CHAPTER 4 Regulation of hematopoiesis
Introduction
Hematopoiesis, including both myelopoiesis and lymphopoiesis, is maintained throughout life by hematopoietic stem cells (HSC). Hematopoietic cells can first be detected in the yolk sac,1 followed by the aorta-gonad-mesonephros (AGM) region of the embryo proper.2,3 HSC then migrate from the AGM region to the fetal liver,4 where they undergo extensive self-renewal to generate a sufficiently large pool of HSC to sustain hematopoiesis throughout adult life.5 Prior to birth HSC seed the bone marrow (BM), and the BM becomes the primary site of hematopoiesis throughout adult life.6 To ascertain that sufficient mature blood cells are generated, hematopoietic cell production is a highly regulated process in which the majority of HSC remain quiescent under steady-state conditions, but may be induced to proliferate under conditions of stress. By contrast, the first descendants from HSC, hematopoietic progenitor cells (HPC), proliferate extensively prior to maturing to terminally differentiated cells. Although significant insights have been gained in the processes that regulate self-renewal and differentiation of HSC, which will be reviewed here, these processes remain incompletely understood. This chapter will highlight a number of the processes that are responsible for coordinating self-renewal and differentiation of HSC and HPC to ensure the controlled generation of mature blood elements under steady-state conditions and conditions of stress. Although we do address mechanisms underlying leukemogenesis or syndromes of hematopoietic failure, it follows that if any of the delicately controlled processes that will be discussed fail, insufficient or excess cells can be produced leading to these disease states.
Characteristics of hematopoietic stem and progenitor cells
The minimal definition of an HSC is a cell capable of extensive self-renewal as well as generation of both myeloid and lymphoid progeny. The concept of the hematopoietic stem cell was first proposed in the 1950s after researchers discovered that lethally irradiated mice could be rescued from BM aplasia by transplanting cells from healthy mouse BM or spleen.7 Subsequent studies demonstrated that HSC possess the unique property to self-renew and maintain the hematopoietic system throughout life. The HSC is capable of generating all blood cell lineages. During postnatal life, HSC reside in the BM where they constitute less than 0.01% of the total cell population.8 Many studies over the last two to three decades have developed tools to characterize HSC and their descendants. This has led to the development of a hematopoietic cell hierarchy wherein HSC are at the top of the hierarchy generating progressively more lineage committed cells which coincides with decreased self-renewal and proliferative potential.
HSC are the only cells that reconstitute hematopoiesis long term, and proliferate rarely in vivo. Studies using BrdU labeling in mouse HSC estimate the frequency of HSC division to be somewhere around once every month;9 however, studies using biotin label or histon 2B-GFP transgenic mice have suggested that multiple populations of HSC exist with different division kinetics and that slow dividing HSC only very rarely exist. The division rate of these different populations of HSC ranges between 0.5% per day and once every 145 days.10–12 In larger mammals, felines and non-human primates, both retroviral marking and telomere length have been used to evaluate frequency of HSC division.13,14 HSC divide in felines about once every 8 weeks,13 in non-human primates once every 25–35 weeks,14 and in humans once every year.15 Hematopoietic progenitor cells (HPC), by contrast, cannot reconstitute the hematopoietic system for the life of the recipient. However, in contrast to HSC, HPC proliferate actively to generate the millions of hematopoietic cells generated daily. At the bottom of the hierarchy are the terminally differentiated hematopoietic cells.
Phenotypic characterization
Although HSC in the mouse have been enriched to 100% purity,16 the exact phenotype of human HSC is not known, because of lack of accurate functional assays that allow enumeration of human HSC. The CD34+ population of hematopoietic cells has been shown to possess the majority of hematopoietic repopulating activity in humans, and is known to enrich for hematopoietic progenitors.17–19 Primitive human progenitors that can initiate long-term cultures or can repopulate immunodeficient animals are lineage−, CD34+, CD133+, CD38−, HLA-DRlow, c-Kit+ and Thy1low, and Lin−CD34+CD38− cells contain approximately 0.1% primitive progenitors that can repopulate the hematopoietic system of severe combined immunodeficient (SCID) mice (SCID-repopulating cells (SRC)).17,20–23 Although HPC are also CD34+, they co-express CD38, as well as cell surface proteins associated to specific lineages, such as CD33 (myeloid lineage),24 CD19 and CD10 (B-lymphoid) or CD7 (NK and T-lymphoid).25–27 Recent studies have indicated that some of the cell surface antigens previously thought to be expressed only on more differentiated cells may be present on HSC, making the characterization of human HSC even more difficult. For instance, CD33 which had been thought only to be on myeloid cells is also expressed on cord blood HSC.28
As stem cells are quiescent, they are spared from cell cycle-specific cytotoxic agents such as 5-fluorouracil, a method used frequently to enrich murine BM for HSC.29,30 In addition, stem cells express functional multidrug resistance proteins, such as p-glycoprotein (MDR1),31 and breast cancer related protein (BCRP),32 which extrude toxins from the cell. This allows selection of stem cells based on their ability to, for instance, extrude the dyes Rhodamine or Hoechst 33342.31,33 Combining these functional characteristics of HSC to cell-surface markers further enriches for human HSC, and 1/30 RholoLin−CD34+CD38− cells are SRC.34
Functional characterization
Even if we now can enrich for HSC using fluorescent activated cell sorting procedures, identification of HSC continues to depend on assays that measure stem cell function. Committed HPC can be assessed using colony-forming assays, where colony forming unit (CFU)- granulocyte-macrophage (GM), burst-forming-unit (BFU)-E, CFU-Mix can be enumerated. An additional primitive HPC subset is the high proliferative potential colony-forming cells (HPP-CFC).35 Even though HPP-CFC generate visible myeloid cell colonies and can be replated to generate new HPP-CFC, demonstrating their extensive self-renewal ability, they do not correspond to HSC.
Dexter and colleagues demonstrated in the late 1970s that long-term hematopoiesis could be established in vitro, by plating BM cells in the presence of fetal calf and horse serum. They demonstrated that this leads to the establishment of an adherent feeder of stromal cells, where hematopoietic progenitors proliferate for several weeks while generating more mature progeny.36 Subsequent adaptations of this culture system, wherein hematopoietic supportive stromal feeders are first established, whereupon hematopoietic cells can be seeded, has allowed investigators to quantify primitive hematopoietic progenitors, also termed long-term culture initiating cells (LTC-IC). LTC-IC can generate more committed CFC in a sustained manner (5 to more than 20 weeks).37,38 Although there is evidence in mice that the number of LTC-IC may correlate with repopulating HSC as progeny are only of the myeloid lineage, this assay cannot assess the frequency of true HSC.39 In vitro assays have also been developed to assess the lymphoid potential of human primitive progenitor cells, all of which also require specific microenvironments (BM stroma or stromal cell lines for B-lymphocytes, NK and dendritic cell differentiation, and either thymus derived feeders or other feeders engineered to express Notch ligands for T-cell differentiation).40–44 As is true for the LTC-IC assays described above, only lymphoid differentiation can be assessed in the latter assays, and thus again not true HSC activity. To assess the ability of cells to generate both myeloid and lymphoid progeny, ‘switch’ cultures have been developed in which the ability of single cells to give rise to both myeloid and lymphoid long-term culture initiating cells can be tested.43,45,46 Enumeration of the frequency of single cells that have the ability to generate both myeloid and lymphoid progeny comes close to assessment of HSC; however, it cannot address homing and engraftment, nor the true long-term expansion ability of cells.
In mice, HSC can be assessed by transplantation into irradiated animals. When this is done in competition with a known source of repopulating cells, the ability of putative HSC to compete with other HSC can be assessed, and when this is combined with limiting dilution analyses, the absolute frequency of repopulating cells can be measured.47–50 In general engraftment is evaluated at 4 months following transplantation; it is, however, clear that the cells generating progeny for only 4 months in vivo may not represent long-term repopulating (LTR-)HSC. Therefore, some groups evaluate engraftment at 8–10 months after transplantation to demonstrate presence of LTR-HSC,51 whereas others perform secondary transplantations to allow assessment of self-renewal ability of HSC.52,53 The development of xenogeneic transplant models in immuno-incompetent animals (immunodeficient mice such as severe combined immunodeficient (SCID) mice,54 non-obese diabetic (NOD)-SCID mice22 or NOD-SCID mice also lacking the gamma-c receptor (γc−/−),55 beige-nude-SCID (BNX) mice56 and Rag2−/−γc−/−,57 or preimmune fetal lambs58) has provided in vivo models that allow not only demonstration of multilineage differentiation but also self-renewal and repopulating ability of human cells. As human HSC have to repopulate a xenogeneic microenvironment, which may support homing, growth and differentiation of human HSC with decreased efficiency compared with a syngeneic human microenvironment, it remains to be proven that these assays enumerate all human HSC. Researchers are therefore trying to further improve mouse models for human HSC transplantation by for instance humanizing certain growth factors that poorly cross-react with human cells and/or HLA antigens to increase the efficiency of human cells to repopulate xenogeneic animal models and develop into a fully competent hematopoietic system.59 Finally, testing of the effect of certain manipulations on stem cells can also be done in large animals including non-human primate or canine models.60–63
Hematopoietic stem cell fate decisions: symmetrical vs. asymmetrical
That asymmetrical vs. symmetrical divisions occur in stem cell compartments has most elegantly been demonstrated in the model organisms such as Caenorhabditis elegans and Drosophila. One example is the fate of Drosophila germ stem cells (GSC). In the Drosophila testes, approximately 12 non-dividing somatic hub cells, located at the apical tip, make up the niche to which 5–9 GSC are attached in a characteristic rosette pattern.64 When GSC divide, one spindle pole associates with the GSC-niche interface.65 The daughter cell that remains attached to the hub cell continues to have stem cell properties, whereas the second cell, no longer attached to the hub, differentiates. The location of the GSC in Drosophila directs the fate of the cells, which has been shown to depend in part on Notch, TGFβ, and Jak/Stat signaling.
