[level-membership-for-opthalmology-category]
Chapter 40 Retinitis Pigmentosa and Allied Disorders
 For additional online content visit http://www.expertconsult.com
For additional online content visit http://www.expertconsult.com
RP may be seen in isolation (typical RP) or in association with systemic disease (syndromic RP). The prevalence of typical RP is approximately 1 : 5000 worldwide. In Maine, USA, the prevalence of typical RP has been found to be 1 : 5200, and the birth incidence of those who will eventually be affected with RP has been calculated as 1 : 3500.1 The prevalence of RP has been found to be 1 : 7000 in Switzerland,2 1 : 4016 in China,3 and 1 : 4500 in Israel.4 The highest frequency of occurrence, 1 in 1878, is among the Navajo Indians.5 The prevalence of syndromic RP is less well documented. The prevalence of Usher syndrome (RP with congenital deafness) is estimated to be 1 : 6000.6
Early history
The first description of the fundus findings and the use of the term “retinitis pigmentosa” are attributed to Donders in 1855 and 1857.7–9 However, the diagnosis of complicated night blindness (presumably RP) has probably existed in all cultures for hundreds, if not thousands, of years. Ovelgün, in 1744, made observations on familial complicated hemeralopia (or night blindness), which probably represented RP.9 Shortly after the discovery of the ophthalmoscope by Helmholtz in 1851, van Trigt10 in 1853 and Ruete11 in 1854 described cases that most assuredly were RP.
Typical retinitis pigmentosa
Clinical features
Nyctalopia
Difficulty with vision at night (night blindness) is one of the two hallmark symptoms of RP. Patients with typical RP usually attribute the beginning of night vision difficulties to the first or second decade of life. In some patients, especially those living in an urban setting, night vision problems may not be apparent until ocular disease is at an advanced stage. Tanino and Ohba12 noted that the onset of symptoms of RP, most commonly nyctalopia, occurred at a median age of 10.7 years in autosomal recessive disease and 23.4 years in autosomal dominant disease. The symptom of nyctalopia should not be confused with the symptom of blurred vision with night myopia or uncorrected refractive error. People with RP have a narrowing of the visual field in the dark and may describe getting easily disoriented on dimly lit evenings when others are able to see adequately. Becoming accident-prone, especially at night, is a highly suggestive symptom.
Visual field loss
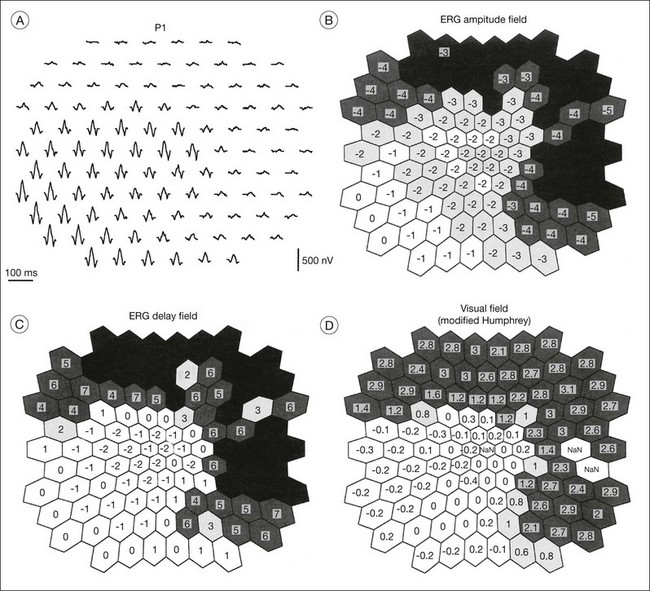
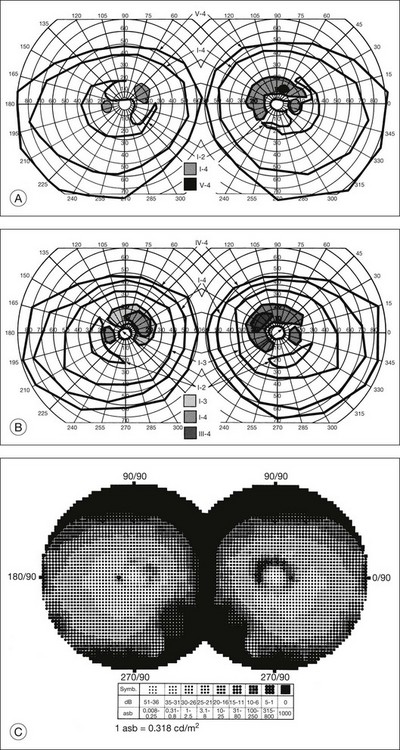
The second hallmark feature or symptom of RP is an insidious, progressive loss of peripheral visual field. In some patients, especially those with severe disease beginning in childhood, this may be manifested as a progressive contraction of the visual field (see also Perimetry under Psychophysical findings). Loss of peripheral vision can be detected in early disease with small, dim test targets (Fig. 40.1). For many types of RP, field deficits are usually found first, and are most severe, in the superior visual field (Fig. 40.2). This reflects the early involvement of the inferior retina in RP. Occasionally the visual field loss will be greater temporally, nasally, or inferiorly (Fig. 40.3). See the section on Perimetry for specifics about testing the visual field.
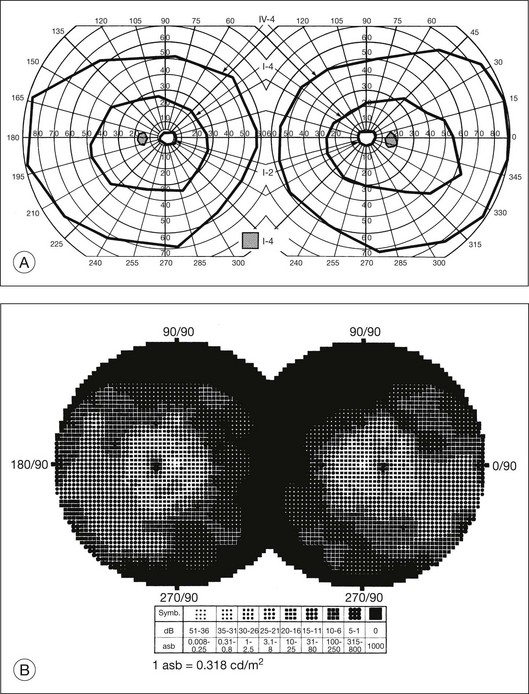
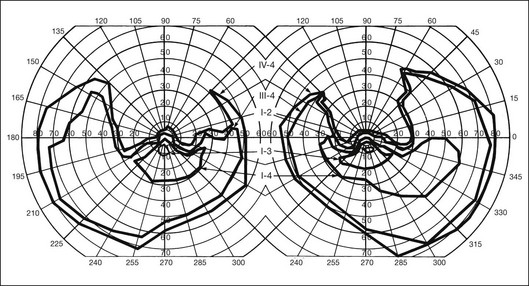
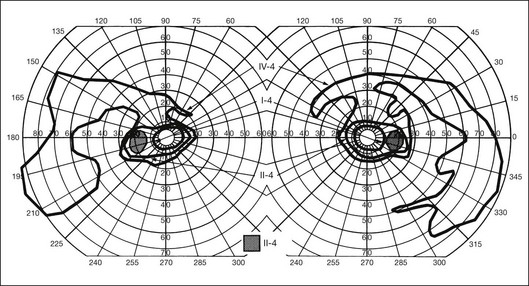
Fig. 40.3 Goldmann kinetic perimetry for an 18-year-old man with autosomal recessive retinitis pigmentosa associated with posterior column ataxia (same patient as Fig. 40.15A and brother of patient in Fig. 40.21). Visual acuity was 20/30, J1, right eye, and 20/50, J2, left eye.
In a group of patients with several types of RP, Berson et al.13 found that overall about 4.6% of the remaining visual field was lost per year. Massof et al.14,15 found that, in most forms of generalized RP, the kinetic visual field shrank approximately 50% over 4.5 years. Massof and Finkelstein15 argue that the time course for field loss in all major genetic and functional classes of RP is nearly identical if one corrects for the critical age, which is defined as the age when visual field loss is first detected by a test target of given size and brightness. In conjunction with this concept, they have proposed a two-stage hypothesis for the natural course of RP. The first stage is a predisposition for retinal degeneration, such as genetic or environmental factors, which would be different for the separate types of RP and possibly among individuals. The second stage involves the actual retinal degeneration, which proceeds on its own internal time course and which would be common to most forms of RP.
In general, there is a strong tendency for the visual field loss to be symmetric between the two eyes.14,15 A notable exception to this is the phenotype expressed in female carriers of X-linked RP. In this situation, the distribution of mutant photoreceptors in the retina is determined by lyonization (X-chromosomal inactivation). This is a random event determining which genes of the two X chromosomes (the normal or mutant copy) are expressed in a particular cell. This leads not only to unusual, irregular patterns of visual field loss in individual eyes but also to quite striking differences in field loss between the two eyes.16 However, even in these cases the visual fields usually correspond quite closely to the fundus appearance, with the greater visual field loss in the eye with greater pigmentary abnormalities.
In typical RP the rate of progression of visual field loss is usually slow and relentless, with changes best appreciated when observed over many years or even decades. However, the visual fields may change dramatically over a few months or years. A patient may not notice what may be a striking loss of peripheral visual field if the central field remains clear. As the visual field reaches the stage of “tunnel vision,” the patient usually becomes acutely aware of subsequent change with time. This often leads the patient to the conclusion that the rate of degeneration is accelerating. In the USA, statutory legal blindness results when the remaining central visual field measures 20° or less horizontally in the better eye, using the III-4-e test target on a Goldmann perimeter.17 In the UK, most RP patients, with normal acuity, would be registered as partially sighted (“gross field defect”). Legal blindness occurs when fields become “very contracted” – a stage at which acuity loss has also usually occurred (www.dh.gov.uk/assetRoot/04/07/48/48/04074848.doc).
Central vision loss
There is a common misconception in RP (documented in older texts) that the central vision will remain good until most, if not all, the peripheral field is lost. Central visual function can, however, be seriously affected early in typical RP, while significant peripheral field remains.18 Cystoid macular edema (CME),19 diffuse retinal vascular leakage,20 macular preretinal fibrosis21 (Fig. 40.4), and retinal pigment epithelial (RPE) defects in the macula or fovea22,23 can occur early in the disease. Macular edema is the topic of a section on treatment of RP later in this chapter.
To a considerable extent, the likelihood of retaining “good vision” (central acuity) to a given age in life depends on the specific inheritance type of RP. Patients with regional (“sector”) RP may retain good visual acuity all their life. Patients with autosomal dominant RP (adRP) are more likely than patients with autosomal recessive (arRP) or X-linked RP to retain good acuity beyond 60 years of age.13 Marmor, in his study of visual loss in adRP and arRP,26 found that when visual acuity began to fail, it generally dropped to 20/200 within 4–10 (median 6) years. Patients with X-linked RP are usually blind (acuity 20/200 or worse) by 30–40 years of age.13,24,25 A good prognostic sign of retention of central acuity is electroretinograph (ERG) amplitudes remaining quite large (e.g., greater than 100 µV).26
Photopsia and other symptoms
Many patients with RP at some time during the course of their disease have light flashes, or photopsias. These are reported as occurring in the midperipheral field of vision, often adjacent to areas of relative or absolute scotomas. These photopsias are described as tiny, blinking or shimmering lights or as a coarse, sparkling graininess to vision. The phenomenon is similar to that reported by patients with ophthalmic migraine except that, although retinal disease involvement expands over the years, the photopsias are generally stationary within the field. Also, unlike ophthalmic migraine, the photopsias may be continuous rather than episodic. As the scotomas become denser over the years, the photopsias decrease and finally disappear. In a retrospective survey of symptoms and findings in 500 patients with RP, Heckenlively et al.27 reported light flashes in 170 (35%). Since 8 patients in this series had retinal detachments, the symptom of light flashes must be considered an indication for careful fundus examination.
The cellular or tissue correlates that underlie photopsias in RP are unknown but may include photoreceptor dysfunction, neurite sprouting, aberrant synapse formation, and secondary remodeling of the retina, all of which occur as sequelae of photoreceptor degeneration (see section on Cell and tissue biology: Histopathology, for further discussion).
Fundus appearance
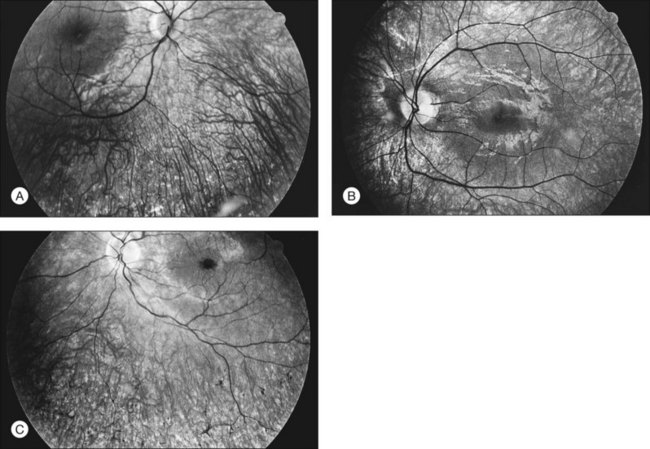
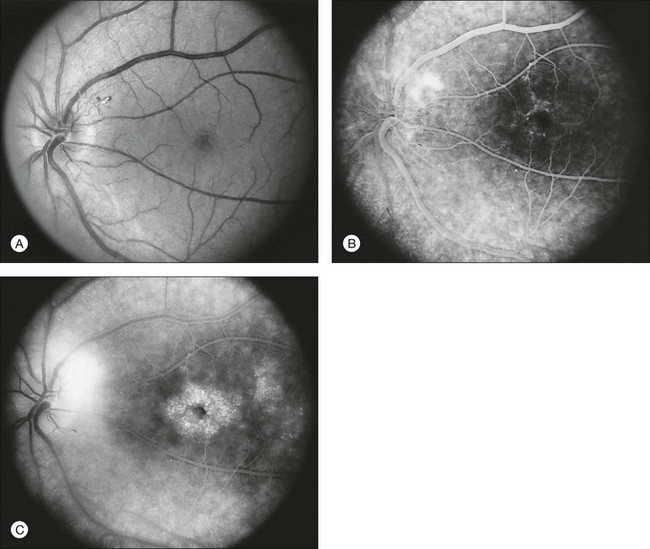
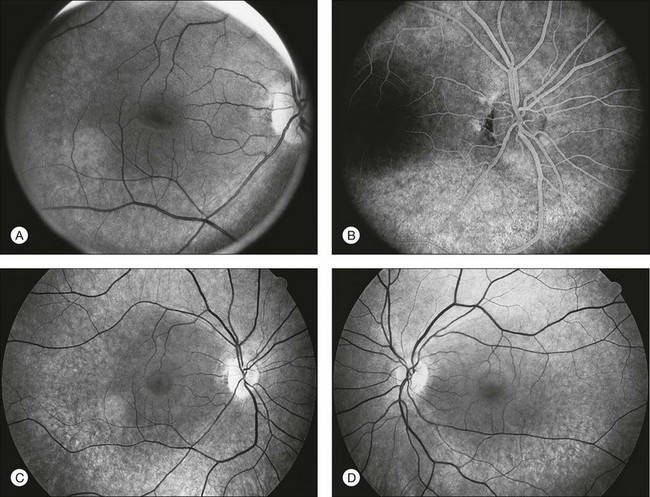
The classically described fundus appearance of RP includes attenuated retinal vessels, mottling and granularity of the RPE, bone spicule intraretinal pigmentation, and optic nerve head pallor (Fig. 40.5). In general, when ophthalmoscopically detectable abnormalities of the fundus are present, there is a high degree of symmetry between the two eyes and fundus pigmentation is often greater in the midperiphery (Fig. 40.6). In advanced disease, atrophy of the RPE and choriocapillaris leads to fundus pallor and larger choroidal vessels become visible (Fig. 40.7). In some forms of RP and allied disorders, sufficient atrophy of the retina and choroid will occur during the late stages of the disease that the fundus appearance overlaps with certain of the primary choroidal atrophies (Fig. 40.7).28 As the disease advances to late stages, vessel attenuation can become so severe that the retinal vessels appear thread-like (Fig. 40.8).
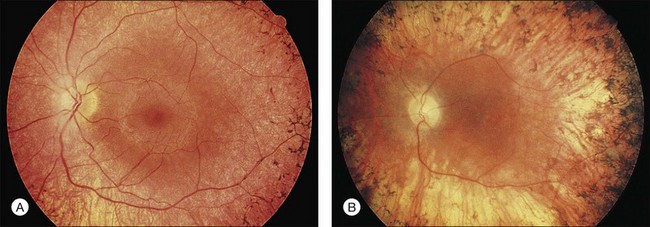
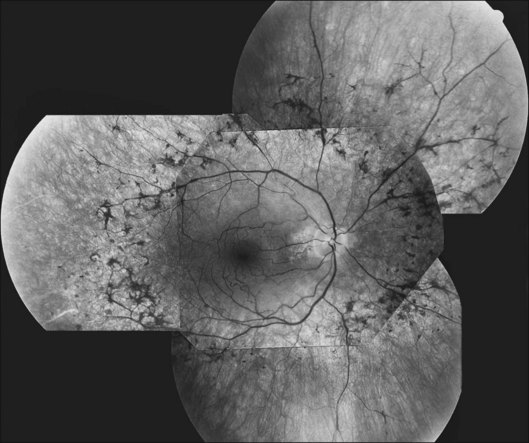
Fig. 40.6 Fundus appearance of the right eye of a 40-year old patient with rhodopsin Pro23His retinitis pigmentosa. Areas of fundus changes correlated well with kinetic visual fields, performed at 39 years of age, as shown in Figure 40.19.
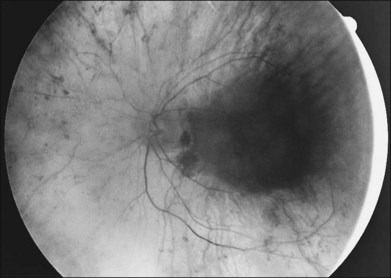
Fig. 40.7 Fundus appearance of the left eye of a 75-year-old patient with advanced rhodopsin Pro23His retinitis pigmentosa. This patient is the father of the patient whose visual field is shown in Figure 40.19 and whose fundus appearance is shown in Figure 40.6.
Almost all forms of RP go through a stage where the retina appears either normal or nearly normal, even though relative scotomas may be evident on visual field testing or areas of decreased retinal threshold may be present on static perimetry. Patients who have very early RP without fundus pigmentary abnormalities are often diagnosed as having RP sine pigmento or paucipigmentary RP. This is no longer considered a specific subtype of RP but a stage through which many, if not most, patients with RP pass. The sine pigmento stage may exist for decades before typical RP signs appear. For patients with high myopia and RP, the typical fundus features of high myopia may delay the appreciation of other retinal abnormalities (Fig. 40.9).

Fig. 40.9 Fundus of a 27-year-old man with high myopia and X-linked retinitis pigmentosa (same patient as in Fig. 40.2). Sparse but definite bone spicules were present in the inferior mid and far periphery. Discs showed a myopic tilt with peripapillary chorioretinal atrophy. The retinal pigment epithelium was diffusely hypopigmented.
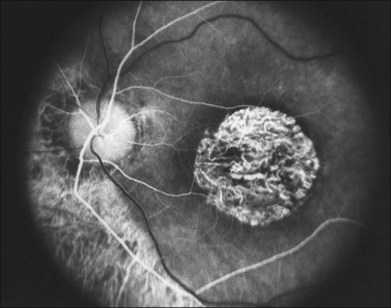
When fundus abnormalities are evident, the earliest features are attenuation of retinal vessels and the appearance of fine mottling or granularity of the RPE in the mid and far periphery (Fig. 40.10). Spectral-domain optical coherence tomography (SD-OCT) imaging demonstrates that retinal thickness is decreased, particularly for outer layers, outside the macula and in areas of abnormal pigmentation with bone spicules.29,30 The macular region may exhibit increased luster, abnormal highlights, or wrinkling, suggesting either macular edema or early epiretinal membrane formation or preretinal fibrosis.19,22 In advanced stages, patients with RP may exhibit atrophic macular lesions (Fig. 40.11) that simulate what some authors have called (perhaps inappropriately so) “macular coloboma.”
Intraretinal, bone spicule pigment formations represent migration of pigment into the retina from disintegration of RPE cells with accumulation in the interstitial spaces surrounding retinal vessels. This process occurs most prominently at the junctions of vessels producing perivascular pigmentary cuffing and spicule-shaped deposits. The loss of pigment within the pigment epithelial cells often produces an overall gray, desaturated appearance to the retina with greater visibility of underlying choroidal vessels through the more transparent pigment epithelium. Eventually, the normal salmon-pink color of the entire mid and far peripheral fundus is replaced by dense bone spicule pigmentary formations. Heckenlively31 has described several patients with advanced RP with preservation of the RPE in a para-arteriolar distribution. Porta and colleagues32 described two additional cases. Van den Born et al.33 reported a large consanguineous family with preserved periarteriolar RP and confirmed the autosomal recessive inheritance. Almost all these patients were hyperopic. The gene for this form of RP (termed RP12) has been found to be the human homolog CRB1 of the Drosophila crumbs gene.
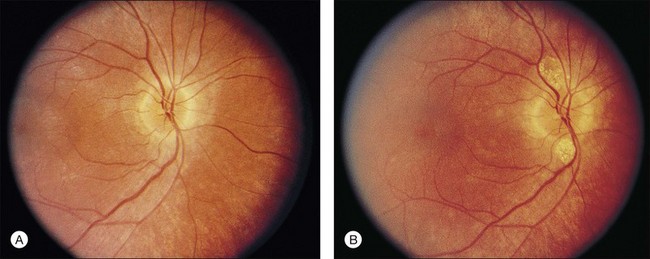
A yellowish-white metallic tapetal-like reflex or sheen can occasionally be observed in women who are carriers for the X-linked form of RP34 (Fig. 40.12) and rarely in young males with early disease35–37 (Fig. 40.13). With time, regional or diffuse mottling and atrophy of the RPE eventually replace the tapetal-like sheen. At least one form of dominant RP can show a tapetal-like reflex limited to the foveal and parafoveal region (Fig. 40.14). No histopathologic assessment has yet identified the source of a tapetal-like reflex. Cideciyan and Jacobson38 suggested that abnormal cones are the sources of the tapetal reflex. Berendschot et al.,39 using spectral fundus reflectance, confirmed that outer segments of peripheral photoreceptor cells are the source of the abnormal reflex but that this abnormality was not confined to cones. The carrier state for X-linked RP appears to be a slowly progressive retinal dystrophy in its own right, with a patchy distribution of affected retina as determined by lyonization.40,41 A number of studies have reported that the ERG is abnormal in most X-linked RP carriers.16,42–45 A tapetal-like reflex is not pathognomonic of X-linked RP but has also been reported in occasional cases of X-linked retinoschisis,46 and Oguchi disease.47
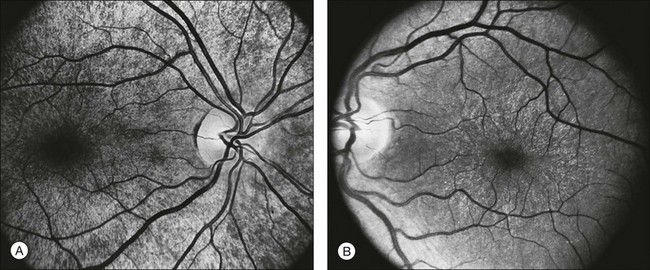
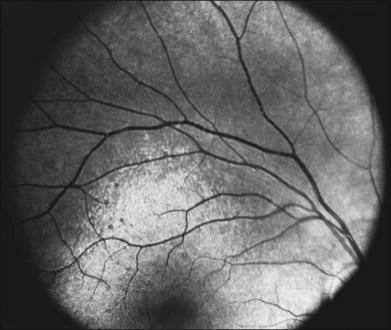
Fig. 40.13 Tapetal-like yellow-white reflex in a 13-year-old boy with X-linked retinitis pigmentosa.
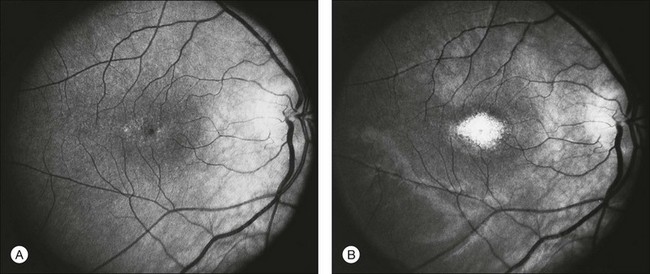
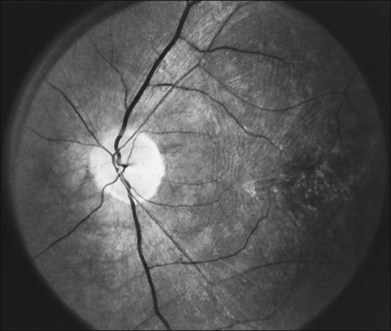
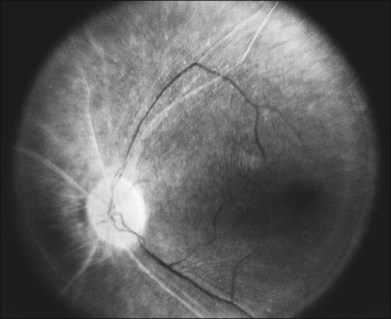
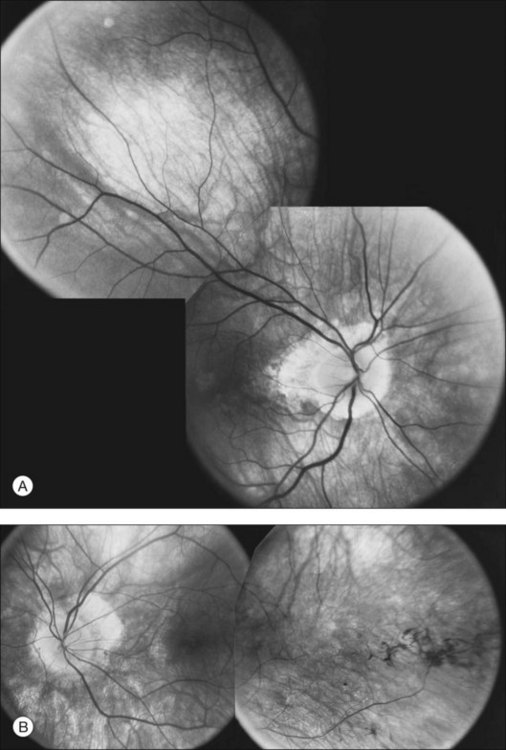
The optic disc may be normal in early RP, show a waxy fullness with hyperemia, or appear waxy and pale (Fig. 40.15A). A “golden ring” or yellowish-white halo can often be seen surrounding the optic disc in early RP (Fig. 40.15B). As disease progresses, this golden ring is replaced with peripapillary mottling, hyperpigmentation, and atrophy of the RPE (Fig. 40.15C). The optic nerve head cup-to-disc ratio has been reported to be significantly smaller in RP patients of all types (0.19 compared to 0.35 in normal subjects).48 In advanced disease, dense optic nerve head pallor results, in part from optic atrophy and in part from gliosis overlying the discs.21 Drusen-like globular excrescences may develop on the optic nerve or adjacent retina (Fig. 40.16). These have been mistaken for astrocytic hamartomas49 and are consistent with disc drusen due to aberrant axoplasmic transport.50,51 In a survey of 117 RP patients,52 10% were found to have clinically apparent optic nerve head drusen. Edwards et al.53 suggested that this phenomenon is particularly common in RP associated with Usher syndrome type 1.
