[level-membership-for-neurology-category]
Chapter 66 Sleep–Wake Disorders*
Complaints about insufficient or nonrestorative sleep are quite common in childhood and adolescence. In a questionnaire survey of 332 children of 11 through 15 years of age, Ipsiroglu et al. [2001] observed that 28 percent of the subjects had snoring, insomnia, or a parasomnia. In another survey of 472 4- to 12-year-old urban and rural children receiving routine pediatric care, Stein et al. [2001] noted a 10 percent prevalence of sleep disorders. Less than one-half of the parents had discussed the sleep problems with the pediatrician. Childhood sleep disorders can have a significant effect on the quality of life. Many disorders are also easily treatable, thus underscoring the importance of their prompt recognition and management. This chapter covers salient aspects of childhood sleep–wake function and common pediatric sleep disorders.
Sleep Physiology and Ontogeny
Sleep–wake regulation is mediated via a complex set of interactions. There is a dynamic balance between the circadian drive for sleep (process C), which serves to enhance alertness, and the homeostatic drive (process S), which facilitates sleep. Adenosine, an extracellular factor that is secreted by neurons in the basal forebrain, plays a key role in sleep induction. (Caffeine is an antagonist of adenosine.) Neurons of the ventrolateral preoptic nucleus that contain gamma-aminobutyric acid (GABA) and galanin serve to inhibit wakefulness-promoting regions of the forebrain and the laterodorsal pontine tegmentum, and are thus also involved in sleep induction. Sleep onset is further facilitated by melatonin, a light-sensitive hormone that is released from the pineal gland. The principal components of the ascending arousal system are the cholinergic neurons of laterodorsal pontine tegmentum and the nucleus basialis of Meynert. Cells of the dorsolateral hypothalamus produce hypocretin (orexin), which is also an important wakefulness-promoting peptide. The hypocretin neurons project widely to the forebrain and brainstem. There is a tendency for mutual inhibition between the wakefulness-promoting and the sleep-promoting circuits. This phenomenon has been characterized as a “flip-flop” switch [Fuller et al., 2006; Lu and Zee, 2010].
The ultimate command center for the once-a-day (circadian) rhythm of sleep and wakefulness is the suprachiasmatic nucleus of the hypothalamus [Miller et al., 1996; Steriade et al., 1993], which has cells with receptors for melatonin. The suprachiasmatic nucleus is strongly influenced by light-mediated impulses received through the retinohypothalamic tract. Clock and Period genes, remarkably preserved across various phyla, were studied initially in Drosophila melanogaster; they also influence the timing of activity of the suprachiasmatic nucleus cells [Challet et al., 2003; Franken and Dijk, 2009; Hamada et al., 2001]. The mammalian target of rapamycin (mTOR) signaling is involved in modulation of photic entrainment of the suprachiasmatic circadian clock [Cao et al., 2010]. The body temperature rhythm is also regulated by the hypothalamus [van Someren, 2000]; a rise in body temperature leads to postponement of sleep. Conversely, individuals are most sleepy around the nadir of body temperature: that is, around 0400 hours. An artificial increase in body temperature in the 1–2 hours before bedtime, such as through vigorous exercise, may provoke sleep-onset insomnia.
Wakefulness can be differentiated from sleep by 27–28 weeks’ postconceptional age in the preterm infant. At this age, sleep is primarily of the active or rapid eye movement (REM) type, which is associated with irregular breathing, phasic electromyographic activity, and low-voltage electroencephalographic (EEG) activity. Cerebral blood flow and metabolism are higher in REM than in non-rapid eye movement (NREM) sleep, which is also termed quiet sleep in newborns. By 40 weeks’ postconceptional age, active (REM) sleep decreases to about 50 percent of the total sleep time, with a corresponding rise in the proportion of quiet (NREM) sleep. By 46–48 weeks’ postconceptional age, sleep spindles appear during stages N2 and N3 of NREM sleep. By 4–6 months of age, NREM sleep has fully differentiated into N1, N2, and N3 sleep, corresponding respectively with lighter to deeper stages in terms of arousal threshold. N3 is also termed slow-wave sleep. It is characterized by the generalized slow-wave activity in the 0.5–4 Hz range on the EEG (Figure 66-1). The bulk of N3 occurs during the first third of the night. Growth hormone release is closely linked to N3 sleep [Van Cauter et al., 2008], with suppression of the latter leading to impaired growth hormone release. The release of cortisol is suppressed during N3 sleep [van Cauter et al., 2008]. REM sleep decreases gradually over time. By the age of 3 years, it constitutes only about 20–25 percent of total sleep.
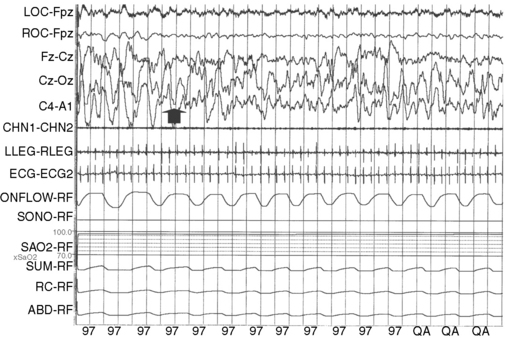
Prior to age of 3 months, the transition from wakefulness is initially into REM sleep. After this age, however, the physiologic transition is from wakefulness into NREM sleep, with REM sleep occurring 90–140 minutes later. The physiologic sleep-onset time in elementary school-age children is usually around 8:00–8:30 pm. Around adolescence, there is a physiologic delay in sleep-onset time, which shifts to around 10:30–11:00 pm [Carskadon et al., 1998]. Teenage girls generally have their final morning awakening about one half-hour earlier than boys. When juxtaposed with early high-school start times of around 7:30 am, it is easy to understand why most teenagers are chronically sleep-deprived.
The International Classification of Sleep Disorders
The International Classification of Sleep Disorders was introduced in 1990, and has undergone subsequent revisions [American Sleep Disorders Association, 2006]. It was created initially through collaborative efforts of the American Sleep Disorders Association, the European Sleep Research Society, the Japanese Society of Sleep Research, and the Latin American Sleep Society. Primary sleep disorders are separated from those due to medical or psychiatric conditions. Primary sleep disorders are further subdivided into:
Dyssomnias are further subdivided into intrinsic, extrinsic, and circadian rhythm sleep disorders. An abbreviated version of the International Classification of Sleep Disorders-2 is presented in Box 66-1.
Box 66-1 The International Classification of Sleep Disorders
Insomnia (Examples)
 Idiopathic insomnia, paradoxical insomnia, adjustment insomnia, psychophysiologic insomnia, insomnia due to mental disorder, inadequate sleep hygiene, insomnia due to substance abuse, insomnia due to medical condition, behavioral insomnia of childhood – sleep-onset association type, behavioral insomnia of childhood – limit-setting type
Idiopathic insomnia, paradoxical insomnia, adjustment insomnia, psychophysiologic insomnia, insomnia due to mental disorder, inadequate sleep hygiene, insomnia due to substance abuse, insomnia due to medical condition, behavioral insomnia of childhood – sleep-onset association type, behavioral insomnia of childhood – limit-setting typeSleep-Related Breathing Disorders (Examples)
 Primary central sleep apnea, Cheyne–Stokes breathing, high-altitude periodic breathing, central sleep apnea due to medical condition, obstructive sleep apnea – adult, obstructive sleep apnea – pediatric, primary apnea – infancy, sleep-related hypoventilation, sleep-related hypoventilation/hypoxemia due to neuromuscular and chest wall disorders
Primary central sleep apnea, Cheyne–Stokes breathing, high-altitude periodic breathing, central sleep apnea due to medical condition, obstructive sleep apnea – adult, obstructive sleep apnea – pediatric, primary apnea – infancy, sleep-related hypoventilation, sleep-related hypoventilation/hypoxemia due to neuromuscular and chest wall disordersHypersomnia of Central Origin not due to a Circadian Rhythm Sleep Disorder (Examples)
 Narcolepsy with cataplexy, narcolepsy without cataplexy, narcolepsy due to medical condition without cataplexy, narcolepsy unspecified, recurrent hypersomnia (including Kleine–Levin syndrome and menstrual-related hypersomnia), idiopathic hypersomnia with long sleep, idiopathic hypersomnia without long sleep, hypersomnia due to medical condition, hypersomnia due to drug or substance, sleep-related breathing, behaviorally induced insufficient sleep syndrome disorder, or other cause of disturbed nocturnal sleep
Narcolepsy with cataplexy, narcolepsy without cataplexy, narcolepsy due to medical condition without cataplexy, narcolepsy unspecified, recurrent hypersomnia (including Kleine–Levin syndrome and menstrual-related hypersomnia), idiopathic hypersomnia with long sleep, idiopathic hypersomnia without long sleep, hypersomnia due to medical condition, hypersomnia due to drug or substance, sleep-related breathing, behaviorally induced insufficient sleep syndrome disorder, or other cause of disturbed nocturnal sleep



(From American Sleep Disorders Association, International Classification of Sleep Disorders, 2nd edn, pocket version: Diagnostic and Coding Manual. Westchester, Illinois: American Academy of Sleep Medicine, 2006.)
Assessment of Sleep–Wake Complaints
Sleep History










The Pediatric Daytime Sleepiness Scale (PDSS) is a simple, validated questionnaire that can be administered to children in the 11–14-year age group [Drake et al., 2003]. It has eight items, each rated on a 0–4 scale. It provides a numerical score for sleepiness; the 50th percentile score on the PDSS is 16, the 75th percentile is 20, and the 90th percentile is 23. Participants who reported low school achievement, high absenteeism, low school enjoyment, low total sleep time and frequent illnesses all had higher levels of sleepiness as measured by this scale [Drake et al., 2003]. Another survey tool that is commonly used in clinical practice is the Children’s Sleep Habits Questionnaire. It is a 45-item, validated questionnaire that is completed by parents of 4–11-year-olds [Owens et al., 2000]. The questions pertain to sleep–wake function in the preceding 2 weeks, such as “the child sleeps too little” or “the child suddenly falls asleep in the middle of active behavior.” The items represent several domains that present as sleep complaints, such as bedtime resistance, sleep-onset delay, sleep duration, sleep anxiety, night awakenings, parasomnias, breathing disturbance, and daytime sleepiness. Responses are rated as rarely (occurring 0–1 time/week; 1 point), sometimes (occurring 2–4 nights/week; 2 points), or usually (occurring 5–7 nights/week; 3 points). Scores of 41 or greater correlate with presence of a sleep disorder. The internal consistency estimate for this questionnaire in a community (nonclinic sample) of 4–10-year-olds is 0.36–0.70. The test-retest reliability over a 2-week period is 0.62–0.79.
Sleep-Related Examination
Height, weight, and body mass index are recorded because obstructive sleep apnea may be associated with poor weight gain during infancy, and with obesity during adolesence [Arens et al., 2010]. The blood pressure should be measured because long-standing and severe obstructive sleep apnea (OSA) can be associated with hypertension. OSA patients may exhibit craniofacial abnormalities such as micrognathia, dental malocclusion, macroglossia, myopathic face and midface hypoplasia, deviated nasal septum, swollen inferior turbinates, tonsillar hypertrophy and mouth breathing [Brooks, 2002; Hoban and Chervin, 2007]. Consultation with a pediatric otolaryngologist may be required to exclude adenoidal hypertrophy. Inattentiveness, irritability, and mood swings may be clues to daytime sleepiness. Obstructive sleep apnea related to brainstem abnormalities like the Chiari type I or II malformations [Gosalakkal, 2008] can lead to hoarseness of voice, decreased gag reflex, and changes in the amplitude of the jaw jerk relative to that of other tendon reflexes. Neuromuscular disorders, like myotonic dystrophy, may be associated with chronic obstructive hypoventilation from a combination of palatal muscle weakness, high-arched palate, and diminished chest wall–abdominal excursion [Givan, 2002; Misuri et al., 2000]. The parent–child interaction should be observed not only for clues indicating parental anxiety and reluctance to set limits on inappropriate behaviors that perpetuate insomnia in toddlers, but also for subtle clues indicating a child maltreatment syndrome. Home videos, if available, may be invaluable in the assessment of restless legs syndrome, parasomnias, and nocturnal seizures.
Nocturnal Polysomnography
This procedure involves the monitoring of multiple physiologic parameters in sleep. It is useful in the evaluation of intrinsic sleep disorders, such as narcolepsy, obstructive sleep apnea, nocturnal spells, and periodic limb movement disorder. It may not be indicated for diagnosing obstructive sleep apnea secondary to severe adenotonsillar enlargement, which can be easily recognized on the basis of clinical findings combined with severe oxygen desaturation on simple overnight oximetry in the home environment. The nocturnal polysomnogram usually consists of simultaneous monitoring of 2–4 channels of the EEG, eye movements, chin and leg electromyogram, nasal pressure, thoracic and abdominal respiratory effort, electrocardiogram, and oxygen saturation [Kotagal and Goulding, 1996; Kotagal and Herold, 2002; Sheldon, 2007]. Patients with Down syndrome, neuromuscular disorders, and obesity may exhibit hypoventilation that is characterized by shallow chest and abdominal wall movement, with resultant CO2 retention. It is therefore important to measure end-tidal CO2 levels concurrently in these patients. Esophageal pH can be monitored simultaneously when there is a suspicion of gastroesophageal reflux. Criteria for the scoring of sleep and sleep-related events such as apnea, arousals, and periodic limb movements have been recently revised [Iber et al., 2007].
