Qualitative Disorders of Platelets and Vasculature
After completion of this chapter, the reader will be able to:
1. Describe the effect of aspirin on the cyclooxygenase pathway.
2. Describe the defect in each of the following hereditary disorders: storage pool disease, gray platelet syndrome, Glanzmann thrombasthenia, and Bernard-Soulier syndrome (BSS).
3. Discuss the mechanisms of action of antiplatelet drugs.
4. Explain the effects of paraproteins on platelet function.
5. Compare and contrast Glanzmann thrombasthenia and BSS.
6. Recognize the clinical presentation of patients with dysfunctional platelets.
7. Distinguish among the following types of inherited platelet disorders: membrane receptor abnormality, secretion disorder, and storage pool deficiency.
8. For each of the inherited platelet disorders listed, name useful laboratory tests and recognize diagnostic results.
9. Identify the most common type of hereditary platelet defect.
10. Discuss the mechanism of the platelet defects associated with myeloproliferative diseases, uremia, and liver disease.
11. Recognize conditions associated with acquired vascular disorders.
Clinical manifestations of bleeding disorders can be divided into two broad, rather poorly defined groups: (1) superficial bleeding (e.g., petechiae, epistaxis, or gingival bleeding), usually associated with a platelet defect or vascular disorder; and (2) deep tissue bleeding (e.g., hematomas or hemarthrosis), usually associated with plasma clotting factor deficiencies.1 This chapter describes platelet and vascular disorders. Bleeding problems resulting from defects in the coagulation mechanism are described in Chapter 41.
Qualitative Platelet Disorders
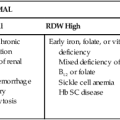
Excessive bruising, superficial (mucocutaneous) bleeding, and a prolonged bleeding time in a patient whose platelet count is normal suggest an acquired or a congenital disorder of platelet function. Congenital disorders have been described that result from abnormalities of each of the major phases of platelet function: adhesion, aggregation, and secretion. Rapid progress in this field began in the 1960s, mostly as a result of the development of instruments and test methods for measuring platelet function.2 Qualitative disorders are summarized in Box 44-1.
Disorders of Adhesion Receptors
Bernard-Soulier (Giant Platelet) Syndrome
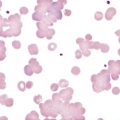
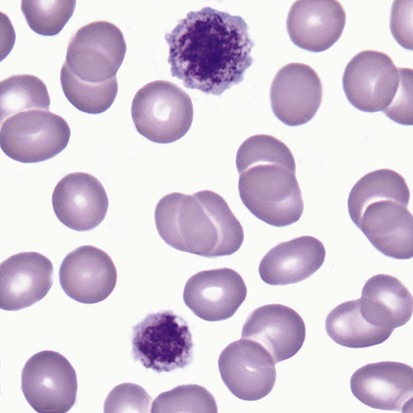
Bernard-Soulier syndrome (BSS) is a rare syndrome that is usually manifested in infancy or childhood with hemorrhage characteristic of defective platelet function: ecchymoses, epistaxis, and gingival bleeding. Hemarthroses and expanding hematomas are only rarely seen. BSS is inherited as an autosomal recessive disorder in which the glycoprotein Ib/IX/V (GP Ib/IX/V) complex is missing from the platelet surface or exhibits abnormal function. Heterozygotes who have about 50% of normal levels of GP Ib, GP V, and GP IX have normal or near-normal platelet function. Homozygotes have a moderate to severe bleeding disorder characterized by a prolonged bleeding time, enlarged platelets, thrombocytopenia, and usually decreased platelet survival. Platelet counts generally range from 40,000/mcL to near normal.3 On peripheral blood films, platelets typically are 5 to 8 µm in diameter, but can be 20 µm (Figure 44-1). Viewed by electron microscopy, BSS platelets contain a larger number of cytoplasmic vacuoles and membrane complexes, and these observations extend to megakaryocytes, in which the appearance of the demarcation membrane system is irregular.2,4–7
Four glycoproteins are required to form the GP Ib/IX/V complex: GP Ibα, GP Ibβ, GP IX, and GP V. In the complex, these proteins are present in the ratio of 2 : 2 : 2 : 1. The gene for GP Ibα is located on chromosome 17, the gene for GP Ibβ is located on chromosome 22, and the genes for GP IX and GP V are located on chromosome 3. For surface expression of the GP Ib/IX complex, it seems that synthesis of three proteins, GP Ibα, GP Ibβ, and GP IX, is required. Only GP V can be expressed alone in significant quantities on the surface of platelets, but the expression seems to be enhanced if the rest of the complex is present. The most frequent forms of BSS involve defects in GP Ibα synthesis or expression. The presence of GP Ibα is essential to normal function because it contains binding sites for von Willebrand factor (VWF) and thrombin. Defects in the GP Ibβ and GP IX genes also are known to result in BSS, however.8–10
BSS platelets have normal aggregation responses to adenosine diphosphate (ADP), epinephrine, collagen, and arachidonic acid but do not respond to ristocetin and have diminished response to thrombin.2,4,6 The lack of response to ristocetin is due to the lack of GP Ib/IX/V complexes and the inability of BSS platelets to bind VWF. Lack of binding to VWF also accounts for the inability of platelets to adhere to exposed subendothelium and the resultant bleeding characteristic of this disorder. This defect in adhesion shows the importance of initial platelet attachment in primary hemostasis. In many respects, this disorder resembles the defect seen in von Willebrand disease (VWD). In contrast to VWD, however, this abnormality cannot be corrected by the addition of normal plasma or cryoprecipitate, which is consistent with a defect that resides in the platelets.
Several unusual variants of BSS have been described in which all or most of the GP Ib/IX/V complex is present, but the mutations affect binding domains, affect interactions between elements of the complex, or result in truncation of a specific protein in the complex. In these cases, the complex fails to bind VWF or does so poorly.9
Platelet-type VWD (pseudo-VWD) is associated with a gain of function and spontaneous binding of plasma VWF. GP Ibα mutations that give rise to platelet-type VWD are 233 Gly →Val or Ser and 239Met→Val.11,12 Loss of residues 421 to 429 in GP Ibα also has been reported to result in platelet-type VWD.13
There is no specific treatment for BSS. Platelet transfusions are the therapy of choice, but patients invariably develop alloantibodies, so that further platelet transfusion is not possible. BSS patients tend to do better if apheresis platelets are used for transfusion, because this tends to limit the number of donors to which the patient is exposed, and the rate of alloimmunization tends to be lower.4,6 Other treatments that have been used include desmopressin acetate (DDAVP) and, more recently, recombinant factor VIIa.
Glanzmann Thrombasthenia
Glanzmann thrombasthenia originally was described as a bleeding disorder associated with abnormal in vitro clot retraction and a normal platelet count. It is inherited as an autosomal recessive disorder and is seen most frequently in populations with a high degree of consanguinity. Heterozygotes are clinically normal, whereas homozygotes have serious bleeding problems. This rare disorder manifests itself clinically in the neonatal period or infancy, occasionally with bleeding after circumcision and frequently with epistaxis and gingival bleeding. Hemorrhagic manifestations include petechiae, purpura, menorrhagia, gastrointestinal bleeding, and hematuria. There are wide variations in the clinical symptoms. Some patients may have minimal symptoms, whereas others may have frequent and serious hemorrhagic complications. The severity of the bleeding episodes seems to decrease with age.14,15
The biochemical lesion responsible for the disorder is a deficiency or abnormality of the platelet membrane GP IIb/IIIa (αIIb/β3) complex, a membrane receptor capable of binding fibrinogen, VWF, fibronectin, and other adhesive ligands. Typically, the platelets of homozygous individuals lack surface-expressed GP IIb/IIIa, whereas the GP IIb/IIIa content of the platelets from heterozygotes has been found to be 50% to 60% of normal.4,16 Binding of fibrinogen to the GP IIb/IIIa complex mediates normal platelet aggregation responses. Failure of such binding results in a profound defect in hemostatic plug formation and the serious bleeding characteristic of thrombasthenia.2,4,6,17–19
More than 70 mutations are known to give rise to Glanzmann thrombasthenia.20–22 The αIIb and β3 genes are present on chromosome 17, and genetic defects are distributed widely over the two genes. αIIb is synthesized in megakaryocytes as pro-αIIb, which complexes β3 in the endoplasmic reticulum. The complex is transported to the Golgi body, where αIIb is cleaved to heavy and light chains to form the complete complex. Uncomplexed αIIb and β3 are not processed in the Golgi body. As with the GP Ib/IX/V complex, it is necessary for both proteins of the GP IIb/IIIa complex to be produced and assembled into a complex for the complex to be expressed on the platelet surface. Gene defects that lead to the absence of production of either protein lead to absence of the complex on the platelet surface. Defects that interfere with or prevent complex formation or affect complex stability have the same effect.
