Resistance to Thyroid Hormone
Thyroid Hormone Action
It is now recognized that entry of thyroid hormones (thyroxine [T4] and triiodothyronine [T3]) into cells is not a passive process. Monocarboxylate transporter 8 (MCT8), a membrane protein, has been shown to mediate cellular thyroid hormone transport, particularly in the central nervous system (CNS). Intracellularly, deiodinase enzymes (DIOs) mediate hormone metabolism, with a high-affinity type 2 enzyme (DIO2) mediating T4-to-T3 conversion in the CNS, including the pituitary and hypothalamus, type 1 deiodinase (DIO1) in peripheral tissues generating T3, and type 3 deiodinase (DIO3) mediating catabolism of thyroid hormones to inactive metabolites. The effects of thyroid hormones on physiologic processes are mediated principally by a receptor protein, the thyroid receptor (TR), belonging to the steroid/nuclear receptor superfamily of ligand-inducible transcription factors, which modulates target gene expression in different tissues (Fig. 23-1). TR binds preferentially to regulatory DNA sequences (thyroid response elements [TREs]) in target gene promoters as a heterodimer with the retinoid X receptor (RXR), although the receptor can bind some TREs as a homodimer or monomer. In the absence of hormone, unliganded receptor homodimers/heterodimers recruit corepressors (e.g., nuclear receptor corepressor [NCoR], silencing mediator for retinoic acid and thyroid receptors [SMRT]) to repress or “silence” gene transcription. Hormone binding results in corepressor dissociation and relief of repression together with ligand-dependent transcriptional activation, mediated by a complex of coactivators (e.g., steroid receptor coactivator 1 [SRC-1], CREB-binding protein [CBP], and CBP-associated factor [pCAF]).1 In humans, two highly homologous thyroid hormone receptors, TRα and TRβ, are encoded by genes on chromosomes 17 and 3, respectively. Two different proteins are generated from the TRα gene locus by alternate splicing: TRα1 is a ubiquitously expressed receptor isoform with particular abundance in the CNS, myocardium, and skeletal muscle; TRα2, which exhibits a modified carboxy-terminal region such that it is unable to bind hormone, is expressed in a variety of tissues (e.g., brain and testis) where it may act as a functional antagonist of TR signaling pathways. The TRβ gene generates two major receptor isoforms, TRβ1 and TRβ2, which differ in their amino-terminal regions. TRβ1, which is widely expressed, is the predominant isoform in liver and kidney; TRβ2 expression is limited principally to the hypothalamus, pituitary, inner ear, and retina.2
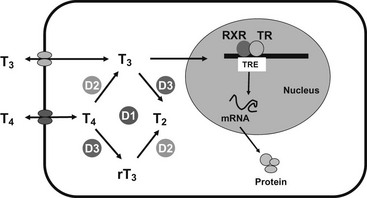
FIGURE 23-1 Transport, deiodination, and nuclear action of thyroid hormones. Transporters are required for passage of T3 and T4 across the plasma membrane, facilitating hormone uptake, efflux, or both. Deiodinases catalyze conversion of T4 to T3 (D1, D2) or inactivation of T4 to rT3 and T3 to T2 (D3). T3 interaction with its nuclear receptor (TR), usually part of a heterodimer with RXR, modulates target gene transcription and protein synthesis.
Differential Diagnosis of Elevated T4, T3 With Nonsuppressed TSH
A number of genetic disorders and clinical contexts are associated with elevated thyroid hormones and nonsuppressed thyroid-stimulating hormone (TSH) levels (Table 23-1). The first step in making a diagnosis is to verify the validity of hormone measurements. Confirmation of elevated free thyroid hormone levels in two-step or equilibrium dialysis assays excludes abnormal circulating binding proteins or antiiodothyronine antibodies. Preservation of linearity when TSH is assayed in dilution suggests this measurement is not artifactual. Many causes (nonthyroidal illness, psychiatric disorder, neonatal period, drugs) can be excluded by clinical context.
Table 23-1
Genetic disorders associated with elevated thyroid hormone levels can also be distinguished on the basis of different patterns of abnormal thyroid function (Table 23-2). The basis for such distinct biochemical profiles in each disorder is described later.

Resistance to Thyroid Hormone
RTH was first described in 1967 in two siblings who were clinically euthyroid despite high circulating thyroid hormone levels. The siblings exhibited several other abnormalities, including deaf-mutism, stippled femoral epiphyses with delayed bone maturation and short stature, and dysmorphic facies, winging of the scapulae, and pectus carinatum.3 It is now clear that some of these features are unique to this kindred in whom the disorder was recessively inherited. The majority of RTH cases that have been subsequently described are dominantly inherited, with a highly variable clinical phenotype. Affected subjects are either asymptomatic or have nonspecific symptoms and may be noted to have a goiter, prompting thyroid function tests that suggest the diagnosis. In these individuals, classified as exhibiting generalized resistance to thyroid hormone (GRTH), the high thyroid hormone levels are thought to compensate for ubiquitous tissue resistance, resulting in a euthyroid state. In contrast, a smaller number of individuals (around 15%) who share the same biochemical phenotype exhibit clinical features of thyrotoxicosis. In adults, these can include weight loss, tremor, palpitations, insomnia, and heat intolerance; in children, failure to thrive, accelerated growth, and hyperkinetic behavior have also been noted. When this clinical entity was first described, patients were thought to exhibit “selective” pituitary resistance to thyroid hormone (PRTH) action, with preservation of normal hormonal responses in peripheral tissues,4 but it is now recognized that peripheral resistance (typically hepatic) to hormone action is present even in these subjects. Less commonly, hypothyroid features such as growth retardation, delayed dentition, and bone age in children or asthenia and hypercholesterolemia in adults have been observed in RTH and may even coexist with thyrotoxic symptoms in the same individual.5 Taken together, these observations suggest that the clinical features of this disorder are influenced by the degree of refractoriness of peripheral tissues to high circulating levels of free thyroid hormones.
The estimated prevalence of RTH is 1 in 50,000 live births; the disorder can be diagnosed neonatally by screening with a combination of TSH and free T4 measurements.6 Over 700 cases of RTH (from more than 250 families) have now been described worldwide, enabling clinical characteristics of this disorder to be defined more precisely.
Goiter
A palpable goiter has been documented in 65% of individuals, particularly adult females. The enlargement is usually diffuse, with multinodular glands being typical of recurrent goiters following partial thyroidectomy. Development of toxic multinodular goiter on the background of RTH has been documented in a single case.7 Interestingly, it has been noted that fewer children with RTH born to affected mothers exhibit thyroid enlargement (35%) compared to offspring of unaffected mothers (87%), suggesting that maternal hyperthyroxinemia with transplacental passage of thyroid hormones during development might protect against goitrogenesis.8 The bioactivity of circulating TSH has been shown to be significantly enhanced in RTH, perhaps accounting for the goiter and markedly elevated serum thyroid hormones, despite the normal immunoreactive TSH levels observed in many cases.9
Cardiovascular System
Palpitations and resting tachycardia have been reported in approximately 75% of those with GRTH and almost all cases of PRTH, with a particular predisposition to atrial fibrillation in older subjects.8 The incidence of these symptoms is notably higher in RTH patients than in unaffected relatives or in the general population, although still less frequent when compared to patients with classic hyperthyroidism.10 In one study, although resting heart rates were comparable to unaffected family members, 30% of RTH subjects showed echocardiographic features of increased myocardial contractility and impaired diastolic relaxation, with a greater incidence of mitral valve prolapse.8 In a prospective study of cardiovascular involvement in a large cohort of children and adults with RTH, resting heart rate was significantly higher. Some indices of cardiac systolic and diastolic function (e.g., stroke volume, cardiac output, maximal aortic flow velocity) were intermediate between values in normal and hyperthyroid subjects. Other parameters (e.g., ejection and shortening fractions of the left ventricle, systolic diameter, and left ventricle wall thickness) were not different, indicating a partially hyperthyroid response of the heart in this disorder.10 Systemic vascular resistance and arterial stiffness are increased in RTH.11,12
Musculoskeletal System
Stippled epiphyses and winged scapulae were noted in the original RTH kindred but have not been observed in other cases. These features may represent a specific manifestation of the known gene deletion (TRβ) or an unrelated genetic abnormality in this consanguineous kindred.3 In contrast, growth retardation and delayed skeletal maturation are more common in childhood RTH patients, with height below the fifth percentile in 18% and delayed bone age (>2 SD) in 29%,8 with no significant differences between GRTH and PRTH cases. Despite these abnormalities, final adult height is often unaffected.13
Basal Metabolic Rate
The basal metabolic rate (BMR) is variably affected in RTH, being normal in some cases.14 In keeping with others,8 we have observed an elevated BMR, particularly in childhood RTH (Gurnell, Chatterjee, and Beck-Peccoz, unpublished observation), which may account for the abnormally low body mass index seen in approximately a third of children.
Central Nervous System
Two studies have documented neuropsychological abnormalities in RTH. First, a history of attention deficit hyperactivity disorder (ADHD) in childhood was elicited more frequently in patients with RTH (75%) compared to their unaffected relatives (15%).15 A second study showed that both children and adults with RTH exhibited problems with language development, manifested by poor reading skills and problems with articulation (e.g., speech delay, stuttering).16 Frank mental retardation (IQ < 60) is relatively uncommon (3%), although 30% of patients show mild learning disability (IQ < 85), probably due to uncompensated CNS hypothyroidism.14 A direct comparison of individuals with ADHD and RTH versus ADHD alone indicates an association with lower nonverbal intelligence and academic achievement in the former group.17 In detailed analysis of one family, RTH cosegregated with lower IQ rather than ADHD,18 so it is possible that low IQ facilitates the manifestation of ADHD. However, two different surveys of unselected children with ADHD failed to detect any cases of RTH by biochemical screening, suggesting that the latter disorder is unlikely to be a common cause of hyperactivity.19,20 Although magnetic resonance imaging (MRI) shows that anomalies of the sylvian fissure or Heschl’s gyri are more frequent in RTH, these features do not correlate with ADHD.21
Hearing and Vision
Significant hearing loss has been documented in 21% of RTH cases, similar to the prevalence reported in congenital hypothyroidism.8 In the majority, audiometric tests indicated a conductive defect, probably related to the increased incidence of recurrent ear infections in childhood RTH (67% in RTH versus 28% in normal controls). Abnormal otoacoustic emissions, consistent with cochlear dysfunction, have also been documented in those with hearing deficit,8,22 and cochlear expression of TRβ has been shown.23 The single kindred with deaf-mutism and recessively inherited RTH harbored a complete deletion of the TRβ gene,3 which correlates with the finding that TRβ knockout (KO) mice are deaf.24,25 Together, these observations underscore the importance of TRβ in auditory development and function. Deletion of the TRβ2 isoform in mice is associated with selective loss of M-cone photoreceptors and abnormal color vision,26 but monochromatic color vision has only been reported in the rare human kindred with recessively inherited RTH and a complete TRβ gene deletion.3 Detailed assessment of 10 subjects with TRβ point mutations and dominantly inherited RTH showed no common color-vision abnormalities (Gurnell and Chatterjee, unpublished observations).