In patients with obvious upper airway obstruction secondary to obesity or neuromuscular disorders like myotonic dystrophy, a therapeutic trial of positive pressure airway breathing can be attempted midway during the course of the sleep recording. A full 16- to 20-channel EEG montage is recommended when parasomnias and seizures are both in the differential diagnosis. Simultaneous video monitoring is standard for all nocturnal polysomnograms. Normative data for common physiologic variables are listed in Table 66-1 [Marcus and Loughlin, 1996]. There is insufficient evidence regarding the utility of ambulatory, in-home polysomnograms, especially in the preschool age group; thus, traditional sleep laboratory monitoring remains the gold standard in pediatric sleep medicine.
| Parameter | Average | Standard deviation |
|---|---|---|
| Total sleep time | 472 min | 42 min |
| Sleep efficiency | 90% | 7 |
| Sleep latency | 24.1 min | 25.6 min |
| REM latency | 87.8 min | 41.2 min |
| REM sleep (% of TST) | 21.1% | 4.9 |
| Stage N3 sleep (% of TST) | 26.3% | 4.8 |
| Stage N2 sleep (% of TST) | 36% | 6.6 |
| Stage N1 sleep (% of TST) | 5.2% | 2 |
| Apnea hypopnea index (events per hour of sleep) | 0.9 | 0.7 |
| End-tidal CO2 > 50 mm (% time) | 25% or less | |
| Oxygen saturation (% time < 90%) | 0.04% | 0.18 |
| Oxygen desaturations > 4% (number/hour of TST) | 0.4 | 0.78 |
REM, rapid eye movement; TST, total sleep time.
(From Montgomery-Downs HE et al. Polysomnographic characteristics in normal preschool and early school aged children. Pediatrics 2006;117:741.)
Multiple Sleep Latency Test
The Multiple Sleep Latency Test (MSLT) assesses how quickly one is able to fall asleep during the daytime and whether transition from wakefulness is into NREM sleep or into REM sleep. The MSLT can reliably detect sleepiness under clinical and experimental conditions [Littner et al., 2005]. The lower age limit at which one can utilize this test is about 6–7 years. Application of the MSLT to children younger than 6 years is unhelpful because healthy pre-school age children tend to take physiologic daytime naps. In order to be able to derive valid conclusions, the MSLT must be preceded the night before by a polysomnogram in which the total sleep time approximates physiologic sleep. The MSLT consists of the provision of four or five daytime nap opportunities at 2-hour intervals: e.g., at 10:00, 12:00, 14:00, and 16:00 hours. The EEG, chin electromyogram, and eye movements are monitored during each nap opportunity. The patient should be dressed in street clothes. The attending parent or guardian should be available to prevent the child from dozing off involuntarily in between the scheduled nap times. At each planned nap opportunity, the lights are turned off and the patient is asked to try to sleep. The time from “lights out” to sleep onset is measured, and represents the sleep latency. A mean sleep latency is also derived for the four naps. Normative values for the mean sleep latency have been established. The nap opportunity is terminated either 15 minutes after sleep onset, or if the patient does not fall asleep, at 20 minutes after “lights out.” The mean sleep latency decreases inversely with an increase in the Tanner stage of sexual development, and ranges between 12 and 18 minutes [Carskadon, 1982]. A mean sleep latency of less than 5 minutes indicates severe daytime sleepiness; a value between 5 and 10 minutes indicates moderate daytime sleepiness. A urine drug screen is obtained in between the naps if illicit drug-seeking behavior is suspected. The occurrence of REM sleep within 15 minutes of sleep onset constitutes a sleep-onset rapid eye movement period (SOREMP). The presence of SOREMPs on two more MSLT nap opportunities in conjunction with a shortened mean sleep latency of less than 5 minutes is highly suggestive of narcolepsy.
In adults, a mean sleep latency of less than 5 minutes in association with two or more SOREMPs is 70 percent sensitive and 97 percent specific for the diagnosis of narcolepsy [Aldrich et al., 1997]. Comparable data on the sensitivity and specificity are not available for children and adolescents. A study by Gozal et al. [2001] suggests that, in prepubertal children, the normal mean sleep latency is 23.7 minutes, plus or minus 3.1 minutes. Palm et al. [1989] also found a mean sleep latency of 26.4 minutes plus or minus 2.8 minutes in 18 prepubertal children. These data suggest that the normal mean sleep latency in children may actually be higher than previously reported, and that normative data for the multiple sleep latency test in childhood might perhaps need revision. One of the merits of the multiple sleep latency test is that it provides reliable and quantitative information about the propensity for sleepiness. It has been validated as a measure of daytime sleepiness following episodes of sleep loss [Rosenthal et al., 1993], sleep disruption [Stepanski et al., 1987], and hypnotic drug and alcohol abuse [Billiard et al., 1987; Papineau et al., 1998]. The effects of treatment of daytime sleepiness with stimulants cannot be reliably measured, however. In addition, although one can control ambient noise and light that might affect sleep during the testing process, one cannot control for internal factors, like anxiety and apprehension, that also affect sleep propensity.
Maintenance of Wakefulness Test
The Maintenance of Wakefulness Test (MWT) is the mirror image opposite to the MSLT; one measures the ability to stay awake in a darkened, quiet environment during the daytime while the patient is seated in a semireclining position [Littner et al., 2005]. EEG, eye movements, and chin electromyography are monitored in a manner identical to that in the MSLT. The patient is provided four or five nap opportunities. The duration of each session is 40 minutes. The average mean sleep latency for normal adults is 35.2 minutes. Normative values have not been established for children. The test helps assess the effect of stimulant medication treatment on daytime sleepiness [Mitler et al., 2000].
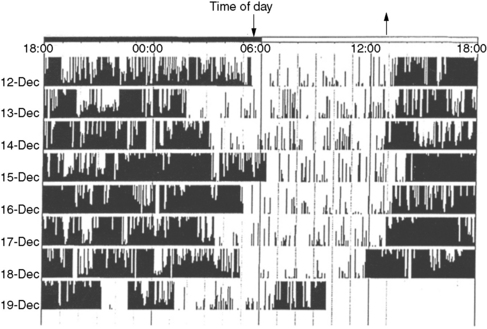
Actigraphy
This technique involves the recording and storing of skeletal muscle activity continuously for 1–2 weeks in the home environment, generally from the nondominant forearm, using a wristwatch-shaped microcomputer device that measures linear acceleration and translates it into a numeric and graphic representation. This numeric representation is sampled frequently – that is, every 0.1 second – and aggregated at a constant interval or epoch length [Acebo and LeBourgeois, 2006]. The device captures signals during periods of muscle activity (generally correlating with wakefulness) and periods of no muscle activity (generally correlating with sleep; Figure 66-2). There is close correlation with polysomnographically determined total sleep time, sleep latency, and sleep efficiency [total sleep time or total time in bed ×100; Kothare and Kaleyias, 2008; van de Water et al., 2010]. Wrist actigraphy should be combined with 1–2 weeks of “sleep logs” that document specific wake-up and sleep-onset times, daytime naps, and so on. Wrist actigraphy is useful in the study of insomnia and circadian rhythm disorders like the delayed sleep phase syndrome [Kothare and Kaleyias, 2008]. In the latter instance, it will depict sleep onset late at night or in the early morning hours and uninterrupted sleep thereafter, with final awakening late in the morning or early afternoon (see Figure 66-1). In patients being evaluated for suspected narcolepsy, 2 weeks of actigraphy before nocturnal polysomnography and the MSLT help exclude the possibility of sleepiness resulting from a circadian rhythm disorder or insufficient sleep at night.
Common Childhood Sleep Disorders
Sleep-Related Breathing Disturbances
In increasing level of severity, the spectrum of sleep-disordered breathing in childhood ranges from snoring without sleep disruption (primary snoring) to the upper airway resistance syndrome (snoring that disrupts sleep continuity but without associated apnea or oxygen desaturation) to classic obstructive sleep apnea and, finally, obstructive hypoventilation (apnea, oxygen desaturation, plus hypercarbia). Between 10 and 12 percent of children snore on a habitual basis [Corbo et al., 2001; O’Brien et al., 2003]. The snoring sound is the result of vibration of the soft palate during inspiration due to narrowing of the oropharynx. Although some guidelines [American Academy of Pediatrics, 2002] suggest that primary snoring may be a “benign condition that does not warrant any specific therapy,” a study of 87 children of 5–7 years of age [O’Brien et al., 2004] found that, compared with age-matched nonsnoring children, those with primary snoring performed worse on neuropsychologic measures of attention and had more social problems and anxious or depressive symptoms. Community-based studies have determined the prevalence of childhood obstructive sleep apnea at 1.1–2.9 percent [Ali et al., 1993; Brunetti et al., 2001].
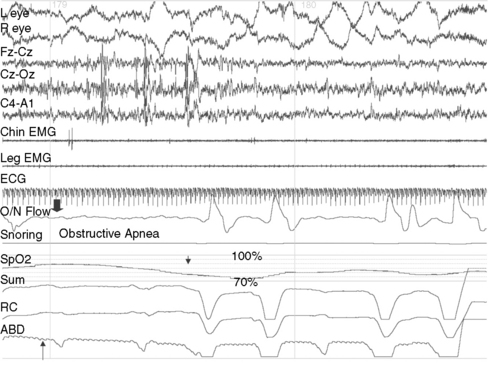
OSA is characterized by partial or complete upper airway occlusion, with impaired air exchange despite persistence of thoracic and abdominal respiratory effort. This generally occurs in association with increased resistance to inspiration and transient oxygen desaturation of 3–4 percent (Figure 66-3). In some instances, there is an additional component of hypoventilation due to shallow abdominal and chest wall motion, which leads to hypercarbia. The most common etiologic factors for childhood OSA are adenotonsillar hypertrophy, craniofacial anomalies like micrognathia or maxillary hypoplasia, neuromuscular disorders such as myotonic dystrophy or congenital nonprogressive myopathies, and obesity [Lumeng and Chervin, 2008]. Repetitive occlusion of the upper airway during sleep, with resultant oxygen desaturation, provokes cortical arousals and suppression of REM and N3 sleep. Nocturnal symptoms of childhood obstructive sleep apnea include habitual snoring, restless sleep with snort arousals, bed wetting, excessive sweating, mouth breathing, choking sounds, and parasomnias such as confusional arousals and sleepwalking. Parental reports of snoring that is interrupted by silent pauses, which then terminate with snorting sounds, are characteristic of OSA. A metabolic syndrome, characterized by insulin resistance, hyperglycemia, hypertension, dyslipidemia, abdominal obesity, and proinflammatory and prothrombotic states, may develop as a consequence of OSA [Arens et al., 2010]. Daytime symptoms of OSA include inattentiveness, impaired academic performance, hyperactivity, and sleepiness [Chervin et al., 2002; Gozal, 2008]. The upper airway resistance syndrome is a form of upper airway obstruction in which no frank apneas or oxygen desaturation are observed during sleep, but the airway narrowing leads to recurrent arousals, fragmented sleep, and daytime sleepiness [Guilleminault et al., 1996]. Some patients exhibit subtle posterior displacement of the tongue, narrow nostrils, or a high-arched palate, but others might not have any craniofacial anomalies. The nocturnal polysomnogram may appear superficially normal, with the exception of snoring and increased EEG arousals of 3 or more seconds (normally less than 10–12 per hour of sleep). Simultaneously obtained intraluminal pressures from an esophageal balloon demonstrate a marked increase in the intrathoracic negative pressure during the upper airway resistance syndrome episodes. Nocturnal polysomnography is not needed to confirm the diagnosis in patients who already manifest the classic symptoms of OSA with marked tonsillar hypertrophy; one night of oximetry in the home environment, which documents recurrent oxygen desaturation, is sufficient for establishing the diagnosis in these patients [Brouilette et al., 2000]. Nocturnal polysomnography is, however, indicated when the diagnosis is uncertain or less obvious. It is also indicated when OSA is suspected in the context of multiple neurologic handicaps, such as in Down syndrome or cerebral palsy, and when the patient needs to be considered for a nonsurgical treatment such as a continuous positive airway pressure device. OSA secondary to adenotonsillar hypertrophy may respond in a modestly favorable manner to adenotonsillectomy, but patients younger than 3 years old and those with severe obstructive sleep apnea should be monitored postoperatively in the intensive care unit for respiratory compromise due to postoperative upper airway edema. Patients should generally be re-evaluated clinically and with polysomnography 2–3 months after adenotonsillectomy. Positive airway pressure breathing devices should be considered if there is significant, residual OSA. Weight-reduction measures are indicated in obese patients. Orthodontic consultation and the use of oral appliances during sleep is indicated in those with retrognathia and tongue prolapse [Rondeau, 1998]. Rapid maxillary distraction is a nonsurgical technique that is used in OSA with associated high-arched palate and consequent narrowing of the nasal passages [Abad and Guillemiault, 2009].