One component of the αIIb/β3 integrin, β3, is a component of the vitronectin receptor, αV/β3, found on endothelial cells, osteoclasts, fibroblasts, monocytes, and activated B lymphocytes, where it acts as a receptor for a variety of adhesive protein ligands. Patients who have β3 gene defects that result in the absence of αIIb/β3 integrin also lack the vitronectin receptor. These patients do not seem to have a more severe form of Glanzmann thrombasthenia.23,24 The vitronectin receptor is thought to play a role in vascularization, but so far no evidence for abnormal blood vessel development has been documented in individuals lacking the vitronectin receptor. It also is unclear whether platelet vitronectin receptors play any significant role in platelet functional processes.20
The typical laboratory features of Glanzmann thrombasthenia are a markedly prolonged bleeding time, a normal platelet count and normal platelet morphology, poor in vitro clot retraction, and a lack of platelet aggregation in response to all platelet-activating agents (including ADP, collagen, thrombin, and epinephrine).2,4,6,18 If the stimulating agent is strong enough (e.g., thrombin), the platelets undergo the release/secretion reaction, even in the absence of aggregation. Ristocetin-induced binding of VWF to platelets and the resulting platelet agglutination are normal. The results of the complete blood count (CBC) are usually normal, unless there is another underlying disorder or the patient has had a recent hemorrhagic episode. Tests for platelet procoagulant activity, previously called the platelet factor 3 test, usually show diminished activity.2,16,25 There seem to be several reasons for this. When normal platelets are activated, procoagulant microvesicles are shed from the platelet surface, and coagulation factors assemble on the microvesicle surfaces during activation of the coagulation cascade. In Glanzmann thrombasthenia, markedly fewer microvesicles are produced. Second, prothrombin binds directly to GP IIb/IIIa. Because this complex is missing in Glanzmann thrombasthenia, significantly less thrombin is generated in response to tissue factor. Finally, Glanzmann thrombasthenia platelets are not as activated by thrombin as are normal platelets.26–29
A subdivision of Glanzmann thrombasthenia into cases with absent (type 1) and subnormal (type 2) in vitro clot retraction has been proposed. In general, individuals with type 2 disease have more residual GP IIb/IIIa complexes (10% to 20% of normal) than those with type 1 disease (0% to 5% of normal), although there is considerable variability within each subdivision.30,31
Thrombasthenia is one of the few forms of platelet dysfunction in which hemorrhage is severe and disabling. Bleeding of all types, including epistaxis, ecchymosis, hemarthrosis, subcutaneous hematoma, menorrhagia, and gastrointestinal and urinary tract hemorrhage, has been reported. Treatment of bleeding episodes in patients with Glanzmann thrombasthenia requires the transfusion of normal platelets. In Glanzmann thrombasthenia, the defective platelets may interfere with the normal transfused platelets, and it may be necessary to infuse more donor platelets than expected to control bleeding. As in BSS or any situation in which repeated transfusions are required, patients with Glanzmann thrombasthenia may become alloimmunized. Strategies to reduce alloimmunization include use of single-donor platelet apheresis products, HLA–matched donor platelets, or ABO-matched donor platelets.32
A variety of treatments have been used successfully to control or prevent bleeding, alone or in combination with platelet transfusion. To a large extent, the site of hemorrhage determines the therapeutic approach used. Hormonal therapy (norethindrone acetate) has been used to control menorrhagia. If the patient is treated with oral contraceptives, excessive bleeding should be reduced. Menorrhagia at the onset of menses is uniformly severe and can be life-threatening, which has led some to suggest that birth control pills be started before menarche. Also, antifibrinolytic therapy (aminocaproic acid or tranexamic acid) can be used to control gingival hemorrhage or excessive bleeding after tooth extraction.31 Recombinant factor VIIa has proved useful to treat severe bleeding in patients with isoantibodies to αIIb/β3 and in patients undergoing invasive procedures.33 Recombinant factor VIIa is thought to enhance thrombus formation at the site of a lesion by stimulating tissue factor–independent thrombus generation and fibrin formation.34
Rarely, a thrombasthenia-like state can be acquired. Such conditions include development of autoantibodies against GP IIb/IIIa, multiple myeloma in which the paraprotein is directed against GP IIIa, and afibrinogenemia. A thrombasthenia-like state also can be induced in individuals with otherwise normal platelet function by the therapeutic antiplatelet drugs ticlopidine and clopidogrel.15,18
Disorders of Platelet Secretion (Release Reactions)
Of the hereditary platelet function defects, disorders involving storage pool defects and the release reaction are the most common. The clinical features of this group of disorders are mucocutaneous hemorrhage and hematuria, epistaxis, and easy and spontaneous bruising. Petechiae are less common than in other qualitative platelet disorders. Hemorrhage is rarely severe but may be exacerbated by ingestion of aspirin or other antiplatelet agents. In most of these disorders, the platelet count is normal, and the bleeding time is usually, although not always, prolonged. Platelet aggregation abnormalities are usually seen but vary depending on the disorder.2,4,35,36
Storage Pool Diseases
Dense Granule Deficiencies
The inheritance of δ-granule (dense granule) deficiency does not follow a single mode, and it is likely that a variety of genetic abnormalities lead to the development of this disorder. Dense granule deficiencies can be subdivided into deficiency states associated with albinism and those in otherwise normal individuals (nonalbinos). In the platelets of nonalbinos, there is evidence for the presence of δ-granule membranes in normal to near-normal numbers, which suggests that the disorder arises from an inability to package the δ-granule contents.37,38 Serotonin accumulates in normal δ-granules by an active uptake mechanism in which plasma serotonin is transported by a specific carrier-mediated system across the plasma membrane into the cytoplasm, and a second carrier-mediated system in the δ-granule membrane transports serotonin from the cytoplasm to the interior of the δ-granules.39 These transport mechanisms are used in the serotonin release assay employed to detect heparin-dependent antiplatelet antibodies (see Chapter 43). In addition to serotonin transport mechanisms, a nucleotide transporter MRP4 (ABCC4) that is highly expressed in platelets and δ-granules has been identified. It would be expected that mutations in the gene for this transporter could affect nucleotide accumulation in δ-granules.40
As an isolated abnormality, δ-granule deficiency does not typically result in a serious hemorrhagic problem. Bleeding is usually mild and most often is limited to easy bruisability. Dense granule deficiency affects the results of platelet aggregation tests. δ-Granules are intracellular storage sites for ADP, adenosine triphosphate, calcium, pyrophosphate, and serotonin. The contents of these granules are extruded when platelet secretion is induced, and secreted ADP plays a major role in the propagation of platelet activation, recruitment, and aggregation and growth of the hemostatic plug. In patients with δ-granule deficiency, addition of arachidonic acid to platelet-rich plasma fails to induce an aggregation response. Epinephrine and low-dose ADP induce a primary wave of aggregation, but a secondary wave is missing. Responses to low concentrations of collagen are decreased to absent, but a high concentration of collagen may induce a near-normal aggregation response.36,39 This aggregation pattern is caused by the lack of ADP secretion and is almost identical to the pattern observed in patients taking aspirin.