It has been shown in both C. Elegans and Drosophila that the plane of the mitotic spindle also appears to predict which daughter cell remains a progenitor/stem cell and which daughter cell differentiates. As the exact niche for HSC is not known, in vivo studies related to symmetrical and asymmetrical divisions of HSC depend on in vitro studies. These studies have shown that when primitive hematopoietic progenitor cells are cultured in vitro, they divide asymmetrically yielding one daughter cell with characteristics of the original cell and the other daughter cell having more differentiated characteristics.66 There is also preliminary evidence that a number of molecules, such as CD53, CD62L/L-selectin, CD63/lamp-3, and CD71/transferrin receptor, distribute asymmetrically which may govern the fate decisions.67
Early during development, HSC undergo symmetrical self-renewing cell divisions to generate the pool of stem cells required throughout adult life. This occurs between e14 and e18 of fetal liver development in mice. It is believed that characteristics intrinsic to the HSC as well as factors provided by the fetal liver (FL) niche must be responsible for the symmetrical divisions of HSC, and hence the net increase in HSC during this period of development. Bowie et al. demonstrated that HSC in the fetal liver are indeed significantly less quiescent than those found early postnatally.68–70 They also demonstrated that this can be explained by a number of cell intrinsic differences between FL HSC and BM HSC, including expression of some but not all of the known transcription factors and cell cycle proteins known to be involved in self-renewal of HSC, as well as differences in response to exogenous cytokines, including SCF and CXCL12. Whether the nature of the cell extrinsic signals in fetal liver stem cell niches differ from those in postnatal BM niches, to favor expansion of HSC, is not yet known but deserves further study as this may aid in developing strategies that allow HSC expansion, even postnatally or in vitro. During postnatal life HSC self-renewal occurs rarely and in an asymmetric fashion, yielding one new HSC and a cell that partakes in extensive proliferation in the transient amplifying pool to generate all mature cells. It is believed that only under stress conditions, HSC may divide symmetrically either yielding two new HSC to recreate the pool of HSC or giving rise to two differentiating cells.71
The hematopoietic stem cell niche
The notion that HSC reside in microenvironments or niches that regulate their behavior (cell quiescence vs. symmetric divisions vs. asymmetric divisions vs. differentiation) was put forward first by Schofield in 1978,72,73 even though it was not until recently that the nature of these niches has become elucidated. The bone marrow microenvironment in which HSC reside in postnatal life consists of both hematopoietic and ‘stromal’ cells.74,75 These stromal cells include endothelial cells, fibroblasts, myocytes, adipocytes and osteoblasts. Stromal cells produce and deposit a complex extracellular matrix (ECM) and produce hematopoietic cytokines that induce or inhibit progenitor proliferation and differentiation.76,77 Hematopoietic cells interact through cell-surface receptors with either immobilized or secreted cytokines, with adhesive ligands present on stromal cells or ECM components, and other hematopoietic cells. The combined effect of cell–cytokine, cell–cell and cell–ECM interactions governs the normal hematopoietic process.
The fact that one can now identify murine HSC based on cell surface antigens to near homogeneity has allowed investigators to determine that HSC can be found either in close proximity with endosteal osteoblasts (the osteoblastic niche)78,79 or near small blood vessels (the vascular niche)16 (Figure 4.1). The osteoblastic niche may be providing a favorable environment to maintain the HSC in a quiescent state, whereas the vascular niche may be providing signals for differentiation and mobilization to the peripheral blood.
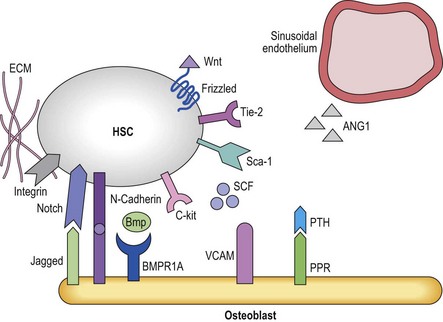
Osteoblastic niche
The development of bone has long been linked to the formation and function of the bone marrow.80 A direct role in vivo has more recently been established by a number of studies. In the adult mouse, HSC reside near the endosteum lining the BM cavity.78,79 The finding that osteoblasts play a role in HSC homeostasis comes from studies in which the osteoblast compartment of the bone was expanded by overexpression of parathyroid hormone (Pth) and its receptors,81 and a second series of studies in which the BMP-receptor-1a was knocked out, both leading to an increase in HSC.82 In both studies an increase in osteoblasts correlated with an increase in HSC.81 Osteoblasts and HSC were found to interact through N-cadherin interactions, although the role of this interaction is unclear.81 As these genetic manipulations increase HSC numbers,83 it is believed that the osteoblastic niche regulates HSC maintenance and self-renewal, perhaps by direct cell–cell interactions via the Notch84 and N-cadherin pathways.85 There is evidence that multiple receptor ligand interactions may affect HSC localized in the osteoblastic niche; many of these will be discussed later in this chapter. They include the morphogens BMP4 and Wnt3a,86,87 interactions between Notch on HSC and Jagged-1 in the niche,88 Tie2 expressed on HSC and angiopoietin-1;83 c-Kit and c-Kit-L;89 or β1 integrins that interact with both VCAM-190 and osteopontin.91,92 Osteoblasts also appear to affect adjacent cells including osteoclasts93 that then affect HSC fate decisions. Likewise, there is evidence that RANK-L produced by osteoclast mediated breakdown of the bone also influences HSC behavior.94 However, other studies demonstrated that ablation of osteoblasts using a Collagen-1a1 conditional knockout mouse leads first to the depletion of more committed progenitors and later to depletion of the HSC compartment, which contradicts the notion that osteoblasts would preserve the HSC pool.95 Like murine HSC, human primitive progenitors can be found in close proximity with the endosteal lining of the marrow cavity,6 although the molecular mechanisms underlying these interactions and the role of endosteal cells in human HSC regulation are unknown.
Vascular niche
In vivo, endothelial cells line the sinusoids of the BM cavity. As in other tissues, entry and exit from the marrow requires that HSC/HPC and mature blood cells pass through the endothelial barrier of the sinusoids. Unlike most sinusoids, BM sinusoids only have a single layer of endothelial cells, which allows for increased permeability.96 Aside from serving as a gatekeeper for cell entry and exit from the BM, it is believed that BM endothelial cells also play a role in regulating hematopoiesis. Aside from endothelial cells, additional cells in the vicinity which may regulate HSC in vivo include pericytes (thought to be the in vivo mesenchymal progenitor cell), megakaryocytes and perivascular reticular cells. Although identification of murine HSC via the SLAM receptors has shown that HSC reside near endothelial cells,16 the exact contribution of the different cell types in the vascular niche to HSC proliferation and differentiation control are, however, less well understood.
Intrinsic regulation of hematopoiesis
HSC behavior is controlled in part by factors exogenous to the HSC, but also in part by cell intrinsic mechanisms. The latter is the result of a complement of transcription factors (TFs) expressed specifically in HSC but not HPC, which can bind to the promoter regions of specific target genes to allow their transcription and ultimate production of the proteins typical for HSC and not HPC. TFs also recruit co-factors that can modify the chromatin surrounding target genes, to allow easier or more difficult transcription of certain regions of the genome, which is part of the epigenetic regulation of gene expression. The latter consist of methylation and acetylation of histones, proteins to which the DNA is bound, as well as of methylation of CpG islands in the promoter regions of target genes, which prevent binding of TFs to allow transcription. The role of these basic processes, important in all steps of development to allow orderly process of lineage commitment and specification, in hematopoiesis has recently been extensively reviewed by Rice et al., and we refer readers to that exhaustive review for more detailed information.97
For many of these genes, aberrant expression is associated with abnormal hematopoiesis, in many instances leukemia development. For instance, recurrent chromosomal translocations in human leukemias that deregulate expression and/or function of genes such as TAL1, LMO2, NOTCH1, and HOX genes98–102 result in aberrant proliferation of HSC/HPC without maturation, findings that have led to the identification of these genes as key regulators of hematopoiesis.
Hematopoietic stem and progenitor cell specific genes
A number of genes have been identified that are indispensable for the creation of HSC and their lineage specific progeny during development. These include, among others, LMO2, TAL1, GATA2, GATA1 and PU.1.103–107 LMO2, TAL1 and GATA2 are expressed already in the hemangioblast, or the mesodermal cell that can generate both endothelium and hematopoietic cells.104–110 Loss of any of these three genes inhibits the emergence of HSC during development. Subsequent commitment to lineage specific hematopoietic cells such as erythroid vs. myeloid and B-cells is then governed by the lineage specific expression of GATA1 and PU.1, respectively.111
Homeobox genes
This is a family of highly preserved genes containing a DNA-binding domain, termed homeodomain, that play important roles in many aspects of development, including normal as well as malignant hematopoiesis.112–117 Aside from the typical HOX genes, there is a second family of homeodomain-containing genes, the three amino acid loop extension (TALE) family of transcription factors, which include PBX1 and MEIS1.118,119 HOX proteins interact with PBX1 to form a complex with MEIS1, and regulate gene expression.120–122 Mice deficient in either PBX1 or MEIS1 are embryonic lethal.123,124 MEIS1 may play a role in expansion of fetal liver HSC as MEIS1−/− fetal liver cells have impaired competitive repopulation abilities.124,125
The HOX family consists of four clusters (A–D) which are located on different chromosomes. Within each cluster, HOX genes are further subclassified from the 3′ to 5′ end in 13 paralog groups, based on sequence homology. Different HOX genes are expressed in HSC and HPC, with HSC being characterized by presence of HOXB3, HOXB4 and HOXA4,125,126 whereas HOXA9 is more highly expressed in HPC that will give rise to granulopoiesis and T- and B-cell lymphopoiesis.113 Although these genes are very important regulators of hematopoiesis, loss of a single homeobox gene does not always lead to hematopoietic defects due to redundancy between different HOX genes. For instance loss of HOXB4 or HOXB3 alone does not lead to defective hematopoiesis, and even combined loss of HOXB4 and HOXB3 only results in a moderate decrease in hematopoietic cell output. By contrast, aberrant expression of HOX genes has been associated with different types of leukemia, including HOXA9 and HOXC13, pointing to their role in HSC and HPC self-renewal and differentiation. When the HSC specific HOXB4 gene is force expressed in murine and human HSC, significant increased proliferation of cells with HSC characteristics in vitro is observed. When grafted in competitive repopulation assays in vivo, HOXB4 transduced cells out-compete normal HSC, without significant skewing of hematopoiesis or frank leukemia development.112,117,127 As increased self-renewal of HSC may also be possible by simple HOXB4 protein transfection, this strategy is being contemplated to induce HSC expansion clinically.128 Kyba et al. also demonstrated that forced expression of HOXB4 in murine embryonic stem cell (ESC) aids in the generation and engraftment of competent HSC from ESC, possibly by inducing the expression of the chemokine receptor CXCR4.129,130 Although HOXB4 does not seem to promote leukemia, deregulation of other HOX family members is linked to hematopoietic malignancies such as leukemia. A number of HOX genes are found fused with NUP98 in human leukemia,131,132 and expression of HOXA9 is associated with a poor prognosis in patients with acute myelogenous leukemia.