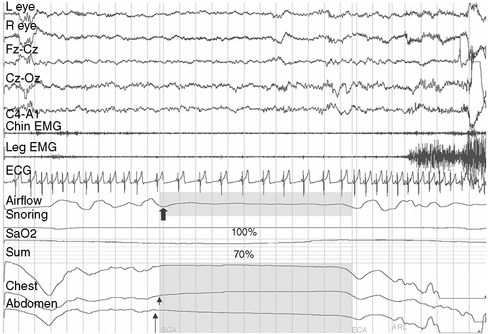
Central Hypoventilation Syndromes
Defective automatic control of breathing during sleep as a result of brainstem dysfunction is characteristic of this category of disorders, which presents in infancy or childhood. Developmental malformations of the brainstem are common, but may be visible only on microscopic examination. Head injury, bulbar poliomyelitis, syringobulbia, Chiari types I and II malformation [Zolty et al., 2000], and inborn errors of metabolism (e.g., Leigh’s syndrome) are some underlying conditions. Polysomnography may reveal central sleep apnea, oxygen desaturation, shallow respiratory effort, and elevated levels of end tidal carbon dioxide (Figure 66-4). Except for surgical decompression in Chiari malformation and syringobulbia-associated hypoventilation, the management is similar to that of primary congenital central alveolar hypoventilation, which is discussed below.
Congenital central alveolar hypoventilation is a disorder in which there is no obvious structural or biochemical etiology for the defective central control of breathing during sleep [Gozal, 2004; Gozal and Harper, 1999]. Onset of symptoms is during infancy or early childhood. The respiratory rate and depth are initially normal during wakefulness, but shallow breathing (hypoventilation), hypercarbia, and oxygen desaturation appear during sleep. The dysfunction is worse in NREM sleep, as compared to REM sleep. Ventilatory challenge with inhalation of a mixture of 5 percent CO2 and 95 percent O2 fails to evoke the physiologic, three- to fivefold increase in minute volume. Common sites of neuronal loss and gliosis include the arcuate nucleus in the medulla, the ventrolateral nucleus of the tractus solitarius, nucleus ambiguus, nucleus retroambigualis, the chemosensitive ventral medullary surface, and the nucleus parabrachialis in the dorsolateral pons. Between 15 and 20 percent of congenital central alveolar hypoventilation patients have coexisting Hirschsprung’s disease or neural crest tumors, such as neuroblastoma or ganglioneuroma [Roshkow et al., 1988; Swaminathan et al., 1989]. It has been hypothesized that congenital central alveolar hypoventilation is a neural crest disorder because, during fetal development, brainstem neurons that regulate chemosensitivity are derived from the neural crest. The proto-oncogene, receptor tyrosine kinase, is involved in this process [Bolk et al., 1996]. Mutations in the PHOX2B gene have been identified in over 90 percent of patients with central hypoventilation syndrome [Traochet et al., 2005]. They generally consist of expansions of the polyalanine tail in this gene, though expansions of non-polyalanine repeats have also been implicated [Weese-Mayer et al., 2010]. Tumors of the neural crest manifest increased prevalence in those with non-polyalanine expansions. Mutations in the PHOX2B gene are transmitted in an autosomal-dominant manner. Mosaicism occurs in 5–10 percent of the parents, which emphasizes the importance of molecular testing in parents of the affected child [Weese-Mayer et al., 2010]. A knockout mouse model has been developed, which also have an inability to respond to hypercarbia, depletion of neurons in the retrotrapezoid nucleus and the parafacial respiratory group of neurons [Amiel et al., 2009]. There is no definitive and satisfactory treatment for congenital central alveolar hypoventilation, though acetazolamide and theophylline enhance chemoreceptivity of the brainstem respiratory neurons to a modest degree. Home ventilation via tracheostomy and diaphragmatic pacing are other therapeutic modalities. Patients may die during infancy or childhood. It is recommended that patients with congenital central hypoventilation syndrome undergo an annual hospital admission for a comprehensive, multidisciplinary evaluation that includes 72-hour Holter monitoring, echocardiogram, evaluation of autonomic dysregulation affecting other organ systems, and imaging for neural crest tumors when appropriate [Weese-Mayer et al., 2010].
Narcolepsy
Chronic daytime sleepiness, hypnagogic hallucinations (vivid dreams at sleep onset), sleep-onset paralysis, cataplexy (sudden loss of skeletal muscle tone in response to emotional triggers like laughter, fright, or surprise), and fragmented night sleep are the characteristic clinical features of narcolepsy. The incidence of narcolepsy in the United States is 1.37 per 100,000 persons per year (1.72 for men and 1.05 for women). It is highest in the second decade, followed by a gradual decline. The prevalence is approximately 56 per 100,000 persons [Silber et al., 2002]. A meta-analysis of 235 pediatric cases derived from three studies [Challamel et al., 1994] found that 34 percent of all narcolepsy subjects experienced onset of symptoms before the age of 15 years, 16 percent before the age of 10 years, and 4.5 percent before age 5 years (Figure 66-5). Although the age of onset is generally in the latter half of the first decade or the second decade, rare cases with onset of extreme sleepiness and cataplexy during infancy have also been reported [Nevsimalova et al., 1986; Sharp and D’Cruz, 2001]. Childhood daytime sleepiness may be overlooked by parents, schoolteachers, and physicians alike. Sleepy children may be mistaken for being “lazy.” They may exhibit mood swings and inattentiveness as a consequence of sleepiness-related prefontal cortical dysfunction. Cataplexy is present in about two-thirds of patients. The cataplectic muscle weakness generally lasts for a few seconds to minutes, and is associated with muscle atonia, absence of muscle stretch reflexes, and full preservation of consciousness. Because cataplexy can be subtle, the examiner might need to ask leading questions about sudden muscle weakness in the lower extremities, neck, or trunk in response to laughter, fright, excitement, or anger. Cataplexy is associated with abrupt hyperpolarization of the spinal alpha motor neurons. Fragmentation of night sleep is also common in narcolepsy. Children younger than 7 or 8 years may not be able to provide a reliable history of cataplexy, hypnagogic hallucinations, or sleep paralysis. Patients with narcolepsy exhibit less circadian clock-dependent alertness during the daytime and also less circadian clock-dependent sleepiness at night [Broughton et al., 1998]. Secondary narcolepsy is a rare entity, but may develop as a sequel to closed head injury and in patients with deep midline brain tumors, lymphomas, and encephalitis [Autret et al., 1994]. Obesity and precocious puberty may accompany the onset of narcolepsy-cataplexy in childhood [Perriol et al., 2010; Kotagal et al., 2004; Plazzi et al., 2006].
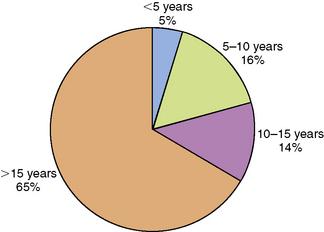
Fig. 66-5 Graph depicting the age of onset of narcolepsy.
(Data derived from Challamel MJ et al. Narcolepsy in children. Sleep 1994;17S:17.)
Stores et al. [2006] assessed the psychosocial difficulties of 42 children with narcolepsy (mean age 12.4, range 7.3–17.9), 18 subjects with excessive daytime sleepiness unrelated to narcolepsy (EDS; mean age 14.2, range 5.1–18.8), and 23 unaffected controls (mean age 11.3, range 6–16.8). They found significantly higher scores on the Strengths and Difficulties Questionnaire in the narcolepsy and EDS groups. The domains of this questionnaire included prosocial, peer problems, hyperactivity, conduct problems, emotional problems, and adverse impact on the family. As compared to healthy controls, both the narcolepsy and the EDS groups scored higher on the Child Depression Inventory. Children with narcolepsy and EDS also had more absences from school (means 6.4 and 5.3 days, respectively), as compared to controls (mean 1.3), and displayed more problems on a composite educational difficulties score, suggesting that sleepiness in general, rather than narcolepsy per se adversely influences the psychosocial and emotional health of the patients.
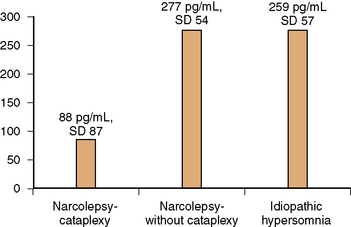
The presence of the histocompatibility antigen DQB1*0602 in close to 100 percent of persons with narcolepsy, as compared with a 12–32 percent prevalence in the general population, indicates genetic susceptibility, which by itself is insufficient to precipitate the clinical syndrome. Monozygotic twins have been reported to remain discordant for narcolepsy [Langdon et al., 1986; Mignot et al., 1999, 2001]. In genetically susceptible individuals, acquired life stresses like minor head injury, systemic illnesses such as infectious mononucleosis, and bereavement may play a role in triggering the disorder, and have been reported in close to two-thirds of subjects – “the two-hit hypothesis” [Billiard et al., 1986]. The key pathophysiologic event in human narcolepsy-cataplexy is hypocretin ligand deficiency [Nishino et al., 2000, 2001]. Hypocretin (orexin) is a peptide that is produced by neurons of the dorsolateral hypothalamus. Hypocretins 1 and 2 (synonymous with orexins A and B) are peptides that are synthesized from preprohypocretin. They have corresponding receptors. Whereas the hypocretin type 1 receptor binds only to hypocretin1, the hypocretin type 2 receptor binds to both type 1 and 2 ligands [Hungs et al., 2001]. Hypocretin-producing neurons have widespread projections to the forebrain and brainstem. Hypocretin promotes alertness and increases motor activity and basal metabolic rate [John et al., 2000]. Of significance is the postmortem examination finding in human narcolepsy of an 85–95 percent reduction in the number of hypocretin-containing neurons of the hypothalamus, whereas melanin-concentrating hormone neurons that are intermingled with hypocretin neurons remain unaffected, thus suggesting a targeted neurodegenerative process [Thannickal et al., 2000]. It is hypothesized that an immune-mediated degenerative process affecting the hypocretin-producing cells of the hypothalamus (perhaps linked to HLA DQB1*0602 by mechanisms yet unknown) provokes a decrease in forebrain noradrenergic activation, which in turn decreases alertness. A corresponding decrease of noradrenergic activity in the brainstem leads to disinhibition of brainstem cholinergic systems, thus triggering cataplexy and other phenomena of REM sleep, such as hypnagogic hallucinations and sleep paralysis. The decrease in hypocretin-1 ligand levels that is characteristic of human narcolepsy-cataplexy is also reflected in the cerebrospinal fluid (CSF). Using a radioimmunoassay, Nishino et al. [2000, 2001] found that the mean CSF level of hypocretin-1 in healthy controls was 280.3 ± 33.0 pg/mL; in neurologic controls it was 260.5 ± 37.1 pg/mL; and in those with narcolepsy-cataplexy, hypocretin-1 was either undetectable or less than 100 pg/mL. Low to absent CSF hypocretin-1 levels were found in 32 of 38 narcolepsy-cataplexy patients, who were all also HLA DQB1*0602-positive. Narcolepsy patients who were HLA DQB1*0602 antigen-negative tended to have normal to high CSF hypocretin-1 levels. In another study of narcolepsy with cataplexy, narcolepsy without cataplexy, and idiopathic hypersomnia, Kanbayashi et al. [2002] found nine narcolepsy-cataplexy subjects who were HLA DQB1*0602-positive but CSF hypocretin-1-deficient. In contrast, narcolepsy without cataplexy and idiopathic hypersomnia patients exhibited normal CSF hypocretin levels (Figure 66-6). The hypocretin assay may be most useful when an HLA DQB1*0602-positive patient with suspected narcolepsy-cataplexy is already receiving central nervous system stimulants at the time of initial presentation, and in whom the discontinuation of these medications for the purpose of obtaining polysomnography and an MSLT is inconvenient or impractical.
Management of narcolepsy requires a combination of lifestyle changes and pharmacotherapy. A planned daytime nap of 20–30 minutes at school and another one in the afternoon upon return home may enhance alertness. The patient should observe regular sleep onset and morning wake-up times, avoid alcohol, and exercise regularly. To minimize the risk of accidents, the patient should avoid sharp, moving objects. Daytime sleepiness is treated pharmacologically with modafinil (100–400 mg/day in two divided doses), or regular or extended-release preparations of methylphenidate (20–60 mg/day in two divided doses) and dextroamphetamine (10–30 mg/day in two divided doses) [Littner et al., 2001]. Cataplexy is treated using clomipramine (25 mg at bedtime), protryptiline (2.5–5 mg/day in two divided doses), or sodium oxybate (gamma-hydroxybutyrate). Sodium oxybate is administered in two divided doses or at night, and seems to work by stabilizing nocturnal sleep architecture. There are insufficient data on its use in childhood. Emotional and behavioral problems that commonly accompany childhood narcolepsy may require the addition of fluoxetine (10–30 mg every morning) or sertraline (25–50 mg twice daily) and supportive psychotherapy. Teenagers should be counseled against driving if they have uncontrolled sleepiness. They should also avoid work and social situations in which they could endanger themselves or others. There have been anecdotal reports of improvement in narcolepsy-cataplexy following the infusion of intravenous immunoglobulin G [Dauvilliers, 2009], but randomized controlled studies have not been carried out. The Narcolepsy Network (www.narcolepsynetwork.org) is a private, nonprofit resource for patients, families, and health professionals. There is also a National Narcolepsy Registry, more information on which can be found at www.ninds.nih.gov/disorders/narcolepsy. Experimental treatment efforts are focused on hypocretin replacement therapy, gene therapy, and cell transplantation [Ritchie et al., 2010].
