Introduction to Leukocyte Neoplasms
After completion of this chapter, the reader will be able to:
1. Name two viruses that play a pathogenic role in lymphoid malignancies.
2. Define oncogene and tumor suppressor gene and explain the difference between them, including the mechanisms by which each produces cancer.
3. Explain why oncogenes are described as acting in a dominant fashion.
4. Give two examples of oncogenes and two examples of tumor suppressor genes.
5. Describe how genetic abnormalities can affect molecular pathways within cells.
6. Identify the two basic subgroups of chemotherapeutic agents.
7. Explain why localized treatments are rarely used for leukocyte neoplasms.
8. Describe examples of supportive care therapies that have been developed from molecular biology techniques, including their functions and purposes.
9. Name two examples of targeted therapeutics for chronic myelogenous leukemia and explain their methods of action.
10. Describe the differences between small molecule inhibitors and monoclonal antibodies.
11. Name three sources of hematopoietic stem cells.
12. Compare and contrast allogeneic and autologous bone marrow transplants.
Classification Schemes for Leukocyte Neoplasms
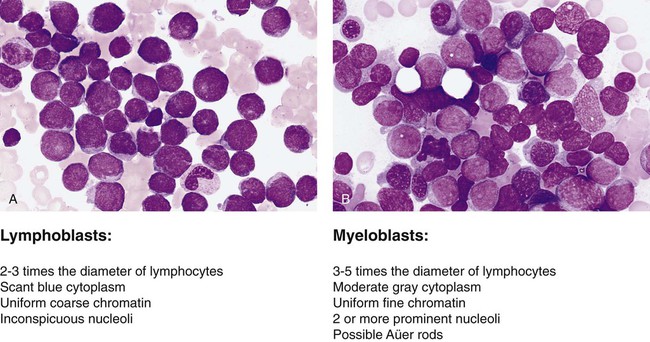
The French-American-British (FAB) classification of the acute leukemias was devised in the 1970s and 1980s. The FAB schemas were based largely on morphologic characteristics and relied heavily on examination of routine histologic stain preparations to distinguish lymphoid neoplasms from myeloid neoplasms (Figure 29-1). Although these types of diagnostic criteria have not been abandoned, pathologists are now moving toward more precise classification of many of the leukocyte neoplasms based on recurring chromosomal and genetic lesions found in many patients. These lesions are related to disruptions of oncogenes, tumor suppressor genes, and other regulatory elements that control proliferation, maturation, apoptosis, and other vital cell functions. In 2001 the World Health Organization (WHO) published new classification schemes for nearly all of the tumors of hematopoietic and lymphoid tissues.1 In some cases WHO melded the older morphologic schemes with the newer schemes. For instance, in the WHO classification scheme for acute myeloid leukemias (AMLs) there are some remnants of the old FAB classification, but new classifications were introduced for leukemias associated with consistently recurring chromosomal translocations. This 2001 WHO classification of hematologic malignancies has undergone a recent revision.2
Chromosomal Abnormalities in Hematologic Neoplasms
In part because of the ease of access to bone marrow compared with other organs of the body, researchers have learned a great deal about the biology of cancer by studying hematopoietic neoplasms. The study of chromosomal translocations in hematologic malignancies has taught how a single mutation, or series of mutations in stepwise fashion, can lead to malignant transformation by disrupting the molecular machinery of the cell. The first two genetic lesions found in any kind of human cancer were identified as chromosomal translocations occurring in leukocyte malignancies. These were the t(9;22) translocation in chronic myelogenous leukemia (CML)3 and the t(8;14) translocation in Burkitt lymphoma.4
Tumor Suppressor Genes
Tumor suppressor genes are so named because they code for proteins that resist malignant transformation. These genes do not act in a dominant fashion as in the case of oncogenes; rather, cells are transformed into a malignant phenotype only after both alleles of these genes have been lost or otherwise inactivated, the so-called two-hit mechanism proposed by Knudson.5 Although tumor suppressor genes are harder to isolate and identify, numerous such genes have now been identified, and many have been found to be associated with autosomal dominant familial cancer predisposition syndromes. Some well-known examples include the RB1 tumor suppressor gene involved in familial retinoblastoma, the TP53 gene in Li-Fraumeni syndrome, the WT1 gene in Wilms tumor, and the NF1 gene in familial neurofibromatosis type 1. Perhaps more importantly, many of these tumor suppressor genes and their protein products are altered in many sporadic cancers, including hematologic cancers.
Molecular Pathways Perturbed by Cellular Transformation
The list of chromosomal and molecular aberrations known to occur in the various leukocyte neoplasms continues to grow on an almost daily basis. Indexing this list is far beyond the scope of this chapter, but some condensed lists are provided in Chapter 31. More complete indices can be found in other publications such as the WHO reclassification scheme,1 its 2008 revision, or other hematology textbooks.6–10
Therapy for Leukocyte Neoplasms
Chemotherapy
1. Cycle-specific agents, which kill cells that are moving through the cell cycle, regardless of whether the cells are in G1, G2, S, or M phase (e.g., alkylating agents, cisplatin).
