Chapter 128 Retinoblastoma
“If most solid tumors of childhood are indeed correctly attributable to mutations in germ and/or somatic cells … then childhood cancer cannot be prevented. … the main effort against childhood cancer must be that of early diagnosis and treatment”1
Introduction
The quote from retinoblastoma pioneer Alfred G. Knudson Jr is as true today as it was in 1976.1 We now know a great deal more about the molecular basis of the mutations in cancer cells than Knudson knew when this was written, but we have made distressingly little progress towards Knudson’s admonishment to clinicians that early diagnosis and treatment is essential for the successful management of this disease. This chapter on retinoblastoma drills in on the failure to diagnose issues facing pediatricians who must listen and take action when parents report seeing “something in my child’s eye.” We continue to emphasize what Nancy Mansfield taught us about the importance of recognizing signs and symptoms of classic post-traumatic stress disorder in children undergoing prolonged treatment for intraocular retinoblastoma and multiple anesthesias in that process.
Clinical advances
Since the fourth edition of this publication, in 2006, the most significant addition to the clinical management of children with retinoblastoma, is an adaptation2–4 of the Japanese experience in the use of intra-arterial (regional) chemotherapy.5,6 Like many other new treatment approaches, the initial excitement has been tempered significantly as significant complications (including central retinal artery occlusion, local orbital recurrence and metastatic disease) have been observed. The pros and cons of this new approach to the treatment of retinoblastoma are discussed later in this chapter. Although RB1 gene mutation testing was recommended for everyone with a diagnosis of retinoblastoma regardless of age or circumstances of presentation (unilateral, bilateral, sporadic, or familial) Woo and Harbour reviewed 676 published second primary tumors (SPTs)7 the leading cause of death in heritable retinoblastoma patients.8,9
Basic science advances
In the previous edition, we cited as advances: (1) the definition of the three-dimensional structure of the retinoblastoma protein (pRB) providing new insights into how it works; (2) the fact that pRb can simultaneously bind to multiple proteins, acting as a coupling factor to recruit proteins to specific genes; and (3) an improved understanding of low-penetrance retinoblastoma.10
Genetics of retinoblastoma
Clinical genetics
About 60% of all patients with retinoblastoma have a nonheritable form of the disease with a normal life expectancy if they are cured of the eye cancer. The average age at diagnosis is about 24 months, the eye tumor is unilateral, and the risk of other cancers is virtually indistinguishable from the normal population.11 In contrast, the other 40% of patients, those with the RB1 germ line mutation, have a heritable cancer predisposition syndrome. The inheritance of a single inactive allele of the RB1 gene confers the predisposition to cancer (a dominant trait) but a second inactivating mutation must occur in at least one retinoblast for retinoblastoma to appear. Because the tumor requires inactivation of both copies of the RB1 gene, it is a recessive trait. However, in pedigrees, the tumor appears to be dominant because so many retinoblasts are at risk that the probability that at least one will get the required mutation to develop a tumor is at least 90–95%. A person with the cancer predisposing syndrome phenotype (RB1+/−) will develop retinoblastoma with a 90–95% probability. Only 7–10% of retinoblastoma patients have a positive family history (someone else in the family with retinoblastoma). Hence, about 30% of patients with retinoblastoma have a new germinal mutation. The average age at diagnosis is younger than the nonheritable form – ranging from newborn to 12 months – and they are predisposed to a variety of cancers throughout life.12 About 85% of heritable retinoblastoma patients develop multiple, bilateral eye tumors. Within the age range of 2–5 years, about 2–3% of these patients will develop a midline intracranial tumor usually involving the pineal or suprasellar region that histologically resembles retinoblastoma and has variably been referred to as a pinealoma, primitive neuroectodermal tumor (PNET), and trilateral retinoblastoma. It is important to recognize that the intracranial tumor is a primary cancer and not a metastasis from the eye tumor. The intracranial tumor is usually diagnosed before, together with, or within 2 years of the retinoblastoma, but it can be detected over 10 years later. Hereditary retinoblastoma patients have an elevated risk of osteosarcomas, soft tissue sarcomas and other mesenchymal tumors through their teenage years, melanomas and brain tumors through middle age, and epithelial malignancies such as lung and bladder cancer into later life. This elevated risk is much greater in the field of radiation treatment.
Genetic terminology
The genetic terminology used in the retinoblastoma field can be confusing. Clinicians erroneously may use the term unilateral to refer to the nonheritable form of retinoblastoma. However, this nomenclature is inappropriate, since about 15% of heritable cases develop tumors in one eye only.13 Consequently, epidemiologic studies that report tumor laterality but no information about the RB1 gene status probably are “contaminated” with unilateral, heritable patients and may give the erroneous impression that nonheritable patients have an elevated second cancer risk.8 The term sporadic is also commonly misused. Sporadic refers to a lack of family history but is not equivalent to nonheritable disease. About 90–93% of all retinoblastomas (heritable and nonheritable) are sporadic with no family history, meaning that they are present because of new mutations. In contradistinction, virtually all cases in a single family are hereditary, with the very rare exception of fortuitous and unfortunate familial aggregation of nonhereditary patients in the same family. Hence, nonheritable patients contain somatic or nongermline RB1 gene mutations (i.e., present only in somatic cells of the retina) are somatic mosaics. Hereditary patients carry germ line RB1 mutations (i.e., present in virtually all cells in the body, both somatic and germ line).
Molecular genetics of retinoblastoma
Because of the autosomal dominant inheritance pattern for retinoblastoma, the RB1 gene was assumed for many years to act in a dominant fashion.14 A major paradigm shift in the genetic understanding of retinoblastoma, and cancer in general, began with an enigmatic paper published in 1971 by Alfred Knudson, who proposed that retinoblastoma was caused by two mutational events: “In the dominantly inherited form of the disease, one mutation is inherited via the germ line and the second occurs in somatic cells. In the nonhereditary form, both mutations occur in somatic cells.”13 The major implication of this “two-hit theory” was that the RB1 gene functions in a recessive manner at the cellular level – an unprecedented suggestion at the time. Today, it is known that many cancer-causing genes are recessive or tumor suppressor genes.
The Knudson hypothesis languished for another decade due to a lack of scientific methods for identifying the RB1 gene. An early clue to the location of the RB1 gene was the recognition in the 1960s that a portion of a group D chromosome (13, 14 and 15) was occasionally deleted in retinoblastoma. Shortly after the Knudson paper, new chromosome banding techniques allowed chromosome 13 to be identified as the target of deletions.15 The smallest common deleted region was later mapped to chromosome 13q14.1 to q14.3.16 An enzyme with a measurable activity, esterase D, had been mapped to chromosome 13, and proved to be critical for linkage analysis in the era before recombinant DNA technology was readily available. Using classic deletion mapping, Sparkes and coworkers studied five patients with retinoblastoma and found that, in all five, esterase D activity was only 50% of normal. These data suggested that the loci for retinoblastoma and esterase D genes were both within the deleted segment of chromosome 13q.17 Based on the fact that the chromosome 13 deletion and the esterase D locus were tightly linked to retinoblastoma in multiple families with clinically and pathologically indistinguishable disease, Murphree and Benedict argued that there was probably a single RB1 locus.18 Further, retinoblastoma tissue from a nonhereditary unilateral patient was also found to contain a 13q14 deletion, suggesting that all forms of retinoblastoma involve the same gene on chromosome 13q14.19 The current consensus is that there is indeed a single retinoblastoma locus, RB1, that is mutated in all forms of retinoblastoma.
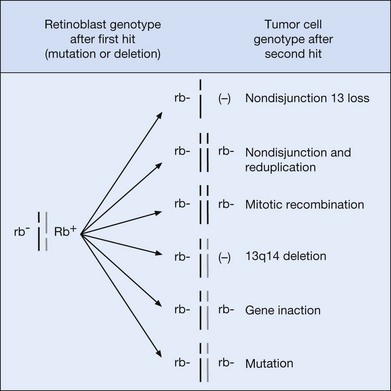
Meanwhile, a body of scientific work was accumulating to support the notion that RB1 is a recessive gene that is lost during tumorigenesis. Benedict and coworkers examined a familial retinoblastoma patient and found a 50% reduction of esterase D in normal cells and a complete absence of the enzyme activity in retinoblastoma cells.20 Dryja and colleagues used DNA fragments from 13q14 to show that homozygous deletions could occasionally be identified in retinoblastoma tissue.21 In two landmark papers, Cavenee et al. proved beyond doubt the recessive nature of the RB1 gene and popularized the use of loss of heterozygosity analysis. By comparing DNA from normal and tumor tissue, they found that the region around the RB1 gene was frequently “reduced to homozygosity” in retinoblastoma tissue. In other words, one copy of the region around RB was lost during tumorigenesis. In 1985, Cavenee et al. went a step further by showing that, in heritable cases, the germ line copy of 13q (carrying the mutant RB1) that was passed among affected family members was always the one that was retained in the tumor.22 The chromosomal mechanisms involved in reduction to homozygosity on 13q14 are shown in Figure 128.1.
Another breakthrough in the hunt for RB1 was the cloning of the esterase D gene.23,24 In the early days of recombinant DNA technology, cloning a gene usually required an activity that could be assayed, and esterase D provided such an activity. Once the esterase D gene was cloned, its DNA sequence was harnessed and could be used to probe adjacent stretches of chromosomal DNA to identify nearby genes. An intensive search ensued for the RB1 gene, and as luck would have it, the esterase D gene proved to be located very nearby the RB1 gene. A few short months following the cloning of esterase D, the search culminated in the discovery of a large gene that contained deletions in many retinoblastomas.25,26 In addition, the mRNA transcript from this gene was either missing or abnormal in size in most retinoblastomas. Even though some early workers questioned whether this gene was indeed RB1,27 further work has confirmed that this is the gene that is mutated in retinoblastoma. For example, re-introduction of RB1 gene into retinoblastoma cells and other RB1-deficient tumors28 suppressed the neoplastic phenotype, indicating that the RB1 gene was indeed a tumor suppressor. Thus, by the early 1990s, there was a basic molecular under standing of how retinoblastoma is inherited. Transmission of an inactive copy of RB1 imparts the predisposition to retinoblastoma. Inactivation of the second copy because of the random background mutation rate leads to tumor development.
The RB1 gene
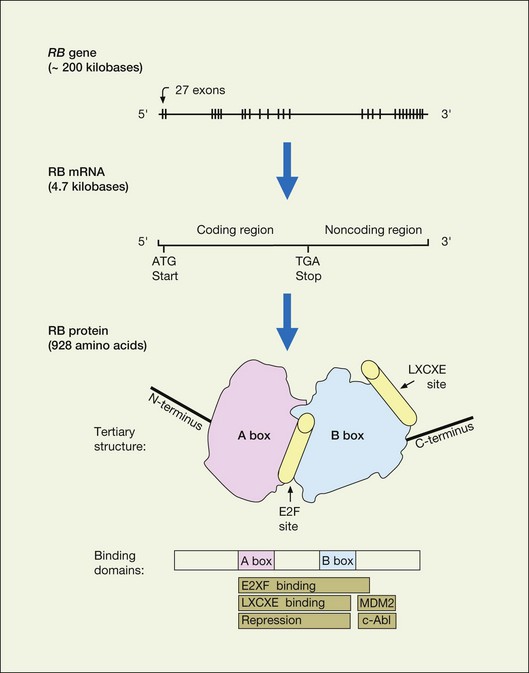
The RB1 gene (Fig. 128.2) contains 27 exons spanning over 200 kilobases of DNA. The 5′ end of the gene is oriented toward the centromere of chromosome 13. The promoter region lacks a typical TATA box but contains a CpG-rich rich region, or CpG island. Germ line mutations in the RB1 gene are distributed throughout the gene with mutational hotspots at CpG dinucleotides.29,30 Less than 10% of retinoblastoma patients have a constitutional chromosome 13q abnormality (usually a deletion) that can be detected by karyotyping.31,32 Deletions that are more extensive can be associated with the 13q-syndrome, with features such as growth and mental retardation, facial dysmorphism, microcephaly, skeletal anomalies, and genitourinary abnormalities. About 15–20% of germinal mutations are too small to detect by cytogenetics but can be detected with techniques for analyzing gross DNA rearrangements (e.g., Southern blot). The remainder of RB1 gene mutations are small alterations involving one or a few nucleotides that can only be detected by high resolution methods and direct sequencing.33,34 The majority of these mutations are frameshift and nonsense mutations, inframe and missense mutations, splicing mutations, and mutations in noncoding regions that result in a truncated, unstable protein product (Table 128.1).35 The possibility of using truncated pRb protein mutants as the basis for genetic testing to detect germ line RB1 mutations was proposed several years ago,29 and recently shown to be feasible.36 Interestingly, most new germ line RB1 mutations are of paternal origin, suggesting that the gene is more susceptible to mutation during spermatogenesis rather than oogenesis.37
Mutation of the second RB1 allele is typically due to chromosomal aberrations, usually recognized as loss of heterozygosity (LOH) by polymorphism analysis.22 These mutations occur at a much higher rate than the first, germ line mutation (10−3 as compared with 10−7 for the first mutation).38 The most common mechanism leading to LOH is mitotic recombination (50%),39 followed by nondisjunction with or without subsequent reduplication (~40%). Other mechanisms include small deletions and gene conversions. Therapeutic exposure to ionizing radiation induces DNA damage and increases the risk of LOH, and hence tumorigenesis, within the field of radiation.
