Seizures in the Critically Ill
Seizures complicate the course of about 3% of adult patients admitted to intensive care units (ICUs) for non-neurologic conditions1 and occur more frequently in specialized neuroscience ICUs.2 The medical and economic impact of seizures in these patients confers a significance on these events out of proportion to their incidence. Seizures are often the first indication of a central nervous system (CNS) complication in these patients, making their rapid etiologic diagnosis mandatory. Furthermore, because epilepsy is a common disorder (affecting about 2% of the general population), patients with preexisting seizure disorders will occasionally require ICU admission for intercurrent conditions. The intensivist usually manages the initial treatment of these patients, so he or she must be familiar with the indications and risks of the potential therapies as they affect the already critically ill patient. In addition, the patient who develops status epilepticus (SE), whether already in the ICU or not, will often require the care of a critical care specialist in addition to a neurologist.
History
The first recorded description of SE is by Gavasetti in 1586.3,4 Sir Thomas Willis described the complications of untreated SE in 1667:
… as to what further belongs to the prognostication of the Disease, if it end not about the time of ripe age, neither can be driven away by the use of medicines, there happens yet a diverse event in several sick Patients, for it either ends immediately in Death, or is changed into some other Disease, to wit, the Palsie, stupidite, or melancholly, for the most part incurable. As to the former, whenas the fits are often repeated, and every time grow more cruell, the animal functions are quickly debilitated; and from thence, by the taint, by degrees brought on by the Spirits, and the Nerves serving the Praecordia, the vital function is by little and little enervated, till at length, the whole body languishing, and the pulse is loosned, and at length ceasing, at last the vital flame is extinguished.5
Attempts at treating SE in the nineteenth century included bromide,6 morphine,7 and ice applications. Barbiturates were introduced in 1912, followed by the identification and use of phenytoin in 1937; these were the first rational treatments for SE.8 Paraldehyde gained brief prominence in the next decades.9 The most recent major improvement is the use of benzodiazepines, pioneered by the French in the 1960s.10
Epidemiology
Few data are available concerning the epidemiology of seizures in ICU patients. A 10-year retrospective study of all ICU patients at the Mayo Clinic reported approximately 7 patients with seizures per 1000 ICU admissions.11 In a 2-year prospective study of a medical ICU, we acquired approximately 35 patients with seizures per 1000 admissions.1 These analyses are not strictly comparable, because the patient populations and methods of detection differed. The incidence of seizures is probably higher in pediatric ICUs than in medical ICUs.12–14
Certain ICU patients appear to be at increased risk for seizures, but the degree of that increased risk has not been quantified. Patients with renal failure or with an altered blood-brain barrier who receive imipenem-cilastatin are an obvious example, but other patients receiving this antibiotic (or γ-aminobutyric acid [GABA] antagonists such as penicillin) occasionally seize. Cefepime has emerged as a cause of nonconvulsive seizures and SE, especially in patients with renal insufficiency.15 Transplant patients, especially those receiving cyclosporine, appear to have an increased risk for convulsions. Patients who rapidly become hypo-osmolar from any cause are also at risk. Nonketotic hyperglycemia patients have a high likelihood of partial seizures; this is a rare instance of a metabolic disorder producing focal neurologic syndromes.16 Less commonly, diabetic ketoacidosis may also produce partial seizures.17
The epidemiology of SE is somewhat better understood. Estimates of the incidence of generalized convulsive SE in the United States range from 50,000 cases/year18 to 250,000 cases/year.19 Some portion of this discrepancy may be due to differences in definitions. The larger estimate comes from the only population-based data available and may be more accurate. Similarly large variations occur in mortality rate estimates, from 1% to 2% in the former study to 22% in the latter. This disagreement stems, at least in part, from a conceptual discordance: the smaller number attempts to determine mortality rate that the authors directly attribute to SE, while the larger figure reflects the overall mortality rate for SE patients, in whom death was frequently a consequence of the cause of the underlying disease rather than SE itself. In the latter study, for example, anoxia was the cause of SE in adults with the highest mortality rate. In many of the reports surveyed in the earlier review, these patients were not included.
Towne and colleagues demonstrated that 8% of an unselected series of comatose medical ICU patients had unsuspected nonconvulsive status epilepticus (NCSE).20 In septic ICU patients, Oddo and colleagues showed that about 30% have periodic epileptiform activity or NCSE when recorded for 24 hours or longer.21
Hospital-based series of SE patients are usually subject to considerable selection bias regarding cause. The data in Table 65.1, based upon 20 years of experience in San Francisco, are of great interest because almost all patients with SE in the city of San Francisco who began to seize outside the hospital are included.22–24
Table 65.1
Etiology of Status Epilepticus at San Francisco General Hospital
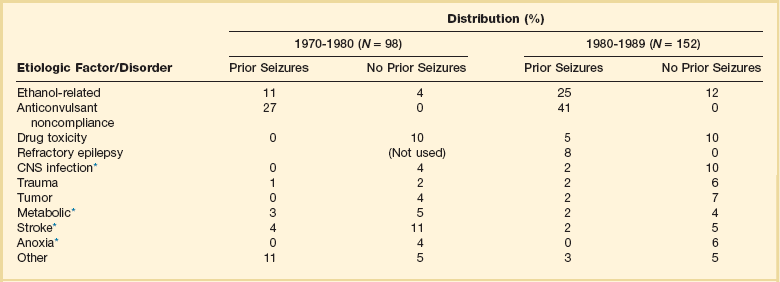
*Conditions most likely to result in admission to intensive care unit (ICU).
