Chapter 69A Simple cysts and polycystic liver disease
Clinical and radiographic features
Overview
The entity common to simple cysts and polycystic liver disease (PCLD) is a malformation, lined by a nontypical biliary-type epithelium that does not communicate with the biliary tree. This differentiates these diseases from hydatid cysts (see Chapter 68), cystic hepatobiliary neoplasms (Chapter 79B), and cystic dilation of the intrahepatic bile ducts (Chapter 46).
This chapter focuses on pathology, prevalence, manifestation, and diagnosis of PCLD. Differential diagnosis from other cystic lesions is addressed at the end of this chapter, and therapeutic management is addressed in Chapter 69B.
Simple Cysts of the Liver
Pathology and Pathogenesis
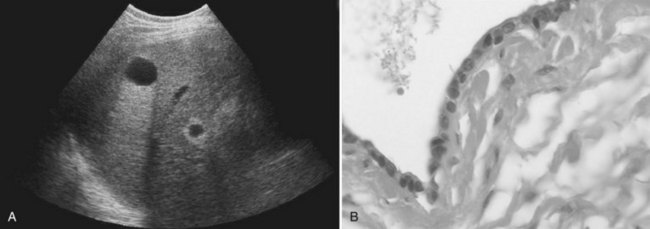
Microscopically, the cysts appear to be bordered by a single layer of cuboid or columnar epithelial cells, resembling biliary epithelial cells (Fig. 69A.1). The cells are uniform, without any atypia. Stroma is absent in small cysts and is reduced to a thin layer of connective tissue in large cysts. Simple cysts of the liver are regarded as a congenital malformation, likely aberrant bile ducts that have lost communication with the biliary tree and dilate progressively. Liver cyst epithelial cells retain differentiated secretory function; they secrete fluid and generate a positive luminal pressure that may be greater than 30 cm H2O, which accounts for most symptoms. The composition of the fluid, which contains water and mineral electrolytes without bile acids and bilirubin, is close to that of the normal secretion of the epithelium of the bile ducts. It is not toxic to the peritoneum, which is the rationale for cyst fenestration (see Chapter 69B).
Prevalence and Etiology
Simple cysts had been considered rare. By 1971, only 350 cases had been reported (Flagg & Robinson, 1967; Moreaux & Bloch, 1971), and early estimates from autopsy studies were less than 1% (Larsen, 1961). However, the development of imaging techniques revealed that simple cysts are much more frequent. Prevalence in ultrasound studies is 3% to 5% (Caremani et al, 1993; Gaines & Sampson, 1989); however, in a more recent spiral computed tomographic (CT) study of an adult population, prevalence was 18% (Carrim & Murchison, 2003). These discrepancies are explained by the usual small size of most cysts, which are discovered only by accurate imaging techniques. An association between simple hepatic cysts and simple renal cysts has been documented (Carrim & Murchison, 2003), but this association has not yet been explained.
The incidence of simple cysts is age and gender related. Simple cysts are uncommonly identified before age 40 years, but their incidence increases sharply thereafter. Simple cysts also are larger in adults older than 50 years (Larsen, 1961). The female/male ratio is 1.5 : 1 for all simple cysts shown at necropsy or imaging; however, this statistic jumps to 9 : 1 in symptomatic or complicated simple cysts (Moreaux & Bloch, 1971). Huge cysts affect women older than 50 years almost exclusively. Based on these observations, the diagnosis of simple cysts should be retained with caution, when a large or symptomatic cystic mass is observed in a male patient or in a young woman.
Manifestations and Diagnosis
US is the best imaging modality for recognizing simple cysts, which appear as a circular or oval, completely anechoic lesion with sharp, smooth borders and strong posterior wall echoes that indicate a well-defined tissue-fluid interface (Liang et al, 2005). Echoes beyond the cyst are accentuated compared with echoes at a similar depth transmitted through normal adjacent liver tissue (see Fig. 69A.1). Accentuation of the echoes beyond the cyst indicates that the lesion is filled with fluid. This so-called acoustic posterior enhancement is observed only when the deep tissue transmits US; it is not seen when a total reflector, such as gas or bone, lies behind the cyst or when the cyst is very posterior. Neither wall nor septation is apparent, but a false image of septation may be seen when two cysts are adjacent. In some patients, intracystic echoes or material can be detected after intracystic bleeding.
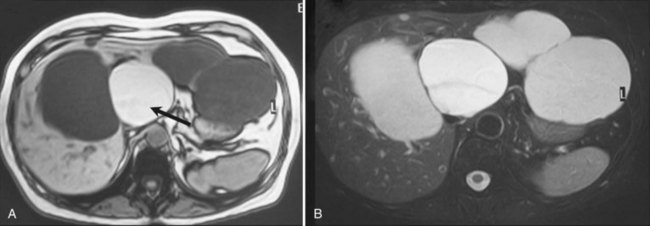
Other imaging procedures have less utility in diagnosis compared with US and are generally not required. CT confirms the presence of one or several round or oval, water-dense lesions but no septations or intracystic formations; dynamic CT shows that such lesions are avascular. In small cysts, recognition of water density may be difficult; the average density of the cyst is obscured by the density of the adjacent liver tissue, but an apparent increase in density may be observed after intravenous injection of contrast medium. On magnetic resonance imaging (MRI), the cyst is homogeneous, very hypointense on T1-weighted images, and very hyperintense on T2-weighted images, as are the gallbladder contents (Fig. 69A.2). If the patient lives or has been living in an area where hydatid disease is endemic, or when any doubt exists about a possible hydatid cyst, serologic tests for this parasitic infection should be performed, although their accuracy is fairly low. MRI should also be performed (see Chapter 68).
Course and Complications
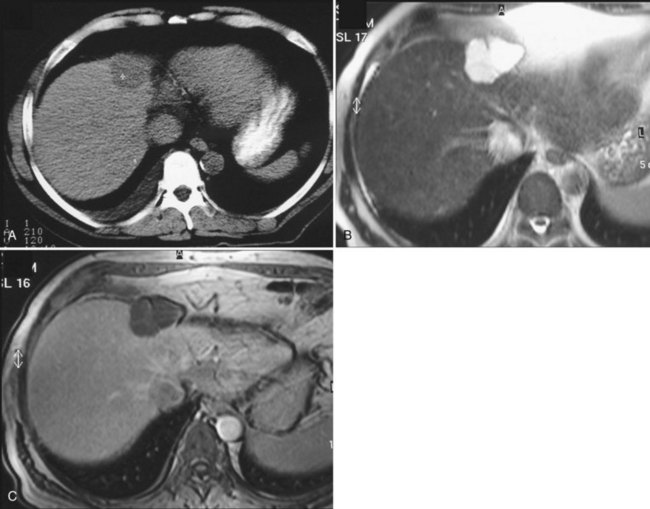
Complications are uncommon, but intracystic bleeding is the most frequent observed (Frisell et al, 1979; Moreaux & Bloch, 1971) and is presumed to result from the erosion of an artery adjacent to the cyst. The clinical manifestations consist of sudden, severe pain and an increase in the size of the cyst. The pain resolves in a few days; in some cases, pain is mild or absent. There is no evidence of anemia, and the biologic liver tests show only a moderate elevation of γ-glutamyltransferase (GGT). On US, echoic material corresponding to clots is seen within the cyst (Fig. 69A.3); this material usually is mobile, sliding in the inferior part of the cyst. Hemorrhagic cysts also become hyperintense on T1-weighted MRI, whereas uncomplicated cysts are hypointense (see Fig. 69A.2). After gadolinium injection, an enhancement of the periphery of the cyst is frequently seen when hemorrhage is recent, which corresponds to compressed liver parenchyma (Fig. 69A.4). Contrast US may also prove useful in the future by showing that neither the cyst wall nor the intracystic material is enhanced, unlike cystadenoma (see Fig. 69A.4; Chapter 79B), but larger studies are required (Akiyama et al, 2008). Communication with the bile duct lumen may develop, presumably also by an erosive mechanism; however, bacterial infection in contrast is exceptional.
