Chapter 523 Renal Tubular Acidosis
Normal Urinary Acidification
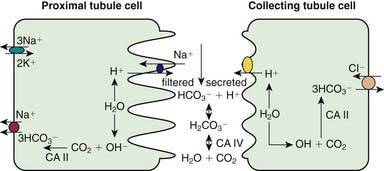
Kidneys contribute to acid-base balance by reabsorption of filtered bicarbonate (HCO3−) and excretion of hydrogen ion (H+) produced every day. Hydrogen ion secretion from tubule cells into the lumen is key in the reabsorption of HCO3−, formation of titratable acidity (H+ bound to buffers such as HPO42−), and formation of ammonium ions (NH4+). Because loss of filtered HCO3− is equivalent to addition of H+ to the body, all filtered bicarbonate should be absorbed before dietary H+ can be excreted. About 90% of filtered bicarbonate is absorbed in the proximal tubule and the remaining 10% in the distal segments, mostly thick ascending limb and outer medullary collecting tubule (CT) (Fig. 523-1). In the proximal tubule and thick ascending limb of the loop of Henle (TAL) H+ from water is secreted by the Na+-H+ exchanger on the luminal membrane. H+ combines with filtered bicarbonate resulting in the formation of H2CO3, which splits into water and CO2 in the presence of carbonic anhydrase (CA) IV. CO2 diffuses freely back into the cell, combines with OH− (from H2O) to form HCO3− in the presence of CA II, and returns to systemic circulation via the Na+-3HCO3− cotransporter situated at the basolateral membrane of the cell. In the CT, H+ is secreted into lumen by H+ATPase (adenosine triphosphatase) and H-CO3− is returned to systemic circulation by HCO3–-Cl− exchanger located on the basolateral membrane. The H+ secreted proximally and distally in excess of the filtered HCO3− is excreted in the urine either as titratable acid or as NH4+.
523.1 Proximal (Type II) Renal Tubular Acidosis
Rajasree Sreedharan and Ellis D. Avner
Pathogenesis
Proximal RTA can be inherited and persistent from birth or occur as a transient phenomenon during infancy. Although rare, it may be primary and isolated. Proximal RTA usually occurs as a component of global proximal tubular dysfunction or Fanconi syndrome, which is characterized by low molecular weight proteinuria, glycosuria, phosphaturia, aminoaciduria, and proximal RTA. The causes of proximal RTA and Fanconi syndrome are outlined in Table 523-1. Many of these causes are inherited disorders. In addition to cystinosis and Lowe syndrome, autosomal recessive and dominant pRTA are addressed further in this section. Other inherited forms of Fanconi syndrome include galactosemia (Chapter 81.2), hereditary fructose intolerance (Chapter 81.3), tyrosinemia (Chapter 79.2), and Wilson disease (Chapter 349.2). Dent disease, or X-linked nephrolithiasis, is discussed in Chapter 525.3. In children, an important form of secondary Fanconi syndrome is exposure to ifosfamide, a component of many treatment regimens for Wilms tumor and other solid tumors.
Table 523-1 COMMON CAUSES OF RENAL TUBULAR ACIDOSIS
PROXIMAL RENAL TUBULAR ACIDOSIS
Primary
Secondary
DISTAL RENAL TUBULAR ACIDOSIS
Primary
Secondary
HYPERKALEMIC RENAL TUBULAR ACIDOSIS
Primary
Secondary
523.2 Distal (Type I) Renal Tubular Acidosis
Rajasree Sreedharan and Ellis D. Avner
Pathogenesis
As with proximal RTA, distal RTA can be sporadic or inherited. It can also occur as a complication of inherited or acquired diseases of the distal tubules. Primary or secondary causes of distal RTA can result in damaged or impaired functioning of one or more transporters or proteins involved in the acidification process, including the H+/ATPase, the HCO3−/Cl− anion exchangers, or the components of the aldosterone pathway. Because of impaired hydrogen ion excretion, urine pH cannot be reduced to <5.5, despite the presence of severe metabolic acidosis. Loss of sodium bicarbonate distally, owing to lack of H+ to bind to in the tubular lumen (see Fig. 523-1), results in increased chloride absorption and hyperchloremia. Inability to secrete H+ is compensated by increased K+ secretion distally, leading to hypokalemia. Hypercalciuria is usually present and can lead to nephrocalcinosis or nephrolithiasis. Chronic metabolic acidosis also impairs urinary citrate excretion. Hypocitraturia further increases the risk of calcium deposition in the tubules. Bone disease is common, resulting from mobilization of organic components from bone to serve as buffers to chronic acidosis.