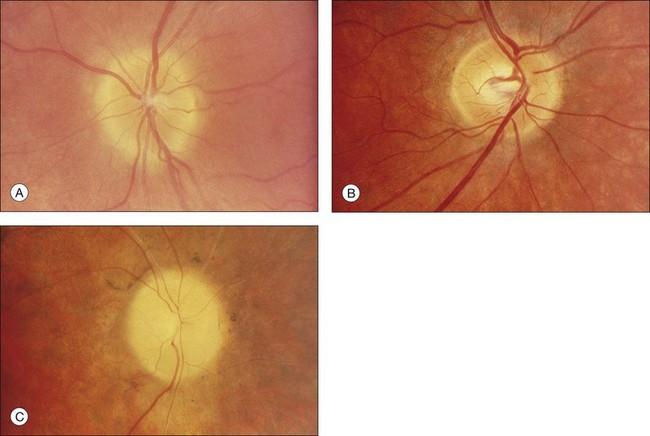
Fig. 40.15 Optic discs in an 18-year-old man with autosomal recessive retinitis pigmentosa as part of posterior-column ataxia and retinitis pigmentosa syndrome (A) (same patient as in Fig. 40.3), a 32-year-old man (B), and his 55-year-old father (C), both of whom have autosomal dominant retinitis pigmentosa. The disc in A shows peripapillary chorioretinal pallor, or the “golden ring” sign, that is characteristic of early retinitis pigmentosa. Note that the disc is nearly normal in the son (B), but shows severe waxy pallor in the father (C).
Flynn et al.54 found that the presence or absence of macular RPE defects is of significant prognostic importance with regard to retention of visual acuity over the next 5 years. Specifically, the absence of macular lesion was associated with only one line of acuity loss over the 5-year period, whereas bull’s-eye or geographic atrophy lesions were associated with a predicted acuity loss of three to four lines. The authors emphasize the importance of this type of information for prognostic counseling of patients with RP regarding the likelihood of preservation of visual acuity. Other maculopathies associated with RP include cellophane maculopathy, macular hole (complications of CME),55 and a single report of RP with central serous retinopathy.56
Vitreous abnormalities
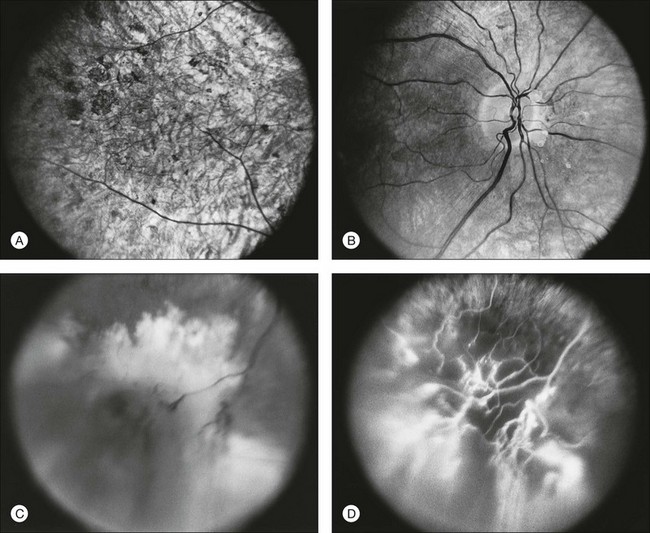
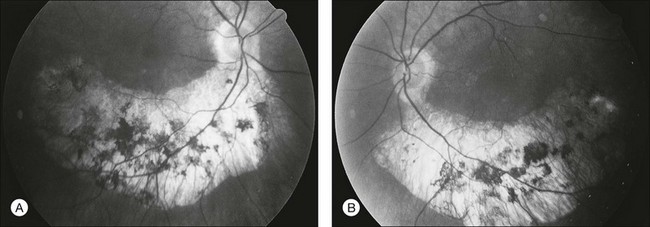
The most common abnormality of the vitreous in RP is the presence of fine, dust-like pigmented cells released from degeneration of the RPE. In patients with RP, complete posterior detachment of the vitreous, “cotton-ball” opacities, interwoven filaments in the retrocortical space, and spindle-shaped vitreous condensations are seen more frequently than in normal subjects.57,58 A single case is reported of vitreous opacity so dense that vitrectomy was required.59 Vitreous abnormalities related to peripheral retinal ischemia and preretinal neovascularization have been reported in rare cases of adRP and arRP.60 Uncommonly, a peripheral Coats-like retinal vasculopathy with lipid exudations and serous retinal elevation can be seen in RP61–63 (Fig. 40.17). Unlike idiopathic Coats syndrome, the retinal vasculopathy seen in RP is usually bilateral, shows no sex predilection, and typically occurs in older patients. Coats-like disease is seen in 3.6% of RP cases64 and has been reported with adRP,62,63 arRP33 but not X-linked inheritance63 (although see Fig. 40.17). Coats-like changes that occur in children64,65 may change over a relatively short period of months to years, must be monitored carefully, and may need to be considered for laser or cryocoagulation. Vitreous hemorrhage related to preretinal neovascularization has also been described in rare cases.60,66 Retinal edema in some cases may be caused by inflammation resulting from the degeneration of outer retina.60,67
Anterior-segment abnormalities
Cataracts are frequent anterior-segment complications of RP. The prevalence of cataracts in RP patients aged 20–39 years varied with the inheritance type, being roughly 52% for autosomal dominant, 39% for recessive, and 72% for X-linked cases.68 The incidence of cataracts in RP is highly age-dependent.68,69 The most frequent type of cataract is a posterior subcapsular lens opacity, which occurs in 35–51% of patients.69,70 One study has suggested that posterior subcapsular opacities are most commonly associated with autosomal dominant inheritance (51%),69,70 whereas another has suggested a more common association with X-linked inheritance.71 Although keratoconus has been claimed to be more frequent among patients with RP,72 the occurrence of keratoconus in typical juvenile- or adult-onset RP is extremely rare in our experience. Glaucoma, especially primary open angle glaucoma, is also allegedly more frequent in RP.73 Sector RP appears to be associated with hyperopia and angle closure glaucoma.74 Because of the other abnormalities of the disc and fundus and because of the already abnormal visual fields, the diagnosis and management of glaucoma are quite challenging in RP patients.
Psychophysical findings
Perimetry
Assessment of visual field is an important aspect of the management of RP. This is an invaluable tool in the diagnosis of RP, as well as being the most reliable method of quantifying real change in visual deficit. It should be remembered when assessing the results of visual field testing that the reproducibility of visual fields is significantly less in patients with RP than in normal subjects. Kinetic visual fields tested on separate days by the same examiner varied by 5–6% in normal subjects but by 11–13% in patients with RP (intraobserver variation); variability between examiners for the same patient was 4–6% for normal subjects but 10–16% for patients with RP (interobserver variation).76
In the great majority of cases of RP, the earliest defects of the visual field, on kinetic perimetry, are relative scotomas in the midperiphery, between 30 and 50° from fixation (Fig. 40.18). These enlarge, deepen, and coalesce to form a ring of visual field loss. These midperipheral depressions of retinal sensitivity are readily documented as decreased retinal thresholds on static perimetry. As ring scotomas enlarge toward the far periphery, islands of relatively normal visual field remain, usually temporal but occasionally inferior or nasal (Fig. 40.19). The peripheral islands or remnants of visual field are often lost before the central field contracts sufficiently to qualify the individual as legally blind.
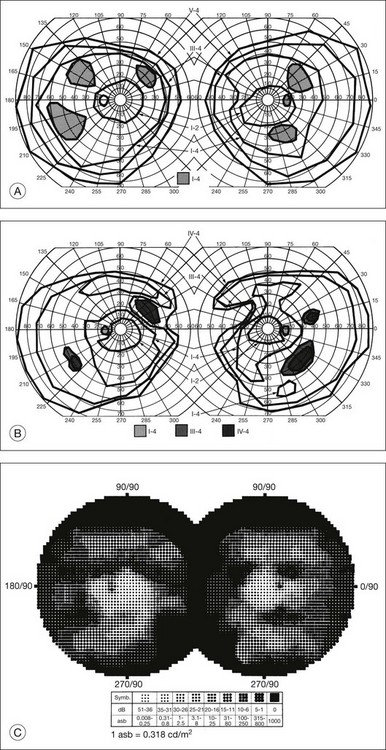
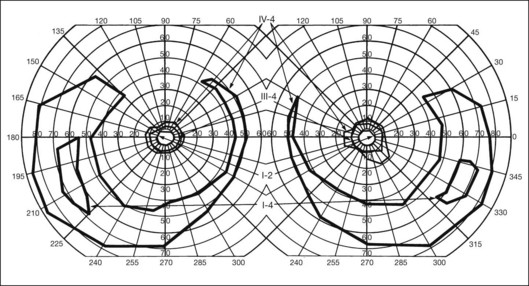
Fig. 40.19 Goldmann kinetic perimetry for a 36-year-old patient with retinitis pigmentosa from the Pro23His mutation of rhodopsin. Dense ring scotomas have broken through to the periphery superiorly, leaving a central field of 25° in diameter and only an incomplete annulus of peripheral vision in each eye. The fundus appearance of this patient at 40 years of age is shown in Figure 40.6.
Kinetic visual field testing has traditionally been performed with the manual Goldmann perimeter. Recent advances have occurred in both kinetic and static perimetry. Semiautomated kinetic perimetry,77 as implemented on the Octopus 101 and 900 perimeters (Haag-Streit, Köniz, Switzerland) (Fig. 40.20) automatically documents the testing and enables quantification of field by measurement of isopter area. A new fast thresholding algorithm, known as German adaptive thresholding estimation (GATE),78 has enabled another advance in perimetry, that of full-field static perimetry to assess the entire visual field in a time frame comparable to that required to test the central 30° field with the Standard 4–2-1 strategy (Fig. 40.20) (Weleber et al., 2012, in preparation). The static threshold data from such testing can be modeled as sensitivity values into a three-dimensional hill of vision from which the magnitude and extent of the total visual field can be determined as a volumetric measurement (Weleber et al. 2011, in preparation).
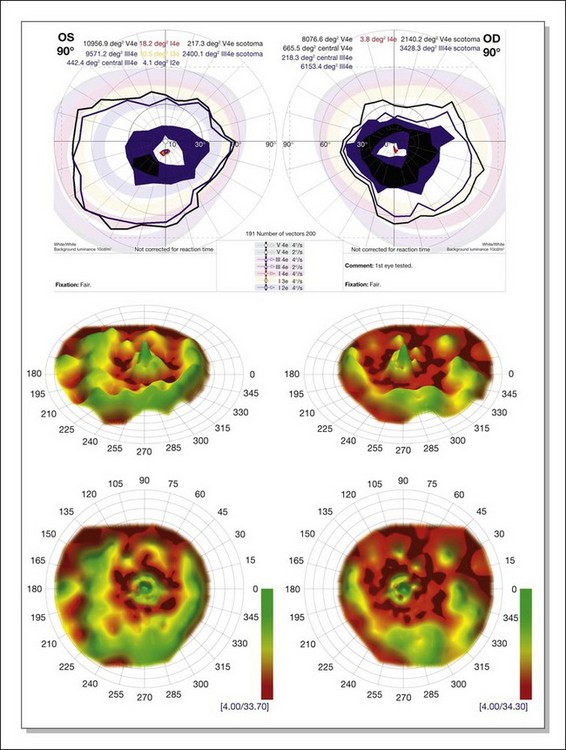
Fig. 40.20 Semiautomated kinetic perimetry in a 27-year-old woman with retinitis pigmentosa (top). The shaded areas depict the normal range for the isopters for the test targets presented. Three-dimensional (3D) model of the hill of vision for full-field static perimetry for this same subject (middle in tilted view, below in face view) using the size V test target and the German adaptive thresholding estimation thresholding algorithm.78 The volume of this model, which was 40.2 dB-sr OD and 28.0 DB-sr OS (compared to a normal 112.5 dB-sr for a normal), reflects the magnitude and extent of the visual field for this subject. Thus, this patient has 35.7% OD and 24.9% OS of expected field volume for his age. Note that the 3D model provides much more topographical information on the location of the defects in the field of vision than does the kinetic perimetry. This is the same patient whose fundus autofluorescence image for the left eye is shown in Figure 40.29A.
Two-color static perimetry has been shown to be a useful tool in assessing the dark-adapted visual fields of patients with RP. Using this technique, Massof and Finkelstein79–81 found that their patients with RP could be divided into two groups: (1) type I RP, which is associated with early diffuse loss of rod sensitivity relative to cone sensitivity and childhood-onset nyctalopia; and (2) type II RP, associated with regional and combined loss of both rod and cone retinal sensitivity and adult-onset nyctalopia. No patients have been observed to change from one type to the other.82 No families have been reported that contain both types I and II in the same sibship.15 An excess of type II RP over the type I form has been reported in simplex disease. Analysis of subgroups within simplex and multiplex RP suggest that a sizable proportion of type II patients may represent sporadic or acquired disease, and not true RP.83
Arden et al.84 studied a series of cases of adRP both with ERGs and psychophysically with scotopic static thresholds. They found that the preservation of sizable rod responses on the ERG correlated perfectly with the Massof type II classification. Absence of rod ERG did not always indicate type I disease, and both types were seen in this group. Lyness et al.85 reported clinical, psychophysical, and ERG findings in 104 patients (from 44 families) with adRP. Subgroup D (13 patients in four families) had diffuse loss of rod function and night blindness before the age of 10. Subgroup R (28 patients in 13 families) had regional loss of rod function, and most of these patients were unaware of night blindness until after the age of 20. Both groups had regional loss of cone functions. In the R group the rod-mediated ERG was usually present and substantial, whereas in the D group the rod ERG was undetectable. Although not identical, the D group bears similarities to Massof type I, and the R group bears similarities to type II. These studies suggest that the two types or groups may represent different pathophysiologic subtypes of RP. It should be emphasized, however, that a large proportion of patients (27 of the 44 families studied by Lyness et al.85) could not be incorporated into the D- versus R-type classification, partly because many patients with RP have almost undetectable ERG responses at presentation, which is quite common.
The techniques of two-color scotopic static perimetry have been further developed to evaluate rod and cone retinal sensitivities in different regions of the retina.86,87 Such testing allows the definition of which class of photoreceptor (rod or cone) is the determinant of retinal threshold in a particular region or area of the retina. This has allowed the characterization of the relative losses of rods and cones in various types of RP associated with specific mutations.88,89 Equally important to the definition of early retinal abnormalities in retinal degenerations, two-color perimetry can be used for monitoring disease therapy or for testing hypothesis of disease pathophysiology. For example, the abnormal, thickened Bruch’s membrane, which acts as a diffusion barrier between the retina and choroid, has been proposed as a major factor leading to night blindness in Sorsby’s fundus dystrophy.90 The return of rod function after oral vitamin A supplements (50 000 IU/day) was elegantly recorded using these techniques.90
Dark adaptometry
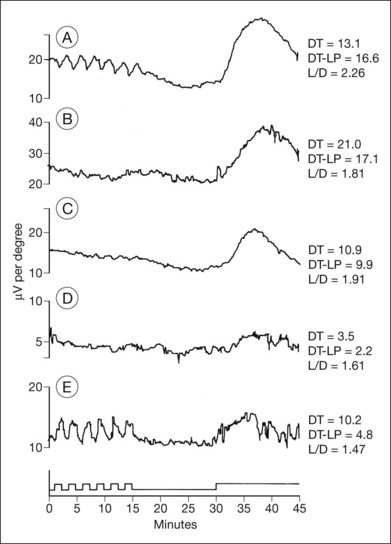
Normal subjects, when placed in the dark after exposure to a strong adapting light, will rapidly reduce their retinal psychophysical threshold using their cone system, reaching a plateau in about 5 minutes. Thereafter rod adaptation slowly increases, and rods determine retinal thresholds for another 3 log units of sensitivity before a second plateau occurs at about 30 minutes of dark adaptation. Patients with RP, when tested with dark adaptometry, may show elevation of the cone segment, the rod segment, or both, to varying degrees (Fig. 40.21). Also, in some patients there may be a delay in reaching what eventually for them is a relatively good final dark adaptation rod threshold91 (Fig. 40.22).
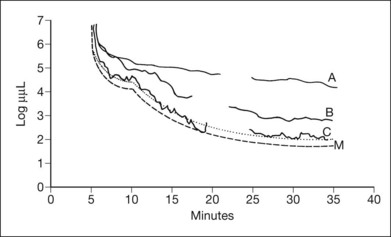
Fig. 40.21 Dark adaptometry showing marked elevation of both cone and rod thresholds with little or no cone–rod break in an 18-year-old woman, broken solid curve (A), with autosomal recessive retinitis pigmentosa and large-fiber sensory peripheral neuropathy (sister of patient shown in Figs 40.3 and 40.15A). Mild elevation of cone threshold with greater elevation of rod threshold in a 15-year-old young woman with Bardet–Biedl syndrome, broken solid curve (B). Borderline normal cone and rod segments in a 15-year-old young woman with type 2 Usher syndrome, broken solid curve (C). Dashed curve (M) is mean for normals aged 10–20 years.
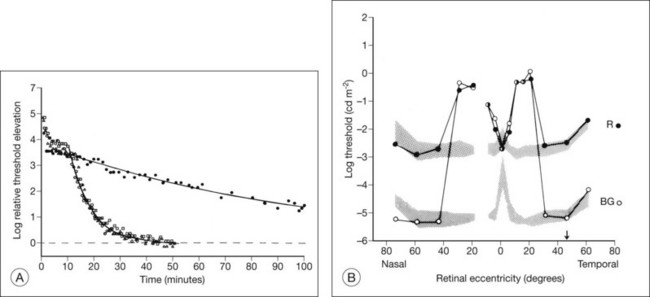
Time course analysis of dark adaptometry has been performed in patients with adRP and prolonged dark adaptation is associated with rhodopsin mutations Thr17Met, Pro23His, Gly106Arg, and Thr58A.88,89,92 The most characteristic feature of the Pro23His genotype was prolonged dark adaptation, which was present in all patients regardless of their stage of disease.89 Jacobson et al.88 also reported prolonged rod dark adaptation in patients with Thr58Arg and Thr17Met mutations of rhodopsin. Indeed, prolonged rod dark adaptation appears to be a characteristic finding of virtually all rhodopsin mutations. These studies suggest that each of these mutations produces specific abnormalities in the rate of reactions within the rods that limit the recovery of scotopic sensitivity. For patients with regional forms of RP from other causes, zones of more normally functioning retina may allow reasonably good final rod thresholds (Fig. 40.22). Minor abnormalities in cone thresholds are more likely to result in complaints of poor dark adaptation than mild to moderate elevation of rod thresholds. One study has indicated that abnormalities in the normal interactions between rods and cones may underlie the symptoms of poor night vision in some patients rather than isolated rod deficits.93
Retinal densitometry (fundus reflectometry)
Retinal densitometry is a technique whereby, to deduce effective photoreceptor photopigment density, measurements are made of the difference between light shone into the eye and light reflected out of the eye. These measurements can be made and compared at different retinal loci, at different times, or against a retinal standard. From these data, estimates can be made of the rates of photopigment regeneration. Retinal densitometry has been performed on patients with RP by several investigators using a variety of techniques.94–97 Rhodopsin levels were found to be reduced in all patients, but rhodopsin bleaching and regeneration kinetics have been normal. Van Meel and van Norren98 found subnormal cone pigment levels in all patients studied and found prolonged cone pigment regeneration in eight of 12 patients with RP.
The losses of retinal sensitivity in RP in some patients are believed to relate to loss of quantum catch by the reduction of rhodopsin levels in the retina.94,95 A reduction of rhodopsin level to one-tenth of normal, for example, would be expected to be associated with a 10-fold elevation in the rod threshold. These calculations are only estimates, however. In vitamin A deficiency, the elevation of the rod threshold is much greater than predicted by the reduction in rhodopsin levels.95 Perlman and Auerbach99 described a group of patients with RP who, when studied with retinal densitometry, were found to have elevations of rod thresholds greater than what would be expected purely on the basis of reductions of rhodopsin levels. Kemp et al.100 found that 4 patients with type II (regional) adRP had elevations of retinal threshold that could be explained solely by loss of quantum catch from reductions of rhodopsin levels in the retina. However, 5 patients with type I (diffuse) adRP had elevations of threshold that were much greater than expected on the basis of loss of rhodopsin and quantal absorption for the reduction of rhodopsin levels observed.100 These studies provide evidence suggesting that the two classifications represent different pathophysiologic entities. Others have studied patients with defined mutations of rhodopsin. Kemp et al.89 have shown that, for patients with the Pro23His mutation of rhodopsin, rhodopsin levels are decreased below normal by amounts that indicate that rod sensitivity loss is determined by the reduced ability to absorb light. Similar findings were reported for patients with rhodopsin Thr17Met, Thr58Arg, and Gln344ter.88
Fulton and Hansen97 have reported reflectometry results on a mother and three daughters whose RP appeared to have a sectoral distribution. Elevations of retinal threshold were greater than those predicted by reductions of rhodopsin seen on densitometry measurements, suggesting that the dysfunction was central to the photoreceptor outer segments.
Electrophysiology
Karpe101 in 1945 first reported that the ERG was “extinguished” in RP. The standing, or resting, potential of the eye was first described by DuBois-Reymond in 1849. Riggs102 in 1954 was the first to report that the resting potential was decreased in pigmentary degeneration of the eye. Arden et al.103 in 1962 developed the currently widely used technique for measurement of the light-induced rise of the resting potential of the eye (the electro-oculogram (EOG)), which is considered a function of the RPE. Fast oscillations of the resting potential can be recorded, and these offer another measure of RPE function.104 Gouras105 in 1970 based clinical ERG on the use of Ganzfeld stimulation. Standardized conditions and assessment protocols have now been established for electrodiagnostic investigations.106,107
Electrodiagnostic responses in an RP patient can range from normal to undetectable. In general, more sizable responses are seen with younger patients or at earlier stages of disease. Most patients with advanced RP have undetectable responses (typically less than 10 µV) to single-flash, nonaveraged techniques.105 Computer averaging, however, can usually detect small ERG responses in even moderately advanced stages.13,108 In studying the natural course of RP, Berson et al.13 found that patients lost an average of 16–18.5% per year of remaining ERG amplitude to bright white flashes (a mixed rod–cone response).
Many studies have compared ERG responses in the different RP inheritance types. Tanino and Ohba12 and Berson,109 using conventional techniques, found that the ERG is more likely to be recordable in older patients with adRP than arRP. Berson et al.13 have also shown that progressive ERG loss of amplitude over a 3-year period is least in adRP. Birch and Fish110 found that the rate of progression of rod loss, as indicated by the half-saturation coefficient of the rod stimulus–response curve, varied among the inheritance groups. This ranged from 0.3 log unit/year in elevation of rod threshold in X-linked RP to 0.12 log unit/year elevation in adRP.
Agreement exists that the cone-mediated implicit times can be significantly prolonged in various forms of RP. Berson111 has also stressed the importance of delays in the implicit time for cone-mediated 30-Hz flicker responses. Berson and colleagues112,113 found that the cone-mediated b-wave implicit time in their patients was characteristically prolonged in adRP with reduced penetrance but not in completely penetrant adRP. Sandberg et al.114 suggested that this was due to an abnormal contribution of extrafoveal cones in adRP with incomplete penetrance. This may be due to abnormal cone light adaptation or a disturbance of normal rod–cone interactions.
Histopathologic study has indicated that, in RP, a postreceptor deficit may contribute to macular dysfunction.115 ERG study has confirmed this. Falsini et al.116 have assessed 8–10-Hz focal ERGs in 22 simplex and recessive RP cases. They found a loss of fundamental and second harmonic components and an increase in the fundamental-to-second harmonic ratio. This was interpreted as suggesting that both inner and outer retinal abnormalities contribute to visual deficit. Cideciyan and Jacobson117 have documented electronegative b-waves in some cases of RP. This finding was seen with abnormalities of on/off components of cone ERGs and reduced oscillatory potentials117 and was interpreted as indicative of significant inner retinal abnormalities in these RP cases.
Models of analysis of the ERG have been developed that attempt to isolate from the ERG the photoreceptor and bipolar contributions, specifically the original components of Granit’s model of the cat ERG, termed P3 and P2118 (Fig. 40.23A). These analysis methods have been developed to quantify the visual deficit attributable to outer and inner retinal disease (Fig. 40.23B–D). P3 is the corneal-negative response of photoreceptor outer segments that contributes to the a-wave of the Ganzfeld ERG. The analysis involves subjecting the leading edge of the a-wave of the ERG at a range of intensities to a mathematical curve-fitting algorithm119–121 (Fig. 40.23C) using equations that describe the kinetics of rod phototransduction activation.122 The method of Hood and Birch123 allows for estimation of two variables: S, which is a sensitivity factor, and Rmp3, which is the maximum amplitude. Such analysis can determine whether phototransduction is affected in a given disease.