Between 6% and 12% of epilepsy patients present with SE,25 and about 20% of seizure patients will experience an episode of SE within 5 years of their first seizure.11
Nosology and Semiology
Numerous systems have evolved for the classification of seizures; the most frequently used today is that of the International League Against Epilepsy26 (Box 65.1). This schema allows classification based primarily on clinical criteria, without inferences about cause. Although a more recent proposal for terminology has been published, it is not yet widely accepted.27 It is important because of its predictive value for cause, prognosis, and treatment decisions in ICU patients. Simple partial seizures arise focally in the cerebral cortex, without taking over either the limbic system or subcortical nuclei. The patient remains aware of the environment during the ictus, and except for the seizure itself appears unchanged. Bilateral limbic system dysfunction results in a complex partial seizure; the patient’s awareness and ability to interact with the environment are diminished (but not always completely abolished). Automatisms are movements that the patient seems to make without being aware of them; typical automatisms include swallowing, masticatory movements, and fumbling with nearby items. Secondary generalization implies invasion of either the other hemisphere (with loss of consciousness) or, more commonly, subcortical structures, with the development of a generalized convulsion.
Primary generalized seizures seem to arise from the entire cerebral cortex and the diencephalon at the same time; there are no visible focal phenomena. Consciousness is lost from the start of the seizure. True absence seizures are usually confined to childhood; they consist of the abrupt onset of a blank stare usually lasting 5 to 15 seconds, without lateralizing phenomena, from which the patient abruptly returns to normal. Atypical absence is usually seen in children who have the Lennox-Gastaut syndrome. Myoclonic seizures begin with brief, bilaterally synchronous jerks without an initial change in consciousness, followed by a generalized convulsion. They occur in several of the genetic epilepsies, but in the ICU are more commonly the consequences of anoxia or metabolic disturbances.28 Clonic seizures involve repetitive movements; they may be generalized (synchronous movements of all extremities and both sides of the face) or partial (e.g., one side of the face and the arm of the same side). Tonic seizures are episodes of tonic extension of the arms, legs, and trunk; they must be distinguished from decerebrate rigidity and from tetanic spasms.29 Tonic-clonic seizures begin with tonic extension, followed by a brief phase of rapid vibration of the extremities, evolving into bilaterally synchronous clonus, and concluding with a postictal phase in which incontinence is common and brief apnea is occasionally noted. They may be primarily generalized or, more commonly, occur as the manifestation of spread of a partial seizure. Only those seizures that are known to involve progression through the tonic and clonic stages should be called tonic-clonic.
SE is classified by a somewhat similar system, with alterations to match the observable clinical phenomena (Box 65.2).30 Again, the ability to use clinical observation without inferences about cause is important. Generalized convulsive SE (GCSE) is the type most commonly encountered in ICUs, and poses the greatest risk to the patient. GCSE may be either primarily generalized, as in the intoxicated patient, or may represent secondary generalization, as in the patient with a brain abscess who develops GCSE. Tonic SE is usually seen in children or adolescents with a history of severe CNS dysfunction. Nonconvulsive SE (NCSE) in the ICU is most commonly the consequence of partially treated GCSE. Some authors use this as a general term for any SE involving altered consciousness without convulsive movements. Although conceptually useful, this blurs the distinctions among absence SE, partially treated GCSE, and complex partial SE (CPSE), which have different causes and treatments. Epilepsia partialis continua (EPC) is a special form of partial SE in which a small area of the body makes repetitive movements, sometimes for months or years following a CNS insult.
Pathophysiology
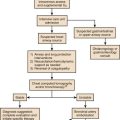
The causes and effects of SE at the cellular, brain, and systemic levels are interrelated, but their individual analysis is useful for understanding them and their therapeutic implications. One must first understand the consequences of a single seizure and then contrast this information with the effects of prolonged or frequent seizures merging into SE. Longer durations of SE produce more profound alterations with an increasing likelihood of permanence, and of becoming refractory to treatment. Figure 65.1 illustrates the variety of processes involved in a single seizure and in the transition to SE.31
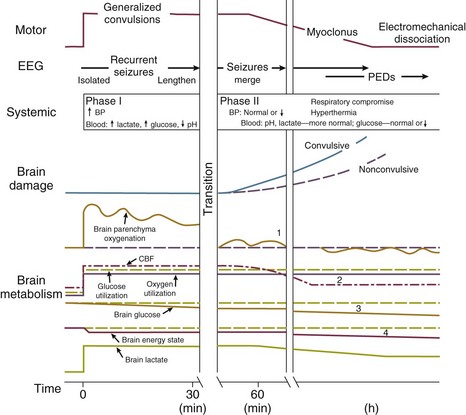
Many other important biophysical and biochemical alterations occur during and after SE. The intense neuronal activity activates immediate-early genes and produces heat shock proteins, providing strong indications of the deleterious effects of SE and insight into the mechanisms by which neurons protect themselves.32 Wasterlain’s group summarized the many mechanisms through which SE damages the nervous system.33
The electrical phenomena of SE at the whole brain level, as seen in the scalp electroencephalogram (EEG), reflect the seizure type that initiates SE (Fig. 65.2). Thus, absence SE begins with a generalized 3-Hz wave-and-spike EEG pattern. During the course of SE, there will usually be some slowing of this rhythm, but the wave-and-spike characteristic persists. In contrast, GCSE goes through the sequence of EEG changes outlined in Table 65.2. The initial high-frequency discharge becomes progressively less well formed over minutes; this pattern implies that neuronal activity is less synchronous. Whether this indicates that inhibitory systems are attempting to terminate SE, a progressive decay in the ability of synaptic mechanisms to maintain synchrony, or global deterioration in neuronal function remains to be determined.
