CHAPTER 14 Sideroblastic anemia
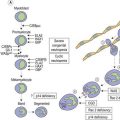
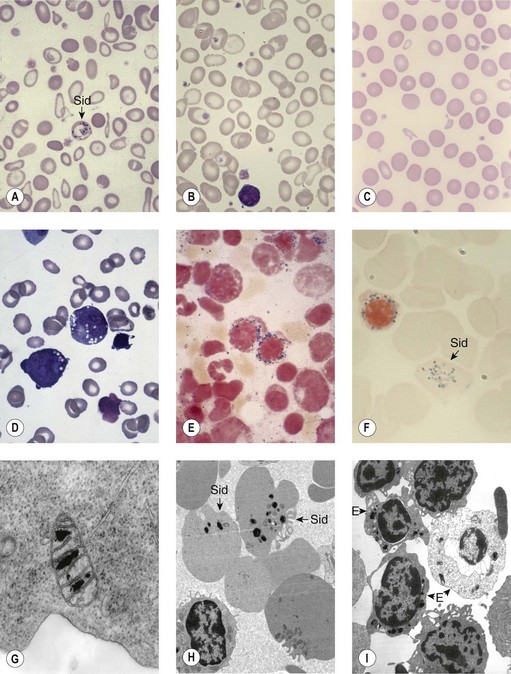
Sideroblastic anemias (SAs) are heterogeneous disorders characterized by accumulation of iron in the mitochondria of developing erythroblasts and varying degrees of red cell hypochromia. The abnormal, perinuclear mitochondria are visualized after Perls’ staining of bone marrow smears for iron. These are seen as a complete or partial ring of Prussian blue staining siderotic granules, in a substantial proportion of erythroblasts (Fig. 14.1).1,2 Recent studies further associate this excess iron with mitochondrial ferritin.3 SAs are inherited or acquired. The acquired forms may be secondary and reversible, or primary in nature.4
Hereditary sideroblastic anemia
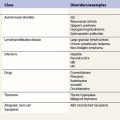
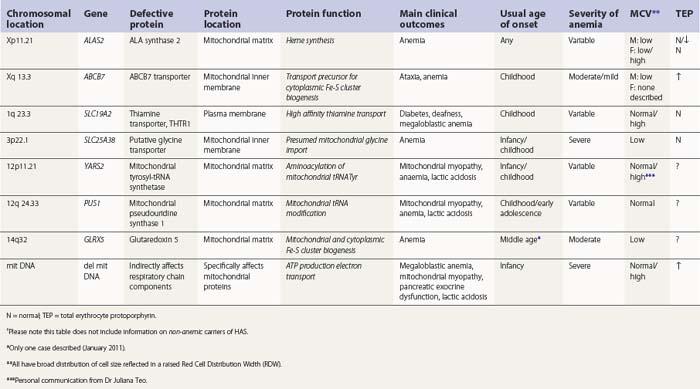
Hereditary SAs (HSAs) are very rare and all patterns of inheritance have been observed (Table 14.1). The anemia may be isolated or syndromic, and severity varies markedly. Some patients die in infancy or childhood; others have a normal life span. Although several genetic defects are now implicated a significant proportion of cases remain unexplained.5 The diagnosis is often achievable from clinical presentation and cell morphology but genetic diagnosis clarifies treatment options. This helps generate confident management plans which will avoid secondary complications and allows risk assessment for recurrence within families.
X-linked inheritance
ALA synthase 2 defect
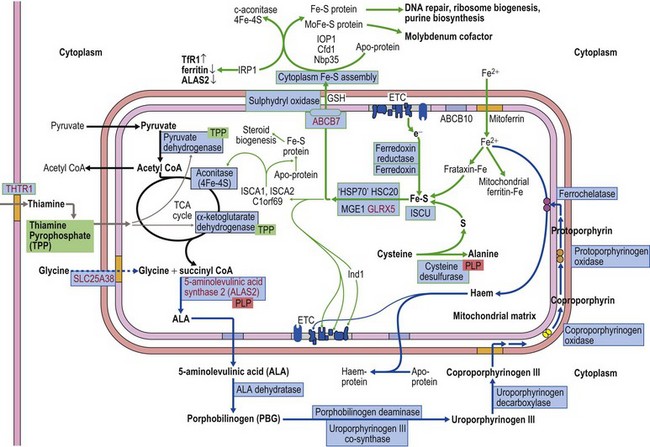
The common form of X-linked SA (XLSA) is caused by mutations in the erythroid-specific, 5′-aminolevulinate synthase gene (ALAS2) at Xp11.21. Within the mitochondrial matrix, ALAS2 homodimer uses co-factor pyridoxal phosphate to catalyze the first step of heme synthesis.6 Decreased ALAS2 activity leads to decreased protoporphyrin production and decreased heme. Iron continues to enter the erythroblast unchecked, accumulates in the mitochondria and becomes most visible in the late erythroblasts on staining.
