CHAPTER 206 Skull Tumors and Fibrous Dysplasia
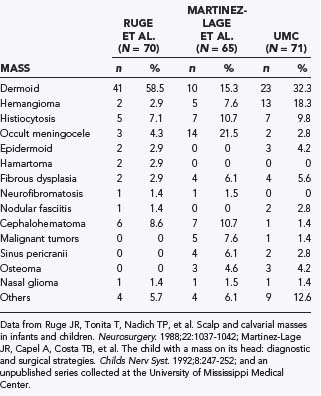
Because such disparate disciplines have been reporting the clinical series, epidemiologic studies have varied considerably in the incidence of the different types of lesions. Variations are noted in the clinical findings associated with the lesion, location, proximity to the skull base, multiplicity of lesions, and degree of intracranial involvement. Therefore, reports on the incidence and types of scalp and skull masses may differ not only because of the focus of a particular institution but also because of the specialty of the physicians collecting the series (Table 206-1).
Inclusion Cysts
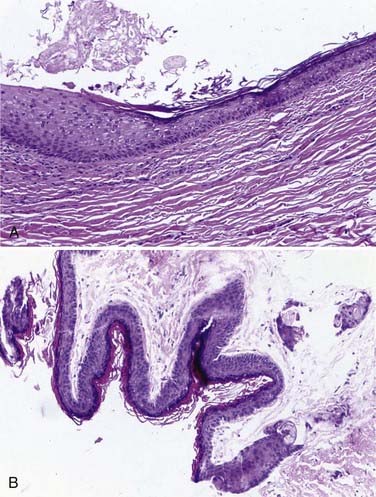
Approximately 50% to 60% of all scalp and skull lesions found in pediatrics are classified as either dermoids or epidermoids (see Table 206-1).1,2 These lesions involve the calvarial bone (especially the outer table), and in some cases the inner table is eroded and the outer leaf of the dura is involved. Dermoids occur more frequently, at a range of 15% to 58%, whereas epidermoids are noted in 3% to 5% of patients. Pathologic analysis differentiates strictly between the two lesions (Fig. 206-1). Epidermoids are composed of only keratin-producing epithelial cells and are characterized by cystic contents with white cheesy keratin debris. Dermoid cysts, in contrast, have walls that are more complex, including the cutaneous adnexal cells of the lower layers of the skin combined with sebaceous and sweat secretions produced by exocrine glands.
Although inclusion cysts may be found anywhere in the scalp, dermoid cysts develop more frequently along the midline in the area covered by the anterior and posterior fontanelles, and epidermoids tend to occur laterally at suture sites between the pterion and the asterion along the squamosal suture.3
On plain radiographs, a lucent center is identifiable and accompanied by an eroding outer table or by involvement of the full thickness of the skull and a rim of hypersclerosis. Computed tomography (CT) will confirm that the lesion is extradural by displaying evidence of bone erosion. Magnetic resonance imaging (MRI) often shows the cyst to have a hypodense or hyperdense core, and the cyst wall may also enhance (Fig. 206-2).4,5
In managing these lesions surgically, a curvilinear incision around the lesion is preferable to an incision on the vertex of the lesion. A curvilinear incision minimizes the risk of incising into the cyst and thereby contaminating the surgical field. Ideally, the cyst should be excised without rupturing it. Although separating the cyst from the surrounding subcutaneous tissue and bone is usually quite simple, a small fibrous or vascular pedicle arising from the suture may require curettage or cauterization. These lesions can occasionally be manifested as a dumbbell configuration affixed to the underlying dura. If the bone defect is very large, reconstruction with split-thickness cranial bone may be necessary. Sharp and scalloped bony margins should be smoothed with a bur, drill, or curet. Incomplete removal of these lesions may lead to recurrence.6–8
Langerhans Cell Histiocytosis