Polycomb genes
Upstream regulators of HOX genes are among others the mixed-lineage leukemia (MLL), a common target of chromosomal translocations in human acute leukemias, which induces gene expression during development and the Polycomb gene (PcG) family, responsible for suppression of gene expression. Both of these are believed to regulate gene expression by complex epigenetic mechanisms. The Polycomb group (PcG) represents a gene family of transcriptional repressors first identified in Drosophila. They play a key role in many developmental processes by regulating self-renewal and proliferation, senescence and cell death,133–137 and this via interactions with the initiation transcription machinery138,139 as well as chromatin-condensation proteins and histones.140,141 The PcG proteins are organized in Polycomb regulatory complexes (PRC). Two PRC complexes have been identified, consisting of EZH, EED and SUZ12 (PRC2) whereas BMI1 and RAE28 are part of the PRC1 complex.142 A number of PcG genes are expressed in differentiating hematopoietic cells, whereas BMI1 and RAE28 are found highly expressed in HSC.136,143–145 The role of BMI1 in HSC has been elucidated using both knock-out studies and by forced expression in HSC. HSC from BMI1−/− mice fail to long-term repopulate lethally irradiated recipients suggesting that BMI1 plays a role in HSC self-renewal. In addition, the HSC compartment in BMI1−/− senesces significantly faster than in WT mice. Similar defects are also seen in other stem cell compartments of BMI1−/− mice.146–148 By contrast, forced expression of BMI1 enhances self-renewal in vivo.136,149,150 Similar findings were seen in RAE28−/− fetal liver HSC, which have impaired engraftment potential.144 MEL18, another PcG gene, which is expressed in a reciprocal fashion with BMI1, by contrast inhibits HSC self-renewal.151,152 There is also evidence for a role of PcG proteins part of the PRC2 complex in HSC proliferation, including EED, EZH2 and SUZ12.153–156 Aside from being upstream regulators of HOX genes, several of the PcG genes may act by modifying the expression of cell cycle regulators p16INK4a/p19ARF.151
Cell cycle regulators
In postnatal life, the majority of HSC are in a quiescent state which is associated with the finding that many inhibitors of the cell cycle are highly expressed in HSC. Cell proliferation is driven by cyclin-dependent kinases (CDKs) and their corresponding cyclins. CDK activity is blocked by a number of cyclin-dependent kinase inhibitors (CDKIs). CDKIs are separated into two groups, the CIP/KIP (p21CIP1, p27KIP1, p57KIP2) and INK4 (p14ARF, p15INK4b, p16INK4a, p18INK4c, p19ARF) families, based on the specific CDKs they inhibit. Loss of p21CIP1 results in an increased pool of HSC. However, when p21CIP1−/− HSC are stressed, such as after multiple serial transplantations, HSC exhaustion occurs.157 siRNA mediated knockdown of p21CIP1 in human CD34+CD38− cells also leads to a modest increase in SRC.158 Whereas loss of p21CIP1 leads to expansion of the HSC compartment (albeit with eventual exhaustion), loss of p27KIP1 leads to minimal increases in HSC but a significant expansion of the HPC pool.159 Members of the INK4/ARF family also play a role in HSC control. As discussed above, p16INK4a/p19ARF expression is regulated by the PcG gene BMI1: loss of BMI1 leads to increased expression of p16INK4a/p19ARF,160 and the associated senescence of HSC seen in BMI1−/− mice can be ascribed to the higher levels of p16INK4a/p19ARF.161 As is seen in p21CIP1−/− mice, the HSC compartment in mice wherein p18INK4c has been knocked out is increased, but unlike p21CIP1−/− HSC, p18INK4c−/− HSC are not exhausted over the lifetime of the animal, but demonstrate a competitive advantage compared with WT HSC.162,163 Moreover, loss of p18INK4c can compensate for the early exhaustion and senescence seen in the HSC compartment of p21CIP1−/− mice.157,162 The mechanism underlying these differences remains to be elucidated. Finally, loss of Rb, which plays a central role in the regulation of the G(1)-S phase of the cell cycle, has minimal or no effects on HSC function.164 Concluding, although loss or gain of cell cycle regulators have some effect on HSC behavior, these effects are relatively mild.
Extrinsic regulation of hematopoiesis
Classical cytokines
Over the last three decades, a large number of hematopoietic cytokines and growth factors and their receptors have been identified. Stem and progenitor cells are thought to express most cytokine receptors, a phenomenon also known as stem cell priming.165 Tyrosine kinase receptors include c-Kit, the receptor for steel factor or stem cell factor (SCF),166 and the fetal liver tyrosine kinase receptor-3 (FLT3) which binds to FLT3-L.167 All HSC express c-Kit and SCF, its ligand, is expressed on osteoblasts in vivo. Although SCF plays a role in self-renewing cell divisions, loss of either SCF or its receptor does not cause complete aplasia,168,169 and c-Kit can also not support long-term self-renewal of HSC in vitro.170,171 Likewise, loss of FLT3 or its ligand FLT3-L does not lead to aplasia,172,173 and although FLT3-L may improve self-renewal cell divisions of HSC in vitro, it can again not prevent differentiation of HSC.170,174,175 Combined loss of SCF and FLT3-L or the two receptors causes near aplasia,172 suggesting that the combination of the two cytokines plays a role in HSC self-renewal in vivo. The second family is the cytokine receptor family, which lacks endogenous tyrosine kinase activity but recruits and activates non-receptor tyrosine kinases such as Jak/Stat and Ras/MAPK.176,177 Ligands for these receptors include interleukin (IL)-3176 and thrombopoietin (TPO), both of which are active on primitive progenitors. However, IL-3 functions chiefly to induce differentiation, as addition of high concentrations to ex vivo cultured HSC/HPC induces terminal differentiation and loss of primitive HPC.178,179 TPO, initially discovered as a cytokine important for thrombopoiesis, also pays an important role in HSC biology.180,181 Like SCF, TPO is expressed by osteoblasts.181 In TPO−/− mice, postnatal HSC frequency and function are normal, but significantly more HSC proliferation is seen, which leads eventually to HSC exhaustion, as is also seen in p16INK4a/p19ARF−/− mice.180 The third family consists of the gp130 family, which includes receptors for IL-6,182 IL-11,183 oncostatin-M184 and leukemia inhibitory factor (LIF).185
Morphogens
Hedgehog signaling
The hedgehog (Hh) family consists of three ligands, Sonic Hh (Shh), Indian Hh (Ihh), and desert Hh (Dhh), which bind to patched (Ptch). This leads to the activation of the intracellular signaling molecule, smoothened (Smo), which is normally inhibited by Ptch when not bound to Hh. As in many developmental processes, Shh and Ihh are involved in hematopoietic emergence and specification from zebrafish to mouse.186–188 In vitro studies with human HSC found that Ihh expanded HSC,189 and in vivo murine studies have shown that Hh may play a role in HSC expansion.190 However, the latter studies also demonstrate that sustained activation of the Hh signaling pathway, causing extensive HSC cycling, leads to exhaustion of the HSC pool. A recent study has also shown that complete loss of Hh signaling, via conditional knockout of Smo in HSC, does not affect hematopoiesis, suggesting that although Hhs may affect HSC behavior, it is not required for maintenance of adult hematopoiesis.191,192
Wnt signaling
The Wnt family consists of 19 different secreted glycoproteins that signal through a number of receptors including 10 transmembrane receptors of the frizzled (Fzd) family, retinoid orphan receptor 2 (Ror2), and two co-receptors, LRP5/6. Wnt proteins are commonly grouped according to the downstream signaling cascade that they activate, most commonly but not limited to ‘canonical’ or β-catenin dependent and ‘non-canonical’ or β-catenin independent proteins. Canonical Wnts bind to cell-surface-expressed frizzled receptors. This causes complex formation between cytoplasmic β-catenin193 and the lymphoid enhancing binding/T-cell transcription factor (LEF/TCF) family of transcription factors. As a result of this complex formation, β-catenin–LEF/TCF translocates to the nucleus,194 where LEF/TCF induces transcription of a number of genes, among which are HOX genes as well as cell cycle regulatory genes.
There is a significant body of evidence that canonical Wnt signaling plays a role in postnatal hematopoiesis. Forced expression of Dickkopf1 (Dkk1), an inhibitor of canonical Wnt signaling, in osteoblasts in vivo significantly reduces the number and repopulation ability of HSC harvested from these mice.87 Addition of Wnt3a to ex vivo cultures of murine HSC leads to improved maintenance and expansion of HSC, which can be mimicked by forced expression of an active form of β-catenin and can be inhibited by forced expression of axin, an inhibitor of β-catenin.86,195 Consistent with this, e12.5 FL cells from Wnt3a−/− mice contain significantly fewer HSC, which leads to severely reduced reconstitution capacity as measured in secondary transplantation assays.196 However, other studies question the role of β-catenin in hematopoiesis in vivo. β-catenin−/− mice have no aberrations in hematopoiesis, nor do mice in which, aside from β-catenin, γ-catenin has also been knocked out,197,198 whereas mice expressing a constitutively active form of β-catenin have a decreased number of HSC.199,200 To confuse matters further, loss of adenomatous polyposis coli (APC), which together with axin and GSK3-β phosphorylates and ubiquitinates β-catenin leading to proteasomal degradation of β-catenin, causes a severe hematopoietic phenotype, with a severe reduction in the HSC and HPC pool, due to increased apoptosis, as well as increased proliferation.201 It should be noted that APC may also have effects on HSC via mechanisms independent of the canonical Wnt signaling pathway. However, the role of canonical Wnt signaling in adult hematopoiesis remains not fully understood.