Other Associated Disorders
Rarely, cases of RTH have been described where coexistent autoimmune thyroid disease has also been documented,27–29 raising the possibility of a pathogenic association between these disorders. Coexistence of Pendred syndrome and RTH has been documented in a single case.30 Pituitary enlargement, as a consequence of impaired negative-feedback regulation of TSH secretion, is another potential association with RTH. While pituitary hyperplasia has been reported in a single case, it occurred in the context of massively elevated TSH levels with suboptimal thyroxine replacement therapy following inappropriate thyroid ablation and regressed once TSH levels normalized.31 Only a few cases of RTH associated with pituitary adenomas have been described,32,33 suggesting that pituitary hyperplasia or adenoma formation are uncommon clinical sequelae in RTH, provided the altered set-point of the HPT axis is not perturbed. A greater frequency of recurrent upper respiratory tract and pulmonary infections has been reported in RTH, and affected individuals have reduced serum immunoglobulin levels.8 A retrospective study of a large Azorean kindred has shown a higher rate of miscarriage in mothers affected by RTH, with unaffected offspring being of lower birth weight, suggesting that intrauterine exposure to high TH levels does have adverse fetal effects.34
Differential Diagnosis
Differentiation of RTH, particularly the form associated with hyperthyroid features from a TSH-secreting pituitary tumor, can be difficult (Fig. 23-2). Similar abnormalities in thyroid function tests in first-degree relatives strongly suggest RTH, together with normal pituitary imaging and serum α-glycoprotein subunit levels.
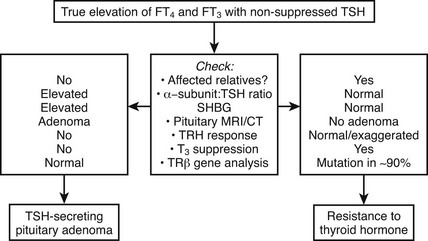
In addition to clinical features, the measurement of various tissue markers of thyroid hormone action has been suggested to be a useful method for evaluating the differing responses of various target organs and tissues to elevated circulating thyroid hormones (Table 23-3). These measurements are most useful in assessing the tissue effects of marked thyroid hormone excess (as typically found in overt thyrotoxicosis) but may be less discriminatory in borderline hyperthyroidism or in hypothyroidism. To improve the sensitivity and specificity of these parameters, it has been suggested that individuals with RTH be assessed by measuring tissue responses dynamically following the administration of graded supraphysiologic doses of T3 (50, 100, and 200 µg/day, each given for a period of 3 days), with comparison of any change in indices from baseline values to those observed in normal subjects.35
Table 23-3
Tissue Indices of Thyroid Hormone Action
| Pituitary: | Thyroid-stimulating hormone (TSH) |
| General: | Basal metabolic rate (BMR) |
| Hepatic: | Sex hormone–binding globulin (SHBG), ferritin, cholesterol |
| Muscle: | Creatine kinase, ankle jerk relaxation time |
| Bone: | Height, bone age, bone density, osteocalcin, alkaline phosphatase, pyridinium crosslinks, type 1 collagen telopeptide |
| Cardiac: | Sleeping pulse rate, systolic time interval, diastolic isovolumic relaxation time |
| Hematologic: | Soluble interleukin 2 receptor (sIL-2R) |
| Lung: | Angiotensin-converting enzyme (ACE) |
Molecular Genetics
Following the cloning of TRα and TRβ, RTH was shown to be tightly linked to the TRβ gene locus in a single family.36 This prompted analysis of the TRβ gene in other cases, and a large number of receptor defects have since been associated with this disorder. Eighty percent of RTH is familial, dominantly inherited, and associated with heterozygous mutations in the TRβ gene14,37–39; de novo receptor mutations occur in the remaining 20% of sporadic cases. Over 100 different defects including point mutations, in-frame deletions, and frameshift insertions have been documented to date, which localize to three mutation clusters within the ligand-binding domain of the receptor (Fig. 23-3). Within each cluster, some codon changes (e.g., R243W, R338W, R438H) representing transitions in mutation-prone CpG dinucleotides occur more frequently and are overrepresented.40
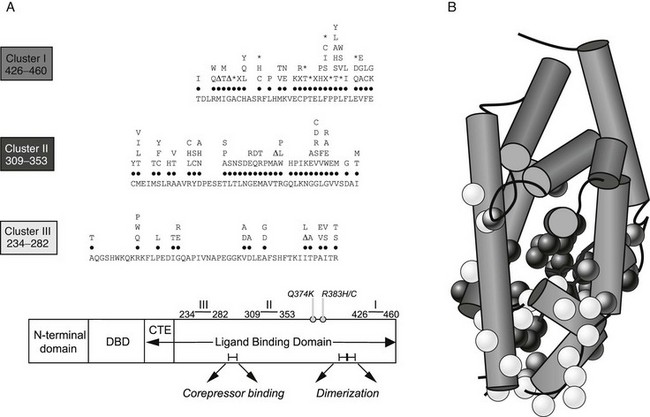
FIGURE 23-3 A, A schematic representation of the domains of TR-β showing that with two exceptions (Q374K, R383H/C), RTH receptor mutations localize to three clusters within the ligand-binding domain (LBD). The receptor defects described include different missense substitutions at each codon, in-frame codon deletions (Δ), premature termination codons (X), and frameshift mutations (*). The mutations shown include those listed in a public database (HGMD) together with our unpublished data. No RTH receptor mutations have been described in the zinc-finger DNA-binding domain (DBD) or its carboxy-terminal extension (CTE), which together mediate interaction with DNA, or regions in the LBD important for corepressor binding or dimerization with RXR. B, The crystal structure of the TR-β ligand-binding domain (LBD) (Protein Data Bank accession no. 1BSX) composed of 12 α helices is shown, with the location of missense mutations associated with RTH superimposed. As predicted from their functional properties, the majority of mutations involve residues surrounding the ligand-binding (T3) cavity.
Based on the supposition that PRTH was associated with selective pituitary resistance, it had been hypothesized that this disorder might be associated with defects in DIO2 or the TRβ2 receptor isoform, but a number of reports have documented TRβ mutations in PRTH.38,41,42 Receptor mutations found in individuals with PRTH have also been associated with GRTH in unrelated kindreds. Furthermore, even within a single family, the same receptor mutation can be associated with abnormal thyroid function and thyrotoxic features consistent with PRTH in some individuals but similar biochemical abnormalities and a lack of symptoms indicative of GRTH in other members. Overall, these findings indicate that GRTH and PRTH represent the phenotypic spectrum associated with a single genetic entity.
Non-TRβ RTH
In a small but significant number of cases (10% to 15%), clear-cut biochemical evidence of RTH is not associated with a mutation in the coding region of TRβ—so-called non-TRβ RTH. One explanation for this is somatic mosaicism, with occurrence of a TRβ mutation whose expression is selective, being detectable in some tissues but not peripheral blood leukocyte DNA.43 Alternatively, defects in other proteins involved in TR signaling have also been postulated. This latter hypothesis is supported by the description of kindreds with thyroid function tests and resistance to exogenous T3 similar to subjects with TRβ RTH, but in whom linkage and sequence analyses have excluded defects in TRβ and TRα genes.44,45 While it is theoretically possible that defects at any point in the pathway of thyroid hormone action could manifest as an RTH phenotype, evidence exists to favor some candidate genes such as RXR or the cofactors (e.g., corepressors, coactivators, TR-associated proteins) that regulate thyroid hormone–dependent gene transcription.
Mice harboring a deletion of the SRC-1 gene show abnormalities in thyroid function tests suggestive of RTH, together with subtle evidence of resistance in other steroid receptor axes.46 Similar findings were noted in mice doubly heterozygous for knockouts of the SRC-1 and transcriptional intermediary factor 2 (TIF-2) coactivator genes.47 To date, no homologous human disorder has been described, with linkage studies and direct sequence analysis of several cofactor genes (e.g., SRC-1, SRC-3, SMRT) in non-TRβ RTH kindreds or individuals failing to identify any abnormality.49 In one case, wild-type TRβ was found to exhibit aberrant binding to a unique 84-kD protein from patient but not control fibroblast nuclear extracts, suggesting abnormal receptor interaction with a cofactor44 whose identity has not been elucidated. It is known that patients with Rubinstein-Taybi syndrome, a disorder associated with heterozygous defects in the nuclear receptor coactivator CBP, exhibit a number of somatic abnormalities (broad thumbs, mental retardation, short stature) yet have normal circulating free T4 and TSH levels,50 indicating that mutations in this cofactor are not a cause of non-TRβ RTH. Several lines of evidence favor RXR as a candidate gene in non-TRβ RTH. First, knockout mice lacking the RXRγ isoform, whose tissue expression is limited but includes pituitary thyrotrophs, exhibit thyroid hormone resistance together with an increased metabolic rate.51 Second, the administration of RXR-selective agonists in humans inhibits pituitary TSH secretion, resulting in central hypothyroidism.52 Finally, in two kindreds with non-TRβ RTH, possible linkage to the RXRγ gene locus was noted,45,48 but in another study, no RXRγ gene mutations were identified in four non-TRβ RTH subjects.53 Together, these observations suggest that defects in pituitary-expressed RXRγ might also impair negative feedback in the pituitary-thyroid axis and manifest as RTH. Finally, it is tempting to speculate that a combination of “less functionally deleterious” mutations or even polymorphisms in several genes involved in thyroid hormone action could result in an RTH phenotype, representing an oligogenic basis for the disorder.
Properties of Mutant Receptors
Consonant with their location in the hormone-binding domain, the majority of receptor mutants identified in RTH exhibit moderate or markedly reduced T3 binding; consequently, their ability to activate or repress target gene expression is impaired.54,55 A subset of RTH mutations associated with markedly abnormal thyroid function in vivo and altered transcriptional function in vitro (but little impairment in ligand binding) have been described. Such natural mutations involve residues that mediate receptor interaction with transcriptional coactivators.39,56 In the first RTH family described, with the recessively inherited form of the disorder, the two affected siblings were found to be homozygous for a complete deletion of both alleles of the TRβ receptor gene.57 Importantly, the obligate heterozygotes in this family harboring a deletion of one TRβ allele were completely normal with no evidence of thyroid dysfunction. This suggested that simple deficiency of a functional β receptor, as a consequence of the single deleted TRβ allele, was insufficient to generate the resistance phenotype. This led to the hypothesis that the heterozygous mutant receptors in dominantly inherited RTH were not simply functionally impaired but also capable of inhibiting wild-type receptor action. Studies confirmed that when coexpressed, the mutant proteins are able to inhibit the function of their wild-type counterparts in a dominant-negative manner.58,59 Further clinical and genetic evidence supporting this notion have been provided by two rare examples of RTH. In the first, a childhood case, severe resistance with marked developmental delay and growth retardation associated with cardiac hyperthyroidism was ultimately fatal due to heart failure following septicemia; this individual was homozygous for a mutation (Δ337T) in both alleles of the TRβ gene.60 In the second more recently reported case, the affected subject also exhibited a particularly severe clinical phenotype and was found to be either homozygous or hemizygous for a TRβ mutation (I280S).61 Presumably, the extreme phenotype observed in both cases reflected not only the absence of normally functioning TRβ but the added dominant-negative inhibitory effect of mutant β receptors.