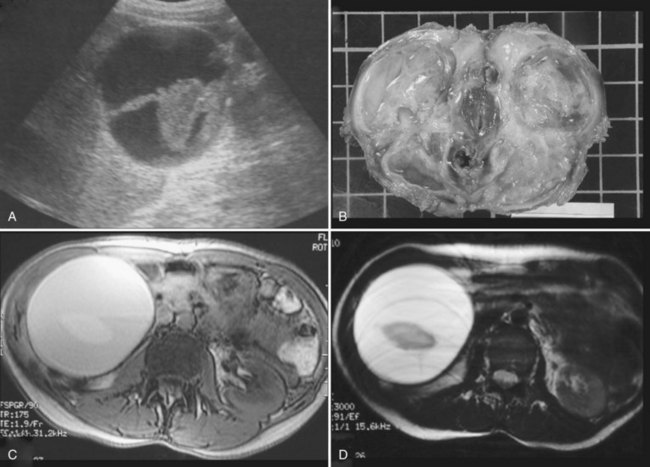
Spontaneous rupture into the peritoneal cavity (Fig. 69A.5; Akriviadis et al, 1989; Salemis et al 2007)—or, more rarely, into the pleural cavity or the duodenum (Williamson et al, 1978)—is exceptional. In contrast to hydatid cysts, peritoneal perforation of simple cysts is self-limited. Severe hemoperitoneum has been described only in patients with PCLD undergoing dialysis.
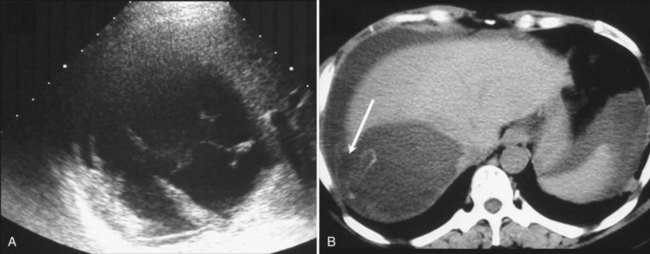
Because simple cysts are tense, they may compress the bile ducts or the inferior vena cava (Fig. 69A.6), and although anecdotal, there have been reports of pulmonary embolism after clot formation (Buyse et al, 2004). As discussed in Chapter 69B, compression of biliary branches or hepatic veins is a potential source of intraoperative or postoperative complications.
Polycystic Liver Disease
PCLD—or, more precisely, diseases—is a term for genetic disorders responsible for the progressive development of multiple cysts in the liver. PCLD was initially considered to exclusively develop in the context of ADPKD (Moschcowitz, 1906). However, identification of families with PCLD but no ADPKD proved that PCLD may also occur alone (Berrebi et al, 1982; Tahvanainen, et al, 2003). These two forms of PCLD—those with and without ADPKD—are linked to distinct gene mutations. Both, however, have an autosomal dominant transmission and an almost identical clinical course. Of note, one third of patients with isolated PCLD may have a few kidney cysts, usually one or two (van Keimpema et al, 2010), as does the general population of adult patients (Carrim & Murchison, 2003). Distinction between the two forms of PCLD is addressed below.
Genetics
Polycystic Liver Disease and Autosomal Dominant Polycystic Kidney Disease
PCLD associated with ADPKD is linked to mutations in either the PKD1 or PKD2 gene. PKD1 (Ward et al, 1996) is located on the short arm of chromosome 16 and encodes polycystin-1; its mutation accounts for 85% of mutations in ADPKD patients. Most of the rest are related to a mutation in PKD2 (Mochizuki et al, 1996), which is located on chromosome 4 and encodes polycystin-2. PKD1 mutation is associated with more renal cysts, larger cysts, more prevalent hypertension, and faster progression to end-stage renal disease than PKD2 mutation.
Isolated Polycystic Liver Disease
Isolated PCLD—that is, PCLD without ADPKD—is associated with heterozygous mutation in either the Protein Kinase C Substrate 80K-H (PRKCSH) or SEC63 genes. Both genes are located in the endoplasmic reticulum and function in the early secretory route of the cell. PRKCSH is located on the short arm of chromosome 19, which encodes the protein hepatocystin (Drenth et al, 2004). Hepatocystin functions as the β-subunit of α-glucosidase II and is involved in the protein-folding processes and quality control of newly synthesized glycoproteins (Trombetta et al, 2001). SEC63 is located on the long arm of chromosome 6 and encodes the SEC63 protein involved in protein transport across the endoplasmic reticulum membrane (Brodsky et al, 1995). However, these mutations only explain 25% to 40% of cases (Davila et al, 2004; van Keimpema et al, 2010), which suggests that one or more other genes is involved.
The Two-Hit Theory
PCLDs have an autosomal dominant phenotype but are considered to be a recessive disease at the molecular level because they require a second somatic mutation. According to this theory, a germline mutation is present in either PKD1 or PKD2. However, cell proliferation and cyst formation occurs in individual cells that have in addition received a second loss-of-function somatic mutation in the other gene copy (Everson et al, 2004). This second hit would explain why affected cells are few and why cysts develop from place to place; it may also account for the extreme heterogeneity of the disease. This theory has been designed for PCLD associated with ADPKD but has not been investigated for isolated PCLD. It is unclear as to why these various mutations may affect both the liver and kidney or only the liver.
Pathology and Pathogenesis
Liver cysts in PCLD are macroscopically and microscopically similar to simple cysts of the liver, and their number and distribution are highly variable. They are lined by a single-layered epithelium that has phenotypic and functional characteristics of biliary epithelium (Perrone et al, 1995) and retain in particular a secretory capacity and responsiveness to secretin (Everson et al, 1990). The cysts result from abnormal remodeling of the ductal plate and mainly arise from dilation of biliary microhamartomas, also known as von Meyenburg complexes, which have lost their communication with the biliary tree (Melnick, 1975). Some cysts also arise from peribiliary glands surrounding large intrahepatic bile ducts (Fig. 69A.7; Kida et al, 1992).