Idiopathic Hypersomnia
Idiopathic hypersomnia is associated with a complaint of excessive daytime sleepiness, occurring daily for at least 3 months. Nocturnal polysomnography should have excluded other conditions leading to hypersomnia. In contrast with the mean sleep latency on the MSLT of less than 5 minutes that is typical of narcolepsy, the mean sleep latency in idiopathic hypersomnia is generally in the 5- to 10-minute range. Two or more SOREMPs that are typically seen on the MSLT in patients with narcolepsy are also not present. Patients do not exhibit cataplexy, hypnagogic hallucinations, or sleep paralysis [Frenette and Kushida, 2009]. Two subtypes of idiopathic hypersomnia have been identified. In idiopathic hypersomnia with long sleep, there is prolonged nocturnal sleep lasting 10 or more hours. In idiopathic hypersomnia without long sleep, the major nocturnal sleep period is 6–10 hours in length. In a review of 42 patients evaluated at the University of Michigan over a 10-year period, Basetti and Aldrich [1997] observed that idiopathic hypersomnia began at a mean age of 19 ± 8 years (range 6–43). Almost half of the subjects described restless sleep with frequent arousals. Habitual dreaming was present in about 40 percent of the subjects. They mention that there is substantial clinical overlap between narcolepsy and idiopathic hypersomnia. In a subset of children, idiopathic hypersomnia may represent a transitional phase en route to the development of classic narcolepsy [Kotagal and Swink, 1996].
Restless Legs Syndrome
Restless legs syndrome is an autosomal-dominant, sensorimotor disorder in which the subject complains of a peculiar “creepy or crawling” feeling in the extremities [Picchietti and Picchietti, 2008]. The discomfort appears in the evening and night-time hours, is exacerbated by immobility, and is momentarily relieved by movement of the limb [Allen et al., 2003].There is also an urge to move the limbs. This discomfort interferes with sleep initiation and maintenance, and may be accompanied by daytime fatigue, inattentiveness, or sleepiness. A large population-based survey of over 10,000 families in the United Kingdom and the United States found a prevalence of definite restless legs syndrome in 1.9 percent of 8–11-year-olds and in 2 percent of 12–17-year-olds [Picchietti et al., 2007]. Childhood restless legs syndrome may be synonymous with “growing pains” in some children [Walters, 2002]. On nocturnal polysomnography, the patient may or may not demonstrate periodic limb movements, which are defined as a series of four or more rhythmic, electromyographically recorded movements of the legs lasting 0.5–10 seconds that occur 5–90 seconds apart, generally during stages N1 or N2 of sleep. The favorable response to dopamine receptor agonists like pramipexole and carbidopa-levodopa suggests that central nervous system dopamine deficiency is a pathophysiologic feature. There may be an associated systemic iron deficiency in the form of low levels of serum ferritin [Kotagal and Silber, 2004] because iron is a co-factor for tyrosine hydroxylase, which is essential in the synthesis of dopamine. There also appears to be an association between restless legs syndrome and attention-deficit disorder [Chervin et al., 2002; Picchietti et al., 1999]; in a community-based questionnaire survey of 866 children aged 2–13.9 years, Chervin et al. [2002] found an odds ratio for significant hyperactivity and restless legs of 1:9. Besides dopamine agonists, oral iron, clonazepam, gabapentin, gabapentin enacarbil [Bogan et al., 2010], and pregabalin [Allen et al., 2010] have also been used to treat restless legs syndrome, but there has been no randomized controlled trial in childhood to determine efficacy definitely.
Periodic Hypersomnia (Kleine–Levin Syndrome)
Periodic hypersomnia is generally seen in adolescents, with a male predominance. Patients develop 1- to 2-week periods of hypersomnolence, characterized by sleeping 18–20 hours per day, cognitive and mood disturbances in association with compulsive hyperphagia, and hypersexual behavior, with an intervening 2–4 months of normal alertness and behavior [Brown and Billiard, 1995; Frenette and Kushida, 2009]. Incomplete forms of the syndrome have also been recognized. The hyperphagia may be in the form of binge eating, and can actually be associated with a 2–5 kg increase in body weight. Nocturnal polysomnography during the sleepy periods exhibits decreased sleep efficiency, shortened latency to REM sleep, and decreased percentage of time spent in N3 sleep. The MSLT reveals moderately shortened mean sleep latency in the 5–10-minute range, but lacks the two or more SOREMPs that are typically seen in narcolepsy. The episodic hypersomnia gradually diminishes, ultimately resolving completely over 2–5 years or evolving into classic depression, thus bringing up the issue of whether the disorder is a variant of depression. A disturbance of hypothalamic function has been hypothesized but not established. The association of Kleine–Levin syndrome with histocompatibility antigen DQB1*0201, and the occasional precipitation after systemic infections, as well as the relapsing and remitting nature, are suspicious for an autoimmune etiology [Dauvilliers et al., 2002]. There is no satisfactory treatment, although lithium has been reported to be effective in a case report [Poppe et al., 2003]. Modafinil may reduce the duration of the symptomatic episodes [Huang et al., 2010].
Delayed Sleep Phase Disorder
Circadian rhythm disorders are characterized by alteration in the timing of sleep onset and offset relative to the societal norms. Delayed sleep phase disorder [DSPD] is the most common circadian rhythm sleep disorder. The basis for all circadian rhythm disorders is abnormal functioning of a molecular feedback loop between CLOCK and BMAL1 (transcription regulating proteins) and the expression of Period genes Per1, Per2, Per3, and Cryptochrome genes Cry1 and Cry2 [Wulff et al., 2009]. DSPD was described initially in a medical student who was unable to wake up in time to attend his morning classes [Weitzman et al., 1981]. Despite hypnotic use, the patient was unable to fall asleep before the early morning hours and was mistaken initially for having insomnia. Sleep in the laboratory was, however, quantitatively and qualitatively normal. The delay in the timing of occurrence of the sleeping phase of the 24-hour sleep–wake cycle was due to a constitutional inability to prepone sleep, which in turn is secondary to altered function of the circadian timekeeper, i.e., the suprachiasmatic nucleus. The disorder typically has onset in adolescence, with a male predominance. The increased frequency of HLA DR1 and the occasional familial clustering of delayed sleep phase syndrome suggest a genetic predisposition [Garcia et al., 2001]. The condition must, however, be differentiated from school avoidance seen in adolescents with delinquent and antisocial behavior, because these individuals can fall asleep at an earlier hour at night in the controlled sleep laboratory setting. Maintenance of sleep logs and wrist actigraphy for 1–2 weeks is helpful in establishing the diagnosis of delayed sleep phase syndrome (see Figure 66-2). “Bright light” therapy is helpful in advancing the sleep-onset time to an earlier hour [Cole et al., 2002]. It consists of the provision of 2700–10,000 lux of bright light via a “light box” for 20–30 minutes immediately upon awakening in the morning. The light box is kept at a distance of 18–24 inches from the face. The phototherapy leads to a gradual advancement (shifting back) of the sleep-onset time at night. Bright light therapy may be combined with melatonin that is administered about 5–5.5 hours before the required bedtime in a dose of 0.5–1 mg. Another therapeutic option is one of progressively delaying bedtime by 3–4 hours per day until it becomes synchronized with socially acceptable sleep–wake time (chronotherapy), and then adhering to this schedule. Over time, however, all DSPD patients remain at risk for drifting back to progressively later and later bedtimes. Daytime stimulants, such as modafinil (100–400 mg/day in two divided doses), may improve the level of daytime alertness. The physician may also need to write a letter to the school, requesting a mid-morning school start time on medical grounds.
The Relationship between Sleep and Epilepsy
The onset of sleep may be associated with increased interictal spiking, as well as with an increased propensity for clinical seizures. Frontal and temporal lobe seizures are especially prone to occur and to generalize secondarily during sleep [Bourgeois, 1996; Kotagal and Yardi, 2008]. Interictal epileptiform discharges may be observed only during sleep [Shinnar et al., 1994]. Nocturnal seizures are most likely to occur during stage N2, followed by N1, N3, and REM sleep, in that order.
The syndrome of electrical status epilepticus during slow-wave sleep (ESES) is characterized by continuous spike-and-wave discharges during 85 percent or more of nocturnal NREM sleep, along with cognitive and behavioral regression [Nickels and Wirrell, 2008]. There are two different forms of ESES – Landau–Kleffner syndrome and the syndrome of continuous spike-wave stupor. Landau–Kleffner syndrome is characterized by regression in language function, typically in the form of auditory verbal agnosia, in association with continuous epileptiform activity during both REM and NREM sleep [Landau and Kleffner, 1957]. Continuous spike-wave stupor, by contrast, is associated with global regression of cognition. The sleep EEG findings are, however, similar to those of the Landau–Kleffner syndrome. Prolonged daytime and night-time EEG may be needed to establish the diagnosis of either entity. Intravenous lorazepam or diazepam may transiently suppress the epileptiform discharges. Long-term maintenance with valproic acid may be modestly effective. Liukkonen et al. followed 32 children with ESES longitudinally (Liukkonen et al., 2010]. Treatment was with valproic acid alone or valproic acid combined with ethosuximide. Ten of 32 children regained their pre-ESES cognitive levels. Unfavorable cognitive outcome was predicted by younger age at ESES diagnosis, lower IQ at initial diagnosis, and no response to drug treatment.
Sleep deprivation leads to activation of seizures. Ellingson et al. [1984] observed that clinical seizures occurred after sleep deprivation in 19 of 788 (2.4 percent) otherwise healthy subjects. Patterns of epileptiform abnormality are also considerably influenced by sleep [Kotagal and Yardi, 2008]. For example, the three-per-second spike-and-wave complexes of absence seizures are replaced in sleep by single spike-and-waves or by polyspike-and-wave complexes. The hypsarrhythmia of infantile spasms is replaced during sleep by brief periods of generalized voltage attenuation. Patients with Lennox–Gastaut syndrome may manifest long runs of generalized spike-and-wave discharges.
Conversely, seizures also influence sleep by suppressing REM sleep, with a corresponding increase in slow-wave sleep. This effect of slowing of the background EEG frequency may persist for days after a seizure. Frequent nocturnal seizures also tend to disrupt sleep continuity, with an increased number of arousals. Patients with Lennox–Gastaut syndrome frequently exhibit tonic seizures during sleep. Vagus nerve stimulation has been found to result in shortened sleep latency, reduced N1 sleep, and increased N3 sleep [Kotagal and Yardi, 2008]. OSA has also been observed after vagus nerve stimulation [Khurana et al., 2007].
Disorders of sleep can also adversely affect seizure control. Patients with OSA have been documented to manifest poor seizure control, which improves after correction of OSA with positive airway pressure breathing, adenotonsillectomy, or tracheostomy [Britton et al., 1997]. In some instances, the daytime somnolence from sleep apnea may be mistaken as a side effect of antiepileptic therapy. In general, antiepileptic drugs lead to stabilization of sleep with a decreased number of nocturnal arousals. Phenobarbital therapy is associated with suppression in the proportion of time spent in REM sleep and increased stage N3 sleep. Both phenytoin and carbamazepine also increase N3 at the expense of N1 and N2 sleep. Benzodiazepines increase stage N3 sleep at the expense of N1 sleep. Lamotrigine can increase the proportion of time spent in REM sleep, with fewer sleep-stage shifts [Kotagal and Yardi, 2008]. Felbamate has been reported to cause insomnia in about 11 percent of subjects [Cilio et al., 2001].