Advances in rapid genetic testing have allowed for preimplantation genetic diagnosis.40 This procedure allows a couple in which one parent has a known germline mutation to undergo in vitro fertilization where the prior to implantation, the embryo’s are tested for the mutation using single cell polymerase chain reaction and rapid DNA sequencing of the exon in question. Only those embryos which are mutation free are implanted back into the mother. This has been successfully performed although due to the expertise and technology required, it is not a widely available technique.
Low penetrance retinoblastoma
The term penetrance refers to the frequency that a heritable disease is manifest in offspring of affected individuals. The word expressivity refers to the variability of clinical manifestations in affected individuals. For example, patients with heritable retinoblastoma who develop only unilateral eye disease manifest reduced expressivity. In general, reduced penetrance and expressivity tend to segregate in the same families.41 Overall, the penetrance of retinoblastoma is about 80–90%, but this represents a heterogeneous group of high-penetrance and low-penetrance families.41 The diseased-eye ratio (the ratio of the number of eyes containing tumors to the number of mutation carriers in a family) was devised to quantitatively identify low penetrance retinoblastoma families by taking into account both penetrance and expressivity.35 Most low penetrance families have diseased-eye ratios <1.5, whereas families with complete penetrance typically have diseased-eye ratios of ≥1.5. Initially, investigators postulated that low penetrance retinoblastoma may be due to immunologic factors, DNA methylation, epigenetic mechanisms, delayed mutation, host resistance factors, a second retinoblastoma locus, or modulator genes.41–44 However, recent research has shown that most low-penetrance retinoblastoma results from mutations at the RB1 gene locus that result in an pRb protein with reduced activity.45–47 One of the most common low penetrance mutations is a missense alteration at codon 661 (exon 20).45,48 Other reported low penetrance mutations include 3-bp deletion in exon 16 that results in the deletion of Asn480,50 a 4-kb deletion involving exons 24 and 25,54 and a splicing mutation at the last base of exon 21.47 Insights gained from these low penetrant RB1 mutants have added much to our understanding of how the retinoblastoma protein works.10
RB1 gene mutations in other tumors
Not surprisingly, RB1 gene mutations occur frequently in tumors linked to retinoblastoma, such as osteosarcomas, soft tissue sarcomas and other mesenchymal tumors.49–51 However, investigators were surprised to find that the RB1 gene is also mutated frequently in some common adult malignancies such as breast, lung, and prostate cancer.52–54 As our understanding of the RB1 gene and its protein product have progressed, it has become clear that RB1 is disrupted in most human cancers, either by mutation of the gene or, more commonly, by functional inactivation of the protein.55
The role of the retinoblastoma protein in tumor suppression
The retinoblastoma protein
The RB1 gene encodes a 4.7 kilobase messenger RNA transcript that produces a protein of 110 kD (kilodaltons) and 928 amino acids (Fig. 128.2). The pRb protein is phosphorylated in a cell cycle-dependent manner and localizes to the nucleus.56–58 The hypophosphorylated form predominates in quiescent and differentiating cells, whereas the hyperphosphorylated species accumulates in cycling cells as they enter DNA synthesis (S) phase (Fig. 128.3).59–63 The hypophosphorylated form of pRb binds several viral oncoproteins, including SV40 large T antigen,64 adenoviral E1a,65 and human papillomavirus E7.66 When bound to pRb, these oncoproteins stimulate cell division. Taken together, these findings provide evidence that hypophosphorylated pRb is important in negatively regulating the cell cycle, and that this inhibitory activity can be thwarted either by phosphorylation or viral oncoprotein binding. Further work has shown that the major cell cycle function of pRb is to inhibit the transition of cells out of gap 1 (G1) phase into S phase.67 However, pRb may also have roles in other cell cycle phases.68,69

A major breakthrough in understanding how pRb regulates the cell cycle was the observation that pRb binds to members of the E2F transcription factor family (referred to here as E2F).70–72 Further work has shown that pRb function is largely dependent on interactions with E2F.73 E2F sites are found in the promoters of many genes that are important for cell cycle progression, and pRb represses transcription of these genes through its interaction with E2F.74–77 Since E2F (but not pRb) has a DNA binding domain, the pRb-E2F association would explain how pRb is brought to specific DNA elements to exert its effect. Most E2Fs have a transactivation domain that stimulates expression of genes containing E2F binding sites in their promoters. pRb binds E2F within the transactivation domain,78,79 thereby masking its activity. Since E2F activates genes involved in cell division,74,80 inhibition of E2F provided a mechanistic explanation for how pRb inhibited cell division. However, the picture became more complicated with the recognition that pRb has intrinsic or “active” transcriptional repressor activity and is able to block the expression of genes when artificially brought to promoters by proteins other than E2F.81 These findings suggest a complex relationship between pRb and E2F. In some situations, pRb inhibits genes by simply masking the E2F transactivation domain, whereas in other situations E2F serves as a “courier” to deliver pRb to specific genes for active repression.
The importance of active repression by pRb was pointed out in several studies that showed that this activity is required for pRb to inhibit the G1-to-S phase transition of the cell cycle.82,83 But how does pRb actively represses transcription? In a series of landmark papers, several groups showed that pRb binds to and recruits to promoters proteins that alter chromatin structure, such as histone deacetylases,83–85 SWI-SNF ATPases,86–88 DNA methyltransferases,89 polycomb complexes,90 and histone methylases.91 Alteration of local chromatin structure into a restricted conformation prevents access by the transcriptional machinery, thereby inhibiting expression, whereas dynamic reorganization of chromatin into an open configuration allows gene transcription. Depending on the nature of the chromatin-remodeling complex that pRb recruits, the cell cycle inhibition can be temporary, as occurs during the quiescent period between cell divisions, or permanent, as occurs during cell differentiation and senescence.92
Studies of the tertiary (three-dimensional) structure of the pRb protein have provided insights into how the protein performs these complex functions (Fig. 128.2). The central region of the pRb protein contains the A box and B box, which are highly conserved from human to plants. These regions interact with each other along an extended interdomain interface to form the A–B pocket. The pocket is critical for the tumor suppressor function of pRb, and is disrupted by most germ line mutations in hereditary retinoblastoma patients and somatic mutations in tumors.29,93 The pocket is required for binding to E2F, chromatin remodeling enzymes, viral oncoproteins, and other molecules. Many pRb-binding proteins contain an LxCxE (leucine – variable amino acid – cysteine – variable amino acid – glutamic acid) binding motif, which has been shown by crystallographic studies to be located in the B box.4 The B box does not assume an active confirmation unless bound to the A box, thereby providing an explanation for why both boxes of the pocket domain are required for pRb activity. Interestingly, E2F does not contain the LxCxE motif and binds pRb at a distinct site (Fig. 128.3). Recent crystallographic studies confirmed that the E2F binding site, located at the interface between the A and B boxes, is distinct from the LxCxE site.94 These findings provide a structural explanation for how pRb simultaneously binds E2F and chromatin remodeling proteins, many of which interact with pRb through the LxCxE site.88 The current molecular view is that pRb orchestrates the assembly of multi-protein complexes, which are then recruited to specific promoters by E2F, where they control access of the transcriptional machinery. Thus, pRb regulates in a dynamic and integrated manner the expression of specific genes involved in cell division, differentiation and apoptosis.95
The carboxy-terminal end of the pRb protein is also critical for its function. This region is required, along with the pocket, for binding to E2F.96 In addition, most of the phosphorylation sites that seem to be critical for regulating pRb activity are located in the carboxy-terminus.97 In fact, recent work has shown that phosphorylation of pRb on the C-terminus initiates a novel intramolecular interaction that progressively strips pRb of activity as the cell moves through the cell cycle.98 The carboxy-terminal region also contains binding sites for the oncoproteins c-abl and MDM2.99,100 The tyrosine kinase activity of c-abl is blocked when it is complexed with pRb,99 and this interaction appears to be important for Rb-mediated growth suppression.101 Besides directly blocking c-abl, the C-terminal region also appears to participate in the assembly of multimeric complexes containing pRb, E2F, c-abl and potentially other proteins.102 The importance of the pRb-MDM2 interaction is less clear. MDM2 interacts with the p53 tumor suppressor protein and opposes its proapoptotic activity by repressing p53 transcriptional activation and by mediating its degradation.103,104 While initial results demonstrated that MDM2 blocks pRb function, more recent studies have shown that pRb can form a trimeric complex with MDM2 and p53 and thereby block the antiapoptotic activity of MDM2 by preventing the degradation of p53.105
The function of the amino-terminal end of the pRb protein remains less clear. This region contains phosphorylation sites that may regulate pRb activity. Additionally, this region interacts with several proteins, including a replication-licensing factor (MCM7),106 a novel G2/M cycle-regulated kinase,107 and other proteins.108 However, the function of these interactions has not been established. RB1 ± mice that develop pituitary tumors due to loss of Rb failed to be “rescued” by expression of a mutant form of pRb that lacked the amino-terminus, although the onset of tumors was delayed.109 In other experiments, tumor suppression by pRb was actually enhanced when the amino-terminal region was removed.110–112 Thus, the amino-terminus appears to contribute only weakly to the overall tumor suppressor activity of pRb.
Not only was the RB gene the first identified tumor suppressor gene, but the Rb pathway was the first, and still one of the most important, tumor suppressor pathways to be elucidated in human cancer.55 As described above, the pRb protein is critical for regulation of the cell cycle, as well as senescence, differentiation and apoptosis, all of which are deregulated during cancer formation.67,92,113–120 While the pRb pathway is deregulated in virtually all cancers,55 the RB1 gene is mutated in only a limited spectrum of cancers. In these other cancers, the pRb protein is inactivated by maintaining it in a hyperphosphorylated state. This occurs by deregulating the pRb pathway, which controls the phosphorylation state of pRb through kinases and kinase inhibitors.
The RB1 tumor suppressor pathway
Cell cycle progression normally occurs when pRb is inactivated by phosphorylation that is catalyzed by cyclin-dependent kinases (CDKs) in complex with their cyclin partners.59,121,122 pRb contains 16 potential sites for CDK phosphorylation, and it oscillates between hypophosphorylated and hyperphosphorylated forms in cycling cells. At least three different cyclin/CDK complexes phosphorylate pRb during the cell cycle (Fig. 128.3). Cyclin D-ck4/6 phosphorylates pRb early in G1, cyclin E-CDK2 phosphorylates the protein near the end of G1, and cyclin A-CDK2 is thought to maintain phosphorylation of pRb during S phase.55 Phosphorylation of specific sites appears to regulate distinct pRb functions, suggesting complex regulation of pRb by these phosphorylation events. For example, binding of E2F, LxCxE proteins, and c-abl are regulated by distinct sets of phosphorylation sites in the carboxy-terminus.123,124
pRb is phosphorylated sequentially by different CDKs during the cell cycle. In fact, successive phosphorylation of pRb by cyclin D-CDK4/6 and cyclin E-CDK2 appears to be necessary to completely hyperphosphorylate pRb.122 Recently, a mechanistic explanation was suggested for how cyclin D-CDK4/6 and cyclin E-CDK2 may regulated distinct pRb functions.98 Cyclin D-CDK4/6 appears to phosphorylate specific sites in the carboxy-terminal region of pRb, and this phosphorylation triggers an intramolecular interaction between the phosphorylated C-terminal region and a positively charged “lysine patch” encircling the LxCxE binding site in the B box of the pocket. This interaction displaces LxCxE proteins such as histone deacetylase from the pocket, thereby blocking the ability of pRb to arrest the cell cycle.82,98 However, pRb can still bind to E2F in this partially phosphorylated state. Under hyperproliferative conditions, the intramolecular interaction between the carboxy-terminal region of pRb and the pocket also can recruit cyclin E/CDK2 to the pocket, where it phosphorylates serine-567, an otherwise inaccessible site buried within the pocket.107 Ser-567 makes critical contacts between domain A and B,4 and this phosphorylation disrupts the A–B interface and disrupts pRb binding to E2F. The sensitive location of Ser-567 is further illustrated by the fact that it is the only CDK phosphoacceptor site in pRb that is a target of naturally occurring missense mutations in tumors.125 The more complete inactivation of pRb, indicated by phosphorylation of serine-567, leads to release of E2F and increased apoptosis.118 Taken together, these findings suggest that the normal cell cycle may be regulated by partial phosphorylation of pRb that is catalyzed by cyclin D-CDK4/6, whereas the more complete phosphorylation that requires cyclin E-CDK2 may serve as a checkpoint for an abnormal hyperproliferative state that would trigger cell death.