Historically, this entity has been referred to variously as histocytosis X or eosinophilic granuloma; Hand-Schüller-Christian disease and Letterer-Siwe disease are sometimes classified as systemic forms of histiocytosis related to Langerhans cell histiocytosis. Accounting for 7% to 10% of all reported skull lesions, this entity is a non-neoplastic lesion believed to be an inflammatory response to an unknown stimulus.9 It may be a solitary lesion classically referred to as an eosinophilic granuloma, or it may extend to multiple lesions, especially in children younger than 2 years. In its systemic manifestations, the process can disseminate into the central nervous system and through the viscera, thereby generating skin lesions, pulmonary infiltrates, hepatosplenomegaly, pancytopenia, and lymphadenopathy. An intermediate chronic form of this entity features lesions involving the calvaria in the granulomatous process, as well as exophthalmos and diabetes insipidus (obviously affecting the neurohypophysial structures). A recent study has associated degenerative central nervous system histiocytosis with this chronic process.10
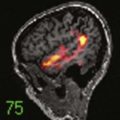
Skull radiographs show an osteolytic lesion with unequal involvement of the outer and inner tables, accompanied by a hyperostotic or sclerotic rim (Fig. 206-3A). CT demonstrates this expansile lesion to best advantage, especially with reconstructed bone formats. Even though the lesion usually enhances moderately, it can have a hemorrhagic core that is hyperdense on non–contrast-enhanced images. Although MRI will confirm the aforementioned findings in soft tissue, it defines the bone changes less clearly than CT does (Fig. 206-3B).
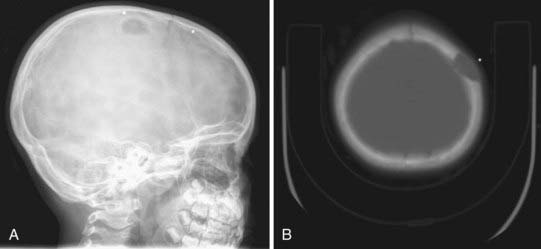
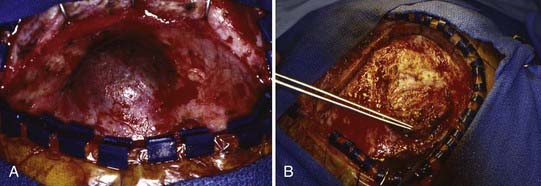
Management of these lesions involves wide surgical resection to normal bone margins (Fig. 206-4). The skull lesions can be attached to the outer leaf of the dura or to subcutaneous tissue, from which the lesion can usually be safely removed by curettage. These friable lesions are quite vascular and expansile within the outer and inner tables of the calvarial bone. Despite occasional location over the venous sinuses, vascular invasion is not seen in radiologic studies of these lesions. Persistence or recurrence of these lesions may require further treatment, including low-dose chemotherapy or, rarely, low-dose radiation therapy.
Hemangioma
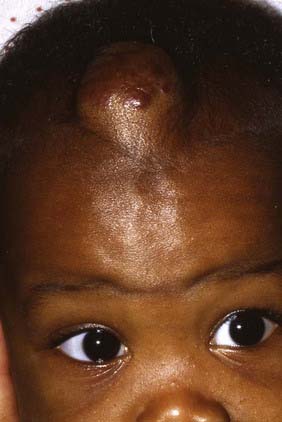
Hemangiomas account for approximately 3% to 20% of reported skull and scalp lesions. They are classified into capillary and cavernous forms. An unusual osseous form that has been noted in adults is rarely seen in children. Hemangiomas tend to occur in the midline but may arise in the diploic spaces, where they extend outward and rarely involve the inner table of the skull, unless their blood supply originates from the dura.11
These lesions may initially be noted primarily as a simple cosmetic deformity or secondarily after trauma as a source of bleeding (Fig. 206-5). In some cases, compression, necrosis, and infection have been identified. In older children, hemangiomas have been accompanied by focal headaches, presumably caused by stretching of the surrounding pericranium.