Sleep in Neurologically Compromised Children
Children with spastic quadriparetic cerebral palsy may exhibit daytime irritability in conjunction with fragmented sleep and frequent night-time awakenings, and oxygen desaturation from OSA due to upper airway collapse or adenotonsillar hypertrophy [Kotagal et al., 1994]. They may also have impaired central arousal mechanisms and be unable to compensate for apnea by changes in body position, making OSA especially problematic for them. Jouvet and Petre-Quadens [1966] have described prolonged initial REM latency and suppression in the proportion of time spent in REM sleep in patients with severe mental retardation. Decreased rapid eye movements and spindle density, as well as the presence of “undifferentiated” sleep, correlate with low levels of intelligence [Zucconi, 2000]. Patients with Leigh’s syndrome manifest recurrent central apneas or hypoventilation as a consequence of brainstem involvement [Cummiskey et al., 1987]. Joubert’s syndrome is associated with periods of hyperpnea and panting respiration. Rett’s syndrome is characterized by deep, sighing respiration during wakefulness and normal breathing patterns during sleep, consistent with impairment of the voluntary, cortical control of breathing, and a normal automatic or brainstem control of respiration [Zucconi, 2000]. Patients with Down syndrome have complicated sleep problems. They may develop OSA from the combination of macroglossia, midface hypoplasia, and hypotonic upper airway musculature [Levanon et al., 1999; Kotagal, 2007]. Superimposed on this may be an element of hypoventilation and consequent retention of carbon dioxide due to shallow movement of intercostal and diaphragm muscles. Sleep architecture is disrupted, with decreased sleep efficiency, increased arousals, suppression of N3, and REM sleep. Patients with achondroplasia may develop OSA in infancy as a result of macroglossia and midface hypoplasia. They may also develop central sleep apnea as a consequence of cervicomedullary compression. There is no correlation between the diameter of the foramen magnum and severity of the sleep apnea, but patients need close follow-up of their neurologic and respiratory function. Zucconi et al. [1996] found sleep-related breathing disorders in close to 75 percent of achondroplasia patients. Certain lysosomal storage disorders, such as the mucopolysaccharidoses like Hurler’s syndrome, have been associated with severe OSA that resolves with surgical reduction of macroglossia or bone marrow transplantation. Patients with Niemann–Pick type C disease frequently manifest cataplexy from brainstem involvement, fragmented night sleep, and daytime sleepiness, occasionally in conjuction with reduced CSF levels of hypocretin-1 [Vankova et al., 2003]. Circadian rhythm disorders, like irregular sleep–wake rhythms, are common in patients with severe mental retardation and cerebral palsy; this is especially true if there is associated blindness with impaired light perception that might impair melanopsin activation in the retinal ganglionic cells, and consequent melatonin secretion from the pineal gland and the development of circadian rhythms. Many blind patients with complete lack of light perception fall asleep relatively early in the evening and are wide awake in early morning hours (3 or 4 am), or exhibit multiple periods of daytime sleepiness (irregular sleep–wake rhythms) [Leger et al., 1999]. Melatonin administration and bright light (if light perception is intact) may be used to reset the sleep schedule in these patients.
Parasomnias
Parasomnias are unpleasant or undesirable events that intrude on to sleep, without altering sleep quality or quantity. These events may occur at the transition to sleep, during REM sleep, or at the time of shifting from N3 sleep into the lighter N2/N1 sleep [Kotagal, 2009]. The incidence of parasomnias is variable, though 30 percent of children have at least one episode of sleepwalking, and sleeptalking is noted in about half the population [D’Cruz and Vaughn, 2001]. Frequently, the patient is unaware of the events, but medical attention is sought owing to disruption of the sleep of other family members or after serious injury to the patient. NREM parasomnias, such as confusional arousals, sleepwalking, and sleep terrors, are most commonly seen during N3 sleep and are thus prevalent in the first third of night, when this type of sleep is most abundant. NREM parasomnias occur during attempted transition from N3 into N2 or N1 sleep, with consequent partial arousal and nonepileptic behaviors. Confusional arousals are seen most often in toddlers, whereas sleepwalking and sleep terrors may occur throughout the first decade. Children with confusional arousals may sit up in bed, moan or whimper inconsolably, and utter words like “no” or “go away.” Autonomic activation, characterized by flushing, sweating, and piloerection, is minimal to absent. The events generally last 5–30 minutes, after which the patient goes right back to sleep and has no recollection of the events whatsoever upon awakening the following morning. Patients with night terrors and sleepwalking may exhibit flushing of the face, increased sweating, inconsolability, and agitation, but again with complete amnesia for the event in the morning. In all of the three NREM parasomnias, a concurrently obtained EEG will show rhythmic activity in the 2–6-Hz range, consistent with partial arousal from slow-wave sleep. A low dose of clonazepam (0.25–0.5 mg at bedtime) is helpful in preventing most NREM parasomnias. In some NREM parasomnias, the partial arousals may be triggered by underlying sleep apnea, restless legs syndrome, or gastroesophageal reflux. If so, treatment of these underlying disorders will help alleviate parasomnia. Environmental safety measures, such as installing a deadbolt lock on the door, may be necessary in patients with habitual sleepwalking. NREM parasomnias can sometimes be confused with nocturnal seizures, especially those with frontal lobe onset [Zucconi et al., 1998]. In general, however, the seizure events are shorter in duration (often 20–30 seconds), and are accompanied by more prominent head segment automatisms, such as lip smacking, eye deviation, or chewing. Further, the patient may have partial recall of the event and headache upon awakening in the morning. In contrast to the 6-channel EEG montage that is utilized in standard video polysomnography, the study of suspected parasomnias in the sleep laboratory calls for a full 16-channel EEG montage to help distinguish seizures from parasomnias. Most NREM parasomnias subside spontaneously by 10–12 years of age.
Autism and Sleep
Sleep–wake problems occur in 40–80 percent of children with autism. Problems with sleep often develop early around the same time as onset of developmental regression [Miano and Ferri, 2010]. There does not seem to be increased predeliction by gender. Sleep initiation and maintenance difficulty are the most common complaints [Richdale, 1999; Malow et al., 2006]. There seem to be many pathophysiologic mechanisms underlying the insomnia. To begin with, the child with autism has a difficult time with self-regulation of emotions [Johnson et al., 2009]. Children with autism may experience difficulties with comprehension of parental directions and also with verbalizing their apprehensions [Goldman et al., 2011]. The memory of exciting or upsetting experiences from earlier in the day may linger right into bedtime, and at times even through the night. Sometimes, parents inadvertently contribute to sleep initiation and maintenance difficulty by resorting to behaviors such as rocking the child to sleep or holding the child in their arms till sleep has ensued, at which point the child is placed in bed. As a consequence, there is habit-forming to these extraneous stimuli. When the child awakens in the middle of the night, it is hard to fall back to sleep without once again being held or rocked. This entity is termed sleep-onset association disorder [Johnson et al., 2009]. Comorbid seizures can disrupt sleep, as might also agents like benzodiazepines, phenobarbital, and felbamate that are used for their treatment. Further, a genetic susceptibility region for autism has been localized to chromosome 15q. This region also expresses a variety of GABA-related genes, the disruption in function of which might predispose to insomnia [McCauley et al., 2004]. An alteration in the timing of release of melatonin, with low levels at night and higher levels in the daytime, has also been reported [Ritvo et al., 1993]. Systemic disturbances, such as gastroesophageal reflux, could also contribute to sleep initiation and maintenance difficulty.
The clinician should try to determine if the daily schedule of the autistic child is unduly rigorous and thus leading to excessive excitement and anxiety. If so, simplifying the afternoon and evening regimen, and adhering to a consistent and simple evening schedule, with 30–60 minutes of quiet time before bedtime, may be employed. Postponing bed onset time by about an hour, avoiding daytime naps, and use of graduated extinction techniques may be useful in managing the sleep onset association disorder. The help of a child psychologist might be required in this regard. Complete extinction (removing reinforcement to reduce a behavior) and various forms of graduated extinction are commonly utilized by psychologists. Controlled-release formulations of melatonin, provided at bedtime in a dose of 3–6 mg, are used empirically. Clonidine, 0.1–0.2 mg in two divided doses, can be used for its anxiolytic effect. Risperidone, administered in low doses (0.5–1 mg/ day) may have a calming effect on the child as well, but higher doses should be avoided, as the dopamine antagonist effect of risperidone can precipitate restless legs syndrome, with consequent rebound deterioration in sleep. The combination of behavioral techniques and melatonin administration has also been recommended [Miano and Ferri, 2010].
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Abad V.C., Guilleminault C. Treatment options for obstructive sleep apnea. Curr Treat Options Neurol. 2009;11(5):358.
Acebo C., LeBourgeois M.K. Actigraphy. Respir Care Clin N Am. 2006;12(1):23.
Aldrich M.S., Chervin R.D., Malow B.A. Value of the multiple sleep latency test (MSLT) for the diagnosis of narcolepsy. Sleep. 1997;20(8):620.
Ali N.J., Pitson D.J., Stradling J.R. Snoring, sleep disturbance and behavior in 4–5 year olds. Arch Dis Child. 1993;68:360.
Allen R., Chen C., Soalta A., et al. A randomized, double-blind 6-week dose-ranging study of pregabalin in patients with restless legs syndrome. Sleep Med. 2010;11(6):512.
Allen R.P., Picchietti D., Hening W.A., et al. Restless legs syndrome: Diagnostic criteria, special considerations, and epidemiology. A report from the restless legs syndrome diagnosis and epidemiology workshop at the National Institutes of Health. Sleep Med. 2003;4:101.
American Academy of Pediatrics Policy Statement. Clinical practice guideline: Diagnosis and management of childhood obstructive sleep apnea syndrome. Pediatrics. 2002;109:704.
American Academy of Sleep Medicine. ICSD-2 – International classification of sleep disorders. Diagnostic and coding manual. 2nd edition. American Academy of Sleep Medicine; 2006.
Amiel J., Dubreuil V., Ramanantsoa N., et al. PHOX2B in respiratory control: lessons from CCHS and its mouse model. Respir Physiol Neurobiol. 2009;168(1–2):125.
Arens R., Mazumdar H. Childhood obesity and obstructive sleep apnea syndrome. J Appl Physiol. 2010;108(2):436.
Autret A., Lucas F., Henry-Lebras F., et al. Symptomatic narcolepsies. Sleep. 1994;17(Suppl 1):21.
Basetti C., Aldrich M.S. Idiopathic hypersomnia: A series of 42 patients. Brain. 1997;120:1423.
Billiard M., Besset A., de Lustrac C., et al. Dose response effects of zopiclone on night sleep and on night time and day time functioning. Sleep. 1987;10(Suppl 1):27.
Billiard M., Seignalet J., Besset A., et al. HLA DR2 and narcolepsy. Sleep. 1986;9:149.
Bogan R.K., Cramer Borneman M.A., Kushida C.A., et al. Long-term maintenance treatment of restless legs syndrome with gabapentin enacarbil: a randomized controlled study. Mayo Clin Proc. 2010;85(6):512.
Bolk S., Augrist M., Schwartz S., et al. Congenital central hypoventilation syndrome: Mutation analysis of the receptor tyrosine kinase (RET). Am J Med Genet. 1996;63:603.
Bourgeois B. The relationship between sleep and epilepsy in children. Semin Pediatr Neurol. 1996;3(1):29.
Britton T.C., O’Donoghue M., Duncan J.S. Exacerbation of epilepsy by sleep apnea. J Neurol Neurosurg Psychiatry. 1997;63:808.
Brooks L.J. Genetic syndromes affecting breathing during sleep. In: Lee-Chiong T.L., Sateia M., Carskadon M.A., editors. Sleep medicine. Philadelphia: Hanley and Belfus; 2002:305.
Broughton R., Krupa S., Boucher B., et al. Impaired circadian waking arousal in narcolepsy-cataplexy. Sleep Res Online. 1998;1(4):159.
Brouillette R.T., Morielli A., Leimanis A., et al. Nocturnal pulse oximetry as an abbreviated testing modality for pediatric obstructive sleep apnea. Pediatrics. 2000;105:405.
Brown L.W., Billiard M. Narcolepsy, Kleine-Levin syndrome, and other causes of sleepiness in children. In: Ferber R., Kryger M., editors. Principles and practice of sleep medicine in the child. Philadelphia: WB Saunders; 1995:125.
Brunetti L., Rana S., Losppalluti M.L., et al. Prevalence of obstructive sleep apnea syndrome in a cohort of 1207 children of Southern Italy. Chest. 2001;120:1930.
Cao R., Li H.Y., Lee B., et al. Mammalian target of rapamycin signaling modulates photic entrainment of the suprachiasmatic circadian clock. J Neurosci. 2010;30(18):6302.
Carskadon M.A. The second decade. In: Guillleminault C., editor. Sleeping and waking disorders. Indications and techniques. Menlo Park, CA: Addison Wesley; 1982:99.
Carskadon M.A., Wolfson A.R., Acebo C., et al. Adolescent sleep patterns, circadian timing, and sleepiness at a transition to early school days. Sleep. 1998;21:871.
Challamel M.J., Mazzola M.E., Nevsimalova S., et al. Narcolepsy in children. Sleep. 1994;17S:17.
Challet E., Caldelas I., Graff C., et al. Synchronization of the molecular clockwork by light- and food-related cues in mammals. Biol Chem. 2003;384:711.
Chervin R.D., Archbold K.H., Dillon J.E., et al. Associations between symptoms of inattention, hyperactivity, restless legs, and periodic leg movements. Sleep. 2002;25:213.
Cilio M.R., Kartashov A.I., Vigevano F. The long-term use of felbamate in children with severe refractory epilepsy. Epilepsy Res. 2001;47:1.
Cole R.J., Smith J.S., Alcala Y.C., et al. Bright-light mask treatment of delayed sleep phase syndrome. J Biol Rhythms. 2002;17:89.
Corbo G.M., Forastiere F., Abigail N., et al. Snoring in 9 to 15 year old children: Risk factors and clinical relevance. Pediatrics. 2001;180:1149.
Cummiskey J., Guilleminault C., Davis R., et al. Automatic respiratory failure: Sleep studies and Leigh’s disease. Neurology. 1987;37:1876.