Proteins called cyclin dependent kinase inhibitors (CDKIs), which inhibit the kinases that phosphorylate Rb (Fig. 128.3) represent another layer of complexity in the regulation of pRb. The p16INK4a protein is a CDKI that specifically inhibits CDK4, which catalyzes the early phosphorylation of pRb.126 A tumor suppressor protein in its own right, p16INK4a is mutated or inactivated in many types of cancer, including cutaneous and uveal melanoma.127,128 Loss of p16INK4a allows cyclin D-CDK4 to act unopposed in phosphorylating pRb, which results in constitutive functional inactivation of pRb. Since pRb and p16INK4a act in the same pathway, and mutation of either gene results in a similar deregulation of the cell cycle, both genes are rarely mutated in the same tumor.129 Other CDKIs, such as p21 and p27 have more broad roles in regulating the cell cycle.130
The RB-E2F regulatory network
The complexity of the pRb tumor suppressor pathway is greatly increased by the fact that there are two other members of the pRb pocket protein family – p107 and p130 – as well as seven or more members of the E2F family.95 The genes for p107 (RBL1) and p130 (RBL2) have been cloned and mapped to human chromosomal regions 20q11.2 and 16q12.2, respectively.131,132 The p107 and p130 proteins share extensive homology with pRb within the pocket domain and are involved in cell cycle regulation.133 While it is still not clear why so many pRb and E2F family members are necessary, recent work suggests that specific pRb family members interact preferentially with specific E2F family members during precise phases of the cell cycle. For example, pRb interacts primarily with E2F1–3 and is most active at the G1-to-S phase transition. In contrast, p130 interacts primarily with E2F4 and E2F5 and is most active in G0 – the quiescent phase of the cell cycle.95 A mechanistic picture is emerging in which the pRb-E2F network regulates gene expression through complex and context-dependent interactions between pocket proteins, E2F family members, and cofactors such as chromatin remodeling.
Molecular pathogenesis of retinoblastoma
There is now overwhelming evidence that supports the hypothesis that mutational inactivation of the RB1 gene is the initiating event in retinoblastoma. However, there remain many unanswered questions about the molecular pathogenesis of this tumor. Since pRb is important for regulating normal cell growth and differentiation throughout the body, why does germ line mutation of the RB1 gene predispose primarily to the rare eye tumor? This question was once quite enigmatic but is now becoming clearer. Indeed, carriers of RB1 mutations are predisposed to a whole range of tumors, but the age of susceptibility is different for each tumor type. Whereas retinoblastomas occur mostly from birth to 5 years of age, mesenchymal tumors tend to occur in the teenage years, and melanomas peak in a slightly older age group. A recent report clearly shows that common epithelial tumors also occur at increased frequency in retinoblastoma survivors, but this effect is only seen in individuals beyond 40 years old.134 Thus, it seems likely that loss of RB1 predisposes to a broad range of neoplasms, each requiring different numbers and types of “hits” for tumorigenesis to become manifest; the fewer “hits” required, the earlier in life the tumor begins to appear in patients.
While mutation of the RB1 gene is clearly necessary for retinoblastoma formation, is inactivation of RB alone sufficient for tumorigenesis? Some investigators have argued additional mutations in other genes must be required based on the following observations. First, deletion of the RB1 gene in normal cells leads to apoptosis, rather than tumor formation, because loss of pRb triggers a p53-mediated apoptotic response.135 This presumably explains why most cancers contain mutations in both the pRb and p53 pathways.92,136 Second, retinoblastomas cannot be produced in mice unless both pRb and p53 are inactivated.137 This led to a search for mutations in p53 or other pro-apoptotic genes. Interestingly, however, p53 is rarely mutated in human retinoblastoma,138 and no other apoptotic genes have been convincingly linked to retinoblastoma. Finally, cytogenetic alterations on other chromosomes (e.g., 6p) are frequently observed in retinoblastomas,139 potentially suggesting the presence of other retinoblastoma-associated genes located at those chromosomal regions.140
Recent work has shown that MDM2 and MDMx may play an important role in attenuating the normal apoptotic events that should occur in an RB1−/− state. MDM2 targets p53 for ubiquitin mediated proteolysis and is also a downstream target of p53. This creates an auto-feedback loop that maintains p53 at a low level. Small molecule inhibition of MDM2 has been shown to lead increased levels of p53 in retinoblastoma cell lines leading to p53 mediated apoptosis.141,142
An alternative explanation for retinoblastoma pathogenesis is simply that retinal progenitor cells pass through a window of susceptibility prior to terminal differentiation in which loss of pRb leads to a differentiation defect, rather than an apoptotic response, which leads to an accumulation of proliferating embryonic retinal cells. This hypothesis does not require the conjectural existence of additional “retinoblastoma genes” and satisfactorily accounts for developmental and clinical observations. pRb is indeed required in a cell-autonomous manner for appropriate cell cycle exit and differentiation of retinal progenitor cells.143 Further, the topographic distribution of retinal tumors parallels the pattern of retinal differentiation. The retina differentiates in a posterior-to-anterior wave from the posterior pole to the ora serrata.144 Interestingly, the chronological development of retinoblastomas follows this same pattern, with earlier tumors occurring posteriorly and later tumors occurring more peripherally.145 Retinal progenitor cells that retain the capacity to differentiate into photoreceptors, neurons, and glia can be identified in the retina until after birth,146 suggesting a window of susceptibility from fetal week 12 until 4–5 years of age for the bi-allelic loss of the RB1 gene to have the possibility of producing retinoblastoma.
Another controversy regards the cell of origin of retinoblastoma. Retinoblastomas derive from cells of the immature neuroepithelial inner layer of the optic cup that have the potential to differentiate into rod and cone photoreceptors and Müller cells.147 Several studies have documented the presence of cone-specific markers, such as transducin, cone photopigments (red and green opsin), and cone phosphodiesterase in retinoblastomas.148 However, since cone differentiation may represent a “default” pathway in the absence of normal signaling,149 it is unclear whether retinoblastomas arise from neuroblasts that are already committed to the cone lineage, or whether retinoblasts that lose pRb are unable to differentiate along their appropriate lineage and are subsequently directed into the cone pathway. Recent work has identified an additional association between cone cell precursors and mature retinoblastoma tumors. Cone cell precursors, but not mature cone cells, express pRb. In addition, cone cell precursors also express MDM2, which is a transcriptional target of Trβ2 that is also transiently expressed in cone cell precursors. Taken together, loss of pRb in a cone cell precursor that is already expressing MDM2 as part of its developmental program could plausibly lead to transformation and would be consistent with this being a potential cell of origin.
Retinoblastoma: the disease
Overview of retinoblastoma
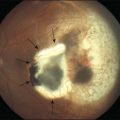
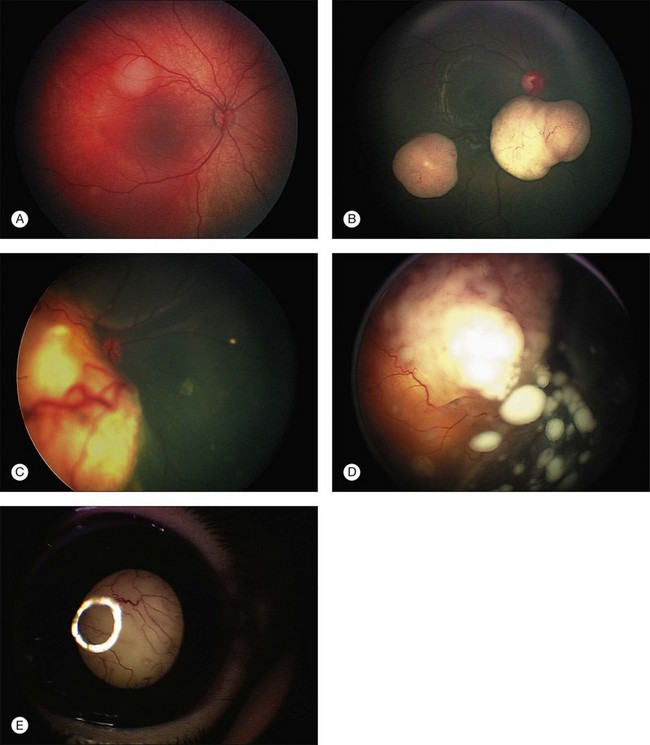
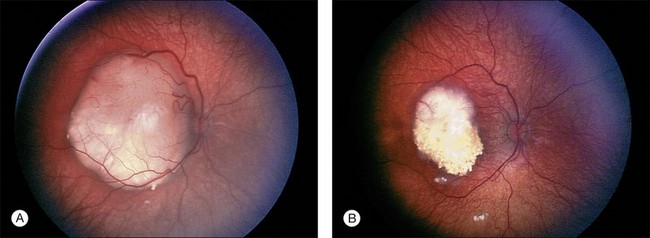
Retinoblastoma may be present at birth. In most cases, it is diagnosed between birth and 5 years of age. The tumor may involve one or both eyes. Each tumor arises independently. Early tumors <3 mm in greatest diameter may appear as discrete round, gray-white masses without intrinsic vascularity usually in the posterior pole of the eye. In dim light when the pupil dilates naturally, in the early days of the tumor natural history, parents may see light reflected by the tumor causing a “cat’s eye” reflection in the pupil or a yellow glow rather than the expected “red eye” on flash photographs (Fig. 128.4). Less commonly, more advanced intraocular retinoblastoma may manifest as the lack of a red pupillary reflex and may appear black compared with the red glow from the other pupil when viewed through a direct ophthalmoscope. This is caused by either blood in the vitreous from tumor necrosis or when there is a total retinal detachment and the retina is pushed up behind the lens. When the tumor destroys central vision early in the course of the disease, and binocularity is lost, the presenting sign may be strabismus.
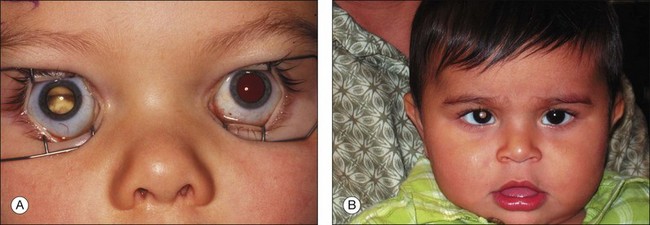
Epidemiology
Since the last edition of this book was published, striking differences between the relative prevalence of unilateral nonheritable and heritable disease has come to wide attention in part due to the important work of Ian McGrath and colleagues at the International Network of Cancer Treatment and Research (see the INCTR website at: http://www.inctr.org/ accessed December 9, 2011). Orjuela and her colleagues at Columbia University in New York and at the Instituto National de Pediatria in Mexico City have suggested deficiencies in maternal diet during pregnancy.150 While the worldwide incidence of the heritable form of the disease is relatively constant, such is not the case for unilateral sporadic non-heritable retinoblastoma.
In the USA, retinoblastoma is the sixth most common solid childhood tumor. In developing tropical and subtropical regions of the world, including Central Africa, Southern Asia, and Central America retinoblastoma is at or near the top of the list as the single most common solid tumor of childhood.151–168 The difference is due for the most part to an increased incidence of unilateral non-heritable disease.
The overall incidence (heritable and nonheritable cases combined) in developed countries is most commonly reported as the number of cases of retinoblastoma diagnosed in a selected period per live births for that period.169 Although this index does not correctly reflect the population at risk, since children continue to be at risk until 5 years of age and beyond,170 it does present some uniformity for comparison of the frequency of the disease across populations.
Earlier studies of the incidence of retinoblastoma in the USA report rates of less than 1 in 20 000. From 1970 on, the prevalence (number of cases per number of persons in the at-risk age group) is consistently reported at around 11 cases per million children under 5 years of age,171 or equivalent to an incidence of 1/18 000 live births.172 Because there is no complete retinoblastoma registry in North America, the exact incidence is unknown. However, the estimate retinoblastoma specialists usually agree upon is about 300 new cases per year in North America. If the estimate were based on the 2007 Census data for the number of annual births in the USA (4 242 000), the incidence would be 1 : 17 000. This number fits closely with studies reported from New Zealand169 and Sweden,173 both taken from national registries, as well as a regional study from Australia,174 all show incidence rates of 1/17 000–1/18 000 live births.