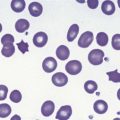
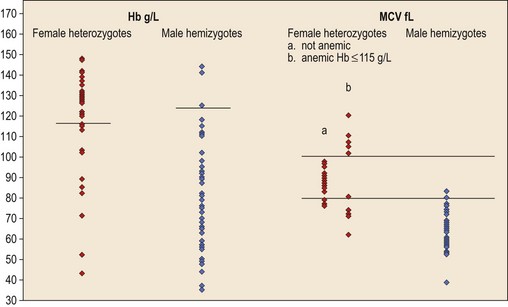
In male patients the severity of the anemia is variable. It is microcytic-hypochromic and may respond to pyridoxine treatment (Fig. 14.1A, B).7,8 Female carriers have the potential to produce two RBC populations, one microcytic-hypochromic and one normal, depending on which erythroblast X-chromosome is active (Fig. 14.1C). Carriers are usually unaffected but some are anemic due to skewed X chromosome inactivation against that carrying the normal allele, accounting for almost one third of probands.9,10 Anemic carriers of mild or moderately severe mutations have the same presentation as male hemizygotes (Fig. 14.2). Patients can present at any age with symptoms of anemia or iron overload.4,11
Physical examination is unremarkable although mild hepatosplenomegaly may be present. Red cell size and hemoglobin content are broadly distributed and variable proportions of microcytic-hypochromic and normocytic-normochromic cells are seen on the blood film (Fig. 14.1A,B). Anisocytosis, poikilocytosis, elliptocytosis and target cells may be present. An occasional cell with basophilic stippling or Pappenheimer bodies and an occasional late erythroblast may be seen; the numbers of these increase after splenectomy. White cell and platelet counts are normal.
In the bone marrow erythropoiesis is expanded but ineffective; iron overload from increased dietary iron absorption may develop. Transferrin saturation is usually increased; even mildly anemic patients may present with secondary iron overload in the absence of blood transfusion.12 Total erythrocyte protoporphyrin (TEP) is low/normal. Severe anemia requiring regular blood transfusions is a rare occurrence. Phlebotomy has been successful for preventing iron overload, reversing iron overload and maintaining normal iron levels in pyridoxine-responsive patients (see below). This may additionally improve the hemoglobin level.13 Iron chelation may be required for correcting iron overload. Splenectomy is not recommended because this has led to thrombotic complications.4
At least 120 cases from 70–80 families are reported (January 2011). More than 50 different mutations are involved, scattered across seven exons encoding the catalytic and C-terminal domains.4,5 Missense mutations predominate; null mutations, or those predicted to be very severe, are found only in female heterozygotes. ALAS2 variations causing anemia in one member of a family may occasionally be silent in another.14
Pyridoxal phosphate (vitamin B6) is required for ALAS2 activity and stability. Many patients respond partially to pharmacologic doses of oral pyridoxine but some do not or barely do so.13 Responsive patients remain on maintenance doses of about 25–200 mg oral pyridoxine/day for life.4 Complete correction of hematological changes with pyridoxine, although rare, has been reported.15 Pyridoxine responsiveness is most associated with mutations involving amino acids fairly close to the pyridoxal phosphate binding site that cause partial loss of enzyme function or stability.16,17 Refractoriness is associated with mutations that cause irreversible disruption of activity or cause loss of in vivo activity through faulty interaction with components required for intramitochondrial processing.18 Iron overload, or its complications, also contribute to pyridoxine refractoriness. There may be some response to folate due to secondary deficiency.