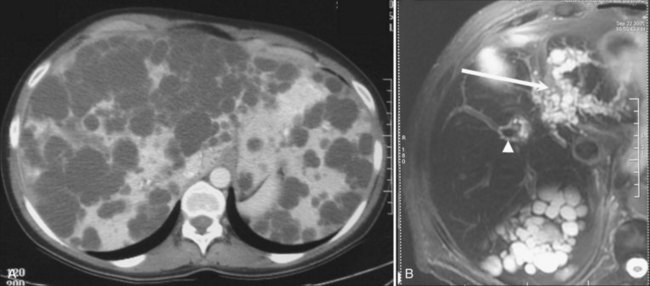
The mechanisms of cyst expansion are still not fully explained but appear similar to those involved in the development of renal cysts. Growth results from a combination of proliferation of epithelial cells, remodeling of the extracellular matrix required for the cyst to invade the surrounding liver parenchyma (Murray et al, 1996), and neovascularization that results in increased density of the vascular bed surrounding the cysts (Bello-Reuss et al, 2001; Nichols et al, 2004).
As in the kidney tubules, the microvilli and long cilia normally on cholangiocytes are believed to play a dominant role. These mechanosensory organelles may be bent by bile flow and can also react to hormonal, morphogen, or growth factor stimuli; they may even function as sensors of cell injury. These cilia and microvilli modulate concentrations of intracellular levels of cyclic adenosine monophosphate (cAMP) and Ca2+ that are mediators of proliferation and secretory activity of cholangiocytes. In PCLD, these cilia progressively disappear from the luminal surface of the epithelium as cysts enlarge (Alvaro et al, 2008). Their absence could impair Ca2+-dependent counterregulation of cAMP, which is a key determinant of cholangiocyte proliferation. It is unclear whether the disappearance of these cilia is a primary event or a result of increased pressure. However, polycystin-1 and polycystin-2 are localized to the cilia, and their absence as a result of a PKD1 or a PKD2 mutation could result in ciliary dysfunction. In patients with isolated PCLD, it has been postulated that polycystins could be the target of aberrant maturation of newly synthesized glycoprotein caused by PRKCSH or SEC63 mutations.
Two other targets of the cell cycle have been identified. One is the mammalian target of the rapamycin (mTOR)-mediated signaling cascade (Shillingford et al, 2006), which explains growing interest from the industry for mTOR inhibitors (see Chapter 69B). The other is through micro-RNAs, small, noncoding RNAs that inhibit target messenger RNA transcripts via sequence-specific base paring; micro-RNA-15a in particular is downregulated in cyst epithelium, resulting in an overexpression of Cdc25A, which activates cyclin-dependent kinases and progression of the cell cycle (Lee et al, 2008).
Fluid secretion by the epithelium lining the cyst may also contribute to cyst expansion. Ciliary dysfunction could also play a role because it has also been shown to result in maintenance of an immature phenotype by biliary epithelial cells with postnatal expression of developmental proteins. A variety of cytokines and growth factors present in increased concentration in the cysts have been implicated in cyst growth by autocrine-paracrine signaling (Alvaro et al, 2008; Fabris et al, 2006; Nichols et al, 2004). Angiogenic factors in particular could play a role by promoting vascularization of the cyst and also by directly stimulating cholangiocytes. Furthermore, the epithelium stains positive for insulin growth factor 1 (IGF-1) and its receptor, as well as for estrogen receptors (Alvaro et al, 2008), and is particularly sensitive to this signaling.
All these observations and hypotheses account for the rationale of using vasopressin antagonists or somatostatin analogues known to inhibit cAMP; sirolimus, an inhibitor of mTOR signaling; or vascular endothelial growth factor inhibitors in the treatment of PCLD (see Chapter 69B). The presence of estrogen receptors also provides a rationale for the influence of the patient’s estrogen status on the clinical course of the disease. In particular, women with ADPKD are more likely to develop hepatic cysts than their male counterparts (Chapman, 2003), and nulliparous women are less likely to develop hepatic cysts than their multiparous counterparts (Gabow et al, 1990). Oral contraceptives and postmenopausal hormonal substitutive therapy increases both the number and size of liver cysts (Sherstha et al, 1997).
Epidemiology
PCLD Associated with ADPKD
ADPKD occurs in approximately 1 in every 800 live births and is characterized by progressive development and enlargement of fluid-filled cysts, ultimately leading to renal failure in 50% of affected individuals. PCLD is the most frequent extrarenal associated condition, but liver cysts develop later than renal cysts. The prevalence of liver cysts also increases with age. This was recently evaluated by MRI in a young adult population with relatively preserved kidney function and was found to be 58% in patients aged 15 to 24 years, 85% in those aged 25 to 34 years, and 94% in those aged 35 to 46 years (Bae et al, 2006). Overall prevalence is the same in men and women, but as for simple cysts, women have more severe liver disease than men; multiparity and use of oral contraceptive drugs or estrogen replacement therapy also tends to worsen the condition (Bae et al, 2006; Sherstha et al, 1997; Torres et al, 2007). Prevalence of liver cysts is also correlated with kidney volume and renal cyst volume (Bae et al, 2006).
Isolated Polycystic Liver Disease
Isolated PCLD is considered rare, with an estimated incidence of less than 0.01%. Compared with PCLD associated with ADPKD, the overall incidence of isolated PCLD is in fact only slightly lower (Karhunen & Tenhu, 1986); however, in clinical series dealing with symptomatic patients, it is two to five times less frequent (Hoevenaren et al, 2008; Schnelldorfer et al, 2009), which may reflect the milder course of isolated PCLD. As for PCLD associated with ADPKD, cysts increase with age, and although the disease is transmitted equally in men and women, 85% of symptomatic patients are women (van Keimpema et al, 2010).
Manifestations and Diagnosis
Symptoms
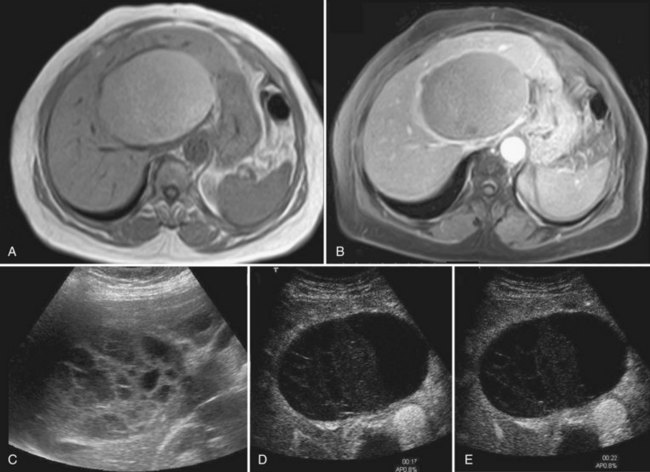
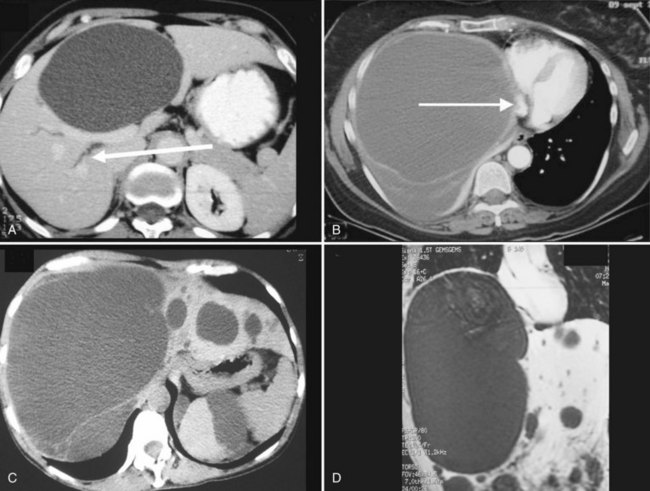
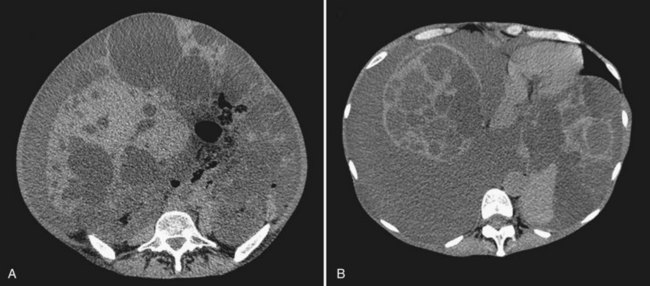
In most patients, liver cysts are small and sparse and remain clinically silent. Even with massive organ enlargement, significant hepatic complications remain fairly uncommon, although patients or others may have noted a protuberant abdomen. Some patients develop symptoms that may include abdominal pain or discomfort, early satiety, shortness of breath, and leg edema. These result from compression of the stomach, diaphragm, and inferior vena cava by single large cysts (see Fig. 69A.6) or, much more frequently, because of enlargement of the entire liver (Fig. 69A.8C). As symptoms have progressively evolved over a prolonged period, patients have often become used to these discomforts and are either unaware of them or minimize them. At clinical examination, the liver may reach an enormous size, and its inferior border is frequently palpated in the iliac fossa. The cysts may be so tense that they can be confused with solid tumors.