The incidence of heritable retinoblastoma among the various populations of the world is remarkably constant, providing strong evidence that environmental influences play little role in the etiology of the hereditary form of this tumor.169,175 Buckley provided evidence that environmental factors play little role in the hereditary form of cancer in very young children.176 Bunin and colleagues from nine North American centers have demonstrated that medical radiation exposure (from lower GI series) prior to conception significantly increased the risk that a child develop sporadic retinoblastoma from a new germline mutation has an extensive epidemiological study underway evaluating the role, if any, of environmental exposure of the father in sporadic heritable retinoblastoma.31
In marked contrast to the uniform incidence of heritable retinoblastoma worldwide, as noted above, there are striking geographic differences in the incidence of the nonheritable, unilateral form of retinoblastoma from one region of the world to another. The differences in overall incidence are due entirely to the excess of nonheritable, unilateral cases of retinoblastoma. There exists a possibility that the increased incidence of nonheritable retinoblastoma in the poorer, tropical and subtropical regions of the world is due to a viral etiology (possibly human papilloma virus, HPV),177 although convincing proof is still lacking. This group has also found that nonheritable retinoblastoma seems to be statistically more common when pregnant mothers eat a diet deficient in fruits and vegetables. A major prospective epidemiological study has been funded to allow this group to evaluate possible dietary influences during pregnancy on the increased incidence of nonheritable retinoblastoma in developing subtropical countries.
Advanced paternal age is unequivocally associated with new sporadic germ line mutations and sporadic heritable retinoblastoma.178–184 Excess cancer in relatives185 is also a common finding in solid childhood tumors, particularly retinoblastoma. Some 80–85% of new heritable tumors preferentially retained the paternal allele (i.e., the mutated allele) and lost the normal maternal allele as the result of a chromosomal error at mitosis.180,186 These data suggest that new germ line RB1 mutations arise more frequently during spermatogenesis than during oogenesis.
Natural history of intraocular retinoblastoma
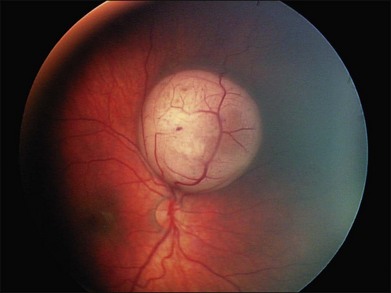
Regardless of when the cellular events that lead to retinoblastoma take place, the earliest physical appearance of the tumor is fixed by the fact that all cells in the tiny tumor focus are identical, i.e., they are daughter cells of the original “founder” cell with the same genes and the same growth rate. A new tumor will expand symmetrically as a round or hemispherical lesion that is homogenous. In its earliest manifestation, retinoblastoma resembles bacterial colonies on an agar plate (Fig. 128.5). Because tumor growth begins with a single immortalized retinoblast, all intraocular retinoblastomas are initially confined to the retina.
More than 25 years ago, aliquots of aqueous from eyes containing intraocular tumors (primary or metastatic) were shown to induce angiogenesis on the chick allantoic membrane even if the aliquot was taken before tumor was clinically apparent.187 Tumor angiogenesis is essential if tumors are to expand. Conversely, avascularity severely restricts the potential growth of tumors.188 Intraretinal lesions of retinoblastoma reach that growth restriction at a diameter of between 1 and 2 mm if they remain dependent on diffusion of nutrients and oxygen from the choroid. Vessels grow into the tumor only in response to growth factors generated from upregulated growth factor genes and downregulated angiogenesis inhibitory genes. A physical marker for the “turned-on” angiogenesis in any particular tumor may rest in our ability to accurately estimate the number of new vessels existing in a tumor. This relative tumor vascular density (RTVD) can be demonstrated by CD-34 staining for vascular endothelium in special stained histopathology sections. The RTVD is statistically greater in ocular tumors of patients with metastatic disease.189,190 In the February 2004 report from Marback and colleagues, tumor vessel density was also greater in tumors that have invaded the choroid and optic nerve, previously identified histological risk factors for metastatic disease.189
Normal cells adhere to each other and the extracellular matrix through a series of specialized molecules. An important component of the “progression of malignancy” process of any tumor cell is loss of this cellular adhesion or anchorage dependence. A tumor suppressor gene, PTEN, is mutated in many advanced tumors.191 When a normal wild-type copy of the gene is introduced into tumor cells the exogenous PTEN inhibits tumor cells’ ability to grow anchorage independently. The product of this tumor suppressor gene was able to revert one of the typical properties of tumor cells to normal. The controls that would ordinarily shunt a cell to apoptosis or the “programmed cell death pathway” if it lost its attachment to the extracellular matrix may be eliminated in the “progression of malignancy” process.
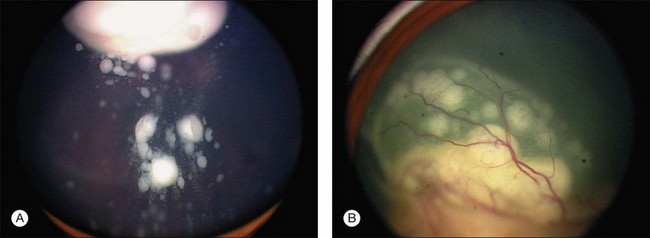
Tumor cells that invade adjacent tissue (the vitreous) or spaces (the subretinal space), a feature that defines group C disease eyes, now enter a different, low-oxygen, low-nutrient microenvironment. The shape and structure of early vitreous seeds are limited to a thickness of two tumor cells surrounding an inner core of necrotic oxygen-starved tumor. Large avascular vitreous and subretinal masses are the final end-result of continued “progression” of malignancy (Fig. 128.6). Tumor cells in these masses thrive in this new low oxygen environment. In advanced group D and E eyes it is common to find large avascular masses (see discussion of retinoblastoma classification, below).
The ability to metastasize requires the acquisition of a number of cell capabilities that are not present in early descendants of the original “founder” cell. The function and structure of the eye are, without question, two of the main reasons that most retinoblastomas are diagnosed and treated before they metastasize. Most retinoblastomas attract attention and are treated, long before they have reached a size or contain enough doublings for any single cell to have achieved all of the enabling mutations required for metastasis. Simple access to the blood stream is required but not sufficient for metastasis. To gain access to distant sites via the blood stream, a tumor cell must have the ability to digest and move through extracellular matrix,192 and to digest and penetrate the basement membrane, adventitia and endothelium of the vessel wall. In order to escape the high flow vascular channels, a tumor cell must be able to adhere to the vascular endothelium at a location outside the eye, digest its way through all layers of the vessel wall, survive in the extravascular environment, recruit a blood supply, and establish its own microenvironment. Because all these abilities are achieved only through a series of spontaneous mutations that require many tumor cell doublings, most patients who develop metastatic disease have had continued presence of viable tumor cells, usually in an only eye where every treatment has failed to control their growth for an extended period of time.
In developed countries metastatic disease is rare at initial diagnosis. Only 18 cases were found with metastatic disease at diagnosis between 1968 and 1993 at the Essen Retinoblastoma Center in Germany.190 For this reason, efforts to salvage an eye that has had multiple episodes of tumor regrowth over a period of more than 6–12 months should raise increasing concerns about the escalating risk of metastatic disease in that child.
Classification of intraocular retinoblastoma
Reese–Ellsworth classification
The Reese–Ellsworth classification system, originally published in 1964, was a major advance in our collective understanding of retinoblastoma.193,194 It is a group classification system and as such, only deals with organ-confined intraocular disease. In a staging system evaluating the whole child, a group classification would fit entirely within stage I disease. The Reese–Ellsworth classification was developed just as the indirect ophthalmoscope was being introduced into clinical practice. Anterior tumors, which can now be easily recognized and treated with cryotherapy or radioactive plaque, cause the eye to be classified in a more advanced group when using the Reese–Ellsworth classification. The classification does not take into account retinal detachment and subretinal tumor seeding. Also, vitreous seeding of any amount places the eye in group 5b (the last of 10 subgroups) with the poorest prognosis. Today, local vitreous seeding can often be successfully treated with brachytherapy.
International Classification for Intraocular Retinoblastoma
In 1989 Kingston and colleagues introduced the systemic chemotherapy combination currently in use in most centers (carboplatin, etoposide, vincristine) in combination with external beam radiotherapy as the primary treatment of retinoblastoma for Reese–Ellsworth group Vb eyes.195,196 In January 1990, Murphree was the first to use chemotherapy plus local consolidation without external beam radiotherapy for all Reese–Ellsworth groups. Initially the early stage Reese–Ellsworth eyes in Los Angeles were treated with carboplatin alone combined with transpupillary diode laser hyperthermia (chemothermotherapy). Advanced intraocular disease was treated with CEV (carboplatin, etoposide, vincristine) systemic chemotherapy combined with sequential aggressive local therapy. These authors introduced the term “chemoreduction” to describe the concept of reducing the volume of the tumor with systemic chemotherapy followed by local consolidation in 1992.197
Efforts at developing a revised classification that reflected the move away from external beam radiotherapy to primary chemotherapy for intraocular retinoblastoma began as early as 1994, when Murphree and Hungerford co-chaired a meeting of retinoblastoma specialists from around the world for a full day of discussions on the shape of a new classification system at the International Congress of Ophthalmology in Toronto. The system that grew out of those discussions took into account zone of disease similar to retinopathy of prematurity. However, the classification that this large group developed was too complicated and cumbersome to be useful. Eventually, the ABC classification system was described at the European Congress of Ophthalmology meeting in Istanbul in 2001. In May 2003, discussions at the Xth International Retinoblastoma Symposium led to a joint international effort in which a number of major retinoblastoma centers signed onto the effort. The case for a new group classification has recently been published.195 The process of validating the International Classification is currently underway.
The International Classification System (Box 128.1) is based both on the natural history of retinoblastoma and on the likelihood of salvaging the eye when systemic chemotherapy is used as the primary treatment. Letters “A” through “E” instead of numbers were chosen to designate each classification group to avoid confusion with the Reese–Ellsworth system. The risk of loss of the eye due to retinoblastoma is graduated from “very low” for Group A to “very high” for Group E.
Box 128.1
International Classification for Intraocular Retinoblastoma195
In this classification, the letter “A” is assigned to those eyes for which both the likelihood of curing the tumor and retaining excellent vision are both high. In intraocular group A eyes, the lesions are small and are away from critical visual structures (foveola and optic nerve). Groups A and B contain all eyes in which the tumor remains confined to the retina. In groups C and D eyes, the tumor has spread into the vitreous and subretinal space. In the case of group C eyes the spread is local. In the case of group D eyes the seeding is diffuse (Fig. 128.7). Group E eyes have been destroyed by the tumor and are rarely salvageable. In this system, the morbidity of the treatment increases from group A to group E, and the probability of salvaging the eye and useful vision decreases from A to E. Within limits, when chemotherapy is the primary treatment of retinoblastoma, absolute volume of the tumor is relatively less important than whether the tumor has become dispersed within the eye to either the vitreous or the subretinal fluid or both.
Disease prognosis
Retinoblastoma survival rates
The overall survival rate from retinoblastoma in developed countries results from a mixture of causes of death. In the first 4 years of life, most of the deaths will be from metastatic retinoblastoma.198 Later deaths are increasingly likely to be the result of genetically predisposed second primary tumors such as osteosarcoma or fibrosarcoma. Thus, the overall survival rate will differ depending upon the time period examined. The survival rate in 1980 from one major retinoblastoma center in the USA was 92%.198 The similar overall 5-year cumulative survival rate was 91% in SEER (Surveillance, Epidemiology, and End Results) data from 1974 to 1985.171
It is important to differentiate between survival from retinoblastoma alone or survival from both retinoblastoma and second primary neoplasms. For example, Abramson and colleagues reported that 86%, not 92%, of bilateral retinoblastoma patients survive 15 years.198 By 5 years after diagnosis more children die of second malignant neoplasms than from retinoblastoma. In the Netherlands the presence of a National Registry shows the striking difference in overall survival between the subgroup of genetically predisposed patients and those who have no genetic predisposition.199 A major controversy has arisen concerning the percentage of patients who will eventually succumb to second malignant neoplasms (SMNs). Abramson and colleagues had previously reported that as many as 90% of patients carrying the retinoblastoma predisposition allele develop a SMN within 30 years of diagnosis of the original retinoblastoma.200 In a more recent report of 1000 patients with nearly complete ascertainment from the New York and Boston group, 6% of bilateral patients had died of SMNs by 40 years after diagnosis, if they had not been treated with external beam radiation (EBR). In contrast, 35% died of SMNs if EBR had been used in their treatment.