Functional studies of mutant receptors indicate that although they are transcriptionally impaired and dominant-negative inhibitors, their ability to bind DNA and form heterodimers with RXR is preserved.54,55 Conversely, it has been shown that the introduction of additional artificial mutations that abolish DNA binding or heterodimer formation abrogates the dominant-negative activity of mutant receptors in vitro.55,62,63 Mice heterozygous for a TRβ mutation lacking DNA binding do not exhibit RTH.64 It has also been suggested that the ability of mutant receptors in RTH to repress or “silence” basal gene transcription is likely to be an important factor contributing to their dominant-negative potency. Non-T3-binding mutants exhibit constitutive silencing function, particularly when bound to DNA as homodimers, which cannot be relieved by ligand. Conversely, RTH mutants with impaired homodimerization properties are weaker dominant-negative inhibitors.65 With the identification of corepressors, these observations have been extended to show that some RTH mutants either bind corepressor more avidly when unliganded or fail to dissociate fully from corepressor upon T3 binding.66 Furthermore, artificial mutations that abolish corepressor binding abrogate the dominant-negative activity of RTH receptor mutants.66 It has also been suggested that corepressors mediate basal activation of negatively regulated gene promoters (e.g., thyrotropin-releasing hormone [TRH], TSH-α, TSH-β) by unliganded TR.67 An unusual RTH receptor mutant (R383H) exhibits both delayed T3-dependent corepressor release and impaired hormone-dependent negative transcriptional regulation.68 Given the pivotal role of negatively regulated target genes in the pathogenesis of RTH, aberrant corepressor recruitment or release may well prove to be the critical receptor abnormality in this disorder.
Together, these observations allow a model to be constructed (Fig. 23-4) in which occupancy of target gene binding sites by mutant receptor-corepressor complexes mediates dominant-negative inhibition by RTH mutants. Mapping of the three clusters of RTH receptor mutations identified hitherto on the crystal structure of the ligand-binding domain of TRβ69 provides insights into structure-function relationships in TR (see Fig. 23-3). As expected from their impaired ligand-binding properties, most mutations are located around the hormone-binding cavity, and receptor regions mediating DNA binding, dimerization, and corepressor interaction are devoid of naturally occurring mutations, possibly because they lack dominant-negative activity and therefore elude discovery—being biochemically and clinically silent.
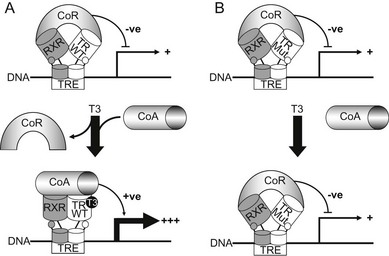
FIGURE 23-4 A model for dominant-negative inhibition by mutant receptors in RTH. Left panel (A) depicts current understanding of wild-type TR action on target genes. The unliganded TR-RXR heterodimer or homodimer (not shown) recruits a corepressor (CoR) complex to inhibit or silence basal gene transcription. Receptor occupancy by T3 promotes corepressor dissociation and derepression, followed by binding of coactivators (CoA), which leads to target gene activation. Right panel (B) shows RTH mutant receptor action. In comparison to wild-type TR, the primary defect in mutant receptors may be impaired hormone-dependent corepressor dissociation and coactivator recruitment. For the majority of receptor mutants, this functional alteration is a consequence of their reduced ability to bind ligand, but a subset of mutants exhibit enhanced corepressor binding, delayed corepressor release, or impaired coactivator recruitment, with relative preservation of hormone binding. Mutant receptor-CoR complexes compete with their wild-type counterparts for occupancy of promoter thyroid response elements (TREs), resulting in inhibition of target gene expression. In this model, DNA binding, dimerization, and corepressor interaction are functional properties preserved in mutant receptors and required for their dominant-negative activity.
Pathogenesis of Variable Resistance
Genetic and functional evidence suggests that the ability to exert a dominant-negative effect on target genes within the HPT axis is a fundamental property of RTH receptor mutants, generating the abnormal thyroid function characteristic of the disorder. Indeed, some studies indicate that for a subset of RTH mutants, there is a correlation between their functional impairment in vitro and the degree of central pituitary resistance, as measured by the magnitude of elevation in serum-free T4 in vivo.70,71 On this biochemical background, the heterogeneous clinical phenotype may be due to differing degrees of peripheral resistance in different individuals as well as variable resistance in different tissues within a single subject. A number of factors may contribute to such variable tissue resistance.
One important contributory element may be the differing tissue distributions of receptor isoforms. The hypothalamus/pituitary, and liver express predominantly TRβ2 and TRβ1 receptors, respectively, and TRα1 is the major species detected in myocardium. Mutations in the TRβ gene are likely to be associated with pituitary and liver resistance, as exemplified by nonsuppressed TSH and normal sex hormone–binding globulin (SHBG) levels seen in patients, whereas the tachycardia and cardiac hyperthyroidism in some cases may represent retention of myocardial sensitivity to high circulating thyroid hormones acting via a normal α receptor. Another factor that may influence the degree of tissue resistance is the relative expression of mutant versus wild-type TRβ alleles. One study has suggested that both alleles are equally expressed72; another showed marked differences in the relative levels of wild-type and mutant receptor messenger RNA in skin fibroblasts from two RTH cases.73 In one of these individuals, a temporal variation in expression of the mutant allele in fibroblasts appeared to correlate with the degree of skeletal tissue resistance. The dominant-negative inhibitory potency of mutant receptors has been shown to differ depending on target gene promoter context55,74 and is a further variable that may influence the degree of resistance. Finally, factors unrelated to the TRβ gene might influence the phenotype. For example, a deleterious R316H mutation was associated with normal thyroid hormone levels in some members of one kindred75 but clearly abnormal thyroid function in an unrelated family,38 suggesting that other genetic variables can modulate the effect of receptor mutations.
While the absence or presence of overt thyrotoxic features allows patients to be classified as either GRTH or PRTH—a clinical definition that will probably remain useful as a guide to the most appropriate form of treatment—studies indicate that there is some overlap of features between the two forms of the disorder. For example, there are no significant differences in age, sex ratio, frequency of goiter, thyroid function, or clinical features between patients with GRTH or PRTH.76 Importantly, features such as tachycardia, hyperkinetic behavior, and emotional disturbance have been documented in individuals with GRTH.76 Conversely, serum SHBG, a hepatic index of thyroid hormone action, is almost invariably normal in patients with PRTH, suggesting that tissue resistance is not solely confined to the HPT axis in this group of patients.77
Attempts to correlate the phenotype of RTH with the nature of the underlying TRβ mutation have been confounded by three factors: (1) the relative imprecision of clinical criteria used to define GRTH and PRTH; (2) the apparent temporal variation in hyperthyroid features in some RTH cases, such that thyrotoxic symptoms and signs can develop and disappear spontaneously when individuals are followed over several years76; and (3) the relatively small number of patients with any given mutation that have been identified. Nevertheless, some interesting correlations have emerged from the published literature. The first patient reported to have PRTH4 was found to harbor an R338W receptor mutation,41 and the same phenotype has been described in the majority of individuals with this or similar substitutions at this codon.38,42 Interestingly, when tested in vitro, this mutant exhibits dominant-negative activity with the negatively regulated pituitary TSH α subunit gene promoter, but it is a relatively poor inhibitor of wild-type receptor action in other promoter contexts.55 When introduced into other RTH receptor–mutant backgrounds, this mutation weakens their dominant-negative potency on positively regulated reporter genes.78 A patient with the R383H receptor defect, which is impaired mainly in regulation of TRH and TSH genes, exhibited predominant central resistance following T3 administration,79 and the R429Q mutation, with similar functional properties, may also occur more frequently in association with PRTH. Some receptor mutants (R338W or L, V349M, R429Q, I431T) associated with PRTH are either more deleterious80 or exert a greater dominant-negative inhibitory effect in a TRβ2 than a TRβ1 context.81A receptor mutation that selectively fails to bind NCoR but not SMRT is associated with PRTH.82
Animal Models
The generation of various receptor knockout mice has greatly enhanced our understanding of the physiologic roles of individual TR isoforms, particularly in regulation of the HPT axis. For example, homozygous TRβ gene deletion (TRβ KO), leading to absence of both TRβ1 and TRβ2 isoforms, increased circulating thyroid hormone levels approximately threefold,24 whereas deletion of the TRα1 isoform had no effect.83 When only the TRβ2 isoform was deleted, the biochemical phenotype was similar to that of TRβ KO, suggesting that TRβ2 is the key isoform mediating feedback regulation of hypothalamic TRH and pituitary TSH output.84 Importantly, many of the features in the recessively inherited cases of RTH associated with a deletion encompassing the human TRβ gene were recapitulated in TRβ KO mice.24 Homozygous animals exhibited elevated serum thyroid hormones and an inappropriately elevated TSH, while their heterozygous littermates were biochemically normal. To explore the properties of mutant TRβs in RTH in vivo, several groups have developed transgenic mice in which dominant-negative TRβ mutants have been overexpressed either ubiquitously85 or selectively in tissues.86,87 These models have provided valuable insights into mutant receptor function and pathophysiologic mechanisms that mediate RTH. For example, selective targeting of an RTH mutant to the pituitary using a tissue-specific promoter generated transgenic mice with elevated TSH but only marginally raised T4 levels. This suggests that the additional dominant-negative effect of mutant receptors on the hypothalamic TRH gene might be required to produce the full biochemical phenotype.87 In contrast, ubiquitous transgenic mutant TRβ expression resulted in an animal model with more generalized tissue resistance; these mice displayed decreased body weight, hyperactivity, and learning deficit, which are recognized features of the human syndrome.85 An important limitation of these animal models is that expression of the mutant receptor transgene is not controlled by the TRβ gene promoter, so the pattern of mutant receptor expression or the resulting phenotype might not correspond with that of human RTH. Transgenic mice in which either a frameshift mutation involving 14 carboxy-terminal amino acids (TRβ PV)88 or an in-frame deletion of a threonine residue (Δ337T)89 have been introduced into the TRβ gene locus have also been generated. Both these TRβ mutations have been identified in human RTH, and the mutant receptors exhibit markedly impaired transcriptional activation and potent dominant-negative activity in vitro. Extensive characterization of the phenotype of TRβ PV88,90 and Δ337T mice has indicated that these animal models recapitulate the human RTH phenotype, with heterozygous mice exhibiting mild to moderate resistance and homozygous littermates severe resistance in the HPT axis. Interestingly, when compared with TRβ KO mice, thyroid hormone and TSH levels were significantly more elevated in the Δ337T knock-in animals, supporting the notion that dominant-negative inhibition by the mutant receptor antagonizes residual TRβ1 activity in the HPT axis. Both heterozygous and homozygous Δ337T mice exhibited abnormalities of vestibulomotor function which correlated with an overall reduction in cerebellar size and in the area of the Purkinje cell layer.