Not all ALAS2 mutations cause SA. Mutations leading to deletion or substitution of the C-terminal 19 amino acids generate truncated ALAS2 protein of increased activity and cause X-linked, dominant erythropoietic protoporphyria, not anemia.19
Female carriers of severe/null mutations that prevent erythrocyte production go undetected unless they become anemic because of either skewed but incomplete X-chromosome inactivation against the normal allele or some mechanism selecting against cells expressing the normal allele. In these cases red cells produced by residual effective erythropoiesis from expression of normal ALAS2 are macrocytic, the number of ring sideroblasts may be very low and the cause of the ineffective erythropoiesis may go unsuspected or misdiagnosed (Fig. 14.2).10,20
ABCB7 defect
A second XLSA is associated with slow/non-progressive, cerebellar ataxia (XLSA/A). This is usually diagnosed in infants and young children who present with delayed walking ability.21 There is mild to moderate microcytic-hypochromic anemia (Hb 8–15 g/dl) with red cell appearances indistinguishable to those of ALAS2 deficiency. However, in contrast, there are features of iron deficiency (normal/decreased serum iron, normal/low transferrin iron saturation, increased serum transferrin receptor concentration and variably increased TEP predominantly of the zinc form) rather than iron overload.22–24 Elevated serum ferritin did, however, develop in two patients, despite low/normal transferrin saturations, so monitoring iron levels is warranted.24 Ferrokinetic studies have not been reported and bone marrow reports do not give a consistent picture. Down-regulation of ABCB7 in non-erythroid cells decreases cell proliferation;25 erythroid hypoplasia may play a role therefore in the anemia. Rational management and treatment of the unusual distribution of iron in these patients awaits a better understanding of the molecular aetiology.
Brain magnetic resonance imaging (MRI) shows selective cerebellar atrophy (Fig. 14.3A). Dysmetria, finger-nose and heel-shin ataxia, dysarthria, intention tremor and diminished deep tendon reflexes are usually present to varying degrees. Nystagmus, strabismus and abnormal plantar responses may or may not be present. Early difficulty in sitting and walking is often followed by some improvement over time. Intelligence is normal and there is no sensory loss.21,26
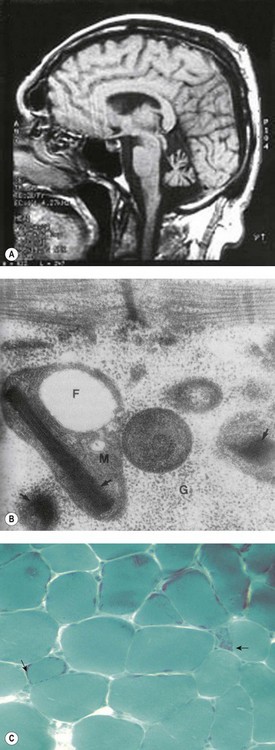
Fig. 14.3 (A–C) Syndromic sideroblastic anaemia: (A) MRI scan of the brain of a patient with XLSA/A showing cerebellar atrophy.26 (B, C) Mitochondrial inclusions and ragged red fibers in a muscle biopsy of a patient with myopathy with lactic acidosis and sideroblastic anemia.32,34
(These figures are reproduced by permission of (A) BMJ Publishing Group Ltd @ 2001, license number 2382030149869, (B) John Wiley and Sons Inc. @ 2005, license number 2382570556744 and (C) BMJ Publishing Group Ltd @ 2007, license number 2382021149604.)
The defect lies in ABCB7 at Xq13.3 encoding an ATP-dependent ‘half-transporter’ consisting of N-terminal transmembrane transporter and C-terminal ATPase domains. Within the inner mitochondrial membrane, ABCB7 homodimer exports some component essential for cytoplasmic Fe-S cluster formation.27 Four separate families with 12 affected patients have been reported. In each of the three families studied a different missense mutation was detected (I400M, V411L, E433K) affecting a different amino acid within the transmembrane transport domain. Yeast cells lacking homologous Atm1p, show reduced cytoplasmic Fe-S cluster generation, normal mitochondrion Fe-S, and mitochondrial iron overload. This phenotype was partially reversed by the intracellular expression of human ABCB7 but less so by the E433K mutant, implicating loss of cytoplasmic Fe-S cluster generation in the XLSA/A syndrome.23
Cellular iron homeostasis in humans is regulated at the translational level through the interaction of two iron regulatory proteins (IRP1 and IRP2) with mRNA hairpin-like secondary structure elements known as iron response elements (IRE). IRP1 is formed by loss of Fe-S from cytoplasmic (c-) aconitase. ALAS2 mRNA has a 5′ IRE and protein levels are controlled at the translation stage by iron.28 Low cytoplasm Fe-S decreases ALAS2 activity through conversion of c-aconitase (4Fe-4S) to IRP1 that binds to the 5′ IRE and blocks translation. This explains microcytic-hypochromic anemia and mitochondrial iron loading with ABCB7 deficiency; however, additionally, excess mitochondrial iron is somehow not available to ferrochelatase that uses zinc instead to form zinc-protoporphyrin.
Autosomal recessive sideroblastic anemia
Putative deficit of mitochondrial glycine import
Mutations in SLC25A38 at 3p22.1 cause non-syndromic HSA with more than 30 probands (January 2011) from more than 25 families29 (author’s unpublished data). This gene, expressed highly in erythroblasts encodes a putative transporter of glycine, located on the mitochondrial inner membrane. ALAS2 has a high KM for its substrate glycine and maximal activity requires millimolar concentrations. Decreased glycine supply within the erythroblast mitochondrion leads to decreased ALAS2 activity and decreased heme. Iron uptake, normally inhibited by heme, continues and excess accumulates.