Table 65.2
Electrographic-Clinical Correlations in Generalized Convulsive Status Epilepticus
| Stage | Typical Clinical Manifestations* | Electroencephalographic Features |
| 1 | Tonic-clonic convulsions; hypertension and hyperglycemia common | Discrete seizures with interictal slowing |
| 2 | Low- or medium-amplitude clonic activity, with rare convulsions | Waxing and waning of ictal discharges |
| 3 | Slight, but frequent, clonic activity, often confined to the eyes, face, or hands | Continuous ictal discharges |
| 4 | Rare episodes of slight clonic activity; hypotension and hypoglycemia become manifest | Continuous ictal discharges punctuated by flat periods |
| 5 | Coma without other manifestations of seizure activity | Periodic epileptiform discharges on a flat background |
*The clinical manifestations may vary considerably, depending on the underlying neuropathophysiologic process (and its anatomy), systemic diseases, and medications. In particular, stages of the electrographic progression may be sufficiently brief to be overlooked. Partially treating status epilepticus may dissociate the clinical and electrographic features.
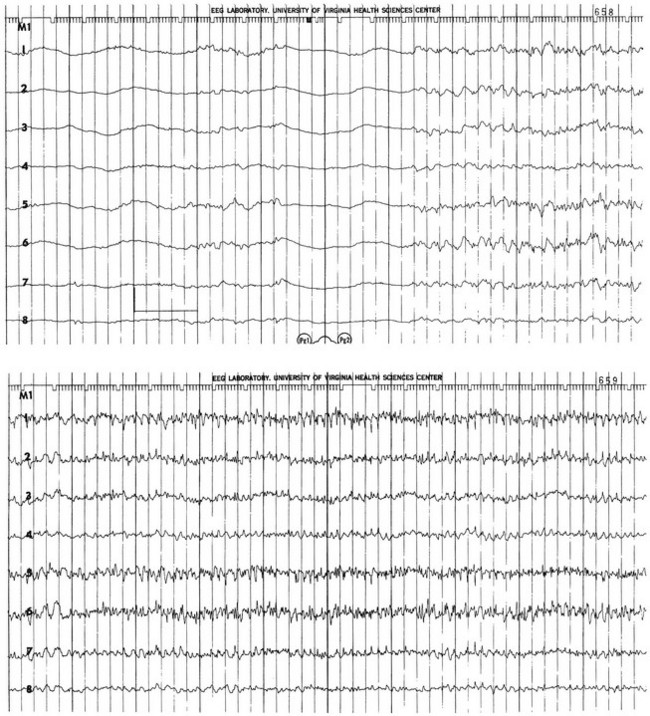
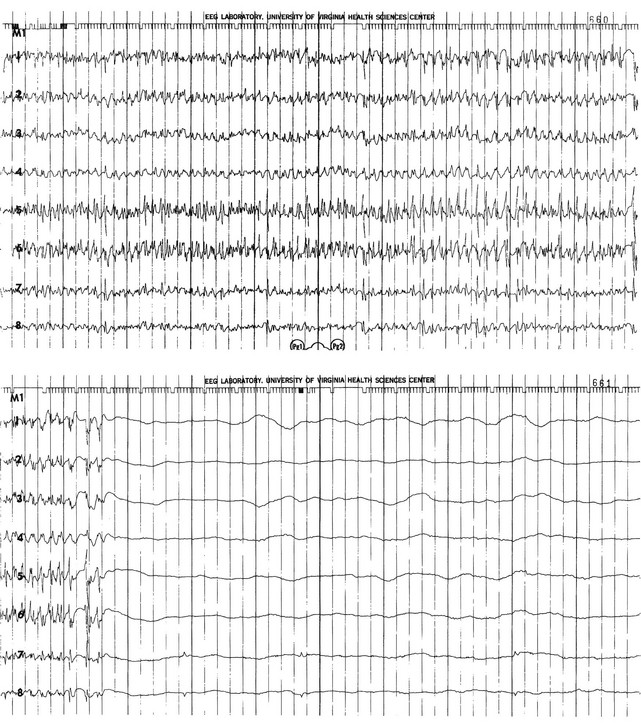
SE can produce cerebral edema, which follows ictal damage to the blood-brain barrier.
In addition to damaging the CNS, GCSE produces serious, often life-threatening, systemic effects.34 Pressures in the systemic arterial system (under sympathetic control) and in the pulmonary arterial system (raised via efferents from pontine and medullary centers) are dramatically elevated from the moment of seizure onset. Epinephrine and cortisol release prompts further elevations of systemic arterial pressure, and also produces hyperglycemia. Increased muscular work raises the circulating lactate concentration. Respiration becomes ineffective; both airway obstruction and diaphragmatic contraction impede air movement. The consequent hypoxia further elevates lactate levels. Ventilatory failure impairs CO2 excretion while CO2 production increases markedly, adding a respiratory component to the acidosis. In GCSE, the arterial blood pH frequently falls below 7.0. The muscular work accelerates heat production; when coupled with decreased dermal blood flow (produced by sympathetic stimulation), GCSE can quickly raise the core temperature to 40° C or higher.
Clinical Manifestations
Recognition of Seizures
ICU patients often have altered awareness in the absence of seizures, reflecting their underlying condition, complications of those conditions (such as septic encephalopathy35), and drugs that depress alertness (intentionally or not). Although clonic motor activity in these patients remains visible, it may be difficult to tell whether a subsequent further decline in alertness reflects a seizure or some other process. In this situation, an EEG is required to make the diagnosis of a complex partial seizure. Although the detailed interpretation of EEGs is beyond the scope of this chapter, the intensivist can easily learn to recognize basic seizure types and other important EEG abnormalities in critically ill patients.36
Many sorts of abnormal movements occur in patients with severe metabolic disturbances or anoxic brain damage. Some of them can be distinguished from seizures by observation; such movements are frequently evoked or exacerbated by sensory stimuli and can sometimes be suppressed by changing the patient’s posture. However, Hirsch and colleagues have demonstrated that seizures in ICU patients may be induced by external stimuli37; if any doubt about the nature of such movements persists, an EEG should be performed.