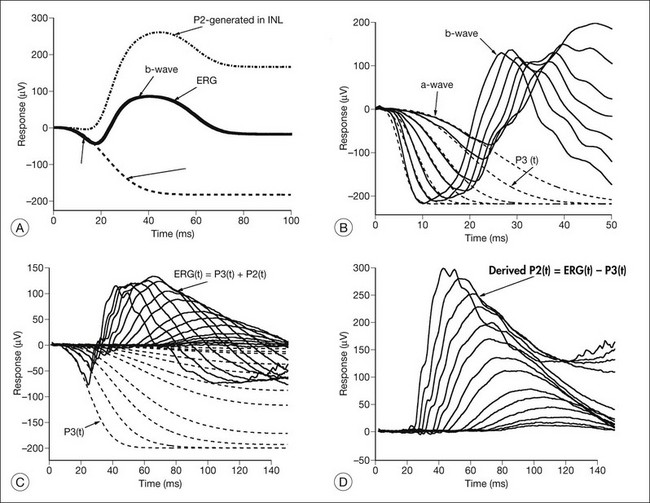
P2 is the bipolar cell-derived component of the rod-isolated b-wave. The P2 component of the rod b-wave, derived from bipolar cells, can be isolated from the full-frequency ERG, using an algorithm whereby the P3 is first calculated for a given series of intensities, after which the P3 is subtracted from the series of rod responses124,125 (Fig. 40.23D). This gives a much better estimate of the ON bipolar contribution to the b-wave, especially for patients in whom the ON bipolar cells are more affected than the photoreceptors.124
Application of these techniques to 15 patients with RP has shown that, for P3 analysis, all patients had significantly reduced values of Rmp3, indicating effective loss or damage of rods, and a wide spectrum of values for S.126 Three patients had normal values for S. Four RP patients had subnormal S values but normal local retinal psychophysical thresholds in some regions of their retina. The rest had abnormal S values and abnormal visual fields with globally reduced thresholds. The rod b-wave amplitudes to a range of intensities for these RP patients were fitted with the Michaelis–Menten equation, generating the parameters Kbw (the semisaturation intensity, which is a sensitivity function) and Vmax (the maximum amplitude). For all patients, the changes in Kbw and Vmax followed the changes seen in S and Rmp3.126 Hood and Birch123 studied rod phototransduction in 11 patients with RP and four with cone–rod dystrophy (CRD). Their findings, which were similar to those above, supported the conclusion that the activation stage of rod phototransduction is affected in some forms of RP and CRD but not all.
Supernormal and delayed derived P2 responses were found by Hood et al.127 when they applied modeling analysis to an unusual retinal dystrophy, first reported by Gouras et al.128 in 1983 as “cone dystrophy, nyctalopia, and supernormal rod responses.” Although Gouras et al.128 initially attributed this dystrophy to an abnormality in receptor cyclic guanosine monophosphate (cGMP) activity, P3 and P2 modeling by Hood et al.127 showed that the delays in the rod and cone b-wave were not caused by the speed or amplification of phototransduction but resulted from delays beyond the outer segments involving a delay in activation of the inner nuclear layer activity. This disorder was subsequently clinically and electrophysiologically defined as the enhanced S-cone syndrome (ESCS)129,130 and was later shown to result from mutation of the gene NR2E3.131
Paradigms have been developed to study the kinetics of rod transduction deactivation.132 This technique uses a bright conditioning flash followed by a probe flash. The recovery of the amplitude of the response to the probe, which is presented at varying intervals after the conditioning flash, provides a measure of the return to baseline of the response to the conditioning flash and, hence, the kinetics of transduction deactivation. The authors conclude that the lifetime of activated rhodopsin in normal human rods is 2.3 seconds. This technique has been applied to humans with the Pro23His rhodopsin mutation, demonstrating a reduction of the gain of activation and marked slowing of deactivation of rhodopsin (by a factor of at least 5).132 Animal models of RP, for example, transgenic mice expressing the mutant Pro23His rhodopsin gene, have also demonstrated increases in the half-maximal recovery period, T50%, indicating abnormally prolonged recovery from the conditioning flash.133
Analysis of the a-wave leading edge has also recently shown that in RP, the efficiency of cone phototransduction is affected very early, even in some patients in whom the sensitivity of rod phototransduction is normal. X-linked inheritance seemed to be associated with greater phototransduction abnormalities in cones and rods at a younger age than seen in other modes of inheritance.134
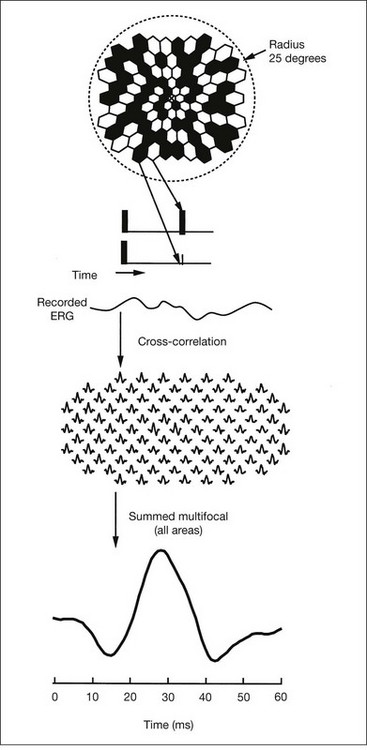
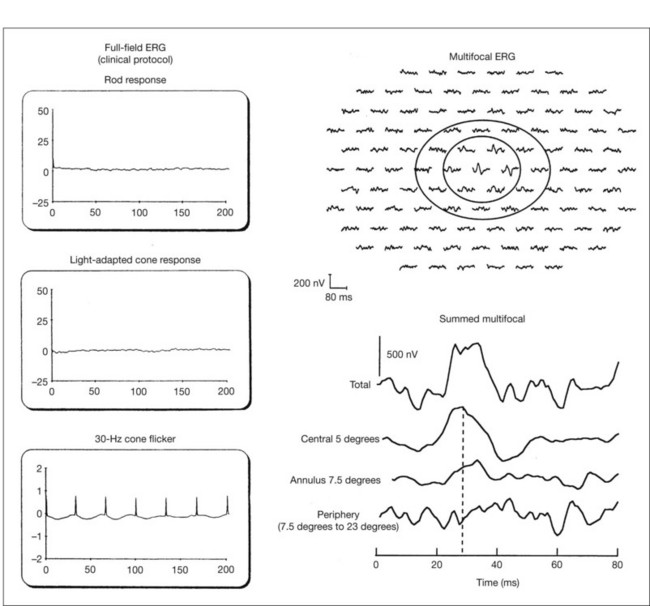
Multifocal ERG techniques have been developed whereby, through patterned stimulation of the retina cross-correlated with recording of the ERG responses from a corneal electrode, very small ERG responses can be obtained from precisely localized regions of the retina135–137 (Fig. 40.24). Multifocal ERGs can be seen in advanced RP where the 30-Hz flicker response is barely detectable138 (Fig. 40.25). Hood and Li137 have shown that, in RP, amplitudes of ERG responses do not correspond as well as b-wave implicit times with the retinal sensitivity as measured by static perimetry (Fig. 40.26). This is most evident with the regional or sectoral types of RP. Thus the b-wave implicit time appears to reflect early involvement of the disease process in regions of the retina that still have relatively good retinal sensitivity. Robson et al.139 compared pattern ERG changes in RP patients with abnormalities of fundus autofluorescence (FAF). In such patients, pattern ERG responses suggested that visual loss was attributable to dysfunction rather than cell death.
In general, the EOG is abnormal in diffuse hereditary rod–cone degenerations of the retina.140 In typical RP, the slow and fast light-induced oscillations of the resting potential are usually decreased simultaneously early in the disease (Fig. 40.27). In some patients the fast oscillations of the resting potential are diminished earlier than the slow oscillation.104 In others the fast oscillations of the EOG are more preserved than the slow oscillation, a finding that also occurs in Best vitelliform macular dystrophy.104,141 In most cases of RP the EOG is abnormal when the ERG is abnormal.
Imaging modalities in RP
Fundus photography/fluorescein angiography
Documentation by fundus photography can assist in monitoring changes in patients with RP. Fluorescein angiography in patients with RP will demonstrate hyperfluorescence in areas of RPE atrophy and can highlight areas of CME (Fig. 40.28). However, fluorescein angiography has largely been supplanted by OCT for detecting cystoid maculopathy. In addition, concerns about light exposure accelerating certain forms of RP in animal models have prompted many ophthalmologists to exercise caution in obtaining excessive photographs.142
In many cases of mild to moderate RP, fluorescein angiography reveals transmission defects of the RPE with later diffuse leakage.143 In 78 of 82 patients with RP, Newsome144 found extravasation of dye from perifoveal capillaries in only a few patients but frequent abnormalities of the blood–retinal barrier at the level of the RPE. Such angiographically significant CME can often be seen at an early stage of disease in young patients who still retain good vision (20/25 or better). Macular edema is a significant cause of early loss of visual acuity in RP.144,145
Autofluorescence
FAF utilizes a scanning laser ophthalmoscope to stimulate intrinsically autofluorescent molecules of lipofuscin to visualize the RPE.146 Studies from patients with RP have shown that lack of signal on FAF correlate well with areas of RPE atrophy, while areas of increased FAF can be seen in areas with persistent macular edema as well as within areas of surviving retina.147 Most RP patients demonstrate a perifoveal ring (Fig. 40.29) of increased autofluorescence within the macula, which denotes the border between functional and dysfunctional retina (see Chapter 4, Autofluorescence).139,147–149 The border of the parafoveal ring of increased autofluorescence has been shown to correlate with functional status as measured by pattern ERG, multifocal ERG, scotopic fine matrix mapping, and microperimetery.150,151 In addition, areas outside the ring have been correlated with the loss of outer nuclear layer thickness and disruption of the inner-segment–outer-segment (IS/OS) junction on OCT.152,153 Near-infrared autofluorescence (NIA) has also been used to image melanin present in the apical tips of the RPE.154 Similar to FAF, rings with increased NIA are seen in patients with RP.155 Combined NIA and blue light FAF imaging suggest that the presence of NIA may correlate better with preserved cone function, while blue light FAF indicates only preservation of RPE cells.155
Optical coherence tomography
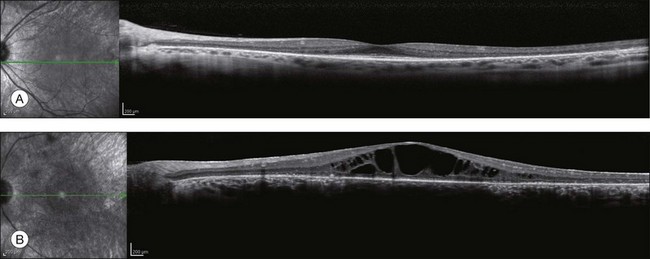
OCT has become one of the most utilized imaging modalities for studying retinal disease in the past several years. Ultrahigh-resolution OCT and SD-OCT have been used to study retinal structures in patients with RP.156,157 In these patients, such studies have demonstrated decreased thickness of the outer nuclear layer and loss of the external limiting membrane and IS/OS junctions. Loss of outer nuclear layer thickness or of the IS/OS has been correlated with visual defects measured by visual fields, microperimetry, or multifocal ERG.29,158–160 SD-OCT is especially useful for detecting CME (Fig. 40.30) or epiretinal membranes, which are common features in patients with RP.161 The ability to detect CME by OCT ring often eliminates the need for fluorescein angiography.
Adaptive optics scanning laser ophthalmoscopy
Traditional imaging modalities cannot resolve individual retina cells due to the optical limits imposed by the cornea and lens which create higher-order aberrations resulting in image blur.162 A combination of adaptive optics with flood illumination or scanning laser ophthalmoscopy can compensate for these factors and provide imaging of individual cone photoreceptors.163 Adaptive optics studies from patients with both CRD and RP have demonstrated increased cone spacing as well as qualitative areas where cone profiles could not be identified.164–166 Recent advances have now enabled imaging of rod photoreceptors and foveal cones.167 The ability to quantify cone photoreceptors has already been studied in one treatment trial for RP168 and will undoubtedly play a role in future studies.
Classification
Subdivision by inheritance type
The most useful subclassification of RP both in clinical and research setting is still subdivision on the basis of mode of inheritance. Typical RP can be inherited as an arRP, adRP, or X-linked recessive trait (X-linked RP). Although mitochondrial inheritance is associated with pigmentary retinopathy, typical nonsyndromic RP has yet to be reported with this type of inheritance. The percentage of each inheritance type has been found to vary from author to author and with country of origin of the study. Despite the fact that typical RP is always genetic, a lack of family history of retinal disease is often reported. Studies around the world have found that no other affected family member can be identified in 15–63% of cases. Such cases are labeled “simplex RP.” Averaging results from different studies suggests that 35% of RP patients in the USA, 42% in the UK, and 48% in China are simplex. It is assumed that large proportions of these cases represent recessive inheritance. Jay169 has estimated that no more than 70% of simplex cases are autosomal recessive.
Subdivision by distribution of retinal involvement or fundus appearance
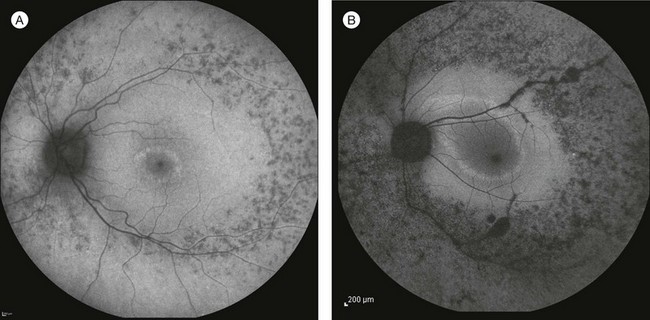
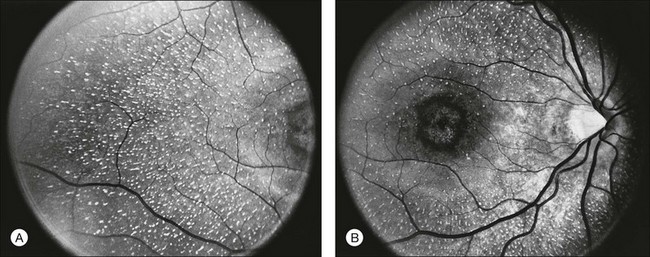
A number of recognizable fundus appearances have been seen in certain cases of RP. RP sine pigmento – RP without signs of intraretinal pigmentation – is one. In almost all cases these represent early RP. In the early stages of RP, fine, whitish, punctate lesions in the mid and far periphery at the level of the RPE can be seen. This fundus appearance is similar to that seen in retinitis punctata albescens.170 White lesions or dots deep in the retina can be seen with RP in younger members of families with older affected members who have typical findings of RP. A myriad of tiny, irregularly shaped, gray-white, deep retinal lesions (Fig. 40.31) associated with lifelong stationary night blindness, minimally attenuated retinal vessels, and absence of pigment clumps or bone spicules are characteristic of fundus albipunctatus. Bilateral central scotomas can be present in later years. History of slow progressive visual loss with the macular atrophic degeneration is suggestive of the fleck retinal degeneration known as retinitis punctata albescens.171 However, differentiation of fundus albipunctatus from retinitis punctata albescens can be difficult. Macular atrophic lesions have been reported by Miyake and colleagues to occur also in fundus albipunctatus.172–174
Sector and sectoral retinitis pigmentosa
Sector RP, first described by Bietti175 in 1937, refers to a specific subtype of RP. This is characterized by pigmentary changes limited to one or two quadrants, visual field defects usually only in the regions of retinal pigmentation, relatively good ERG responses, and minimal or no extension of the retinal area involved with time.
Patients may be minimally symptomatic. The area of retinal involvement is usually an arcuate swathe of retina just below the macula. In later years this involved region of the fundus may show almost a total regional atrophy of choroid and retina.176 Occasionally the nasal retina177 or inferior and nasal retina is involved.176 Rarely, sector RP has been reported as affecting the temporal or superior quadrants.178 True sector RP can be either autosomal dominant or autosomal recessive.176 Although sector RP has been reported with mutations of the rhodopsin gene179,180 and with mutations of USH1C,181 sporadic or isolated cases of sector retinal degeneration are common and may possibly result from nongenetic causes.
Massof and Finkelstein79 have shown in autosomal dominant sector RP that the absolute retinal thresholds are elevated throughout the retina, including the fovea. Rods and cones appear to be equally affected. Over a period of years, the visual field defects worsen. Overall, however, visual prognosis is good. Using a combination of testing modalities, Fleckenstein et al.151 studied the FAF associated with various forms of retinal dystrophy, including one case with sector RP. Microperimetry disclosed that the ring of hyperfluorescence sharply delineated the areas of severe impairment of sensitivity. In cases of true sector RP, the ERG demonstrates relative preservation of amplitudes, with mild to moderate subnormalities of both rod- and cone-mediated responses with normal implicit times.177 One form of sector RP appears to be associated with angle closure glaucoma.74
Most cases of RP that begin or present in a sectoral distribution are, in fact, merely the sectoral presentation of what will become with time a more diffuse disease. One notable example of this is the brother of the patient shown in Figures 40.3 and 40.15A, who presented with a strikingly sectoral phenotype in association with rhodopsin Pro23His RP. Although he had poor night vision from age 17, he began to notice areas of blindness in his upper visual fields at age 30 years. The diagnosis of RP was made at age 42 years. At age 50, his best-corrected visual acuity was 20/20 J1 in each eye. His visual fields (Fig. 40.32) showed dense superior loss but good preservation of inferior field. Fundus appearance (Fig. 40.33) showed inferior and nasal pigmentary changes in the right eye and well-demarcated inferior sectoral changes in the left eye. The Ganzfeld ERG at age 51 showed small but measurable rod responses, markedly subnormal scotopic bright flash responses, and modestly subnormal photopic cone responses; rod and cone implicit times were normal. The 45-minute dark-adapted rod psychophysical threshold was elevated about 0.5 log unit above normal in each eye. His acuity remains normal, and he was able to drive a car both during the day and at night until his mid-60s. This patient illustrates the phenotypic variability that can be evident with the age of onset of symptoms, varying from early adulthood to the fifth decade of life. The normal implicit times for rod and cone responses for his case are unusual but are consistent with the sectoral nature of the expression of his disease.
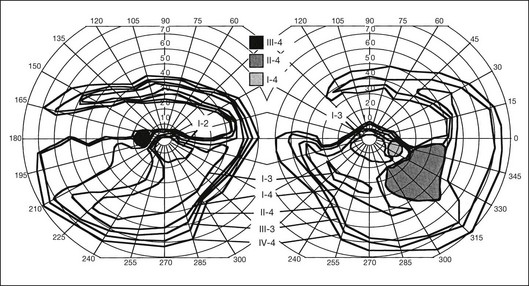
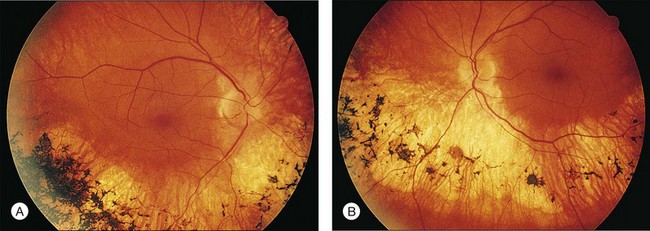
Fig. 40.33 Fundus appearance of the right (A) and left (B) eyes of the 51-year-old patient with autosomal dominant sectoral retinitis pigmentosa from the Pro23His mutation of rhodopsin. Note the discrete border between affected and normal-appearing retina and how well the fundus appearance correlates with the visual field in Figure 40.32.
Pericentral retinitis pigmentosa
Pericentral RP is a special phenotype whereby the loss of visual field typically occurs between 5 and 15° (Fig. 40.34) from fixation rather than between 20 and 40° from fixation, as is seen more commonly. Retinal diseases may commence inferiorly (Fig. 40.35), with commensurate superior field loss leading to the misconclusion that this is a form of sector RP. Pericentral RP is an important subtype because, as the areas of depressed field deepen, coalesce, and enlarge, they encroach more on the central region of seeing field and, thus, create greater disability at an earlier stage of disease.182 Eventually, as the central region of retina becomes progressively smaller or if the macula develops cystoid edema or atrophic changes, the visual acuity can decrease rapidly from relatively good acuity to less than 20/400. Pericentral RP can occur in many genetic forms of RP and can even be seen in relatives whose other affected family members have a different pattern or a more limited form of disease (Fig. 40.36), suggesting that modifying genes, other ocular conditions (e.g., high myopia), or environmental factors may contribute to this phenotype. Pericentral RP can occur as an autosomal recessive or dominant trait. Selmer et al.183 reported a Norwegian family with autosomal dominant pericentral retinal dystrophy associated with a novel mutation of the gene TOPORS.
Unilateral or extremely asymmetrical retinitis pigmentosa
Extremely asymmetrical RP of genetic etiology can occur in two instances. One is the carrier state for X-linked RP. Lyonization, or X-chromosomal inactivation,40 occurs close in time to lateralization during embryogenesis. Thus, if the number of cells undergoing inactivation of the X-chromosomes that contain the normal gene for RP is uneven at the time of lateralization and, by chance occurrence, a greater number of those cells are directed to one side of the developing embryo, the carrier will express an extremely asymmetrical phenotype with asymmetrical field loss (Fig. 40.37) and pigmentary changes (Fig. 40.38). The second mechanism by which unilateral RP can occur as a genetic trait is through somatic mosaicism of a dominant gene for RP. This mechanism has been reported as the cause of unilateral RP in a patient with somatic mosaicism of RP1.184
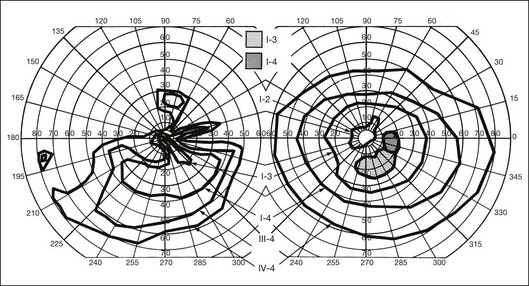
Fig. 40.37 Goldmann kinetic perimetry for the 64-year-old carrier of X-linked retinitis pigmentosa, whose fundus appearance is shown in Figure 40.38. She had mild, lifelong difficulty with night vision that was stable. The vision in her left eye began to fail as a young adult. At 64 years of age, the visual acuity was 20/25 OD and 20/60 OS, with −5.00 +1.00 axis 120° OD and −9.75 +0.75 axis 105° OS. Color vision was normal on the right and markedly disturbed on the left (AOHRR plates). Note the inferior arcuate defect in the right eye and severe irregular superior field loss in the left eye. The electroretinogram was moderately subnormal (OD) and severely abnormal (OS).
Complicated retinitis pigmentosa
Systemic associations
Most cases of RP are not associated with manifestations outside the eye. Patients with RP have been reported to have a greater than normal risk for thyroid disease.185 Of 670 respondents to a questionnaire mailed to patients with RP, 7.3% reported mild and 1.3% reported severe thyroid conditions.186,187 Several studies have reported immunologic abnormalities for patients with RP.188 We suspect, however, that many of the psychiatric and other systemic associations with RP are either coincidental or epiphenomena.
However, approximately 30% of all patients with RP might experience mild to severe hearing loss as adults.189 It is unknown what proportion of these are in fact cases of Usher syndrome and what proportion is coincidental or a feature of other genes associated with RP. Zito et al.190 reported RP with a sensorineural and conductive hearing loss, recurrent ear infections, sinusitis, and chronic recurrent respiratory tract infections in a family with a 845–846delTG mutation of RPGR. This and other reports of similar systemic features in X-linked RP from mutation of RPGR may be related to the ubiquitous expression of elsewhere in the body. Kenna et al.191 reported a large nonconsanguineous Irish family with a progressive pigmentary retinopathy (RP), deafness beginning in the third decade of life, and a subclinical myopathy, identified by skeletal muscle biopsy, electromyography, and electrocardiography. Mansergh et al.192 identified a C12258A mutation in the mitochondrial gene MTTS2 in this family; RetNet (http://www.sph.uth.tmc.edu/Retnet/home.htm) lists this mutation as affecting the serine tRNA 2 (AGU/C), nt 12207–12265.
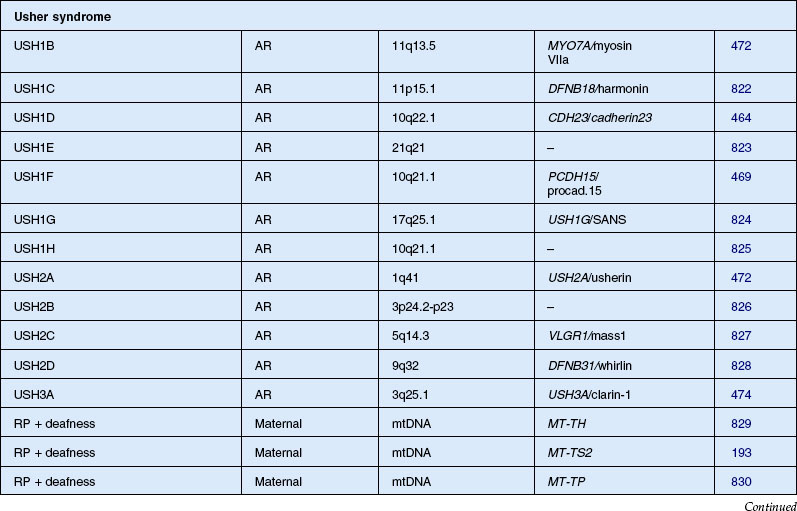
Usher syndrome
Usher syndrome is defined as autosomal recessive deafness (most commonly congenital) with retinopathy indistinguishable from typical RP. Although first described by von Graefe in 1858, credit is given to the British ophthalmologist Charles Usher for the appreciation that this condition was familial and represented a distinct entity.193 He also recognized the existence of at least two types on the basis of severity of hearing loss, age of onset, and rate of vision loss. Usher syndrome is the most common of the syndromes associated with RP and accounts for about 18% of all patients with RP.194 Although the prevalence has been estimated at between 1.8 and 6.2 cases per 100 000,195,196 a more recent study found the population prevalence to be 1 : 6000.6 Usher syndrome accounts for 50% of those persons in the USA who are both deaf and blind.196,197 The Usher Consortium has recommended specific clinical criteria for the diagnosis of Usher syndrome.198
Usher syndrome can be divided clinically into three major groups. The two most frequent forms are type 1, with profound congenital sensorineural deafness and resultant prelingual deafness or severe speech impairment, vestibular symptoms, and childhood-onset retinopathy, and type 2, with congenital partial, nonprogressive deafness, absence of vestibular symptoms, and milder, later-onset retinopathy.199 The least common is type 3 Usher syndrome, which is characterized by progressive deafness starting late in the second to fourth decades, adult-onset retinopathy, and hypermetropic astigmatism.200–202 Another variant, Hallgren syndrome, was defined as congenital progressive deafness, vestibular ataxia, and retinopathy.203 The validity of this as an entity distinct from Usher type 1 has been questioned.199 Approximately 3–6% of those persons with profound prelingual deafness have type 1 Usher syndrome.197 Piazza et al.,204 in a study of 106 patients with Usher syndrome, found that 33% had type 1 and 67% had type 2 disease. No type 3 cases were identified. Most cases of type 3 disease are of Finnish descent.205 As many as 40% of Usher syndrome cases are classified as type 3 in Finnish202 and Ashkenazi Jewish206 populations.