Glutaredoxin 5 defect
Glutaredoxin 5 (GLRX5) is one of two mitochondrial glutaredoxins. GLRX5 is involved in transport of assembled Fe-S clusters from the scaffold protein (ISCU) to both mitochondrial and cytoplasmic Fe-S destinations.27
One GLRX5 mutation has been reported to cause HSA in a single patient.30 The patient presented in middle age with iron overload, diabetes, darkened skin and a microcytic-hypochromic anemia (Hb 8.9 g/dL, MCV 59 fl, MCH 16 pg) unresponsive to folate and pyridoxine. Blood transfusions were required but with subsequent iron chelation the patient became transfusion independent (Hb 11.3 g/dl, MCV 64 fl, MCH 22 pg). In peripheral blood mononuclear cells, a splice-site mutation in its homozygous form decreased GLRX5 mRNA, decreased both cytoplasm and mitochondrial Fe-S cluster proteins and increased IRP1 activity. In erythroblasts this would decrease ALAS2 activity and decrease heme as occurs in the GLRX5-deficient zebrafish.31 No other tissue appears to be affected.
Pseudouridine synthase 1 defect
Several families (6 families, 11 cases (January 2011)) have been described carrying missense (x1) or nonsense (x2) mutations in PUS1 at 12q24.33.5,32–34 This gene encodes pseudouridine synthase 1 required to modify mitochondrial tRNA uridine ensuring accurate translation of mitochondrial mRNA. Patients present in infancy, childhood or early teenage years with hypotonia, fatigue, weakness, exercise intolerance, lactic acidosis and SA (known as myopathy with lactic acidosis and sideroblastic anemia or MLASA). The myopathy usually progresses, with distal muscle wasting particularly affecting the hands. Pallor, short stature, microcephaly, a short philtrum and a slow gait are usually apparent. Marked growth failure, cognitive and behavior problems have also been described. Severity varies among family members.
The red blood cells show great variation in size with fairly normocytic mean values (MCV 71–100 fl); the severity of the anemia varies from mild/moderate (Hb 7–10 g/dl) to transfusion-dependent. There is iron deposition in mitochondria of erythroblasts. Muscle biopsy may show paracrystalline inclusions within the mitochondria ragged red fibers and/or decreased staining for cytochrome oxidase (Fig. 14.3B, C); complex I and IV activities may be decreased.
Mitochondrial tyrosyl-tRNA synthetase defect
A second gene affecting mitochondrial protein synthesis is now also implicated in the MLASA syndrome. Three cases from two unrelated families without PUS1 mutations were found to be homozygous for the same mutation in the YARS2 gene that encoded a functionally abnormal mitochondrial tyrosyl-tRNA synthetase. Decreased activities of skeletal muscle respiratory chain (RC) complexes I, III and IV and specific reduction of mitochondrial-DNA encoded RC subunit synthesis in myotubes generated by transdifferentiation of patient-derived fibroblasts were demonstrated. Outcomes differed between the families but were more severe than that from defective PUS1, with earlier onset of both anaemia (10 weeks, infancy, 7 yr) and the chronic lactic acidosis (3 months, infancy, early childhood). The two most severely affected became dependent on regular blood transfusion in infancy, suffered great difficulty with walking and swallowing and one succumbed to respiratory failure at 17 yr; cognition and IQ are not affected.35
Defective high-affinity thiamine transporter THTR-1
Most cases of thiamine-responsive megaloblastic anemia (TRMA) are associated with sideroblastic erythropoiesis. TRMA typically presents in infancy or childhood as vitamin B12/folate-refractory megaloblastic anemia, sensorineural deafness and diabetes mellitus.36 Additional complications vary including mild leukopenia, thrombocytopenia, nystagmus, optic atrophy, retinitis pigmentosa, short stature, stroke, cardiac anomalies. More recently, patients lacking one of the usual triad of symptoms have been diagnosed.37
TRMA is caused by homozygosity or compound heterozygosity for loss of function or missense mutations in SLC19A2 on 1q 23.3, encoding the high-affinity, plasma-membrane, thiamine transporter THTR-1.38,39 More than 20 mutations have been described. Passive thiamine transport and/or co-expression of THTR-2 maintain cell viability. Pharmacologic doses of thiamine (usually 20–75 mg/day orally) correct the anemia and decrease or delay the need for insulin but achieve little or no improvement in hearing. Follow-up studies have now been reported with unexplained differences in long-term outcomes regarding maintenance of response to thiamine.37,40