Studies with TRβ PV mice have provided insights into the molecular basis for dominant-negative activity in vivo, confirming that mutant receptor homodimers and heterodimers compete with wild-type TRβ for binding to target gene TREs. The interplay of receptor isoform predominance (e.g.,TRβ1 in liver, TRα1 in heart) together with the promoter context of target gene TREs can influence the degree of dominant-negative inhibition observed in different tissues.90 Interestingly, crossing TRβ PV mice with SRC-1 KO animals enhanced the degree of resistance in the HPT axis in heterozygous TRβ PV mice, providing evidence that coactivator “availability” can also modulate mutant TRβ action in vivo.91 Metastatic thyroid carcinoma was an unexpected finding in older, homozygous TRβ PV mutant mice but has not been observed in their heterozygous counterparts.92 Mice harboring a TRβ mutation (R429Q) associated with PRTH exhibit greater dysregulation of negative versus positively regulated target genes, providing a mechanism for correlation of some TRβ genotypes with phenotype.93
Finally, it is appropriate to include a brief description of mice harboring mutations in the TRα gene locus. Selective knockout of the TRα1 isoform was associated with low or normal serum thyroid hormones, a decreased heart rate, and lower body temperature—a phenotype quite dissimilar to RTH.83 Knock-in mice with heterozygous point mutations in TRα that correspond to naturally occurring TRβ mutations in RTH have either normal or mildly reduced thyroid hormone levels,95–98 with additional features including bradycardia, growth retardation, CNS abnormalities,95,98 and insulin resistance,96 which may provide important clues to the probable phenotype of a homologous human disorder.97
Management
The management of RTH is difficult, since variable resistance makes it difficult to maintain euthyroidism in all tissues. In general, the presence or absence of hyperthyroid features is a useful guide to the need for therapy. In most individuals, the receptor defect is compensated by high circulating thyroid hormone levels, leading to a euthyroid state not associated with abnormalities other than a small goiter. Attempts to treat the biochemical abnormality with surgery or radioiodine are usually unsuccessful, with recrudescence of the goiter (often nodular in nature) and disruption of the thyroid axis.14 Certain circumstances such as hypercholesterolemia in adults or developmental delay and growth retardation in young children may warrant the administration of supraphysiologic doses of L-T4 to overcome a higher degree of resistance in certain tissues. Although successful in some cases,14 such therapy needs careful monitoring of indices of thyroid hormone action (e.g., SHBG, heart rate, BMR, bone markers) to avoid the adverse cardiac effects or excess catabolism associated with thyroxine overtreatment. Inappropriate thyroid ablation also renders the RTH patient hypothyroid, with elevated TSH levels and risk of thyrotroph hyperplasia,31 and is another context in which supraphysiologic thyroxine replacement is indicated. Alternate-day administration of L-T3 in supraphysiologic dosage led to significant regression of goiter without inducing thyrotoxic symptoms in one case.99
In contrast, a general reduction in thyroid hormone levels may be of benefit in the management of patients with thyrotoxic symptoms. However, the administration of conventional antithyroid drugs usually causes a further rise in serum TSH levels that stimulates thyroid enlargement and may also induce thyrotroph hyperplasia, with a theoretic risk of developing autonomous neoplasms at either site. Accordingly, agents that inhibit pituitary TSH secretion but are relatively devoid of peripheral thyromimetic effects are administered to reduce thyroid hormone levels. The most widely used example is the thyroid hormone analog 3,5,3′-triiodothyroacetic acid (TRIAC), which has been shown to be beneficial in both childhood and adult cases.100–102 This compound has a number of interesting properties that make it an attractive therapeutic option in RTH: (1) it exerts predominantly pituitary and hepatic thyromimetic effects in vivo,103 target tissues that are relatively refractory to thyroid hormones in RTH; (2) it has greater affinity, potency, and activity than T3 for TRβ104; and (3) it exhibits a higher affinity for TRβ than TRα in vitro.105 A daily dose of 1.4 to 2.8 mg is generally used, and one study has suggested that twice daily administration might be optimal in inhibiting TSH secretion.106 TRIAC has been successfully used in pregnancy to control maternal thyrotoxic symptoms but may have induced fetal goiter.107 Treatment with TRIAC is not always effective, however,108 and dextrothyroxine is another agent that has been shown to be useful in some cases.109,110 If these compounds fail, the dopaminergic agent, bromocriptine,111 or the somatostatin analog, octreotide,112 may be administered, but past experience indicates that TSH secretion escapes the inhibitory effects of both bromocriptine100,111 and octreotide.113 In view of the spontaneous variation in thyrotoxic symptoms in RTH, periodic cessation of thyroid hormone–lowering therapy and reevaluation of the clinical status of the patient is advisable. In rare circumstances such as severe thyrotoxic cardiac failure associated with RTH, thyroid ablation followed by subphysiologic thyroxine replacement may be indicated.
The treatment of thyrotoxic features (e.g., failure to thrive) in childhood RTH also requires careful monitoring to ensure that any reduction in thyroid hormone levels is not associated with growth retardation or adverse neurologic sequelae. Indeed, control of cardiac and sympathomimetic manifestations with β-blockade may be the safest course in this context. One study reported that L-T3 therapy improved hyperactivity in nine children with ADHD and RTH, including three individuals who were unresponsive to methylphenidate.114
The therapeutic potential of TRIAC in RTH could be limited by the nature of the TRβ mutation, with some mutant receptors being less responsive, such that the dose of TRIAC required to activate mutant TRβ function is associated with unwanted TRα-mediated toxicity (e.g., tachycardia). Accordingly, the identification of compounds with higher affinity and selective agonist activity for mutant TRβ than for normal TRβ or TRα would represent a major therapeutic advance: TRβ-selective thyromimetics (e.g., GC1, KB2115) are being developed and may have utility in treating some abnormalities (e.g., dyslipidemia) in RTH.115,116 HY1, an analog of GC1, is five times more potent with an R320C TRβ mutant than wild-type TRβ, suggesting that it may indeed be feasible to design hormone analogs that selectively overcome abnormal mutant receptor function without further activating normal β or α receptors.117 Rational molecular design has also led to the development of TR isoform–selective antagonists that may also be useful in, for example, controlling TRα-mediated toxic symptoms in RTH.118
Disorder of Thyroid Hormone Transport
Worldwide, at least 50 families have been reported in which males are affected by severe psychomotor retardation associated with a particular combination of abnormal serum thyroid hormone levels. In 1944, long before thyroid involvement was suspected, the first description of this syndrome of X-linked mental retardation (XLMR) in a large family was published by Allan, Herndon, and Dudley.119 Since then, this disorder has been typically referred to as the Allan-Herndon-Dudley syndrome (AHDS). Only 60 years later was it realized that patients with AHDS also have abnormal thyroid function tests.120–122
Usually, patients with AHDS are born at term after an uncomplicated pregnancy, with a normal birthweight, body length, and head circumference. During the first 6 months, general hypotonia is noticed. During development, truncal hypotonia persists, resulting in poor head control, whereas distal hypotonia progresses into spasticity. Growth is relatively normal, but final body length is reduced, and body weight is usually extremely low with obvious signs of muscle wasting. There is also progressive microcephaly. In the first 2 years of life, brain MRI shows delayed myelination, but this normalizes in subsequent years. Based on the delayed myelination, this combination of clinical features has also been referred to as Pelizaeus-Merzbacher-like disorder (PMLD).123
Although the clinical phenotype is somewhat milder in some families, AHDS patients are usually incapable of sitting, standing, or walking independently and do not develop any speech. They are severely mentally retarded, with IQ values below 40. Feeding is a problem in AHDS patients; they have swallowing difficulties, and aspiration is a frequent cause of pneumonia. Patients with AHDS usually have a friendly nature. Recent reviews provide a detailed description of the clinical features of patients with AHDS.123–126
In addition to severe psychomotor retardation, AHDS patients have a characteristic combination of abnormal serum thyroid hormone levels.125 This is also observed in a subset of patients with PMLD.123 Both T4 and free T4 levels are either low-normal or clearly reduced, whereas serum T3 and free T3 are markedly elevated, and rT3 is always low. Consequently, circulating T3/T4 and T3/rT3 ratios are markedly elevated. Although within the normal range, mean serum TSH levels in AHDS patients are about twice that in healthy controls. Serum SHBG levels are markedly elevated, and several studies have reported raised serum lactate in young patients.127,128
Molecular Genetics
In all male patients with the characteristic combination of psychomotor retardation and abnormal serum thyroid hormone levels, different mutations in the monocarboxylate transporter 8 (MCT8) gene have been identified (Table 23-4). Both MCT8 and the highly homologous protein, MCT10, have been shown to be specific and active thyroid hormone transporters, although MCT10 is also capable of transporting aromatic amino acids.129–131 These proteins belong to a wider family of MCT transporters, so named because MCTs 1 to 4 have been shown to facilitate transport of monocarboxylates such as lactate and pyruvate.132 The function of most other MCTs is as yet unknown.

MCT8 and MCT10 have identical gene structures; both consist of six exons and five introns, with a particularly long first intron (~100 kb). The MCT10 gene is located on chromosome 6q21-q22, encoding a protein of 515 amino acids. The MCT8 gene is located on chromosome Xq13.2 and has two possible translation start sites, generating either 613 (long) or 539 (short) amino acid protein products (Fig. 23-5). The significance of the N-terminal extension in the long form of the MCT8 protein (shown in light gray in Fig. 23-5) remains to be elucidated. All in vitro studies of the function of wild-type and mutated MCT8 have been carried out in the short protein context. Like MCT10, MCT8 has 12 putative transmembrane domains (TMDs), with both the N-terminal and C-terminal ends of the protein being located intracellularly. The amino acids that are identical and occupy corresponding positions in MCT8 and MCT10 are highlighted (see Fig. 23-5), showing that homology between these proteins is particularly high in their TMDs.
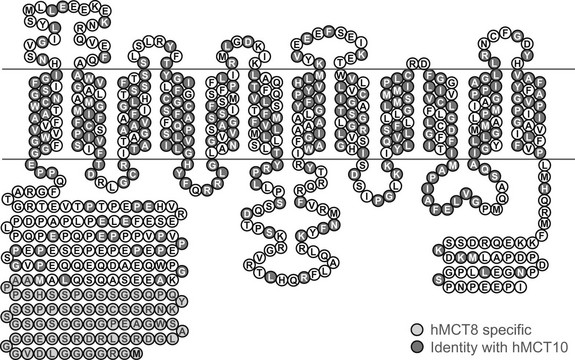
FIGURE 23-5 Predicted topology of human MCT8, showing the 12 putative transmembrane domains and the extended N-terminal domain (shaded light gray) in the long form of the protein. Residues that are conserved in identity and location with human MCT10 are also highlighted (shaded dark gray).
MCT8 mutations have now been identified in over 50 families with AHDS, and the majority of them are listed in Table 23-4. These mutations include (1) rather large deletions affecting one or more exons; (2) smaller deletions or insertions which result in a shift in the reading frame, leading to altered peptide sequence and/or truncation of proteins; (3) nonsense mutations resulting in truncated proteins; (4) three-nucleotide changes causing single codon deletions or insertions; (5) single nucleotide changes causing amino acid substitutions; and (6) a splice site mutation resulting in deletion of 94 amino acids and 3 TMDs.