One of the earliest signs of type 1 Usher syndrome is vestibular dysfunction, which in infancy can manifest as a delay in motor development in children and in adulthood as a nonprogressive ataxia.199 Occasional ataxia is present in type 2 disease and has been attributed to cerebellar atrophy.199 Because of vestibular dysfunction, almost all children with type 1 Usher syndrome fail to walk before the age of 18 months.207 Patients with type 1 Usher syndrome almost invariably report onset of nyctalopia later, by age 15 years, whereas type 2 and type 3 patients report onset of nyctalopia over a greater range, up to the early 30s.199,208 Visual acuity appears to be better retained in older patients with type 2 Usher syndrome, as compared to type 1.204 The cumulative percentage of patients retaining 20/40 visual acuity or better at age 29 was 69% for type 1 and 94% for type 2. For retention of 20/80 visual acuity at age 29, the figures were 89% for type 1 and 98% for type 2. Approximately 77% of type 1 patients and 95% of type 2 patients maintained 20/200 visual acuity at age 40. Foveal lesions were seen on fluorescein angiogram more frequently and at an earlier age in type 1 (14 of 35 with a mean age of 34.4 years) as compared with type 2 (19 of 71 with a mean age of 42.9 years). No difference in the prevalence of posterior subcapsular cataracts was noted between the two types in this study (roughly 50% in each).204 No comparable figures are available for type 3 disease. The ERG is often profoundly abnormal to undetectable by nonaveraging techniques in all types.
The dual impairments of deafness and severe visual impairment in all Usher syndrome patients necessitate a great deal of educational and sociopsychologic interventions to help them to maintain independence and productivity.209,210
Cochlear implantation in profoundly deaf children has been successful in allowing these children to enter the world of the hearing. In order that the parents understand the importance of cochlear implantation, we believe that all infants with profound congenital deafness should be screened for Usher syndrome. Usher syndrome can only be reliably diagnosed in infancy and early childhood through the ERG.207 Because receptive and expressive language is most closely correlated with the precocity of cochlear implantation, the diagnosis of Usher syndrome should be established as early as feasible to optimize speech therapy.207
Differential diagnosis – phenocopies of retinitis pigmentosa
Many other inherited retinal conditions may be confused with RP. These can be conditions confined to the retina or have associated systemic manifestations that may or may not be apparent at the time of examination. Misdiagnosis is common and causes the greatest problem in pediatric practice. Such phenocopies can usually be differentiated from RP on detailed retinal investigation and a thorough survey for systemic signs in the patient and relatives. This differentiation is important because it has a great bearing on the genetic and prognostic counseling given to the family. A great deal of anxiety and social problems can be avoided, for example, in families with BBS if problems of obesity are predicted for a child who might otherwise be labeled as having an eating disorder as a result of the psychologic stress of blindness. When confronted with inherited retinal disease, especially when associated with systemic manifestations, the reader is encouraged to consult the phenotype catalog of Online Mendelian Inheritance in Man (OMIM), which is available at http://www.ncbi.nlm.nih.gov/omim.
Cone–rod and cone dystrophy
CRD is characterized by early loss of visual acuity and color vision, with subsequent progressive peripheral visual field loss. There is little literature on the prevalence of CRD, although it has been suggested that the disease may be relatively common.211 Moreover, some patients otherwise labeled as having RP may have a greater involvement of cones than rods. One study of 278 RP patients with recordable ERGs and fields large enough for rod threshold measurements reported that 41% had a cone–rod-type ERG deficit.27 Macular pigmentation and atrophy precede variable degrees of peripheral pigmentary abnormality in CRD. In early disease, before peripheral field deficits or peripheral retinal abnormalities are apparent, a diagnosis of macular or cone dystrophy may be made.211 Peripheral retinal bone spicule pigmentation, in later disease, may resemble that seen in classic RP. Often the midperipheral retina is relatively spared or affected late in the evolution of disease.212,213
With such potential for diagnostic confusion, it has been suggested that a diagnosis of CRD should be made on the basis of marked reduction or absence of cone ERG responses in the presence of quantitatively less reduction in rod responses.214 However, this definition includes a range of phenotypes, and early attempts have subclassified CRD on the basis of fundus appearance215 or visual field deficits.216 Two studies have attempted to subclassify CRD on the basis of dark-adapted static threshold profiles217 or differences in ERG responses.218 Yagasaki and Jacobson217 tested 14 autosomal recessive and simplex CRD cases using full-field ERGs, dark adaptometry, and modified perimetric techniques. They suggested that a subclassification could be based on three distinct patterns of visual field loss. Type 1 case had central rod and cone functional loss, eccentric fixation, mild peripheral photoreceptor dysfunction, and slow progression. Type 2 was described as more severe, with a central scotoma, eccentric fixation, more cone than rod functional loss in the periphery, and relatively normal midperipheral fields. Subjects classified as type 3 had central fixation, no measurable cone function, and patchy rod function loss. Szlyk and coworkers218 studied 33 CRD patients, and reviewed the records of a further 150, and described four functionally distinct subtypes. Subjects were subdivided into type 1 (less rod than cone dysfunction) and type 2 (equal cone and rod dysfunction) on the basis of quantitative ERG responses. These groups were further subdivided into type a (cone thresholds more elevated centrally, rod thresholds more peripherally) or b (matching areas of cone and rod threshold elevation mostly peripherally) on the basis of pattern of field loss and threshold elevation.
There is a tendency for the visual field defects in CRD to begin in the pericentral region between 5 and 30° from fixation.216 Many nonretinal conditions have been documented in association with CRD in certain patients, including optic nerve head atrophy and telangiectasia,215,216 high myopia,219 and macular coloboma.220 Also, there have been associations with spinocerebellar ataxia,221 dental ameliogenesis imperfecta,222 alopecia,223 and hypertrichosis.224 In the vast majority of cases, however, CRD is identified as the only genetic disease.
Autosomal dominant, recessive, and X-linked inheritance patterns of inheritance have been described, as well as simplex cases implying genetic heterogeneity.225 Recent molecular genetic studies have identified 16 different genomic regions, each of which contains a CRD-causing gene (Table 40.1).
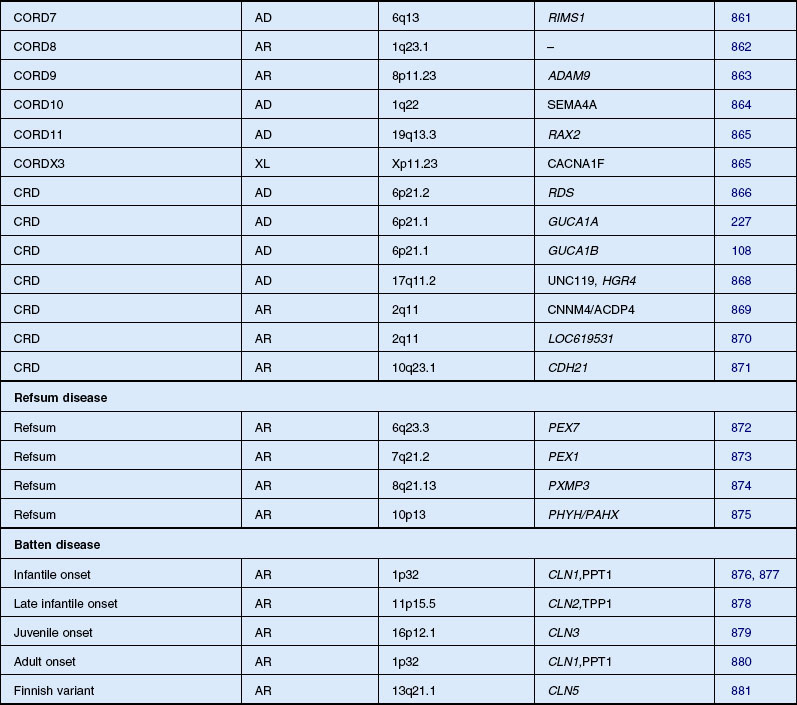
In the early stage of disease, an isolated cone dystrophy may be difficult to distinguish from a CRD. CRD can be associated with mutations in the gene GUCY2D for guanylate cyclase-activating protein-1 (GCAP-1)226 and cone dystrophy (COD3) has been associated with the functionally related gene GUCA1A encoding GCAP-1.227,228 GCAPs play an important role in regulating the function of RETGC-1 in a calcium-dependent manner, but the reason why defects of these proteins result in degeneration limited to cones is difficult to explain. One suggested explanation focuses on the interactions of the defect of GCAPs at different levels of calcium.228
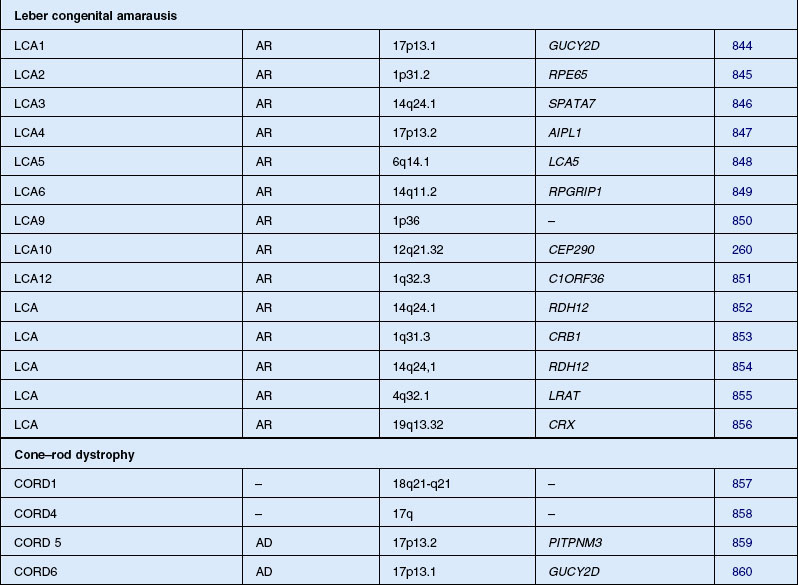
Leber congenital amaurosis/severe early childhood onset retinal dystrophy (SECORD)
Leber congenital amaurosis (LCA) is not a single entity but a group of disorders due to mutation of at least 16 different genes. The disorder is characterized by severe visual impairment or blindness from infancy. The vast majority of cases are autosomal recessive, although rare dominant families have been reported, all of which, when molecularly characterized, have been found to result from 1- and 2-bp deletions of CRX.229,230 Cases of LCA are often confused with early-onset RP, especially sporadic cases. In 1869, Theodor Leber originally described children with the condition as severely visually impaired before age 1 year, with nystagmus, poor pupillary reflexes, either normal or abnormal fundus appearance, and an autosomal recessive inheritance pattern.231 In 1954, Franceschetti and Dieterlé232 described a profoundly abnormal or absent ERG that has since become a requirement for the diagnosis of LCA. Eye-rubbing – the oculodigital sign – is a common association.233
Foxman and colleagues234 and Heckenlively and Foxman235 have subdivided LCA into two types. Uncomplicated LCA is described as congenital blindness, nystagmus, and high hyperopia with extinguished ERG responses.235–237 Complicated LCA is used to group together cases of LCA associated with other ocular or systemic features.235 Fulton et al.238 studied 36 patients with LCA and did not find that hyperopia predicted an uncomplicated course. More likely, it is the lifelong extreme defect of vision combined with the hyperopia normally present during early infancy that interferes with the emetropization process, leaving the child with high hyperopia.
In 1916, Leber wrote that in his experience the disorder that he originally described in infants (that now carries his name) merged into a continuum with children who presented later in early childhood, without the history of nystagmus or poor papillary responses from birth.239 These cases typically presented at age 4–5 and had extremely poor vision by 30 years of age. Several names have been used for children presenting this second phenotype, including juvenile and early-onset RP,234 childhood-onset severe retinal dystrophy,240 early-onset severe retinal dystrophy,241 and SECORD.242
Most patients with LCA or SECORD show either a normal fundus appearance or only subtle RPE granularity and mild vessel attenuation. Retinal abnormalities that have been described in LCA include so-called macular coloboma,243,244 “salt and pepper” retinopathy,245 retinitis punctata albescens,246 and nummular pigmentation.247 LCA is often associated with keratoconus.248 Elder249 evaluated 35 children with LCA and found a prevalence of keratoconus of 29%. The author suggested that keratoconus is not a consequence of the eye-rubbing frequently seen in LCA but may be due to other genetic factors.249
Systemic associations are commonly made. Several reports describe the association of LCA with deafness,250 renal anomalies,251 infantile cardiomyopathy (this case was later reclassified by Russell-Eggitt et al.252 as Alström syndrome), hepatic dysfunction,253 and skeletal abnormalities.247,253 Neurologic abnormalities are the most common association and have been reported in 17–37% of LCA cases.245,254 Nickel and Hoyt255 have suggested that mental retardation is secondary to the visual impairment, although we believe this is unlikely to account for the severe intellectual impairment that can occur.256 More recent studies suggest that up to 20% of LCA children without associated anomalies will develop mental retardation.257,258 It is unknown whether these cases represent undiagnosed systemic disorders or a genetic subtype or subtypes of LCA. To date, none of the molecularly defined types of LCA are associated with mental retardation or neurodevelopmental degeneration. Walia et al.259 studied 169 patients with LCA and 27 patients with early childhood-onset RP and found that the visual acuity varied widely among those with mutations of RPE65, RDH12, and CRB1 whereas AIPL1, CUCY2D, CRX, and RPGRIP gene mutations were associated with severely decreased visual acuity commencing within the first year of life. Patients with either RPE65 or CRB1 mutations had progressive vision loss with age; those with onset of symptoms after infancy had better visual acuity. The gene CEP290 is not only a frequent cause of LCA260 but also is associated with the Joubert spectrum of diseases and with nephronophisis.
Unfortunately, unless other diagnoses are considered and the correct tests are performed, several other entities can be confused with LCA,261 including IRD,262,263 congenital stationary night blindness,264 early infantile neuronal ceroid lipofuscinosis (INCL; Hagberg–Santavuori syndrome),265,266 and any of several renal–retinal syndromes (Loken–Senior syndrome and Saldino–Mainzer syndrome).251,267–269 There is the suggestion that the original report by Leber may have included cases of INCL.270 Although, in the past, LCA has been considered synonymous with infantile blindness and a “flat ERG,” LCA should be thought of as a clinical/electrophysiologic sign rather than a distinct pathologic entity. In the context of systemic anomalies, such misdiagnosis can be avoided if LCA is considered a diagnosis of exclusion.
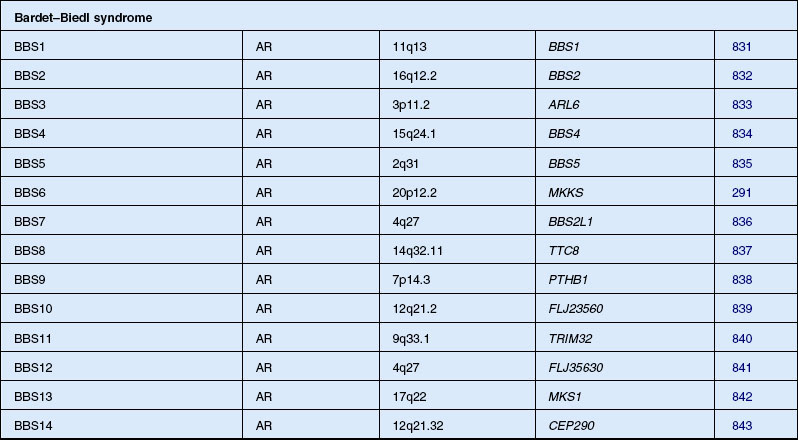
Bardet–Biedl syndrome
Bardet271 in 1920 described a patient with retinopathy, polydactyly, and congenital obesity. Biedl272 in 1922 added the fourth and fifth cardinal features, mental retardation and hypogenitalism, of the disorder now known as BBS. A similar syndrome to BBS had been described by Laurence and Moon273 in 1866, and Hutchinson274 in 1900. As well as retinopathy and mental retardation, they described paraplegia as a prominent feature without polydactyly or obesity. Some authors classified all these cases together under the term “Laurence–Moon–Bardet–Biedl syndrome” until Ammann275 in 1970 reasserted the separation of the two syndromes. The controversy continued, however, with some authors still recommending the combined term.276,277 Most ophthalmologists today consider the features of Laurence–Moon within the spectrum of BBS. Beales et al. refined the diagnostic criteria in 1999 and proposed that the phenotype in the patients be renamed “polydactyly–obesity–kidney–eye syndrome.”278 The prevalence has been placed at 1 in 160 000 in Switzerland.275 Farag and Teebi279 have found that BBS is more prevalent among the consanguineous Arab population of Kuwait and among the Bedouin, where the estimated minimum prevalence was 1 in 13 500.280 Because of a founder effect, the prevalence of BBS in Newfoundland is approximately 1 in 17 500.281
Importantly, the retinopathy in BBS differs from typical RP in that visual acuity fails early in the course of the disease and usually the fundus shows little pigmentary dispersion until later stages. Macular lesions and atrophy of the RPE or choriocapillaris often develop early and prominently as the disease progresses.282 These macular abnormalities may include macular wrinkling, preretinal membrane formation, and leakage on fluorescein angiogram from paramacular capillaries. When detectable, the ERG may show a rod–cone loss or, in many cases, even within the same family, a cone–rod loss pattern, the latter of which has led authors to call the retinal dystrophy in BBS a cone–rod retinal degeneration.214,283,284 The absence of pigmentary deposits has also led to the retinopathy in BBS being called RP sine pigmento, or, when patchy whitish RPE lesions are evident, retinitis punctata albescens.282 The onset of night blindness is recognized by a mean age of 8.5 years and legal blindness by a mean age of 15.5 years.194 Approximately 73% of patients reach legal blindness status by age 20 years and 86% by the age of 30 years.282
Incomplete manifestation of the five cardinal features is the rule rather than the exception in BBS. Prosperi et al.285 estimated from previous reports that 40–45% of cases are incomplete. Another study of 102 cases showed only 24 with the complete syndrome.286 Schachat and Maumenee283 reviewed BBS and related disorders and suggested that at least four of the five cardinal features, of which retinopathy must be one, must be present to establish the diagnosis conclusively. Pigmentary retinopathy is reported in 90–100%281,283 of cases, with ERG responses being abnormal in all cases. Most agree that mental retardation, which has been reported in 85–87% of cases,283 is not an essential feature of this syndrome. Green et al.281 found only 13 of 32 patients had mental retardation. When encountered, mental retardation is mild in slightly over 50% of cases.282 Indeed, intelligence has been reported to be above normal in some patients. Although the tendency for obesity seems nearly universal, some patients have been able to control their weight by dieting and exercise. Polydactyly is present in 75% of cases, is postaxial, and may involve any or all extremities.283 Syndactyly or brachydactyly is present in 14.4% of patients.282 Both have been considered as equivalents of polydactyly with reference to determining the number of cardinal features present.281,282,287 Hypogenitalism is present in roughly half of patients over the age of 15. Infertility is particularly prominent in male BBS patients,284 although in our experience rare patients do remain fertile and father children. Vaginal atresia,288 urogenital sinuses, uterine and ovarian hypoplasia,288 and congenital hydrometrocolpos289 have been described in female BBS patients. Many of these patients, however, have also met the cardinal diagnostic features (hydrometrocolpos and polydactyly) for McKusick–Kaufman syndrome, which is now recognized as having significant phenotypic overlap with the much more frequent disorder BBS.290–292
Of nonocular abnormalities not considered cardinal features of BBS, renal abnormalities are the most common, having been reported in 19 of 21 autopsied patients.293 Hurley et al.294 have observed radiologic abnormalities of the renal parenchyma or collecting system in 11 of 11 patients. Since renal disease can be severe enough to lead to uremia and death, Churchill et al.276 have argued for renal disease to be considered the sixth cardinal feature of BBS. Pagon et al.295 reported a BBS child with renal failure and hepatic fibrosis. Cardiac abnormalities were identified on ultrasound in 50% of patients from Bedouin families.296 Deafness is uncommonly associated with BBS, occurring in approximately 5% of patients.283 Occasional cases with Hirschsprung disease have been described.297 Croft and Swift298 suggested that even heterozygotes have an increased frequency of obesity, hypertension, diabetes mellitus, and renal disease.
Only subtle phenotypic differences between the different linkage types have been reported, the most striking of which was the finding of taller affected offspring compared with their parents in BBS1 families. Affected subjects in the BBS2 and BBS4 groups were significantly shorter than their parents. Carmi et al.299 have shown that polydactyly of all four limbs seems associated with BBS3, whereas it is confined to the hands in BBS4. Early-onset obesity seems common in families with BBS4, whereas obesity is not associated with BBS2.299 No confirmation of these correlations between phenotype and linkage loci has been reported with limited studies of molecularly confirmed genetic types of BBS.
Refsum syndromes
Infantile Refsum disease
IRD was first reported in 1982 by Scotto et al.300 in infants presenting with craniofacial malformation, severe hypotonia, psychomotor retardation, bleeding episodes, liver dysfunction by 6 months of age, and severe deafness within the first year of life. The ophthalmic manifestations were reported in 1984262 to include nystagmus, poor vision, retinal degeneration with white flecks evident in the midperipheral retina (leopard spots) that fade and are replaced by coarse pigment clumping, optic atrophy, and eventually cataract. The ERG is profoundly abnormal early in the disease and can be electronegative.262,263 Abnormalities of general peroxisomal biogenesis and function, similar to that seen with Zellweger syndrome and neonatal adrenoleukodystrophy, were identified in IRD and it is now classified as a milder variant of Zellweger syndrome (reviewed by Pennesi and Weleber263). The disease is often fatal by the second or third decade of life.262,263
Adult-onset Refsum disease
This autosomal recessive disease, also called heredopathia atactica polyneuritiformis, was first described in 1946301 with the ophthalmic features reviewed by Refsum in 1977.302 This disease has been recently reviewed by Pennesi and Weleber.263 The earliest symptoms in adult-onset Refsum syndrome are ataxia, weakness in the extremities, and nyctalopia presenting in later childhood. Progressive peripheral neuropathy and peripheral muscle wasting usually follow this and cardiac conduction defects occur in early adulthood. Other common findings include paresthesia, anosmia, deafness, dry skin and ichthyosis, epiphyseal dysplasia, spondylitis, and kyphoscoliosis.
Phytanic acid levels in blood and urine are always very high due to a deficiency of phytanic acid oxidation. Protein levels in the cerebrospinal fluid are also characteristically elevated. Rather than a deficit of peroxisome biogenesis, as is seen in IRD, a specific peroxisomal enzyme, phytanoyl-coenzyme A hydroxylase, is deficient and inactivating mutations of the gene PAHX, also called PHYH, as well as mutations of the PTS2 receptor have been identified in Refsum syndrome patients.303,304
Restriction of dietary phytanic acid (which is present in dairy products and ruminant fat) can limit progression of disease and often improve the ichthyosis, neurologic deficits, and cardiac disease. There is limitation without improvement of visual or auditory deficits.305,306
Neuronal ceroid lipofuscinosis (Batten’s disease)
The NCLs are a group of progressive neurodegenerative disorders characterized by accumulation of complex storage material within lysosomes. An extensive review by Mole and Williams is available at the website for GeneReview.307 This class of conditions is the commonest neurodegenerative disorder to affect children, with a collective incidence worldwide of about 1 : 12 500 live births.308 Characteristically, severe psychomotor deterioration eventually leads to a vegetative state, seizures, visual failure from retinal degeneration, and premature death.309 Four classic forms exist: three childhood-onset forms, which are all autosomal recessive, and one adult-onset form, which may be autosomal recessive or dominant:
1. An infantile-onset form (INCL, CLN1), also called Haltia–Santavuori disease, Hagberg–Santavuori disease, or simply the Finnish form. This usually manifests at 8–24 months of age with severe psychomotor retardation, blindness, and microcephaly.310
2. A late infantile-onset form (LINCL, CLN2), also called Jansky–Bielschowsky disease. This condition manifests at 2–4 years of age with ataxia, loss of speech, regression of development milestones, seizures, and later loss of vision.311–313
3. A juvenile-onset form (JNCL, CLN3), also called Batten–Mayou syndrome, Spielmeyer–Vogt disease, or Spielmeyer–Sjögren syndrome, which manifests at 4–8 years of age with visual acuity loss that progresses to loss of virtually all useful vision over a year or two.309,314,315 The fundus appearance on presentation includes attenuated retinal vessles, a bull’s-eye maculopathy, and fine pigmentary changes (Fig. 40.39).
4. The adult-onset disorder (ANCL, CLN4), also called Kufs disease,316 usually manifests as a motor disturbance without visual symptoms or findings. Although Kufs disease is believed to be an autosomal recessive trait, autosomal dominant inheritance has been described.317
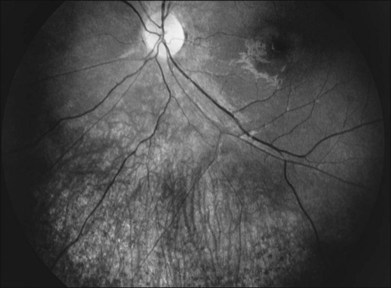
In addition, as many as 15 atypical forms have been described, some of which may be allelic to certain of the classical forms.318 One of the variant forms (vLINCL, CLN5) occurs essentially only in the Finnish population and shows linkage to a site (13q22) distinct from the three classic forms of childhood NCL.319 In Europe, the term “Batten disease” is often used collectively for all forms of NCL.320,321
INCL has an incidence of 1 in 13 000–20 000 in Finland, 1 in 50 000 in Scandinavia, and 1 in 100 000 worldwide.309,322 LINCL has a frequency of 0.46 per 100 000 in Germany.323 JNCL has an incidence of 1 in 21 000 in Finland and a frequency of 0.71 per 100 000 in Germany.323
The visual failure in the three classic childhood forms (numbers 1–3 above) involves central vision first and eventually results in profound visual loss, often with complete blindness, within a few years. The ERG becomes abnormal early in the course of all these disorders and within a few years is usually totally abolished to standard single-flash recording techniques. Goebel309 states that the ERG becomes flat (undetectable to standard techniques) for LINCL between ages 3 and 4 years and for JNCL between 5 and 7 years of age. Visual symptoms and abnormalities on electrophysiologic testing are rare and even then occur late in the course of Kufs disease.316
Recent studies have examined the ERG in patients with INCL, LINCL, and JNCL.265,266 The ERG is abnormal early in the course of all three disease types. For patients with INCL, rod responses were severely subnormal; the scotopic ERG to the 0.6 log cd-s/m2 stimulus was normal for the a-wave and profoundly subnormal for the b-wave, indicating that the earliest manifestation of this disease appears not to affect phototransduction directly. Instead, this result was interpreted as an effect on neurotransmission from proximal photoreceptors to ON bipolar cells. This appeared to occur from one of three possible sites: (1) a disturbance of proximal photoreceptor function that interfered with presynaptic neurotransmission; (2) a disturbance of the postsynaptic plate region; or (3) some other effect on the bipolar cells, with subsequent reduction of the generation of the b-wave. The ERGs of three patients with LINCL were different in showing nearly normal rod amplitudes, mildly prolonged rod implicit times, and severely subnormal, prolonged cone responses. Patients with more advanced stages of LINCL had a greater deficit of the b-wave than a-wave, suggesting development of loss of effective transmission of the signal from photoreceptor inner segments to bipolar cells. Unlike the ERG in either INCL or JNCL, the rod responses in early LINCL were only mildly subnormal and prolonged but with much more preserved amplitude, even though cone responses were severely subnormal and prolonged. Three patients with JNCL had a third ERG phenotype (Fig. 40.40) with essentially no discernible rod responses and severely subnormal cone responses with normal to prolonged implicit times. The maximum scotopic a-waves in all three JNCL cases were subnormal, indicating loss of effective outer segments. The b-wave responses, however, were even more subnormal, creating an electronegative configuration. Oscillatory potentials were profoundly subnormal. The greater disturbance of the b-wave than that of the a-wave for patients with JNCL is consistent with the inner retinal localization of the CLN3 gene product, which appears localized to mitochondria of Müller cells, inner retinal neurons, and the inner segments (but not the outer segments) of photoreceptors.324
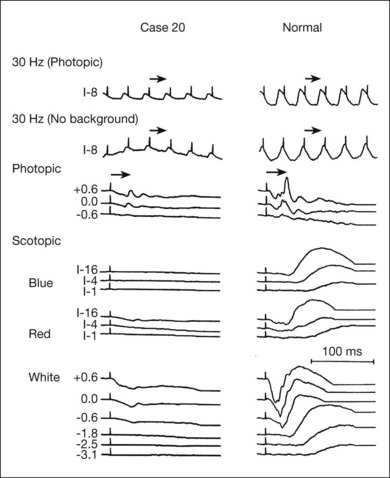
All forms of NCL show accumulation of storage material that is autofluorescent, sudanophilic, and periodic acid–Schiff-positive within lysosomes in neurons and other cells. Because of its osmophilic nature and appearance on light microscopy, the storage material resembles ceroid and lipofuscin, but actually it is a complex mixture of lipoproteins and other hydrophobic peptides. The lipoprotein deposits within cells on electron microscopy take on characteristic patterns that are used for diagnosis and classification. Granular inclusions are seen in INCL, Kufs disease, and some atypical forms of JNCL. Curvilinear inclusions predominate in LINCL (with occasional to rare fingerprint inclusions). Fingerprint inclusions are seen in JNCL (with occasional to rare curvilinear inclusions).325,326
Historically the diagnosis of this group of disorders has been established by looking for inclusion bodies in cells from brain biopsy or full-thickness rectal biopsy.327,328 Skin or bulbar conjunctival biopsies typically show fingerprint inclusions (Fig. 40.41), which have been valuable in establishing the diagnosis.324,325 Buffy-coat leukocytes can be used but may include a wider range of inclusions that may represent other storage disorders, such as the mucopolysaccharidoses.329,330 Fingerprint and tubular inclusions on electron microscopy of lymphocytes have been seen in children with LINCL and JNCL. These can also be seen in carriers of these conditions.331,332 Rapola et al.328 were unable to determine reliably the presence or absence of electron microscopic inclusions in lymphocytes from patients with INCL. Muscle biopsy may represent the best approach to ultrastructural diagnosis for ANCL.333 However, since skeletal muscle fibers show only the curvilinear type of inclusions on electron microscopy, one cannot use muscle biopsy to differentiate the specific types of NCL.334 Brain biopsies are no longer justifiable for establishment of the diagnosis of NCL.