Additional modes of inheritance not yet explained
There remain families with HSA where the mode of inheritance and/or pathogenesis remain unresolved. For instance, in one pedigree there was moderately severe microcytic-normocytic anemia, autosomal dominant inheritance of red cell dimorphism and increased total erythrocyte protoporphyrin and in which two family members had ring sideroblasts. In another pedigree there was maternally inherited mild SA, normocytic-macrocytic RBC and increased free erythrocyte protoporphyrin.41,42
Mitochondrial inheritance
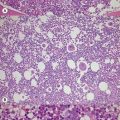
Pearson’s syndrome is a rare, often fatal disorder of infancy characterized by macrocytic anemia, pancreatic exocrine dysfunction and lactic acidosis.43,44 Patients present with failure to thrive and persistent diarrhea. Neutropenia and thrombocytopenia may be present, and hepatic and renal functions may be impaired. It is caused by one of more than 19 deletion mutations in mitochondrial DNA of varying sizes including the common 4977bp lying between positions 5500 and 16000bp. Loss of genes encoding mitochondria-specific tRNA disrupts mitochondrial biogenesis and function and the degree to which a tissue is affected depends on the proportion of mitochondrial DNA that is the deleted form. All hemopoietic lineages can be affected and prominent vacuoles in both myeloid and erythroid cells are seen on bone marrow smears (Fig. 14.1D). Erythropoiesis is megaloblastic with a high percentage of ring sideroblasts. Heteroplasmy renders maternal inheritance difficult to determine and most cases are described as of ‘sporadic’ occurrence. Treatment is supportive and although prognosis is poor some patients survive to develop Kearns–Sayre syndrome, a mitochondrial encephalomyopathy.45
Acquired sideroblastic anemia
Acquired SAs are the most common types subdivided into:
Secondary acquired sideroblastic anemia
Drugs and toxic agents reported to cause sideroblastic erythropoiesis include alcohol, isoniazid, progesterone, D,L-penicillamine, chloramphenicol and zinc supplement excess that antagonizes copper absorption. Copper deficiency in parenteral food and through trientene chelation also causes SA.4 Lead poisoning may lead to SA.46 Hemodialysis-induced pyridoxine deficiency can precipitate SA even without ALAS2 mutation.47 The resultant red cell and bone marrow appearances depend on the pathways affected; a hemolytic component may be involved causing reticulocytosis, a feature which is unusual in primary SA.
Primary acquired sideroblastic anemia
Primary acquired SA, referred to as refractory anemia with ring sideroblasts (RARS), is a myelodysplastic syndrome (MDS). It affects mainly elderly people or people of late middle age and is characterized by ineffective hemopoiesis, peripheral blood cytopenias and an increased risk of developing leukemia.48–50 Patients present with progressive anemia and associated symptoms such as breathlessness, fatigue and pallor. Organomegaly is not usually present except in cases of iron overload.
The blood count shows normocytic or macrocytic anemia refractory to treatment with vitamin B12 or folate, normal neutrophils and platelets. The red cell size distribution is increased but reticulocyte numbers remain normal or only slightly increased. Red cell anisocytosis may be marked and some hypochromia is usually present giving rise to a dimorphic appearance of the red cells in some patients. The bone marrow shows erythroid hyperplasia with ineffective dyserythropoiesis such as increased proportion of early erythroblasts. Abnormalities in other cell lineages are absent and blast cells remain below 5%. More than 15% erythroblasts are ring sideroblasts (providing the patient is not iron deficient) occurring at all stages of erythroid differentiation (Fig. 14.1E).
There is no association with any particular cytogenetic change except for rearrangements involving Xq13.50 Prognosis is good if there is no karyotypic abnormality, trilineage dysplasia or transfusion dependence. Life expectancy of patients over 70 years may not be reduced. The overall risk of transformation to leukemia is between 5% and 10% at 10 years from diagnosis in those with a normal karyotype who are not dependent on blood transfusion.
Treatment consists of alleviation of symptoms of anemia and avoidance or treatment of iron overload resulting from blood transfusion. More vigorous interventions may play a role when shown to be efficacious and safe.52,53 Regular monitoring of the blood count and iron stores is required.