The larger deletions and frameshift and nonsense mutations are obviously deleterious for MCT8 function. The functional consequences of single amino acid substitutions, deletions, or insertions have been investigated in cells transfected with wild-type or mutated MCT8. Most mutations were found to result in an almost complete loss of thyroid hormone transport by MCT8. However, the extent to which these mutations affect MCT8 function depends on the cell type used for functional studies, for reasons which need to be fully explored.133–136
Animal Models
Studies in humans and animals have indicated that MCT8 is expressed in a variety of tissues, including brain, liver, kidney, heart, skeletal muscle, and thyroid. The distribution of MCT8 expression in mouse brain has been examined in detail by Heuer et al.,137 indicating that MCT8 is predominantly found in neurons in different brain areas, including hippocampus, cerebral cortex, striatum, hypothalamus, and cerebellum. Significantly, MCT8 is also expressed in capillary endothelial cells, the choroid plexus, and tanycytes which line the third ventricle.137,138 MCT8 expression in neurons coincides with expression of DIO3, which catalyzes the degradation of T3. DIO2, which catalyzes the conversion of T4 to T3, is largely expressed in adjacent astrocytes. Another transporter involved specifically in brain T4 uptake, OATP1C1, is expressed in capillaries and the choroid plexus.137,138
Based on studies by Heuer et al.137and Bernal,139 a pathway which regulates T3 supply to neuronal target cells is shown in Fig. 23-6. The steps involved in this process include: (1) transport of T4 by OATP1C1 through the blood-brain barrier, (2) uptake of T4 into astrocytes by an unidentified transporter, (3) conversion of T4 to T3 by DIO2 in astrocytes, (4) release of T3 from the astrocytes by another unidentified transporter, and (5) neuronal uptake of T3 mediated by MCT8. Neurons may also express DIO3 to catabolize and terminate T3 action, but this schema is an oversimplification. It ignores the importance of thyroid hormone transport across the blood-brain barrier by MCT8 and across the blood-CSF barrier by both MCT8 and OATP1C1, as well as the importance of other target cells for thyroid hormone in the brain, such as oligodendrocytes.
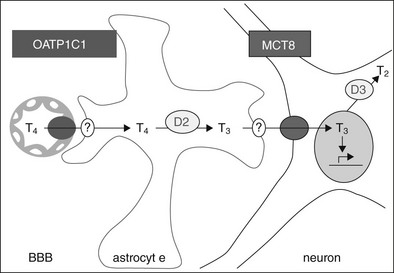
FIGURE 23-6 A pathway which regulates T3 supply to neuronal target cells. OATP1C1 mediates T4 transport through the blood-brain barrier (BBB), with T4 uptake into astrocytes via an unidentified transporter, conversion to T3 by DIO2, and release of T3 by another unidentified transporter. Neuronal uptake of T3 is mediated by MCT8, and the cells also express DIO3 which catabolizes and may therefore terminate hormone action.
MCT8 KO mice have been studied by Trajkovic et al.,140 Dumitrescu et al.,141 and Wirth et al.142 In contrast to the severe neurologic features in male patients with MCT8 mutations, neither hemizygous MCT8 null male mice nor homozygous MCT8 KO female mice show an obvious neurologic phenotype. However, they do exhibit the same biochemical thyroid abnormalities as patients with MCT8 mutations: a large decrease in T4, a large increase in serum T3, and slightly elevated TSH levels. In addition, MCT8 KO mice show the following features: (1) normal brain T4 uptake but impaired brain T3 uptake, (2) decreased brain T4 and T3 content, (3) increased DIO2 and decreased DIO3 activities in brain, (4) normal liver T4 and T3 uptake, (5) increased kidney T4 and T3 uptake, (6) increased kidney T4 and T3 content, and (7) increased type 1 deiodinase (DIO1) activity in both liver and kidney.
The paradoxical increase in renal T4 and T3 uptake in MCT8 KO mice is unexplained, but the combination of increased renal T4 content and DIO1 expression may lead to enhanced renal T4-to-T3 conversion, thus contributing to the observed decrease in serum T4 and increase in serum T3 levels.143 There is also evidence suggesting that thyroidal hormone synthesis is affected by MCT8 inactivation, leading to preferential T3 secretion (Trajkovic et al., unpublished observations). Since MCT8 is expressed in the hypothalamus, its inactivation is associated with impaired feedback action of thyroid hormone, presumably contributing to slightly increased serum TSH levels.140 The lack of an obvious neurologic phenotype in MCT8 KO mice remains to be explained.
Management
Two types of treatment have been evaluated in MCT8 deficiency. One is normalization of serum T4 and T3 levels with a block-and-replace regimen using a combination of PTU and T4. Treatment of a 16-year-old boy with markedly low body mass (25 kg) and tachycardia did not result in major neurologic improvement but had a marked beneficial effect on body weight and heart rate.144 Perhaps neurologic benefit can only occur if such therapy is initiated soon after birth.
An alternative treatment could involve the administration of a thyroid hormone analog that is taken up by the brain independently of MCT8. Promising results using diiodothyropropionic acid (DITPA)145 have been observed in MCT8 KO mice. Clinical trials in patients with MCT8 mutations have been initiated, and the results are eagerly awaited.
Disorder of Thyroid Hormone Metabolism
Three families (A, B, C) with affected individuals exhibiting a disorder of thyroid hormone metabolism have been described.146,147 In each instance, childhood growth retardation was a common feature that brought probands to clinical attention. Affected individuals showed a distinctive abnormal pattern of thyroid function tests, with elevated free T4, low free T3, raised reverse T3, and normal or slightly high TSH levels. This pattern was suggestive of a defect in iodothyronine metabolism. Consistent with this hypothesis, affected siblings in family A required higher-than-normal amounts of exogenous T4 to suppress TSH levels, whereas their response to T3 administration was normal.146 We have identified additional adult and childhood cases of this disorder with a similar pattern of thyroid function tests and growth retardation in childhood (Gurnell, Beck-Peccoz, and Chatterjee, unpublished observations).
Molecular Genetics
The biochemical phenotype in affected cases suggested a defect in T4-to-T3 conversion, but linkage studies ruled out defects at DIO loci or in genes involved in posttranslational modification (ubiquitination/de-ubiquitination) of DIO2. However, affected subjects in family A shared homozygous haplotypes at the SECISBP2/SBP2 locus.146 Subsequent analyses of this gene have identified a homozygous missense mutation (R540Q) in cases from family A, compound heterozygous abnormalities (nonsense mutation K438X plus missplicing of ~50% of transcripts from the other allele) in the proband from family B, and a homozygous stop mutation (R128X) in the propositus from family C.147
There are 25 known human proteins which contain the amino acid selenocysteine (Sec), and SECIS-binding protein 2 (SBP2) is a factor required for the cotranslational incorporation of Sec during their biosynthesis. The mechanism of selenoprotein synthesis and role of SBP2 is further described in Fig 23-7. Recent studies also indicate that the architecture of SBP2 is complex, with alternative splicing of the gene generating multiple transcripts and internal methionine residues directing synthesis of shorter protein isoforms.148 Indeed, there is evidence (albeit in vitro) to suggest that usage of methionines downstream of a homozygous null mutation (e.g., in proband of family C) might generate shorter but functional SBP2 protein to ameliorate the phenotype of this disorder.147
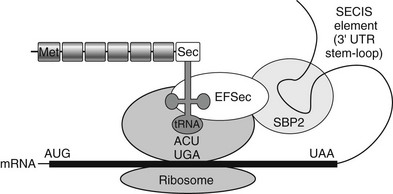
FIGURE 23-7 Mechanism of selenoprotein biosynthesis. The 3′-untranslated region of selenoprotein mRNA contains a stem-loop RNA structure (SECIS element) which interacts with a protein complex that includes SBP2 and Sec-specific elongation factor (EFSec) to enable ribosomal recruitment of selenocysteyl-transfer RNA to the UGA codon and selenocysteine (Sec) incorporation into the nascent polypeptide. Failure of this mechanism results in miscoding of the UGA as a stop codon, terminating protein synthesis.
Pathogenesis
Selenocysteine is a component of all three deiodinase enzymes, and DIO2 activity was indeed reduced in fibroblasts from family A.146 However, based on the biochemical phenotype of DIO null mice, it is likely that the abnormal thyroid function test pattern in this disorder is likely to be mediated by combined, partial deficiency of all three DIOs rather than lack of a single enzyme.
Targeted disruption of the murine SBP2 gene has not been described, so no animal model of this disorder exists, but cellular knockdown of SBP2 has been achieved and is associated with variable but global reduction in levels of many selenoproteins.149 Consistent with this notion, deficiencies of other selenoproteins have been documented in affected cases: glutathione peroxidase (GPx) activity in cells (mainly GPx type1) and serum (mainly GPx type 3) is markedly reduced, and circulating levels of hepatic selenoprotein P (SEPP) are low.146,147 Such deficiency of the major circulating selenoproteins (SEPP, GPx3) is also likely to be the basis of low serum selenium levels recorded in cases.146 Furthermore, functional characterization of the mutant SBP2 protein identified in family A indicates that its ability to bind Sec insertion sequence (SECIS) elements in other selenoprotein genes (e.g., GPx4) is impaired,150 suggesting the possibility of more widespread selenoprotein deficiencies in patients with this disorder.
Management
Trials of oral selenium supplementation, either in the form of selenomethionine-rich yeast which can be incorporated generally into circulating proteins or sodium selenite, a substrate for selenocysteine incorporation, have been undertaken in affected cases. Although selenomethionine treatment raised circulating selenium concentrations, there was no change in thyroid function, GPx3 activity or SEPP levels.147,151 However, T3 treatment was clearly beneficial for growth in one published report147 and our childhood case (Gurnell and Chatterjee, unpublished observations).
References
1. Horlein, AJ, Heinzel, T, Rosenfeld, MG. Gene regulation by thyroid hormone receptors. Curr Opin Endocrinol Diabetes. 1996;3:412–416.
2. Lazar, MA. Thyroid hormone receptors: multiple forms, multiple possibilities. Endocr Rev. 1993;14:184–193.
3. Refetoff, S, De Wind, LT, De Groot, LJ. Familial syndrome combining deaf-mutism, stippled epiphyses, goiter and abnormally high PBI: possible target organ refractoriness to thyroid hormone. J Clin Endocrinol Metab. 1967;27:279–294.
4. Gershengorn, MC, Weintraub, BD. Thyrotropin-induced hyperthyroidism caused by selective pituitary resistance to thyroid hormone. A new syndrome of inappropriate secretion of TSH. J Clin Invest. 1975;56:633–642.
5. Magner, JA, Petrick, P, Menezes-Ferreira, M, et al. Familial generalized resistance of thyroid hormones: a report of three kindreds and correlation of patterns of affected tissues with the binding of [125I]triiodothyronine to fibroblast nuclei. J Endocrinol Invest. 1986;9:459–469.