In addition to occurring as an isolated problem, δ-granule deficiency is found in association with several disorders. Hermansky-Pudlak syndrome is an autosomal recessive disorder characterized by tyrosinase-positive oculocutaneous albinism, defective lysosomal function in a variety of cell types, ceroid-like deposition in the cells of the reticuloendothelial system, and a profound platelet δ-granule deficiency.41 Several of the mutations responsible for Hermansky-Pudlak syndrome have been mapped to chromosome 19. Mutations in at least seven genes individually can give rise to Hermansky-Pudlak syndrome. These genes encode for proteins that are involved in intracellular vesicular trafficking and are active in the biogenesis of organelles.42 Whereas the bleeding associated with most δ-granule deficiencies is rarely severe, Hermansky-Pudlak syndrome seems to be an exception. Although most bleeding episodes in Hermansky-Pudlak syndrome are not severe, lethal hemorrhage has been reported, and in one series hemorrhage accounted for 16% of deaths in patients with Hermansky-Pudlak syndrome. A unique morphologic abnormality has been described in the platelets of four families with Hermansky-Pudlak syndrome. This abnormality consists of marked dilation and tortuosity of the surface-connecting tubular system (the so-called Swiss cheese platelet).2,6,25,43
Chédiak-Higashi syndrome is a rare autosomal recessive disorder characterized by partial oculocutaneous albinism, frequent pyogenic bacterial infections, giant lysosomal granules in cells of hematologic (see Figure 28-5) and nonhematologic origin, platelet δ-granule deficiency, and hemorrhage. The Chédiak-Higashi syndrome protein gene is located on chromosome 13, and a series of nonsense and frameshift mutations all result in a truncated Chédiak-Higashi syndrome protein that gives rise to a disorder of generalized cellular dysfunction involving fusion of cytoplasmic granules. The disorder progresses to an accelerated phase in 85% of patients with Chédiak-Higashi syndrome and is marked by lymphocytic proliferation in the liver, spleen, and marrow and macrophage accumulation in tissues. During this stage, the pancytopenia worsens, which leads to hemorrhage and ever-increasing susceptibility to infection; the result is death at an early age. Initially, the bleeding time is increased because of δ-granule deficiency and consequent defective platelet function. During the accelerated phase, however, the thrombocytopenia also contributes to the prolonged bleeding time. Bleeding episodes vary from mild to moderate but worsen as the platelet count decreases.6,25
Wiskott-Aldrich syndrome is an X-linked recessive disease characterized by the triad of severe eczema, recurrent infections owing to immune deficiency, and life-threatening thrombocytopenia. Individuals with this disorder lack the ability to make antipolysaccharide antibodies, which results in a propensity for pneumococcal sepsis. Bleeding episodes are typically moderate to severe. A milder form without immune deficiency is known as hereditary X-linked thrombocytopenia (see Chapter 43). In Wiskott-Aldrich syndrome, a combination of ineffective thrombocytopoiesis and increased platelet sequestration and destruction accounts for the thrombocytopenia. As with all X-linked recessive disorders, it is found primarily in males.6,9,16,25,44 The gene that is mutated in Wiskott-Aldrich syndrome codes for a 502-amino-acid protein, WASP, that is found exclusively in hematopoietic cells. WASP is primarily involved in signal transduction.
Wiskott-Aldrich platelets are also structurally abnormal. The number of δ-granules is decreased, and the platelets are small, a feature of diagnostic importance. Other than in Wiskott-Aldrich syndrome, such small platelets are seen only in TORCH (Toxoplasma, other agents, rubella virus, cytomegalovirus, herpesvirus) infections. Diminished levels of stored adenine nucleotides are reflected in the lack of δ-granules observed on transmission electron micrographs. The platelet aggregation pattern in Wiskott-Aldrich syndrome is typical of a storage pool deficiency. The platelets show a decreased aggregation response to ADP, collagen, and epinephrine and lack a secondary wave of aggregation in response to these agonists. The response to thrombin is normal, however.6,25 The most effective treatment for the thrombocytopenia seems to be splenectomy, which would be consistent with peripheral destruction of platelets. Platelet transfusions may be needed to treat hemorrhagic episodes. Bone marrow transplantation also has been attempted with some success.25,45
Thrombocytopenia with absent radii (TAR) syndrome (see Chapter 43) is a rare autosomal recessive disorder characterized by the congenital absence of the radial bones (the most pronounced skeletal abnormality), numerous cardiac and other skeletal abnormalities, and thrombocytopenia (90% of cases). It is mentioned here because the platelets have structural defects in δ-granules with corresponding abnormal aggregation responses. Marrow megakaryocytes may be decreased in number, immature, or normal.6,46
α-Granule Deficiency: Gray Platelet Syndrome
The α-granules are the storage site for proteins produced by the megakaryocyte (e.g., platelet-derived growth factor, thrombospondin, and platelet factor 4) or present in plasma and taken up by platelets and transported to α-granules for storage (e.g., albumin, immunoglobulin G [IgG], and fibrinogen). There are 50 to 80 α-granules per platelet, which are primarily responsible for the granular appearance of platelets on stained blood films. Gray platelet syndrome, a rare disorder first described in 1971, is characterized by the specific absence of morphologically recognizable α-granules in platelets. The disorder is inherited in an autosomal recessive fashion. Clinically, gray platelet syndrome is characterized by lifelong mild bleeding tendencies, prolonged bleeding time, moderate thrombocytopenia, fibrosis of the marrow, and large platelets whose gray appearance on a Wright-stained blood film is the source of the name of this disorder.2,4,6,47
In electron photomicrographs of platelets and megakaryocytes, the platelets appear to have virtually no α-granules, although they do contain vacuoles and small α-granule precursors that stain positive for VWF and fibrinogen. The other types of granules are present in normal numbers. The membranes of the vacuoles and the α-granule precursors have P-selectin (CD62) and GP IIb/IIIa, and these proteins can be translocated to the cell membrane on stimulation with thrombin. This indicates that these structures are α-granules that cannot store the typical α-granule proteins. This may provide an explanation for the observation that, in gray platelet syndrome, the plasma levels of platelet factor 4 and α-thromboglobulin are increased. Most patients develop early-onset myelofibrosis, which can be attributed to the inability of megakaryocytes to store newly synthesized platelet-derived growth factors.38
Treatment of severe bleeding episodes may require platelet transfusions. Few other treatments are available for these patients. Cryoprecipitate has been used to control bleeding. Desmopressin acetate was found to shorten the bleeding time and has been used as successful prophylaxis in a dental extraction procedure. Some authors believe that desmopressin acetate should be the initial therapy of choice.2,4,38,48,49
Other Storage Pool Diseases
A rare disorder in which both α- and δ-granules are deficient is known as α-δ storage pool deficiency. It seems to be inherited as an autosomal dominant characteristic. In these patients, other membrane abnormalities also have been described.9
Quebec platelet disorder is an autosomal dominant bleeding disorder that results from a deficiency of multimerin (a multimeric protein that is stored complexed with factor V in α-granules) and shows protease-related degradation of many α-granule proteins, even though α-granule structure is maintained. Thrombocytopenia may be present, although it is not a consistent feature.9
Thromboxane Pathway Disorders: Aspirin-like Effects
Platelet secretion requires the activation of several biochemical pathways. One such pathway is the one leading to thromboxane formation. A series of phospholipases catalyze the release of arachidonic acid and several other compounds from membrane phospholipids. Arachidonic acid is converted to intermediate prostaglandins by cyclooxygenase and to thromboxane A2 by thromboxane synthase. Thromboxane A2 and other substances generated during platelet activation cause mobilization of ionic calcium from internal stores into the cytoplasm, occupancy of several activation receptors, and initiation of a cascade of events resulting in secretion and aggregation of platelets (see Chapter 13).50
Several acquired or congenital disorders of platelet secretion can be traced to structural and functional modifications of arachidonic acid pathway enzymes. Inhibition of cyclooxygenase occurs on ingestion of drugs such as aspirin and ibuprofen. As a result, the amount of thromboxane A2 produced from arachidonic acid depends on the degree of inhibition. Thromboxane A2 is required for storage granule secretion and maximal platelet aggregation in response to epinephrine, ADP, and low concentrations of collagen.6,14,25,51
Hereditary absence or abnormalities of the components of the thromboxane pathway are usually termed aspirin-like defects because the clinical and laboratory manifestations resemble those that follow aspirin ingestion. Platelet aggregation responses are similar to those in δ storage pool disorders (see earlier). Unlike in storage pool disorders, however, ultrastructure and granular contents are normal. Deficiencies of the enzymes cyclooxygenase and thromboxane synthase are well documented, and dysfunction or deficiency of thromboxane receptors is known.38
Inherited Disorders of Other Receptors and Signaling Pathways
The α2β1 (GP Ia/IIa) integrin is one of the collagen receptors in the platelet membrane. A deficiency of this receptor has been reported in a patient who lacked an aggregation response to collagen, whose platelets did not adhere to collagen, and who had a lifelong mild bleeding disorder.52 A deficiency in another collagen receptor, GP VI, also has been reported in patients with mild bleeding. The platelets of these patients failed to aggregate in response to collagen, and adhesion to collagen also was impaired.53 A family with gray platelet syndrome and defective collagen adhesion has been described. Affected members of the family have a severe deficiency of GP VI.54
Platelets seem to contain at least three receptors for ADP. P2X1 is linked to an ion channel that facilitates calcium ion influx. P2Y1 and P2Y12 (P2TAC) are members of the seven-transmembrane domain (STD) family of G protein–linked receptors. P2Y1 is thought to mediate calcium mobilization and shape change in response to ADP. Pathology of the P2Y1 receptor has not yet been reported. P2Y12 is thought to be responsible for macroscopic platelet aggregation and is coupled to adenylate cyclase through a G-inhibitory (Gi) protein complex.55 Some patients have been reported to have decreased platelet aggregation in response to ADP but normal platelet shape change and calcium mobilization. These patients have an inherited deficiency of the P2Y12 receptor.56–58 Bleeding problems seem to be relatively mild in these patients, but the only treatment for severe bleeding is platelet transfusion.