During therapeutic cooling for patients in a coma after cardiac arrest, seizures may be difficult to detect clinically, especially when neuromuscular junction blockade is used.38 EEG monitoring should be performed.39
Manifestations of Status Epilepticus
The neurologic manifestations of SE depend on the type of SE and, for the partial forms, the area of cortex from which the abnormality arises. Box 65.2 summarizes the types of SE encountered in clinical practice. This section will focus on the varieties of SE seen most frequently among ICU patients.
Diagnostic Approach
The Intensive Care Unit Patient with New-Onset Seizures
In addition to routine biochemical studies, screening for drugs of abuse should be performed on patients with unexplained seizures. Cocaine has emerged as a prominent cause of seizures in many urban hospitals.40 One area of controversy involves the importance of divalent cation disturbances in adult seizures. Hypocalcemia is rarely a cause of seizures beyond the neonatal period, and its discovery should not be the end of the diagnostic workup. Hyperparathyroidism has been linked anecdotally to seizures, with the inference that parathormone is neurotoxic. Similarly, hypomagnesemia has an unwarranted reputation as a cause of seizures, especially in the malnourished alcoholic patient.
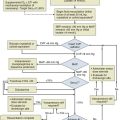
In our prospective study of neurologic complications in medical ICU patients, 38 of 61 patients (62%) with seizures had a vascular, infectious, or neoplastic explanation for their fits.1 Computed tomography (CT) or magnetic resonance imaging (MRI) should be performed on all ICU patients with new seizures, with a few exceptions. Hypoglycemia and nonketotic hyperosmolar hyperglycemia will commonly produce seizures (even partial seizures), and such patients might be treated for their metabolic disturbance and observed if there is no other indication of neurologic disease. With currently available technology, there are almost no patients who cannot be transported to undergo CT scanning. Although MRI is preferable diagnostically in most situations, the magnetic field precludes infusion pumps and other metallic devices (nonferromagnetic ventilators are available). The decision whether to administer contrast agent for a CT or MRI scan depends on the clinical setting and on the appearance of the plain scan.
The Patient Presenting with or Developing Status Epilepticus
In contrast to the ICU patient with a single or a few seizures, the SE patient will require concomitant diagnostic and therapeutic efforts. The first issue is to make a diagnosis of SE. Because most seizures stop within 5 to 7 minutes,41 it is reasonable to begin treatment after 5 minutes of continuous seizure activity or after the second or third seizure occurring without recovery between the spells. The available treatments are discussed later.
SE has a limited differential diagnosis. GCSE might rarely be confused with decerebrate posturing, but the evolutionary nature of the former and the stimulus sensitivity of the latter make their clinical distinction straightforward. Generalized tetanus patients are awake during their spasms, and almost always flex their arms rather than extending them.20 The distinction of seizures from movement disorders and psychiatric conditions is discussed earlier.
EEG monitoring is frequently useful in SE,42 but treatment should not be delayed to obtain it when the diagnosis is apparent. A variety of EEG findings may be present, depending on the type of SE and its duration (see Table 65.2). The most typical pattern early in SE is that of rhythmic, high-frequency (>12 Hz) activity that increases in amplitude and decreases in frequency, finally terminating abruptly and leaving postictal low-amplitude slowing in its wake. CPSE patients often lack such organized discharges, but may instead have waxing and waning rhythmic activity in one or several head regions. Such a pattern requires a high index of suspicion in order to correctly diagnose CPSE; a diagnostic trial of an intravenous benzodiazepine is often necessary. Patients who develop refractory SE or experience seizures during neuromuscular blockade will require continuous EEG monitoring. The technology to perform such monitoring outside specialized epilepsy centers is only now becoming available.
Management Approach
The Intensive Care Unit Patient with New-Onset Seizures
The ICU patient with CNS disease who has even a single seizure should usually be started on a chronic anticonvulsant regimen, with the decision to continue medication reviewed prior to hospital discharge. It is now apparent that initiating anticonvulsant therapy after the first unprovoked (e.g., not drug- or withdrawal-related) seizure helps delay the onset of subsequent seizures, but does not change their eventual incidence.43 Starting treatment after the first seizure in a critically ill patient who has a condition predictive of seizure recurrence may be even more important if the patient’s problems include coagulopathies, myocardial ischemia, or other conditions that would be seriously complicated by a convulsion.
The Neurocritical Care Society recently published extensive guidelines for the treatment of SE,44 which should be consulted for detailed information about the drugs discussed briefly here.
Hypersensitivity to PHT is the major adverse effect of concern to the intensivist. This allergy may be manifested solely as fever, but more commonly includes a rash and eosinophilia. Febrile reactions appear to be more common with intravenous than with enteral loading. The Stevens-Johnson syndrome occurs rarely. The diagnosis and management of adverse reactions to PHT and other anticonvulsants have been reviewed.45 PHT is associated with a number of long-term adverse effects in patients with subarachnoid or intracerebral hemorrhage.46
Status Epilepticus
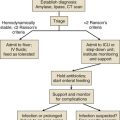
The patient in GCSE has an obvious medical emergency; unfortunately, the NCSE and CPSE patients also require emergent treatment but are less straightforward to recognize. In a patient with any of these three conditions, the clinician must move quickly to stop seizures in order to prevent further brain destruction.47 A suggested management protocol for these conditions is presented in Box 65.3, and Figure 65.3 shows a management algorithm for SE. Patients with simple partial SE or EPC appear to be at substantially less risk of developing widespread cerebral damage and also appear less likely to respond to the aggressive approach outlined in Box 65.3. In this group, correction of underlying problems, if possible (such as nonketotic hyperosmolar hyperglycemia), is most important. Of the available anticonvulsants, PB seems most likely to be efficacious. These patients are often loaded with PHT in the hope that this agent will prevent secondary generalization, but the actual value of this practice is unknown.