6. Snyder, D, Sesser, D, Skeels, M, et al. Thyroid disorders in newborn infants with elevated screening T4. Thyroid. 1997;2(suppl 1):S–29. [(abstract)].
7. Taniyama, M, Ishikawa, N, Momotani, N, et al. Toxic multinodular goiter in a patient with generalized resistance to thyroid hormone who harbours the R429Q mutation in the thyroid hormone receptor β gene. Clin Endocrinol. 2001;54:121–124.
8. Brucker-Davis, F, Skarulis, MC, Grace, MB, et al. Genetic and clinical features of 42 kindreds with resistance to thyroid hormone. Ann Intern Med. 1995;123:572–583.
9. Persani, L, Asteria, C, Tonacchera, M, et al. Evidence for the secretion of thyrotropin with enhanced bioactivity in syndromes of thyroid hormone resistance. J Clin Endocrinol Metab. 1994;78:1034–1039.
10. Kahaly, JG, Matthews, CH, Mohr-Kahaly, S, et al. Cardiac involvement in thyroid hormone resistance. J Clin Endocrinol Metab. 2002;87:204–212.
11. Pulcrano, M, Palmieri, EA, Ciulla, DM, et al. Impact of resistance to thyroid hormone on the cardiovascular system in adults. J Clin Endocrinol Metab. 2009;94:2812–2816.
12. Owen, PJD, Chatterjee, VK, John, R, et al. Augmentation index in resistance to thyroid hormone (RTH). Clin Endocrinol. 2009;70:650–654.
13. Weiss, RE, Refetoff, S. Effect of thyroid hormone on growth: lessons from the syndrome of resistance to thyroid hormone. Endocrinol Metab Clin North Am. 1996;25:719–730.
14. Refetoff, S, Weiss, RE, Usala, SJ. The syndromes of resistance to thyroid hormone. Endocr Rev. 1993;14:348–399.
15. Hauser, P, Zametkin, AJ, Martinez, P, et al. Attention deficit-hyperactivity disorder in people with generalized resistance to thyroid hormone. N Engl J Med. 1993;328:997–1001.
16. Mixson, AJ, Parrilla, R, Ransom, SC, et al. Correlation of language abnormalities with localization of mutations in the β-thyroid hormone receptor in 13 kindreds with generalized resistance to thyroid hormone: identification of four new mutations. J Clin Endocrinol Metab. 1992;75:1039–1045.
17. Stein, MA, Weiss, RE, Refetoff, S. Neurocognitive characteristics of individuals with resistance to thyroid hormone: comparisons with individuals with attention-deficit hyperactivity disorder. J Dev Behav Pediatr. 1995;16:406–411.
18. Weiss, RE, Stein, MA, Duck, SC, et al. Low intelligence but not attention deficit hyperactivity disorder is associated with resistance to thyroid hormone caused by mutation R316H in the thyroid hormone receptor β gene. J Clin Endocrinol Metab. 1994;78:1525–1528.
19. Weiss, RE, Stein, MA, Trommer, B, et al. Attention-deficit hyperactivity disorder and thyroid function. J Paediatr. 1993;123:539–545.
20. Valentine, J, Rossi, E, O’Leary, P, et al. Thyroid function in a population of children with attention deficit hyperactivity disorder. J Paediatr Child Health. 1997;33:117–120.
21. Leonard, CM, Martinez, P, Weintraub, BD, et al. Magnetic resonance imaging of cerebral anomalies in subjects with resistance to thyroid hormone. Am J Med Genet. 1995;60:238–243.
22. Brucker-Davis, F, Skarulis, MC, Pikus, A, et al. Prevalence and mechanism of hearing loss in patients with resistance to thyroid hormone. J Clin Endocrinol Metab. 1996;81:2768–2772.
23. Bradley, DJ, Twole, HC, Young, WS. A and b thyroid hormone receptor (TR) gene expression during auditory neurogenesis: evidence for TR isoform specific transcriptional regulation in vivo. Proc Natl Acad Sci U S A. 1994;91:439–443.
24. Forrest, D, Hanebuth, E, Smeyne, RJ, et al. Recessive resistance to thyroid hormone in mice lacking thyroid hormone receptor beta: evidence for tissue-specific modulation of receptor function. EMBO J. 1996;15:3006–3015.
25. Forrest, D, Erway, LC, Ng, L, et al. Thyroid hormone receptor beta is essential for development of auditory function. Nat Genet. 1996;13:354–357.
26. Ng, L, Hurley, JB, Bierks, B, et al. A thyroid hormone receptor that is required for the development of green cone photoreceptors. Nat Genet. 2001;27:94–98.
27. De Meirleir, K, Golstein, J, Jonckheer, MH, et al. Hypothyroidism with normal thyroid hormone levels as a consequence of autoimmune thyroiditis and peripheral resistance to thyroid hormone. Acta Clin Belg. 1980;35:107–109.
28. Aksoy, DY, Gurlek, A, Ringkananont, U, et al. Resistance to thyroid hormone associated with autoimmune thyroid disease in a Turkish family. J Endocrinol Invest. 2005;28:379–383.
29. Fukata, S, Brent, GA, Sugawara, M. Resistance to thyroid hormone in Hashimoto’s thyroiditis. N Engl J Med. 2005;352:517–518.
30. Borck, G, Seewi, O, Jung, A, et al. Genetic causes of goiter and deafness: Pendred syndrome in a girl and cooccurrence of Pendred syndrome and resistance to thyroid hormone in her sister. J Clin Endocrinol Metab. 2009;94:2106–2109.
31. Gurnell, M, Rajanayagam, O, Barbar, I, et al. Reversible pituitary enlargement in the syndrome of resistance to thyroid hormone. Thyroid. 1998;8:679–682.
32. Watanabe, K, Kameya, T, Yamauchi, A, et al. Thyrotropin-producing adenoma associated with pituitary resistance to thyroid hormone. J Clin Endocrinol Metab. 1993;76:1025–1030.
33. Safer, JD, Colan, SD, Fraser, LM, et al. A pituitary tumour in a patient with thyroid hormone resistance: a diagnostic dilemma. Thyroid. 2001;11:281–291.
34. Anselmo, J, Cao, D, Karrison, T, et al. Fetal loss associated with excess thyroid hormone exposure. J Am Med Assoc. 2004;292:691–695.
35. Sarne, DH, Refetoff, S, Rosenfield, RL, et al. Sex hormone-binding globulin in the diagnosis of peripheral tissue resistance to thyroid hormone: the value of changes after short term triiodothyronine administration. J Clin Endocrinol Metab. 1988;66:740–746.
36. Usala, SJ, Bale, AE, Gesundheit, N, et al. Tight linkage between the syndrome of generalized thyroid hormone resistance and the human c-erbA β gene. Mol Endocrinol. 1988;2:1217–1220.
37. Parrilla, R, Mixson, AJ, McPherson, JA, et al. Characterization of seven novel mutations of the c-erbA β gene in unrelated kindreds with generalized thyroid hormone resistance: evidence for two “hot spot” regions of the ligand binding domain. J Clin Invest. 1991;88:2123–2130.
38. Adams, M, Matthews, CH, Collingwood, TN, et al. Genetic analysis of twenty-nine kindreds with generalised and pituitary resistance to thyroid hormone. J Clin Invest. 1994;94:506–515.
39. Collingwood, TN, Wagner, R, Matthews, CH, et al. A role of helix 3 of the TRβ ligand binding domain in coactivator recruitment identified by characterization of a third cluster of mutations in resistance to thyroid hormone. EMBO J. 1998;17:4760–4770.
40. Weiss, RE, Weinberg, M, Refetoff, S. Identical mutations in unrelated families with generalized resistance to thyroid hormone occur in cytosine-guanine-rich areas of the thyroid hormone receptor β gene. J Clin Invest. 1993;91:2408–2415.
41. Mixson, AJ, Renault, JC, Ransom, S, et al. Identification of a novel mutation in the gene encoding the β-triiodothyronine receptor in a patient with apparent selective pituitary resistance to thyroid hormone. Clin Endocrinol. 1993;38:227–234.
42. Sasaki, S, Nakamura, H, Tagami, T, et al. Pituitary resistance to thyroid hormone associated with a base mutation in the hormone-binding domain of the human 3,5,3′-triiodothyronine receptor β. J Clin Endocrinol Metab. 1993;76:1254–1258.
43. Mamanasiri, S, Yesil, S, Dumitrescu, AM, et al. Mosaicism of a thyroid hormone receptor-beta gene mutation in resistance to thyroid hormone. J Clin Endocrinol Metab. 2006;91:3471–3477.
44. Weiss, RE, Hayashi, Y, Nagaya, T, et al. Dominant inheritance of resistance to thyroid hormone not linked to defects in the thyroid hormone receptor alpha or beta genes may be due to a defective cofactor. J Clin Endocrinol Metab. 1996;81:4196–4203.
45. Pohlenz, J, Weiss, RE, Macchia, PE, et al. Five new families with resistance to thyroid hormone not caused by mutations in the thyroid hormone receptor β gene. J Clin Endocrinol Metab. 1999;84:3919–3928.
46. Weiss, RE, Xu, J, Ning, G, et al. Mice deficient in the steroid receptor coactivator 1 (SRC-1) are resistant to thyroid hormone. EMBO J. 1999;18:1900–1904.
47. Weiss, RE, Gehin, M, Xu, J, et al. Thyroid function in mice with compound heterozygous and homozygous disruptions of SRC-1 and TIF-2 coactivators: evidence for haploinsufficiency. Endocrinology. 2002;143:1554–1557.
48. Refetoff, S, Sadow, PM, Reutrakul, S, et al. Resistance to thyroid hormone in the absence of mutations in the thyroid hormone receptor genes. In: Beck-Peccoz, ed. Syndromes of hormone resistance on the hypothalamic-pituitary-thyroid axis. ed 1. Boston: Kluwer Academic Publishers; 2004:89–107.
49. Hamon, B, Hamon, P, Bovier-Lapierre, M, et al. A child with resistance to thyroid hormone without thyroid hormone receptor gene mutation: a 20-year follow-up. Thyroid. 2008;18:35–44.
50. Olson, DP, Koenig, RJ. Thyroid function in Rubinstein-Taybi syndrome. J Clin Endocrinol Metab. 1997;82:3264–3266.
51. Brown, NS, Smart, A, Sharma, V, et al. Thyroid hormone resistance and increased metabolic rate in the RXR-γ-deficient mouse. J Clin Invest. 2000;106:73–79.
52. Sherman, SI, Gopal, J, Haugen, BR, et al. Central hypothyroidism associated with retinoid X receptor-selective ligands. N Engl J Med. 1999;340:1075–1079.
53. Romeo, S, Menzaghi, C, Rocco, B, et al. Search for genetic variants in the retinoid X receptor-γ gene by polymerase chain reaction-single strand conformation polymorphism in patients with resistance to thyroid hormone without mutations in the thyroid hormone receptor β gene. Thyroid. 2004;14:355–358.
54. Meier, CA, Dickstein, BM, Ashizawa, K, et al. Variable transcriptional activity and ligand binding of mutant b1 3,5,3′-triiodothyronine receptors from four families with generalised resistance to thyroid hormone. Mol Endocrinol. 1992;6:248–258.