Congenital defects of the α2-adrenergic (epinephrine) receptor associated with decreased platelet activation and aggregation in response to epinephrine are known. The receptors that mediate aggregation in response to epinephrine, ADP, and collagen are STD receptors, as are the protease-activated receptors (PARs) for thrombin. So far, defects in the PAR receptors have not been described.9
A group of intracellular defects that affect platelet function includes defects in which all elements of the thromboxane pathway are normal, but insufficient calcium is released from the dense tubular system, and the cytoplasmic concentration of ionic calcium in the cytoplasm never reaches levels high enough to support secretion. This group of disorders is often referred to as calcium mobilization defects. These represent a heterogeneous group of disorders in which the defects reside in the various intracellular signaling pathways, including defects in G protein subunits and phospholipase C isoenzymes.25,59,60
Scott syndrome is a rare autosomal recessive disorder of calcium-induced membrane phospholipid scrambling (necessary for coagulation factor assembly) and thrombin generation on platelets. Platelets secrete and aggregate normally but do not transport phosphatidylserine and phosphatidylethanolamine from the inner leaflet to the outer leaflet of the plasma membrane. This phospholipid “flip” normally occurs during platelet activation and is essential for the binding of vitamin K–dependent clotting factors. In the membrane of resting platelets, phosphatidylserine and phosphatidylethanolamine are restricted to the inner leaflet of the plasma membrane, and phosphatidylcholine is expressed on the outer leaflet. This asymmetry is maintained by the enzyme aminophospholipid translocase.61 When platelets are activated, the asymmetry is lost, and phosphatidylserine and phosphatidylethanolamine flip to the outer leaflet and facilitate the assembly of clotting factor complexes. The phospholipid flip is mediated by a calcium-dependent enzyme, scramblase.62 In Scott syndrome, platelet plug formation (including adhesion, aggregation, and secretion) occurs normally, but clotting factor complexes do not assemble on the activated platelet surface, and thrombin generation is absent or much reduced. Because lack of thrombin generation leads to inadequate fibrin, the platelet plug is not stabilized, and a bleeding diathesis results.63,64
Lastly, Stormorken syndrome is a condition in which platelets are always in an “activated” state and express phosphatidylserine on the outer leaflet of the membrane without prior activation. It has been postulated that patients with this syndrome have a defective aminophospholipid translocase.65
Acquired Defects of Platelet Function
Drug-Induced Defects
Drugs That Inhibit the Prostaglandin Pathway
Unlike inherited disorders of platelet function, which are rare, acquired disorders of platelet function are commonly encountered. The most frequent cause of acquired platelet dysfunction is drug ingestion, with aspirin and other drugs that inhibit the platelet prostaglandin synthetic pathways being the most common culprits. A single 200-mg dose of acetylsalicylic acid (aspirin) can irreversibly acetylate 90% of the platelet cyclooxygenase (see Figure 13-19). In platelets the acetylated cyclooxygenase (cyclooxygenase-1, or COX-1) is completely inactive. Platelets lack a nucleus and cannot synthesize new enzymes. The inhibitory effect is permanent for the circulatory life span of the platelet (7 to 10 days). Endothelial cells synthesize new cyclooxygenase, and endothelial cell cyclooxygenase seems to be less sensitive to aspirin than the platelet enzyme, at least at low dosages. This has led to the view that low dosages of aspirin may be better than higher dosages, because platelet thromboxane production is inhibited whereas endothelial cells recover prostacyclin production. Others argue that inhibition of platelet function is the more important effect and that higher dosages of aspirin are better for this purpose. For these reasons, there are wide-ranging opinions as to the optimal dosage of aspirin. What is lost in these arguments is that endothelial cells also produce another potent platelet inhibitor, nitric oxide (NO), and its production is not affected by aspirin. Although aspirin may inhibit a proaggregatory mechanism (thromboxane production) and an antiaggregatory mechanism (prostacyclin production), the NO platelet inhibitory mechanism is not affected. It may be necessary to define a test system to determine the optimal dosage of aspirin on an individual basis, because some patients have, or develop, aspirin resistance, and a dosage that previously was sufficient to inhibit platelet function effectively may no longer able to produce that effect. Finally, unlike the practice with almost all other therapeutic agents, a single dose of aspirin is usually prescribed in a “one dose fits all” fashion (e.g., 325 mg) without regard to the patient’s weight, age, health status, or other measurable parameters. This practice is based on the assumption that the biologic effect will be the same in all patients. Evidence is emerging, however, that there are considerable interindividual differences in the response to a single dose of aspirin.5,25,66–68 One study has shown that patients who do not respond well to aspirin have worse outcomes than patients who respond well.69
Individuals known to have a defect in their hemostatic mechanism, such as a storage pool deficiency, thrombocytopenia, a vascular disorder, or VWD, may experience a marked increase in bleeding time after aspirin ingestion, and such individuals should be advised to avoid the use of aspirin and related agents.25
The list of drugs affecting the prostaglandin pathway that converts arachidonic acid to thromboxane is long and beyond the scope of this chapter. Many of these drugs inhibit cyclooxygenase, but, unlike with aspirin and closely related compounds, the inhibition is reversible. These drugs are said to be competitive inhibitors of cyclooxygenase, and as the blood concentration of the drug decreases, platelet function is recovered. This group of drugs includes ibuprofen and related compounds, such as ketoprofen and fenoprofen, naproxen, and sulfinpyrazone. In contrast to aspirin, most of these agents have little effect on the bleeding time test. Except for their potential to irritate the gastric mucosa, these drugs have not been reported to cause clinically important bleeding.14,17,25,70 Interestingly, ibuprofen appears to have a prothrombotic effect when ingested within 2 hours of aspirin, because it blocks the acetylation site for aspirin on COX-1. Patients taking aspirin should be cautioned to avoid ibuprofen and related drugs near the time of aspirin ingestion.