Dauvilliers Y., Mayer G., Lecendreux M., et al. Kleine Levin syndrome. An autoimmune hypothesis based on clinical and genetic analyses. Neurology. 2002;59:1739.
Dauvilliers Y., Abril B., Mas E., et al. M. Normalization of hypocretin-1 in narcolepsy after immunoglobulin G treatment. Neurology. 2009;73(16):1333.
Dauvilliers Y., Abril B., Mas E., et al. Normalization of hypocretin-1 in narcolepsyafter intravenous immunoglobulin treatment. Neurology. 2009;73:1333-1334.
D’Cruz F.O., Vaughn B.V. Parasomnias – An update. Semin Pediatr Neurol. 2001;8:251.
Drake C., Nickel C., Burduvali E., et al. The Pediatric Daytime Sleepiness Scale (PDSS): Sleep Habits and School Outcomes in middle school children. Sleep. 2003;26(4):455.
Ellingson R.J., Wilken K., Bennet D.R. Efficacy of sleep deprivation as an activating procedure in epilepsy patients. J Clin Neurophysiol. 1984;1:83.
Franken P., Dijk D.J. Circadian clock genes and sleep homeostasis. Eur J Neurosci. 2009;29(9):1820-1829.
Frenette E., Kushida C.A. Primary hypersomnias of central origin. Semin Neurol. 2009;29(4):354.
Fuller P.M., Gooley J.J., Saper C.B. Neurobiology of the sleep-wake cycle: sleep architecture, circadian regulation, and regulatory feedback. J Biol Rhythms. 2006;21(6):482.
Garcia J., Rosen G., Mahowald M. Circadian rhythms and circadian rhythm disorders in children and adolescents. Semin Pediatr Neurol. 2001;8:229.
Givan D.C. Sleep in children with neuromuscular disorders. In: Lee-Chiong T.L., Sateia M., Carskadon M.A., editors. Sleep medicine. Philadelphia: Hanley and Belfus; 2002:297.
Goldman S.E., Malow B.A. Autism and other neurodevelopmental disorders. In: Kothare S.V., Kotagal S., editors. Sleep in Childhood Neurological Disorders. New York: Demos Medical; 2011:143-152.
Gosalakkal J.A. Sleep disordered breathing in Chairi malformation type I. Pediatr Neurol. 2008;39(3):207-208.
Gozal D. New concepts in abnormalities of respiratory control in children. Curr Opin Pediatr. 2004;16:305.
Gozal D., Harper R.M. Novel insights into congenital hypoventilation syndrome. Curr Opin Pulm Med. 1999;5:335.
Gozal D., Wang M., Pope D.W. Objective sleepiness measures in pediatric obstructive sleep apnea. Pediatrics. 2001;108:693.
Gozal D. Obstructive sleep apnea in children: implications for the developing central nervous system. Semin Pediatr Neurol. 2008;15:100.
Guilleminault C., Pelayo R., Leger D., et al. Recognition of sleep-disordered breathing in children. Pediatrics. 1996;98:871.
Hamada T., LeSauter J., Venuti J.M., et al. Expression of Period genes: Rhythmic and nonrhythmic compartments of the suprachiasmatic nucleus pacemaker. J Neurosci. 2001;21:7742.
Hoban T.F., Chervin R.D. Sleep-related breathing disorders of childhood: description, and clinical picture, diagnosis and treatment approaches. Sleep Med Clin. 2007;2:445.
Huang Y.S., Lakkis C., Guilleminault C. Kleine Levin syndrome: current status. Med Clin North Am. 2010;94(3):557.
Hungs Fan J., Lin X., Maki R.A., et al. Identification and functional analysis of mutations in the hypocretin (orexin) genes of narcoleptic canines. Genome Res. 2001;11:531.
Iber C., Ancoli-Israel S., Chesson A., et alAmerican Academy of Sleep Medicine. The AASM Manual for the Scoring of Sleep and Associated Events: Rules, Terminology, and Technical Specifications, ed 1. Westchester, Illinois: American Academy of Sleep Medicine; 2007.
Ipsiroglu O.S., Fatemi A., Werner I., et al. Prevalence of sleep disorders in school children between 11 and 15 years of age (German). Wien Klin Wochenschr. 2001;113:235.
John J., Wu M.-F., Siegel J.M. Systemic administration of hypocretin-1 reduces cataplexy and normalizes sleep and waking durations in narcoleptic dogs. Sleep Res Online. 2000;3:23.
Johnson K.P., Giannotti F., Cortesi F. Sleep patterns in autistic spectrum disorders. Child Adolesc Psychiatric Clin N Am. 2009;18:917.
Jouvet M., Petre-Quadens O. Paradoxical sleep and dreaming in the mentally deficient. Acta Neurol Psychiatr Belg. 1966;66:116.
Kanbayashi T., Inoue Y., Chiba S., et al. CSF hypocretin-1 (orexin-A) concentrations in narcolepsy with and without cataplexy and idiopathic hypersomnia. J Sleep Res. 2002;11:91.
Kotagal P., Yardi N. The relationship between sleep and epilepsy. Semin Pediatr Neurol. 2008;15:42.
Kotagal S. Sleep in children at risk. Sleep Med Clin. 2007;2:477.
Kotagal S. Parasomnias in childhood. Sleep Med Rev. 2009;13(2):157.
Kotagal S., Gibbons V.P., Stith J. Sleep abnormalities in patients with severe cerebral palsy. Dev Med Child Neurol. 1994;36:304.
Kotagal S., Goulding P.M. The laboratory assessment of daytime sleepiness in childhood. J Clin Neurophysiol. 1996;13:208.
Kotagal S., Herold D.L. The laboratory assessment of childhood sleep-wake disorders. Am J End Technol. 2002;42:73.
Kotagal S., Krahn L.E., Slocumb N. A putative link between childhood narcolepsy and obesity. Sleep Med. 2004;5(2):147.
Kotagal S., Silber M.H. Childhood-onset restless legs syndrome. Ann Neurol. 2004;56:803.
Kotagal S., Swink T.D. Excessive daytime sleepiness in a 13-year old. Semin Pediatr Neurol. 1996;3:170.
Kothare S.V., Kaleyias J. The clinical and laboratory assessment of the sleepy child. Semin Pediatr Neurol. 2008;15(2):61.
Khurana D.S., Reumann M., Hobdell E.F., et al. Vagus nerve stimulation in children with refractory epilepsy: unusual complications and relationship to sleep disordered breathing. Childs Nerv Syst. 2007;23:1309.
Landau W.M., Kleffner F.R. Syndrome of acquired aphasia with convulsive disorder in children. Neurology. 1957;7:523.
Langdon N., Lock C., Welsh K., et al. Immune factors in narcolepsy. Sleep. 1986;9:143.
Leger D., Prevot E., Phillip P., et al. Sleep disorders in children with blindness. AnnNeurol. 1999;46(4):648.
Levanon A., Tarasiuk A., Tal A. Sleep characteristics in children with Down syndrome. J Pediatr. 1999;134:755.
Littner M., Johnson S.F., McCall W.V., et al. Practice parameters for treatment of narcolepsy: An update for 2000. Sleep. 2001;24:451.
Littner M.R., Kushida C., Wise M., et al. Practice parameters for clinical use of the multiple sleep latency test and maintenance of wakefulness test. Sleep. 2005;28(1):113.
Liukkonen E., Kantola-Sorsa E., Paetau R., et al. Long term outcome of 32 children with encephalopathy with status epilepticus during sleep, or ESES syndrome. Epilepsia. 2010. in press
Lu B.S., Zee P.C. Neurobiology of sleep. Clin Chest Med. 2010;31(2):309.
Lumeng J.C., Chervin R.D. Epidemiology of pediatric obstructive sleep apnea. Proc Am Thorac Soc. 2008;5:242.
Malow B.A., Mrazek M.L., McGrew S.G., et al. Characterizing sleep in children with autism spectrum disorders: a multidimensional approach. Sleep. 2006;29(12):1563.
Marcus C.L., Loughlin G.M. Obstructive sleep apnea in children. Semin Pediatr Neurol. 1996;3:23.
McCauley J.L., Olson M., Delahanty R., et al. A linkage disequilibrium map of the 1-Mb 15q12 GABA receptor subunit cluster and association to autism. Am J Med Genet. 2004;131:51.
Miano S., Ferri R. Epidemiology and management of insomnia in children with autistic spectrum disorders. Paediatr Drugs. 2010;12(2):75.
Mignot E., Lin L., Rogers W., et al. Complex HLA-DR and -DQ interactions confer risk of narcolepsy-cataplexy in three ethnic groups. Am J Hum Genet. 2001;68:686.
Mignot E., Young T., Lin L., et al. Nocturnal sleep and daytime sleepiness in normal subjects with HLA-DQB1*0602. Sleep. 1999;22:347.
Miller J.D., Morin L.P., Schwartz W.J., et al. New insights into the mammalian circadian clock. Sleep. 1996;19:641.
Misuri G., Lanini B., Gigliotti F., et al. Mechanisms of CO2 retention in patients with neuromuscular disease. Chest. 2000;117:447.
Mitler M.M., Doghramji K., Shapiro C. The maintenance of wakefulness test. Normative data by age. J Psychosom Res. 2000;9:63.
Nevsimalova S., Roth B., Zouhar A., et al. Narkolepsie-kataplexie a periodicka hypersomnie se zacatkem v kojeneckem veku. Cs Pediatr. 1986;41:324.
Nickels K., Wirrell W. Electrical status epielticus in sleep. Semin Pediatr Neurol. 2008;15:50.
Nishino S., Ripley B., Overeem S., et al. Hypocretin (orexin) deficiency in human narcolepsy. Lancet. 2000;355:39.
Nishino S., Ripley B., Overeem S., et al. Low cerebrospinal fluid hypocretin (orexin) and altered energy homeostasis in human narcolepsy. Ann Neurol. 2001;50:381.
O’Brien L.M., Holbrook C.R., Mervis C.B., et al. Sleep and neurobehavioral characteristics in 5–7 year old hyperactive children. Pediatrics. 2003;111:554.
O’Brien L.M., Mervis C.B., Holbrook C.R., et al. Neurobehavioral implications of habitual snoring in children. Pediatrics. 2004;114:44.
Owens J.A., Spirito A., McGuinn M. The Children’s Sleep Habits Questionnaire (CHSQ): Psychometric properties of a survey instrument for school-aged children. Sleep. 2000;23:1.
Palm L., Persson E., Elmquist D., et al. Sleep and wakefulness in normal pre-adolescents. Sleep. 1989;12:299.
Papineau K.L., Roehts T.A., Petrucelli N., et al. Electrophysiological Assessment (the multiple sleep latency test) of the biphasic effects of ethanol in humans. Alcohol Clin Exp Res. 1998;2:231.
Perriol M.P., Cartigny M., Lamblin M.D., et al. Childhood-onset narcolepsy, obesity and puberty in four consecutive children: a close temporal link. J Pediatr Endocrinol Metab. 2010;23(3):257.
Picchietti D.L., Allen R.P., Walters A.S., et al. Restless legs syndrome: prevalence and impact in childhood and adolesence – The Peds REST study. Pediatrics. 2007;120:253.
Picchietti D.L., Underwood D.J., Farris W.A., et al. Further studies on periodic limb movement disorder and restless legs syndrome in children with attention deficit-hyperactivity disorder. Mov Disord. 1999;14:1000.
Picchietti M.A., Picchietti D.L. Restless legs syndrome and periodic limb movement disorder in children and adolescents. Semin Pediatr Neurol. 2008;15:91-99.
Plazzi G., Parmeggiani A., Mignot E., et al. Narcolepsy-cataplexy associated with precocius puberty. Neurology. 2006;66(10):1577.
Poppe M., Friebel D., Reuner U., et al. The Kleine-Levin syndrome – Effects of treatment with lithium. Neuropediatrics. 2003;34:113.
Richdale A.L. Sleep problems in autism: prevalence, cause, and intervention. Dev Med Child Neurol. 1999;41(1):60.
Ritchie C., Okuro M., Kanbayashi T., et al. Hypocretin ligand deficiency in narcolepsy: recent basic and clinical insights. Clin Neurol Neurosci. 2010;10(3):180.
Ritvo E.R., Ritvo R., Yuwiler A., et al. Elevated daytime melatonin concentrations in autism: a pilot study. Eur Child Adolesc Psychiatry. 1993;2(2):75.
Rondeau B.H. Dentist’s role in the treatment of snoring and sleep apnea. Funct Orthodont. 1998;15:4.
Rosenthal L., Roehrs T.A., Rosen A., et al. Level of sleepiness and total sleep time following various time in bed conditions. Sleep. 1993;16:226.
Roshkow J.E., Haller J.O., Berdon W.E., et al. Hirschsprung’s disease, Ondine’s curse, and neuroblastoma – Manifestations of neurocristopathy. Pediatr Radiol. 1988;19:45.
Sharp S.J., D’Cruz O.F. Narcolepsy in a 12 month old boy. J Child Neurol. 2001;16:145.
Sheldon S.H. Diagnostic methods in pediatric sleep medicine. Sleep Med Clin. 2007;2:343.