55. Collingwood, TN, Adams, M, Tone, Y, et al. Spectrum of transcriptional dimerization and dominant negative properties of twenty different mutant thyroid hormone β receptors in thyroid hormone resistance syndrome. Mol Endocrinol. 1994;8:1262–1277.
56. Collingwood, TN, Rajanayagam, O, Adams, M, et al. A natural transactivation mutation in the thyroid hormone β receptor: impaired interaction with putative transcriptional mediators. Proc Natl Acad Sci U S A. 1997;94:248–253.
57. Takeda, K, Sakurai, A, De Groot, LJ, et al. Recessive inheritance of thyroid hormone resistance caused by complete deletion of the protein-coding region of the thyroid hormone receptor-β gene. J Clin Endocrinol Metab. 1992;74:49–55.
58. Sakurai, A, Miyamoto, T, Refetoff, S, et al. Dominant negative transcriptional regulation by a mutant thyroid hormone receptor β in a family with generalised resistance to thyroid hormone. Mol Endocrinol. 1990;4:1988–1994.
59. Chatterjee, VKK, Nagaya, T, Madison, LD, et al. Thyroid hormone resistance syndrome: inhibition of normal receptor function by mutant thyroid receptors. J Clin Invest. 1991;87:1977–1984.
60. Ono, S, Schwartz, ID, Mueller, OT, et al. Homozygosity for a dominant negative thyroid hormone receptor gene responsible for generalized resistance to thyroid hormone. J Clin Endocrinol Metab. 1991;73:990–994.
61. Frank-Raue, K, Lorenz, A, Haag, C, et al. Severe form of thyroid hormone resistance in a patient with homozygous/hemizygous mutation of T3 receptor gene. Eur J Endocrinol. 2004;150:819–823.
62. Nagaya, T, Madison, LD, Jameson, JL. Thyroid hormone receptor mutants that cause resistance to thyroid hormone: evidence for receptor competition for DNA sequences in target genes. J Biol Chem. 1992;267:13014–13019.
63. Nagaya, T, Jameson, JL. Thyroid hormone receptor dimerization is required for dominant negative inhibition by mutations that cause thyroid hormone resistance. J Biol Chem. 1993;268:15766–15771.
64. Shibusawa, N, Hashimoto, K, Nikrodhanond, AA, et al. Thyroid hormone action in the absence of thyroid hormone receptor DNA-binding in vivo. J Clin Invest. 2003;112:588–597.
65. Kitajima, K, Nagaya, T, Jameson, JL. Dominant negative and DNA-binding properties of mutant thyroid hormone receptors that are defective in homodimerization but not heterodimerization. Thyroid. 1995;5:343–353.
66. Yoh, SM, Chatterjee, VKK, Privalsky, ML. Thyroid hormone resistance syndrome manifests as an aberrant interaction between mutant T3 receptors and transcriptional corepressors. Mol Endocrinol. 1997;11:470–480.
67. Tagami, T, Madison, LD, Nagaya, T, et al. Nuclear receptor corepressors activate rather than suppress basal transcription of genes that are negatively regulated by thyroid hormone. Mol Cell Biol. 1997;17:2642–2648.
68. Clifton-Bligh, RJ, de Zegher, F, Wagner, RL, et al. A novel TRβ mutation (R383H) in resistance to thyroid hormone predominantly impairs corepressor release and negative transcriptional regulation. Mol Endocrinol. 1998;12:609–621.
69. Wagner, RL, Huber, BR, Shiau, AK, et al. Hormone selectivity in thyroid hormone receptors. Mol Endocrinol. 2001;15:398–410.
70. Hayashi, Y, Weiss, RE, Sarne, DH, et al. Do clinical manifestations of resistance to thyroid hormone correlate with the functional alteration of the corresponding mutant thyroid hormone b receptors? J Clin Endocrinol Metab. 1995;80:3246–3256.
71. Gurnell, M, Rajanayagam, O, Agostini, M, et al. Three novel mutations at codon 314 in the thyroid hormone receptor β differentially impair ligand binding in the syndrome of resistance to thyroid hormone. Endocrinology. 1999;140:5901–5906.
72. Hayashi, Y, Janssen, OE, Weiss, RE, et al. The relative expression of mutant and normal thyroid hormone receptor genes in patients with generalized resistance to thyroid hormone determined by estimation of their specific messenger ribonucleic acid products. J Clin Endocrinol Metab. 1993;76:64–69.
73. Mixson, AJ, Hauser, P, Tennyson, G, et al. Differential expression of mutant and normal β T3 receptor alleles in kindreds with generalized resistance to thyroid hormone. J Clin Invest. 1993;91:2296–2300.
74. Zavacki, AM, Harney, JW, Brent, GA, et al. Dominant negative inhibition by mutant thyroid hormone receptors is thyroid response element and receptor isoform specific. Mol Endocrinol. 1993;7:1319–1330.
75. Geffner, ME, Su, F, Ross, NS, et al. An arginine to histidine mutation in codon 311 of the c-erbA β gene results in a mutant thyroid hormone receptor that does not mediate a dominant negative phenotype. J Clin Invest. 1993;91:538–546.
76. Beck-Peccoz, P, Chatterjee, VKK. The variable clinical phenotype in thyroid hormone resistance syndrome. Thyroid. 1994;4:225–232.
77. Beck-Peccoz, P, Roncoroni, R, Mariotti, S, et al. Sex hormone-binding globulin measurement in patients with inappropriate secretion of thyrotropin (IST): evidence against selective pituitary thyroid hormone resistance in nonneoplastic IST. J Clin Endocrinol Metab. 1990;71:19–25.
78. Ando, S, Nakamura, H, Sasaki, S, et al. Introducing a point mutation identified in a patient with pituitary resistance to thyroid hormone (Arg 338 to Trp) into other mutant thyroid hormone receptors weakens their dominant negative activities. J Endocrinol. 1996;151:293–300.
79. Safer, JD, O’Connor, MG, Colan, SD, et al. The thyroid hormone receptor-β gene mutation R383H is associated with isolated central resistance to thyroid hormone. J Clin Endocrinol Metab. 1999;84:3099–3109.
80. Wan, W, Farboud, B, Privalsky, ML. Pituitary resistance to thyroid hormone syndrome is associated with T3 receptor mutants that selectively impair beta2 isoform function. Mol Endocrinol. 2005;19:1529–1542.
81. Safer, JD, Langlois, MF, Cohen, R, et al. Isoform variable action among thyroid hormone receptor mutants provides insight into pituitary resistance to thyroid hormone. Mol Endocrinol. 1997;11:16–26.
82. Wu, SY, Cohen, RN, Simsek, E, et al. A novel thyroid hormone receptor-beta mutation that fails to bind nuclear receptor corepressor in a patient as an apparent cause of severe, predominantly pituitary resistance to thyroid hormone. J Clin Endocrinol Metab. 2006;91:1887–1895.
83. Wikstrom, L, Johansson, C, Salto, C, et al. Abnormal heart rate and body temperature in mice lacking thyroid hormone receptor α1. EMBO J. 1998;17:455–461.
84. Abel, ED, Boers, ME, Pazos-Moura, C, et al. Divergent roles for thyroid hormone beta receptor isoforms in the endocrine axis and auditory system. J Clin Invest. 1999;104:291–300.
85. Wong, R, Vasilyev, VV, Ting, Y-T, et al. Transgenic mice bearing a human mutant thyroid hormone β1 receptor manifest thyroid function anomalies, weight reduction and hyperactivity. Mol Med. 1997;3:303–314.
86. Hayashi, Y, Xie, J, Weiss, RE, et al. Selective pituitary resistance to thyroid hormone produced by expression of a mutant thyroid hormone receptor beta gene in the pituitary gland of transgenic mice. Biochem Biophys Res Commun. 1998;245:204–210.
87. Abel, ED, Kaulbach, HC, Campos-Barros, A, et al. Novel insight from transgenic mice into thyroid hormone resistance and the regulation of thyrotropin. J Clin Invest. 1999;103:271–279.
88. Kaneshige, M, Kaneshige, K, Zhu, X, et al. Mice with a targeted mutation in the thyroid hormone beta receptor gene exhibit impaired growth and resistance to thyroid hormone. Proc Natl Acad Sci U S A. 2000;97:13209–13214.
89. Hashimoto, K, Curty, FH, Borges, PP, et al. An unliganded thyroid hormone receptor causes severe neurological dysfunction. Proc Natl Acad Sci U S A. 2001;98:3998–4003.
90. Cheng, S-Y. Multi-factorial regulation of in vivo action of TRβ mutants. Lessons learned from RTH mice with a targeted mutation in the TRβ gene. In: Beck-Peccoz P, ed. Syndromes of hormone resistance on the hypothalamic-pituitary-thyroid axis. ed 1. Boston: Kluwer Academic Publishers; 2004:137–148.
91. Kamiya, Y, Zhang, XY, Ying, H, et al. Modulation by steroid receptor coactivator-1 of target-tissue responsiveness in resistance to thyroid hormone. Endocrinology. 2003;144:4144–4153.
92. Suzuki, H, Willingham, MC, Cheng, SY, et al. Mice with mutation in the thyroid hormone receptor β gene spontaneously develop thyroid carcinoma: A mouse model of thyroid carcinogenesis. Thyroid. 2002;12:963–969.
93. Machado, DS, Sabet, A, Santiago, LA, et al. a thyroid hormone receptor mutation that dissociates thyroid hormone regulation of gene expression in vivo. Proc Natl Acad Sci U S A. 2009;106:9441–9446.
95. Kaneshige, M, Suzuki, H, Kaneshige, K, et al. A targeted dominant negative mutation of the thyroid hormone α1 receptor causes increased mortality, infertility, and dwarfism in mice. Proc Natl Acad Sci U S A. 2001;98:15095–15100.
96. Liu, YY, Schultz, JJ, Brent, GA, et al. A thyroid hormone receptor alpha gene mutation (P398H) is associated with visceral adiposity and impaired catecholamine-stimulated lipolysis in mice. J Biol Chem. 2003;278:38913–38920.
97. Vennstrom, B, Mittag, J, Wallis, K. Severe psychomotor and metabolic damages caused by a mutant thyroid hormone receptor alpha 1 in mice: can patients with a similar mutation be found and treated? Acta Paediatr. 2008;97:1605–1610.
98. Tinnikov, A, Nordstrom, K, Thoren, P. Retardation of post-natal development caused by a negatively acting thyroid hormone receptor alpha1. EMBO J. 2002;21:5079–5087.
99. Anselmo, J, Refetoff, S. Regression of a large goiter in a patient with resistance to thyroid hormone by every other day treatment with triiodothyronine. Thyroid. 2004;14:71–74.
100. Beck Peccoz, P, Piscitelli, G, Cattaneo, MG, et al. Successful treatment of hyperthyroidism due to nonneoplastic pituitary TSH hypersecretion with 3,5,3′-triiodothyroacetic acid (TRIAC). J Endocrinol Invest. 1983;6:217–223.