2. Cycle-nonspecific agents, which kill nondividing cells or cells in the resting state (e.g., steroids and antitumor antibiotics).
Phase-specific agents are effective only if present during a certain phase in the cell cycle (see Chapters 31 and 32). Within a certain dosage range, agents in this category show no increase in killing of cells with a further increase in dosage. Examples are l-asparagine amidohydrolase (G1 phase), antimetabolites such as methotrexate (S phase), and vinca alkaloids (M phase).
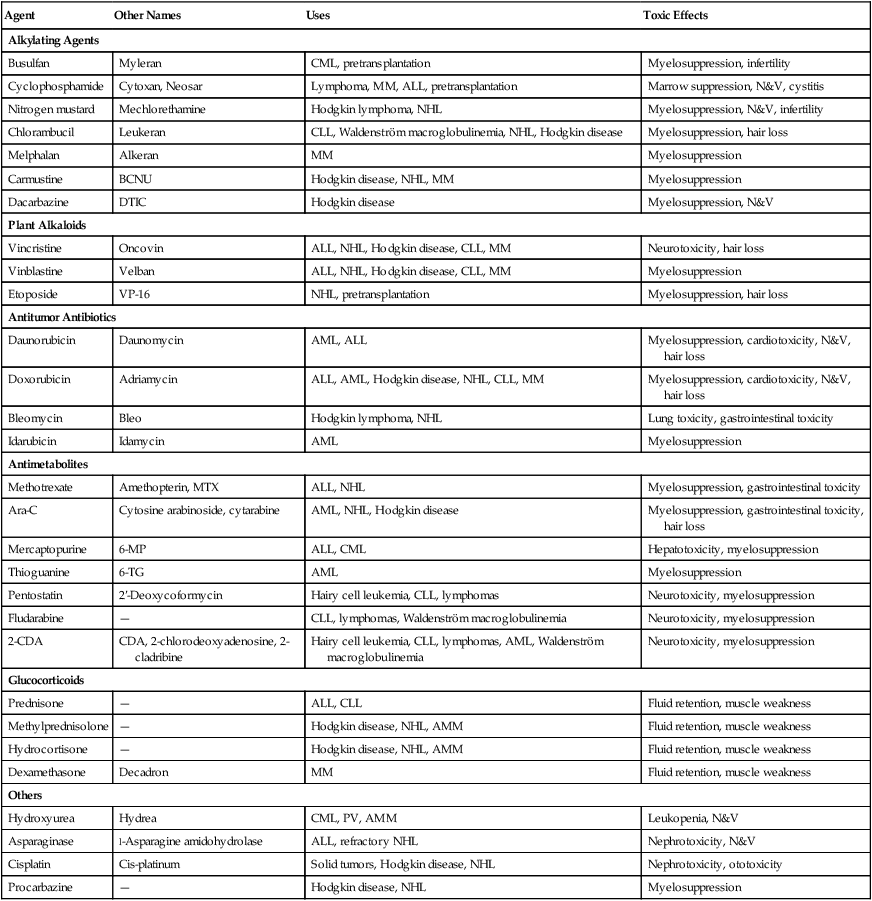
Chemotherapeutic agents affect both neoplastic and normal cells. The effect is most pronounced on rapidly dividing cells, such as those of the mucosa of the gastrointestinal tract and the bone marrow. This limits the dosage and usually determines the maximum tolerated dose for a patient. Chemotherapy agents are categorized in Table 29-1.