The association of chronic alcohol consumption with thrombocytopenia is well known. Chronic, periodic, and even acute alcohol consumption may result in a transient decrease in platelet function, however, and the inhibitory effect seems to be more pronounced when alcohol is consumed in excess. These effects are well known, and most patients who are scheduled to undergo a medical procedure in which there may be hemostatic challenge are advised to abstain from alcohol consumption for about 3 days before the procedure. The impaired platelet function seems to be related at least in part to inhibition of thromboxane synthesis. A reduced platelet count and impaired platelet function may contribute to the increased incidence of gastrointestinal hemorrhage associated with chronic excessive alcohol intake.14,25,71,72
Drugs That Inhibit Membrane Function
Many drugs interact with the platelet membrane and cause a clinically significant platelet function defect that may lead to hemorrhage. Some of these drugs are useful antiplatelet agents, whereas for many other drugs, their effects on the platelet membrane are an adverse side effect.50
The thienopyridine derivatives clopidogrel and prasugrel and their predecessor ticlopidine are antiplatelet agents used to treat patients with arterial occlusive disease for prevention of myocardial infarction, patients with cerebrovascular disease for reduction of the risk of thrombotic stroke, and patients who are intolerant of aspirin. In contrast to the effect of aspirin, the effects of these agents do not reach a steady state for 3 to 5 days, although a steady state can be reached sooner with a loading dose. As prophylactic agents, they have been shown to be as efficacious as aspirin. The mechanism of action seems to be interference with the binding of ADP to the P2Y12 platelet membrane STD receptor.56 The major effect appears to be inhibition of stimulus-response coupling between the platelet ADP receptor and fibrinogen binding to GP IIb/IIIa. As a consequence, platelet activation and aggregation induced by ADP are markedly inhibited, and responses to other aggregating agents, such as collagen, are reduced. Clopidogrel has more effect on the bleeding time than aspirin, although there is little difference in the risk of clinical bleeding.73 Clopidogrel and ticlopidine can produce major side effects in some patients, including long-lasting neutropenia, aplastic anemia, thrombocytopenia, gastrointestinal distress, and diarrhea. Clopidogrel has the same clinical efficacy as ticlopidine, but the incidence and severity of these side effects are much lower.14,74 Clopidogrel has become the drug of choice for this class of antiplatelet agents. Clopidogrel and aspirin are increasingly being used in combination to prevent arterial thrombosis, primarily based on the synergistic action of these two drugs, which inhibit platelet function by different mechanisms. Similar to aspirin effects, the effects of the thienopyridines are not readily reversible; platelet function after cessation of drug intake is about 50% of normal at 3 days, and complete recovery of function occurs at 7 to 10 days.75
A newer group of antiplatelet agents targets the platelet membrane GP IIb/IIIa (αIIb/β3) receptor, interfering with the ability of this receptor to bind fibrinogen and inhibiting platelet aggregation in response to all of the usual platelet aggregating agents. Results of platelet function studies on platelets from patients receiving therapeutic dosages of these drugs essentially mimic those of a mild form of Glanzmann thrombasthenia. At present, three of these agents are approved for use in the United States. Two different types of agents are included in this group. The first such agent approved for clinical use in the United States was the Fab fragment of the mouse/human chimeric monoclonal antibody 7E3 (c7E3 Fab; abciximab [ReoPro]), which binds to GP IIb/IIIa, prevents the binding of fibrinogen, and prevents platelet aggregation. Numerous studies have shown the efficacy of this drug as an antiplatelet and antithrombotic agent. The second type of agent in this group targets a GP IIb/IIIa recognition site for an arginine–glycine–aspartic acid (RGD) sequence found in fibrinogen and several adhesive proteins. These agents bind to the recognition site, prevent the binding of fibrinogen, and consequently prevent platelet aggregation. These compounds are relatively easily synthesized and contain the RGD sequence recognized by the receptor (tirofiban) or a structure that mimics the RGD sequence (eptifibatide) and are bound by the RGD recognition site on GP IIb/IIIa. When the receptor site is occupied by the drug, GP IIb/IIIa is no longer functional as the aggregation receptor. The goal of therapy with these drugs is to induce a controlled thrombasthenia-like state. At present these agents are primarily used in patients undergoing percutaneous coronary intervention and are administered concurrently with heparin and other antiplatelet agents. Currently, the use of these agents is limited by the need to administer them by constant intravenous infusion. An orally active agent of this type is under development and is in clinical trials.76–78
Dipyridamole is an inhibitor of platelet phosphodiesterase, the enzyme responsible for converting cyclic adenosine monophosphate (cAMP) to AMP (see Figure 13-20). Elevation of cytoplasmic cAMP is inhibitory to platelet function, and inhibition of phosphodiesterase allows the accumulation of cAMP in the cytoplasm. Dipyridamole alone does not inhibit platelet aggregation in response to the usual platelet agonists, but promotes inhibition of agents that stimulate cAMP formation, such as prostacyclin, stable analogues of prostacyclin, and NO. At one time, dipyridamole, alone or in combination with aspirin, was widely used. By the 1990s, interest in dipyridamole had waned. There has been a resurgence of interest in dipyridamole, however, as a combination agent compounded with aspirin (Aggrenox).
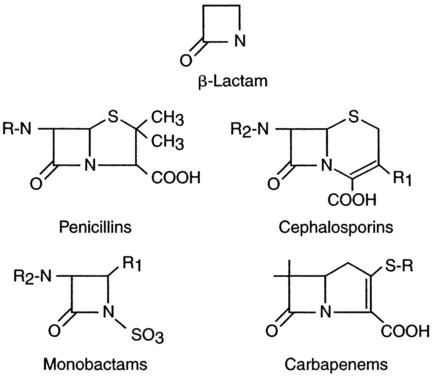
Antibiotics are well known for their ability to interfere with platelet function. Most of the drugs with this effect contain the β-lactam ring and are either a penicillin or a cephalosporin (Figure 44-2). These drugs can prolong the bleeding time, but this effect is seen only in patients receiving large parenteral doses and is a problem only for hospitalized patients. One postulated mechanism for the antiplatelet effect of these drugs is that they associate with the membrane via a lipophilic reaction and block receptor-agonist interactions or stimulus-response coupling. They also may inhibit calcium influx in response to thrombin stimulation, reducing the ability of thrombin to activate platelets. Although these drugs may prolong the bleeding time and in vitro aggregation responses to certain agonists, their association with a hemostatic defect severe enough to cause clinical hemorrhage is uncertain and is not predicted by the bleeding time test results.5,17,51,74
Nitrofurantoin is an antibiotic that is not related to the β-lactam drugs but may prolong the bleeding time and inhibit platelet aggregation when high concentrations are present in the blood. This drug is not known to cause clinical bleeding, however.74
The dextrans, another class of commonly used drugs, can prolong the bleeding time, inhibit platelet aggregation, and impair platelet procoagulant activity when given as an intravenous infusion. These drugs have no effect on platelet function, however, when added directly to platelet-rich plasma. Dextrans are partially hydrolyzed, branched-chain polysaccharides of glucose. The two most commonly used are dextran 70 (molecular mass of 70,000 to 75,000 D) and dextran 40 (molecular mass of 40,000 D), also known as low-molecular-weight dextran. Both drugs are effective plasma expanders and are commonly used for this purpose. Because of their effects on platelets, they have been extensively used as antithrombotic agents. There does not seem to be any increased risk of hemorrhage associated with the use of these agents, but their efficacy in preventing postoperative pulmonary embolism is equal to that of low-dose subcutaneous heparin.14,44,71,74
Hydroxyethyl starch, or hetastarch, is a synthetic glucose polymer with a mean molecular mass of 450,000 D that also is used as a plasma expander. It has effects similar to those of the dextrans. The mechanism of action of these drugs has not been clearly elucidated but is presumed to involve interaction with the platelet membrane.14,44,71
Miscellaneous Drugs That Inhibit Platelet Function
Several other agents of diverse chemical structure and function are known to inhibit platelet function. The mechanisms by which they induce platelet dysfunction are largely unknown. Nitroglycerin, nitroprusside, propranolol, and isosorbide dinitrate are drugs used to regulate cardiovascular function that seem to be able to cause a decrease in platelet secretion and aggregation. Patients taking phenothiazine or tricyclic antidepressants may have decreased secretion and aggregation responses, but these effects are not associated with an increased risk for hemorrhage. Local and general anesthetics may impair in vitro aggregation responses. The same is true of antihistamines. Finally, some radiographic contrast agents are known to inhibit platelet function.74