Shinnar S., Kang H., Berg A.T., et al. EEG abnormalities in children with a first unprovoked seizure. Epilepsia. 1994;35:471.
Silber M.H., Krahn L.E., Olson E.J., et al. Epidemiology of narcolepsy in Olmstead County, Minnesota. Sleep. 2002;25:197.
Stein M.A., Mendelsohn J., Obermeyer W.H., et al. Sleep and behavior problems in school-aged children. Pediatrics. 2001;107:E60.
Stepanski E., Lamphere J., Roehrs T., et al. Experimental sleep fragmentation in normal subjects. Int J Neurosci. 1987;33:207.
Steriade M., Contreras D., Curro Dossi R., et al. The slow (< 1 Hz) oscillation in reticular thalamic and thalamocortical neurons: Scenario of sleep rhythm generation in interacting thalamic and neocortical networks. J Neurosci. 1993;13:3284.
Stores G., Montgomery P., Wiggs L. The psychosocial problems of children with narcolepsy and those with excessive daytime sleepiness of uncertain origin. Pediatrics. 2006;118(4):e1116-e1123.
Swaminathan S., Gilsanz V., Atkinson J., et al. Congenital central hypoventilation syndrome associated with multiple ganglioneuromas. Chest. 1989;96:423.
Thannickal T.C., Moore R.Y., Nienhuis R., et al. Reduced number of hypocretin neurons in human narcolepsy. Neuron. 2000;27:469.
Trochet D., O’Brien L.M., Gozal D., et al. PHOX2B genotype allows for prediction of tumor risk in congenital central hypoventilation syndrome. Am J Hum Genet. 2005;76:421.
Van Cauter E., Spiegel K., Tasali E., et al. Metabolic consequences of sleep and sleep loss. Sleep Med. 2008;9(Suppl 1):S23.
Van de Water A.T., Holmes A., Hurley D.A. Objective measurements of sleep for non-laboratory settings as alternatives to polysomnography – a systematic review. J Sleep Res. 2010. in press
Van Someren E.J. More than a marker: Interaction between the circadian regulation of temperature and sleep, age-related changes, and treatment possibilities. Chronobiol Int. 2000;17:313.
Vankova J., Stepanova I., Jech R., et al. Sleep disturbances and hypocretin deficiency in Niemann-Pick disease type C. Sleep. 2003;26(4):427.
Walters A.S. Is there a subpopulation of children with growing pains who really have restless legs syndrome? A review of the literature. Sleep Med. 2002;3:93.
Weese-Mayer D.E., Berry-Kravis E.M., Ceccherini I., et al. An official ATS clinical policy statement: congenital central hypoventilation syndrome: genetic basis, diagnosis, and management. Am J Res Crit Care Med. 2010;181(6):626.
Weitzman E.D., Czeisler C.A., Coleman R.M., et al. Delayed sleep phase syndrome. A chronobiological disorder with sleep-onset insomnia. Arch Gen Psychiatry. 1981;38:737.
Wulff K., Porcheret K., Cussans E., et al. Sleep and circadian rhythm disturbances: multiple genes and multiple phenotypes. Curr Opin Genet Dev. 2009;19(3):237.
Zolty P., Sanders M.H., Pollack I.F. Chiari malformation and sleep-disordered breathing: A review of diagnostic and management issues. Sleep. 2000;23:637.
Zucconi M. Sleep disorders in children with neurological disease. In: Loughlin G.M., Carroll J.L., Marcus C.L., editors. Sleep and breathing in children. A developmental approach. New York: Marcel Dekker; 2000:363.
Zucconi M., Ferini-Strambi L., Oldani A., et al. Nocturnal motor behaviors in children: A video-polysomnographic study. Sleep. 1998;21(Suppl):148.
Zucconi M., Weber G., Castronovo V., et al. Sleep and upper airway obstruction in children with achondroplasia. J Pediatr. 1996;129:743.
* Portions of this chapter have appeared in Kotagal S. Sleep disorders in childhood. Neurol Clin 2003;21:961;81.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 66 Sleep–Wake Disorders*
Complaints about insufficient or nonrestorative sleep are quite common in childhood and adolescence. In a questionnaire survey of 332 children of 11 through 15 years of age, Ipsiroglu et al. [2001] observed that 28 percent of the subjects had snoring, insomnia, or a parasomnia. In another survey of 472 4- to 12-year-old urban and rural children receiving routine pediatric care, Stein et al. [2001] noted a 10 percent prevalence of sleep disorders. Less than one-half of the parents had discussed the sleep problems with the pediatrician. Childhood sleep disorders can have a significant effect on the quality of life. Many disorders are also easily treatable, thus underscoring the importance of their prompt recognition and management. This chapter covers salient aspects of childhood sleep–wake function and common pediatric sleep disorders.
Sleep Physiology and Ontogeny
Sleep–wake regulation is mediated via a complex set of interactions. There is a dynamic balance between the circadian drive for sleep (process C), which serves to enhance alertness, and the homeostatic drive (process S), which facilitates sleep. Adenosine, an extracellular factor that is secreted by neurons in the basal forebrain, plays a key role in sleep induction. (Caffeine is an antagonist of adenosine.) Neurons of the ventrolateral preoptic nucleus that contain gamma-aminobutyric acid (GABA) and galanin serve to inhibit wakefulness-promoting regions of the forebrain and the laterodorsal pontine tegmentum, and are thus also involved in sleep induction. Sleep onset is further facilitated by melatonin, a light-sensitive hormone that is released from the pineal gland. The principal components of the ascending arousal system are the cholinergic neurons of laterodorsal pontine tegmentum and the nucleus basialis of Meynert. Cells of the dorsolateral hypothalamus produce hypocretin (orexin), which is also an important wakefulness-promoting peptide. The hypocretin neurons project widely to the forebrain and brainstem. There is a tendency for mutual inhibition between the wakefulness-promoting and the sleep-promoting circuits. This phenomenon has been characterized as a “flip-flop” switch [Fuller et al., 2006; Lu and Zee, 2010].
The ultimate command center for the once-a-day (circadian) rhythm of sleep and wakefulness is the suprachiasmatic nucleus of the hypothalamus [Miller et al., 1996; Steriade et al., 1993], which has cells with receptors for melatonin. The suprachiasmatic nucleus is strongly influenced by light-mediated impulses received through the retinohypothalamic tract. Clock and Period genes, remarkably preserved across various phyla, were studied initially in Drosophila melanogaster; they also influence the timing of activity of the suprachiasmatic nucleus cells [Challet et al., 2003; Franken and Dijk, 2009; Hamada et al., 2001]. The mammalian target of rapamycin (mTOR) signaling is involved in modulation of photic entrainment of the suprachiasmatic circadian clock [Cao et al., 2010]. The body temperature rhythm is also regulated by the hypothalamus [van Someren, 2000]; a rise in body temperature leads to postponement of sleep. Conversely, individuals are most sleepy around the nadir of body temperature: that is, around 0400 hours. An artificial increase in body temperature in the 1–2 hours before bedtime, such as through vigorous exercise, may provoke sleep-onset insomnia.
Wakefulness can be differentiated from sleep by 27–28 weeks’ postconceptional age in the preterm infant. At this age, sleep is primarily of the active or rapid eye movement (REM) type, which is associated with irregular breathing, phasic electromyographic activity, and low-voltage electroencephalographic (EEG) activity. Cerebral blood flow and metabolism are higher in REM than in non-rapid eye movement (NREM) sleep, which is also termed quiet sleep in newborns. By 40 weeks’ postconceptional age, active (REM) sleep decreases to about 50 percent of the total sleep time, with a corresponding rise in the proportion of quiet (NREM) sleep. By 46–48 weeks’ postconceptional age, sleep spindles appear during stages N2 and N3 of NREM sleep. By 4–6 months of age, NREM sleep has fully differentiated into N1, N2, and N3 sleep, corresponding respectively with lighter to deeper stages in terms of arousal threshold. N3 is also termed slow-wave sleep. It is characterized by the generalized slow-wave activity in the 0.5–4 Hz range on the EEG (Figure 66-1). The bulk of N3 occurs during the first third of the night. Growth hormone release is closely linked to N3 sleep [Van Cauter et al., 2008], with suppression of the latter leading to impaired growth hormone release. The release of cortisol is suppressed during N3 sleep [van Cauter et al., 2008]. REM sleep decreases gradually over time. By the age of 3 years, it constitutes only about 20–25 percent of total sleep.
Prior to age of 3 months, the transition from wakefulness is initially into REM sleep. After this age, however, the physiologic transition is from wakefulness into NREM sleep, with REM sleep occurring 90–140 minutes later. The physiologic sleep-onset time in elementary school-age children is usually around 8:00–8:30 pm. Around adolescence, there is a physiologic delay in sleep-onset time, which shifts to around 10:30–11:00 pm [Carskadon et al., 1998]. Teenage girls generally have their final morning awakening about one half-hour earlier than boys. When juxtaposed with early high-school start times of around 7:30 am, it is easy to understand why most teenagers are chronically sleep-deprived.
The International Classification of Sleep Disorders
The International Classification of Sleep Disorders was introduced in 1990, and has undergone subsequent revisions [American Sleep Disorders Association, 2006]. It was created initially through collaborative efforts of the American Sleep Disorders Association, the European Sleep Research Society, the Japanese Society of Sleep Research, and the Latin American Sleep Society. Primary sleep disorders are separated from those due to medical or psychiatric conditions. Primary sleep disorders are further subdivided into:
Dyssomnias are further subdivided into intrinsic, extrinsic, and circadian rhythm sleep disorders. An abbreviated version of the International Classification of Sleep Disorders-2 is presented in Box 66-1.
Box 66-1 The International Classification of Sleep Disorders
Insomnia (Examples)
Idiopathic insomnia, paradoxical insomnia, adjustment insomnia, psychophysiologic insomnia, insomnia due to mental disorder, inadequate sleep hygiene, insomnia due to substance abuse, insomnia due to medical condition, behavioral insomnia of childhood – sleep-onset association type, behavioral insomnia of childhood – limit-setting typeSleep-Related Breathing Disorders (Examples)
Primary central sleep apnea, Cheyne–Stokes breathing, high-altitude periodic breathing, central sleep apnea due to medical condition, obstructive sleep apnea – adult, obstructive sleep apnea – pediatric, primary apnea – infancy, sleep-related hypoventilation, sleep-related hypoventilation/hypoxemia due to neuromuscular and chest wall disordersHypersomnia of Central Origin not due to a Circadian Rhythm Sleep Disorder (Examples)
Narcolepsy with cataplexy, narcolepsy without cataplexy, narcolepsy due to medical condition without cataplexy, narcolepsy unspecified, recurrent hypersomnia (including Kleine–Levin syndrome and menstrual-related hypersomnia), idiopathic hypersomnia with long sleep, idiopathic hypersomnia without long sleep, hypersomnia due to medical condition, hypersomnia due to drug or substance, sleep-related breathing, behaviorally induced insufficient sleep syndrome disorder, or other cause of disturbed nocturnal sleep(From American Sleep Disorders Association, International Classification of Sleep Disorders, 2nd edn, pocket version: Diagnostic and Coding Manual. Westchester, Illinois: American Academy of Sleep Medicine, 2006.)
Assessment of Sleep–Wake Complaints
Sleep History
The Pediatric Daytime Sleepiness Scale (PDSS) is a simple, validated questionnaire that can be administered to children in the 11–14-year age group [Drake et al., 2003]. It has eight items, each rated on a 0–4 scale. It provides a numerical score for sleepiness; the 50th percentile score on the PDSS is 16, the 75th percentile is 20, and the 90th percentile is 23. Participants who reported low school achievement, high absenteeism, low school enjoyment, low total sleep time and frequent illnesses all had higher levels of sleepiness as measured by this scale [Drake et al., 2003]. Another survey tool that is commonly used in clinical practice is the Children’s Sleep Habits Questionnaire. It is a 45-item, validated questionnaire that is completed by parents of 4–11-year-olds [Owens et al., 2000]. The questions pertain to sleep–wake function in the preceding 2 weeks, such as “the child sleeps too little” or “the child suddenly falls asleep in the middle of active behavior.” The items represent several domains that present as sleep complaints, such as bedtime resistance, sleep-onset delay, sleep duration, sleep anxiety, night awakenings, parasomnias, breathing disturbance, and daytime sleepiness. Responses are rated as rarely (occurring 0–1 time/week; 1 point), sometimes (occurring 2–4 nights/week; 2 points), or usually (occurring 5–7 nights/week; 3 points). Scores of 41 or greater correlate with presence of a sleep disorder. The internal consistency estimate for this questionnaire in a community (nonclinic sample) of 4–10-year-olds is 0.36–0.70. The test-retest reliability over a 2-week period is 0.62–0.79.