The cause is unknown. It appears to be a clonal disorder arising from a hemopoietic stem cell abnormality that is yet to be defined. Whether the mitochondrial iron overload is caused by the mutation causing the clonal proliferation, is due to a somatic mutation in the cell triggered by subsequent mutation to proliferate or occurs entirely as a secondary phenomenon remains to be seen. Characteristic metabolic changes are beginning to be identified confirming RARS as a distinct MDS category.54,55 Mitochondrial ferritin is upregulated even at the earliest stage of differentiation. Genes encoding heme synthesis enzymes are up-regulated and ABCB7 down-regulated.
Some RARS patients have an associated thrombocytosis (>450 × 109/l). These have been included in the WHO Classification as MDS/myeloproliferative neoplasms in the entity ‘RARS associated with marked thrombocytosis’ (RARS-T). An association between acquisition of JAK2 V617F and the development of thrombocytosis suggests ‘transformation’ from RARS.55
Differential diagnosis of primary sideroblastic anemia
The diagnosis of sideroblastic anemia relies on the visual observation firstly of peripheral blood red cell hypochromia and then of ring sideroblasts in the marrow after staining for iron using a Perls’ Prussian blue reaction (Fig. 14.1F). Electron microscopy may be required to confirm the subcellular location of the iron in difficult or unusual cases (Fig. 14.1G–I). For syndromic cases, histochemical staining of sections from biopsied muscle and measurement of respiratory complex activity by assay or by staining are often required and brain MRI is essential for some. There is considerable overlap of peripheral blood morphology and bone marrow features between different SA types. Sex, age of onset, severity of anemia, MCV, TEP, the ratio of zinc : free erythrocyte protoporphyrin (EPP) and presence of additional syndrome-associated pathology are therefore additional important differentiating features (Table 14.1). Almost half inherited or congenital SA remain unexplained and further genetic causes remain to be found. With their high rate of heme synthesis, erythroid cells are particularly sensitive to defects of mitochondrial iron metabolism. The genes identified in SA affect mitochondrial proteins directly or indirectly (Fig. 14.4, Table 14.1) and the syndromic cases highlight the importance of pathways of iron homeostasis to other conditions.
Acknowledgments
The chapter structure and a few sentences remain those of Professor SN Wickramasinghe, a source of great inspiration who sadly could not contribute more. Thanks to Ruth Pitman and Kevin Barnes for the electron microscopic images in Fig. 14.1, and to Dr Edward Fitzsimons for Fig. 14.1D. I would also like to thank Dr Briedgeen Kerr for her help in researching the data for Figure 14.2 and for her permission to use it here in this way. Thanks also to Professors Massimo Zeviani and Simon Hammans for agreeing to my use of their published images and to Dr Juliana Teo and Professor John Christodoulou for permission to refer to their unpublished data.
1 Bessis MC, Breton-Gorius J. Ferritin and ferruginous micelles in normal erythroblasts and hypochromic hypersideremic anemias. Blood. 1959;14:423-432.
2 Wickramasinghe SN, Fulker MJ, Losowsky MS, Hall R. Microspectrophotometric and electron microscopic studies of bone marrow in hereditary sideroblastic anaemia. Acta Haematol. 1971;45:236-244.
3 Cazzola M, Invernizzi R, Bergamaschi G. Mitochondrial ferritin expression in erythroid cells from patients with SA. Blood. 2003;101:1996-2000.
4 Bottomley SS. Sideroblastic anemias. In: Greer JP, Foerster J, Rodgers GM, et al, editors. Wintrobe’s Clinical Hematology. 12th ed. Philadelphia: Lippincott Williams & Wilkins; 2008:835-855.
5 Bergmann AK, Campagna DR, McLoughlin EM, et al. Systematic molecular genetic analysis of congenital sideroblastic anemia: evidence for genetic heterogeneity and identification of novel mutations. Pediatr Blood Cancer. 2010;54:273-278.
6 Ferreira GC, Gong J. 5-Aminolevulinate synthase and the first step of heme biosynthesis. J Bioenerg Biomembr. 1995;27:151-159.
7 Harris JW, Whittington RM, Weisman RJr, Horrigan DL. Pyridoxine responsive anemia in the human adult. Proc Soc Exp Biol Med. 1956;91:427-432.
8 May A, Bishop DF. The molecular biology and pyridoxine responsiveness of X-linked sideroblastic anaemia. Haematologica. 1998;83:56-70.
9 Cazzola M, May A, Bergamaschi G, et al. Familial-skewed X-chromosome inactivation as a predisposing factor for late-onset X-linked sideroblastic anemia in carrier females. Blood. 2000;96:4363-4365.
10 Aivado M, Gatterman N, Rong A, et al. X-linked sideroblastic anemia associated with a novel ALAS2 mutation and unfortunate skewed X-chromosome inactivation patterns. Blood Cells Mol Dis. 2006;37:40-45.