Disorders That Affect Platelet Function
Myeloproliferative Neoplasms
Chronic myeloproliferative neoplasms (MPNs) include polycythemia vera, chronic myelogenous leukemia, essential thrombocythemia, and myelofibrosis with myeloid metaplasia (see Chapter 34). Platelet dysfunction is a common finding in patients with these disorders. Hemorrhagic complications occur in about one third, thrombosis occurs in another third, and although it is uncommon, both develop in some patients. These complications are serious causes of morbidity and mortality. Although the occurrence of hemorrhage or thrombosis in MPN patients is largely unpredictable, certain patterns have emerged. Hemorrhage and thrombosis are less common in chronic myelogenous leukemia than in the other MPNs. Bleeding seems to be more common in myelofibrosis with myeloid metaplasia, but thrombosis is more common in the other MPNs. Abnormal platelet function has been postulated as a contributing cause. This hypothesis is supported by the observation that bleeding is usually mucocutaneous in nature, and thrombosis may be arterial or venous. In patients with these disorders, thrombosis may occur in unusual sites, including the mesenteric, hepatic, and portal circulations. Patients with essential thrombocythemia may develop digital artery thrombosis and ischemia of the fingers and toes, occlusions of the microvasculature of the heart, and cerebrovascular occlusions that result in neurologic symptoms.74
The most common abnormalities are decreased aggregation and secretion in response to epinephrine, ADP, and collagen.79 Possible causes of the platelet dysfunction include loss of platelet surface membrane α-adrenergic (epinephrine) receptors, impaired release of arachidonic acid from membrane phospholipids in response to stimulation by agonists, impaired oxidation of arachidonic acid by the cyclooxygenase and lipoxygenase pathways, a decrease in the contents of δ-granules and α-granules, and loss of a variety of platelet membrane receptors for adhesion and activation. There seems to be no correlation between a given MPN and the type of platelet dysfunction observed, with the exception that most patients with essential thrombocythemia lack an in vitro platelet aggregation response to epinephrine. This observation may be helpful in the differential diagnosis.14,17,74,80
Multiple Myeloma and Waldenström Macroglobulinemia
Platelet dysfunction is observed in approximately one third of patients with IgA myeloma or Waldenström macroglobulinemia, a much smaller percentage of patients with IgG multiple myeloma, and only occasionally in patients with monoclonal gammopathy of undetermined significance (see Chapter 37). Platelet dysfunction results from coating of the platelet membranes by paraprotein and does not depend on the type of paraprotein present. In addition to interacting with platelets, the paraprotein may interfere with fibrin polymerization and the function of other coagulation proteins. Almost all patients with malignant paraprotein disorders have clinically significant bleeding, but thrombocytopenia is still the most likely cause of bleeding in these patients. Other causes of bleeding include hyperviscosity syndrome, complications of amyloidosis (e.g., acquired factor X deficiency), and, in rare instances, presence of a circulating heparin-like anticoagulant or fibrinolysis.16,71,74
Cardiopulmonary Bypass Surgery
Cardiopulmonary bypass induces thrombocytopenia and a severe platelet function defect that assumes major importance in surgical bleeding after bypass. The function defect most likely results from platelet activation and fragmentation in the extracorporeal circuit. Causes of platelet activation include adherence and aggregation of platelets to fibrinogen (adsorbed onto the surfaces of the bypass circuit material), mechanical trauma and shear stresses, blood conservation devices, bypass pump-priming solutions, hypothermia, complement activation, and exposure of platelets to the blood-air interface in bubble oxygenators. Some degree of platelet degranulation typically is found after bypass surgery, which indicates that platelet activation and secretion has occurred during the operation. Platelet membrane fragments or “microparticles” are found consistently in the blood of bypass patients, providing additional evidence of the severe mechanical stress encountered by platelets during bypass procedures. The severity of the platelet function defect closely correlates with the length of time on bypass. After an uncomplicated bypass procedure, normal platelet function returns in about 1 hour, although the platelet count does not return to normal for several days. Thrombocytopenia is caused by hemodilution, accumulation of platelets on the surfaces of the bypass materials, sequestration or removal of damaged platelets by the liver and reticuloendothelial system, and consumption associated with normal hemostatic processes after surgery.71,74
Liver Disease
Moderate to severe liver disease is reported to be associated with a variety of hemostatic abnormalities, including reduction in clotting proteins, reduction of proteins in the natural anticoagulant pathways, dysfibrinogenemia, and excessive fibrinolysis. Mild to moderate thrombocytopenia is seen in approximately one third of patients with chronic liver disease in association with hypersplenism or as a result of alcohol toxicity.4,74
In chronic alcoholic cirrhosis, the thrombocytopenia and platelet abnormalities may result from the direct toxic effects of alcohol on bone marrow megakaryocytes. The severe bleeding diathesis associated with end-stage liver disease has many causes, such as markedly decreased or negligible coagulation factor production, excessive fibrinolysis, dysfibrinogenemia, thrombocytopenia, and (occasionally) disseminated intravascular coagulation. Upper gastrointestinal tract bleeding is a relatively common feature of cirrhosis, particularly alcoholic cirrhosis, and recombinant factor VIIa has been shown to be effective treatment in some patients.81
Uremia
Uremia is commonly accompanied by bleeding caused by platelet dysfunction. In uremia, guanidinosuccinic acid (GSA) is present in the circulation in higher than normal levels as a result of inhibition of the urea cycle. GSA is dialyzable, and dialysis (peritoneal dialysis or hemodialysis) is usually effective in correcting the prolonged bleeding time and the abnormal platelet function characteristic of uremia. NO diffuses into platelets, activates soluble guanylate cyclase, and inhibits platelet adhesion, activation, and aggregation.82 Because GSA is an NO donor, NO is present in the circulation at higher than normal levels in uremia. Abnormal platelet function is far more common than clinically significant bleeding in uremic patients.2,50,52
Platelet aggregation pattern abnormalities are not uniform, and any combination of defects may be seen. There is evidence of a deficient release reaction, such as lack of primary ADP-induced aggregation, and subnormal platelet procoagulant activity. The bleeding time is characteristically prolonged in uremia and seems to correlate with the severity of renal failure in these patients. There does not seem to be any significant correlation, however, between the bleeding time and the risk of clinically significant bleeding. Anemia is an independent cause of prolonged bleeding time, and the severity of anemia in uremic patients correlates with the severity of renal failure. Many uremic patients are treated with recombinant erythropoietin to increase their hematocrit. Maintenance of the hematocrit at greater than 30% also may help to normalize the bleeding times.2,71,73
Bleeding is uncommon in uremic patients and is seen more often with concurrent use of drugs that interfere with platelet function or in association with heparin use in hemodialysis. Platelet concentrates often are used to treat severe hemorrhagic episodes in patients with uremia but usually do not correct the bleeding. Other therapies that are sometimes effective include cryoprecipitate, desmopressin acetate, and conjugated estrogen.2,71,73
Hereditary Afibrinogenemia
Hereditary afibrinogenemia has been documented in more than 150 families. Although it is not truly a platelet function disorder, platelets do not exhibit normal function in the absence or near-absence of fibrinogen. In most patients, the bleeding time is prolonged, and because fibrinogen is essential for normal platelet aggregation, platelet aggregation test results are abnormal. Abnormal results on platelet retention and adhesion studies involving the use of glass beads also have been documented. In addition, results of all clot-based tests (including partial thromboplastin time, prothrombin time, reptilase time, thrombin time, and whole-blood clotting time) are abnormal. Addition of fibrinogen to samples or infusion of fibrinogen into the patient results in correction of the abnormal test results.2,83