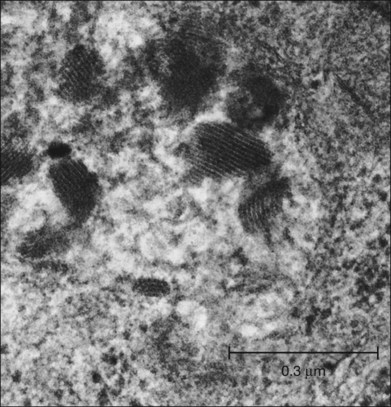
Differential diagnosis: pseudoretinitis pigmentosa
Retinal inflammatory diseases
Rubella retinopathy
The most common ocular manifestation of congenital rubella, rubella retinopathy,335 is occasionally confused with RP. This confusion is especially likely in children with congenital deafness and a pigmentary retinopathy that are erroneously thought to represent both congenital rubella retinopathy and deafness rather than Usher syndrome. Uncommonly, the converse occurs, and the pigmentary disturbance of rubella retinopathy in a child with deafness triggers the suspicion or even the presumptive diagnosis of RP. Rubella retinopathy is one of the most characteristic manifestations of congenital rubella.336 Rubella retinopathy may take any of several forms. In some, the macula is the only site of abnormality, with a speckling of fine pigment granules. In others, pigmentary changes can extend further into the peripheral retina. Usually the correct diagnosis can be established by a combination of clinical features and ERG, which is only mildly abnormal in rubella retinopathy but will be, almost invariably, severely abnormal in Usher syndrome. Rubella retinopathy is a disease that is capable of mild progressive increase in pigmentary changes337 or the development of clinically significant subretinal neovascularization.338
Syphilis
Congenital or acquired syphilis can present as a pigmentary retinopathy that may appear similar in some respects to advanced RP,339 but careful examination usually establishes the correct diagnosis. Interstitial keratitis is commonly seen in congenital syphilis, and the pigmentary retinal changes are more varied and patchy. Usually the pigment deposits are clumps or large patches of black pigment associated with chorioretinal scars; typical bone spicule pigment formations are uncommon. The posterior pole may be as involved as the periphery. There may be evidence of past or present overlying vitreous reaction or uveitis. Acquired syphilis can also present with a diffuse retinal involvement.339
Autoimmune paraneoplastic retinopathy
Several reports have documented the existence of severe panretinal degeneration as a remote effect of cancer, usually either small-cell (oat-cell) carcinoma of the lung340,341 or small-cell undifferentiated cervical carcinoma.342 In several cases the vision loss preceded the diagnosis of the cancer.271 Cancer-associated retinopathy (CAR) can progress rapidly or slowly with loss of vision over a period of months or years. ERG responses are usually severely abnormal. The fundus appearance may be normal even when the ERG is extinguished and the vision severely decreased. The first autoantigen in the human retina identified with a CAR is recoverin.343,344 Antibodies to retinal enolase have also been reported in CAR.345,346
A paraneoplastic form of night blindness associated with “shimmering lights” and a characteristic ERG has been reported with malignant melanoma and has been called melanoma-associated retinopathy (MAR).347,348 The scotopic ERG to maximum-intensity flash has a “negative” configuration, identical to that seen with congenital stationary night blindness. The a-wave amplitude is normal, and the b-wave amplitude is severely subnormal, distinguishing this form of paraneoplastic retinopathy from CAR. Alexander et al.349 found a defect in the ON response of the ERG, reflecting abnormal rod neurotransmission to depolarizing bipolar cells.
It has been suggested that in MAR there is a selective loss of function subserved by magnocellular cells with preservation of midget type 1 parvocellular cell function.350 We have seen a patient with acquired night blindness and a negative-configuration ERG typical for congenital stationary night blindness associated with an anaplastic squamous cell carcinoma of the nasopharynx (WT Shults and RG Weleber, unpublished observations, 1991). Other acquired cases of visual symptoms, field loss, and electronegative-configuration ERGs have been reported in the absence of either melanoma or other discernible cancer but with serum antiretinal antibodies that specifically labeled the inner plexiform layer of cadaver retina by indirect immunoperoxidase testing.351 Presumably, these patients have produced an antibody to an antigen that is similar (by molecular mimicry) to one found in normal retina, producing the disturbance of retinal function and electrophysiology.
Drug toxicity (see Chapter 89, Drug toxicity)
Thioridazine
This phenothiazine has been linked to severe, blinding retinal toxicity. In general, phenothiazines bind to melanin and concentrate in the uveal tract and RPE.352,353 The retinal toxicity of thioridazine is believed to result from the presence of a piperidyl group. Thioridazine can cause a pigmentary retinopathy that can be confused with RP (in its early stages) or choroideremia (in its later stages).354 In most patients with thioridazine retinopathy the drug has been taken at doses higher than 800 mg/day.355
The earliest symptoms include blurring of central vision, poor night vision, and a brownish discoloration to the vision. Early toxicity produces fine, deep retinal pigment posterior to the equator, which becomes coarser as the condition progresses. Large “signal” plaques of pigment deposition occur late. The characteristic appearance of advanced thioridazine retinopathy is diffuse, patchy atrophy of choriocapillaris, RPE, and overlying retina, simulating the fundus appearance of early choroideremia.356 The ERG may show decreased amplitudes of both cone- and rod-mediated responses; responses of both a-waves and b-waves are subnormal.
Chlorpromazine
This drug has been reported to produce a pigmentary retinopathy when taken at high doses for prolonged periods.357 The pigmentary retinopathy does not significantly affect retinal function, and the retinopathy appears to regress when the drug is stopped.
Chloroquine
Similar to the phenothiazines, chloroquine, if taken over a prolonged period, binds to melanin and causes a toxic retinal degeneration. Very few cases have been reported with patients taking a total dose of less than 300 g.358 Early in the course of toxicity the ERG, EOG, and visual fields can be normal, although in late, severe toxicity, they can become markedly abnormal.359,360 In advanced chloroquine retinopathy, unlike RP, the dark adaptometry may be either normal or only minimally abnormal.359 The typical picture of advanced chloroquine toxicity is that of a bull’s-eye maculopathy, although pigmentary changes with bone spicule formations can occur in the mid and far periphery.279,360
Hydroxychloroquine
This drug has mostly supplanted chloroquine usage for rheumatoid arthritis and systemic lupus erythematosus. However, toxicity can occur with chronic usage and the current recommendation is periodic ophthalmological examination with static permetric visual fields (Humphrey field analyzer 10–2 or 24–2) when the duration of usage is over 5 years, particularly if the dosage is greater than 6.5 mg/kg per day.361 The multifocal ERG appears to be a useful diagnostic tool to help differentiate pericentral visual field loss due to hydroxychloroquine from that due to optic nerve or other causes.362
Quinine
Retinopathy due to oral ingestion of quinine is usually a consequence of large doses taken either during a suicide attempt or to abort a pregnancy. Often, the diagnosis is aided by the history of acute loss of vision and the finding of characteristic pallor and edema of the retina early, with later development of attenuation of retinal vessels and optic nerve head pallor.363,364 At this stage, quinine toxicity may be misdiagnosed as RP sine pigmento.363 The ERG in quinine toxicity is usually of a “negative” configuration, with far greater depression of the scotopic b-wave than a-wave.365,366
Pigmented paravenous retinochoroidal atrophy
Pigmented paravenous retinochoroidal atrophy (PPRCA) was first described in 1937 as retinochoroiditis radiata.367 PPRCA is a pigmentary retinopathy that is currently poorly understood but probably represents an acquired response pattern to an infectious or inflammatory disease.176,368–373 It has been reported in association with meningoencephalitis,370 tuberculosis,176,368,370,371 syphilis,176,372 and rubeola.374,375 In most, the fundus appearance is first noticed on a routine examination. Three small pedigrees with PPRCA have been described.376–378 These may, however, represent examples of infectious factors clustering in families rather than true genetic cases. As the name implies, the pigmentary changes are closely associated in distribution with retinal veins (Fig. 40.42). Most cases are relatively stable over time, although progression has been reported in one case.379,380 ERG responses are only mildly to moderately abnormal, if at all,176,368,381–383 but the EOG is usually affected, often significantly.382
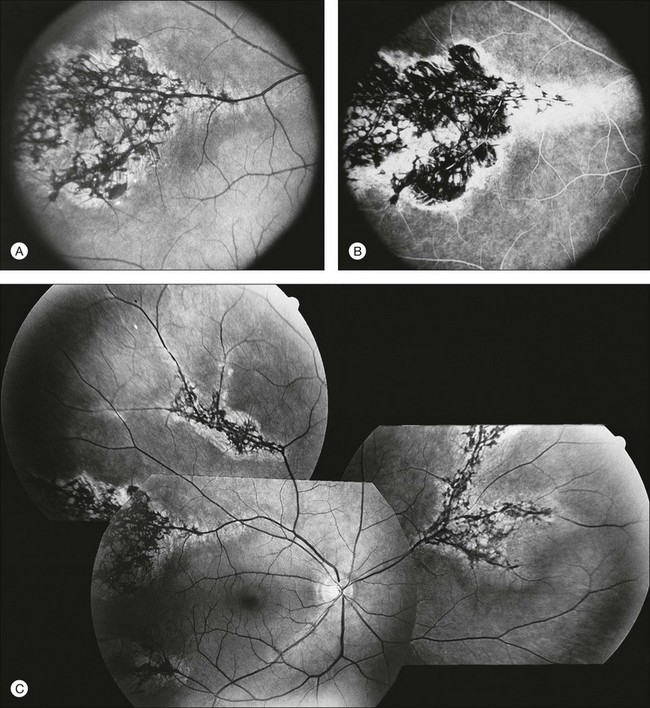
Traumatic retinopathy
Traumatic retinopathy is probably the commonest acquired retinopathy that is confused with RP and may, with DUSN, account for many previously misnamed cases of “unilateral RP.” The RPE and retina have only a limited range of responses to a traumatic insult. One of these is regional or generalized loss of the RPE with migration of melanin into the retinal layers where it accumulates along vessels, especially at branching points, creating a bone spicule pattern. Thus patients with past traumatic injury to an eye can present with a fundus appearance in the traumatized eye that mimics RP.384
Diffuse unilateral subacute neuroretinitis
DUSN is the term now used for the disorder previously called “unilateral wipe-out syndrome,”385 and “unilateral RP.”176 True unilateral inherited RP does not exist, except as an example of extreme lyonization of retinal involvement in a carrier of X-linked RP. DUSN is believed to result from the panretinal degeneration that takes place in eyes that have been infected by any of several possible worms. The raccoon nematode (Baylisascaris procyonis)386,387 has been incriminated, but other worms such as Toxocara canis388 have been suspected. Cases have been seen mainly in the USA and Caribbean, but a few cases have also been reported from Brazil388 and Germany.389 Cuhna de Souza et al.390 described the first instance of bilateral acute neuroretinitis with documentation of the nematode in both eyes.
At the time of initial visual disturbance, the retina may either appear normal or show early signs of retinal degeneration (mottling, edema, narrowing of retinal vessels). Occasionally, one can see an elevated gliotic mass in the mid or far periphery that may represent the encased worm. The visual field often shows abnormalities early in the course of the disease, but these are usually patchy. With time, the visual function usually deteriorates in the affected eye but remains normal in the fellow eye. Eventually the retina develops pigmentary clumping and scarring reminiscent of advanced RP (Fig. 40.43). The pigmentary abnormalities within the retina in some patients may take the form of accumulation of medium to coarse clumps of pigment rather than bone spicules. ERG recordings show clinically significant abnormalities in the affected eye only. Retinal laser photocoagulation has been used in cases where the fundal view is clear.385 Vitrectomy with surgical removal of the subretinal nematode has been achieved,388 and oral tiabendazole or ivermectin is indicated when vitritis obscures retinal detail.391
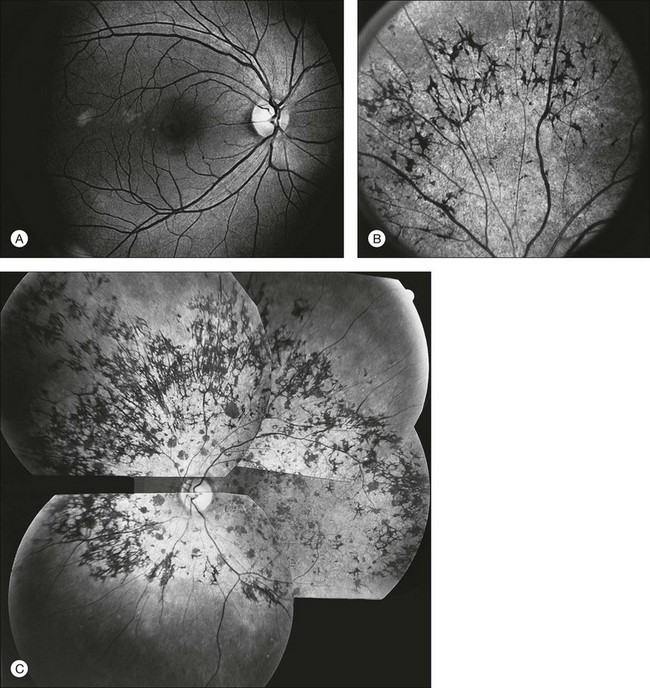
Grouped pigmentation of the retina
Grouped pigmentation of the retina, also called “bear-track” pigmentation, is a benign congenital condition in which round and irregularly shaped lesions representing RPE hypertrophy are scattered throughout the retina392 (Fig. 40.44). The condition does not appear to be hereditary. Usually the differentiation of this condition from more significant pigmentary retinopathies presents little difficulty. Retinal function and electrophysiologic tests are normal.
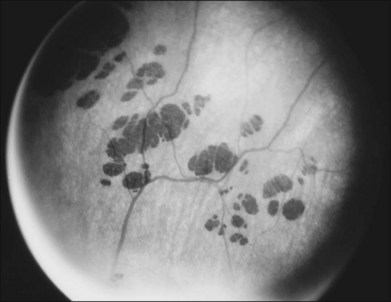
Fig. 40.44 Grouped pigmentation (“bear tracks”) of the retina.
(Reproduced with permission from Buettner H. Congenital hypertrophy of the retinal pigment epithelium. Am J Ophthalmol 1975;79:177–89.)
Grouped pigmentation of the retina should be differentiated from multiple patches of congenital hypertrophy of the RPE, which are associated with familial adenomatous polyposis or Gardner syndrome.393
Basic science
Molecular biology
Molecular genetics
Using molecular genetic analysis, 14 specific genes have been associated with adRP, 29 with arRP (with three more loci), two specific genes associated with X-linked RP (along with three more loci), and one with digenic inheritance. Further genetic mutations are now being identified. For those interested in the most recent gene assignments and localizations, a list of cloned and mapped genes causing retinal degenerations or allied disorders has been compiled by Dr. Stephen P. Daiger and is accessible on the internet at the following address: www.sph.uth.tmc.edu/Retnet/.
Autosomal dominant RP genes
In rod photoreceptors, rhodopsin is the light-absorbing, conjugated photopigment found in outer-segment discs. It is composed of an apoprotein opsin molecule covalently bound to 11-cis-retinal (Figs 40.45 and 40.46).394 Incident light induces isomerization of the retinal and decay through a sequence of spectrally defined photoproducts. These light-induced changes result in a number of conformational changes in the opsin molecule, exposing G-protein-binding sites. A cascade of reactions (the phototransduction cascade; Fig. 40.47) is then set in motion and results in closing cGMP-dependent cation channels and relative transmembrane hyperpolarization.395
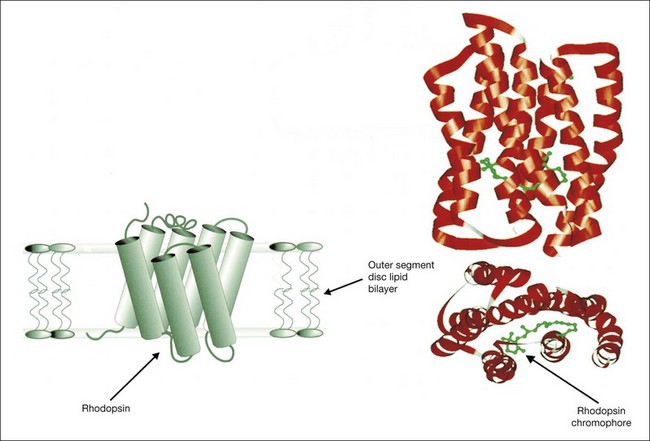
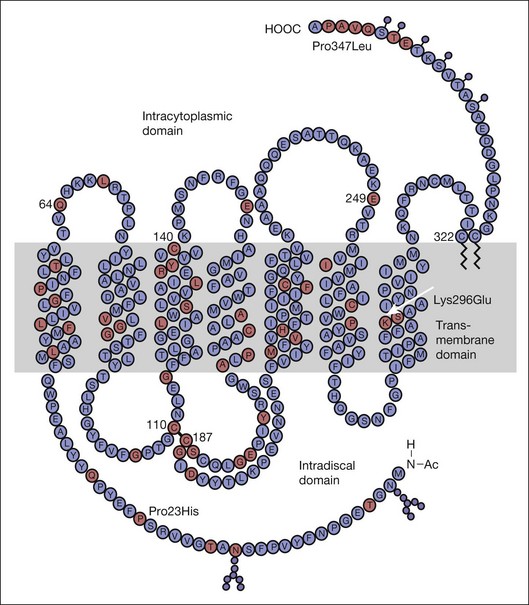
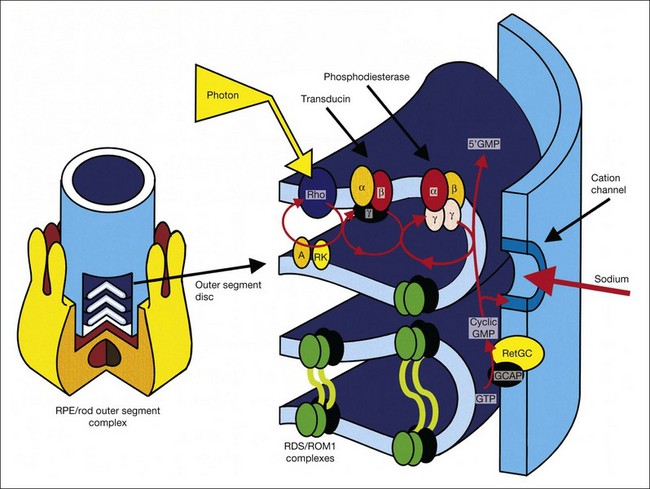
McWilliam et al.396 were the first to detect linkage of adRP with an anonymous marker on the long arm of chromosome 3q. Shortly thereafter, Dryja et al.397 reported a mutation of codon 23 (Pro23His) of the rhodopsin gene mapping to chromosome 3q (Fig. 40.45). A mutation in the third exon of rhodopsin, Met207Arg, has been found in the Irish family originally linked to chromosome 3q.398
It has been estimated that rhodopsin mutation accounts for approximately 25% of adRP.399 In the USA, rhodopsin Pro23His is by far the most common of all rhodopsin mutations, being found in 12–15% of all American families with adRP.400 It is interesting that the Pro23His mutation has rarely been reported anywhere else in the world.401 In fact, the mutation rate of the rhodopsin gene appears to vary with ethnic background. For instance, frequencies of RHO mutations in Asian populations, such as Japanese (5.9%),402 Chinese (2.0%–5.6%),403 Indian (2.0%),404 and Korean (2.0%),405 are lower than that in the USA,400 and Europe.406
From the occurrence of a rare, intron 1 polymorphism in all patients with Pro23His adRP, it has been suggested that all persons with this mutation may have originated from a single-mutation founder.407 The phenotype associated with rhodopsin Pro23His is best described as Massof type II79 or R-type RP.84,85 Most rhodopsin adRP phenotypes have in fact been described as type II or R-type disease,408–410 with a minority of mutations associated with type I or D-type disease.411,412
Pro347Leu is the next most frequent mutation of rhodopsin, accounting for approximately 5% of 148 adRP families in one series.413 This form of adRP is a more severe, diffuse phenotype, with comparatively smaller visual fields with more ERG deficit than that seen in cases of Pro23His adRP.414 The ophthalmologic findings of patients with Gly106Arg mutation of the rhodopsin gene408 appear to be variable in expression, with patients rarely having normal cone and low-normal rod ERGs, a finding quite unusual among patients with RP. Sieving et al.415 have reported the evaluations of seven affected members of a large family with a Gly90Asp rhodopsin mutation and described the condition for this family as autosomal dominant congenital nyctalopia, to distinguish it from typical RP.