11 Anderson KE, Sassa S, Bishop DF, Desnick RJ. Disorders of heme biosynthesis: X-linked sideroblastic anemia and the porphyrias. 8th ed. Scriver CR, Beaudet AL, Sly WS, et al, editors. The Metabolic and Molecular Bases of Inherited Disease. vol. 2. New York, NY: McGraw-Hill; 2001:2991-3062.
12 Peto TE, Pippard MJ, Weatherall DJ. Iron overload in mild sideroblastic anaemias. Lancet. 1983;1(8321):375-378.
13 Cotter PD, May A, Li L, et al. Four new mutations in the erythroid-specific 5-aminolevulinate synthase (ALAS2) gene causing X-linked sideroblastic anemia: increased pyridoxine responsiveness after removal of iron overload by phlebotomy and coinheritance of hereditary hemochromatosis. Blood. 1999;93:1757-1769.
14 Cazzola M, May A, Bergamaschi G, et al. Absent phenotypic expression of X-linked sideroblastic anemia in one of 2 brothers with a novel ALAS2 mutation. Blood. 2002;100:4236-4238.
15 Cotter PD, May A, Fitzsimons EJ, et al. Late-onset X-linked sideroblastic anemia. Missense mutations in the erythroid δ-aminolevulinate synthase (ALAS2) gene in two pyridoxine-responsive patients initially diagnosed with acquired refractory anemia and ringed sideroblasts. J Clin Invest. 1995;96:2090-2096.
16 Shoolingin-Jordan PM, Al-Daihan S, Alexeev D, et al. 5-Aminolevulinic acid synthase: mechanism, mutations and medicine. Biochim Biophys Acta. 2003;1647:361-366.
17 Astner I, Schulze JO, van den Hevel J, et al. Crystal structure of 5-aminolevulinate synthase, the first enzyme of heme biosynthesis, and its link to XLSA in humans. EMBO J. 2005;24:3166-3177.
18 Furuyama K, Fujita H, Nagai T, et al. Pyridoxine refractory X-linked sideroblastic anemia caused by a point mutation in the erythroid 5-aminolevulinate synthase gene. Blood. 1997;90:822-830.
19 Whatley SD, Ducamp S, Gouya L, et al. C-terminal deletions in the ALAS2 gene lead to gain of function and cause a previously undefined type of human porphyria, X-linked dominant protoporphyria, without anemia or iron overload. Am J Hum Genet. 2008;83:408-414.
20 Cortesao E, Vidan J, Pereira J, et al. Onset of X-linked sideroblastic anemia in the fourth decade. Haematologica. 2004;89:1261-1263.
21 Pagon RA, Bird TD, Detter JC, Pierce I. Hereditary sideroblastic anaemia and ataxia: an X linked recessive disorder. J Med Genet. 1985;22:267-273.
22 Allikmets R, Raskind WH, Hutchinson A, et al. Mutation of a putative mitochondrial iron transporter gene (ABCB7) in X-linked sideroblastic anemia and ataxia (XLSA/A). Hum Mol Genet. 1999;8:743-749.
23 Bekri S, Kispal G, Lange H, et al. Human ABC7 transporter: gene structure and mutation causing X-linked sideroblastic anemia with ataxia with disruption of cytosolic iron-sulfur protein maturation. Blood. 2000;96:3256-3264.
24 Maguire A, Hellier K, Hammans S, May A. X-linked cerebellar ataxia and sideroblastic anaemia associated with a missense mutation in the ABC7 gene predicting V411L. Br J Haematol. 2001;115:910-917.
25 Cavadini P, Biasiotto G, Poli M, et al. RNA silencing of the mitochondrial ABCB7 transporter in HeLa cells causes an iron-deficient phenotype with mitochondrial iron overload. Blood. 2007;109:3552-3559.
26 Hellier KD, Hatchwell E, Duncombe AS, et al. X-linked sideroblastic anaemia with ataxia: another mitochondrial disease? J Neurol Neurosurg Psychiatry. 2001;70:65-69.
27 Lill R. Function and biogenesis of iron-sulphur proteins. Nature. 2009;460:831-838.
28 Dandekar T, Stripecke R, Gray NK, et al. Identification of a novel iron-responsive element in murine and human erythroid δ-aminolevulinic acid synthase mRNA. EMBO J. 1991;10:1903-1909.
29 Guernsey DL, Jiang H, Campagna DR, et al. Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat Genet. 2009;41:651-653.
30 Camaschella C, Campanella A, De Falco L, et al. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood. 2007;110:1353-1358.