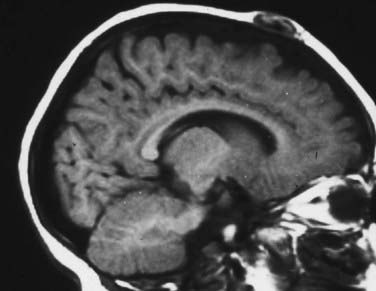
Scalp hemangiomas are best imaged with MRI, which should confirm that the lesions are confined extradurally, especially when they are located in the area of the anterior fontanelle (Fig. 206-6). Either plain radiographs or CT scans provide the clearest images of the bony form of these lesions with sunburst-radiating trabeculae. The vertical orientation of this trabecular honeycombed arrangement of the skull is especially well visualized on tangential cross sections (Fig. 206-7).
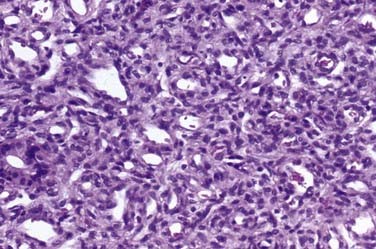
Occult Meningoceles
Cutis aplasia tends to be found over the cranial vertex along the sutural lines (Fig. 206-8). It may vary from less than 1 cm in size to very large scalp lesions (>10 cm). The resulting scalp defect may include the absence of bone formation and even a dural defect. Although smaller lesions (<3 cm) will spontaneously re-epithelialize, larger lesions (especially those with bone and dural defects) will require surgical reconstruction. Involvement of a plastic surgeon for skin rotation flaps, skin grafts, or tissue expanders is highly recommended for these larger lesions.
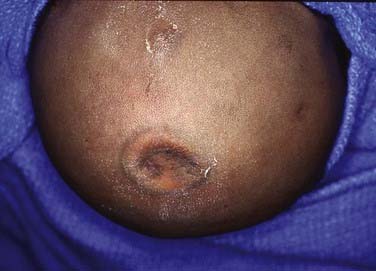
Cephalohematomas
This lesion is usually resorbed in 4 to 6 weeks, but occasionally it may progress to ossification wherein the outer layers of the cephalohematoma calcify around the fluid-filled central cavity of lysed blood cells. Even in cases of ossification, the cranium will continue to remodel as skull growth progresses (Fig. 206-9). In extremely rare cases, severe deformity might require reconstruction of the cranium either by simply removing the outer table of calcification or by performing autologous bone cranioplasties.
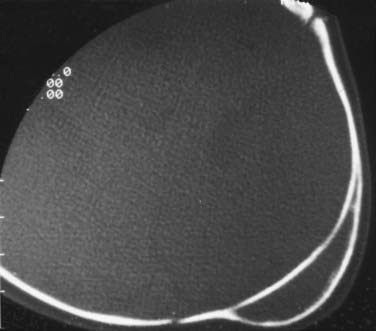
In some cases, cephalohematomas have been associated with infections, and a significant scalp abscess can occur in the subgaleal space (Fig. 206-10). These abscesses might have developed loculations and cannot simply be aspirated. In such cases, incision and dissection of the membranes become necessary, and placement of a drain is recommended. Organisms cultured from these infections have included staphylococci, as well as gram-negative organisms presumably acquired during vaginal delivery.
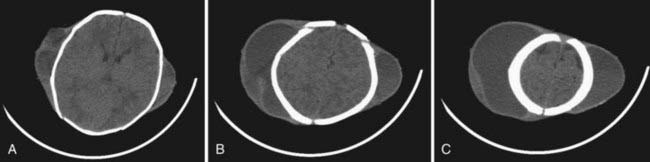
Aneurysmal Bone Cysts
Characteristic fluid-fluid levels within the cyst are seen frequently, and the cortical bone surrounding the lesion is expanded but not destroyed (Fig. 206-11). Despite displacing surrounding central nervous system tissue, these lesions do not penetrate the dura.