Sandberg et al.416 have reported clinical details on patients with 27 different dominant rhodopsin mutations. They suggested that severity of disease, measured by visual acuity, visual field diameter, dark-adapted sensitivity, and ERG amplitudes, increased with increasing codon number (Fig. 40.46). Berson et al.417 have suggested that the rate of progression of RP from mutation of rhodopsin is correlated with the site of mutation and that other modifying factors, genetic or environmental, may influence phenotypic expression. A “constant equivalent light” model has been proposed for the severe adRP phenotype associated with rhodopsin codon 296 mutation. Lys296 is the chromophore-opsin attachment site and is also involved in holding the opsin in an inactive conformation. Theoretically this would lead to overstimulation of the phototransduction cascade, a situation similar to constant light exposure. This situation is known to lead to photoreceptor cell death.418 This explanation has been questioned, however, because the opsin appears to be inactivated by phosphorylation and arrestin binding in Lys296Glu transgenic mice.419
The peripherin/rds gene was first associated with RP in 1991,420,421 and over 39 sequence variants have been associated with a range of autosomal dominant phenotypes, including RP, macular dystrophy, CRD, central areolar choroidal dystrophy, fundus flavimaculatus, and pattern dystrophy.422 Most are missense mutations, but it is unlikely that peripherin/rds mutations account for more than 5% of adRP.423 These reports do however represent the first instance of which an animal model of RP has been found to have a directly correlated human disease counterpart. Much of what we understand to be the role of peripherin/RDS in human photoreceptors (Figs 40.48–40.50) comes from initial studies on this naturally occurring mouse model (rds), which has a 10-kb insert in codon 230 of the disease gene, creating a null allele.424 From this work and more recent human studies, it has been found that the peripherin/rds molecule accumulates in the rims of photoreceptor outer-segment discs and plays an important role in disc morphogenesis and stability.425
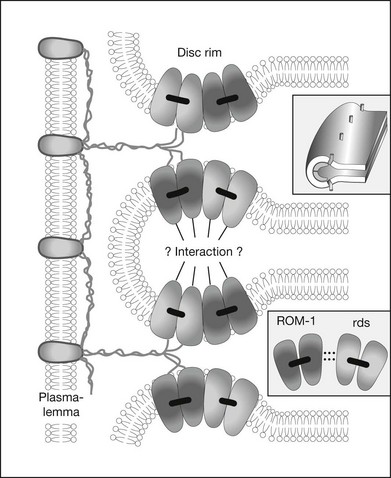
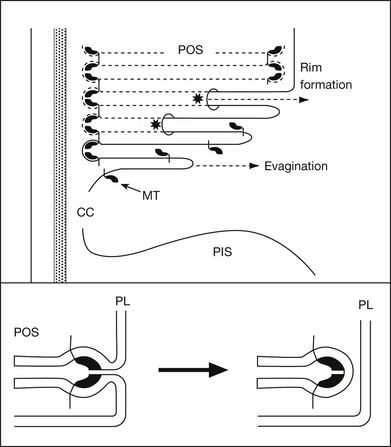
Bessant et al.426 reported a large family with adRP previously linked to chromosome 14q11 that is associated with a Ser50Thr mutation of NRL. The gene product NRL is a retinal-specific DNA-binding transcription factor that interacts with the cone–rod homeobox transcription factor CRX to promote and regulate transcription of rhodopsin and other retinal genes. The Ser50Thr NRL mutation, when coexpressed with CRX in transfection experiments, produced a marked increase in transactivation of the rhodopsin promoter, producing excessive transcription of rhodopsin. Other families with adRP have been reported with NRL mutations.427
A rare but remarkable regional (type II) phenotype with graded disease severity (variable expressivity) has been reported associated with mutations in PIM1, on chromosome 7p.428 This encodes a protein kinase involved in transcriptional control. The gene FSCN2, which encodes a photoreceptor-specific paralog of the actin-bundling protein fascin, with presumptive cytoskeletal formation functions, has been reported with autosomal dominant RP.429 A common FSCN2 mutation, 208delG, appears to account for 3.3% of adRP in Japan.429
Autosomal recessive RP genes
The third phototransduction cascade protein (Fig. 40.48), cGMP phosphodiesterase (PDE), has been associated with a number of phenotypes.430 PDE is a holoenzyme consisting of two large subunits α and β, which become enzymatically active when the two γ subunits are removed by activated transducin. Many nonsense and missense mutations of the α and β subunits of PDE (gene symbols PDE6A and PDE6B, respectively) have been associated with arRP, with the PDE6A mutations representing null alleles. No mutation of the PDEγ subunit gene, PDEG, has yet been associated with human retinal disease. This might be due to the fact that PDEG has an exceptionally low mutation rate, that in humans the gene is expressed in other vital tissues and is lethal in utero, or the function of the mutant PDEγ may be taken over by some other protein. Most PDEβ subunit mutations associated with arRP are found in the C-terminal half of the molecule, which contains the catalytic domain. These mutations presumably directly affect the enzymatic activity of the protein. The His258Asp mutation, however, is near the N-terminal portion adjacent to the PDEγ-binding domain. This mutation has been reported in a Danish family associated with an autosomal dominant congenital stationary night blindness. It has been postulated that this mutation impedes complete inactivation of PDE in dark-adapted rod photoreceptors. This would result in rod desensitization and an inability to transduce at low light levels.431
Activated PDE hydrolyses intracellular cGMP. This second-messenger molecule is generated from guanosine triphosphate by membrane-bound and soluble guanylate cyclase.432 Increased intracellular cGMP leads to relative opening of membrane-bound cGMP-gated cation channels and cell hyperpolarization. The cGMP-gated cation channels are composed of three subunits, α, β, and γ. Mutations (usually null alleles) of the gene for the α subunit (CNGA1) have been associated with arRP.433
Recovery of active rod opsin, for further phototransduction, involves the regeneration of the 11-cis retinaldehyde chromophore from the all-trans isomer. In part, this process involves a cellular retinaldehyde-binding protein (CRALBP), which binds and promotes 11-cis retinol oxidation to 11-cis retinaldehyde in the RPE.434,435 A homozygous R150Q mutation of the gene for CRALBP (RLBP1) has been associated with childhood-onset arRP.435 Nyctalopia was evident by 3–4 years of age. Optic atrophy, vascular attenuation, macular atrophy, and peripheral white dots without classic bone spicule pigmentation were reported. Mutations of RLBP1 have also been reported in autosomal recessive retinitis punctata albescens,436 Bothnia dystrophy,437 and Newfoundland rod–cone dystrophy.438
The tubby-like protein (TULP) gene family includes TUB, the human homolog of the mouse tub gene, TULP1, and TULP2, and TULP3. All four are expressed in the retina but have unknown functions.439 Mice, homozygous for a tub mutation, develop maturity-onset obesity, insulin resistance, cochlear degeneration, and a progressive retinal dystrophy. No mutations of TUB have yet been associated with human retinal disease, but mutations of TULP1, which is retina-specific, have been associated with arRP.440 Homozygous splice-site mutation IVS14+1, G→A, associated with severe early-onset retinal degeneration, has been reported in two families from the same Dominican Republic village.440 Two North American individuals have been reported with compound heterozygous mutations, one patient with missense mutations Arg420Pro and Phe491Leu, and another with Ile459Lys and splice site mutation IVS14+1, G→A.240
One of the most studied of animal models for RP is the Royal College of Surgeons (RCS) rat, which is characterized by the inability of outer segments shed by photoreceptors to be recognized and phagocytized by RPE, leading to retinal degeneration. Mutations in the receptor tyrosine kinase gene Mertk have been found to underly the defect in the RCS rat.441 Patients with arRP have been reported with mutations of the human ortholog MERTK.442
X-linked RP genes
Positional cloning in X-linked RP at the RP3 locus has led to the discovery of mutations in RPGR (RP GTPase regulator).443 A number of mutations have now been described. Detailed clinical assessments of families with a number of mutations444,445 have revealed severe, early-onset RP in hemizygotes and variable disease in heterozygotes, as has been typically described in X-linked RP. All the affected males have shown evidence of both rod and cone ERG loss consistent with the expression of the gene product of RPGR in both classes of photoreceptor. On the basis of poorer ERG responses in hemizygotes and severe symptoms and clinical findings in heterozygotes, Andréasson et al.446 have suggested that microdeletion mutation of exons 8 through 10 is a more severe disease than that seen with splice site mutations. Despite a strong association between the tapetal-like reflex and the X-linked RP carrier state, this has been documented in few patients carrying an RPGR mutation.445
Although studies suggested that 70–90% of X-linked pedigrees are linked to the RP3 locus, mutations of RPGR were initially identified in only about 10–20% of families with X-linked RP.443,446,447 Recently, however, another retina-enriched transcript, called the ORF15 transcript, was discovered, which utilizes exons 1–13 as before but utilizes exon 14 through part of intron 15 as a large terminal exon (termed the ORF15 exon). This ORF15 exon has accounted for the remainder of families linked to RP3.448 Mutations of RPGR can result in other phenotypes, including CRD,449 cone dystrophy,373 and even macular dystrophy.450
A disease gene has also been identified at the RP2 locus.451 Most of the mutations cause truncation of the gene product.452 Mutations of this gene, termed RP2, account for approximately 8–10% of families of X-linked RP.453 The gene product has homology with human cofactor C, a protein involved in the ultimate step of β-tubulin folding, suggesting that this gene may act as a chaperone or in some way be involved in the structure or function of the cilium of photoreceptors.451,452 Together, mutations of RP2 and RPGR account for the vast majority of cases of X-linked RP and the RP phenotypes associated with these genes have been reported as clinically indistinguishable from one another or, at most, to show modest differences.454
Digenic inheritance and RP
In rod photoreceptors, two peripherin/rds molecules form homodimers that bind noncovalently to homodimers of the glycoprotein ROM-1455,456 (Fig. 40.49). In cones, peripherin/rds homodimers form tetramers with homodimers of either ROM-1 or a homolog.457 These tetramers may represent the “extracellular marginal templates” described by Corless and Fetter458 (Fig. 40.50) that are crucial to the creation and structural integrity of the disc rim.
In a few families, three rds point mutations – Leu185Pro, Arg13Trp, and Leu45Phe459 – have been seen in association with ROM-1 mutations. The affected compound heterozygotes had a severe RP phenotype. This type of inheritance has been termed digenic and highlights the important structural relationship between RDS and ROM-1 in the architecture of the rod photoreceptor. Despite studies around the world, very few RP families have been found to exhibit this phenomenon. Putative mutations of ROM-1 in isolation have been reported in some small families.460 As yet, isolated ROM-1 mutations are not convincingly associated with retinal disease.423
Usher syndrome molecular genetics
Molecular genetic linkage analysis has shown that not only are the three clinical subtypes of Usher syndrome genetically distinct but also that there is notable genetic heterogeneity. Currently 15 chromosomal loci have been linked to Usher syndrome and 12 of the associated disease genes have been identified (Table 40.1). The most severe and most common subtype, USH1B, has been associated with mutations of myosin VIIA (MYO7A).461 Mutation of MYO7A can also cause nonsyndromal deafness of either recessive (DFNB2) or dominant (DFNA22) type.
Four other genes have been discovered for type 1 Usher syndrome. The gene for USH1C encodes harmonin, a novel PDZ domain containing protein that functions in the organization and assembly of larger protein complexes for cell adhesion, signal transduction, and intracellular transport.462 Mutations in the ear-specific exons of the gene for harmonin also cause nonsyndromic recessive deafness (DFNB18).463 The gene for USH1D is CDH23, which encodes cadherin 23, a novel member of a class of intercellular adhesion proteins.464 Mutations in CDH23 also cause autosomal recessive nonsyndromic deafness (DFNB12).465 CDH23 is expressed in the cochlea and retina and mutations in the mouse ortholog cause disorganization of inner-ear stereocilia and deafness in the waltzer mouse animal model.466 Two of the harmonin PDZ domains interact with MYO7A and CDH23, indicating that these three proteins form a complex important for organization, maintenance, and function of stereocilia in the cochlea.467,468 The gene for USH1F is PCDH15, which encodes protocadherin 15.469 Mutations in PCDH15 have also been reported in nonsyndromic recessive deafness (DFNB23).470 PCDH15 localizes to stereocilia in inner-ear hair cells and to the photoreceptors of the retina and is presumed to be essential for homeostasis. Mutation of the murine homolog causes vestibular dysfunction and deafness in the Ames waltzer mouse.471 USH1G encodes the protein SANS, which is a novel scaffold protein gene that is expressed in retina and cochlea and is associated with harmonin within stereocilia.472 Thus, it appears that many of the genes for type 1 Usher syndrome (MYO7A, harmonin, CDH23, and SANS) function together in a complex essential for determining and maintaning cohesion of the stereocilia of the cochlea.472
The commonest gene association with type 2 Usher syndrome (USH2A) has recently been identified,473 and encodes a protein called usherin that has motifs that show homology to thrombospondin, laminin epidermal growth factor, and fibronectin type III. This indicates that the gene product may function as a cellular adhesion molecule or as a component of the basal lamina and extracellular matrix. Although discovered first in patients with type 2 Usher syndrome, a Cys759Phe mutation of USH2A was found in approximately 4.5% of 224 patients with arRP without hearing loss.
The gene for type 3 Usher syndrome (USH3A) was first reported as encoding a 120-amino-acid protein in 2001 by Joensuu et al.474 A common termination mutation, Y100X, accounts for the majority of mutant alleles in Finnish cases.474 The gene product, clarin-1, is believed to have a role in hair cell and photoreceptor cell synapses.475 Pennings et al. have reported that type USH3A mutations can produce disease that mimics either type 1 or 2 Usher syndrome and that the fundus appearance may mimic retinitis punctata albescens or RP sine pigmento.476
Protein chemistry
Considerable information has now accumulated on the molecular consequences of retinal dystrophy gene mutation. In turn that has led to a much deeper understanding of the molecular physiology of the human retina. Readers are directed to a number of significant reviews on the subject.477,478
Some general principles can be applied to the molecular consequences of retinal gene mutation. Deficiency of gene product seems to be the most likely explanation for recessive disease. In two recessive RP pedigrees with rhodopsin mutations (a nonsense and splice site mutation) for example, the aberrant alleles theoretically encode nonfunctional rhodopsin.479,480 Haploinsufficiency is unlikely to be important in dominant rhodopsin disease though, because carriers of at least one apparently null allele have been shown to be phenotypically normal.480 Mutant rhodopsin expression in COS cells has shown that mutations in the intradiscal domain adversely influence normal rhodopsin folding.481 Autosomal dominant mutations within the rhodopsin transmembrane helices still cause misfolding, but to a much lesser effect, and have a more profound effect on retinal binding.482 In vitro studies have shown that mutant rhodopsin can accumulate within the rough endoplasmic reticulum of rod photoreceptors.483 This could lead to inhibition of transport of the wild-type rhodopsin to the cytoplasmic membrane and loss of function and might explain pathogenesis in some cases of retinal dystrophy with rhodopsin mutations. However, different results in rhodopsin expression in in vitro studies in nonmammalian models and mammals have suggested that the true relevance of this mechanism in humans is as yet unproved. Truncation of the rhodopsin protein due to mutation of codon,345,484 for example, is associated with expression of mutant and wild-type rhodopsin in the plasma membrane of tissue culture cells but leads to accumulation of the mutant in the rough endoplasmic reticulum in transgenic mice. The effect in humans is unknown.
Abnormal pre-mRNA splicing
Proteins encoding three RP genes, PRPF8 (RP13), PRPF31 (RP11), and PRPF3 (RP18), have been associated with RNA splicing. RNA splicing is an essential process that removes intron sequences from pre-mRNA. This is carried out by the spliceosome, a high-molecular-weight ribonucleoprotein complex.485 The vast majority of pre-mRNA introns (designated the U2 type) are spliced by the major spliceosome, which is composed of five uridine-rich small nuclear ribonucleoproteins, or snRNPs, termed U1, U2, U4, U5, and U6. Each snRNP is the central component of a protein complex that also contains seven Sm or Sm-like proteins, which are assembled by the so-called SMN (survival of motor neurons) protein. In addition to the Sm and Sm-like proteins, snRNPs also contain 3–10 particle-specific proteins.486 The major spliceosome also contains a poorly defined number of other non-snRNP protein factors. The minor spliceosome is responsible for splicing a rare class of pre-mRNA introns, called the U12 type.485 Minor spliceosomes contain the U5 snRNP and four nonabundant snRNPs: U11, U12, U4atac, and U6atac.
The PRPF8 gene (chromosome 17p13.3) encodes precursor RNA processing factor 8 (PRPF8). The yeast homolog, Prp8p, is a U5 snRNP factor that is first required for assembly of the U4/U6 and U5 tri-snRNP487 and then for facilitating the binding of the tri-snRNP to the 50 and 30 splice sites.488 It functions as the catalytic core of the spliceosome, either by facilitating formation of the core and/or by stabilizing RNA interactions.489 Seven different missense mutations of PRPF8, all clustered within a 14-codon stretch in the last exon, have been identified in five RP13-linked families.490 PRPF8 mutations are associated with early-onset adRP with diffuse retinal involvement and a severe prognosis in comparison with other forms of adRP.490,491 Individuals experience night blindness beginning between 4 and 10 years of age and constricted visual field as teenagers.491 Classic midequatorial bone spicule pigmentation is seen by middle age.491 Typically, individuals are registered as blind or partially sighted by age 30.490 PRPF31 (chromosome 19q13.4) encodes protein 61K, the putative ortholog of the yeast pre-mRNA splicing factor Prp31p.492 Protein 61K is specific to the U4/U6 snRNP and is required for pre-mRNA splicing in vitro.493 Protein 61K participates in the formation of the tri-snRNP, possibly by physically tethering the U5 snRNP to the U4/U6 snRNP.
Haploinsufficiency appears to be the mechanism underlying the photoreceptor degeneration in the majority of alleles of the PRPF31 gene associated with RP because most of these alleles are predicted to be protein truncation mutations. Families with PRPF31 mutations are unique in showing bimodal expressivity of the phenotype, with asymptomatic carriers who have both affected parents and affected children.494,495 Symptomatic individuals experience night blindness and loss of visual field in their teenage years and are typically registered as blind by their 30s.494 Evans et al. suggested that the bimodal expressivity seen in these pedigrees may be explained by a second allelic genetic factor influencing phenotype. This idea was taken further by McGee et al.,496 who confirmed that penetrance of the PRPF31 mutations may be influenced in trans by otherwise silent PRPF31 alleles or by a closely linked locus on the wild-type chromosome, because a statistically significant correlation was found between the development of RP in a carrier and the inheritance of the region around RP from the noncarrier parent.496 Vithana et al.492 have demonstrated that asymptomatic patients inherit a different wild-type allele to the one inherited by their symptomatic siblings. The third of the three known pre-mRNA splicing factors associated with adRP is encoded by the PRPF3 gene (chromosome 1q21.1).497 PRPF3 protein associates with the U4/U6 snRNP during pre-mRNA splicing.498 Mutations in its yeast ortholog, Prp3p, cause instability of the U4/U6 snRNP and prevent assembly of the tri-snRNP, thereby preventing splicing.499 Two different missense mutations have been identified in three individuals and three families.499 Affected individuals suffer night blindness toward the end of the first decade of life.497 Visual field defects are seen by the fourth decade of life but the central field remains relatively spared. A few patients progress to total blindness by age 80.
Several mechanisms have been proposed to account for the fact that mutations in these three, ubiquitously expressed, splicing factor genes only cause adRP and not a more widespread abnormality. The loss of one functional copy of these splice factor genes may affect only rods because of their unusually high requirement for protein synthesis and, consequently, for pre-mRNA splicing.500 Alternatively, the adRP-associated mutations may decrease the rate at which spliceosome activation occurs,501 making activation the rate-limiting step in rod photoreceptors but not other cells. Finally, it has also been suggested that the retina-specific phenotype may occur as a result of an unidentified retina-specific splicing cofactor that interacts with all three of these splicing factor proteins.496
RPGR interactome
An interactome is a complex representation of functional interactions between molecules either within a cell or within the organism as a whole. Often such interactomes reveal important interactions between molecules that at first would not appear to be functionally related. In this way, the functional consequences of genetic mutation can be predicted without the need for lengthy experimentation. An RPGR interactome has also been proposed.502 RPGR is a component of centrioles, ciliary axonemes, and microtubular transport complexes, although its precise function is unknown. It colocalizes with RPGRIP1 at the axonemes of connecting cilia in rod and cone photoreceptors503 by binding to RPGRIP1. This localization is lost in Rpgrip1 knockout (KO) mice. Rpgr KO mice develop a slow retinal degeneration, with features resembling a cone–rod degeneration – cone photoreceptors degenerate faster than rods and there is partial mislocalization of cone opsins.504 Some residual RpgrORF15 expression has been reported in this model.505 RPGR has been shown to co-immunoprecipitate in retinal extracts with a number of different axonemal, basal body, and microtubular transport proteins.505 These include nephrocystin-5 and calmodulin, which localize to photoreceptor-connecting cilia; the microtubule-based transport proteins, kinesin II (KIF3A, KAP3 subunits), dynein (DIC subunit), SMC1, and SMC3; and two regulators of cytoplasmic dynein, p150Glued and p50-dynamitin, which tether cargoes to the dynein motor. Inhibition of dynein by overexpressing p50-dynamitin abrogates the localization of RPGRORF15 to basal bodies. RPGRORF15 can be co-immunoprecipitated from retinal extracts with other basal body proteins, including NPM, IFT88, 14–3-3ε, and γ-tubulin.502, 505 RPGRORF15 and RPGRIP1 co-localize at centrosomes in a wide variety of nonciliated cells and at basal bodies in ciliated cells. Both proteins are core components of centrioles and basal bodies.502 In summary, RPGRORF15 appears to have a role in microtubule-based transport to and from the basal bodies and within photoreceptor axonemes, perhaps concerned with movement of cargoes between IS and OS. RPGRORF15 is predominantly expressed in photoreceptor connecting cilia and basal bodies but expression has also been reported in OS in some species,506 although this has been disputed.503 RPGR is also expressed in the transitional zone of motile cilia in the epithelial lining of human bronchi and sinuses (RPGRex1–19 only) and within the human and monkey cochlea.503,507–509
Ush interactome
The USH genes encode proteins of different classes and families, including motor proteins, scaffold proteins, cell adhesion molecules, and transmembrane receptor proteins. In vitro direct interaction studies between USH proteins and also localization studies in mouse models have however suggested functional relationships between these proteins such that an “USH interactome” has been proposed.510 In these studies, it should be noted, however, that although all the USH mouse models show severe hearing loss and vestibular dysfunction, only the Ush2A–/– mouse, shows signs of RP.511 From such work, a “multiprotein scaffold complex” model has been proposed for harmonin, whirlin, and sans. There is also evidence that harmonin and whirlin can bind all other components of the USH network, including cadherin 23, protocadherin 15, usherin, VLGR1, and myosinVIIA.467,507,512
Myosin VIIa has also been found to interact with sans 11 and protocadherin 15.468 Such findings suggest that the USH proteins form a transmembrane network that regulates hair bundle morphogenesis, particularly the growing stereocilia or the kinocilium, and may also have a role in the mechanoelectrical signal transduction and synaptic function of mature hair cells.513 In the USH interactome model, it is proposed that the extracellular interstereocilia links are anchored intracellularly by the scaffold proteins, harmonin and whirlin, through direct binding to the actin cytoskeleton or through other proteins, including myosin VIIa.467,507,514,515
Additionally, it has been shown that the association of other proteins with the USH complex (through harmonin) may function in determining cell polarity and cell–cell interactions.516 Also Myo7a is believed to use long filaments of actin as tracks along which to transport other USH complex molecules.517 In photoreceptors, proteins of the USH interactome are localized in the connecting cilium and may participate in the ciliary transport, but are also arranged at the interface between the inner segment and the connecting cilium where they control cargo passage. USH protein complexes may also provide mechanical stabilization to membrane calycal processes and the connecting cilium.518 Studies of coexpression of USH1 and USH2 proteins also show accumulation at synaptic endings of photoreceptor cells, indicating that they are organized in an USH protein network there.519
Bardet–Biedl syndrome and the “BBSome”
Recently it has been proposed that many of the proteins encoded by Bardet–Biedl genes form complexes, e.g., BBS1, BBS2, BBS4, BBS5,BBS7, BBS8 and BBS9 – the “BBSome.”520 The complex is important in the function of primary cilia, nonmotile projections found in numerous cell types. In particular, BBSome complexes are thought to play a central role in vesicular trafficking of membrane proteins within cilia. The BBSome binds to Rab8, a GTP/GDP exchange factor involved in docking and fusion of rhodopsin carrier vesicles in the connecting cilium of photoreceptors.521 It is also known that other Bardet–Biedl genes, e.g., BBS3 (Arl6), form functional relationships with this BBSome.522 Others appear to function as chaperones, e.g., BBs6, BBS10, and BBS12, and in protein ubiquitination (BBS11/TRIM32).523
Abnormal intracellular trafficking
An increasingly important consequence of retinal disease gene mutation has been the concept of abnormal intracellular trafficking.400,524 For instance, another rod damage in rhodopsin mutants is often associated with mutant rhodopsin accumulating in the outer segment.400,525,526 It is thought that this interferes with normal phototransduction and triggers apoptotic cell death. It has been shown in transgenic mice that mutant rhodopsin is only transported to the rod outer segment when the C-terminal region of the molecule is intact.527 This observation has been corroborated in human eyes.528
Cell death pathways
A prevalent proposition in the last decade has been that, since many different gene mutations result in a similar clinical outcome (RP), there must be a “final common cell death pathway” precipitated by these mutations.529 Apoptosis, the first cell death biochemical pathway linked to RP, characteristically appears histologically as a rounding up of the cell, reduction of cellular volume, chromatin condensation, and finally engulfment by resident phagocyte. Apoptosis is triggering of intrinsic and (to some extent) extrinsic pathways involving activation of caspases which can be subclassified into both initiator (2, 8, 9, 10) and effector (3, 6, 7) molecules. Studies with caspase inhibitors, however, have often produced only marginal neuroprotection in RP models.530,531 Other studies have highlighted that caspase-independent inducers of cell death such as apoptosis-inducing factor, calpains, and poly(ADP-ribose) polymerases 1 are activated during retinal degeneration.532
Previously, necrosis had not been considered a significant part of retinal degeneration. This passive, unregulated form of cell death is now considered to be inducible by regulated signal transduction pathways such as those mediated by RIP kinases.532 Such necroptosis may play a role in RP.532,533 Apoptosis and necroptosis mechanisms may act together or may form alternate pathways.
Cell and tissue biology
Histopathology
Early histopathologic studies found that the first degenerative abnormalities in RP occur in the photoreceptors. As more and more animal model histopathologic correlates of specific human genetic mutations become available, our knowledge of both outer and inner retinal changes in RP is leading to new concepts for therapy.477,534
Photoreceptor abnormalities
The earliest histologic sign of retinal dystrophy is shortening of rod outer segments (Fig. 40.51).535 With progression of disease, rod outer segments become more severely shortened, and eventually whole cells are lost. This is reflected in reduced numbers of nuclei in the outer nuclear layer. Rod cell loss is usually seen initially in the midperiphery. In certain cases cell loss is initially seen in the inferior retina. This is a typical finding in cases with rhodopsin mutation Pro23His.179 In such cases there is a corresponding altitudinal field defect. Interestingly, a similar pattern of photoreceptor loss is seen in transgenic Pro23His mutant mice. In this model, if mutant mice were dark-reared, the resultant retinal degeneration was more uniform across the retina and was significantly milder.536 This suggests that limiting light exposure in patients with this mutation may moderate the progression of disease.
Using immunocytochemistry and antirhodopsin antibodies, extensive studies have been undertaken of cytopathologic abnormalities in photoreceptors. In normal rods, rhodopsin is concentrated in the outer segments. In a number of rhodopsin-mutant animal models, abnormal localization of mutant rhodopsin has been seen,537 and this has also been seen in human eyes.538,539 Such observations have led to interesting hypotheses as to how mutant protein may damage photoreceptors.540 It has been proposed that certain mutant rhodopsin molecules accumulate in the endoplasmic reticulum or Golgi apparatus in the inner segment and lead to cell death by interfering with the function of these organelles. Clearly, though, there are species differences in the cytopathologic effects of particular mutations. In transgenic Pro347Ser mice, rhodopsin is seen to accumulate in extracellular vesicles attached to rod inner segments.541 Alternatively, in the transgenic Pro347Ser pig, rhodopsin accumulates in the inner segments.542,543 Such aberrant accumulation of rhodopsin in the inner segment has not, however, been seen in human cases of Pro347Ser.
Clarke et al.544,545 studied 11 different animal models of outer retinal dystrophy. These authors noted that the kinetics of photoreceptor death fit an exponential model. This in turn suggests three important features: (1) the risk of death is constant throughout the lifetime of each mutant photoreceptor; (2) each mutant photoreceptor is at the same risk of death as every other mutant photoreceptor (other things being equal); (3) the time at which death will occur is random and independent of the time of death of other photoreceptors. The regional variation in photoreceptor death in RP is not inconsistent with the feature of equal risk because local geographic factors may be superimposed on the equal risk of death of all the photoreceptors, resulting in a higher risk in certain parts of the retina. In the animal model studies, the same region of the retina was always examined, eliminating any effect of regional variation. The identification of these three characteristics of outer retinal dystrophies is important for at least two reasons. First, exponential kinetics refutes the major alternative model of photoreceptor death, the cumulative damage model, in which the risk of death increases with time (akin to the risk of death in an aging population of people) and the cell death kinetics are sigmoidal, not exponential. Second, the recognition that mutant photoreceptors are at a constant risk of death requires that biochemical mechanisms proposed to underlie the photoreceptor death must explain both the exponential kinetics as well as other major characteristics of the disease. For example, this may mean that, for all the different gene mutations, probably only a few pathways triggering apoptotic cell death are relevant to photoreceptor degeneration.546
Outer retinal disease
After photoreceptor cell death, the RPE becomes detached from Bruch’s membrane and migrates into the neurosensory retina. The subretinal space is abolished. Accumulation of RPE cells in a cuff around retinal vessels leads to bone spicule pigmentation.547 Bruch’s membrane thickening has also been recorded under degenerate RP retina. In late-onset RP, material has been seen to accumulate between the RPE and the inner collagenous layer of Bruch’s membrane.548–550 This material was periodic acid–Schiff-positive and contained lipid, calcium, and iron. Choriocapillaries underlying areas of debrided RPE undergo atrophy.551
Inner retinal pathology
Reactive changes have been reported in all cell types in the inner retina subsequent to photoreceptor cell death. Müller cells undergo reactive gliosis.552,553 In advanced RP, entangled, thickened Müller cell processes result in extensive scarring in the remaining retina. Glial fibrillary acidic protein-positive astrocytes undergo reactive hyperplasia. This growth of astrocytes contributes to the pallor of the optic nerve head and epiretinal membrane formation.554,555 Microglia migration into the outer retina has been documented in the RCS rat model of retinal dystrophy,556 but the human and pathophysiologic significance of this is unknown.