There is also evidence that the non-canonical Wnts may play a role in hematopoiesis. Austin et al. and Van Den Berg et al. demonstrated that Wnt5a increases proliferation in vitro of primitive murine and human hematopoietic progenitors, respectively.202,203 Wnt5a appears to also affect HSC. Although exposure of human CD34+ cells to Wnt5a in vitro did not significantly affect their proliferation or differentiation, administration of Wnt5a containing conditioned medium to mice transplanted with human umbilical cord blood CD34+CD38−Lin− cells increased the repopulation ability of these cells. Although suggestive that Wnt5a affects HSC, this study did not address whether the effect of Wnt5a was directly on HSC.204 More recently, evidence was provided for a direct effect of Wnt5a on HSC, as culture of highly enriched murine HSC with Wnt5a alone under serum free conditions increased in short term repopulating HSC, possibly by increasing the maintenance of a quiescent state of the ex vivo cultured HSC.204 As no clear-cut increase in β-catenin was observed, the authors concluded that this occurred via non-canonical signaling.
TGFβ superfamily
The TGFβ family consists of 35 ligands, including TGFβs, activins, nodal and BMP, which regulate cell proliferation, apoptosis and differentiation.205 The TGFβ family, and members of the BMP family in particular, are important during development for the specification of mesoderm. The TGFβ and BMP signaling pathways activate the Smad signaling cascade and the complex is translocated to the nucleus where they function as transcriptional regulators by binding directly to DNA or to other transcription factors.206
Tgfβ1−/− mice are embryonic lethal with defects in yolk sac formation, including erythroid cell development, with a similar phenotype also seen in TgfβrII−/− mice suggesting an important role of TGFβ in hematopoietic development.207–209 By contrast, Tgfβr−/− mice have increases in early erythroid cells suggesting that TGFβ is not important in early hematopoietic development.207–209 The addition of TGFβ to in vitro HSC cultures inhibits proliferation of HSC and primitive progenitors but not more mature progenitor populations.209 Neutralization of TGFβ by blocking antibodies in BM cultures leads to increased repopulation of murine HSC.210,211 How TGFβ influences hematopoietic cells is not fully understood, even though there is evidence that it regulates cell cycle progression and apoptosis.212
As Bmp4−/− mice are embryonically lethal, its role in mammalian development as well as postnatal hematopoiesis is unclear.213 BMP4 is thought to play a role in initiation of mammalian HSC as it is highly expressed in the AGM region where the first HSC are found214 as well as in the ICM region of zebrafish, where HSC are born. Studies in which BMP4 was added to ex vivo expansion cultures of human HSC demonstrated that BMP4 acts in a dosage dependent manner to increase human SRC ex vivo.215 On the other hand, BMP4 was found to have no effect on mouse HSC cultured ex vivo.216 Forced expression of the inhibitory Smad, Smad-7, in BM cells resulted in reduced proliferation of KLS in vitro; however in vivo, Smad7-overexpressing HSC demonstrated increased self-renewal and engraftment ability.217 Smad5−/− mice, activated by BMP, die early in development with defects in yolk sac development, but they have increased numbers of HPP-CFC in the yolk sac.218 As most mice in which BMPs or Smads, activated by BMPs, are embryonic lethal the role of BMP signaling in adult hematopoiesis remains incomplete.219
Notch pathway
Differentiation into multiple cell types from a population of initially equivalent cells is a fundamental process in the development of all multicellular organisms.220–223 Studies initially in Drosophila and later in mammalian cells have shown that intercellular signaling through the Notch/LIN-12 transmembrane receptors is imperative for normal growth and differentiation during the development of all species. The Notch family is made up of five ligands, Jagged-1/2 and Delta-1/3/4. Binding of the Notch receptor to its ligand causes proteolysis of the Notch receptor.
Both Notch and Notch ligands are expressed in primitive hematopoietic cells as well as in the hematopoietic microenvironment.88,224,225 Overexpression of the activated form of Notch in murine HSC causes lymphoid leukemia.226 The human homolog of one of the Notch ligands, Jagged-1, is expressed in human-marrow-derived stromal cells that support growth of primitive human hematopoietic progenitors in vitro.227 Another Notch family member, Delta-1, when engineered in immobilized form, expanded human HPC in ex vivo cultures.84,228 Also, an increase of HSC is found in vivo when the parathyroid-hormone-related receptor is activated on osteoblasts, which results in increased expression of Jagged-1 by the osteoblast. This increase in Jagged-1 correlates with an increase in Notch-1 activation on HSC.88 Although in a transgenic Notch reporter mouse, BM c-Kit+ cells express the active Notch reporter,229 Notch-1 deficient HSC engraft Jagged-1 deficient mice and reconstitute the hematopoietic system normally.224 Studies have also shown that other ligands for Notch, Delta-4 and delta like (Dlk) act as regulators of primitive hematopoietic progenitors.230,231
Angiopoietin-like proteins
The interaction between Tie2, a receptor tyrosine kinase, and its ligand angiopoietin-1 has been found to play a role in HSC maintenance in the osteoblastic niche. Tie2 is expressed on HSC that are in the quiescent state, and angiopoietin is expressed by osteoblasts lining the bone marrow cavity.83 Arai et al. found that angopoietin-1 enhanced HSC binding to the osteoblast and promoted HSC quiescence enabling maintenance of HSC by its niche.83 They also found that injection of recombinant angiopoietin-1 protein in mice resulted in protection from myeloablation after irradiation,83 suggesting an important role of Tie2/angiopoietin in maintaining the HSC pool within the BM.
Insulin-like growth factors
Insulin-like growth factor (IGF)-1 and -2 hormones are produced by osteoblasts in the bone and are regulated by PTH signaling.232 HSC cultured with cells expressing IGF-1 and -2 leads to expansion of HSC.233 Although IGFs are produced by osteoblast in the BM and are important in maintaining bone, it is unclear whether they directly interact with HSC in vivo.
Conclusions
Regulation of hematopoiesis takes place in the BM, where it is believed that HSC reside in ‘niches’ that control the fate of stem cells by presenting factors that control self-renewal vs. differentiation. It is noteworthy that factors that appear to play a role in the extrinsic control of HSC are not solely the classical cytokines and hematopoietic growth factors; they also include such molecules as angiopoietin-like proteins,83,234 IGFs,233 members of the TGFβ family and Wnts,195,204 as well as cell–cell interactions via Notch and N-cadherin. Although many signals from the microenvironment have been identified that affect HSC, it remains clear that intrinsic regulation also plays a key role in cell fate decisions. There is also mounting evidence that HSC and their microenvironment may differ at different stages of development. For instance, the proliferative behavior of HSC found in fetal liver differs from that of adult HSC. Further elucidation of the differences in both intrinsic and extrinsic control of FL vs. BM HSC may aid in developing methods for HSC expansion.69,70,180,181
1 Yoder MC, Hiatt K, Mukherjee P. In vivo repopulating hematopoietic stem cells are present in the murine yolk sac at day 9.0 postcoitus. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(13):6776-6780.
2 Medvinsky A, Dzierzak E. Definitive hematopoiesis is autonomously initiated by the AGM region. Cell. 1996;86(6):897-906.
3 de Bruijn MF, Speck NA, Peeters MC, Dzierzak E. Definitive hematopoietic stem cells first develop within the major arterial regions of the mouse embryo. EMBO J. 2000;19(11):2465-2474.
4 Dzierzak E, Medvinsky A. Mouse embryonic hematopoiesis. Trends Genet. 1995;11(9):359-366.
5 Morrison SJ, Hemmati HD, Wandycz AM, Weissman IL. The purification and characterization of fetal liver hematopoietic stem cells. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(22):10302-10306.
6 Charbord P, Tavian M, Humeau L, Peault B. Early ontogeny of the human marrow from long bones: an immunohistochemical study of hematopoiesis and its microenvironment. Blood. 1996;87(10):4109-4119.
7 Jacobson LO, Simmons EL, Marks EK, Eldredge JH. Recovery from radiation injury. Science (New York, NY). 1951;113(2940):510-511.
8 Morrison SJ, Uchida N, Weissman IL. The biology of hematopoietic stem cells. Annual Review of Cell and Developmental Biology. 1995;11:35-71.
9 Cheshier SH, Morrison SJ, Liao X, Weissman IL. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(6):3120-3125.
10 Wilson A, Laurenti E, Oser G, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135(6):1118-1129.
11 Nygren JM, Bryder D. A novel assay to trace proliferation history in vivo reveals that enhanced divisional kinetics accompany loss of hematopoietic stem cell self-renewal. PloS one. 2008;3(11):e3710.
12 Foudi A, Hochedlinger K, Van Buren D, et al. Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nature Biotechnology. 2009;27(1):84-90.
13 Abkowitz JL, Catlin SN, McCallie MT, Guttorp P. Evidence that the number of hematopoietic stem cells per animal is conserved in mammals. Blood. 2002;100(7):2665-2667.
14 Shepherd BE, Kiem HP, Lansdorp PM, et al. Hematopoietic stem-cell behavior in nonhuman primates. Blood. 2007;110(6):1806-1813.
15 Shepherd BE, Guttorp P, Lansdorp PM, Abkowitz JL. Estimating human hematopoietic stem cell kinetics using granulocyte telomere lengths. Experimental Hematology. 2004;32(11):1040-1050.
16 Kiel MJ, Yilmaz OH, Iwashita T, et al. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121(7):1109-1121.
17 Huang S, Terstappen LW. Lymphoid and myeloid differentiation of single human CD34+, HLA−DR+, CD38− hematopoietic stem cells. Blood. 1994;83(6):1515-1526.
18 Osawa M, Hanada K, Hamada H, Nakauchi H. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science (New York, NY). 1996;273(5272):242-245.
19 Tavian M, Coulombel L, Luton D, et al. Aorta-associated CD34+ hematopoietic cells in the early human embryo. Blood. 1996;87(1):67-72.
20 Sharma Y, Astle CM, Harrison DE. Heterozygous Kit mutants with little or no apparent anemia exhibit large defects in overall hematopoietic stem cell function. Experimental Hematology. 2007;35(2):214-220.
21 Baum CM, Weissman IL, Tsukamoto AS, et al. Isolation of a candidate human hematopoietic stem-cell population. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(7):2804-2808.