Previous history of self-resolving acute abdominal pain is not unusual and probably witnesses spontaneous bleeding within one or more cysts. However, these episodes may remain clinically silent. As the disease progresses, nutritional deficits may develop and be associated with ascites or pleural effusion (Fig. 69A.9). Body weight is an inaccurate marker because muscular loss correlates with the progressive increase in the weight of the liver, which may reach more than 10 kg. The clinician should instead focus on the thickness of the muscles of the abdominal wall or psoas muscle on imaging studies.
Typically, patients referred to surgeons are women between 35 and 50 years of age with liver cysts diagnosed 10 years previously whose symptoms have become incapacitating for 6 to 18 months. Clinical manifestations, as well as complications and management, are the same whether PCLD is isolated or associated with ADPKD (Hoevenaren et al, 2008; Qian et al, 2003).
Biology
The only abnormality may be an increase in GGT or alkaline phosphatase, but this is infrequent. Although the liver may be diffusely involved by liver cysts, the hepatic parenchymal volume is preserved (Everson et al, 1988). Therefore no sign of liver failure is evident; when present, a superimposed cause of chronic liver disease is likely. This lack of influence on liver function explains how polycystic livers from brain-dead donors have even been used in the context of emergency liver transplantation (Glanemann et al, 2000).
Imaging
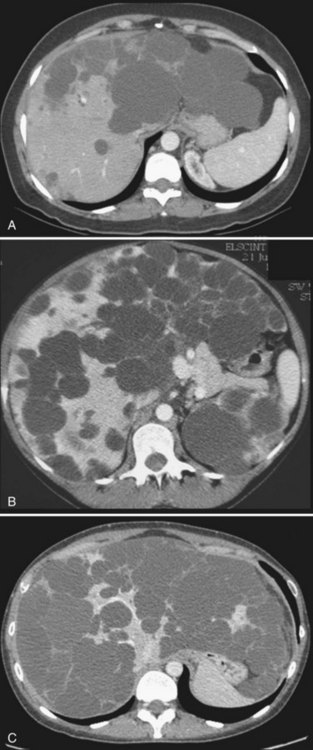
US shows multiple, fluid-filled, round or oval cysts with sharp margins in the liver and, when associated with ADPKD, in the kidneys. Coalescent cysts may give a false image of septation. On CT scan, the cysts have fluid attenuation values of −5 to +20 Hounsfield units (HU) and have distinct margins with the parenchyma. Calcification of the cysts has been reported but is rare (Coffin et al, 1990; Kutcher et al, 1977). Considering the high prevalence of associated ADPKD, injection of contrast agents should be used with extreme caution. Cysts do not show contrast enhancement, and it may be extremely difficult to identify vascular and biliary structures or even normal landmarks of the liver. Similar to simple cysts, uncomplicated cysts of PCLD are very hyperintense on T2-weighted MRI and very hypointense on T1-weighted images; cysts complicated by hemorrhage become hyperintense on T1-weighted images (see Fig. 69A.2). Identifying peribiliary cysts is difficult but may be suspected when tubular structures run parallel to portal branches, mimicking a biliary dilatation (see Fig. 69A.7; Gupta et al, 1999).
With respect to treatment (see Chapter 69B), attention should be paid to several factors:
1 The severity of cystic involvement (see Fig. 69A.8). In a small subset of patients, symptoms are not related to the size of the entire liver but to one or two large cysts. These patients should be identified because even though they may have multiple cysts, management is the same as for those with simple cysts.
2 The distribution of the cysts. This is frequently uneven; some areas are relatively spared, in particular segments V or VI, which is the rationale for liver resection.
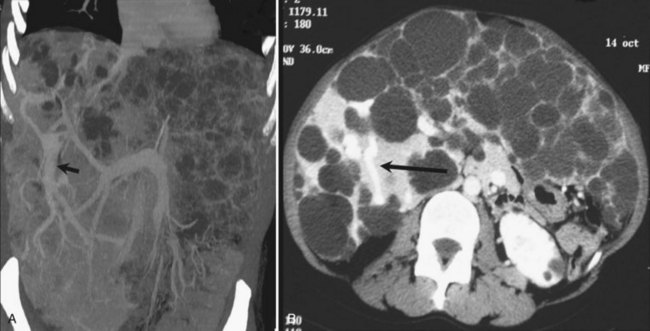
3 Peribiliary cysts or cysts located around the biliary confluence and those located in the vicinity of the termination of the hepatic veins. These may be a common finding. Right hepatic vein compression is frequent and results in an easily identifiable collateral circulation between the right and middle hepatic vein (Fig. 69A.10), which can be a source of intraoperative bleeding.
Natural History
In patients with ADPKD, the number and size of liver cysts are greater in women than in men, and they increase with patient age and correlate with the size of the kidney (Bae et al, 2006) and the severity of kidney dysfunction. Pregnancy, multiparity, and use of female steroid hormones further increase the risk of severe hepatic cystic disease (Sherstha et al, 1997). The natural course is highly variable from one individual to the other or within members of the same family, but the progression is fairly linear for a given patient. Hence, age at the time of diagnosis, first symptoms, incapacitating symptoms, and kidney involvement are useful markers to predict the future need for transplantation (see Chapter 69B). As the lifespan of patients with ADPKD has lengthened with dialysis and transplantation, liver-related symptoms have become more frequent.
In patients with isolated PCLD, liver involvement also correlates with female gender and number of pregnancies (Qian et al, 2003; Van Keimpema et al, 2010). Apart from kidney symptoms, which are absent, the course is the same as that of PCLD associated with ADPKD (Hoevenaren et al, 2008).