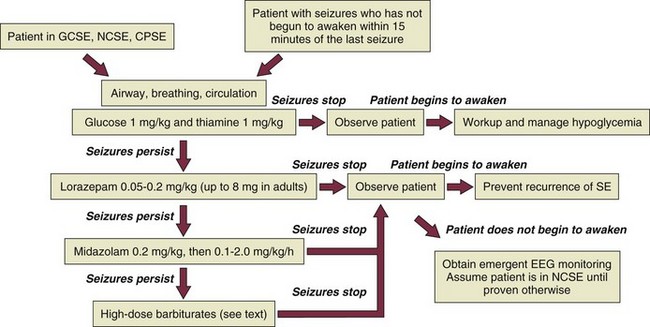
Figure 65.3 Management algorithm for status epilepticus.
Specific Agents
Benzodiazepines
LRZ is emerging as the agent of first choice for terminating SE. A study in the Veterans Affairs medical system compared LRZ, DZ followed by PHT, PHT alone, and PB as first-line agents and demonstrated that LRZ is the definitive agent of first choice.48 The advantages of LRZ over PHT include (1) its longer duration of action against SE (4-14 hours as opposed to 20 minutes) and (2) a higher initial response rate.49 One study concluded that children receiving PHT for SE were far more likely to require intubation for ventilatory failure than comparable children receiving LRZ50; the same was true for all ages in the San Francisco Prehospital Status Study.51 PHT and LRZ remain the only agents in this class with FDA indications for SE. In Europe, midazolam (MDZ) or clonazepam is often used initially. MDZ is exceptionally useful for refractory SE, but it is hampered by tachyphylaxis,52 which occurs because the GABAA receptors bearing benzodiazepine-sensitive subunits are removed from the neuronal cell membrane and replaced with receptors bearing benzodiazepine-insensitive ones.53 Respiratory depression is the major adverse effect of all agents in this class when administered intravenously.
Data from the Veterans Affairs cooperative trial indicate that the use of other conventional agents after failure of the first one is very unlikely to terminate SE.49
Hydantoins
PHT is highly insoluble, and it must be dissolved in sodium hydroxide and propylene glycol at a pH greater than 11 to remain in solution. Therefore, extravasation can produce severe necrosis. The drug can also cause thrombophlebitis, which may result in the “purple glove syndrome.”54
Hydantoins should not be used in absence SE, as they may worsen the condition.
Barbiturates
PB has long been one of the major anti-SE agents. Some advocate it as a first-line drug,55 but it is rarely used in this role. It has classically been a third-line agent for control of SE, after a benzodiazepine and PHT. Its utility in SE is diminished by the length of time required to obtain a therapeutic effect in patients who have already failed with a benzodiazepine and PHT. It remains an important agent in patients with simple partial SE, and in preparing patients to be withdrawn from high-dose pentobarbital.
Pentobarbital and thiopental are commonly reserved for the control of refractory SE, although thiopental is not currently available in the United States. Although these agents will be effective if used in large enough doses, side effects often limit their use56 or may even be fatal.57 They are important when other rapidly available modalities have failed (see Box 65.3).
Valproate
Intravenous valproate, given in a dose of 20 to 30 mg/kg, has gained popularity for the treatment of SE because it does not produce respiratory depression or marked sedation. It has been successful in case series58,59 but has not been directly compared to the other available agents. Hypotension may occur with large doses.60 This drug should be avoided if the patient has an inborn error of metabolism affecting the liver, as it may precipitate fulminant hepatic failure.61 Valproate has a number of drug interactions that limit its utility in the ICU.62
Isoflurane and Desflurane
The inhalational anesthetics isoflurane and desflurane are effective in controlling refractory SE, but the difficulties involved in delivering the gases (such as the requirement for an anesthesia machine) and the need for a gas scavenging system have essentially confined their use to the operating suite or the recovery area.63 Advances in delivery systems may make its use in the ICU more reasonable, but long-term use is potentially hepatotoxic.
Propofol
Propofol has been reported effective in refractory SE in doses up to 250 µg/kg/minute64 but has not been directly compared with other drugs. It theoretically offers a lower risk of respiratory depression and more rapid recovery of consciousness after the agent is stopped. We use it in SE patients who have failed or become resistant to MDZ.65 One should observe for evidence of the propofol infusion syndrome and stop the drug should a metabolic acidosis or evidence of muscle injury develop.
Ketamine
Although only anecdotes and small case series are available, ketamine appears to be a useful agent for the termination of refractory SE.66 Its NMDA blocking effect distinguishes it from the other agents discussed here, and it carries theoretical advantages in terms of brain protection.67 Its intrinsic sympathomimetic effect makes it a useful choice in hypotensive patients, and it does not markedly impair ventilation. The appropriate dose in SE has not been established; we use a loading dose of 1 to 5 mg/kg, with an infusion rate of 10 to 50 µg/kg/minute.