22 Larochelle A, Vormoor J, Hanenberg H, et al. Identification of primitive human hematopoietic cells capable of repopulating NOD/SCID mouse bone marrow: implications for gene therapy. Nature Medicine. 1996;2(12):1329-1337.
23 Sutherland HJ, Eaves CJ, Eaves AC, et al. Characterization and partial purification of human marrow cells capable of initiating long-term hematopoiesis in vitro. Blood. 1989;74(5):1563-1570.
24 Terstappen LW, Huang S, Safford M, et al. Sequential generations of hematopoietic colonies derived from single nonlineage-committed CD34+CD38− progenitor cells. Blood. 1991;77(6):1218-1227.
25 Galy A, Travis M, Cen D, Chen B. Human T. B, natural killer, and dendritic cells arise from a common bone marrow progenitor cell subset. Immunity. 1995;3(4):459-473.
26 Miller JS, Alley KA, McGlave P. Differentiation of natural killer (NK) cells from human primitive marrow progenitors in a stroma-based long-term culture system: identification of a CD34+7+ NK progenitor. Blood. 1994;83(9):2594-2601.
27 LeBien TW. B-cell lymphopoiesis in mouse and man. Current Opinion in Immunology. 1998;10(2):188-195.
28 Pearce DJ, Taussig DC, Bonnet D. Implications of the expression of myeloid markers on normal and leukemic stem cells. Cell Cycle (Georgetown, Tex). 2006;5(3):271-273.
29 Randall TD, Weissman IL. Phenotypic and functional changes induced at the clonal level in hematopoietic stem cells after 5-fluorouracil treatment. Blood. 1997;89(10):3596-3606.
30 Hodgson GS, Bradley TR. Properties of haematopoietic stem cells surviving 5-fluorouracil treatment: evidence for a pre-CFU-S cell? Nature. 1979;281(5730):381-382.
31 Chaudhary PM, Roninson IB. Expression and activity of P-glycoprotein, a multidrug efflux pump, in human hematopoietic stem cells. Cell. 1991;66(1):85-94.
32 Morita Y, Ema H, Yamazaki S, Nakauchi H. Non-side-population hematopoietic stem cells in mouse bone marrow. Blood. 2006;108(8):2850-2856.
33 Goodell MA, Brose K, Paradis G, et al. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. Journal of Experimental Medicine. 1996;183(4):1797-1806.
34 McKenzie JL, Takenaka K, Gan OI, et al. Low rhodamine 123 retention identifies long-term human hematopoietic stem cells within the Lin-CD34+CD38− population. Blood. 2007;109(2):543-545.
35 McNiece IK, Robinson BE, Quesenberry PJ. Stimulation of murine colony-forming cells with high proliferative potential by the combination of GM-CSF and CSF-1. Blood. 1988;72(1):191-195.
36 Dexter TM, Allen TD, Lajtha LG. Conditions controlling the proliferation of haemopoietic stem cells in vitro. Journal of cellular physiology. 1977;91(3):335-344.
37 Fraser CC, Szilvassy SJ, Eaves CJ, Humphries RK. Proliferation of totipotent hematopoietic stem cells in vitro with retention of long-term competitive in vivo reconstituting ability. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(5):1968-1972.
38 Hao QL, Thiemann FT, Petersen D, et al. Extended long-term culture reveals a highly quiescent and primitive human hematopoietic progenitor population. Blood. 1996;88(9):3306-3313.
39 Ploemacher RE, van der Sluijs JP, van Beurden CA, et al. Use of limiting-dilution type long-term marrow cultures in frequency analysis of marrow-repopulating and spleen colony-forming hematopoietic stem cells in the mouse. Blood. 1991;78(10):2527-2533.
40 Miller JS, McCullar V, Verfaillie CM. Ex vivo culture of CD34+/Lin-/DR- cells in stroma-derived soluble factors, interleukin-3, and macrophage inflammatory protein-1alpha maintains not only myeloid but also lymphoid progenitors in a novel switch culture assay. Blood. 1998;91(12):4516-4522.
41 Schmitt TM, Zuniga-Pflucker JC. Induction of T cell development from hematopoietic progenitor cells by delta-like-1 in vitro. Immunity. 2002;17(6):749-756.
42 Berardi AC, Meffre E, Pflumio F, et al. Individual CD34+CD38lowCD19-CD10- progenitor cells from human cord blood generate B lymphocytes and granulocytes. Blood. 1997;89(10):3554-3564.
43 Punzel M, Wissink SD, Miller JS, et al. The myeloid-lymphoid initiating cell (ML-IC) assay assesses the fate of multipotent human progenitors in vitro. Blood. 1999;93(11):3750-3756.
44 Whitlock CA, Robertson D, Witte ON. Murine B cell lymphopoiesis in long term culture. J Immunol Methods. 1984;67(2):353-369.
45 Whitlock CA, Witte ON. Long-term culture of B lymphocytes and their precursors from murine bone marrow. Proceedings of the National Academy of Sciences of the United States of America. 1982;79(11):3608-3612.
46 Hao QL, Smogorzewska EM, Barsky LW, Crooks GM. In vitro identification of single CD34+CD38− cells with both lymphoid and myeloid potential. Blood. 1998;91(11):4145-4151.
47 Spangrude GJ, Heimfeld S, Weissman IL. Purification and characterization of mouse hematopoietic stem cells. Science (New York, NY). 1988;241(4861):58-62.
48 Lemischka IR, Raulet DH, Mulligan RC. Developmental potential and dynamic behavior of hematopoietic stem cells. Cell. 1986;45(6):917-927.
49 Jordan CT, McKearn JP, Lemischka IR. Cellular and developmental properties of fetal hematopoietic stem cells. Cell. 1990;61(6):953-963.
50 Szilvassy SJ, Fraser CC, Eaves CJ, et al. Retrovirus-mediated gene transfer to purified hemopoietic stem cells with long-term lympho-myelopoietic repopulating ability. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(22):8798-8802.
51 Zhong RK, Astle CM, Harrison DE. Distinct developmental patterns of short-term and long-term functioning lymphoid and myeloid precursors defined by competitive limiting dilution analysis in vivo. J Immunol. 1996;157(1):138-145.
52 Iscove NN, Nawa K. Hematopoietic stem cells expand during serial transplantation in vivo without apparent exhaustion. Curr Biol. 1997;7(10):805-808.
53 Jordan CT, Lemischka IR. Clonal and systemic analysis of long-term hematopoiesis in the mouse. Genes and Development. 1990;4(2):220-232.
54 McCune JM, Namikawa R, Kaneshima H, et al. The SCID-hu mouse: murine model for the analysis of human hematolymphoid differentiation and function. Science (New York, NY). 1988;241(4873):1632-1639.
55 Ishikawa F, Yasukawa M, Lyons B, et al. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor γ chain(null) mice. Blood. 2005;106(5):1565-1573.
56 Nolta JA, Hanley MB, Kohn DB. Sustained human hematopoiesis in immunodeficient mice by cotransplantation of marrow stroma expressing human interleukin-3: analysis of gene transduction of long-lived progenitors. Blood. 1994;83(10):3041-3051.
57 Hiramatsu H, Nishikomori R, Heike T, et al. Complete reconstitution of human lymphocytes from cord blood CD34+ cells using the NOD/SCID/gammacnull mice model. Blood. 2003;102(3):873-880.
58 Srour EF, Zanjani ED, Cornetta K, et al. Persistence of human multilineage, self-renewing lymphohematopoietic stem cells in chimeric sheep. Blood. 1993;82(11):3333-3342.
59 Pearson T, Greiner DL, Shultz LD. Creation of ‘humanized’ mice to study human immunity. Current Protocols in Immunology, edited by John E Coligan 2008; Chapter 15:Unit 15 21.
60 Norol F, Drouet M, Pflumio F, et al. Ex vivo expansion marginally amplifies repopulating cells from baboon peripheral blood mobilized CD34+ cells. British Journal of Haematology. 2002;117(4):924-934.
61 Andrews RG, Bryant EM, Bartelmez SH, et al. CD34+ marrow cells, devoid of T and B lymphocytes, reconstitute stable lymphopoiesis and myelopoiesis in lethally irradiated allogeneic baboons. Blood. 1992;80(7):1693-1701.
62 Tisdale JF, Hanazono Y, Sellers SE, et al. Ex vivo expansion of genetically marked rhesus peripheral blood progenitor cells results in diminished long-term repopulating ability. Blood. 1998;92(4):1131-1141.
63 Horn PA, Thomasson BM, Wood BL, et al. Distinct hematopoietic stem/progenitor cell populations are responsible for repopulating NOD/SCID mice compared with nonhuman primates. Blood. 2003;102(13):4329-4335.
64 Kiger AA, Jones DL, Schulz C, et al. Stem cell self-renewal specified by JAK-STAT activation in response to a support cell cue. Science (New York, NY). 2001;294(5551):2542-2545.
65 Tulina N, Matunis E. Control of stem cell self-renewal in Drosophila spermatogenesis by JAK-STAT signaling. Science (New York, NY). 2001;294(5551):2546-2549.
66 Giebel B, Zhang T, Beckmann J, et al. Primitive human hematopoietic cells give rise to differentially specified daughter cells upon their initial cell division. Blood. 2006;107(5):2146-2152.
67 Giebel B, Beckmann J. Asymmetric cell divisions of human hematopoietic stem and progenitor cells meet endosomes. Cell Cycle (Georgetown, Tex). 2007;6(18):2201-2204.
68 Bowie MB, Kent DG, Copley MR, Eaves CJ. Steel factor responsiveness regulates the high self-renewal phenotype of fetal hematopoietic stem cells. Blood. 2007;109(11):5043-5048.
69 Bowie MB, McKnight KD, Kent DG, et al. Hematopoietic stem cells proliferate until after birth and show a reversible phase-specific engraftment defect. Journal of Clinical Investigation. 2006;116(10):2808-2816.
70 Bowie MB, Kent DG, Dykstra B, et al. Identification of a new intrinsically timed developmental checkpoint that reprograms key hematopoietic stem cell properties. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(14):5878-5882.