Complications
Within Cysts
Bacterial infection is the most severe complication and, although very rare, can be severe enough to be life threatening (Robson & Fenster, 1964; Telenti et al, 1990). Symptoms may include fever, chills, and right upper quadrant pain or less typical symptoms. Cyst infection has mainly been observed in patients with ADPKD under dialysis or in those taking immunosuppressive drugs after renal transplantation (Bourgeois et al, 1983). It is exceptional, if it exists at all, in patients with isolated PCLD (Hoevenaren et al, 2008; Qian et al, 2003; van Keimpema et al, 2010). Differentiating infection within kidney or liver cysts is difficult. The infected cysts can be recognized by US and CT, which show echoic material within the cysts, corresponding to pus (Telenti et al, 1990). Thickened, irregular cyst wall, hyperdense content, and fluid-fluid or air-fluid levels may also be seen. Positron emission tomography (PET) with 18F-fluorodeoxyglucose labeled leucocytes or 111In-labeled leukocyte scintigraphy may be used to document and localize infection (Kjaer et al, 2004; Desouza et al, 2009). Treatment of an infected cyst should include aspiration and drainage in addition to antibiotics provided the infection has been documented (Telenti et al, 1990).
Complications from Cyst Compression
Cholestasis secondary to compression of the biliary bifurcation by large cysts (Ergün et al, 1980; Howard et al, 1976) or by peribiliary cysts may occur. In contrast, jaundice is rare and may result from progressive or acute enlargement of cysts, the latter in cases of intracystic bleeding. Mild dilation of the common bile duct is also not unusual (Ishikawa et al, 1996). As a result of portal or hepatic vein compression, portal hypertension with ascites and variceal bleeding have been described (McGarrity et al, 1986; Ratcliffe et al, 1984; Sato et al, 2002). Severe forms may also occur in the few patients with ADPKD who have associated congenital hepatic fibrosis or in patients with autosomal recessive PKD, in which liver cysts are rare.
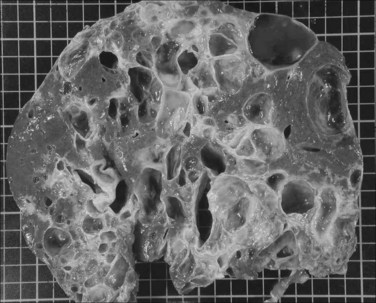
Compression of adjacent organs by the enlarged liver is responsible for most symptoms, as previously discussed; less common is the compression of the right atrium, which may result in hypotentive episodes (Lasic et al, 2004), or the inferior vena cava. The parenchymal impact of hepatic or portal vein compression is frequent. In our experience with liver resection or transplantation, this is present in more than 75% of the patients and includes centrolobular fibrosis, sinusoidal congestion, peliosis, and nodular regenerative hyperplasia (Fig. 69A.11).
Complications from Associated Conditions
ADPKD is well known for other extrarenal manifestations other than those of the liver. These include, most notably, intracranial aneurysms, but dolichoectasias, thoracic aortic and cervicocephalic artery dissections, and coronary artery aneurysms are also possible. These vascular diseases have directly been linked to PKD1 or PKD2 mutations in experimental models (Bichet et al, 2006).
The incidence of cerebral aneurysms is 8%, three to four times higher than in the general population (Pirson et al, 2002). It has been linked to a mutation in the 5′ region of PKD1, rather than the 3′ region; therefore the main risk factor is a family history of intracerebral aneurysms or subarachnoid hemorrhage (SAH) (Rossetti et al, 2003). In this context, the incidence is 16%. Most of these aneurysms are small (<6 mm in diameter) and do not seem to be at increased risk of growth or rupture compared with those of the general population (Gibbs et al, 2004). Widespread screening is not indicated and should be restricted to individuals with a family history of aneurysm or SAH, previous aneurysm rupture, preparation for major elective surgery, high-risk occupations (such as airline pilots), and patient anxiety despite adequate information (Pirson et al, 2002). The ideal method of detection is MRI.
Mitral valve prolapse and pericardial effusion are frequently also found in 25% and 35% of patients with ADPKD, respectively. These are generally well tolerated and do not require therapeutic intervention (Lumiaho et al, 2001; Qian et al, 2007). Cerebral aneurysms and mitral valve abnormalities have been reported with the same or a slightly lower incidence in patients with isolated PCLD (Hoevenaren et al, 2008; Qian, et al 2003).
Staging
Gigot’s Classification
Gigot’s classification relies on CT imaging and was designed to identify the indications and limits of fenestration in the treatment of PCLD (Gigot et al, 1997): type I shows a limited number (<10) of large cysts (>10 cm); type II shows diffuse involvement of liver parenchyma by multiple medium-sized cysts with remaining large areas of noncystic liver parenchyma on preoperative CT; type III shows massive, diffuse involvement of liver parenchyma by small and medium-sized liver cysts and only a few areas of normal liver parenchyma between cysts. The drawback of this classification is that some patients may seem to have either one or the other type depending on the CT slice because the distribution of cysts is uneven.
Qian’s Classification
Qian’s classification has been used in the context of familial screening and relies on cyst number and the presence of symptomatic hepatomegaly (Qian et al, 2003): grade 0 refers to 0 cysts; grade 1, 1 to 10 cysts; grade 2, 11 to 20 cysts; grade 3, more than 20 cysts; and grade 4, more than 20 cysts and symptomatic hepatomegaly.
Schnelldorfer’s Classification
Schnelldorfer’s classification aims at differentiating patients who could benefit from resection or transplantation. It is more complex but also more reliable (Schnelldorfer et al, 2009). It takes into account the presence of symptoms (absent, mild, or severe), the number and size of the cysts, the presence of relative normal areas of liver parenchyma (≥2 sectors, ≥1 sector, or <1 sector), and whether an isosectoral portal or hepatic vein occlusion of the preserved sector is present.
Differentiation Between Simple Cysts, Isolated PCLD, and PCLD Associated with ADPKD
The distinction between isolated PCLD and PCLD associated with ADPKD relies on the number of kidney cysts and familial history. In adults with a positive family history, the diagnosis of ADPKD is established by radiologic evidence of at least two unilateral or bilateral cysts in patients younger than 30 years, at least two cysts in each kidney in those 30 to 59 years, and at least four cysts in each kidney in those 60 years or older (Ravine et al, 1994). Of note, one third of patients with isolated PCLD may have a few kidney cysts, but most usually have one or two (van Keimpema et al, 2010), as does the general population of adult patients (Carrim & Murchinson 2003). In the absence of a family history, the diagnosis of ADPKD is retained if more than five kidney cysts in either one or both kidneys are visible on CT.
Unlike ADPKD, no clinical guidelines have established what distinguishes PCLD from simple cysts that are frequently multiple. Some surgical series have used a threshold of five or six cysts, whereas others have required involvement of more than 50% of the parenchyma. These differences should be taken into account when analyzing the results of these series. More recently, in the case of family screenings, it has been proposed that sporadic cases of PCLD be defined by the presence of more than 15 to 20 cysts, but when there is a positive familial history the presence of four cysts is sufficient (Hoevenaren et al, 2008; Qian et al, 2003). Genetic testing is neither required nor useful in clinical practice for either form of PCLD because not all mutations have been identified.