101. Crino, A, Borrelli, P, Salvatori, R, et al. Anti-iodothyronine autoantibodies in a girl with hyperthyroidism due to pituitary resistance to thyroid hormones. J Endocrinol Invest. 1992;15:113–120.
102. Radetti, G, Persani, L, Molinaro, G, et al. Clinical and hormonal outcome after two years of TRIAC treatment in a child with thyroid hormone resistance. Thyroid. 1997;7:775–778.
103. Bracco, D, Morin, O, Schutz, Y, et al. Comparison of the metabolic and endocrine effects of 3,5,3′-triiodothyroacetic acid and thyroxine. J Clin Endocrinol Metab. 1993;77:221–228.
104. Schueler, PA, Schwartz, HL, Strait, KA, et al. Binding of 3,5,3′-triiodothyronine (T3) and its analogues to the in vitro translational products of c-erbA protooncogenes—differences in the affinity of the alpha forms and beta forms for the acetic-acid analogue and failure of the human testis and kidney alpha-2 products to bind T3. Mol Endocrinol. 1990;4:227–234.
105. Takeda, T, Suzuki, S, Liu, R-T, et al. Triiodothyroacetic acid has unique potential for therapy of resistance to thyroid hormone. J Clin Endocrinol Metab. 1995;80:2033–2040.
106. Ueda, S, Takamatsu, J, Fukata, S, et al. Differences in response of thyrotropin to 3,5,3′-triiodothyronine and 3,5,3′-triiodothyroacetic acid in patients with resistance to thyroid hormone. Thyroid. 1996;6:563–570.
107. Asteria, C, Rajanayagam, O, Collingwood, TN, et al. Prenatal diagnosis of thyroid hormone resistance. J Clin Endocrinol Metab. 1999;84:405–410.
108. Kunitake, JM, Hartman, N, Henson, LC, et al. 3,5,3′-triiodothyroacetic acid therapy for thyroid hormone resistance. J Clin Endocrinol Metab. 1989;69:461–466.
109. Hamon, P, Bovier-LaPierre, M, Robert, M, et al. Hyperthyroidism due to selective pituitary resistance to thyroid hormones in 15-month-old boy: efficacy of D-thyroxine therapy. J Clin Endocrinol Metab. 1988;67:1089–1093.
110. Dorey, F, Strauch, G, Gayno, JP. Thyrotoxicosis due to pituitary resistance to thyroid hormones. Successful control with D-thyroxine; a study in three patients. Clin Endocrinol. 1990;32:221–227.
111. Dulgeroff, AJ, Geffner, ME, Koyal, SN, et al. Bromocriptine and TRIAC therapy for hyperthyroidism due to pituitary resistance to thyroid hormone. J Clin Endocrinol Metab. 1992;75:1071–1075.
112. Williams, G, Kraenzlin, M, Sandler, L, et al. Hyperthyroidism due to non-tumoural inappropriate TSH secretion: effect of long-acting somatostatin analogue (SMS 201-995). Acta Endocr (Copenh). 1986;113:42–46.
113. Beck-Peccoz, P, Mariotti, S, Guillausseau, PJ, et al. Treatment of hyperthyroidism due to inappropriate secretion of thyrotropin with the somatostatin analog SMS 201-995. J Clin Endocrinol Metab. 1989;68:208–214.
114. Weiss, RE, Stein, MA, Refetoff, S. Behavioral effects of liothyronine (L-T3) in children with attention deficit hyperactivity disorder in the presence and absence of resistance to thyroid hormone. Thyroid. 1997;7:389–393.
115. Chiellini, G, Apriletti, JW, Yoshihara, HA, et al. A high-affinity subtype-selective agonist ligand for the thyroid hormone receptor. Chem Biol. 1998;5:299–306.
116. Berkenstam, A, Kristensen, J, et al. The thyroid hormone mimetic compound KB2115 lowers plasma LDL cholesterol and stimulates bile acid synthesis without cardiac effects in humans. Proc Natl Acad Sci U S A. 2008;105:663–667.
117. Koh, JT, Putnam, MC. Towards the rational design of hormone analogues which complement receptor mutations. In: Beck-Peccoz P, ed. Syndromes of hormone resistance on the hypothalamic-pituitary-thyroid axis. ed 1. Boston: Kluwer Academic Publishers; 2004:119–136.
118. Schapira, M, Raaka, BM, Das, S, et al. Discovery of diverse thyroid hormone receptor antagonists by high-throughput docking. Proc Natl Acad Sci U S A. 2003;100:7354–7359.
119. Allan, W, Herndon, CN, Dudley, FC. Some examples of the inheritance of mental deficiency: apparently sex-linked idiocy and microcephaly. Am J Mental Defic. 1944;48:325–334.
120. Dumitrescu, AM, Liao, XH, Best, TB, et al. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet. 2004;74:168–175.
121. Friesema, EC, Grueters, A, Biebermann, H, et al. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet. 2004;364:1435–1437.
122. Schwartz, CE, May, MM, Carpenter, NJ, et al. Allan-Herndon-Dudley syndrome and the monocarboxylate transporter 8 (MCT8) gene. Am J Hum Genet. 2005;77:41–53.
123. Vaurs-Barriere, C, Deville, M, Sarret, C, et al. Pelizaeus-Merzbacher-like disease presentation of MCT8 mutated male subjects. Ann Neurol. 2009;65:114–118.
124. Brockmann, K, Dumitrescu, AM, Best, TT, et al. X-linked paroxysmal dyskinesia and severe global retardation caused by defective MCT8 gene. J Neurol. 2005;252:663–666.
125. Friesema, EC, Jansen, J, Heuer, H, et al. Mechanisms of disease: psychomotor retardation and high T3 levels caused by mutations in monocarboxylate transporter 8. Nat Clin Pract Endocrinol Metab. 2006;2:512–523.
126. Holden, KR, Zuniga, OF, May, MM, et al. X-linked MCT8 gene mutations: characterization of the pediatric neurologic phenotype. J Child Neurol. 2005;20:852–857.
127. Herzovich, V, Vaiani, E, Marino, R, et al. Unexpected peripheral markers of thyroid function in a patient with a novel mutation of the MCT8 thyroid hormone transporter gene. Horm Res. 2007;67:1–6.
128. Namba, N, Etani, Y, Kitaoka, T, et al. Clinical phenotype and endocrinological investigations in a patient with a mutation in the MCT8 thyroid hormone transporter. Eur J Pediatr. 2008;167:785–791.
129. Friesema, EC, Ganguly, S, Abdalla, A, et al. Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J Biol Chem. 2003;278:40128–40135.
130. Friesema, EC, Jansen, J, Jachtenberg, JW, et al. Effective cellular uptake and efflux of thyroid hormone by human monocarboxylate transporter 10. Mol Endocrinol. 2008;22:1357–1369.
131. Friesema, ECH, Kuiper, GGJM, Jansen, J, et al. Thyroid hormone transport by the human monocarboxylate transporter 8 and its rate-limiting role in intracellular metabolism. Mol Endocrinol. 2006;20:2761–2772.
132. Halestrap, AP, Meredith, D. The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers Arch. 2004;447:619–628.
133. Jansen, J, Friesema, EC, Kester, MH, et al. Functional analysis of monocarboxylate transporter 8 mutations identified in patients with x-linked psychomotor retardation and elevated serum triiodothyronine. J Clin Endocrinol Metab. 2007;92:2378–2381.
134. Jansen, J, Friesema, EC, Kester, MH, et al. Genotype-phenotype relationship in patients with mutations in thyroid hormone transporter MCT8. Endocrinology. 2008;149:2184–2190.
135. Kinne, A, Roth, S, Biebermann, H, et al. Surface translocation and T3 uptake of mutant MCT8 proteins are cell type-dependent. J Mol Endocrinol. 2009. [(ahead of print)].
136. Visser, WE, Jansen, J, Friesema, EC, et al. Novel pathogenic mechanism suggested by ex vivo analysis of MCT8 (SLC16A2) mutations. Hum Mutat. 2009;30:29–38.
137. Heuer, H, Maier, MK, Iden, S, et al. The monocarboxylate transporter 8 linked to human psychomotor retardation is highly expressed in thyroid hormone-sensitive neuron populations. Endocrinology. 2005;146:1701–1706.
138. Roberts, LM, Woodford, K, Zhou, M, et al. Expression of the thyroid hormone transporters MCT8 (SLC16A2) and OATP14 (SLCO1C1) at the blood-brain barrier. Endocrinology. 2008;149:6251–6261.
139. Bernal, J. Thyroid hormones and brain development. Vitam Horm. 2005;71:95–122.
140. Trajkovic, M, Visser, TJ, Mittag, J, et al. Abnormal thyroid hormone metabolism in mice lacking the monocarboxylate transporter 8. J Clin Invest. 2007;117:627–635.
141. Dumitrescu, AM, Liao, XH, Weiss, RE, et al. Tissue-specific thyroid hormone deprivation and excess in monocarboxylate transporter (MCT) 8-deficient mice. Endocrinology. 2006;147:4036–4043.
142. Wirth, EK, Roth, S, Blechschmidt, C, et al. Neuronal 3′,3,5-triiodothyronine (T3) uptake and behavioral phenotype of mice deficient in Mct8, the neuronal T3 transporter mutated in Allan-Herndon-Dudley syndrome. J Neurosci. 2009;29:9439–9449.
143. Trajkovic, M, Visser, TJ, Darras, WM, et al. Consequences of MCT8 deficiency for renal transport and metabolism of thyroid hormones in mice. Endocrinology. 2009. [In press].
144. Wemeau, JL, Pigeyre, M, Proust-Lemoine, E, et al. Beneficial effects of propylthiouracil plus L-thyroxine treatment in a patient with a mutation in MCT8. J Clin Endocrinol Metab. 2008;93:2084–2088.
145. Di Cosmo, C, Liao, XH, Dumitrescu, AM, et al. A thyroid hormone analog with reduced dependence on the monocarboxylate transporter 8 for tissue transport. Endocrinology. 2009;150:4450–4458.
146. Dumitrescu, AM, Liao, X, Abdullah, MSY, et al. Mutations in SECISBP2 result in abnormal thyroid hormone metabolism. Nat Genet. 2005;37:1247–1252.
147. Di Cosmo, C, McLellan, N, Liao, X, et al. Clinical and molecular characterization of a novel selenocysteine insertion sequence-binding protein 2 (SBP2) gene mutation (R128X). J Clin Endocrinol Metab. 2009;94:4003–4009.
148. Papp, LV, Wang, J, Kennedy, D, et al. Functional characterization of alternatively spliced human SECISBP2 transcript variants. Nucleic Acids Research. 2008;36:7192–7206.
149. Squires, J, Stoytchev, I, Forry, EP, et al. SBP2 binding affinity is a major determinant in differential selenoprotein mRNA translation and sensitivity to nonsense-mediated decay. Mol Cell Biol. 2007;27:7848–7855.
150. Bubenik, JL, Driscoll, DM. Altered RNA binding activity underlies abnormal thyroid hormone metabolism linked to a mutation in selenocysteine insertion sequence-binding protein 2. J Biol Chem. 2007;282:34653–34662.