31 Wingert RA, Galloway JL, Barut B, et al. Deficiency of glutaredoxin 5 reveals Fe-S clusters are required for vertebrate haem synthesis. Nature. 2005;436:1035-1039.
32 Inbal A, Avissar N, Shaklai M, et al. Myopathy, lactic acidosis, and sideroblastic anemia: a new syndrome. Am J Med Genet. 1995;55:372-378.
33 Casas KA, Fischel-Ghodsian N. Mitochondrial myopathy and sideroblastic anaemia. Am J Med Genet. 2004;125A:201-204.
34 Fernandez-Vizarra E, Berardinelli A, Valente L, et al. Nonsense mutation in pseudouridylate synthase 1 (PUS1) in two brothers affected by myopathy, lactic acidosis and sideroblastic anaemia (MLASA). J Med Genet. 2007;44:173-180.
35 Riley LG, Cooper S, Hickey P, et al. Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia-MLASA syndrome. Am J Hum Genet. 2010;87:52-59.
36 Rogers LE, Porter S, Sidbury JBJr. Thiamine-responsive megaloblastic anemia. J Pediatr. 1969;74:494-504.
37 Bergmann AK, Sahai I, Falcone JF, et al. Thiamine-responsive megaloblastic anemia: identification of novel heterozygotes and mutation update. J Pediatr. 2009;155:888-892.
38 Fleming JC, Tartaglini E, Steinkamp MP, et al. The gene mutated in thiamine-responsive anemia with diabetes and deafness (TRMA) encodes a functional thiamine transporter. Nat Genet. 1999;22:305-308.
39 Neufeld EJ, Fleming JC, Tartaglini E, Steinkamp MP. Thiamine-responsive megaloblastic anemia syndrome: a disorder of high-affinity thiamine transport. Blood Cells Mol Dis. 2001;27:135-138.
40 Ricketts CJ, Minton JA, Samuel J, et al. Thiamine-responsive megaloblastic anaemia syndrome: long-term follow-up and mutation analysis of seven families. Acta Paediatr. 2006;95:99-104.
41 van Waveren Hogervorst GD, van Roermund HPC, Snijders PJ. Hereditary sideroblastic anaemia and autosomal inheritance of erythrocyte dimorphism in a Dutch family. Eur J Haematol. 1987;38:405-409.
42 Tuckfield A, Ratnaike S, Hussein S, Metz J. A novel form of hereditary sideroblastic anaemia with macrocytosis. Br J Haematol. 1997;97:279-285.
43 Pearson HA, Lobel JS, Kocoshis SA, et al. A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J Pediatr. 1979;95:976-984.
44 Rotig A, Cormier V, Blanche S, et al. Pearson’s marrow-pancreas syndrome: a multisystem mitochondrial disorder in infancy. J Clin Invest. 1990;86:1601-1608.
45 McShane MA, Hammans SR, Sweeney M, et al. Pearson syndrome and mitochondrial encephalomyopathy in a patient with a deletion of mtDNA. Am J Hum Genet. 1991;48:39-42.
46 Domingo-Claros A, Alonso E, Banda Ed Ede L. Schizophrenia and refractory anaemia with ring sideroblasts. Br J Haematol. 2004;125:543.
47 Gill H, Yip T, Lo W-K. Anemia in a patient newly transferred from peritoneal dialysis to hemodialysis. Am J Kid Dis. 2010;56:xxxvii-xxxvix.
48 Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100:2292-2302.
49 Cazzola M, Malcovati L. Myelodysplastic syndromes – coping with ineffective hematopoiesis. New Engl J Med. 2005;352(6):536-538.
50 Malcovati L, Germing U, Kuendgen A, et al. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J Clin Oncol. 2007;25:3503-3510.
51 Dewald GW, Brecher M, Travis LB, Stupca PJ. Twenty-six patients with hematologic disorders and X chromosome abnormalities. Frequent idic(X)(q13) chromosomes and Xq13 anomalies associated with pathologic ringed sideroblasts. Cancer Genet Cytogenet. 1989;42:173-185.
52 Bowen D, Culligan D, Jowitt S, et al. Guidelines for the diagnosis and therapy of adult myelodysplastic syndromes. Br J Haematol. 2003;120:187-200.
53 Hellström-Lindberg E, Malcovati L. Supportive care, growth factors, and new therapies in myelodysplastic syndromes. Blood Rev. 2008;22:75-91.
54 Boultwood J, Pellagatti A, Nikpour M, et al. The role of the iron transporter ABCB7 in refractory anemia with ring sideroblasts. PLoS One. 2008;3:e1970.
55 Malcovati L, Della Porta MG, Pietra D, et al. Molecular and clinical features of refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Blood. 2009;114:3538-3545.