Differential Diagnosis
Single Cysts
Hydatid cysts, liver abscesses, cystadenoma, and cystadenocarcinoma are the main differential diagnoses, especially when simple cysts have become atypical. These entities are addressed separately in Chapters 68, 66, 67, and 79B, respectively.
Embryonal sarcomas (see Chapter 82) are mostly observed in children between the ages of 2 and 15 years and are exceptional in adults. The tumor is usually large and poorly differentiated with abundant myxoid or necrotic content. Although the tumor may appear as cystic on CT scan and MRI because of the very high water content of the myxoid stroma, US shows that it is, in fact, solid. Late-phase contrast imaging on CT and MRI shows late enhancement, which is incompatible with a cystic tumor.
Other Primary Tumors
Hepatocellular carcinoma and cholangiocarcinoma may occasionally be cystic (see Chapter 50A, Chapter 50B, Chapter 50C, Chapter 50D, Chapter 80 ). This cystic appearance corresponds to necrosis that may be either spontaneous, particularly for rapidly growing tumors, or treatment induced. However, with few exceptions, these tumors remain mostly solid. Giant hemangiomas may occasionally present as partly cystic; this corresponds to noncirculating areas. Hemangiomas are hypointense on T1-weighted MRI images and hyperintense on T2-weighted images, the same as any cystic lesion, but the kinetics of contrast enhancement is typical (see Chapters 17 and 79A).
In the context of acute pancreatitis or pancreatic duct rupture, intrahepatic pseudocysts are exceptional, with fewer than 20 reported cases (Balzan et al, 2005). They may be located in the left liver along the lesser omentum, or in the right liver along the right portal vein. They have a subcapsular location, hemorrhagic or necrotic content, and a fibrous wall that enhances at the late phase of the injection. Pancreatic MRI is occasionally useful to document rupture of the pancreatic duct (see Chapter 54).
Liver hematomas are rarely spontaneous and usually appear in the context of trauma or liver surgery. They have also been described in association with hepatocellular carcinoma, liver adenoma, or, very rarely, metastases. When occurring at the end of a pregnancy, HELLP syndrome (hemolysis, elevated liver enzymes, and low platelet count) should be suspected. Clinical symptoms are usually obvious, with pain and associated symptoms from intraabdominal hemorrhage if capsular rupture has occurred. When the clots are fresh, these hematomas are spontaneously echogenic on US, hyperdense on CT scan, hyperintense on T1-weighted MRI sequences, and hypointense on T2-weighted MRI sequences. At a later phase, clots liquefy and the content appears cystic (see Chapter 17).
Ciliated Hepatic Foregut Cysts
Although extremely rare, ciliated hepatic cysts warrant special consideration. Such cysts are located within the liver parenchyma and have a characteristic four-layer border that consists of a pseudostratified, ciliated columnar epithelium covering a subepithelial connective tissue, smooth muscle bundles, and an outer fibrous capsule. The lining epithelium may secrete fluids of different composition, from watery to viscous or mucous, which explains why its echogenicity may be variable and why calcifications may be present, although this is exceptional. The second characteristic of these cysts is that they are subcapsular, usually located on the anterior surface of the liver close to the insertion of the falciform ligament. Two thirds are localized in the left lobe of the liver in segment IV, but right lobe cysts also have been described (Takahiro et al, 2003). The final characteristic of these cysts is that they are classically unilocular and usually less than 4 cm in diameter. Typically these cysts do not communicate with the biliary system, although this has been reported, particularly in children.
Prevalence and Etiology
Although recognized since 1857, ciliated hepatic cysts are exceptional, with approximately 100 cases reported in the literature, most in the past 20 years (Sharma et al, 2008). There is no gender predominance, and patient age ranges from newborn with prenatal diagnosis (Guérin et al, 2010) to 82 years, with a mean age of 48 years.
Manifestation and Diagnosis
Three characteristics that appear on imaging are 1) their predominant location in segment IV, 2) their small size (4 ± 2 cm), and 3) their subcapsular location. Most (>90%) are unilocular. Two thirds of the cysts are hypoechoic on US, hypodense on CT, and do not enhance after injection of contrast material (Fig. 69A.12). They are anechoic as formal biliary cysts in less than 5% (Horii et al, 2003). Some ciliated cysts may also have an atypical, solid tumor appearance on CT and a noncystic appearance on US (hyperechoic), probably as a result of mucinous material, calcium, and cholesterol crystals within the cyst. On MRI the lesion is highly hyperintense on T2-weighted images but highly variable on T1 sequences; they are hyperintense in 52%, isointense in 24%, and hypointense in 14%. When atypical on imaging, the differential diagnosis includes cystadenoma or cystadenocarcinoma and may be difficult, especially because carbohydrate antigen (CA) 19-9 may be expressed by the epithelium of the ciliated cysts, and its serum level may be slightly elevated (Wu et al, 1998). Fine needle aspiration cytology may help by showing ciliated, pseudostratified, tall columnar epithelial cells suspended in a mucous background. The presence of these cells is virtually diagnostic of this condition, and the positive predictive value of aspiration is 76% (Sharma et al, 2008).
Course, Complications, and Treatment
Ciliated hepatic foregut cysts are usually believed to follow a benign course. Compression of adjacent structures is extremely rare, with a single case report of portal compression resulting in portal hypertension (Harty et al, 1998) and another case report of jaundice (Vick et al, 1999). Malignant transformation into squamous cell carcinoma has been reported in four patients in recent years (de Lajarte-Thirouard et al, 2002; Furlanetto & Dei Tos, 2002; Vick et al, 1999; Zhang et al, 2009), and one other patient had extensive squamous metaplasia (Ben Mena et al, 2006), yielding a risk of malignancy of less than 5%. Because this transformation has occurred in patients with cysts larger than 7 cm, most authors agree that excision is indicated when the cyst is larger than 4 to 5 cm, shows abnormalities in its wall, enlarges, or is symptomatic, although other authors recommend broader indications.
Multiple Cysts
Biliary hamartomas, also known as von Meyenburg complexes, also correspond to an anomalous arrangement of the ductal plate; long recognized by pathologists and surgeons, they are a frequent finding during liver surgery (see Chapters 47 and 48). Biliary hamartomas may be observed within an otherwise normal liver (6% of the population) but may also be associated with Caroli disease (see Chapter 46) and congenital hepatic fibrosis; in PCLD, they are particularly prevalent—virtually constant. Identification of biliary hamartomas is important because they may be mistaken for liver metastases. These lesions are asymptomatic and are discovered incidentally. They range in size between 2 and 10 mm and may appear on US as hypoechogenic, hyperechogenic, or heterogenous depending on their size, associated biliary dilation, and fibrous stroma (Zheng et al, 2005). They are hypodense on CT and do not enhance; on MRI, they are very hypointense on T1-weighted images, very hyperintense on T2-weighted images, and are best seen on cholangio-MRI sequences (Fig. 69A.13; see Chapters 16 and 17).