Levetiracetam
Levetiracetam is now available for intravenous use, but its role in SE remains to be determined. The effective dose in adults is reported to be between 1 and 6 g/day.68 Oral administration may be useful for maintenance.69
Lacosamide
Lacosamide, which is available for both intravenous and oral use, is typically started at 200 mg twice daily and may be increased to 400 mg twice daily if needed.70
The management of “super-refractory” SE has long been an area of contention. These patients may require general anesthesia for a month or longer, while the stimulus for epileptogenicity comes under control or remits. Some of these patients are now recognized to have an autoimmune or paraneoplastic cause for their condition, which requires immunologic therapy.71 Although these patients sometimes seem hopeless, it is important to remember that 35% will return to their premorbid level of function.72
Controversial Management Issues
EPC is usually diagnosed in a patient who has an isolated repetitive movement (usually of the hand or face), often following an infectious or vascular insult, or in the setting of nonketotic hyperglycemia.73 The movement may persist for months or years. Most patients receive anticonvulsants to prevent spread of the discharge, but these agents seldom affect EPC itself. Attempts at treatment with high-dose barbiturates result in short-term suppression of the movement, but it usually returns as the drug levels decline.
Prognosis
Wijdicks and Sharbrough reported that 34% of patients experiencing a seizure in any ICU at the Mayo Clinic died during that hospitalization.11 In our prospective study of neurologic complications in medical ICU patients,10 having even one seizure while in the unit for a non-neurologic reason doubled the patient’s in-hospital mortality rate. This effect on prognosis appeared to be due to the effect of the cause of the ictus, rather than the seizure itself.
Most studies of SE outcome have concentrated on mortality rates in GCSE. Hauser18 summarized the data available in 1990, showing mortality rates for SE to vary from 1% to 53%. The few studies that attempted to distinguish the mortality rate due to SE from that of the underlying disease attributed rates of 1% to 7% to SE and 2% to 25% to its cause. The Virginia Commonwealth University population-based studies have analyzed the mortality risks for various aspects of GCSE.12 SE lasting longer than 1 hour was associated with a 10-fold increase in mortality rate when compared to SE lasting less than 1 hour. Other causes associated with marked increases in mortality rate were anoxia, intracranial hemorrhages, tumors, infections, and trauma.
Very few findings are available regarding the functional status of GCSE survivors, and none reliably allows a distinction between the effects of SE and its causes. A review of intellectual impairment as an outcome of SE concluded intellectual abilities probably did decline as a consequence of SE.74 Survivors of SE frequently have memory and behavioral disorders out of proportion to any structural damage produced by the cause of their seizures. This observation is supported by a wealth of experimental data and argues strongly for the rapid and effective control of SE. The prognosis of CPSE is less certain, but case reports of severe memory deficits following prolonged CPSE have appeared.75
The effect of the treatment of SE on the risk of subsequent epilepsy is uncertain. Experimental studies suggest that SE does lower the threshold for subsequent seizures.76
References
1. Bleck, TP, Smith, MC, Pierre-Louis, JC, et al. Neurologic complications of critical medical illnesses. Crit Care Med. 1993; 21:98–103.
2. Varelas, PN, Mirski, M. Treatment of seizures in the neurologic intensive care unit. Curr Treat Options Neurol. 2007; 9:136–145.
3. Gavassetti, M. Libri duo. Alter de rebus praeter naturum: Alter de indicationibus curativus. Venice, 1586. Quoted by Hunter RA: Status epilepticus. History, incidence and problems. Epilepsia. 1959/1960; 1:162–188.
4. Hunter, RA. Status epilepticus. History, incidence and problems. Epilepsia. 1959/1960; 1:162–188.
5. Willis, T. Pathologiae cerebri et nervosi generis specimen. In quo agitur de morbis convulsivis et de scorbuto. 1667. Translated by S. Pordage. London, Dring, 1681, p 18. Quoted by Hunter RA: Status epilepticus. History, incidence and problems. Epilepsia. 1959/1960; 1:162–188.
6. Wilks, S. Bromide and iodide of potassium in epilepsy. Med Times Gaz (Lond). 1861; 2:635–636.
7. Gowers, WR. Epilepsy and Other Chronic Convulsive Diseases: Their Causes, Symptoms, and Treatment. London: J&A Churchill; 1881.
8. Bleck, TP, Klawans, HL. Mechanisms of epilepsy and anticonvulsant action. In: Klawans HL, Goetz CG, Tanner CM, eds. Textbook of Clinical Neuropharmacology. New York: Raven Press; 1992:23–30.
9. Weschler, IS. Intravenous injection of paraldehyde for control of convulsions. JAMA. 1940; 114:2198.
10. Gastaut, H, Naquet, R, Poiré, R, Tassinari, CA. Treatment of status epilepticus with diazepam (Valium). Epilepsia. 1965; 6:167–182.
11. Wijdicks, EFM, Sharbrough, FW. New-onset seizures in critically ill patients. Neurology. 1993; 43:1042–1044.
12. Hussain, N, Appleton, R, Thorburn, K. Aetiology, course and outcome of children admitted to paediatric intensive care with convulsive status epilepticus: A retrospective 5-year review. Seizure. 2007; 16(4):305–312.
13. Valencia, I, Lozano, G, Kothare, SV, et al. Epileptic seizures in the pediatric intensive care unit setting. Epileptic Disord. 2006; 8:277–284.
14. Saengpattrachai, M, Sharma, R, Hunjan, A, et al. Nonconvulsive seizures in the pediatric intensive care unit: Etiology, EEG, and brain imaging findings. Epilepsia. 2006; 47:1510–1518.
15. Chatellier, D, Jourdain, M, Mangalaboyi, J, et al. Cefepime-induced neurotoxicity: An underestimated complication of antibiotherapy in patients with acute renal failure. Intensive Care Med. 2002; 28:214–217.