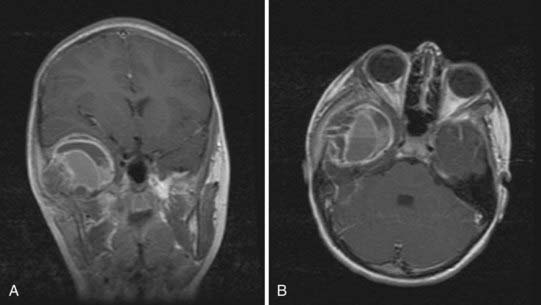
FIGURE 206-11 Coronal (A) and axial (B) magnetic resonance images of an aneurysmal bone cyst showing the large vascular channel.
Surgical management of these lesions should include preoperative angiography accompanied by preoperative embolization to minimize intraoperative bleeding of the very vascular lesions. After complete resection of the lesion, primary reconstruction of the cranial defect is ideal. To reduce the recurrence rate of these lesions, some authors have recommended cryosurgery or low-dose irradiation. Recent reports of medical treatment with methylprednisolone and calcitonin have suggested that a new medical option might be used for lesions in areas difficult to excise, such as in the spinal atlas-axis joint.12
Cranial Fasciitis
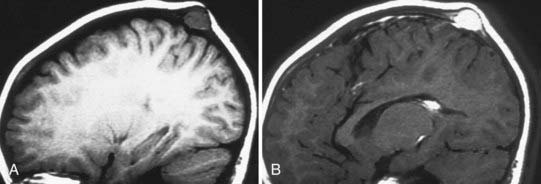
This unusual skull and scalp lesion occurs rarely in the pediatric population and is generally a painless mass.13,14 Cranial fasciitis tends to grow rapidly and has been characterized histologically by inflammatory cells and focal multinucleated cells separated by bundles of ionized collagen tissue. These lesions must be differentiated from fibrous histiocytoma and fibrous sarcoma, which rarely occur in pediatric patients. The lesions often erode the cranium and may extend to the dura. Symptoms are generally related to a mass effect.
A distinct finding on MRI is hyperintense areas with central nonenhancement on T2-weighted images (Fig. 206-12). The lesions are readily excisable, and reconstruction of the underlying skull may be required, depending on the age of the child (Fig. 206-13). The masses have been described as firm and fleshy but are readily dissectible from the dura by blunt dissection.
Melanotic Neuroectodermal Tumor (Melanotic Progonoma)
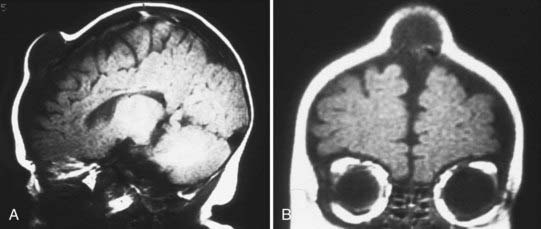
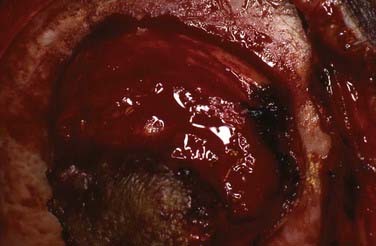
These lesions tend to occur in infancy and have been thought to be derived from neural crest cells. Two thirds of the lesions will be found in the midface region, whereas the remainder may be found in either the mandible or the cranial vault. Radiologically, these lesions resemble osteogenic sarcoma (Fig. 206-14). At the time of surgery, patients tend to have discolored bone that is somewhat yellowish and very thick. The lesion must be resected to normal bone margins, and reconstruction of the cranial defect is required. In rare cases, these lesions have been reported as malignant.15 Histologically, there is an epithelial-like arrangement of melanotic cells interwoven with small cells resembling neuroblasts.