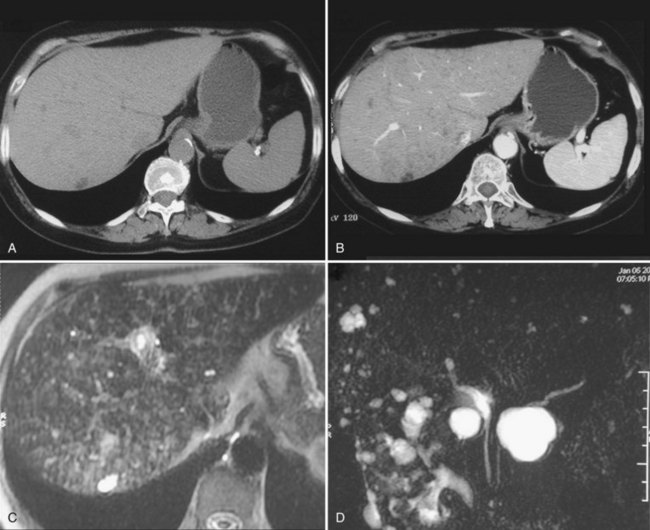
Peribiliary cysts arise from cystic enlargement of peribiliary glands located in and around the walls of extrahepatic and large intrahepatic bile ducts. These cysts may have a diameter up to 2 cm and are mainly observed in the context of severe chronic liver disease, including cirrhosis with portal hypertension or PCLD and after liver transplantation. They are usually asymptomatic, but obstructive jaundice has been reported. When known, imaging features are easily recognized. At US, the cysts are located along the main bile ducts or within portal tracts beside portal veins. At cholangio-MRI, they may mimic an enlarged bile duct, but smaller biliary branches upstream are not dilated. Increases in size and diameter are possible but rare (Motoo et al, 2001).
Akiyama T, et al. Levovist ultrasonography imaging in intracystic hemorrhage of simple liver cyst. World J Gastroenterol. 2008;14:805-807.
Akriviadis EA, et al. Spontaneous rupture of nonparasitic cyst of the liver. Gastroenterology. 1989;97:213-215.
Alvaro D, et al. Morphological and functional features of hepatic cyst epithelium in autosomal dominant polycystic kidney disease. Am J Pathol. 2008;172:321-332.
Bae KT, et al. Magnetic resonance imaging evaluation of hepatic cysts in early autosomal-dominant polycystic kidney disease: the Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease cohort. Clin J Am Soc Nephrol. 2006;1:64-69.
Balzan S, et al. Right intrahepatic pseudocyst following acute pancreatitis: an unusual location after acute pancreatitis. J Hepatobiliary Pancreat Surg. 2005;12:135-137.
Bello-Reuss E, Holubec K, Rajaraman S. Angiogenesis in autosomal-dominant polycystic kidney disease. Kidney Int. 2001;60:37-45.
Ben Mena N, et al. Ciliated hepatic foregut cyst with extensive squamous metaplasia: report of a case. Virchows Arch. 2006;449:730-733.
Berrebi G, Erickson RP, Marks BW. Autosomal dominant polycystic liver disease: a second family. Clin Genet. 1982;21:342-347.
Bichet D, et al. Cardiovascular polycystins: insights from autosomal dominant polycystic kidney disease and transgenic animal models. Trends Cardiovasc Med. 2006;16:292-298.
Bourgeois M, et al. Infection of hepatic cysts following kidney transplantation in polycystic disease. World J Surg. 1983;7:629-631.
Brodsky JL, Goeckeler J, Schekman R. BiP and Sec63p are required for both co- and posttranslational protein translocation into the yeast endoplasmic reticulum. Proc Natl Acad Sci U S A. 1995;92:9643-9646.
Buyse S, et al. Acute pulmonary embolism: a rare complication of a large non-parasitic hepatic cyst. Eur J Gastroenterol Hepatol. 2004;16:1241-1244.
Caremani M, et al. Echographic epidemiology of non-parasitic hepatic cysts. J Clin Ultrasound. 1993;21:115-118.
Carrim ZI, Murchison JT. The prevalence of simple renal and hepatic cysts detected by spiral computed tomography. Clin Radiol. 2003;58:626-629.
Chapman AB. Cystic disease in women: clinical characteristics and medical management. Adv Ren Replace Ther. 2003;10:24-30.
Coffin B, et al. Calcified hepatic and renal cysts in adult dominant polycystic kidney disease. Dig Dis Sci. 1990;35:1172-1175.
Davila S, et al. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat Genet. 2004;36:575-577.
de Lajarte-Thirouard AS, et al. Squamous cell carcinoma arising in a ciliated hepatic forgut cyst. Pathol Res Pract. 2002;198:697-700.
Desouza RM, et al. Differentiation between infection in kidney and liver cysts in autosomal dominant polycystic kidney disease: use of PET-CT in diagnosis and to guide management. Transplant Proc. 2009;41:1942-1945.
Drenth JP, et al. Molecular characterization of hepatocystin, the protein that is defective in autosomal dominant polycystic liver disease. Gastroenterology. 2004;126:1819-1827.
Ergün H, Wolf BH, Hissung SL. Obstructive jaundice caused by polycystic liver disease. Radiology. 1980;136:435-436.
Everson GT, Scherzinger A, Berger-Leff N. Polycystic liver disease: quantitation of parenchymal and cyst volumes from computed tomography images and clinical correlates of hepatic cysts. Hepatology. 1988;8:1627-1634.
Everson GT, et al. Functional similarities of hepatic cystic and biliary epithelium: studies of fluid constituents and in vivo secretion in response to secretin. Hepatology. 1990;11:557-565.
Everson GT, et al. Polycystic disease of the liver. Hepatology. 2004;40:774-782.
Fabris L, et al. Effects of angiogenic factor overexpression by human and rodent cholangiocytes in polycystic liver diseases. Hepatology. 2006;43:1001-1012.
Flagg RS, Robinson DW. Solitary nonparasitic hepatic cysts: report of oldest known case and review of the literature. Arch Surg. 1967;95:964-973.
Frisell J, et al. Compression of the inferior caval vein: a rare complication of a large non-parasitic liver cyst. Acta Med Scand. 1979;205:541-542.
Furlanetto A, Dei Tos AP. Squamous cell carcinoma arising in a ciliated hepatic foregut cyst. Virchows Arch. 2002;441:296-298.
Gabow PA, et al. Risk factors for the development of hepatic cysts in autosomal dominant polycystic kidney disease. Hepatology. 1990;11:1033-1037.
Gaines PA, Sampson MA. The prevalence and characterization of simple hepatic cysts by ultrasound examination. Br J Radiol. 1989;62:335-337.
Gibbs GF, et al. Follow-up of intracranial aneurysms in autosomal-dominant polycystic kidney disease. Kidney Int. 2004;65:1621-1627.
Gigot JF, et al. Adult polycystic liver disease: is fenestration the most adequate operation for long-term management? Ann Surg. 1997;225:286-294.
Glanemann M, et al. Polycystic liver grafts for orthotopic liver transplantation. Transplant Proc. 2000;32:134-135.
Guérin F, et al. Prenatal and postnatal ciliated hepatic foregut cysts in infants. J Pediatr Surg. 2010;45:E9-14.
Gupta S, et al. CT of liver cysts in patients with autosomal dominant polycystic kidney disease. Acta Radiol. 1999;40:444-448.
Harty MP, et al. Ciliated hepatic foregut cyst causing portal hypertension in an adolescent. AJR Am J Roentgenol. 1998;170:688-690.