The overall survival rate in developing countries depends upon whether or not there was a delay in receiving medical attention. These delays are likely to be cultural, e.g., there is no tradition of seeking medical help until a process is far advanced. For example, in a report from Malaysia in 1980, only 6 of 20 patients survived retinoblastoma. All were initially seen with advanced disease and were older at initial diagnosis.201 In comparison, in Great Britain where there is a tradition of early access to medical care, only 26 of 317 patients managed at Moorfield’s and St Bartholomew’s Hospitals in London between 1970 and 1984 developed orbital disease indicative of late diagnosis.202 In Argentina, delayed diagnosis was related to lack of access to medical care and initial consultation with a pediatrician instead of an ophthalmologist.203 In the 1960s, Nigeria had an extremely high mortality rate because of late medical attention and lack of treatment facilities.204 In Japan 50 years ago, only 50% of patients with unilateral disease and 17% of patients with bilateral disease survived for 5 years.205
Factors affecting survival
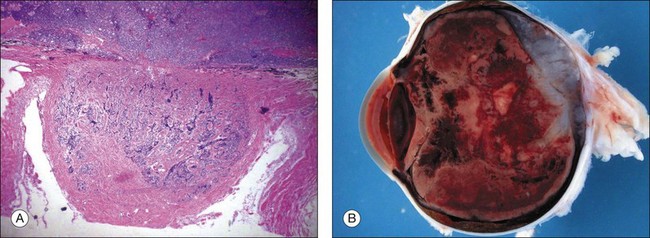
Traditionally there have been histologic features in enucleated eyes that have seemed to correlate with increased risk of metastatic disease.206 Invasion by the tumor into the optic nerve posterior to the lamina cribrosa combined with massive invasion of the choroid is a combination that most centers would agree needs preventive or adjuvant chemotherapy (Fig. 128.8). Recent reports have questioned the value of retrolaminar involvement of the optic nerve and choroidal invasion in predicting risk to metastatic disease.207 Marback and colleagues have also questioned the effectiveness of invasion past the lamina cribrosa as a reliable predictor of increased risk of metastatic disease.189
There appear to be no inherent differences in the risk for metastatic disease between patients that are genetically predisposed to retinoblastoma and those without genetic predisposition.22 Delay in diagnosis is a major factor adversely affecting survival of both heritable and nonheritable disease.208
Factors affecting salvage of eye and vision
As suggested by Knudson in 1976,1 early detection of signs of retinoblastoma with timely referral and treatment beginning when the tumors are small is perhaps the most important factor that increases the likelihood of salvage of the eye. A significant delay in initial diagnosis is associated with a decreased likelihood of retaining the eye. Haik and his colleagues evaluated the cause of the delay in diagnosis.209 According to these authors, the first delay in diagnosis of retinoblastoma occurs before the observation of the first symptom by the child’s parents or other observer. A second delay occurs between the observation of the first symptom and the visit to the primary care physician. These authors maintain that educating parents on signs to look for could possibly reduce this second source of delay. In addition, more than 50% of all primary care physicians made a referral to a pediatric ophthalmologist within 1 week, when there was no family history of disease, and 75% of the primary physicians referred the family to a pediatric ophthalmologist when there was a positive family history. However, 47% of the patients without a positive family history and 25% of patients with a positive family history had a delay averaging 4–5 months before the primary care physicians referred them to an ophthalmologist.209
For patients with macular tumors, post-treatment vision is often 20/200 or worse, but can be better. In a series of 17 patients with large macular tumors, post-external beam radiotherapy treatment vision ranged from 5/200 to 20/50.210 The authors pointed out that the ophthalmoscopic appearance and size of the tumors gave little reliable guidelines to visual outcome. In another series of 11 patients following treatment of macular retinoblastomas, two patients eventually recovered 20/20 vision despite having subretinal edema in the macula at the time of diagnosis.211 We have had experience with a patient who had a macular lesion completely filling the space within the vascular arcades. After treatment this tumor mass shrunk away from the disc and the fovea to a site 4 DD temporal to the fovea. Vision returned with excellent central fixation. In this case, and in some other cases, the tumor obviously had mushroomed over the surface of the retina and did not destroy the foveal cones.
Diagnosis of retinoblastoma
Signs and symptoms
Leukocoria is the phenomenon created when light entering the eye is reflected back out through the pupil by the yellow or yellow-white tumor (Fig. 128.4). The observer sees this reflex or reflected light diffusely filling the pupil, giving rise to the term “leukocoria” or white pupil. Retinoblastomas in the posterior pole of the globe that have reached 3 mm in basal diameter are sufficiently large to generate a leukocoric reflex. In the USA more than half of all retinoblastomas are suspected or diagnosed after observation of a leukocoria, in the affected eye usually by a close family member.
In a retrospective review of 1265 patients with retinoblastoma from New York Hospital, leukocoria was the presenting sign in 56% of all cases.212 It is interesting and informative to note that in the New York series observation of leukocoria correlated with the presence of advanced (Reese–Ellsworth group Va or Vb) disease. Yet we know that with a large pupil diameter, even small tumors may create leukocoria. The red-reflex test, as used in a standard manner by pediatricians, has been documented to be positive in the presence of undilated pupils only when advanced or large retinoblastoma is present.213
Flash photography can easily document the presence of leukocoria. However, the camera must be either an inexpensive cameras that lacks the “eliminate-red eye” pre-flash or must have that feature turned off. The family often takes holiday or baby photographs in dim illumination when the child’s pupil is naturally dilated. The flash illuminates the interior of the eye, and the film records the reflected glow from the tumor (leukocoria) well before the pupil has a chance to constrict to the bright flash. Parents are urged to take and develop large numbers of flash photographs of their infants with cameras known to record “red-eye” in the first few weeks and months of life and become aware of the possibility of an abnormal pupil. If they observe such an abnormality, they should immediately take the photograph to their pediatrician demonstrating the leukokoria and insist on a referral to an ophthalmologist, preferably one who specializes in pediatrics. A number of parents in the last 4–5 years have searched the internet for “white pupil” and have found descriptions of the association between what they observe in their child and the possible presence of retinoblastoma. (Examples of websites where information is available for parents are: www.retinoblastoma.com and www.retinoblastoma.net, both accessed on December 9, 2011.)
The American Academy of Pediatrics (AAP) in May 2002 published a new policy statement regarding how their members should perform the red reflex test. This was subsequently updated in 2008.213A (Their recommendations can be found on the AAP website at: www.aap.org, under Policy Statements; red reflex testing.) This policy statement is a tremendous step forward in the effort to assure early detection for all infants and toddlers with intraocular retinoblastoma.
Small tumors in the foveola or near the foveola can significantly reduce the visual acuity in that eye. Strabismus is the initial sign in one of every five patients with retinoblastoma and almost always is a result of either a tumor or of tumor-associated subretinal fluid in the macula.212 Decreased visual acuity caused by destruction or obscuration of the fovea is the immediate cause of the strabismus. If vision is decreased only because the tumor occludes the visual axis but does not destroy the fovea, there is a good possibility that the vision will improve after treatment. Before treatment begins it may not be clear whether or not the fovea has been destroyed by the tumor. Strabismus in the first 6 months of life always requires an immediate retinal examination to rule out retinoblastoma.
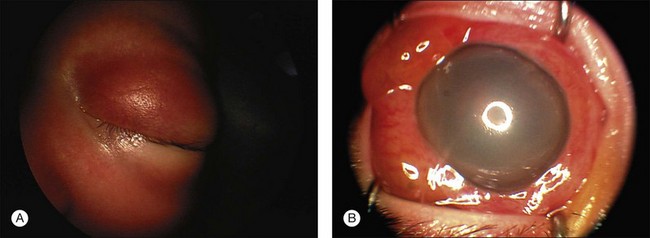
Other less common signs and symptoms include a red, painful eye with glaucoma, cloudy cornea, poor vision, and an aseptic orbital cellulitis (Fig. 128.9). In five cases of retinoblastoma presenting with the clinical picture of aseptic orbital cellulitis,214 the imaging studies were misinterpreted as showing extraocular extension of the tumor into the optic nerve. We have had similar experiences in patients with retinoblastoma presenting as orbital cellulitis. Systemic treatment with 3 days of oral corticosteroids results in dramatic reduction of the swelling. In one case, pre-steroid and post-steroid CT scans were dramatically different. The edema in the perineural soft tissues responsible for the false-positive interpretation of tumor invasion into the nerve may disappear after steroid treatment.
Diseases simulating retinoblastoma (pseudoretinoblastoma)
In 500 consecutive referrals for retinoblastoma, simulating lesions were found in 212 of the 500 patients.215 there were a total of 23 different conditions that referring pediatricians or ophthalmologists were unable to distinguish from retinoblastoma. The three most common causes of pseudoretinoblastoma were persistent hyperplastic primary vitreous (28%), Coats disease (16%), and presumed ocular toxocariasis (16%) (Fig. 128.10). Congenital cataract and retinopathy of prematurity were less frequently mistaken for possible retinoblastoma in this series than in other, earlier reports. Our experience in Los Angeles shows many fewer cases referred as possible retinoblastoma that turn out to be pseudoretinoblastoma. Other diagnoses that we have seen simulate retinoblastoma include Familial Exudative Vitreoretinopathy (FEVR), Norrie disease, and congenital retinal folds.
In 1996, de Potter and colleagues216 evaluated the role of MRI in differentiating solid intraocular tumors from intraocular lesions with primary retinal detachment. They reported that solid intraocular tumors appeared hyperintense on T1-weighted images and hypointense on T2-weighted images. Also, these lesions showed minimal to marked enhancement on contrast-enhanced T1-weighted images with fat suppression techniques. In secondary serous or exudative retinal detachment from intraocular lesions (Coats disease, persistent hyperplastic primary vitreous, phthisis bulbi, and retinopathy of prematurity), MRI showed hyperintensity of the subretinal space on both T1- and T2-weighted images and no enhancement in the subretinal space on contrast-enhanced sequences.
Diagnostic workup
Regardless of whether calcium can be demonstrated by ocular ultrasound on the child in the office, we routinely order an MRI scan of the brain and orbits (with gadolinium enhancement and fat suppression) for two reasons. First, to evaluate whether there is extraocular extension and optic nerve invasion by the tumor ideally before the staging examination under anesthesia, and second, the MRI of the brain should have specific cuts directed through the region of the pineal gland to eliminate the possibility of the “trilateral” retinoblastoma syndrome first described in 1983,217,218 and discussed later in this chapter. The MRI scan can demonstrate infiltrative spread into the optic nerve, subarachnoid seeding, and involvement of the brain. MRI and CT are both useful in determining the extent of recurrent disease, extraocular spread, and defining second primary tumors.219
Unlike MRI, CT scanning involves exposing the child to a low dose of radiation. This exposure should be avoided if possible in cases of heritable retinoblastoma because of the generalized cancer predisposition syndrome. The CT findings were described in 80 cases of retinoblastoma.219 All cases of exophytic retinoblastoma demonstrated calcification, either in a solid mass or in multifocal locations. Soft tissue components of retinoblastoma showed enhancement with contrast material in all cases. As these authors pointed out, calcification can occur in any of the retinoblastoma-simulating lesions where significant ocular disruption or phthisis is evident, but this dystrophic calcification usually is deposited along the lines of normal structures. If the ultrasound does not demonstrate the presence of calcium at the EUA, then a subsequent CT scan may be necessary if the diagnosis is in doubt.
Staging examination under anesthesia
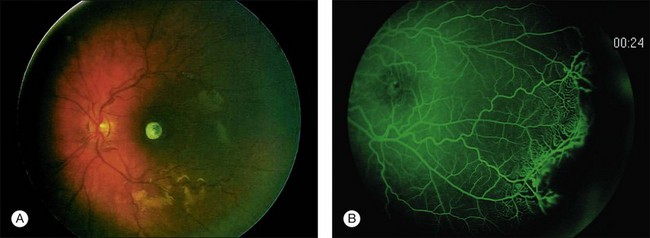
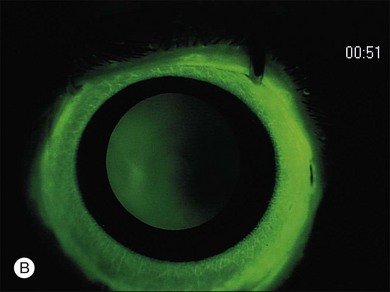
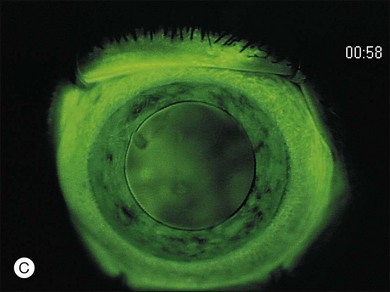
Wide-angle retinal photography or RetCam 120 digital imaging is extremely useful in documenting the size and location of the tumors as well and confirming and documenting the features that allow group classification of the eye. Printed images of the tumor help the family accept the reality of a tumor and give them something to keep and show to other family members. Comparison images are useful in follow-up examinations under anesthesia to demonstrate to the family the response or lack of response to treatment. In addition the RetCam 120 also allows the use of fluorescein angiography in the diagnosis and management of retinoblastoma. A fluorescein angiogram is especially useful in confirming the presence of neovascularization of the iris in retinoblastoma eyes with glaucoma and/or chronic retinal detachment (Fig. 128.11). We also find fluorescein angiogram of value if there is a questionable area of recurrent retinoblastoma in a previously treated lesion or scar. In this case, an actively growing recurrent lesion will leak, stain with fluorescein, and accumulate dye. Inactive lesions do not stain. The diagnosis of an early “presumed” retinoma may be facilitated by fluorescein angiography. A newly diagnosed, untreated eye that contains a lesion that appears to be type II or type III regression and does not leak or stain on fluorescein angiography may well be a retinoma (see a more thorough discussion of retinoma later in the chapter.