71 Attar EC, Scadden DT. Regulation of hematopoietic stem cell growth. Leukemia. 2004;18(11):1760-1768.
72 Schofield R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells. 1978;4(1–2):7-25.
73 Quesenberry PJ, Crittenden RB, Lowry P, et al. In vitro and in vivo studies of stromal niches. Blood Cells. 1994;20(1):97-104. discussion 104–6
74 Lemischka IR. Microenvironmental regulation of hematopoietic stem cells. Stem Cells (Dayton, Ohio). 1997;15(Suppl. 1):63-68.
75 Clark BR, Keating A. Biology of bone marrow stroma. Annals of the New York Academy of Sciences. 1995;29(770):70-78.
76 Yoder MC, Williams DA. Matrix molecule interactions with hematopoietic stem cells. Experimental Hematology. 1995;23(9):961-967.
77 Verfaillie C, Hurley R, Bhatia R, McCarthy JB. Role of bone marrow matrix in normal and abnormal hematopoiesis. Critical Reviews in Oncology/Hematology. 1994;16(3):201-224.
78 Gong JK. Endosteal marrow: a rich source of hematopoietic stem cells. Science (New York, NY). 1978;199(4336):1443-1445.
79 Nilsson SK, Johnston HM, Coverdale JA. Spatial localization of transplanted hemopoietic stem cells: inferences for the localization of stem cell niches. Blood. 2001;97(8):2293-2299.
80 Patt HM, Maloney MA. Bone formation and resorption as a requirement for marrow development. Proc Soc Exp Biol Med. 1972;140(1):205-207.
81 Zhang J, Niu C, Ye L, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425(6960):836-841.
82 Park C, Afrikanova I, Chung YS, et al. A hierarchical order of factors in the generation of FLK1- and SCL-expressing hematopoietic and endothelial progenitors from embryonic stem cells. Development (Cambridge, England). 2004;131(11):2749-2762.
83 Arai F, Hirao A, Ohmura M, et al. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118(2):149-161.
84 Varnum-Finney B, Brashem-Stein C, Bernstein ID. Combined effects of Notch signaling and cytokines induce a multiple log increase in precursors with lymphoid and myeloid reconstituting ability. Blood. 2003;101(5):1784-1789.
85 Haug JS, He XC, Grindley JC, et al. N-cadherin expression level distinguishes reserved versus primed states of hematopoietic stem cells. Cell Stem Cell. 2008;2(4):367-379.
86 Willert K, Brown JD, Danenberg E, et al. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423(6938):448-452.
87 Fleming HE, Janzen V, Lo Celso C, et al. Wnt signaling in the niche enforces hematopoietic stem cell quiescence and is necessary to preserve self-renewal in vivo. Cell Stem Cell. 2008;2(3):274-283.
88 Calvi LM, Adams GB, Weibrecht KW, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425(6960):841-846.
89 Kent D, Copley M, Benz C, et al. Regulation of hematopoietic stem cells by the steel factor/KIT signaling pathway. Clin Cancer Res. 2008;14(7):1926-1930.
90 Hidalgo A, Sanz-Rodriguez F, Rodriguez-Fernandez JL, et al. Chemokine stromal cell-derived factor-1alpha modulates VLA-4 integrin-dependent adhesion to fibronectin and VCAM-1 on bone marrow hematopoietic progenitor cells. Experimental Hematology. 2001;29(3):345-355.
91 Nilsson SK, Johnston HM, Whitty GA, et al. Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood. 2005;106(4):1232-1239.
92 Stier S, Ko Y, Forkert R, et al. Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. Journal of Experimental Medicine. 2005;201(11):1781-1791.
93 Porter RL, Calvi LM. Communications between bone cells and hematopoietic stem cells. Arch Biochem Biophys. 2008;473(2):193-200.
94 Adams GB, Chabner KT, Alley IR, et al. Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature. 2006;439(7076):599-603.
95 Visnjic D, Kalajzic Z, Rowe DW, et al. Hematopoiesis is severely altered in mice with an induced osteoblast deficiency. Blood. 2004;103(9):3258-3264.
96 Tavassoli M. Structure and function of sinusoidal endothelium of bone marrow. Progress in Clinical and Biological Research. 1981;59B:249-256.
97 Rice KL, Hormaeche I, Licht JD. Epigenetic regulation of normal and malignant hematopoiesis. Oncogene. 2007;26(47):6697-6714.
98 Goldfarb AN, Greenberg JM. T-cell acute lymphoblastic leukemia and the associated basic helix-loop-helix gene SCL/tal. Leukemia and Lymphoma. 1994;12(3–4):157-166.
99 Rabbitts TH, Bucher K, Chung G, et al. The effect of chromosomal translocations in acute leukemias: the LMO2 paradigm in transcription and development. Cancer Research. 1999;59(Suppl. 7):1794s-1798s.
100 Abramovich C, Humphries RK. Hox regulation of normal and leukemic hematopoietic stem cells. Current Opinion in Hematology. 2005;12(3):210-216.
101 Jundt F, Schwarzer R, Dorken B. Notch signaling in leukemias and lymphomas. Current Molecular Medicine. 2008;8(1):51-59.
102 McGonigle GJ, Lappin TR, Thompson A. Grappling with the HOX network in hematopoiesis and leukemia. Front Biosci. 2008;13:4297-4308.
103 Scott EW, Simon MC, Anastasi J, Singh H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science (New York, NY). 1994;265(5178):1573-1577.
104 Tsai FY, Keller G, Kuo FC, et al. An early haematopoietic defect in mice lacking the transcription factor GATA-2. Nature. 1994;371(6494):221-226.
105 Warren AJ, Colledge WH, Carlton MB, et al. The oncogenic cysteine-rich LIM domain protein rbtn2 is essential for erythroid development. Cell. 1994;78(1):45-57.
106 Robb L, Lyons I, Li R, et al. Absence of yolk sac hematopoiesis from mice with a targeted disruption of the SCL gene. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(15):7075-7079.
107 Shivdasani RA, Mayer EL, Orkin SH. Absence of blood formation in mice lacking the T-cell leukaemia oncoprotein tal-1/SCL. Nature. 1995;373(6513):432-434.
108 Patterson LJ, Gering M, Eckfeldt CE, et al. The transcription factors Scl and Lmo2 act together during development of the hemangioblast in zebrafish. Blood. 2007;109(6):2389-2398.
109 Gering M, Rodaway AR, Gottgens B, et al. The SCL gene specifies haemangioblast development from early mesoderm. Embo J. 1998;17(14):4029-4045.
110 Gering M, Yamada Y, Rabbitts TH, Patient RK. Lmo2 and Scl/Tal1 convert non-axial mesoderm into haemangioblasts which differentiate into endothelial cells in the absence of Gata1. Development (Cambridge, England). 2003;130(25):6187-6199.
111 Maeno M, Mead PE, Kelley C, et al. The role of BMP-4 and GATA-2 in the induction and differentiation of hematopoietic mesoderm in Xenopus laevis. Blood. 1996;88(6):1965-1972.
112 Sauvageau G, Thorsteinsdottir U, Eaves CJ, et al. Overexpression of HOXB4 in hematopoietic cells causes the selective expansion of more primitive populations in vitro and in vivo. Genes and Development. 1995;9(14):1753-1765.
113 Lawrence HJ, Helgason CD, Sauvageau G, et al. Mice bearing a targeted interruption of the homeobox gene HOXA9 have defects in myeloid, erythroid, and lymphoid hematopoiesis. Blood. 1997;89(6):1922-1930.
114 Sauvageau G, Thorsteinsdottir U, Hough MR, et al. Overexpression of HOXB3 in hematopoietic cells causes defective lymphoid development and progressive myeloproliferation. Immunity. 1997;6(1):13-22.
115 Thorsteinsdottir U, Sauvageau G, Humphries RK. Hox homeobox genes as regulators of normal and leukemic hematopoiesis. Hematology/oncology clinics of North America. 1997;11(6):1221-1237.
116 Thorsteinsdottir U, Sauvageau G, Hough MR, Dragowska W, Lansdorp PM, Lawrence HJ, et al. Overexpression of HOXA10 in murine hematopoietic cells perturbs both myeloid and lymphoid differentiation and leads to acute myeloid leukemia. Molecular and cellular biology. 1997;17(1):495-505.
117 Antonchuk J, Sauvageau G, Humphries RK. HOXB4-induced expansion of adult hematopoietic stem cells ex vivo. Cell. 2002;109(1):39-45.
118 Shen WF, Montgomery JC, Rozenfeld S, et al. AbdB-like Hox proteins stabilize DNA binding by the Meis1 homeodomain proteins. Molecular and Cellular Biology. 1997;17(11):6448-6458.
119 Shen WF, Rozenfeld S, Lawrence HJ, Largman C. The Abd-B-like Hox homeodomain proteins can be subdivided by the ability to form complexes with Pbx1a on a novel DNA target. Journal of Biological Chemistry. 1997;272(13):8198-8206.
120 Swift GH, Liu Y, Rose SD, et al. An endocrine-exocrine switch in the activity of the pancreatic homeodomain protein PDX1 through formation of a trimeric complex with PBX1b and MRG1 (MEIS2). Molecular and Cellular Biology. 1998;18(9):5109-5120.
121 Shen WF, Rozenfeld S, Kwong A, et al. HOXA9 forms triple complexes with PBX2 and MEIS1 in myeloid cells. Molecular and Cellular Biology. 1999;19(4):3051-3061.
122 Jacobs Y, Schnabel CA, Cleary ML. Trimeric association of Hox and TALE homeodomain proteins mediates Hoxb2 hindbrain enhancer activity. Molecular and Cellular Biology. 1999;19(7):5134-5142.
123 DiMartino JF, Selleri L, Traver D, et al. The Hox cofactor and proto-oncogene Pbx1 is required for maintenance of definitive hematopoiesis in the fetal liver. Blood. 2001;98(3):618-626.
124 Hisa T, Spence SE, Rachel RA, et al. Hematopoietic, angiogenic and eye defects in Meis1 mutant animals. Embo J. 2004;23(2):450-459.
125 Pineault N, Helgason CD, Lawrence HJ, Humphries RK. Differential expression of Hox, Meis1, and Pbx1 genes in primitive cells throughout murine hematopoietic ontogeny. Experimental Hematology. 2002;30(1):49-57.
126 Argiropoulos B, Humphries RK. Hox genes in hematopoiesis and leukemogenesis. Oncogene. 2007;26(47):6766-6776.
127 Thorsteinsdottir U, Sauvageau G, Humphries RK. Enhanced in vivo regenerative potential of HOXB4-transduced hematopoietic stem cells with regulation of their pool size. Blood. 1999;94(8):2605-2612.
