard, FM, Jr. Mechanical ventilation for respiratory failure in myasthenia gravis. Two-year experience with 22 patients. Mayo Clin Proc. 1983;58:597.
82. Tether, JE. Management of myasthenic and cholinergic crisis. Am J Med. 1955;19:740.
83. Griggs, RC, Donohoe, KM. Emergency management of neuromuscular disease. In: Henning RJ, Jackson DL, eds. Handbook of Critical Care Neurology and Neurosurgery. New York: Praeger; 1985:211.
84. Sellman, MS, Mayer, RF. Treatment of myasthenic crisis in late life. South Med J. 1985;78:1208.
85. Okun, MS, Charriez, CM, Bhatti, MT, et al. Asystole induced by edrophonium following beta blockade. Neurology. 2001;57:739.
86. Ing, EB, Ing, SY, Ing, T, et al. The complication rate of edrophonium testing for suspected myasthenia gravis. Can J Ophthalmol. 2000;35:141.
87. Van Dyk, HJ, Florence, L. The Tensilon test. A safe office procedure. Ophthalmology. 1980;87:210.
88. Gould, L, Zahir, M, Gomprecht, RF. Cardiac arrest during edrophonium administration. Am Heart J. 1971;81:437.
89. Mayor, GH, Willis, PW, 3rd. Cardiac arrest after edrophonium therapy of supraventricular tachycardia. South Med J. 1976;69:1437.
90. Rossen, RM, Krikorian, J, Hancock, EW. Ventricular asystole after edrophonium chloride administration. JAMA. 1976;235:1041.
91. Youngberg, JA. Cardiac arrest following treatment of paroxysmal atrial tachycardia with edrophonium. Anesthesiology. 1979;50:234.
92. Oh, SJ, Cho, HK. Edrophonium responsiveness not necessarily diagnostic of myasthenia gravis. Muscle Nerve. 1990;13:187.
93. Oh, SJ, Kim, DE, Kuruoglu, R, et al. Diagnostic sensitivity of the laboratory tests in myasthenia gravis. Muscle Nerve. 1992;15:720.
94. Dirr, LY, Donofrio, PD, Patton, JF, et al. A false-positive edrophonium test in a patient with a brainstem glioma. Neurology. 1989;39:865.
95. Moorthy, G, Behrens, MM, Drachman, DB, et al. Ocular pseudomyasthenia or ocular myasthenia “plus”: a warning to clinicians. Neurology. 1989;39:1150.
96. Ragge, NK, Hoyt, WF. Midbrain myasthenia: fatigable ptosis, “lid twitch” sign, and ophthalmoparesis from a dorsal midbrain glioma. Neurology. 1992;42:917.
97. Seybold, ME. Myasthenia gravis. A clinical and basic science review. JAMA. 1983;250:2516.
98. Chatzistefanou, KI, Kouris, T, Iliakis, E, et al. The ice pack test in the differential diagnosis of myasthenic diplopia. Ophthalmology. 2009;116:2236–2243.
99. Larner, AJ, Thomas, DJ. Can myasthenia gravis be diagnosed with the “ice pack test”? A cautionary note. Postgrad Med. 2000;76:162–163.
100. Larner, AJ. The place of the ice pack test in the diagnosis of myasthenia gravis. Int J Clin Pract. 2004;58:887.
101. Singer, JI, Back, K. Postural guarding and hypertension as initial manifestations of Guillain-Barré syndrome. Am J Emerg Med. 1989;7:177.