B-scan ultrasonography is useful to measure the height of each discrete tumor in millimeters. Ultrasonography is almost as good as CT in its ability to detect the presence of calcium within retinoblastoma tumors (Fig. 128.12). In 1973, Coleman described the reliability of ultrasound in retinoblastoma diagnosis.220,221 In 1975, Sterns et al. reported two configurations, solid and cystic.222 However, these authors were not always able to differentiate between vitreous hemorrhage and retinoblastoma. In one series of 38 eyes, retinoblastoma was the ultrasound diagnosis in 25 and pseudoglioma in 11, and all diagnoses were correct.223 Ultrasound should be able to detect a tumor as small as 2 mm in diameter.224
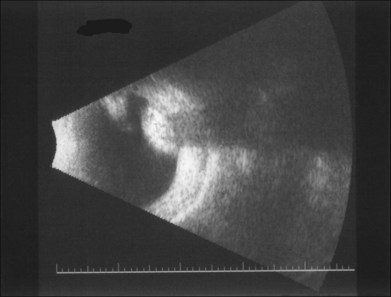
However, some problems do exist with using ultrasound to diagnose retinoblastoma. Atypical retinoblastoma, i.e., retinoblastoma with no necrosis and thus no calcification may lead to errors in diagnosis if only ultrasound is used. For this reason, the rule-of-thumb is that ultrasonography is useful if it demonstrates the presence of calcium in the lesion. If it fails to detect calcium, that finding does not rule out retinoblastoma. In that case a CT scan is necessary. In rare cases of retinoblastoma, the CT scan may fail to detect intralesional calcium. Orbital plain films are of no value and should not be part of the workup. Following enucleation wrapped implants may occasionally be the site of secondary calcification.225 Three-dimensional ultrasound of newly diagnosed retinoblastoma is beginning to be explored.226 Fine-needle aspiration biopsy has been described as a diagnostic tool,227 but today the indications for this procedure are extremely rare. Its use is contraindicated except by persons in ocular oncology centers who have experience with the procedure. In 2002, Karcioglu published a survey of major ocular oncology centers that had treated 3651 patients since 1986.228 During those 17 years, fine-needle biopsy was performed only eight times (once in every 456 patients with retinoblastoma). The biopsy was usually done in older children with a diagnosis of uveitis in which retinoblastoma could not be ruled out. Six of the eight patients were 4 years of age or older.228 The use of fine-needle biopsy should be limited to those exceptional cases in which a diagnosis cannot be arrived at by any other means. With increased experience, atypical cases can often be diagnosed correctly without the need for introducing a needle into an eye with possible retinoblastoma.
Specular microscopy of the corneal endothelium may be useful in differentiating retinoblastoma cells from keratic precipitates on the corneal endothelial surface.229 These authors demonstrated a well-demarcated aggregate of dark and bright reflections against a background of normal-appearing, high-density endothelium. In contrast, keratic precipitates appear as bright reflections when fresh, and, as they resolve, the bright reflections develop a peripheral dark rim.
Treatment methods and techniques
Developing a customized treatment plan
Unilateral nonheritable retinoblastoma
The ABC classification of intraocular retinoblastoma195 helps clinicians decide when to treat unilateral retinoblastoma conservatively. Nearly all centers will attempt to salvage unilateral group A and B eyes. E eyes should be enucleated.
If the likelihood of salvaging group D eyes with diffusely disseminated intraocular tumor is ≤40%, a strong case can be made for recommending enucleation primarily and sparing the child the trauma of treatment to retain an eye that will likely be of no visual use. Shields and colleagues initially described their experience treating unilateral retinoblastoma with primary chemotherapy.230 The eyes that did well without needing either enucleation or external beam radiotherapy were eyes that would have been classified as intraocular groups A–C. Group D eyes did not do well. If the child and not the eye is taken into consideration in the decision-making process, strong consideration should be made for enucleation of unilateral group D eyes. If the chance of salvaging useful vision is not good, heroic treatment approaches are unwarranted. It makes little sense to subject a child with one completely normal eye to prolonged systemic chemotherapy and possibly external beam radiotherapy.
Advanced intraocular disease (groups D and E)
If the worse eye has features of group E disease, then enucleation is generally recommended. In some centers, if systemic chemotherapy is initiated for the contralateral eye, salvage of a group E is considered. There have been rare case reports of successful salvage of group E eyes.231 If, in a group E eye the neuroimaging study suggests invasion of the tumor into the optic nerve and the other eye has group B or higher and requires systemic chemotherapy, there are two approaches. Many centers delay enucleation of the group E eye until after six cycles of chemotherapy. Others feel that this approach is flawed as it alters the histopathologic features of an eye potentially masking high risk features such as cut end disease or massive choroidal involvement. Those centers advocate immediate enucleation with adjuvant treatment based on histopathology.
Systemic intravenous chemotherapy
The introduction of systemic intravenous (IV) chemotherapy for the primary treatment of intraocular retinoblastoma by Kingston et al.,196 set off a dramatic shift in the management of intraocular retinoblastoma. Although the outcome from the first 14 eyes with Reese–Ellsworth group Vb retinoblastoma treated in London were disappointing, variations on their protocol are widely used today for the treatment of this disease throughout the world.
Pre-1989 chemotherapy for extraocular disease
Before 1989, systemic intravenous chemotherapy was used rarely for the treatment of intraocular retinoblastoma. Most of the experience in that era was with using chemotherapy for extraocular retinoblastoma. Oncologists tested many different drugs. The usual agents employed at that time included cyclophosphamide (at what we know now was an inadequate dose), vincristine and doxorubicin. Pratt et al. described the St Jude experience with chemotherapy for extraocular retinoblastoma between 1962 and 1984.232 A total of 11 of 114 children received chemotherapy for measurable extraocular disease that had been present at diagnosis (7/11 patients) or developed later (4/11 patients). The two patients with only orbital disease had complete response to chemotherapy and subsequent radiotherapy and are the only long-term survivors. Single-agent treatment was ineffective. Only cyclophosphamide and ifosfamide showed any value at all when given alone. Drugs that induced no response as a single agent included vincristine, doxorubicin, cisplatin, and VM-26.
Background of the currently used chemotherapy regimen
Kingston and colleagues at St Bartholomew’s Hospital in London were the first group systematically to explore the effect of the platinum group of drugs in this tumor.196 In their 1987 report, they described 11 children with metastatic retinoblastoma treated between 1970 and 1984 with salvage chemotherapy. Three of the 11 had complete short-term remissions with a regimen of four drugs containing cisplatin. One additional child responded well to a combination of vincristine and cyclophosphamide. Based on these results they suggested that retinoblastoma appeared to be a chemosensitive tumor. In addition, these results led these authors to try a sandwich protocol of carboplatin, etoposide and vincristine with 40–44 Gy EBR as planned primary therapy in 14 group Vb eyes between March, 1989 and April, 1995.
After hearing a report of their work at a meeting in March, 1990, Murphree et al. began the use of a carboplatin-based regimen for the primary treatment of all Reese–Ellsworth stage eyes. Initially in Los Angeles, carboplatin alone was combined with transpupillary hyperthermia for the treatment of smaller localized tumors. The results from thermochemotherapy were encouraging but better results and reduced time in the OR came with the use of triple agent chemotherapy combined with transpupillary laser or transscleral cryotherapy.233
By 1994, the Los Angeles three-drug protocol of primary chemotherapy with carboplatin, etoposide and vincristine (CEV) followed by focal consolidation was being used widely. By 1996, the patient outcomes from London, Los Angeles, Toronto, and Philadelphia were all published in the November issue of the Archives of Ophthalmology.196,233–235
Primary systemic chemotherapy
Systemic chemotherapy has replaced external beam radiotherapy as the most commonly used primary treatment for intraocular retinoblastoma. The chemotherapy agents may vary slightly but most protocols consist of the systemic administration of carboplatin, etoposide, and vincristine (known as CEV or VEC in North American centers, and JOE in Britain), given over a 2-day period every 3 or 4 weeks (the administration of the drugs and the time required for the suppressed bone marrow to recover is considered one cycle). In Los Angeles, we use six cycles of CEV for intraocular group C and D disease and three cycles for intraocular group B disease (Fig. 128.13). There is some concern about etoposide, as it has been associated with the appearance of a secondary acute myelogenous leukemia (AML), in a small cohort of children treated for retinoblastoma (see below).236,237 For this reason, some centers have dropped etoposide from the regimen for less advanced disease (groups A and B).
Subtenon carboplatin
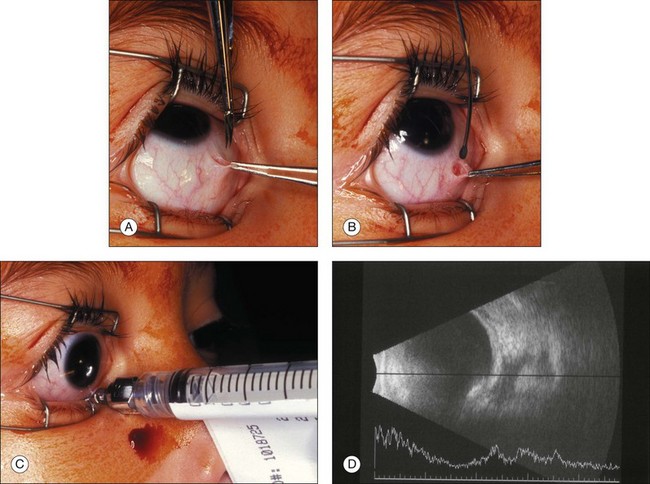
The addition of subtenon carboplatin to augment penetration of systemic carboplatin has been advocated by a number of groups over the past 10 years. Introduced and described as subconjunctival injections238 we modified the technique. Following a transconjunctival and trans-tenon incision, an olive tip cannulas is used to inject the chemotherapy in one of the quadrants of the eye between the rectus muscles (Fig. 128.14).
Complications of primary systemic chemotherapy
The chemotherapeutic agents currently used to treat intraocular retinoblastoma can cause many complications. Most complications are dose-related and are of short duration. As far as is known, these chemotherapeutic agents do not predispose children with heritable retinoblastoma to the same tumors for which they are genetically at risk, unlike the situation with external beam radiotherapy. The use of etoposide has been a source of significant debate. The drug is known to be leukemogenic and there has been concern that exposing patients with the RB1 mutation would increase their risk for secondary AML (sAML). While there have been a series of patients treated with systemic chemotherapy who later developed sAML, the number remain small; the majority of these patients were treated with high-dose multi-agent modalities well above the standard three-agent six-cycle regimen.236,237 Most experts agree that if etoposide poses a risk for second tumors, it remains a small one. Perhaps a bigger concern is the potential risk of hearing loss following carboplatin use. Wilson and colleagues have reported an increase in carboplatin related ototoxicity at St Judes. Similar results have not been duplicated elsewhere.239 The risk of ototoxicity is significant when one considers that many retinoblastoma patients are visually disabled relying on other sensory input such as hearing. As with other intravenous forms of chemotherapy risks of bone marrow suppression, alopecia and central line infection exist; in rare cases these can severe and life-threatening.