128 Amsellem S, Pflumio F, Bardinet D, et al. Ex vivo expansion of human hematopoietic stem cells by direct delivery of the HOXB4 homeoprotein. Nature Medicine. 2003;9(11):1423-1427.
129 Kyba M, Perlingeiro RC, Daley GQ. HoxB4 confers definitive lymphoid-myeloid engraftment potential on embryonic stem cell and yolk sac hematopoietic progenitors. Cell. 2002;109(1):29-37.
130 Kyba M, Perlingeiro RC, Daley GQ. Development of hematopoietic repopulating cells from embryonic stem cells. Methods in Enzymology. 2003;365:114-129.
131 Nakamura T. NUP98 fusion in human leukemia: dysregulation of the nuclear pore and homeodomain proteins. International Journal of Hematology. 2005;82(1):21-27.
132 Slape C, Aplan PD. The role of NUP98 gene fusions in hematologic malignancy. Leukemia and Lymphoma. 2004;45(7):1341-1350.
133 Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer. 2006;6(11):846-856.
134 Dietrich N, Bracken AP, Trinh E, et al. Bypass of senescence by the polycomb group protein CBX8 through direct binding to the INK4A-ARF locus. Embo J. 2007;26(6):1637-1648.
135 Guo WJ, Datta S, Band V, Dimri GP. Mel-18, a polycomb group protein, regulates cell proliferation and senescence via transcriptional repression of Bmi-1 and c-Myc oncoproteins. Molecular Biology of the Cell. 2007;18(2):536-546.
136 Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423(6937):255-260.
137 Loring JF, Porter JG, Seilhammer J, et al. A gene expression profile of embryonic stem cells and embryonic stem cell-derived neurons. Restorative Neurology and Neuroscience. 2001;18(2–3):81-88.
138 Dellino GI, Schwartz YB, Farkas G, et al. Polycomb silencing blocks transcription initiation. Molecular Cell. 2004;13(6):887-893.
139 Wang L, Brown JL, Cao R, et al. Hierarchical recruitment of polycomb group silencing complexes. Molecular Cell. 2004;14(5):637-646.
140 Breiling A, Bonte E, Ferrari S, et al. The Drosophila polycomb protein interacts with nucleosomal core particles in vitro via its repression domain. Molecular and Cellular Biology. 1999;19(12):8451-8460.
141 Ogawa H, Ishiguro K, Gaubatz S, et al. A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G0 cells. Science (New York, NY). 2002;296(5570):1132-1136.
142 Lund AH, van Lohuizen M. Polycomb complexes and silencing mechanisms. Current Opinion in Cell Biology. 2004;16(3):239-246.
143 Raaphorst FM, Otte AP, Meijer CJ. Polycomb-group genes as regulators of mammalian lymphopoiesis. Trends in Immunology. 2001;22(12):682-690.
144 Ohta H, Sawada A, Kim JY, et al. Polycomb group gene rae28 is required for sustaining activity of hematopoietic stem cells. Journal of Experimental Medicine. 2002;195(6):759-770.
145 Lessard J, Sauvageau G. Polycomb group genes as epigenetic regulators of normal and leukemic hemopoiesis. Experimental Hematology. 2003;31(7):567-585.
146 Molofsky AV, Pardal R, Iwashita T, et al. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature. 2003;425(6961):962-967.
147 Molofsky AV, He S, Bydon M, et al. Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16Ink4a and p19Arf senescence pathways. Genes and Development. 2005;19(12):1432-1437.
148 Zhang HW, Ding J, Jin JL, et al. Defects in mesenchymal stem cell self-renewal and cell fate determination lead to an osteopenic phenotype in Bmi-1 null mice. J Bone Miner Res. 2010;25(3):640-652.
149 Park IK, Qian D, Kiel M, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423(6937):302-305.
150 Iwama A, Oguro H, Negishi M, et al. Enhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1. Immunity. 2004;21(6):843-851.
151 Kajiume T, Ninomiya Y, Ishihara H, et al. Polycomb group gene mel-18 modulates the self-renewal activity and cell cycle status of hematopoietic stem cells. Experimental Hematology. 2004;32(6):571-578.
152 Kajiume T, Ohno N, Sera Y, Kawahara Y, Yuge L, Kobayashi M. Reciprocal expression of Bmi1 and Mel-18 is associated with functioning of primitive hematopoietic cells. Experimental hematology. 2009;37(7):857-866. e2
153 Kamminga LM, Bystrykh LV, de Boer A, et al. The Polycomb group gene Ezh2 prevents hematopoietic stem cell exhaustion. Blood. 2006;107(5):2170-2179.
154 De Haan G, Gerrits A. Epigenetic control of hematopoietic stem cell aging: the case of Ezh2. Annals of the New York Academy of Sciences. 2007;1106:233-239.
155 Majewski IJ, Blewitt ME, de Graaf CA, et al. Polycomb repressive complex 2 (PRC2) restricts hematopoietic stem cell activity. PLoS Biology. 2008;6(4):e93.
156 Lessard J, Schumacher A, Thorsteinsdottir U, et al. Functional antagonism of the Polycomb-Group genes eed and Bmi1 in hemopoietic cell proliferation. Genes and Development. 1999;13(20):2691-2703.
157 Cheng T, Rodrigues N, Shen H, et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287(5459):1804-1808.
158 Stier S, Cheng T, Forkert R, et al. Ex vivo targeting of p21Cip1/Waf1 permits relative expansion of human hematopoietic stem cells. Blood. 2003;102(4):1260-1266.
159 Cheng T, Rodrigues N, Dombkowski D, et al. Stem cell repopulation efficiency but not pool size is governed by p27(kip1). Nature Medicine. 2000;6(11):1235-1240.
160 Oguro H, Iwama A, Morita Y, et al. Differential impact of Ink4a and Arf on hematopoietic stem cells and their bone marrow microenvironment in Bmi1-deficient mice. Journal of Experimental Medicine. 2006;203(10):2247-2253.
161 Janzen V, Forkert R, Fleming HE, et al. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443(7110):421-426.
162 Yu H, Yuan Y, Shen H, Cheng T. Hematopoietic stem cell exhaustion impacted by p18 INK4C and p21 Cip1/Waf1 in opposite manners. Blood. 2006;107(3):1200-1206.
163 Yuan Y, Shen H, Franklin DS, et al. In vivo self-renewing divisions of haematopoietic stem cells are increased in the absence of the early G1-phase inhibitor, p18INK4C. Nature Cell Biology. 2004;6(5):436-442.
164 Walkley CR, Orkin SH. Rb is dispensable for self-renewal and multilineage differentiation of adult hematopoietic stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(24):9057-9062.
165 Billia F, Barbara M, McEwen J, et al. Resolution of pluripotential intermediates in murine hematopoietic differentiation by global complementary DNA amplification from single cells: confirmation of assignments by expression profiling of cytokine receptor transcripts. Blood. 2001;97(8):2257-2268.
166 Hamel W, Westphal M. The road less travelled: c-kit and stem cell factor. Journal of Neuro-oncology. 1997;35(3):327-333.
167 Lyman SD, Williams DE. Biology and potential clinical applications of flt3 ligand. Current Opinion in Hematology. 1995;2(3):177-181.
168 McCulloch EA, Siminovitch L, Till JE, et al. The cellular basis of the genetically determined hemopoietic defect in anemic mice of genotype Sl-Sld. Blood. 1965;26(4):399-410.
169 McCulloch EA, Siminovitch L, Till JE. Spleen-Colony Formation in Anemic Mice of Genotype Ww. Science (New York, NY). 1964;144:844-846.
170 Zandstra PW, Conneally E, Petzer AL, et al. Cytokine manipulation of primitive human hematopoietic cell self-renewal. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(9):4698-4703.
171 Audet J, Miller CL, Eaves CJ, Piret JM. Common and distinct features of cytokine effects on hematopoietic stem and progenitor cells revealed by dose-response surface analysis. Biotechnology and Bioengineering. 2002;80(4):393-404.
172 Mackarehtschian K, Hardin JD, Moore KA, et al. Targeted disruption of the flk2/flt3 gene leads to deficiencies in primitive hematopoietic progenitors. Immunity. 1995;3(1):147-161.
173 McKenna HJ, Stocking KL, Miller RE, et al. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood. 2000;95(11):3489-3497.
174 Miller CL, Eaves CJ. Expansion in vitro of adult murine hematopoietic stem cells with transplantable lympho-myeloid reconstituting ability. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(25):13648-13653.
175 Ueda T, Tsuji K, Yoshino H, et al. Expansion of human NOD/SCID-repopulating cells by stem cell factor, Flk2/Flt3 ligand, thrombopoietin, IL-6, and soluble IL-6 receptor. Journal of Clinical Investigation. 2000;105(7):1013-1021.
176 Bagley CJ, Woodcock JM, Stomski FC, Lopez AF. The structural and functional basis of cytokine receptor activation: lessons from the common beta subunit of the granulocyte-macrophage colony-stimulating factor, interleukin-3 (IL-3), and IL-5 receptors. Blood. 1997;89(5):1471-1482.
177 Liu KD, Gaffen SL, Goldsmith MA. JAK/STAT signaling by cytokine receptors. Current Opinion in Immunology. 1998;10(3):271-278.
178 Petzer AL, Zandstra PW, Piret JM, Eaves CJ. Differential cytokine effects on primitive (CD34+CD38-) human hematopoietic cells: novel responses to Flt3-ligand and thrombopoietin. Journal of Experimental Medicine. 1996;183(6):2551-2558.
179 Petzer AL, Hogge DE, Landsdorp PM, et al. Self-renewal of primitive human hematopoietic cells (long-term-culture-initiating cells) in vitro and their expansion in defined medium. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(4):1470-1474.
180 Qian H, Buza-Vidas N, Hyland CD, et al. Critical role of thrombopoietin in maintaining adult quiescent hematopoietic stem cells. Cell Stem Cell. 2007;1(6):671-684.
181 Yoshihara H, Arai F, Hosokawa K, et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell. 2007;1(6):685-697.
182 Peters M, Muller AM, Rose-John S. Interleukin-6 and soluble interleukin-6 receptor: direct stimulation of gp130 and hematopoiesis. Blood. 1998;92(10):3495-3504.