Sinus Pericranii
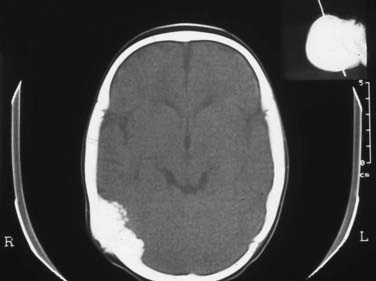
Sinus pericranii are usually median or paramedian lesions between the extracranial and intracranial veins. They tend to be painless and are characterized as being compressible or deflating in the upright position and engorging either during a Valsalva maneuver or in the recumbent position. Although most of these lesions are congenital, some have been acquired by trauma. There have been cases in which they were noted to enlarge with time, and because of such enlargement, cosmetic issues, and concern about the potential for hemorrhage, surgery has been advocated. These lesions are best visualized on CT or CT angiography. They can also be visualized on magnetic resonance venography (Fig. 206-15).
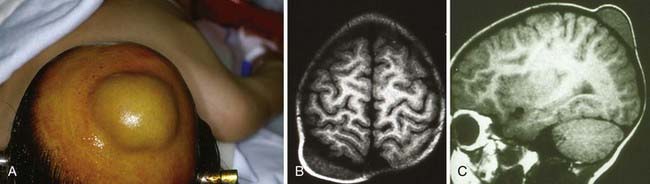
Fibrous Dysplasia
Fibrous dysplasia, a benign skeletal lesion of immature mesenchymal cells, typically involves the long bones, ribs, and skull.16,17 It begins as mutated pluripotent embryonic cells from which the skeletal stem cells develop.18 Lesional bone is formed when cancellous bone is replaced with fibrous, cellular tissue and when collagen is deposited with immature woven bone.19 Affected bone is rich in versican and osteonectin, which are antiadhesion molecules, and lacks the adhesion-promoting molecules osteopontin and bone sialoprotein.20 The affected bone can be monostotic, or involving one bone, or polyostotic, in which multiple bones are affected.
The genetic mutation for fibrous dysplasia is located on chromosome 20q13 at the GNAS locus. The aberrant gene codes for a constitutively active mutant of the Gsα subunit of the G protein that couples intracellular cyclic adenosine monophosphate production to hormone.21,22 As a result, the normal guanosine triphosphatase activity required to deactivate the G protein is diminished. The abnormal allele is present in mosaic patterning.22 Interestingly, this mutation is also found in hypersecreting thyroid tumors, in Leydig cell tumors, and in up to 40% of growth hormone–secreting pituitary adenomas.21,22 Parental imprinting (an epigenetic phenomenon involving post-transcriptional DNA methylamine and histone processing, as well as early embryonic mutation) probably accounts for the phenotypic variance and mosaicism.21
McCune-Albright syndrome, also a result of activation of Gsα, consists of polyostotic fibrous dysplasia, endocrinopathy (particularly precocious puberty), and café au lait spots. A clinical diagnosis can be made with two of these three criteria.23
Radiographically, lesional bone is evident on thin-cut CT and displays a classic ground-glass appearance. Three-dimensional CT and multiplanar views can be helpful in preoperative planning (Fig. 206-16).