Previously it has been suggested that RP has little effect on ganglion cells.557,558 Stone et al.115 studied 41 RP retinas and found significant reductions in ganglion cell numbers in each inheritance type of RP. The most significant losses were in X-linked RP and adRP. Transneuronal degeneration534,559 has been the explanation for this phenomenon. Compression of cell bodies by retinal vessels560 and inner retinal ischemia has also been proposed as causes for ganglion cell loss. Inner retinal ischemia, a consequence of pigment epithelium perivascular cuffing,547 may in part also explain the optic nerve head pallor that is observed clinically.561
More recent publications have expanded on the details of the anomalous remodeling and the misguided attempts at repair that take place within the retina in retinal degenerations following death and loss of the photoreceptors.562,563 The changes appear in response to the stress of injury and cell death and progress through three stages: (1) the primary insult to the photoreceptors, which results in their dysfunction (and produces visual symptoms); (2) death of photoreceptors that ablates the sensory retina, by initial photoreceptor stress, phenotype deconstruction, and cell death of other cell types, through bystander effects or loss of trophic support; and (3) a protracted period of global remodeling and large-scale reorganization of the remaining neural retina.564 These attempts at repair result in three major tranformations: (1) the reactive hypertrophy of Müller cells and the formation of a distal glial seal that essentially isolates the remaining neuroretina from the surviving RPE/choroids; (2) apparent neuronal migration along glial surfaces to ectopic sites; and (3) the establishment of aberrant rewiring through a complex formation of neurite fasicles, new synaptic foci, and aberrant connections within the retina.561 It is also now known that bipolar and horizontal cells in particular undergo significant morphological changes and changes in neurotransmitter receptor expression in response to outer retinal degeneration.564 RP triggers permanent loss of bipolar cell glutamate receptor expression, though spontaneous iGluR-mediated signaling by amacrine and ganglion cells. It has been proposed that rod bipolar cell dendrite switching likely triggers new gene expression patterns and may impair cone pathway function.565 These changes may place constraints on the concepts of cell or tissue transplantation or implantation of artificial retinas for regaining sight in advanced retinal degenerations.
Cellular remodeling and vascular changes
In the slowly degenerating dystrophic retina, peripheral rods sprout long, axon-like processes that can extend as far as the inner limiting membrane.535,566 These neurites have been found in rhodopsin mutant eyes and in cases of X-linked RP. They are found in areas of marked cell death but have yet to be seen in the macula. Neurite processes bypass bipolar and horizontal cell synapses to become closely associated with hypertrophied Müller cell processes (Fig. 40.52). Neurites aggregate to form fascicles that arborize at the inner plexiform layer to form beaded processes under the inner limiting membrane. Although numerous vesicles similar to those seen in synapses are found in neurites, true synaptic connections have yet to be identified. Immunocytochemistry has shown that rhodopsin photopigment accumulates within neurites. It is still unknown whether these processes fulfill any functional role. Rod neurite sprouting does not seem to occur in dystrophic mouse models of RP. This suggests that the process occurs in retinas that undergo protracted degeneration over years, rather than rapid degeneration over weeks to months that is typical of mouse models of retinal dystrophy. Such models therefore may not be suitable in retinal transplantation studies ultimately intended to justify human studies. The environment in which it is intended to re-establish synaptic contact between transplanted rods and second-order neurons must be very different in dystrophic mice as compared with humans.566 Thus, models of RP in larger animals, such as the transgenic pig,542,567 are necessary.
Cone degeneration may occur early or late in RP. The evolution of cone changes is similar to that seen in rods. Cytoplasmic densification, axon elongation, and eventual reduction in number of cell bodies indicative of cell death follow initial cone outer-segment shortening. In the peripheral retina, shortened cone outer segments become surrounded by pyramidal-shaped RPE processes.568 Cones do not, however, undergo neurite sprouting to the extent seen in rods. In some RP eyes, peripheral cones occasionally evolve limited projections.535,566 In an RP eye with a rhodopsin 64ter mutation, enlarged cone axons that extended into the inner plexiform layer were noted.535 If cell death has been extensive, a monolayer of cone somata is still evident in the macula. A cell survival factor specifically elaborated by rods but important for cones has been discovered.569 This factor, termed rod-derived cone viability factor, is a truncated thioredoxin-like protein that holds promise for the generation of potential new treatment possibilities for RP.
Retinal vascular abnormalities can be a prominent feature in cases of RP. Perivascular cuffing547 and arteriolar attenuation are common features of advanced disease. Retinal blood flow decreases in advanced RP.570 This may be caused by this perivascular cuffing, which constitutes migrated pigment epithelium and deposited extracellular matrix, or it may be a phenomenon secondary to reduced metabolic need. Reduced cell density in the degenerating retina may lead to a reduced metabolic demand and therefore reduced flow.570
In a few RP patients, changes in retinal permeability can lead to exudative retinal detachment (Coats-like disease).63 This can be associated with peripheral retinal telangiectasia.571 Peripheral retinal ischemia and preretinal neovascularization have also been reported.60 Histopathologic evaluations of these vascular changes in humans however have only been reported from eyes that were already at the endstage of disease.
Genetic consultation
A diagnosis of RP always implies genetic disease. Therefore, once a diagnosis is suspected, further patient management is carried out within the special context of genetic consultation.572 This can be subdivided into three areas: (1) diagnosis; (2) genetic counseling; and (3) management of therapeutic options. The first steps in the management of a patient with RP are to establish an accurate diagnosis and a mode of inheritance. Both involve the traditional methods of history-taking and clinical examination. Clinical investigations and molecular genetic diagnosis can augment information gathered in this way. The patient whose diagnosis brought the family to the attention of physicians is called the proband or propositus.189
The scope for molecular genetic diagnosis is clearly opening up and a number of centers around the world now offer molecular diagnostic testing for a growing number of RP genes. A list of laboratories can be obtained through www.genetests.org. For other genes, molecular diagnosis can only be obtained through research laboratories.
Counseling family groups
Genetic counseling may be defined as a communication process that deals with the human problems associated with the occurrence, or risk of genetic disease, in a family. After a diagnosis has been made, the first important issue is to determine who should undertake the genetic counseling of an RP patient. Traditionally, this has been done by the ophthalmologist who establishes the diagnosis. With the recent advances in RP research, however, many ophthalmologists now refer such patients to specialist ophthalmologists who are more actively involved in the field. Recently it has been suggested that counseling should be undertaken by clinical medical geneticists, genetic counselors, or genetic nurses. This is in recognition of skills not formally within the training of ophthalmologists. There is increasing awareness of the need for more than just simple passing on of information about disease pathophysiology, mode of inheritance, and therapeutic options during the counseling process. The single most common patient complaint about genetic counseling is that the consultant does not address the problems that really concern the patient,573 which are more often psychologic and social problems than specific details of disease. One of the most psychologically stressful periods in the life of an RP patient consists of the events surrounding the diagnosis of RP. Michie et al.,574 in a survey of patients undergoing counseling, found that 50% expected direct advice, 30% expected help in making decisions, and 50% wanted simple reassurances about how the disease would affect their and their family’s lives. The RP patient is often more concerned about how to cope, now that driving is prohibited because of peripheral field loss, than what actual percentage of field remains. No clinical trials have been undertaken to determine the best way to undertake the counseling of RP patients, but, with the promise of future advances in genetic therapeutics, this is certainly an area that will change dramatically within the next decade.
A major principle of genetic counseling has been that it should be nondirective and supportive. However, it has been conceded that, to some extent, this is an ideal rather than a practical aim.574,575 Most RP patients, for example, those who are given options to choose whether to undergo cataract surgery, act on the recommendation of the consulting physician. Similarly, many patients are influenced by the manner of presentation of risks and options for family planning. Reproductive counseling should always present the various options in as nondirective a manner as possible.
Predictions of severity of disease can be influenced by a number of non-Mendelian factors that are becoming more recognized in ophthalmic genetic variable expressivity (variation in severity)576 and incomplete penetrance (disease gene carriers who are not symptomatic)494 is particularly common in autosomal dominant disease. Digenic inheritance459 (i.e., symptomatic individuals heterozygous for mutations in two separate genes) can also influence the predictions of severity. Other non-Mendelian influences, such as meiotic drive,577 anticipation,578 and imprinting,579 may also be relevant to predictions of disease severity in particular families.
Classically, X-linked traits are only symptomatic in affected males (hemizygotes). However, female carriers of X-linked RP can be symptomatic even with minimal fundus abnormality.580 Affected males cannot transmit the abnormal gene to their sons, but all daughters will be carriers (obligate heterozygotes). The variability and spectrum of clinical expression in heterozygotes are attributed to lyonization, the random inactivation of one of the two X chromosomes, with the extent of disease depending on the relative proportion of activity for the parental X chromosome that carries the mutant gene.40,41 Rarely, because of extremes of lyonization, a carrier for an X-linked disorder, such as X-linked RP or choroideremia, may be moderately to severely affected.
With the advent of molecular diagnosis, the possibility of prenatal diagnosis with the option of selective termination is becoming more of a reality in ophthalmic genetic disease.581–583 Prevailing opinion and current law relating to prenatal diagnosis with subsequent termination of an affected fetus vary from country to country. In the UK, it is considered justifiable if two registered medical practitioners perceive the handicap as severe (the Abortion Act 1967 and Human Fertilization and Embryology Act 1990). A similar situation exists in Germany, with the restriction that termination be undertaken before 22 weeks’ gestation (Strafgesetzbuch para. 218 and 218a abs. 3, amended 1995). No laws in the USA specifically forbid prenatal diagnosis for an ocular disorder with subsequent termination of an affected fetus, but in practice it has been rarely, if ever, done, at least for risk of a retinal dystrophy. In Ireland prenatal diagnosis for selective termination of pregnancy is prohibited. A definition of “severe” handicap is often not given. Childhood-onset recessive RP might be considered “severe,” whereas adult-onset pattern retinal dystrophy might not. Between these two extremes, opinion is divided and greatly depends on the definition of “severe handicap” as understood by the patient, the family, and the attendant practitioners.
In terms of ophthalmic genetic counseling, it is required to explain the severity of probable visual handicaps in terms that are understandable to the patient (e.g., relating loss of ability to read and to the extent of restriction of mobility rather than predicting probable Snellen visual acuity). To some extent comparisons can be made to the experience of another older affected family member. Many visually impaired individuals, despite severe visual deficit, are surprisingly well adjusted and lead highly productive lives. Such families may perceive the handicap as milder than would an outside observer, and support for affected children within these families may be so great that the risk of passing on the disease is not perceived as a large burden. When discussing such matters the practitioner needs to be particularly sensitive to racial, religious, and social influences. For instance, termination is rarely acceptable to Catholics or Muslims.584
No study has yet attempted to assess the requirements for prenatal diagnosis in ophthalmic genetics in the USA, although such studies have been undertaken in Europe. Members of the German Retinitis Pigmentosa Society participated in a questionnaire survey on attitudes toward prenatal diagnosis of RP.585 Of the 414 respondents, 64% thought that prenatal diagnosis was appropriate. A similar study in Sweden found that 60% had a positive attitude toward prenatal diagnosis.586 Both studies found that, although most people would accept an offer of prenatal diagnosis, this did not imply that they would then proceed with the termination of an affected pregnancy. In the former study, over 32% responded that they would disagree with termination if the child were destined to be blind soon after birth, and 61% would decline if the onset of blindness would occur in adulthood. The latter study also found that, although prenatal diagnosis would be used, over 30% would not use this information to decide on termination. These attitudes were expressed even though the fetus is subjected to risk during such tests. However, despite the theoretical possibilities and some evidence to suggest a modicum of patient need, we are not aware of much demand for prenatal diagnosis of RP in the USA or UK.
Support services
Patients can benefit greatly from appropriate referral to local, regional, or national agencies that provide services and support to the visually impaired. These services may be for visually impaired individuals in general or to those specifically afflicted with RP. All patients should be offered referral. Coordination by social services personnel familiar with working with the visually handicapped is appropriate. This is particularly true when dealing with issues such as integrating the visually impaired child into the school system or coordinating vocational rehabilitation training.587
Specific societies for RP patients have been established in many countries. In the USA there is the Foundation Fighting Blindness, which was formerly called the Retinitis Pigmentosa Foundation (7168 Columbia Gateway Drive, Suite 100, Columbia, MD 21046, USA. Tel: +1 (800) 683–5555+. www.blindness.org/). In the UK there is RP Fighting Blindness (PO Box 350, Buckingham, MK18 1GZ, UK. Tel: +44 01280 821334+, Fax: +44 1 712 723 862. info@rpfightingblindness.org.uk). In Canada, there is The Foundation Fighting Blindness Canada (890 Yonge Street, 12th Floor, M4W 3P4, Ontario, Toronto, Tel: ++ 14163604200, Fax: ++14163600060. www.ffb.ca). Addresses of some other RP societies are given below. The most updated addresses of RP societies in other countries may be found at www.retina-international.org/.
For deaf and blind RP patients the following contacts might be appropriate: in the USA, Helen Keller National Center, 141 Middle Neck Road, Sands Point, NY 11050, USA. Tel: +1 516 944 8900. In the UK, Sense, Sense Head Office, 101 Pentonville Road, London N1 9LG. Tel: +44 0845 127 0060. Fax: +44 0845 127 0061. info@sense.org.uk).
For problems of visual handicap in general, in the USA, Commissions for the Blind have been established in each state to help in vocational training of blind adults. State and regional programs for the visually impaired child also exist. In the UK, patients may be referred to the Royal National Institute of Blind People (RNIB), 105 Judd Street, London, WC1H 9NE, UK. Tel: +44 020 7388 1266. www.rnib.org.uk/).
[/level-membership-for-opthalmology-category][not-level-membership-for-opthalmology-category]
Chapter 40 Retinitis Pigmentosa and Allied Disorders
For additional online content visit http://www.expertconsult.com
RP may be seen in isolation (typical RP) or in association with systemic disease (syndromic RP). The prevalence of typical RP is approximately 1 : 5000 worldwide. In Maine, USA, the prevalence of typical RP has been found to be 1 : 5200, and the birth incidence of those who will eventually be affected with RP has been calculated as 1 : 3500.1 The prevalence of RP has been found to be 1 : 7000 in Switzerland,2 1 : 4016 in China,3 and 1 : 4500 in Israel.4 The highest frequency of occurrence, 1 in 1878, is among the Navajo Indians.5 The prevalence of syndromic RP is less well documented. The prevalence of Usher syndrome (RP with congenital deafness) is estimated to be 1 : 6000.6
Early history
The first description of the fundus findings and the use of the term “retinitis pigmentosa” are attributed to Donders in 1855 and 1857.7–9 However, the diagnosis of complicated night blindness (presumably RP) has probably existed in all cultures for hundreds, if not thousands, of years. Ovelgün, in 1744, made observations on familial complicated hemeralopia (or night blindness), which probably represented RP.9 Shortly after the discovery of the ophthalmoscope by Helmholtz in 1851, van Trigt10 in 1853 and Ruete11 in 1854 described cases that most assuredly were RP.
Typical retinitis pigmentosa
Clinical features
Nyctalopia
Difficulty with vision at night (night blindness) is one of the two hallmark symptoms of RP. Patients with typical RP usually attribute the beginning of night vision difficulties to the first or second decade of life. In some patients, especially those living in an urban setting, night vision problems may not be apparent until ocular disease is at an advanced stage. Tanino and Ohba12 noted that the onset of symptoms of RP, most commonly nyctalopia, occurred at a median age of 10.7 years in autosomal recessive disease and 23.4 years in autosomal dominant disease. The symptom of nyctalopia should not be confused with the symptom of blurred vision with night myopia or uncorrected refractive error. People with RP have a narrowing of the visual field in the dark and may describe getting easily disoriented on dimly lit evenings when others are able to see adequately. Becoming accident-prone, especially at night, is a highly suggestive symptom.
Visual field loss
The second hallmark feature or symptom of RP is an insidious, progressive loss of peripheral visual field. In some patients, especially those with severe disease beginning in childhood, this may be manifested as a progressive contraction of the visual field (see also Perimetry under Psychophysical findings). Loss of peripheral vision can be detected in early disease with small, dim test targets (Fig. 40.1). For many types of RP, field deficits are usually found first, and are most severe, in the superior visual field (Fig. 40.2). This reflects the early involvement of the inferior retina in RP. Occasionally the visual field loss will be greater temporally, nasally, or inferiorly (Fig. 40.3). See the section on Perimetry for specifics about testing the visual field.
Fig. 40.3 Goldmann kinetic perimetry for an 18-year-old man with autosomal recessive retinitis pigmentosa associated with posterior column ataxia (same patient as Fig. 40.15A and brother of patient in Fig. 40.21). Visual acuity was 20/30, J1, right eye, and 20/50, J2, left eye.
In a group of patients with several types of RP, Berson et al.13 found that overall about 4.6% of the remaining visual field was lost per year. Massof et al.14,15 found that, in most forms of generalized RP, the kinetic visual field shrank approximately 50% over 4.5 years. Massof and Finkelstein15 argue that the time course for field loss in all major genetic and functional classes of RP is nearly identical if one corrects for the critical age, which is defined as the age when visual field loss is first detected by a test target of given size and brightness. In conjunction with this concept, they have proposed a two-stage hypothesis for the natural course of RP. The first stage is a predisposition for retinal degeneration, such as genetic or environmental factors, which would be different for the separate types of RP and possibly among individuals. The second stage involves the actual retinal degeneration, which proceeds on its own internal time course and which would be common to most forms of RP.
In general, there is a strong tendency for the visual field loss to be symmetric between the two eyes.14,15 A notable exception to this is the phenotype expressed in female carriers of X-linked RP. In this situation, the distribution of mutant photoreceptors in the retina is determined by lyonization (X-chromosomal inactivation). This is a random event determining which genes of the two X chromosomes (the normal or mutant copy) are expressed in a particular cell. This leads not only to unusual, irregular patterns of visual field loss in individual eyes but also to quite striking differences in field loss between the two eyes.16 However, even in these cases the visual fields usually correspond quite closely to the fundus appearance, with the greater visual field loss in the eye with greater pigmentary abnormalities.
In typical RP the rate of progression of visual field loss is usually slow and relentless, with changes best appreciated when observed over many years or even decades. However, the visual fields may change dramatically over a few months or years. A patient may not notice what may be a striking loss of peripheral visual field if the central field remains clear. As the visual field reaches the stage of “tunnel vision,” the patient usually becomes acutely aware of subsequent change with time. This often leads the patient to the conclusion that the rate of degeneration is accelerating. In the USA, statutory legal blindness results when the remaining central visual field measures 20° or less horizontally in the better eye, using the III-4-e test target on a Goldmann perimeter.17 In the UK, most RP patients, with normal acuity, would be registered as partially sighted (“gross field defect”). Legal blindness occurs when fields become “very contracted” – a stage at which acuity loss has also usually occurred (www.dh.gov.uk/assetRoot/04/07/48/48/04074848.doc).
Central vision loss
There is a common misconception in RP (documented in older texts) that the central vision will remain good until most, if not all, the peripheral field is lost. Central visual function can, however, be seriously affected early in typical RP, while significant peripheral field remains.18 Cystoid macular edema (CME),19 diffuse retinal vascular leakage,20 macular preretinal fibrosis21 (Fig. 40.4), and retinal pigment epithelial (RPE) defects in the macula or fovea22,23 can occur early in the disease. Macular edema is the topic of a section on treatment of RP later in this chapter.
To a considerable extent, the likelihood of retaining “good vision” (central acuity) to a given age in life depends on the specific inheritance type of RP. Patients with regional (“sector”) RP may retain good visual acuity all their life. Patients with autosomal dominant RP (adRP) are more likely than patients with autosomal recessive (arRP) or X-linked RP to retain good acuity beyond 60 years of age.13 Marmor, in his study of visual loss in adRP and arRP,26 found that when visual acuity began to fail, it generally dropped to 20/200 within 4–10 (median 6) years. Patients with X-linked RP are usually blind (acuity 20/200 or worse) by 30–40 years of age.13,24,25 A good prognostic sign of retention of central acuity is electroretinograph (ERG) amplitudes remaining quite large (e.g., greater than 100 µV).26
Photopsia and other symptoms
Many patients with RP at some time during the course of their disease have light flashes, or photopsias. These are reported as occurring in the midperipheral field of vision, often adjacent to areas of relative or absolute scotomas. These photopsias are described as tiny, blinking or shimmering lights or as a coarse, sparkling graininess to vision. The phenomenon is similar to that reported by patients with ophthalmic migraine except that, although retinal disease involvement expands over the years, the photopsias are generally stationary within the field. Also, unlike ophthalmic migraine, the photopsias may be continuous rather than episodic. As the scotomas become denser over the years, the photopsias decrease and finally disappear. In a retrospective survey of symptoms and findings in 500 patients with RP, Heckenlively et al.27 reported light flashes in 170 (35%). Since 8 patients in this series had retinal detachments, the symptom of light flashes must be considered an indication for careful fundus examination.
The cellular or tissue correlates that underlie photopsias in RP are unknown but may include photoreceptor dysfunction, neurite sprouting, aberrant synapse formation, and secondary remodeling of the retina, all of which occur as sequelae of photoreceptor degeneration (see section on Cell and tissue biology: Histopathology, for further discussion).
Fundus appearance
The classically described fundus appearance of RP includes attenuated retinal vessels, mottling and granularity of the RPE, bone spicule intraretinal pigmentation, and optic nerve head pallor (Fig. 40.5). In general, when ophthalmoscopically detectable abnormalities of the fundus are present, there is a high degree of symmetry between the two eyes and fundus pigmentation is often greater in the midperiphery (Fig. 40.6). In advanced disease, atrophy of the RPE and choriocapillaris leads to fundus pallor and larger choroidal vessels become visible (Fig. 40.7). In some forms of RP and allied disorders, sufficient atrophy of the retina and choroid will occur during the late stages of the disease that the fundus appearance overlaps with certain of the primary choroidal atrophies (Fig. 40.7).28 As the disease advances to late stages, vessel attenuation can become so severe that the retinal vessels appear thread-like (Fig. 40.8).
Fig. 40.6 Fundus appearance of the right eye of a 40-year old patient with rhodopsin Pro23His retinitis pigmentosa. Areas of fundus changes correlated well with kinetic visual fields, performed at 39 years of age, as shown in Figure 40.19.
Fig. 40.7 Fundus appearance of the left eye of a 75-year-old patient with advanced rhodopsin Pro23His retinitis pigmentosa. This patient is the father of the patient whose visual field is shown in Figure 40.19 and whose fundus appearance is shown in Figure 40.6.
Almost all forms of RP go through a stage where the retina appears either normal or nearly normal, even though relative scotomas may be evident on visual field testing or areas of decreased retinal threshold may be present on static perimetry. Patients who have very early RP without fundus pigmentary abnormalities are often diagnosed as having RP sine pigmento or paucipigmentary RP. This is no longer considered a specific subtype of RP but a stage through which many, if not most, patients with RP pass. The sine pigmento stage may exist for decades before typical RP signs appear. For patients with high myopia and RP, the typical fundus features of high myopia may delay the appreciation of other retinal abnormalities (Fig. 40.9).
Fig. 40.9 Fundus of a 27-year-old man with high myopia and X-linked retinitis pigmentosa (same patient as in Fig. 40.2). Sparse but definite bone spicules were present in the inferior mid and far periphery. Discs showed a myopic tilt with peripapillary chorioretinal atrophy. The retinal pigment epithelium was diffusely hypopigmented.
When fundus abnormalities are evident, the earliest features are attenuation of retinal vessels and the appearance of fine mottling or granularity of the RPE in the mid and far periphery (Fig. 40.10). Spectral-domain optical coherence tomography (SD-OCT) imaging demonstrates that retinal thickness is decreased, particularly for outer layers, outside the macula and in areas of abnormal pigmentation with bone spicules.29,30 The macular region may exhibit increased luster, abnormal highlights, or wrinkling, suggesting either macular edema or early epiretinal membrane formation or preretinal fibrosis.19,22 In advanced stages, patients with RP may exhibit atrophic macular lesions (Fig. 40.11) that simulate what some authors have called (perhaps inappropriately so) “macular coloboma.”
Intraretinal, bone spicule pigment formations represent migration of pigment into the retina from disintegration of RPE cells with accumulation in the interstitial spaces surrounding retinal vessels. This process occurs most prominently at the junctions of vessels producing perivascular pigmentary cuffing and spicule-shaped deposits. The loss of pigment within the pigment epithelial cells often produces an overall gray, desaturated appearance to the retina with greater visibility of underlying choroidal vessels through the more transparent pigment epithelium. Eventually, the normal salmon-pink color of the entire mid and far peripheral fundus is replaced by dense bone spicule pigmentary formations. Heckenlively31 has described several patients with advanced RP with preservation of the RPE in a para-arteriolar distribution. Porta and colleagues32 described two additional cases. Van den Born et al.33 reported a large consanguineous family with preserved periarteriolar RP and confirmed the autosomal recessive inheritance. Almost all these patients were hyperopic. The gene for this form of RP (termed RP12) has been found to be the human homolog CRB1 of the Drosophila crumbs gene.
A yellowish-white metallic tapetal-like reflex or sheen can occasionally be observed in women who are carriers for the X-linked form of RP34 (Fig. 40.12) and rarely in young males with early disease35–37 (Fig. 40.13). With time, regional or diffuse mottling and atrophy of the RPE eventually replace the tapetal-like sheen. At least one form of dominant RP can show a tapetal-like reflex limited to the foveal and parafoveal region (Fig. 40.14). No histopathologic assessment has yet identified the source of a tapetal-like reflex. Cideciyan and Jacobson38 suggested that abnormal cones are the sources of the tapetal reflex. Berendschot et al.,39 using spectral fundus reflectance, confirmed that outer segments of peripheral photoreceptor cells are the source of the abnormal reflex but that this abnormality was not confined to cones. The carrier state for X-linked RP appears to be a slowly progressive retinal dystrophy in its own right, with a patchy distribution of affected retina as determined by lyonization.40,41 A number of studies have reported that the ERG is abnormal in most X-linked RP carriers.16,42–45 A tapetal-like reflex is not pathognomonic of X-linked RP but has also been reported in occasional cases of X-linked retinoschisis,46 and Oguchi disease.47
Fig. 40.13 Tapetal-like yellow-white reflex in a 13-year-old boy with X-linked retinitis pigmentosa.