16. Chung, SJ, Lee, JH, Lee, SA, et al. Co-occurrence of seizure and chorea in a patient with nonketotic hyperglycemia. Eur Neurol. 2005; 54:230–232.
17. Placidi, F, Floris, R, Bozzao, A, et al. Ketotic hyperglycemia and epilepsia partialis continua. Neurology. 2001; 57:534–537.
18. Hauser, WA. Status epilepticus: Epidemiologic considerations. Neurology. 1990; 40(Suppl 2):9–13.
19. DeLorenzo, RJ, Towne, AR, Pellock, JM, et al. Status epilepticus in children, adults, and the elderly. Epilepsia. 1992; 33(Suppl 4):S15–S25.
20. Towne, AR, Waterhouse, EJ, Boggs, JG, et al. Prevalence of nonconvulsive status epilepticus in comatose patients. Neurology. 2000; 54:340–345.
21. Oddo, M, Carrera, E, Claassen, J, et al. Continuous electroencephalography in the medical intensive care unit. Crit Care Med. 2009; 37:2051–2056.
22. Aminoff, MJ, Simon, RP. Status epilepticus: Causes, clinical features and consequences in 98 patients. Am J Med. 1980; 69:657–666.
23. Lowenstein, DH, Alldredge, BK. Status epilepticus in an urban public hospital in the 1980s. Neurology. 1993; 42:483–488.
24. Bleck, TP. Status epilepticus. Univ Rep Epilepsy. 1992; 1:1–7.
25. Ettinger, AB, Shinnar, S. New-onset seizures in an elderly hospitalized population. Neurology. 1993; 43:489–492.
26. Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia. 1981; 22:489–501.
27. Berg, AT, Berkovic, SF, Brodie, MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilepsia. 2010; 51:676–685.
28. Bleck, TP. Metabolic encephalopathy. In: Weiner WJ, Shulman LM, eds. Emergent and Urgent Neurology. 2nd ed. Philadelphia: Lippincott; 1999:223–253.
29. Bleck, TP, Brauner, JS. Tetanus. In: Scheld WM, Whitley RJ, Durack DT, eds. Infections of the Central Nervous System. 2nd ed. New York: Lippincott-Raven; 1997:629–653.
30. Bleck, TP. Status epilepticus. In: Klawans HL, Goetz CG, Tanner CM, eds. Textbook of Clinical Neuropharmacology. 2nd ed. New York: Raven Press; 1992:65–73.
31. Lothman, EW. The biochemical basis and pathophysiology of status epilepticus. Neurology. 1990; 40(Suppl 2):13–23.
32. Lowenstein, DH, Simon, RP, Sharp, FR. The pattern of 72-kDa heat shock protein-like immunoreactivity in the rat brain following fluothyl-induced status epilepticus. Brain Res. 1990; 531:173–182.
33. Wasterlain, CG, Fujikawa, DG, Penix, L, Sankar, R. Pathophysiological mechanisms of brain damage from status epilepticus. Epilepsia. 1993; 34(Suppl 1):S37–S53.
34. Walton, NY. Systemic effects of generalized convulsive status epilepticus. Epilepsia. 1993; 34(Suppl 1):S54–S58.
35. Bleck, TP. Neurologic alterations in sepsis. In: Fein AM, Abraham E, Balk R, et al, eds. Textbook of Sepsis and Multiorgan Failure. Media, PA: Williams & Wilkins; 1997:236–242.
36. Bleck, TP, Hirsch, LJ, Vespa, PM. Electroencephalography in the intensive care unit. In: Engel JE, Pedley T, eds. Epilepsy: A Comprehensive Textbook. 2nd ed. Philadelphia: Lippincott Williams & Wilkins; 2008:855–862.
37. Hirsch, LJ, Pang, T, Claassen, J, et al. Focal motor seizures induced by alerting stimuli in critically ill patients. Epilepsia. 2008; 49(6):968–973.
38. Geocadin, RG, Koenig, MA, Stevens, RD, Peberdy, MA. Intensive care for brain injury after cardiac arrest: Therapeutic hypothermia and related neuroprotective strategies. Crit Care Clin. 2006; 22:619–636.
39. Hovland, A, Nielsen, EW, Klüver, J, Salvesen, R. EEG should be performed during induced hypothermia. Resuscitation. 2006; 68(1):143–146.
40. Rowbotham, MC, Lowenstein, DH. Neurologic complications of cocaine use. Annu Rev Med. 1990; 41:417–422.
41. Lowenstein, DH, Bleck, T, Macdonald, RL. It’s time to revise the definition of status epilepticus. Epilepsia. 1999; 40:120–122.
42. Ross, C, Blake, A, Whitehouse, WP. Status epilepticus on the paediatric intensive care unit—The role of EEG monitoring. Seizure. 1999; 8:335–338.
43. First Seizure Trial Group. Randomized clinical trial of the efficacy of antiepileptic drugs in reducing the risk of relapse after a first unprovoked tonic-clonic seizure. Neurology. 1993; 43:478–483.
44. Brophy, G, Bell, R, Claassen, J, et al. Guidelines for the evaluation and management of status epilepticus. Neurocrit Care. 2012; 17(1):3–23.
45. Smith, MC, Bleck, TP. Toxicity of anticonvulsants. In: Klawans HL, Goetz CG, Tanner CM, eds. Textbook of Clinical Neuropharmacology. 2nd ed. New York: Raven Press; 1992:45–64.
46. Naidech, AM, Kreiter, KT, Janjua, N, et al. Phenytoin exposure is associated with functional and cognitive disability after subarachnoid hemorrhage. Stroke. 2005; 36:583–587.