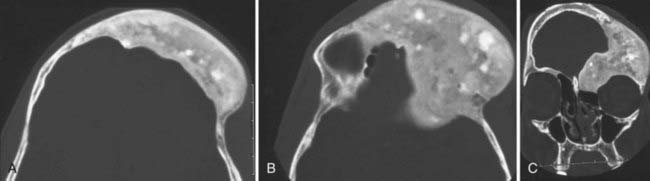
The bone disease usually stabilizes by adolescence and can be managed conservatively, but its sequelae, such as pain and fractures, can last throughout adulthood.24 The bone pain is most often relieved with bisphosphonate therapy, but the effects of other sequelae on bone turnover, bone density, and radiographic changes still remain unclear.25 From a craniofacial standpoint, orbital apical involvement is not uncommon and most often consists of proptosis, dystonia, and distorted facies.26 Although optic nerve encasement is common, blindness is more infrequent.26,27 Growth hormone excess seems to be a risk factor for both nerve encasement and visual loss.27 One series of patients with craniofacial fibrous dysplasia found that the ethmoid bones (71%) were involved most often, followed by the sphenoid (43%), frontal bones (33%), maxilla (29%), temporal bones (24%), parietal bones (14%), and occipital bone (5%).28 Goals of surgery include decompression of any neurological structures and cosmesis. Malignant transformation of fibrous dysplasia is rare, and the possibility that radiation could induce malignant transformation is controversial. In one large retrospective series, the incidence of sarcomas arising from fibrous dysplasia was 2.5%, with just under half of the patients reporting a history of irradiation.29
Bianco P, Kuznetsov SA, Riminucci M, et al. Reproduction of human fibrous dysplasia of bone in immunocompromised mice by transplanted mosaics of normal and Gs alpha–mutated skeletal progenitor cells. J Clin Invest. 1998;101:1737-1744.
Chanson P, Salenave S, Orcel P. McCune-Albright syndrome in adulthood. Pediatr Endocrinol Rev. 2007;4(suppl 4):453-462.
Chauhari AB, Ladapo F, Mordi VP. Congenital inclusion cyst of the subgaleal space. J Neurosurg. 1982;56:540-544.
Cruz AA, Constanzi M, de Castro FA, et al. Apical involvement with fibrous dysplasia: implications for vision. Ophthal Plast Reconstr Surg. 2007;23:450-454.
Cutler CM, Lee JS, Butman JA, et al. Long-term outcome of optic nerve encasement and optic nerve decompression in patients with fibrous dysplasia: risk factors for blindness and safety of observation. Neurosurg. 2006;59:1011-1017.
De Carvalho GTC, Fagundes-Pereyra WJ, Marques JAP, et al. Congenital inclusion cysts of the anterior fontanelles. Surg Neurol. 2001;56:400-405.
DiCaprio MR, Enneking WF. Fibrous dysplasia. Pathophysiology, evaluation, and treatment. J Bone Joint Surg Am. 2005;87:1848-1864.
DiMeglio LA. Bisphosphonate therapy for fibrous dysplasia. Pediatr Endocrinol Rev. 2007;4(suppl 4):440-445.
Di Rocco F, Roujeau T, Puget S, et al. Slow-growing lambdoid melanotic neuroectodermal tumor of infancy. Eur J Pediatr. 2007;166:739-741.
Gibson SE, Prayson RA. Primary skull lesions in the pediatric population—a 25-year experience. Arch Pathol Lab Med. 2007;131:761-766.
Glasauer FE, Levy LF, Auchterlonie WC. Congenital inclusion dermoid cyst of the anterior fontanel. J Neurosurg. 1978;48:274-278.
Howard R. Congenital midline lesions: pits and protuberances. Pediatr Ann. 1998;27:150-160.
Johnson KK, Dannenbaum MJ, Bhattacharjee MB, et al. Diagnosing cranial fasciitis based on distinguishing radiological features. J Neurosurg Pediatr. 2008;2:370-374.
Kershenovich A, Price AV, Koral K, et al. Failure to treat obstructive hydrocephalus with endoscopic third ventriculostomy in a patient with neurodegenerative Langerhans cell histiocystosis. J Neurosurg Pediatr. 2008;2:304-309.
Lania A, Mantovani G, Spada AG. G protein mutations in endocrine diseases. Eur J Endocrinol. 2001;145:543-559.
Lietman SA, Schwindinger WF, Levine MA. Genetic and molecular aspects of McCune-Albright syndrome. Pediatr Endocrinol Rev. 2007;4(suppl 4):380-385.