A high incidence of hemorrhagic manifestations is found in patients with afibrinogenemia (or severe hypofibrinogenemia). Bleeding is the cause of death in about one third of such patients. Cryoprecipitate or fibrinogen concentrates can be used to treat bleeding episodes. Some patients develop antibodies to fibrinogen, and this treatment then becomes ineffective.83
Hyperaggregable Platelets
Patients with a variety of disorders associated with thrombosis or increased risk for thrombosis, including hyperlipidemia, diabetes mellitus, peripheral arterial occlusive disease, acute arterial occlusion, myocardial infarction, and stroke, have been reported to have increased platelet reactivity. Platelets from these patients tend to aggregate at lower concentrations of aggregating agents than do platelets from individuals without these conditions. Spontaneous aggregation (aggregation in response to stirring only) is also an indicator of abnormally increased platelet reactivity and often accompanies increased sensitivity to platelet agonists. The presence of spontaneous aggregation by itself is considered to be consistent with the presence of a hyperaggregable state. Because participation of platelets is necessary for the development of arterial thrombosis, the presence of hyperaggregable platelets is often an indication that an antiplatelet agent should be used as part of a therapeutic or prophylactic regimen for arterial thrombosis.71,84,85
Acquired platelet function defects are seen occasionally in patients with autoimmune disorders, including systemic lupus erythematosus, rheumatoid arthritis, scleroderma, and the immune thrombocytopenias, such as immune thrombocytopenic purpura.74
Purified fibrin degradation products can induce platelet dysfunction in vitro. The pathophysiologic relevance of this observation is uncertain, because the concentrations of fibrin degradation products required are unlikely to be reached in vivo. Patients with disseminated intravascular coagulation may have reduced platelet function, however, as a result of in vivo stimulation by thrombin and other agonists, resulting in in vivo release of granule contents. This has been called acquired storage pool disease; the term exhausted platelets may be more appropriate.86
Vascular Disorders
The pathophysiology of disorders of vessels and their supporting tissues is obscure. Laboratory studies of platelets and blood coagulation usually yield normal results. The diagnosis is often based on medical history and is made by ruling out other sources of bleeding disorders. The usual clinical sign is the tendency to bruise easily or to bleed spontaneously, especially from mucosal surfaces. Vascular disorders are summarized in Box 44-2.
Hereditary Vascular Disorders
Hereditary Hemorrhagic Telangiectasia (Rendu-Osler-Weber Syndrome)
The mode of inheritance of hereditary hemorrhagic telangiectasia is autosomal dominant. The vascular defect of this disorder is characterized by thin-walled blood vessels with a discontinuous endothelium, inadequate smooth muscle, and inadequate or missing elastin in the surrounding stroma. Telangiectasias (dilated superficial blood vessels that create small, focal red lesions) occur throughout the body but are most obvious on the face, lips, tongue, conjunctiva, nasal mucosa, fingers, toes, and trunk and under the tongue. The lesions blanch when pressure is applied. The disorder usually becomes manifest by puberty and progresses throughout life. Telangiectasias are fragile and prone to rupture. Epistaxis is an almost universal finding, and symptoms almost always worsen with age. The age at which nosebleeds begin is a good gauge of the severity of the disorder. Although the oral cavity, gastrointestinal tract, and urogenital tract are common sites of bleeding, bleeding can occur in virtually every organ.87
The laboratory findings relate to the severity of the hemorrhagic tendencies. The bleeding time is usually normal, and the tourniquet test result may be normal or show increased capillary fragility. The diagnosis of hereditary hemorrhagic telangiectasia is based on the characteristic skin or mucous membrane lesions, a history of repeated hemorrhage, and a family history of a similar disorder. Patients with hereditary hemorrhagic telangiectasia do well despite the lack of specific therapy and the seriousness of their hemorrhagic manifestations.2,6,87 There are several other disorders and conditions in which telangiectasias are present, including cherry-red hemangiomas (common in older men and women), ataxia-telangiectasia (Louis-Bar syndrome), and chronic actinic telangiectasia; they also are seen in association with chronic liver disease and pregnancy.87
Hemangioma-Thrombocytopenia Syndrome (Kasabach-Merritt Syndrome)
Kasabach and Merritt originally described the association of a giant cavernous hemangioma (vascular tumor), thrombocytopenia, and a bleeding diathesis. The hemangiomas are visceral or subcutaneous, but rarely both. External hemangiomas may become engorged with blood and resemble hematomas. Other well-recognized features of Kasabach-Merritt syndrome include acute or chronic disseminated intravascular coagulation and microangiopathic hemolytic anemia. A hereditary basis for this syndrome has not been established, but the condition is present at birth. Several treatment modalities are available for the tumors and the coagulopathy and range from corticosteroid therapy to surgery.6,88
Ehlers-Danlos Syndrome and Other Genetic Disorders
Ehlers-Danlos syndrome may be transmitted as an autosomal dominant, recessive, or X-linked trait. It is manifested by hyperextensible skin, hypermobile joints, joint laxity, fragile tissues, and a bleeding tendency, primarily subcutaneous hematoma formation. Eleven distinct varieties of the disorder are recognized. The severity of bleeding ranges from easy bruisability to arterial rupture. The disorder generally can be ascribed to defects in collagen production, structure, or cross-linking, with resulting inadequacy of the connective tissues. Platelet abnormalities have been reported in some patients. Common abnormal laboratory findings include a positive tourniquet test result and prolonged bleeding time.6
Other inherited vascular disorders include pseudoxanthoma elasticum and homocystinuria (autosomal recessive disorders), and Marfan syndrome and osteogenesis imperfecta (autosomal dominant disorders). In addition to vascular defects, Marfan syndrome is characterized by skeletal and ocular defects.6
Acquired Vascular Disorders
Allergic Purpura (Henoch-Schönlein Purpura)
The term allergic purpura or anaphylactoid purpura generally is applied to a group of nonthrombocytopenic purpuras characterized by apparently allergic manifestations, including skin rash and edema. Allergic purpura has been associated with certain foods and drugs, cold, insect bites, and vaccinations. The term Henoch-Schönlein purpura is applied when the condition is accompanied by transient arthralgia, nephritis, abdominal pain, and purpuric skin lesions, which are frequently confused with the hemorrhagic rash of immune thrombocytopenic purpura.2,6,44
General evidence implicates autoimmune vascular injury, but the pathophysiology of the disorder is unclear. Preliminary evidence indicates that the vasculitis is mediated by immune complexes containing IgA antibodies. It has been suggested that allergic purpura may represent autoimmunity to components of vessel walls.2,6
Henoch-Schönlein purpura is primarily a disease of children, occurring most commonly in children 3 to 7 years of age. It is relatively uncommon among individuals younger than age 2 and older than age 20. Twice as many boys as girls are affected. The onset of the disease is sudden, often following an upper respiratory tract infection. The infectious organism may damage the endothelial lining of blood vessels, which results in vasculitis. Attempts have been made to implicate a specific infectious agent, particularly β-hemolytic streptococcus.2,6
Malaise, headache, fever, and rash may be the presenting symptoms. The delay in the appearance of the skin rash often poses a difficult problem in differential diagnosis. The skin lesions are urticarial and gradually become pinkish, then red, and finally hemorrhagic. The appearance of the lesions may be very rapid and accompanied by itching. The lesions have been described as “palpable purpura,“ in contrast to the perfectly flat lesions of thrombocytopenia and most other forms of vascular purpura. These lesions are most commonly found on the feet, elbows, knees, buttocks, and chest. Ultimately, a brownish red eruption is seen. Petechiae also may be present.2,6
As the disease progresses, abdominal pain, polyarthralgia, headaches, and renal disease may develop. Renal lesions are present in 60% of patients during the second to third week of the disorder. Proteinuria and hematuria are commonly present.2,4,6