Hoevenaren IA, et al. Polycystic liver: clinical characteristics of patients with isolated polycystic liver disease compared with patients with polycystic liver and autosomal dominant polycystic kidney disease. Liver Int. 2008;28:264-270.
Horii T, et al. Ciliated hepatic foregut cyst: a report of one case and a review of the literature. Hepatol Res. 2003;26:243-248.
Howard RJ, Hanson RF, Delaney JP. Jaundice associated with polycystic liver disease: relief by surgical decompression of the cysts. Arch Surg. 1976;111:816-817.
Ishikawa I, et al. High incidence of common bile duct dilatation in autosomal dominant polycystic kidney disease patients. Am J Kidney Dis. 1996;27:321-326.
Karhunen PJ, Tenhu M. Adult polycystic liver and kidney diseases are separate entities. Clin Genet. 1986;30:29-37.
Kida T, Nakanuma Y, Terada T. Cystic dilatation of peribiliary glands in livers with adult polycystic disease and livers with solitary nonparasitic cysts: an autopsy study. Hepatology. 1992;16:334-340.
Kjaer A, et al. Fever of unknown origin: prospective comparison of diagnostic value of 18F-FDG PET and 111In-granulocyte scintigraphy. Eur J Nucl Med Mol Imaging. 2004;31:622-626.
Kutcher R, Schneider M, Gordon DH. Calcification in polycystic disease. Radiology. 1977;122:77-80.
Larsen KA. Benign lesions affecting the bile ducts in the postmortem cholangiogram. Acta Pathol Microbiol Immunol Scand. 1961;51:47-62.
Lasic LB, et al. Refractory hypotension and edema caused by right atrial compression in a woman with polycystic kidney disease. Am J Kidney Dis. 2004;43:13-17.
Lee SO, et al. MicroRNA15a modulates expression of the cell-cycle regulator Cdc25A and affects hepatic cystogenesis in a rat model of polycystic kidney disease. J Clin Invest. 2008;118:3714-3724.
Liang P, et al. Differential diagnosis of hepatic cystic lesions with gray-scale and color Doppler sonography. J Clin Ultrasound. 2005;33:100-105.
Lumiaho A, et al. Mitral valve prolapse and mitral regurgitation are common in patients with polycystic kidney disease type 1. Am J Kidney Dis. 2001;38:1208-1216.
McGarrity TJ, Koch KL, Rasbach DA. Refractory ascites associated with polycystic liver disease: treatment with peritoneovenous shunt. Dig Dis Sci. 1986;31:217-220.
Melnick PJ. Polycystic liver: analysis of seventy cases. Arch Pathol Lab Med. 1975;59:162-172.
Mochizuki T, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272:1339-1342.
Moreaux J, Bloch P. Les kystes biliaires solitaires du foie. Arch Fran Mal App Dig. 1971;60:203-224.
Moschcowitz E. Non-parasitic cysts (congenital) of the liver, with a study of aberrant bile ducts. Am J Med Sci. 1906;131:674-699.
Motoo Y, et al. Hepatic peribiliary cysts diagnosed by magnetic resonance cholangiography. J Gastroenterol. 2001;36:271-275.
Murray SL, et al. Matrix metalloproteinase activity in human intrahepatic biliary epithelial cell lines from patients with autosomal dominant polycystic kidney disease. Connect Tissue Res. 1996;33:249-256.
Nichols MT, et al. Secretion of cytokines and growth factors into autosomal dominant polycystic kidney disease liver cyst fluid. Hepatology. 2004;40:836-846.
Perrone RD, et al. Continuous epithelial cell lines from ADPKD liver cysts exhibit characteristics of intrahepatic biliary epithelium. Am J Physiol. 1995;269:G335-G345.
Pirson Y, Chauveau D, Torres V. Management of cerebral aneurysms in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2002;13:269-276.
Qian Q, et al. Increased occurrence of pericardial effusion in patients with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2007;2:1223-1227.
Qian Q, et al. Clinical profile of autosomal dominant polycystic liver disease. Hepatology. 2003;37:164-171.
Ratcliffe PJ, Reeders S, Theaker JM. Bleeding oesophageal varices and hepatic dysfunction in adult polycystic kidney disease. Br Med J. 1984;288:1330-1331.
Ravine D, et al. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet. 1994;343:824-827.
Robson GB, Fenster F. Fatal liver abscess developing in a polycystic liver. Gastroenterology. 1964;47:82.
Rossetti S, et al. Association of mutation position in polycystic kidney disease 1 (PKD1) gene and development of a vascular phenotype. Lancet. 2003;361:2196-2201.
Salemis NS, et al. Spontaneous rupture of a giant nonparasitic hepatic cyst presenting as an acute surgical abdomen. Ann Hepatol. 2007;6:190-193.
Sato Y, et al. Double selective shunting for esophagogastric and rectal varices in portal hypertension due to congenital hepatic polycystic disease. Hepatogastroenterology. 2002;49:1528-1530.
Schnelldorfer T, et al. Polycystic liver disease: a critical appraisal of hepatic resection, cyst fenestration, and liver transplantation. Ann Surg.. 2009;250:112-118.
Sharma S, et al. Ciliated hepatic foregut cyst: an increasingly diagnosed condition. Hepatobiliary Pancreat Dis Int. 2008;7:581-589.
Sherstha R, et al. Postmenopausal estrogen therapy selectively stimulates hepatic enlargement in women with autosomal dominant polycystic kidney disease. Hepatology. 1997;26:1282-1286.
Shillingford JM, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A. 2006;103:5466-5471.
Tahvanainen P, et al. Polycystic liver disease is genetically heterogeneous: clinical and linkage studies in eight Finnish families. J Hepatol. 2003;38:39-43.
Takahiro H, et al. Ciliated hepatic foregut cyst: a report of one case and a review of the literature. Hepatol Res. 2003;26:243-248.
Telenti A, et al. Hepatic cyst infection in autosomal dominant polycystic kidney disease. Mayo Clin Proc. 1990;65:933-942.
Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369:1287-1301.
Trombetta ES, Fleming KG, Helenius A. Quaternary and domain structure of glycoprotein processing glucosidase II. Biochemistry. 2001;40:10717-10722.
van Keimpema L, et al. Patients with isolated polycystic liver disease referred to liver centres: clinical characterization of 137 cases. Liver Int. 2010;31(1):92-98.
Vick DJ, Goodman ZD, Ishak KG. Squamous cell carcinoma arising in a ciliated hepatic foregut cyst. Arch Pathol Lab Med. 1999;123:1115-1117.
Ward CJ, et al. Polycystin, the polycystic kidney disease 1 protein, is expressed by epithelial cells in fetal, adult, and polycystic kidney. Proc Natl Acad Sci U S A. 1996;93:1524-1528.
Williamson RCN, Ramus NI, Shorey BA. Congenital solitary cysts of the liver and spleen. Br J Surg. 1978;65:871-876.
Wu ML, Abecassis MM, Rao MS. Ciliated hepatic foregut cyst mimicking neoplasm. Am J Gastroenterol. 1998;93:2212-2214.
Zhang X, et al. Squamous cell carcinoma arising in a ciliated hepatic foregut cyst: case report and literature review. Pathol Res Pract. 2009;205:498-501.
Zheng RQ, et al. Imaging findings of biliary hamartomas. World J Gastroenterol. 2005;28(11):6354-6359.