47. Alldredge, B, Treiman, DM, Bleck, TP, Shorvon, SD. Treatment of status epilepticus. In: Engel JE, Pedley T, eds. Epilepsy: A Comprehensive Textbook. 2nd ed. Philadelphia: Lippincott Williams & Wilkins; 2008:1357–1363.
48. Treiman, DM, Meyers, PD, Walton, NY, et al. A comparison of four treatments for generalized convulsive status epilepticus. Veterans Affairs Status Epilepticus Cooperative Study Group. N Engl J Med. 1998; 339:792–798.
49. Bleck, TP. Critical care of the patient in status epilepticus. In: Wasterlain C, Treiman D, eds. Status Epilepticus. Boston: MIT Press; 2006:607–613.
50. Chuilli, DA, Ternfrup, TE, Kanter, RK. The influence of diazepam or lorazepam on the frequency of endotracheal intubation in childhood status epilepticus. J Emerg Med. 1991; 9:13–17.
51. Alldredge, BK, Gelb, AM, Isaacs, SM, et al. A comparison of lorazepam, diazepam, and placebo for the treatment of out-of-hospital status epilepticus. N Engl J Med. 2001; 345:631–637.
52. Kumar, A, Bleck, TP. Intravenous midazolam for the treatment of refractory status epilepticus. Crit Care Med. 1992; 20:483–488.
53. Joshi, S, Kapur, J. GABAA receptor plasticity during status epilepticus. In Noebels JL, Avoli M, Rogawski MA, et al, eds. : Jasper’s Basic Mechanisms of the Epilepsies, 4th ed, New York: Oxford, 2012.
54. O’Brien, TJ, Cascino, GD, So, EL, Hanna, DR. Incidence and clinical consequence of the purple glove syndrome in patients receiving intravenous phenytoin. Neurology. 1998; 51:1034–1039.
55. Shaner, DM, McCurdy, SA, Herring, MO, Gabor, AJ. Treatment of status epilepticus: A prospective comparison of diazepam and phenytoin versus phenobarbital and optional phenytoin. Neurology. 1988; 38:202–206.
56. Yaffe, K, Lowenstein, DH. Prognostic factors of pentobarbital therapy for refractory generalized status epilepticus. Neurology. 1993; 43:895–900.
57. Bleck, TP. High-dose pentobarbital treatment of refractory status epilepticus: A meta-analysis of published studies. Epilepsia. 1992; 33:5.
58. Venkataraman, V, Wheless, JW. Safety of rapid intravenous infusion of valproate loading doses in epilepsy patients. Epilepsy Res. 1999; 35:147–153.
59. Sinha, S, Naritoku, DK. Intravenous valproate is well tolerated in unstable patients with status epilepticus. Neurology. 2000; 55:722–724.
60. White, JR, Santos, CS. Intravenous valproate associated with significant hypotension in the treatment of status epilepticus. J Child Neurol. 1999; 14:822–823.
61. Krahenbuhl, S, Brandner, S, Kleinle, S, et al. Mitochondrial diseases represent a risk factor for valproate-induced fulminant liver failure. Liver. 2000; 20:346–348.
62. Spriet, I, Meersseman, W, De Troy, E, et al. Meropenem-valproic acid interaction in patients with cefepime-associated status epilepticus. Am J Health Syst Pharm. 2007; 64:54–58.
63. Bleck, TP. Therapy for status epilepticus. Clin Neuropharmacol. 1983; 6:255–268.
64. Stecker, MM, Kramer, TH, Raps, EC, et al. Treatment of refractory status epilepticus with propofol: Clinical and pharmacokinetic findings. Epilepsia. 1998; 39:18–26.
65. Prasad, A, Worrall, BB, Bertram, EB, Bleck, TP. Propofol and midazolam in the treatment of refractory status epilepticus. Epilepsia. 2001; 42:380–386.
66. Sheth, RD, Gidal, BE. Refractory status epilepticus: Response to ketamine. Neurology. 1998; 51:1765–1766.
67. Mazarati, AM, Wasterlain, CG. N-methyl-D-asparate receptor antagonists abolish the maintenance phase of self-sustaining status epilepticus in rat. Neurosci Lett. 1999; 265:187–190.
68. Falip, M, Carreno, M, Amaro, S, et al. Use of levetiracetam in hospitalized patients. Epilepsia. 2006; 47:2186–2188.
69. Rossetti, AO, Bromfield, EB. Determinants of success in the use of oral levetiracetam in status epilepticus. Epilepsy Behav. 2006; 8:651–654.
70. Goodwin, H, Hinson, HE, Shermock, KM, et al. The use of lacosamide in refractory status epilepticus. Neurocrit Care. 2011; 14:348–353.
71. Bleck, TP. Less common etiologies of status epilepticus. Epilepsy Curr. 2010; 10:31–33.
72. Shorvon, S, Ferlisi, M. The outcome of therapies in refractory and super-refractory convulsive status epilepticus and recommendations for therapy. Brain. 2012; 135(Pt 8):2314–2328.
73. Schomer, DL. Focal status epilepticus and epilepsia partialis continua in adults and children. Epilepsia. 1993; 34(Suppl 1):S29–S36.
74. Dodrill, CB, Wilensky, AJ. Intellectual impairment as an outcome of status epilepticus. Neurology. 1990; 40(Suppl 2):23–27.
75. Treiman, DM, Delgado-Escueta, AV. Complex partial status epilepticus. Adv Neurol. 1983; 34:69–81.
76. Lothman, EW, Bertram, EH. Epileptogenic effects of status epilepticus. Epilepsia. 1993; 34(Suppl 1):S59–S70.