183 Nandurkar HH, Robb L, Begley CG. The role of IL-II in hematopoiesis as revealed by a targeted mutation of its receptor. Stem Cells (Dayton, Ohio). 1998;16(Suppl. 2):53-65.
184 Miyajima A, Kinoshita T, Tanaka M, et al. Role of oncostatin M in hematopoiesis and liver development. Cytokine and Growth Factor Reviews. 2000;11(3):177-183.
185 Taupin JL, Pitard V, Dechanet J, et al. Leukemia inhibitory factor: part of a large ingathering family. International Reviews of Immunology. 1998;16(3–4):397-426.
186 Peeters M, Ottersbach K, Bollerot K, et al. Ventral embryonic tissues and hedgehog proteins induce early AGM hematopoietic stem cell development. Development (Cambridge, England). 2009;136(15):2613-2621.
187 Dyer MA, Farrington SM, Mohn D, et al. Indian hedgehog activates hematopoiesis and vasculogenesis and can respecify prospective neurectodermal cell fate in the mouse embryo. Development (Cambridge, England). 2001;128(10):1717-1730.
188 Baron M. Induction of embryonic hematopoietic and endothelial stem/progenitor cells by hedgehog-mediated signals. Differentiation. 2001;68(4–5):175-185.
189 Kobune M, Ito Y, Kawano Y, et al. Indian hedgehog gene transfer augments hematopoietic support of human stromal cells including NOD/SCID-beta2m−/− repopulating cells. Blood. 2004;104(4):1002-1009.
190 Trowbridge JJ, Scott MP, Bhatia M. Hedgehog modulates cell cycle regulators in stem cells to control hematopoietic regeneration. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(38):14134-14139.
191 Gao J, Graves S, Koch U, et al. Hedgehog signaling is dispensable for adult hematopoietic stem cell function. Cell Stem Cell. 2009;4(6):548-558.
192 Hofmann I, Stover EH, Cullen DE, et al. Hedgehog signaling is dispensable for adult murine hematopoietic stem cell function and hematopoiesis. Cell Stem Cell. 2009;4(6):559-567.
193 Willert K, Nusse R. Beta-catenin: a key mediator of Wnt signaling. Current opinion in genetics and Development. 1998;8(1):95-102.
194 Hsu SC, Galceran J, Grosschedl R. Modulation of transcriptional regulation by LEF-1 in response to Wnt-1 signaling and association with beta-catenin. Molecular and Cellular Biology. 1998;18(8):4807-4818.
195 Reya T, Duncan AW, Ailles L, et al. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423(6938):409-414.
196 Luis TC, Weerkamp F, Naber BA, et al. Wnt3a deficiency irreversibly impairs hematopoietic stem cell self-renewal and leads to defects in progenitor cell differentiation. Blood. 2009;113(3):546-554.
197 Koch U, Wilson A, Cobas M, et al. Simultaneous loss of beta- and gamma-catenin does not perturb hematopoiesis or lymphopoiesis. Blood. 2008;111(1):160-164.
198 Cobas M, Wilson A, Ernst B, et al. Beta-catenin is dispensable for hematopoiesis and lymphopoiesis. Journal of Experimental Medicine. 2004;199(2):221-229.
199 Kirstetter P, Anderson K, Porse BT, et al. Activation of the canonical Wnt pathway leads to loss of hematopoietic stem cell repopulation and multilineage differentiation block. Nature Immunology. 2006;7(10):1048-1056.
200 Scheller M, Huelsken J, Rosenbauer F, et al. Hematopoietic stem cell and multilineage defects generated by constitutive beta-catenin activation. Nature Immunology. 2006;7(10):1037-1047.
201 Qian Z, Chen L, Fernald AA, et al. A critical role for Apc in hematopoietic stem and progenitor cell survival. Journal of Experimental Medicine. 2008;205(9):2163-2175.
202 Van Den Berg DJ, Sharma AK, Bruno E, Hoffman R. Role of members of the Wnt gene family in human hematopoiesis. Blood. 1998;92(9):3189-3202.
203 Austin TW, Solar GP, Ziegler FC, et al. A role for the Wnt gene family in hematopoiesis: expansion of multilineage progenitor cells. Blood. 1997;89(10):3624-3635.
204 Nemeth MJ, Topol L, Anderson SM, et al. Wnt5a inhibits canonical Wnt signaling in hematopoietic stem cells and enhances repopulation. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(39):15436-15441.
205 Mishra L, Derynck R, Mishra B. Transforming growth factor-beta signaling in stem cells and cancer. Science (New York, NY). 2005;310(5745):68-71.
206 Piek E, Heldin CH, Ten Dijke P. Specificity, diversity, and regulation in TGF-beta superfamily signaling. FASEB J. 1999;13(15):2105-2124.
207 Dickson MC, Martin JS, Cousins FM, et al. Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development (Cambridge, England). 1995;121(6):1845-1854.
208 Martin JS, Dickson MC, Cousins FM, et al. Analysis of homozygous TGF beta 1 null mouse embryos demonstrates defects in yolk sac vasculogenesis and hematopoiesis. Annals of the New York Academy of Sciences. 1995;752:300-308.
209 Larsson J, Goumans MJ, Sjostrand LJ, et al. Abnormal angiogenesis but intact hematopoietic potential in TGF-beta type I receptor-deficient mice. EMBO J. 2001;20(7):1663-1673.
210 Soma T, Yu JM, Dunbar CE. Maintenance of murine long-term repopulating stem cells in ex vivo culture is affected by modulation of transforming growth factor-beta but not macrophage inflammatory protein-1 alpha activities. Blood. 1996;87(11):4561-4567.
211 Fortunel NO, Hatzfeld A, Hatzfeld JA. Transforming growth factor-beta: pleiotropic role in the regulation of hematopoiesis. Blood. 2000;96(6):2022-2036.
212 Jacobsen FW, Stokke T, Jacobsen SE. Transforming growth factor-beta potently inhibits the viability-promoting activity of stem cell factor and other cytokines and induces apoptosis of primitive murine hematopoietic progenitor cells. Blood. 1995;86(8):2957-2966.
213 Zhao GQ. Consequences of knocking out BMP signaling in the mouse. Genesis. 2003;35(1):43-56.
214 Marshall CJ, Kinnon C, Thrasher AJ. Polarized expression of bone morphogenetic protein-4 in the human aorta-gonad-mesonephros region. Blood. 2000;96(4):1591-1593.
215 Bhatia M, Bonnet D, Wu D, et al. Bone morphogenetic proteins regulate the developmental program of human hematopoietic stem cells. Journal of Experimental Medicine. 1999;189(7):1139-1148.
216 Utsugisawa T, Moody JL, Aspling M, et al. A road map toward defining the role of Smad signaling in hematopoietic stem cells. Stem Cells (Dayton, Ohio). 2006;24(4):1128-1136.
217 Blank U, Karlsson G, Moody JL, et al. Smad7 promotes self-renewal of hematopoietic stem cells. Blood. 2006;108(13):4246-4254.
218 Liu B, Sun Y, Jiang F, et al. Disruption of Smad5 gene leads to enhanced proliferation of high-proliferative potential precursors during embryonic hematopoiesis. Blood. 2003;101(1):124-133.
219 Canalis E, Economides AN, Gazzerro E. Bone morphogenetic proteins, their antagonists, and the skeleton. Endocr Rev. 2003;24(2):218-235.
220 Weinmaster G. The ins and outs of notch signaling. Molecular and Cellular Neurosciences. 1997;9(2):91-102.
221 Simpson P. Notch signalling in development: on equivalence groups and asymmetric developmental potential. Current Opinion in Genetics and Development. 1997;7(4):537-542.
222 Muskavitch MA. Delta-notch signaling and Drosophila cell fate choice. Developmental Biology. 1994;166(2):415-430.
223 Dale TC. Signal transduction by the Wnt family of ligands. Biochemical Journal. 1998;329(Pt 2):209-223.
224 Mancini SJ, Mantei N, Dumortier A, et al. Jagged1-dependent Notch signaling is dispensable for hematopoietic stem cell self-renewal and differentiation. Blood. 2005;105(6):2340-2342.
225 Milner LA, Kopan R, Martin DI, Bernstein ID. A human homologue of the Drosophila developmental gene, Notch, is expressed in CD34+ hematopoietic precursors. Blood. 1994;83(8):2057-2062.
226 Pear WS, Aster JC, Scott ML, et al. Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. Journal of Experimental Medicine. 1996;183(5):2283-2291.
227 Varnum-Finney B, Purton LE, Yu M, et al. The Notch ligand, Jagged-1, influences the development of primitive hematopoietic precursor cells. Blood. 1998;91(11):4084-4091.
228 Ohishi K, Varnum-Finney B, Bernstein ID. Delta-1 enhances marrow and thymus repopulating ability of human CD34(+)CD38(-) cord blood cells. Journal of Clinical Investigation. 2002;110(8):1165-1174.
229 Duncan AW, Rattis FM, DiMascio LN, et al. Integration of Notch and Wnt signaling in hematopoietic stem cell maintenance. Nature Immunology. 2005;6(3):314-322.
230 Moore KA, Pytowski B, Witte L, et al. Hematopoietic activity of a stromal cell transmembrane protein containing epidermal growth factor-like repeat motifs. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(8):4011-4016.
231 Karanu FN, Murdoch B, Miyabayashi T, et al. Human homologues of Delta-1 and Delta-4 function as mitogenic regulators of primitive human hematopoietic cells. Blood. 2001;97(7):1960-1967.
232 LeRoith D, Werner H, Burguera B, et al. The insulin-like growth factor family of peptides, binding proteins and receptors: their potential role in tissue regeneration. Advances in Experimental Medicine and Biology. 1992;321:21-28. discussion 9–30
233 Zhang CC, Lodish HF. Insulin-like growth factor 2 expressed in a novel fetal liver cell population is a growth factor for hematopoietic stem cells. Blood. 2004;103(7):2513-2521.
234 Zhang CC, Kaba M, Iizuka S, et al. Angiopoietin-like 5 and IGFBP2 stimulate ex vivo expansion of human cord blood hematopoietic stem cells as assayed by NOD/SCID transplantation. Blood. 2008;111(7):3415-3423.