523.3 Hyperkalemic (Type IV) Renal Tubular Acidosis
Rajasree Sreedharan and Ellis D. Avner
Diagnostic Approach
The first step in the evaluation of a patient with suspected RTA is to confirm the presence of a normal anion gap metabolic acidosis, identify electrolyte abnormalities, assess renal function, and rule out other causes of bicarbonate loss such as diarrhea (Chapter 52). Metabolic acidosis associated with diarrheal dehydration is extremely common, and acidosis generally improves with correction of volume depletion. Patients with protracted diarrhea can deplete their total-body bicarbonate stores and can have persistent acidosis despite apparent restoration of volume status. In instances where a patient has a recent history of severe diarrhea, full evaluation for RTA should be delayed for several days to permit adequate time for reconstitution of total-body bicarbonate stores. If acidosis persists beyond a few days in this setting, additional studies are indicated.
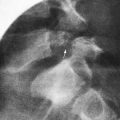
Once the presence of a non–anion gap metabolic acidosis is confirmed, urine pH can help distinguish distal from proximal causes. A urine pH <5.5 in the presence of acidosis suggests proximal RTA, whereas patients with distal RTA typically have a urine pH >6.0. The urine anion gap ([urine Na+ + urine K+] − urine Cl−) is sometimes calculated to confirm the diagnosis of distal RTA. A positive gap suggests a deficiency of ammoniagenesis and, thus, the possibility of a distal RTA. A negative gap is consistent with proximal tubule bicarbonate wasting (gastrointestinal bicarbonate wasting). A urinalysis should also be obtained to determine the presence of glycosuria, proteinuria, or hematuria, suggesting more global tubular damage or dysfunction. Random or 24-hr urine calcium and creatinine measurements will identify hypercalciuria. Renal ultrasonography should be performed to identify underlying structural abnormalities such as obstructive uropathies as well as to determine the presence of nephrocalcinosis (Fig. 523-2).
Alper SL. Genetic diseases of acid-base transporters. Annu Rev Physiol. 2002;64:899-923.
Chan JCM, Scheinman JI, Roth KS. Renal tubular acidosis. Pediatr Rev. 2001;22:277-286.
Fry AC, Karet FE. Inherited renal acidoses. Physiology. 2007;22:202-211.
Fulop M, Mackay M. Renal tubular acidosis, Sjögren syndrome, and bone disease. Arch Intern Med. 2004;164:905-909.
Gahl WA. Cystinosis. In: Avner ED, Harmon WE, Niaudet P, et al, editors. Pediatric nephrology. ed 6. Heidelberg, Germany: Springer-Verlag; 2009:1019-1038.
Gahl WA. Early oral cysteamine therapy for nephropathic cystinosis. Eur J Pediatr. 2003;162(Suppl 1):S38-S41.
Hsu SY, Tsai IJ, Tsau YK. Comparison of growth in primary Fanconi syndrome and proximal renal tubular acidosis. Pediatr Nephrol. 2005;20:460.
Igarashi T. Fanconi syndrome. In: Avner ED, Harmon WE, Niaudet P, et al, editors. Pediatric nephrology. ed 6. Heidelberg, Germany: Springer-Verlag; 2009:1039-1068.
Izzedine H, Launay-Vacher V, Isnard-Bagnis C, et al. Drug-induced Fanconi’s syndrome. Am J Kidney Dis. 2003;41:292-309.
Kalatzis V, Antignac C. New aspects of the pathogenesis of cystinosis. Pediatr Nephrol. 2003;18:207-215.
Magen D, Berger L, Coady MJ, et al. A loss-of-function mutation in NaPi-IIa and renal Fanconi’s syndrome. N Engl J Med. 2010;362(12):1102-1108.
Nicoletta JA, Schwartz GJ. Distal renal tubular acidosis. Curr Opin Pediatr. 2004;16:194-198.
Pongchaiyakul C, Domrongkitchaiporn S, Stichantrakul W, et al. Incomplete renal tubular acidosis and bone mineral density: a population survey in a area of endemic renal tubular acidosis. Nephrol Dial Transplant. 2004:3029-3033.
Prie D, Friedlander G. Genetic disorders of renal phosphate transport. N Engl J Med. 2010;362:2399-2409.
Quigley R. Renal tubular acidosis. In: Avner ED, Harmon WE, Niaudet P, et al, editors. Pediatric nephrology. ed 6. Heidelberg, Germany: Springer-Verlag; 2009:979-1004.
Rodriguez-Soriano J. New insights into the pathogenesis of renal tubular acidosis—from functional to molecular studies. Pediatr Nephrol. 2000;14:1121-1136.
Rose BD, Post TW. Regulation of acid-base balance. In Clinical physiology of acid-base and electrolyte disorders, ed 5, New York: McGraw-Hill; 2001:325-371.
Wappner RS. Lowe syndrome. In GeneClinics: Clinical Genetic Information Resource (database online), http://www.geneclinics.org/.
Wuhl E, Haffner D, Offner G, et al. Long-term treatment with growth hormone in short children with nephropathic cystinosis. J Pediatr. 2001;138:880-887.