Lustig LR, Holliday MJ, McCarthy EF, et al. Fibrous dysplasia involving the skull base and temporal bone. Arch Otolaryngol Head Neck Surg. 2001;127:1239-1247.
Martínez JF, Torroba A, López, et al. Cranial fasciitis of the anterior fontanel. Childs Nerv Syst. 1997;13:626-628.
Martinez-Lage JF, Quiñonez MA, Poza M, et al. Congenital epidermoid cysts over the anterior fontanelle. Childs Nerv Syst. 1985;1:319-323.
Martinez-Lage JR, Capel A, Costa TB, et al. The child with a mass on its head: diagnostic and surgical strategies. Childs Nerv Syst. 1992;8:247-252.
Michienzi S, Cherman N, Holmbeck K, et al. GNAS transcripts in skeletal progenitors: evidence for random asymmetric allelic expression of Gs alpha. Hum Mol Genet. 2007;16:1921-1930.
Ohashi M, Ito T, Hirano T, et al. Percutaneous intralesional injection of calcitonin and methylprednisolone for treatment of an aneurismal bone cyst at C-2. J Neurosurg Pediatr. 2008;2:365-369.
Riminucci M, Fisher LW, Shenker A, et al. Fibrous dysplasia of bone in the McCune-Albright syndrome: abnormalities in bone formation. Am J Pathol. 1997;151:1587-1600.
Riminucci M, Saggio I, Robey PG, et al. Fibrous dysplasia as a stem cell disease. J Bone Miner Res. 2006;21(Suppl 2):125-131.
Ruge JR, Tonita T, Nadich TP, et al. Scalp and calvarial masses in infants and children. Neurosurgery. 1988;22:1037-1042.
Ruggieri P, Sim FH, Bond JR, et al. Malignancies in fibrous dysplasia. Cancer. 1994;73:1411-1414.
Stokes RB, Saunders CJ, Thaller SR. Bregmatic epidermoid inclusion cyst eroding both calvarial tables. J Craniofac Surg. 1996;7:148-150.
Weinstein LS. G(s) alpha mutations in fibrous dysplasia and McCune-Albright syndrome. J Bone Miner Res. 2006;21(Suppl 2):120-124.
Yoon SH, Park S-H. A study of 77 cases of surgically excised scalp and skull masses in pediatric patients. Childs Nerv Syst. 2008;24:459-465.
1 Ruge JR, Tonita T, Nadich TP, et al. Scalp and calvarial masses in infants and children. Neurosurgery. 1988;22:1037-1042.
2 Martinez-Lage JR, Capel A, Costa TB, et al. The child with a mass on its head: diagnostic and surgical strategies. Childs Nerv Syst. 1992;8:247-252.
3 De Carvalho GTC, Fagundes-Pereyra WJ, Marques JAP, et al. Congenital inclusion cysts of the anterior fontanelles. Surg Neurol. 2001;56:400-405.
4 Stokes RB, Saunders CJ, Thaller SR. Bregmatic epidermoid inclusion cyst eroding both calvarial tables. J Craniofac Surg. 1996;7:148-150.
5 Martinez-Lage JF, Quiñonez MA, Poza M, et al. Congenital epidermoid cysts over the anterior fontanelle. Childs Nerv Syst. 1985;1:319-323.
6 Gibson SE, Prayson RA. Primary skull lesions in the pediatric population—a 25-year experience. Arch Pathol Lab Med. 2007;131:761-766.
7 Chauhari AB, Ladapo F, Mordi VP. Congenital inclusion cyst of the subgaleal space. J Neurosurg. 1982;56:540-544.
8 Glasauer FE, Levy LF, Auchterlonie WC. Congenital inclusion dermoid cyst of the anterior fontanel. J Neurosurg. 1978;48:274-278.
9 Yoon SH, Park S-H. A study of 77 cases of surgically excised scalp and skull masses in pediatric patients. Childs Nerv Syst. 2008;24:459-465.