Sleep-Related Examination
Height, weight, and body mass index are recorded because obstructive sleep apnea may be associated with poor weight gain during infancy, and with obesity during adolesence [Arens et al., 2010]. The blood pressure should be measured because long-standing and severe obstructive sleep apnea (OSA) can be associated with hypertension. OSA patients may exhibit craniofacial abnormalities such as micrognathia, dental malocclusion, macroglossia, myopathic face and midface hypoplasia, deviated nasal septum, swollen inferior turbinates, tonsillar hypertrophy and mouth breathing [Brooks, 2002; Hoban and Chervin, 2007]. Consultation with a pediatric otolaryngologist may be required to exclude adenoidal hypertrophy. Inattentiveness, irritability, and mood swings may be clues to daytime sleepiness. Obstructive sleep apnea related to brainstem abnormalities like the Chiari type I or II malformations [Gosalakkal, 2008] can lead to hoarseness of voice, decreased gag reflex, and changes in the amplitude of the jaw jerk relative to that of other tendon reflexes. Neuromuscular disorders, like myotonic dystrophy, may be associated with chronic obstructive hypoventilation from a combination of palatal muscle weakness, high-arched palate, and diminished chest wall–abdominal excursion [Givan, 2002; Misuri et al., 2000]. The parent–child interaction should be observed not only for clues indicating parental anxiety and reluctance to set limits on inappropriate behaviors that perpetuate insomnia in toddlers, but also for subtle clues indicating a child maltreatment syndrome. Home videos, if available, may be invaluable in the assessment of restless legs syndrome, parasomnias, and nocturnal seizures.
Nocturnal Polysomnography
This procedure involves the monitoring of multiple physiologic parameters in sleep. It is useful in the evaluation of intrinsic sleep disorders, such as narcolepsy, obstructive sleep apnea, nocturnal spells, and periodic limb movement disorder. It may not be indicated for diagnosing obstructive sleep apnea secondary to severe adenotonsillar enlargement, which can be easily recognized on the basis of clinical findings combined with severe oxygen desaturation on simple overnight oximetry in the home environment. The nocturnal polysomnogram usually consists of simultaneous monitoring of 2–4 channels of the EEG, eye movements, chin and leg electromyogram, nasal pressure, thoracic and abdominal respiratory effort, electrocardiogram, and oxygen saturation [Kotagal and Goulding, 1996; Kotagal and Herold, 2002; Sheldon, 2007]. Patients with Down syndrome, neuromuscular disorders, and obesity may exhibit hypoventilation that is characterized by shallow chest and abdominal wall movement, with resultant CO2 retention. It is therefore important to measure end-tidal CO2 levels concurrently in these patients. Esophageal pH can be monitored simultaneously when there is a suspicion of gastroesophageal reflux. Criteria for the scoring of sleep and sleep-related events such as apnea, arousals, and periodic limb movements have been recently revised [Iber et al., 2007].
In patients with obvious upper airway obstruction secondary to obesity or neuromuscular disorders like myotonic dystrophy, a therapeutic trial of positive pressure airway breathing can be attempted midway during the course of the sleep recording. A full 16- to 20-channel EEG montage is recommended when parasomnias and seizures are both in the differential diagnosis. Simultaneous video monitoring is standard for all nocturnal polysomnograms. Normative data for common physiologic variables are listed in Table 66-1 [Marcus and Loughlin, 1996]. There is insufficient evidence regarding the utility of ambulatory, in-home polysomnograms, especially in the preschool age group; thus, traditional sleep laboratory monitoring remains the gold standard in pediatric sleep medicine.
| Parameter | Average | Standard deviation |
|---|---|---|
| Total sleep time | 472 min | 42 min |
| Sleep efficiency | 90% | 7 |
| Sleep latency | 24.1 min | 25.6 min |
| REM latency | 87.8 min | 41.2 min |
| REM sleep (% of TST) | 21.1% | 4.9 |
| Stage N3 sleep (% of TST) | 26.3% | 4.8 |
| Stage N2 sleep (% of TST) | 36% | 6.6 |
| Stage N1 sleep (% of TST) | 5.2% | 2 |
| Apnea hypopnea index (events per hour of sleep) | 0.9 | 0.7 |
| End-tidal CO2 > 50 mm (% time) | 25% or less | |
| Oxygen saturation (% time < 90%) | 0.04% | 0.18 |
| Oxygen desaturations > 4% (number/hour of TST) | 0.4 | 0.78 |
REM, rapid eye movement; TST, total sleep time.
(From Montgomery-Downs HE et al. Polysomnographic characteristics in normal preschool and early school aged children. Pediatrics 2006;117:741.)
Multiple Sleep Latency Test
The Multiple Sleep Latency Test (MSLT) assesses how quickly one is able to fall asleep during the daytime and whether transition from wakefulness is into NREM sleep or into REM sleep. The MSLT can reliably detect sleepiness under clinical and experimental conditions [Littner et al., 2005]. The lower age limit at which one can utilize this test is about 6–7 years. Application of the MSLT to children younger than 6 years is unhelpful because healthy pre-school age children tend to take physiologic daytime naps. In order to be able to derive valid conclusions, the MSLT must be preceded the night before by a polysomnogram in which the total sleep time approximates physiologic sleep. The MSLT consists of the provision of four or five daytime nap opportunities at 2-hour intervals: e.g., at 10:00, 12:00, 14:00, and 16:00 hours. The EEG, chin electromyogram, and eye movements are monitored during each nap opportunity. The patient should be dressed in street clothes. The attending parent or guardian should be available to prevent the child from dozing off involuntarily in between the scheduled nap times. At each planned nap opportunity, the lights are turned off and the patient is asked to try to sleep. The time from “lights out” to sleep onset is measured, and represents the sleep latency. A mean sleep latency is also derived for the four naps. Normative values for the mean sleep latency have been established. The nap opportunity is terminated either 15 minutes after sleep onset, or if the patient does not fall asleep, at 20 minutes after “lights out.” The mean sleep latency decreases inversely with an increase in the Tanner stage of sexual development, and ranges between 12 and 18 minutes [Carskadon, 1982]. A mean sleep latency of less than 5 minutes indicates severe daytime sleepiness; a value between 5 and 10 minutes indicates moderate daytime sleepiness. A urine drug screen is obtained in between the naps if illicit drug-seeking behavior is suspected. The occurrence of REM sleep within 15 minutes of sleep onset constitutes a sleep-onset rapid eye movement period (SOREMP). The presence of SOREMPs on two more MSLT nap opportunities in conjunction with a shortened mean sleep latency of less than 5 minutes is highly suggestive of narcolepsy.
In adults, a mean sleep latency of less than 5 minutes in association with two or more SOREMPs is 70 percent sensitive and 97 percent specific for the diagnosis of narcolepsy [Aldrich et al., 1997]. Comparable data on the sensitivity and specificity are not available for children and adolescents. A study by Gozal et al. [2001] suggests that, in prepubertal children, the normal mean sleep latency is 23.7 minutes, plus or minus 3.1 minutes. Palm et al. [1989] also found a mean sleep latency of 26.4 minutes plus or minus 2.8 minutes in 18 prepubertal children. These data suggest that the normal mean sleep latency in children may actually be higher than previously reported, and that normative data for the multiple sleep latency test in childhood might perhaps need revision. One of the merits of the multiple sleep latency test is that it provides reliable and quantitative information about the propensity for sleepiness. It has been validated as a measure of daytime sleepiness following episodes of sleep loss [Rosenthal et al., 1993], sleep disruption [Stepanski et al., 1987], and hypnotic drug and alcohol abuse [Billiard et al., 1987; Papineau et al., 1998]. The effects of treatment of daytime sleepiness with stimulants cannot be reliably measured, however. In addition, although one can control ambient noise and light that might affect sleep during the testing process, one cannot control for internal factors, like anxiety and apprehension, that also affect sleep propensity.
Maintenance of Wakefulness Test
The Maintenance of Wakefulness Test (MWT) is the mirror image opposite to the MSLT; one measures the ability to stay awake in a darkened, quiet environment during the daytime while the patient is seated in a semireclining position [Littner et al., 2005]. EEG, eye movements, and chin electromyography are monitored in a manner identical to that in the MSLT. The patient is provided four or five nap opportunities. The duration of each session is 40 minutes. The average mean sleep latency for normal adults is 35.2 minutes. Normative values have not been established for children. The test helps assess the effect of stimulant medication treatment on daytime sleepiness [Mitler et al., 2000].
Actigraphy
This technique involves the recording and storing of skeletal muscle activity continuously for 1–2 weeks in the home environment, generally from the nondominant forearm, using a wristwatch-shaped microcomputer device that measures linear acceleration and translates it into a numeric and graphic representation. This numeric representation is sampled frequently – that is, every 0.1 second – and aggregated at a constant interval or epoch length [Acebo and LeBourgeois, 2006]. The device captures signals during periods of muscle activity (generally correlating with wakefulness) and periods of no muscle activity (generally correlating with sleep; Figure 66-2). There is close correlation with polysomnographically determined total sleep time, sleep latency, and sleep efficiency [total sleep time or total time in bed ×100; Kothare and Kaleyias, 2008; van de Water et al., 2010]. Wrist actigraphy should be combined with 1–2 weeks of “sleep logs” that document specific wake-up and sleep-onset times, daytime naps, and so on. Wrist actigraphy is useful in the study of insomnia and circadian rhythm disorders like the delayed sleep phase syndrome [Kothare and Kaleyias, 2008]. In the latter instance, it will depict sleep onset late at night or in the early morning hours and uninterrupted sleep thereafter, with final awakening late in the morning or early afternoon (see Figure 66-1). In patients being evaluated for suspected narcolepsy, 2 weeks of actigraphy before nocturnal polysomnography and the MSLT help exclude the possibility of sleepiness resulting from a circadian rhythm disorder or insufficient sleep at night.
Common Childhood Sleep Disorders
Sleep-Related Breathing Disturbances
In increasing level of severity, the spectrum of sleep-disordered breathing in childhood ranges from snoring without sleep disruption (primary snoring) to the upper airway resistance syndrome (snoring that disrupts sleep continuity but without associated apnea or oxygen desaturation) to classic obstructive sleep apnea and, finally, obstructive hypoventilation (apnea, oxygen desaturation, plus hypercarbia). Between 10 and 12 percent of children snore on a habitual basis [Corbo et al., 2001; O’Brien et al., 2003]. The snoring sound is the result of vibration of the soft palate during inspiration due to narrowing of the oropharynx. Although some guidelines [American Academy of Pediatrics, 2002] suggest that primary snoring may be a “benign condition that does not warrant any specific therapy,” a study of 87 children of 5–7 years of age [O’Brien et al., 2004] found that, compared with age-matched nonsnoring children, those with primary snoring performed worse on neuropsychologic measures of attention and had more social problems and anxious or depressive symptoms. Community-based studies have determined the prevalence of childhood obstructive sleep apnea at 1.1–2.9 percent [Ali et al., 1993; Brunetti et al., 2001].
OSA is characterized by partial or complete upper airway occlusion, with impaired air exchange despite persistence of thoracic and abdominal respiratory effort. This generally occurs in association with increased resistance to inspiration and transient oxygen desaturation of 3–4 percent (Figure 66-3). In some instances, there is an additional component of hypoventilation due to shallow abdominal and chest wall motion, which leads to hypercarbia. The most common etiologic factors for childhood OSA are adenotonsillar hypertrophy, craniofacial anomalies like micrognathia or maxillary hypoplasia, neuromuscular disorders such as myotonic dystrophy or congenital nonprogressive myopathies, and obesity [Lumeng and Chervin, 2008]. Repetitive occlusion of the upper airway during sleep, with resultant oxygen desaturation, provokes cortical arousals and suppression of REM and N3 sleep. Nocturnal symptoms of childhood obstructive sleep apnea include habitual snoring, restless sleep with snort arousals, bed wetting, excessive sweating, mouth breathing, choking sounds, and parasomnias such as confusional arousals and sleepwalking. Parental reports of snoring that is interrupted by silent pauses, which then terminate with snorting sounds, are characteristic of OSA. A metabolic syndrome, characterized by insulin resistance, hyperglycemia, hypertension, dyslipidemia, abdominal obesity, and proinflammatory and prothrombotic states, may develop as a consequence of OSA [Arens et al., 2010]. Daytime symptoms of OSA include inattentiveness, impaired academic performance, hyperactivity, and sleepiness [Chervin et al., 2002; Gozal, 2008]. The upper airway resistance syndrome is a form of upper airway obstruction in which no frank apneas or oxygen desaturation are observed during sleep, but the airway narrowing leads to recurrent arousals, fragmented sleep, and daytime sleepiness [Guilleminault et al., 1996]. Some patients exhibit subtle posterior displacement of the tongue, narrow nostrils, or a high-arched palate, but others might not have any craniofacial anomalies. The nocturnal polysomnogram may appear superficially normal, with the exception of snoring and increased EEG arousals of 3 or more seconds (normally less than 10–12 per hour of sleep). Simultaneously obtained intraluminal pressures from an esophageal balloon demonstrate a marked increase in the intrathoracic negative pressure during the upper airway resistance syndrome episodes. Nocturnal polysomnography is not needed to confirm the diagnosis in patients who already manifest the classic symptoms of OSA with marked tonsillar hypertrophy; one night of oximetry in the home environment, which documents recurrent oxygen desaturation, is sufficient for establishing the diagnosis in these patients [Brouilette et al., 2000
[/not-level-membership-for-neurology-category]