Heat and chemotherapy
Therapeutic synergism is said to exist when the combined effect of two treatments is greater than summing the effect of each individually. The synergism that results when heat and chemotherapy are used concurrently in the treatment of cancer is extensively documented.240–255 The exact molecular mechanisms at work when heat enhances the effect of chemotherapy or radiotherapy are unclear in all instances. Possible mechanisms may include “direct cytotoxic effect of heat, and heat-induced alterations of the tumor microenvironment. The presumed cellular effects of hyperthermia include the expression of heat-shock proteins (HSP), induction and regulation of apoptosis, signal transduction, and modulation of drug resistance by hyperthermia.254 At hyperthermia temperatures of 44° and higher, at least for carboplatin, there may be enhanced drug uptake into tumor cells.256
The use of hyperthermia as an adjunct in the treatment of cancer was suggested as early as 1966.257 It is clear that the killing efficacy of cisplatin and carboplatin are synergistic with hyperthermia at 40–42°C and that the thermal effect ratios increase with temperature in that range.255 Of the two drugs, carboplatin has been preferred for the treatment of retinoblastoma because the incidence of renal and ototoxicity from carboplatin is significantly less than from cisplatin.258,259 It is of more than passing interest that etoposide is synergistic with the platinum agents.255
A number of publications have examined hyperthermia combined with chemotherapy in eyes with retinoblastoma. Murphree and colleagues demonstrated in 1992 that carboplatin combined with transpupillary diode hyperthermia to 42°C was effective for the clinical control of posterior pole retinoblastoma confined to the retina.197 In the 1992 presentation, that group introduced the concept of thermochemotherapy to the management of posterior pole intraretinal retinoblastoma, combining low-power, direct hyperthermia with single-agent carboplatin. In 1996 this same group demonstrated that hyperthermia in the rabbit anterior chamber xenograph model increases the cellular uptake of carboplatin by 100%.233 A number of publications confirmed the benefit of the combination in retinoblastoma in both animal models and patients.260–266
Intra-arterial (IA) chemotherapy
Primary systemic intravenous (IV) chemotherapy for intraocular retinoblastoma administers a large volume of medication to the entire body to treat a relatively small organ. As far back as 1953, Kupfer described a case of retinoblastoma treated with nitrogen mustard directly to the peri-ocular circulation.267 Later, in the 1960s and 1970s, Reese and Ellsworth combined external beam radiotherapy with intracarotid chemotherapeutic agents.268–270 They latter abandoned this, not having detected significant benefits of the combined approach over radiotherapy alone. In the 1980s, Kaneko at the National Cancer Institute in Tokyo, Japan developed a new method to administer ocular chemotherapy – he described it as selective ophthalmic arterial infusion (SOAI).5,271–273 With this approach, developed primarily to avoid enucleation, a balloon catheter was inserted in the femoral artery, past the internal carotid and guided just past the origin of the ophthalmic artery. The balloon was then inflated and melphalan injected into the arterial vasculature. Often adjuvant treatments were also administered but more than half of treated eyes were preserved.6
In 2008, Abramson and colleagues modified this technique with direct insertion of the canula just at the ostea of the artery. A phase 1/2 trial of ten patients with Group V retinoblastoma salvaged seven eyes that would have otherwise been enucleated. While the initial series used melphalan additional follow-up reports have infused other agents including carboplatin and topotecan with good results. The technique has been used successfully in unilateral and bilateral cases, as a primary and salvage approach.4,274,275 Following up electroretinogram (ERG) data suggests improved ERG findings in some very advanced cases. Defining an event as “enucleation or need for radiotherapy,” recent 4-year data from this group demonstrated a 81.7% event-free survival for eyes that received intra-arterial chemotherapy as primary treatment, and 58.4% for eyes that had previous treatment failure with intravenous chemotherapy and/or external beam radiation therapy.4
The technique is technically challenging, requiring significant expertise. It has been likened to surgery.274 Many practitioners have raised concerns regarding the potential systemic and CNS risks with this approach including death and stroke; especially in the context of unilateral disease, where enucleation is generally curative with low risk for morbidity and mortality. At the time of this writing, the technique is limited to a handful of centers outside New York, using it as a primary modality. Further data will be necessary to assess whether it will be widely adopted by the ocular oncology community and whether it will replace primary systemic intravenous chemotherapy.
Focal consolidation
Laser
The technique we find useful with the argon 532 nm is essentially the same for both primary treatment of group A lesions and focal consolidation following primary chemotherapy in groups B–D. In general, focal consolidation begins concurrently with the beginning of the 2nd or 3rd cycle of systemic chemotherapy after the tumor volume has been reduced. The goal of the therapy is to completely cover each lesion with 30% overlap during at least three different sessions. We choose initial power setting of 250–300 mW, with a duration of 300–500 msec. The power and time settings are kept low to prevent tumor disruption and hemorrhage that may be associated with excessive energy delivery. The first burns are placed at the edge of the lesion with the spot half on and half off the tumor. The power and/or duration can be adjusted to achieve gentle whitening of the tumor. We do not recommend exceeding 500–600 mW and 700 msec with the 532 mm laser. Once the lesion is outlined, then the entire lesion including any type I regression-associated calcium is covered with overlapping rows of burns (Fig. 128.15). A small to moderate-sized lesion may require 200–400 burns for good coverage. The burns over the thicker areas of the tumor may be virtually invisible compared with those placed at the edge of the lesion. The power or duration should not be increased to compensate for the decreased “take” over the thicker parts of the lesion. Repeat the laser coverage at 2–4-week intervals during and/or after the administration of systemic chemotherapy until the entire lesion has been covered on at least three different occasions (Fig. 128.16).
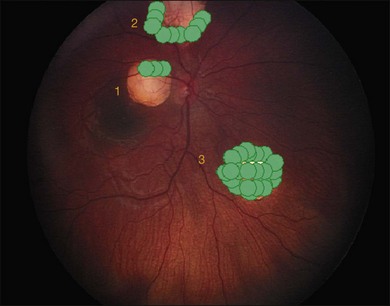
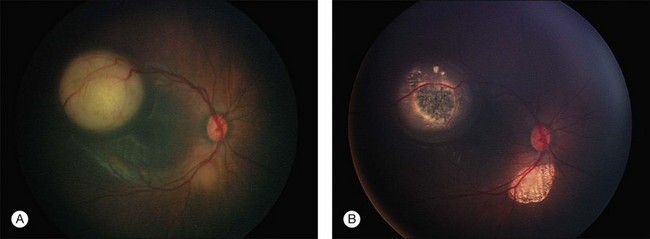
The most significant long-term complication of focal consolidation is due to decreased vision from RPE scar migration or “creep” in lesions near the foveola. Care must be taken when applying laser on the foveal side of a tumor near fixation. Lee and colleagues demonstrated an increase in the size of laser scars following red diode laser application.276 It is reasonable to consider close observation after sufficient primary chemotherapy of a small tumor located near the fovea until documented growth is seen. In some instances, central vision can be spared if regrowth does not occur. Tumors that exist in the maculopapillary bundle can be managed in this fashion, especially if the contralateral eye has been enucleated or has poor visual prognosis.
Cryotherapy
Destruction of the tumor by cryotherapy results from disruption of cellular membranes following the freeze–thaw cycle. It can also have a local vaso-occlusive effect on the tumor and nearby retina/choroid. Cryotherapy is useful and can be used successfully in tumors up to 3.5 mm in diameter and 2.0 mm in thickness; more than one treatment is generally recommended.277–279 The difficulty in using cryotherapy as local treatment for posterior pole tumors is that a surgical procedure is required to open the conjunctiva so that accurate placement of the probe can be obtained. In addition, because the probe tip cannot be visualized while the freezing is taking place, it is possible to freeze the optic nerve. An important consideration is that cryotherapy routinely destroys a great deal of normal retina surrounding the lesion, thereby increasing the visual deficit and the resulting chorioretinal scar.
Complications of cryotherapy include vitreous hemorrhage, subretinal fluid, and retinal holes. Retinal detachment can result from a combination of atrophic retina and vitreous traction. Cryotherapy results in strong adhesions at the margins of scars.117,280,281 Extensive cryotherapy can cause atrophy of the sclera, with formation of a pseudocoloboma of the sclera. We have observed the creation of retinal breaks and rhegmatogenous retinal detachment when the calcium portions of type I regression is included in the ice ball. The presence of retinal detachment in the region of proposed cryotherapy is a relative contraindication. Cryotherapy should not be combined with the administration of subtenon carboplatin because toxic levels of the drug may accumulate inside the eyes.
Radiation therapy
External beam radiotherapy (teletherapy)
External beam radiotherapy (EBR) was the treatment of choice as primary therapy of intraocular retinoblastoma for much of the 20th century. Verhoeff and Reese pioneered the method in the early 1900s. Retinoblastoma is undoubtedly a radiosensitive tumor. Tumor control rates and ocular salvage rates are relatively high.282–284 However, with decades of use, one late effect of the treatment in heritable retinoblastoma, second malignant neoplasms (SMNs), caused particular alarm in retinoblastoma centers. As a result most centers now use EBR as a salvage technique if primary chemotherapy has failed or cases where extraocular spread has developed (cut end disease or eyes inadvertently biopsied).
In a report from the Essen group on the prevention of tumor recurrence, primary teletherapy was most effective at high doses, with 49% of patients who received 40 Gy having recurrent disease compared with only 22% of those who received a dose of 50 Gy.283 A highly accurate alignment of the beam tended to decrease the incidence of recurrent tumors by half: 22% versus 48% recurrent tumors with conventional alignment made to the lateral orbital wall.
Two different techniques for EBR have been compared.284 In this study, lens-sparing electron beam approach was used between 1979 and 1984, and the modified lateral beam technique was used from 1984 to 1987. The latter approach was clearly superior for eyes with RE groups I–III disease, as measured by freedom from recurrent tumors and decreased frequency of enucleation. No apparent difference between methods was noted in groups IV and V retinoblastoma.
A problem that has always been difficult to manage is the eye movement that occurs even under general anesthesia. Schipper285–287 in Utrecht has been a pioneer in devising a method to fixate both the eye and the head for the duration of the treatment. Variations on the Utrecht method have been developed at St Bartholomew’s Hospital in London by Harnett et al.288,289 Both groups use either a 6- or 8-mV linear accelerator with a lateral D-shaped field of 26–32 mm. The D-shaped field is contoured to include the entire retinal surface, the vitreous, and 10 mm of the anterior optic nerve. The eye is fixed to the beam-defining collimator by using a low-vacuum contact lens, which is held in place by a magnetic millimeter scale. Because the eye is fixed in the isocenter of the accelerator, rotation of the gantry directs the beam. In the series reported by Schipper et al.,287 18 of 54 radiated eyes developed clinically detectable cataract, but only five of the 18 eyes so affected required cataract surgery. Lens opacity developed only in lenses in which a posterior portion of >1 mm had to be included in the treatment field. The minimum cataractogenic dose in that series was 8 Gy to the lens. Hungerford et al. noted that, by using the Utrecht device (the contact lens) for fixating the eye, the lens and anterior segment can be effectively spared while the whole retina is treated with a full dose of radiation.290
Intensity modulated radiation therapy
Intensity modulated radiation therapy (IMRT) is an alternate method of delivering radiotherapy to the eye. It is effective in reducing the dose to normal tissues and is available at most large radiation treatment centers. IMRT uses computer controlled tungsten blades selectively to block areas of the treatment volume for a prescribed fraction of each beam’s treatment time to make a radiation field with varying intensities. Figure 128.17 is a three-dimensional reconstruction of a CT scan done on a retinoblastoma patient with his head positioned using a custom-molded headrest and a custom dental impression and custom bolus material over his retinoblastoma-containing right globe. The green arrows represent the central rays of the eight incident IMRT radiation beams converging on the patient’s right retina. Figure 128.18 compares the radiation dose distributions produced by irradiating the entire globe using the 8-beam IMRT arrangement (A) and a single 20 million volt electron beam (B). The IMRT substantially reduces the dose to the frontal lobe posterior to the orbit. Figure 128.19 compares the radiation dose distributions produced by irradiating the retina using the 8-beam IMRT arrangement (A) and a single lateral 6 million volt photon beam. Both methods limit the radiation dose to the lens of the eye. The lateral photon beam, however, gives a substantially higher dose to the sphenoid sinus and contralateral orbit.

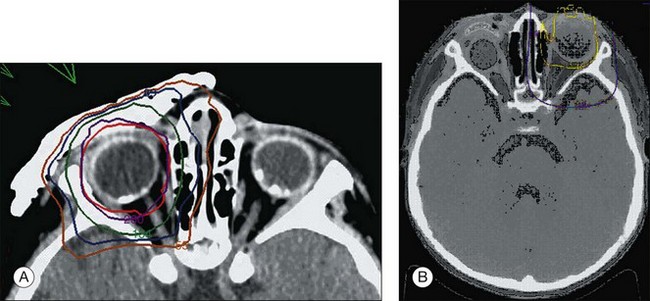
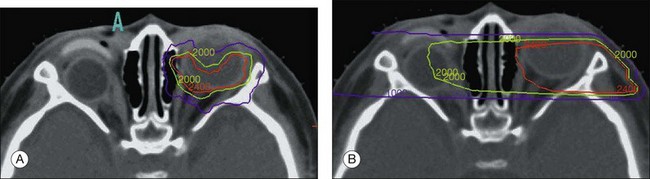
Proton beam radiotherapy
This form of teletherapy uses charged particles with a very sharp Bragg peak curve. This results in dosimetric plans that are much tighter with less scatter to critical adjacent structures including the orbital bones, CNS and pineal region. Recent studies have demonstrated the dosimetric superiority of protons over more traditional EBR and even IMR.291 The challenge remains the high cost of such equipment and the relatively few centers capable of administering fractionated proton beam treatments under anesthesia to the pediatric population.