The optic disc may be normal in early RP, show a waxy fullness with hyperemia, or appear waxy and pale (Fig. 40.15A). A “golden ring” or yellowish-white halo can often be seen surrounding the optic disc in early RP (Fig. 40.15B). As disease progresses, this golden ring is replaced with peripapillary mottling, hyperpigmentation, and atrophy of the RPE (Fig. 40.15C). The optic nerve head cup-to-disc ratio has been reported to be significantly smaller in RP patients of all types (0.19 compared to 0.35 in normal subjects).48 In advanced disease, dense optic nerve head pallor results, in part from optic atrophy and in part from gliosis overlying the discs.21 Drusen-like globular excrescences may develop on the optic nerve or adjacent retina (Fig. 40.16). These have been mistaken for astrocytic hamartomas49 and are consistent with disc drusen due to aberrant axoplasmic transport.50,51 In a survey of 117 RP patients,52 10% were found to have clinically apparent optic nerve head drusen. Edwards et al.53 suggested that this phenomenon is particularly common in RP associated with Usher syndrome type 1.
Fig. 40.15 Optic discs in an 18-year-old man with autosomal recessive retinitis pigmentosa as part of posterior-column ataxia and retinitis pigmentosa syndrome (A) (same patient as in Fig. 40.3), a 32-year-old man (B), and his 55-year-old father (C), both of whom have autosomal dominant retinitis pigmentosa. The disc in A shows peripapillary chorioretinal pallor, or the “golden ring” sign, that is characteristic of early retinitis pigmentosa. Note that the disc is nearly normal in the son (B), but shows severe waxy pallor in the father (C).
Flynn et al.54 found that the presence or absence of macular RPE defects is of significant prognostic importance with regard to retention of visual acuity over the next 5 years. Specifically, the absence of macular lesion was associated with only one line of acuity loss over the 5-year period, whereas bull’s-eye or geographic atrophy lesions were associated with a predicted acuity loss of three to four lines. The authors emphasize the importance of this type of information for prognostic counseling of patients with RP regarding the likelihood of preservation of visual acuity. Other maculopathies associated with RP include cellophane maculopathy, macular hole (complications of CME),55 and a single report of RP with central serous retinopathy.56
Vitreous abnormalities
The most common abnormality of the vitreous in RP is the presence of fine, dust-like pigmented cells released from degeneration of the RPE. In patients with RP, complete posterior detachment of the vitreous, “cotton-ball” opacities, interwoven filaments in the retrocortical space, and spindle-shaped vitreous condensations are seen more frequently than in normal subjects.57,58 A single case is reported of vitreous opacity so dense that vitrectomy was required.59 Vitreous abnormalities related to peripheral retinal ischemia and preretinal neovascularization have been reported in rare cases of adRP and arRP.60 Uncommonly, a peripheral Coats-like retinal vasculopathy with lipid exudations and serous retinal elevation can be seen in RP61–63 (Fig. 40.17). Unlike idiopathic Coats syndrome, the retinal vasculopathy seen in RP is usually bilateral, shows no sex predilection, and typically occurs in older patients. Coats-like disease is seen in 3.6% of RP cases64 and has been reported with adRP,62,63 arRP33 but not X-linked inheritance63 (although see Fig. 40.17). Coats-like changes that occur in children64,65 may change over a relatively short period of months to years, must be monitored carefully, and may need to be considered for laser or cryocoagulation. Vitreous hemorrhage related to preretinal neovascularization has also been described in rare cases.60,66 Retinal edema in some cases may be caused by inflammation resulting from the degeneration of outer retina.60,67
Anterior-segment abnormalities
Cataracts are frequent anterior-segment complications of RP. The prevalence of cataracts in RP patients aged 20–39 years varied with the inheritance type, being roughly 52% for autosomal dominant, 39% for recessive, and 72% for X-linked cases.68 The incidence of cataracts in RP is highly age-dependent.68,69 The most frequent type of cataract is a posterior subcapsular lens opacity, which occurs in 35–51% of patients.69,70 One study has suggested that posterior subcapsular opacities are most commonly associated with autosomal dominant inheritance (51%),69,70 whereas another has suggested a more common association with X-linked inheritance.71 Although keratoconus has been claimed to be more frequent among patients with RP,72 the occurrence of keratoconus in typical juvenile- or adult-onset RP is extremely rare in our experience. Glaucoma, especially primary open angle glaucoma, is also allegedly more frequent in RP.73 Sector RP appears to be associated with hyperopia and angle closure glaucoma.74 Because of the other abnormalities of the disc and fundus and because of the already abnormal visual fields, the diagnosis and management of glaucoma are quite challenging in RP patients.
Psychophysical findings
Perimetry
Assessment of visual field is an important aspect of the management of RP. This is an invaluable tool in the diagnosis of RP, as well as being the most reliable method of quantifying real change in visual deficit. It should be remembered when assessing the results of visual field testing that the reproducibility of visual fields is significantly less in patients with RP than in normal subjects. Kinetic visual fields tested on separate days by the same examiner varied by 5–6% in normal subjects but by 11–13% in patients with RP (intraobserver variation); variability between examiners for the same patient was 4–6% for normal subjects but 10–16% for patients with RP (interobserver variation).76
In the great majority of cases of RP, the earliest defects of the visual field, on kinetic perimetry, are relative scotomas in the midperiphery, between 30 and 50° from fixation (Fig. 40.18). These enlarge, deepen, and coalesce to form a ring of visual field loss. These midperipheral depressions of retinal sensitivity are readily documented as decreased retinal thresholds on static perimetry. As ring scotomas enlarge toward the far periphery, islands of relatively normal visual field remain, usually temporal but occasionally inferior or nasal (Fig. 40.19). The peripheral islands or remnants of visual field are often lost before the central field contracts sufficiently to qualify the individual as legally blind.
Fig. 40.19 Goldmann kinetic perimetry for a 36-year-old patient with retinitis pigmentosa from the Pro23His mutation of rhodopsin. Dense ring scotomas have broken through to the periphery superiorly, leaving a central field of 25° in diameter and only an incomplete annulus of peripheral vision in each eye. The fundus appearance of this patient at 40 years of age is shown in Figure 40.6.
Kinetic visual field testing has traditionally been performed with the manual Goldmann perimeter. Recent advances have occurred in both kinetic and static perimetry. Semiautomated kinetic perimetry,77 as implemented on the Octopus 101 and 900 perimeters (Haag-Streit, Köniz, Switzerland) (Fig. 40.20) automatically documents the testing and enables quantification of field by measurement of isopter area. A new fast thresholding algorithm, known as German adaptive thresholding estimation (GATE),78 has enabled another advance in perimetry, that of full-field static perimetry to assess the entire visual field in a time frame comparable to that required to test the central 30° field with the Standard 4–2-1 strategy (Fig. 40.20) (Weleber et al., 2012, in preparation). The static threshold data from such testing can be modeled as sensitivity values into a three-dimensional hill of vision from which the magnitude and extent of the total visual field can be determined as a volumetric measurement (Weleber et al. 2011, in preparation).
Fig. 40.20 Semiautomated kinetic perimetry in a 27-year-old woman with retinitis pigmentosa (top). The shaded areas depict the normal range for the isopters for the test targets presented. Three-dimensional (3D) model of the hill of vision for full-field static perimetry for this same subject (middle in tilted view, below in face view) using the size V test target and the German adaptive thresholding estimation thresholding algorithm.78 The volume of this model, which was 40.2 dB-sr OD and 28.0 DB-sr OS (compared to a normal 112.5 dB-sr for a normal), reflects the magnitude and extent of the visual field for this subject. Thus, this patient has 35.7% OD and 24.9% OS of expected field volume for his age. Note that the 3D model provides much more topographical information on the location of the defects in the field of vision than does the kinetic perimetry. This is the same patient whose fundus autofluorescence image for the left eye is shown in Figure 40.29A.
Two-color static perimetry has been shown to be a useful tool in assessing the dark-adapted visual fields of patients with RP. Using this technique, Massof and Finkelstein79–81 found that their patients with RP could be divided into two groups: (1) type I RP, which is associated with early diffuse loss of rod sensitivity relative to cone sensitivity and childhood-onset nyctalopia; and (2) type II RP, associated with regional and combined loss of both rod and cone retinal sensitivity and adult-onset nyctalopia. No patients have been observed to change from one type to the other.82 No families have been reported that contain both types I and II in the same sibship.15 An excess of type II RP over the type I form has been reported in simplex disease. Analysis of subgroups within simplex and multiplex RP suggest that a sizable proportion of type II patients may represent sporadic or acquired disease, and not true RP.83
Arden et al.84 studied a series of cases of adRP both with ERGs and psychophysically with scotopic static thresholds. They found that the preservation of sizable rod responses on the ERG correlated perfectly with the Massof type II classification. Absence of rod ERG did not always indicate type I disease, and both types were seen in this group. Lyness et al.85 reported clinical, psychophysical, and ERG findings in 104 patients (from 44 families) with adRP. Subgroup D (13 patients in four families) had diffuse loss of rod function and night blindness before the age of 10. Subgroup R (28 patients in 13 families) had regional loss of rod function, and most of these patients were unaware of night blindness until after the age of 20. Both groups had regional loss of cone functions. In the R group the rod-mediated ERG was usually present and substantial, whereas in the D group the rod ERG was undetectable. Although not identical, the D group bears similarities to Massof type I, and the R group bears similarities to type II. These studies suggest that the two types or groups may represent different pathophysiologic subtypes of RP. It should be emphasized, however, that a large proportion of patients (27 of the 44 families studied by Lyness et al.85) could not be incorporated into the D- versus R-type classification, partly because many patients with RP have almost undetectable ERG responses at presentation, which is quite common.
The techniques of two-color scotopic static perimetry have been further developed to evaluate rod and cone retinal sensitivities in different regions of the retina.86,87 Such testing allows the definition of which class of photoreceptor (rod or cone) is the determinant of retinal threshold in a particular region or area of the retina. This has allowed the characterization of the relative losses of rods and cones in various types of RP associated with specific mutations.88,89 Equally important to the definition of early retinal abnormalities in retinal degenerations, two-color perimetry can be used for monitoring disease therapy or for testing hypothesis of disease pathophysiology. For example, the abnormal, thickened Bruch’s membrane, which acts as a diffusion barrier between the retina and choroid, has been proposed as a major factor leading to night blindness in Sorsby’s fundus dystrophy.90 The return of rod function after oral vitamin A supplements (50 000 IU/day) was elegantly recorded using these techniques.90
Dark adaptometry
Normal subjects, when placed in the dark after exposure to a strong adapting light, will rapidly reduce their retinal psychophysical threshold using their cone system, reaching a plateau in about 5 minutes. Thereafter rod adaptation slowly increases, and rods determine retinal thresholds for another 3 log units of sensitivity before a second plateau occurs at about 30 minutes of dark adaptation. Patients with RP, when tested with dark adaptometry, may show elevation of the cone segment, the rod segment, or both, to varying degrees (Fig. 40.21). Also, in some patients there may be a delay in reaching what eventually for them is a relatively good final dark adaptation rod threshold91 (Fig. 40.22).
Fig. 40.21 Dark adaptometry showing marked elevation of both cone and rod thresholds with little or no cone–rod break in an 18-year-old woman, broken solid curve (A), with autosomal recessive retinitis pigmentosa and large-fiber sensory peripheral neuropathy (sister of patient shown in Figs 40.3 and 40.15A). Mild elevation of cone threshold with greater elevation of rod threshold in a 15-year-old young woman with Bardet–Biedl syndrome, broken solid curve (B). Borderline normal cone and rod segments in a 15-year-old young woman with type 2 Usher syndrome, broken solid curve (C). Dashed curve (M) is mean for normals aged 10–20 years.
Time course analysis of dark adaptometry has been performed in patients with adRP and prolonged dark adaptation is associated with rhodopsin mutations Thr17Met, Pro23His, Gly106Arg, and Thr58A.88,89,92 The most characteristic feature of the Pro23His genotype was prolonged dark adaptation, which was present in all patients regardless of their stage of disease.89 Jacobson et al.88 also reported prolonged rod dark adaptation in patients with Thr58Arg and Thr17Met mutations of rhodopsin. Indeed, prolonged rod dark adaptation appears to be a characteristic finding of virtually all rhodopsin mutations. These studies suggest that each of these mutations produces specific abnormalities in the rate of reactions within the rods that limit the recovery of scotopic sensitivity. For patients with regional forms of RP from other causes, zones of more normally functioning retina may allow reasonably good final rod thresholds (Fig. 40.22). Minor abnormalities in cone thresholds are more likely to result in complaints of poor dark adaptation than mild to moderate elevation of rod thresholds. One study has indicated that abnormalities in the normal interactions between rods and cones may underlie the symptoms of poor night vision in some patients rather than isolated rod deficits.93
Retinal densitometry (fundus reflectometry)
Retinal densitometry is a technique whereby, to deduce effective photoreceptor photopigment density, measurements are made of the difference between light shone into the eye and light reflected out of the eye. These measurements can be made and compared at different retinal loci, at different times, or against a retinal standard. From these data, estimates can be made of the rates of photopigment regeneration. Retinal densitometry has been performed on patients with RP by several investigators using a variety of techniques.94–97 Rhodopsin levels were found to be reduced in all patients, but rhodopsin bleaching and regeneration kinetics have been normal. Van Meel and van Norren98 found subnormal cone pigment levels in all patients studied and found prolonged cone pigment regeneration in eight of 12 patients with RP.
The losses of retinal sensitivity in RP in some patients are believed to relate to loss of quantum catch by the reduction of rhodopsin levels in the retina.94,95 A reduction of rhodopsin level to one-tenth of normal, for example, would be expected to be associated with a 10-fold elevation in the rod threshold. These calculations are only estimates, however. In vitamin A deficiency, the elevation of the rod threshold is much greater than predicted by the reduction in rhodopsin levels.95 Perlman and Auerbach99 described a group of patients with RP who, when studied with retinal densitometry, were found to have elevations of rod thresholds greater than what would be expected purely on the basis of reductions of rhodopsin levels. Kemp et al.100 found that 4 patients with type II (regional) adRP had elevations of retinal threshold that could be explained solely by loss of quantum catch from reductions of rhodopsin levels in the retina. However, 5 patients with type I (diffuse) adRP had elevations of threshold that were much greater than expected on the basis of loss of rhodopsin and quantal absorption for the reduction of rhodopsin levels observed.100 These studies provide evidence suggesting that the two classifications represent different pathophysiologic entities. Others have studied patients with defined mutations of rhodopsin. Kemp et al.89 have shown that, for patients with the Pro23His mutation of rhodopsin, rhodopsin levels are decreased below normal by amounts that indicate that rod sensitivity loss is determined by the reduced ability to absorb light. Similar findings were reported for patients with rhodopsin Thr17Met, Thr58Arg, and Gln344ter.88
Fulton and Hansen97 have reported reflectometry results on a mother and three daughters whose RP appeared to have a sectoral distribution. Elevations of retinal threshold were greater than those predicted by reductions of rhodopsin seen on densitometry measurements, suggesting that the dysfunction was central to the photoreceptor outer segments.
Electrophysiology
Karpe101 in 1945 first reported that the ERG was “extinguished” in RP. The standing, or resting, potential of the eye was first described by DuBois-Reymond in 1849. Riggs102 in 1954 was the first to report that the resting potential was decreased in pigmentary degeneration of the eye. Arden et al.103 in 1962 developed the currently widely used technique for measurement of the light-induced rise of the resting potential of the eye (the electro-oculogram (EOG)), which is considered a function of the RPE. Fast oscillations of the resting potential can be recorded, and these offer another measure of RPE function.104 Gouras105 in 1970 based clinical ERG on the use of Ganzfeld stimulation. Standardized conditions and assessment protocols have now been established for electrodiagnostic investigations.106,107
Electrodiagnostic responses in an RP patient can range from normal to undetectable. In general, more sizable responses are seen with younger patients or at earlier stages of disease. Most patients with advanced RP have undetectable responses (typically less than 10 µV) to single-flash, nonaveraged techniques.105 Computer averaging, however, can usually detect small ERG responses in even moderately advanced stages.13,108 In studying the natural course of RP, Berson et al.13 found that patients lost an average of 16–18.5% per year of remaining ERG amplitude to bright white flashes (a mixed rod–cone response).
Many studies have compared ERG responses in the different RP inheritance types. Tanino and Ohba12 and Berson,109 using conventional techniques, found that the ERG is more likely to be recordable in older patients with adRP than arRP. Berson et al.13 have also shown that progressive ERG loss of amplitude over a 3-year period is least in adRP. Birch and Fish110 found that the rate of progression of rod loss, as indicated by the half-saturation coefficient of the rod stimulus–response curve, varied among the inheritance groups. This ranged from 0.3 log unit/year in elevation of rod threshold in X-linked RP to 0.12 log unit/year elevation in adRP.
Agreement exists that the cone-mediated implicit times can be significantly prolonged in various forms of RP. Berson111 has also stressed the importance of delays in the implicit time for cone-mediated 30-Hz flicker responses. Berson and colleagues112,113 found that the cone-mediated b-wave implicit time in their patients was characteristically prolonged in adRP with reduced penetrance but not in completely penetrant adRP. Sandberg et al.114 suggested that this was due to an abnormal contribution of extrafoveal cones in adRP with incomplete penetrance. This may be due to abnormal cone light adaptation or a disturbance of normal rod–cone interactions.
Histopathologic study has indicated that, in RP, a postreceptor deficit may contribute to macular dysfunction.115 ERG study has confirmed this. Falsini et al.116 have assessed 8–10-Hz focal ERGs in 22 simplex and recessive RP cases. They found a loss of fundamental and second harmonic components and an increase in the fundamental-to-second harmonic ratio. This was interpreted as suggesting that both inner and outer retinal abnormalities contribute to visual deficit. Cideciyan and Jacobson117 have documented electronegative b-waves in some cases of RP. This finding was seen with abnormalities of on/off components of cone ERGs and reduced oscillatory potentials117 and was interpreted as indicative of significant inner retinal abnormalities in these RP cases.
Models of analysis of the ERG have been developed that attempt to isolate from the ERG the photoreceptor and bipolar contributions, specifically the original components of Granit’s model of the cat ERG, termed P3 and P2118 (Fig. 40.23A). These analysis methods have been developed to quantify the visual deficit attributable to outer and inner retinal disease (Fig. 40.23B–D). P3 is the corneal-negative response of photoreceptor outer segments that contributes to the a-wave of the Ganzfeld ERG. The analysis involves subjecting the leading edge of the a-wave of the ERG at a range of intensities to a mathematical curve-fitting algorithm119–121 (Fig. 40.23C) using equations that describe the kinetics of rod phototransduction activation.122 The method of Hood and Birch123 allows for estimation of two variables: S, which is a sensitivity factor, and Rmp3, which is the maximum amplitude. Such analysis can determine whether phototransduction is affected in a given disease.
P2 is the bipolar cell-derived component of the rod-isolated b-wave. The P2 component of the rod b-wave, derived from bipolar cells, can be isolated from the full-frequency ERG, using an algorithm whereby the P3 is first calculated for a given series of intensities, after which the P3 is subtracted from the series of rod responses124,125 (Fig. 40.23D). This gives a much better estimate of the ON bipolar contribution to the b-wave, especially for patients in whom the ON bipolar cells are more affected than the photoreceptors.124
Application of these techniques to 15 patients with RP has shown that, for P3 analysis, all patients had significantly reduced values of Rmp3, indicating effective loss or damage of rods, and a wide spectrum of values for S.126 Three patients had normal values for S. Four RP patients had subnormal S values but normal local retinal psychophysical thresholds in some regions of their retina. The rest had abnormal S values and abnormal visual fields with globally reduced thresholds. The rod b-wave amplitudes to a range of intensities for these RP patients were fitted with the Michaelis–Menten equation, generating the parameters Kbw (the semisaturation intensity, which is a sensitivity function) and Vmax (the maximum amplitude). For all patients, the changes in Kbw and Vmax followed the changes seen in S and Rmp3.126 Hood and Birch123 studied rod phototransduction in 11 patients with RP and four with cone–rod dystrophy (CRD). Their findings, which were similar to those above, supported the conclusion that the activation stage of rod phototransduction is affected in some forms of RP and CRD but not all.
Supernormal and delayed derived P2 responses were found by Hood et al.127 when they applied modeling analysis to an unusual retinal dystrophy, first reported by Gouras et al.128 in 1983 as “cone dystrophy, nyctalopia, and supernormal rod responses.” Although Gouras et al.128 initially attributed this dystrophy to an abnormality in receptor cyclic guanosine monophosphate (cGMP) activity, P3 and P2 modeling by Hood et al.127 showed that the delays in the rod and cone b-wave were not caused by the speed or amplification of phototransduction but resulted from delays beyond the outer segments involving a delay in activation of the inner nuclear layer activity. This disorder was subsequently clinically and electrophysiologically defined as the enhanced S-cone syndrome (ESCS)129,130 and was later shown to result from mutation of the gene NR2E3.131
Paradigms have been developed to study the kinetics of rod transduction deactivation.132 This technique uses a bright conditioning flash followed by a probe flash. The recovery of the amplitude of the response to the probe, which is presented at varying intervals after the conditioning flash, provides a measure of the return to baseline of the response to the conditioning flash and, hence, the kinetics of transduction deactivation. The authors conclude that the lifetime of activated rhodopsin in normal human rods is 2.3 seconds. This technique has been applied to humans with the Pro23His rhodopsin mutation, demonstrating a reduction of the gain of activation and marked slowing of deactivation of rhodopsin (by a factor of at least 5).132 Animal models of RP, for example, transgenic mice expressing the mutant Pro23His rhodopsin gene, have also demonstrated increases in the half-maximal recovery period, T50%, indicating abnormally prolonged recovery from the conditioning flash.133
Analysis of the a-wave leading edge has also recently shown that in RP, the efficiency of cone phototransduction is affected very early, even in some patients in whom the sensitivity of rod phototransduction is normal. X-linked inheritance seemed to be associated with greater phototransduction abnormalities in cones and rods at a younger age than seen in other modes of inheritance.134
Multifocal ERG techniques have been developed whereby, through patterned stimulation of the retina cross-correlated with recording of the ERG responses from a corneal electrode, very small ERG responses can be obtained from precisely localized regions of the retina135–137 (Fig. 40.24). Multifocal ERGs can be seen in advanced RP where the 30-Hz flicker response is barely detectable138 (Fig. 40.25). Hood and Li137 have shown that, in RP, amplitudes of ERG responses do not correspond as well as b-wave implicit times with the retinal sensitivity as measured by static perimetry (Fig. 40.26). This is most evident with the regional or sectoral types of RP. Thus the b-wave implicit time appears to reflect early involvement of the disease process in regions of the retina that still have relatively good retinal sensitivity. Robson et al.139 compared pattern ERG changes in RP patients with abnormalities of fundus autofluorescence (FAF). In such patients, pattern ERG responses suggested that visual loss was attributable to dysfunction rather than cell death.
In general, the EOG is abnormal in diffuse hereditary rod–cone degenerations of the retina.140 In typical RP, the slow and fast light-induced oscillations of the resting potential are usually decreased simultaneously early in the disease (Fig. 40.27). In some patients the fast oscillations of the resting potential are diminished earlier than the slow oscillation.104 In others the fast oscillations of the EOG are more preserved than the slow oscillation, a finding that also occurs in Best vitelliform macular dystrophy.104,141 In most cases of RP the EOG is abnormal when the ERG is abnormal.
Imaging modalities in RP
Fundus photography/fluorescein angiography
Documentation by fundus photography can assist in monitoring changes in patients with RP. Fluorescein angiography in patients with RP will demonstrate hyperfluorescence in areas of RPE atrophy and can highlight areas of CME (Fig. 40.28). However, fluorescein angiography has largely been supplanted by OCT for detecting cystoid maculopathy. In addition, concerns about light exposure accelerating certain forms of RP in animal models have prompted many ophthalmologists to exercise caution in obtaining excessive photographs.142
In many cases of mild to moderate RP, fluorescein angiography reveals transmission defects of the RPE with later diffuse leakage.143 In 78 of 82 patients with RP, Newsome144 found extravasation of dye from perifoveal capillaries in only a few patients but frequent abnormalities of the blood–retinal barrier at the level of the RPE. Such angiographically significant CME can often be seen at an early stage of disease in young patients who still retain good vision (20/25 or better). Macular edema is a significant cause of early loss of visual acuity in RP.144,145
Autofluorescence
FAF utilizes a scanning laser ophthalmoscope to stimulate intrinsically autofluorescent molecules of lipofuscin to visualize the RPE.146 Studies from patients with RP have shown that lack of signal on FAF correlate well with areas of RPE atrophy, while areas of increased FAF can be seen in areas with persistent macular edema as well as within areas of surviving retina.147 Most RP patients demonstrate a perifoveal ring (Fig. 40.29) of increased autofluorescence within the macula, which denotes the border between functional and dysfunctional retina (see Chapter 4, Autofluorescence).139,147–149 The border of the parafoveal ring of increased autofluorescence has been shown to correlate with functional status as measured by pattern ERG, multifocal ERG, scotopic fine matrix mapping, and microperimetery.150,151 In addition, areas outside the ring have been correlated with the loss of outer nuclear layer thickness and disruption of the inner-segment–outer-segment (IS/OS) junction on OCT.152,153 Near-infrared autofluorescence (NIA) has also been used to image melanin present in the apical tips of the RPE.154 Similar to FAF, rings with increased NIA are seen in patients with RP.155 Combined NIA and blue light FAF imaging suggest that the presence of NIA may correlate better with preserved cone function, while blue light FAF indicates only preservation of RPE cells.155
Optical coherence tomography
OCT has become one of the most utilized imaging modalities for studying retinal disease in the past several years. Ultrahigh-resolution OCT and SD-OCT have been used to study retinal structures in patients with RP.156,157 In these patients, such studies have demonstrated decreased thickness of the outer nuclear layer and loss of the external limiting membrane and IS/OS junctions. Loss of outer nuclear layer thickness or of the IS/OS has been correlated with visual defects measured by visual fields, microperimetry, or multifocal ERG.29,158–160 SD-OCT is especially useful for detecting CME (Fig. 40.30) or epiretinal membranes, which are common features in patients with RP.161 The ability to detect CME by OCT ring often eliminates the need for fluorescein angiography.
Adaptive optics scanning laser ophthalmoscopy
Traditional imaging modalities cannot resolve individual retina cells due to the optical limits imposed by the cornea and lens which create higher-order aberrations resulting in image blur.162 A combination of adaptive optics with flood illumination or scanning laser ophthalmoscopy can compensate for these factors and provide imaging of individual cone photoreceptors.163 Adaptive optics studies from patients with both CRD and RP have demonstrated increased cone spacing as well as qualitative areas where cone profiles could not be identified.164–166 Recent advances have now enabled imaging of rod photoreceptors and foveal cones.167 The ability to quantify cone photoreceptors has already been studied in one treatment trial for RP168 and will undoubtedly play a role in future studies.