10 Kershenovich A, Price AV, Koral K, et al. Failure to treat obstructive hydrocephalus with endoscopic third ventriculostomy in a patient with neurodegenerative Langerhans cell histiocystosis. J Neurosurg Pediatr. 2008;2:304-309.
11 Howard R. Congenital midline lesions: pits and protuberances. Pediatr Ann. 1998;27:150-160.
12 Ohashi M, Ito T, Hirano T, et al. Percutaneous intralesional injection of calcitonin and methylprednisolone for treatment of an aneurismal bone cyst at C-2. J Neurosurg Pediatr. 2008;2:365-369.
13 Johnson KK, Dannenbaum MJ, Bhattacharjee MB, et al. Diagnosing cranial fasciitis based on distinguishing radiological features. J Neurosurg Pediatr. 2008;2:370-374.
14 Martínez JF, Torroba A, López, et al. Cranial fasciitis of the anterior fontanel. Childs Nerv Syst. 1997;13:626-628.
15 Di Rocco F, Roujeau T, Puget S, et al. Slow-growing lambdoid melanotic neuroectodermal tumor of infancy. Eur J Pediatr. 2007;166:739-741.
16 Weinstein LS. G(s) alpha mutations in fibrous dysplasia and McCune-Albright syndrome. J Bone Miner Res. 2006;21(suppl 2):120-124.
17 DiCaprio MR, Enneking WF. Fibrous dysplasia. Pathophysiology, evaluation, and treatment. J Bone Joint Surg Am. 2005;87:1848-1864.
18 Riminucci M, Saggio I, Robey PG, et al. Fibrous dysplasia as a stem cell disease. J Bone Miner Res. 2006;21(suppl 2):125-131.
19 Bianco P, Kuznetsov SA, Riminucci M, et al. Reproduction of human fibrous dysplasia of bone in immunocompromised mice by transplanted mosaics of normal and Gs alpha–mutated skeletal progenitor cells. J Clin Invest. 1998;101:1737-1744.
20 Riminucci M, Fisher LW, Shenker A, et al. Fibrous dysplasia of bone in the McCune-Albright syndrome: abnormalities in bone formation. Am J Pathol. 1997;151:1587-1600.
21 Michienzi S, Cherman N, Holmbeck K, et al. GNAS transcripts in skeletal progenitors: evidence for random asymmetric allelic expression of Gs alpha. Hum Mol Genet. 2007;16:1921-1930.
22 Lania A, Mantovani G, Spada AG. G protein mutations in endocrine diseases. Eur J Endocrinol. 2001;145:543-559.
23 Lietman SA, Schwindinger WF, Levine MA. Genetic and molecular aspects of McCune-Albright syndrome. Pediatr Endocrinol Rev. 2007;4(suppl 4):380-385.
24 Chanson P, Salenave S, Orcel P. McCune-Albright syndrome in adulthood. Pediatr Endocrinol Rev. 2007;4(suppl 4):453-462.
25 DiMeglio LA. Bisphosphonate therapy for fibrous dysplasia. Pediatr Endocrinol Rev. 2007;4(suppl 4):440-445.
26 Cruz AA, Constanzi M, de Castro FA, et al. Apical involvement with fibrous dysplasia: implications for vision. Ophthal Plast Reconstr Surg. 2007;23:450-454.
27 Cutler CM, Lee JS, Butman JA, et al. Long-term outcome of optic nerve encasement and optic nerve decompression in patients with fibrous dysplasia: risk factors for blindness and safety of observation. Neurosurgery. 2006;59:1011-1017.
28 Lustig LR, Holliday MJ, McCarthy EF, et al. Fibrous dysplasia involving the skull base and temporal bone. Arch Otolaryngol Head Neck Surg. 2001;127:1239-1247.
29 Ruggieri P, Sim FH, Bond JR, et al. Malignancies in fibrous dysplasia. Cancer. 1994;73:1411-1414.