The platelet count is normal. Tests of hemostasis, including the bleeding time, tourniquet test, and tests of blood coagulation, usually yield normal results in patients with allergic purpura. Anemia generally is not present unless the hemorrhagic manifestations have been severe. The white blood cell count and the erythrocyte sedimentation rate are usually elevated. The disease must be distinguished from other forms of nonthrombocytopenic purpura. Numerous infectious diseases that may be associated with purpura also must be considered in the differential diagnosis. Drugs or chemicals sometimes may be implicated.2,6
In the pediatric age group, the average duration of the initial episode is about 4 weeks. Relapses are frequent, usually after a period of apparent well-being. Except for patients in whom chronic renal disease develops, the prognosis is usually good. Occasionally, death from renal failure has occurred. Management is directed primarily at symptomatic relief, because there currently is no effective treatment. Corticosteroids sometimes have been helpful in alleviating symptoms. Most patients recover without treatment.2,6
Paraproteinemia and Amyloidosis
Platelet function can be inhibited by myeloma proteins. Abnormalities in platelet aggregation, secretion, clot retraction, and procoagulant activity correlate with the concentration of the plasma paraprotein and are likely due to coating of the platelet membrane with the paraprotein. Under these conditions, platelet adhesion and activation receptor functions are inhibited, and the paraprotein coating also inhibits assembly of clotting factors on the platelet surface. High concentrations of paraprotein can cause severe hemorrhagic manifestations as a result of a combination of hyperviscosity and platelet dysfunction. About one third of patients with IgA myeloma and Waldenström macroglobulinemia and approximately 5% of patients with IgG myeloma (usually IgG3) exhibit platelet function abnormalities. Finally, the paraprotein may contribute further to bleeding by inhibiting fibrin polymerization. In these patients, there is poor correlation between abnormal results on laboratory tests (e.g., prothrombin time, activated partial thromboplastin time, thrombin time, bleeding time) and evidence of clinical bleeding. Treatment for the bleeding complications of these disorders is primarily reduction in the level of the paraprotein. This can be accomplished quickly, albeit transiently, by plasmapheresis. Longer-term treatment is usually chemotherapy for the underlying plasma cell malignancy.74,89
Amyloid is a fibrous protein consisting of rigid, linear, nonbranching, aggregated fibrils approximately 7.5 to 10 nm wide and of indefinite length. Amyloid is deposited extracellularly and may lead to damage of normal tissues. Various proteins can serve as subunits of the fibril, including monoclonal light chains (λ more frequently than κ). Amyloidosis, the deposition of abnormal quantities of amyloid in tissues, may be primary or secondary and localized or systemic. A discussion of the clinical spectrum of amyloidosis is beyond the scope of this chapter. Purpura, hemorrhage, and thrombosis may be a part of the clinical presentation of patients with amyloidosis, however. Thrombosis and hemorrhage have been ascribed to amyloid deposition in the vascular wall and surrounding tissues. Platelet function has been shown to be abnormal in a few cases, and in rare cases patients may have thrombocytopenia. Current treatments for amyloidosis are not effective.90
Senile Purpura
Senile purpura occurs more commonly in elderly men than in women and is due to a lack of collagen support for small blood vessels and loss of subcutaneous fat and elastic fibers. The incidence increases with advancing age. The dark blotches are flattened, are about 1 to 10 mm in diameter, do not blanch with pressure, and resolve slowly, often leaving a brown stain in the skin (age spots). The lesions are limited mostly to the extensor surfaces of the forearms and backs of the hands and occasionally occur on the face and neck. With the exception of increased capillary fragility, results of laboratory tests are normal, and no other bleeding manifestations are present.2,6
Drug-Induced Vascular Purpuras
Purpura associated with drug-induced vasculitis occurs in the presence of functionally adequate platelets. A variety of drugs are known to cause vascular purpura, including aspirin, warfarin, barbiturates, diuretics, digoxin, methyldopa, and several antibiotics. Sulfonamides and iodides have been implicated most frequently. The lesions vary from a few petechiae to massive, generalized petechial eruptions. Mechanisms include development of antibodies to vessel wall components, development of immune complexes, and changes in vessel wall permeability. As soon as the disorder is recognized, the offending drug should be discontinued. No other treatment is necessary.6
Miscellaneous Causes of Vascular Purpura
Insufficient dietary intake of vitamin C (ascorbic acid) results in scurvy and decreased synthesis of collagen, with weakening of capillary walls and the appearance of purpuric lesions.6 A diagnosis of purpura simplex (simple vascular purpura) or vascular fragility is made when a cause for purpura cannot be found. The ecchymoses are superficial, bleeding is usually mild, and laboratory test results are most often normal.6 Cutaneous bleeding and bruising through intact skin has been observed in patients in whom no vascular or platelet dysfunction can be detected. Most such cases involve women with emotional problems, and the bruising is often accompanied by nausea, vomiting, or fever. Evidence for a psychosomatic origin is equivocal. Laboratory test results are invariably normal.6
Summary
• Inherited qualitative platelet disorders can cause bleeding disorders ranging from mild to severe.
• BSS is caused by the lack of expression of GP Ib/IX/V complexes on the platelet surface. This receptor complex is responsible for platelet adhesion, and its absence results in a severe bleeding disorder.
• Glanzmann thrombasthenia is caused by the lack of expression of GP IIb/IIIa complexes on the platelet surface. This complex is known as the platelet aggregation receptor, and its absence is associated with a severe bleeding disorder.
• Storage pool disorders result from the absence of intraplatelet α-granules, dense granules, or both. Platelet dysfunction associated with these disorders is generally mild; bleeding symptoms also are usually mild.
• Aspirin-like effects result from defects in elements of the arachidonic acid metabolic pathway. Platelet dysfunction mimics that seen after aspirin ingestion.
• Deficiencies of several of the receptors for platelet-activating substances have been documented, and bleeding symptoms of varying severity are associated with these deficiencies.
• Drugs are the most common cause of acquired platelet dysfunction, and aspirin is the most frequent culprit. Several new classes of antiplatelet agents with different effects than aspirin are now available and gaining in popularity.
• A variety of pathologic conditions can result in platelet dysfunction and range from hematologic malignancies to kidney disease and liver disease.
• Vascular disorders that result in bleeding are uncommon. There are a few well-recognized inherited disorders, however, such as Ehlers-Danlos syndrome and hereditary hemorrhagic telangiectasia, that can result in substantial blood loss.
• Vascular disorders can be acquired, and these are much more common than inherited disorders. Causes range from the effects of aging to drug effects to allergic reaction.
Review Questions
1. The clinical presentation of platelet-related bleeding may include all of the following except:
2. A defect in GP IIb/IIIa causes:
3. Aspirin ingestion blocks the synthesis of:
4. Patients with BSS have which of the following laboratory test findings?
a. Abnormal platelet response to arachidonic acid
b. Abnormal platelet response to ristocetin
5. Which of the following is the most common of the hereditary platelet function defects?
6. A mechanism of antiplatelet drugs targeting GP IIb/IIIa function is:
a. Interference with platelet adhesion to the subendothelium by blocking of the collagen binding site
b. Inhibition of transcription of the GP IIb/IIIa gene
7. The impaired platelet function in MPNs results from:
8. Which is a congenital qualitative platelet disorder?
9. In uremia, platelet function is impaired by higher than normal levels of:
10. The platelet defect associated with increased paraproteins is:
a. Impaired membrane activation owing to protein coating
b. Hypercoagulability owing to antibody binding and membrane activation
c. Impaired aggregation because the hyperviscous plasma prevents platelet-endothelium interaction
d. Hypercoagulability because the increased proteins bring platelets closer together, which lead to inappropriate aggregation