Brachytherapy
The use of local radioactivity in the treatment of retinoblastoma was originally pioneered by Moore and Scott in 1929. Interstitial implants were used initially, and modifications of this technique were reported later. Ophthalmic applications were designed using radium and, later, cobalt 60. In 1977, Rosengren and Tengroth reported on the reasonably successful results of such treatment in 20 patients.292
Primary brachytherapy may be the treatment of choice in isolated group B intraocular retinoblastoma located at or anterior to the equator. The use of radioactive plaques in the primary and secondary management of retinoblastoma has increased as the danger of external beam therapy in genetically predisposed patients has become recognized. The results of plaque therapy in 50 selected patients have been reported.293 Of 51 eyes treated, vitreous seeding was evident in 49. Only two of 18 eyes were salvaged with radioactive plaque after everything else had failed. Thirty-three patients with small tumors did well, with most retaining vision. The difficulty with plaque therapy for posterior pole tumors is the likelihood of permanent visual loss associated with the massive doses of radiation to the retina and other vital structures. However, radioactive plaques have distinct advantages over EBR in cases in which the area of treatment is limited to the tumor and a small marginal rim.
Iodine125 (125I) isotope became a common radiation source used in brachytherapy when cobalt was abandoned decades ago. The seeds are secured in a gold carrier. Gold prevents radiation from penetrating the substance of the plaque and shields normal bone and tissue from most of the radiation. Luxton and colleagues worked out much of the dosimetry planning for the 125I isotope.294 Dosimetry planning is carried out with the help of sophisticated software, such as that available from Bebig Corporation.295 The gold carrier for the iodine was later modified with deeper wells for the seeds creating a conformal treatment plan (Fig. 128.20).296 In primary brachytherapy, the calculated dose to the apex of the tumor as measured by the peak height on ultrasonography is generally in the range of 40–45 Gy.
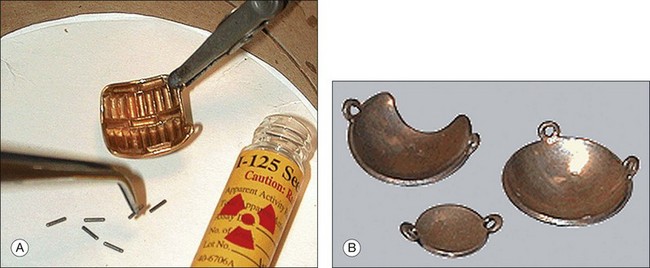
The technique of localizing the plaque when treating retinoblastoma differs from the localization technique for choroidal melanoma, primarily because of absence of pigment in the intraocular tumor. Transillumination techniques used with melanomas are not helpful in localizing retinoblastoma. The surgical approach is otherwise identical. The computerized treatment plan should be available to the surgeon. With the Bebig Corp. Software, the radiation physicist can tell the surgeon the position in clock-hours for the center of the anterior edge of the plaque, the location of the eyelets, and how many millimeters those points are located posterior to the limbus (Fig. 128.21). After the 12, 3, 6, and 9 o’clock locations are marked at the limbus, a 360° peritomy is performed, and traction is placed on appropriate rectus muscles by 4–0 silk sutures passed behind the muscles without needles.
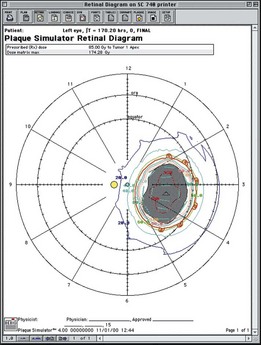
 th-inch lead is fashioned as a shield to be worn during the postoperative course for iodine plaques. Because ruthenium is a beta emitter, the lead is not necessary.
th-inch lead is fashioned as a shield to be worn during the postoperative course for iodine plaques. Because ruthenium is a beta emitter, the lead is not necessary.Care must be taken when combining external beam radiotherapy and brachytherapy sources. This may result in serious complications that can result in destruction of the eye.297 Use of 125I brachytherapy for consolidation immediately following primary systemic chemotherapy is associated with an extremely high risk of aggressive radiation retinopathy that can destroy the eye if traditional doses for isolated brachytherapy are used. We experienced significant rapid radiation retinopathy in 6/6 eyes when 40 Gy was given to the apex of the regressed tumor immediately following completion of CEV chemotherapy. A dose reduction from 40–45 Gy to 20–25 Gy would be appropriate. Perhaps a better alternative is to reserve brachytherapy for treatment of recurrent focal disease several months following the completion of chemotherapy.
Preoperative preparation for enucleation
Surgical technique
Obtaining the longest section of optic nerve is important and can be facilitated using straight mayo or Metzenbaum scissors passed along the nasal wall of the orbit. The optic nerve follows a temporal to nasal route as it plunges to the orbital apex. The tips of the scissors should be felt to plunge through posterior Tenon’s capsule. It is counterintuitive to keep the tips of the scissors pressed nasally but with this technique optic nerve lengths of 12–15 mm can be obtained routinely. A snare, which crushes the optic nerve, should be used in enucleation of eyes with retinoblastoma only with the approval of the ocular pathologist. Likewise, clamping the optic nerve is unwise and should be avoided for similar reasons. Immediately after removing the eye we use a test-tube filled with a frozen-slush saline solution to tamponade the orbital apex. The ice-filled test-tube is placed into the orbital apex and held firmly for 10 minutes (Fig. 128.22). The combination of infusing the orbit with epinephrine-enhanced local anesthetic and using the ice-filled test tube as a tamponade results in little swelling or bruising, eliminates the need for a pressure patch and allows us to remove the patch the morning following outpatient surgery.
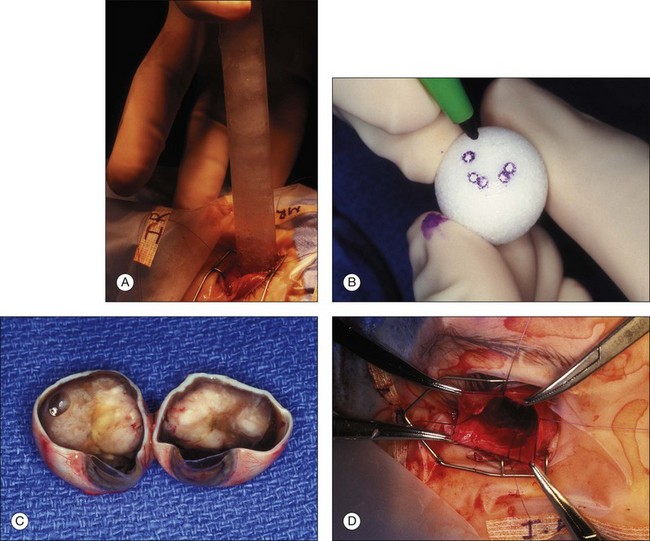
Our experience supports the recommendations of most ocularists is that the implant be as large and as anterior as possible. Thin prostheses allow the best movement. The long-term complications reported in association with pegging the hydroxyapatite implant have convinced us to abandon its use. We currently use an integrated pre-drilled, nonwrapped conical modification of the Medpor implant described in 1994, by Karesh and Dresner.298 Medpor orbital implants are available in three sizes: a 16-mm cone with an 18-mm volume; an 18-mm cone with a 20-mm volume, and a 20-mm cone with a 22-mm volume. We use a 16-mm cone only for infants and very young children. An 18-mm conical implant works well for most enucleations in children 6 months and older. The 20-mm conical implant is reserved for children older than 2.5 years. The material is well tolerated, resists infection, is nonantigenic, and promotes rapid tissue ingrowth because of it porosity. We prefer the conical shape which provides an additional volume posteriorly. The smooth, porous anterior surface of this particular implant is designed to minimize the possibility of late exposure. Many centers prefer hydroxyapatite. The only significant difference with this implant is that is requires a wrap to cover the surface. Sclera is used by many centers but others have abandoned this material in preference for the prewrapped or polymer coated variety.299
Both Medpor and coated hydroxyapatite implants have predrilled suture tunnels which allow for direct attachment of the rectus muscles. The preformed suture tunnels can be difficult to see if not highlighted with a marking pen (Fig. 128.22). Bending the needle to increase its curvature and then passing the hub through the tunnel first, makes the process quick and easy. Recently, we have been carefully dissecting extraocular fat and connective tissue from the outside of the removed eye and reinserting that fat over the surface of the implant after the rectus muscles have been attached. This tissue “cushion” prevents suture Knots from being the source of implant exposure. We have not seen an expose implant since introducing the implant “cushion.”
Tumor harvesting
The enucleated specimen is examined carefully by the surgeon, ocular pathologist, or both looking for evidence of extraocular tumor. If no extraocular tumor is seen on gross examination and on preoperative neuroimaging, the retrobulbar connective tissue and adherent fat can be bluntly dissected from the attached optic nerve stump, placed on a saline soaked gauze pad, and returned to the surgical field for later use as filler between Tenon’s and the anterior implant surface as described above. After the connective tissue has been dissected free, the globe and the attached optic nerve can be photographed. The length of optic nerve obtained is measured with the caliper or millimeter rule. The nerve is then cut leaving a 1-mm stump on the back of the globe. The severed nerve is placed alone in one of the containers of fixative. The globe is stabilized with the fingers and bisected through the cornea with a sterile, single-edge 2-inch razor blade so that half the tumor mass and the entire optic nerve stump remain in one calotte. Photographs of the bisected globe are then taken (Fig. 128.22). The calotte containing the optic nerve stump and half the tumor mass is placed in the second container of formalin without being disturbed. The tumor material in the remaining calotte can be placed into a small Petri dish to be hand-collected as soon after harvesting as possible for tumor karyotyping and/or DNA studies as provided in the consent. Viable tumor is relatively firm and gray-white and can be lifted with forceps. Necrotic tumor is liquid, often contains calcium grains, and is usually yellow-white.
Surgical closure
We have found it very useful to grasp the edge of the opened conjunctiva, which is now easily identified by the purple ink from the marking pen, with five or six hemostats. This elevates the edge of the wound and opens the posterior orbital cavity for easy visualization (Fig. 128.22). After the implant has been inserted and the muscles attached to the pre-drilled tracts as described above closure can begin. During closure of the Tenon’s fascia layer it is helpful if Tenon’s is hydrated with copious saline irrigation. We use a lubricated 5–0 Vicryl suture on a P-3 needle to close Tenon’s fascia with 3–5 horizontal mattress sutures passed 3–4 mm posterior to the conjunctival edge. Alternatively, this step can be closed with a pursestring suture. Before or during Tenon closure, the fat and connective tissue that was previously harvested from the enucleated eye, is placed on top of the implant and spread evenly. A second row of reinforcing vertical interrupted sutures should be placed to provide strength to the closure. This double suturing of Tenon’s fascia provides superior strength to the wound and significantly decreases the risk of exposure and extrusion. The wound should be meticulously inspected to ensure that there are no holes or gaps in the Tenon’s capsule. The conjunctiva is then closed with running 6–0 plain gut or Vicryl suture.
Postoperative care following enucleation
Orbital growth following enucleation will be normal if a large orbital implant is placed in the orbit at the time of surgery and no orbital radiation from an external source is given.300 However, failure to replace the eye with an orbital implant and failure to maintain the presence of an ocular prosthesis will result in orbital growth deficiency.301 Presumably, the severe bony hypoplasia associated with EBR would not be a cosmetic problem in these cases. Imhoff and colleagues demonstrated that the growth of irradiated orbits was significantly impaired (P<0.001) when compared with nonirradiated orbits and that secondary enucleation did not add to the bony growth retardation triggered by external beam radiotherapy. Not surprisingly, these authors observe the growth-impairing effect of EBR to be most profound when the child is irradiated before 6 months of age (P<0.01).302 When reports assume that both enucleation and radiation contribute to retardation of orbital growth and do not differentiate between the two, confusion is the outcome.303 Orbital growth studies in rabbits show a decelerated increase in orbital mass following enucleation that was mitigated by an expandable but not static orbital implant.304 Based on the findings of Fountain et al. growth in enucleated human orbits may be normal if a large but nonexpandable orbital implant is used.300
Retrolaminar optic nerve involvement
If a pathologic evaluation demonstrates optic nerve involvement beyond the lamina cribrosa and massive choroidal invasion, 6 months of adjuvant chemotherapy is recommended by some centers but not all. Chantada and colleagues question the need for this adjuvant therapy.207 The initial series reporting the risk of retrolaminar optic nerve involvement came from New York in 1989.305 It is instructive that currently in New York, this group is not treating retrolaminar retinoblastoma with adjuvant chemotherapy.
The chemotherapy protocol commonly used for adjuvant therapy is the combination of carboplatin, etoposide, and vincristine given at 3-week intervals for six cycles. The Children’s Oncology Group has now completed accrual in a trial assessing the risks and benefits associated with administering adjuvant chemotherapy in eye with so called high risk histopathologic features (see below).306