TABLE 29-1
| Agent | Other Names | Uses | Toxic Effects |
| Alkylating Agents | |||
| Busulfan | Myleran | CML, pretransplantation | Myelosuppression, infertility |
| Cyclophosphamide | Cytoxan, Neosar | Lymphoma, MM, ALL, pretransplantation | Marrow suppression, N&V, cystitis |
| Nitrogen mustard | Mechlorethamine | Hodgkin lymphoma, NHL | Myelosuppression, N&V, infertility |
| Chlorambucil | Leukeran | CLL, Waldenström macroglobulinemia, NHL, Hodgkin disease | Myelosuppression, hair loss |
| Melphalan | Alkeran | MM | Myelosuppression |
| Carmustine | BCNU | Hodgkin disease, NHL, MM | Myelosuppression |
| Dacarbazine | DTIC | Hodgkin disease | Myelosuppression, N&V |
| Plant Alkaloids | |||
| Vincristine | Oncovin | ALL, NHL, Hodgkin disease, CLL, MM | Neurotoxicity, hair loss |
| Vinblastine | Velban | ALL, NHL, Hodgkin disease, CLL, MM | Myelosuppression |
| Etoposide | VP-16 | NHL, pretransplantation | Myelosuppression, hair loss |
| Antitumor Antibiotics | |||
| Daunorubicin | Daunomycin | AML, ALL | Myelosuppression, cardiotoxicity, N&V, hair loss |
| Doxorubicin | Adriamycin | ALL, AML, Hodgkin disease, NHL, CLL, MM | Myelosuppression, cardiotoxicity, N&V, hair loss |
| Bleomycin | Bleo | Hodgkin lymphoma, NHL | Lung toxicity, gastrointestinal toxicity |
| Idarubicin | Idamycin | AML | Myelosuppression |
| Antimetabolites | |||
| Methotrexate | Amethopterin, MTX | ALL, NHL | Myelosuppression, gastrointestinal toxicity |
| Ara-C | Cytosine arabinoside, cytarabine | AML, NHL, Hodgkin disease | Myelosuppression, gastrointestinal toxicity, hair loss |
| Mercaptopurine | 6-MP | ALL, CML | Hepatotoxicity, myelosuppression |
| Thioguanine | 6-TG | AML | Myelosuppression |
| Pentostatin | 2′-Deoxycoformycin | Hairy cell leukemia, CLL, lymphomas | Neurotoxicity, myelosuppression |
| Fludarabine | — | CLL, lymphomas, Waldenström macroglobulinemia | Neurotoxicity, myelosuppression |
| 2-CDA | CDA, 2-chlorodeoxyadenosine, 2-cladribine | Hairy cell leukemia, CLL, lymphomas, AML, Waldenström macroglobulinemia | Neurotoxicity, myelosuppression |
| Glucocorticoids | |||
| Prednisone | — | ALL, CLL | Fluid retention, muscle weakness |
| Methylprednisolone | — | Hodgkin disease, NHL, AMM | Fluid retention, muscle weakness |
| Hydrocortisone | — | Hodgkin disease, NHL, AMM | Fluid retention, muscle weakness |
| Dexamethasone | Decadron | MM | Fluid retention, muscle weakness |
| Others | |||
| Hydroxyurea | Hydrea | CML, PV, AMM | Leukopenia, N&V |
| Asparaginase | l-Asparagine amidohydrolase | ALL, refractory NHL | Nephrotoxicity, N&V |
| Cisplatin | Cis-platinum | Solid tumors, Hodgkin disease, NHL | Nephrotoxicity, ototoxicity |
| Procarbazine | — | Hodgkin disease, NHL | Myelosuppression |
Targeted Therapy
As more has been learned about the specific genetic lesions that cause cancers, researchers have worked to develop targeted therapies that act specifically on the tumor cell and leave normal cells untouched.11 As a result of these advances in translational research, cancer therapy is realizing the dream of targeted therapeutics and is moving away from nonspecific therapies such as chemotherapy and radiation. The Philadelphia chromosomal abnormality, t(9;22) associated with CML was first reported in 1960. The Philadelphia chromosome results from the juxtaposition of two genes, BCR and ABLl, and the resulting fusion protein has elevated tyrosine kinase activity. It took until almost the year 2000 to develop an agent that specifically targets the product of the Philadelphia chromosome. Imatinib mesylate (Gleevec) is a tyrosine kinase inhibitor that inhibits the kinase activity of all proteins that contain ABL. Imatinib is not specific solely for the BCR/ABLl tyrosine kinase; it also inhibits the tyrosine kinase activity of the c-KIT and the platelet-derived growth factor receptor. It is through its activity on these other receptors that imatinib has found usefulness against other tumors as a targeted therapeutic. In CML, however, imatinib has quickly become the standard treatment for patients newly diagnosed with the disease who are not candidates for immediate transplant (see Chapter 34). Resistance to imatinib has developed in some CML cells, and second-generation, more powerful kinase inhibitors, such as dasatinib and nilotinib, have now been cleared by the FDA. Small-molecule inhibitors such as imatinib are typically available in oral form and have relatively few side effects.
Rather than utilizing small-molecule inhibitors, treatment of lymphoid neoplasms has relied more on monoclonal antibodies for targeted therapeutic strategies. Rituximab specifically targets the CD20 antigen present on many non-Hodgkin lymphoma cells. When the antibody binds the lymphoma cell, complement is activated, and the cell lyses. Other possible scenarios leading to cell death with the use of monoclonal antibodies include antibody-mediated cellular cytotoxicity and stimulation of apoptosis.10 In contrast to small-molecule inhibitors, monoclonal antibodies generally must be delivered intravenously or subcutaneously. Monoclonal antibodies such as rituximab have evolved from the original monoclonal antibodies that were derived from mice. Rituximab represents a humanized form of an antibody designed or modified to be “more human” so that the patient’s immune system will not raise an antibody against the monoclonal antibody. Additional modifications are now being developed, such as radioactively labeled monoclonal antibodies that can carry a killing dose of radioactivity directly to the mutated cell.
Stem Cell Transplantation
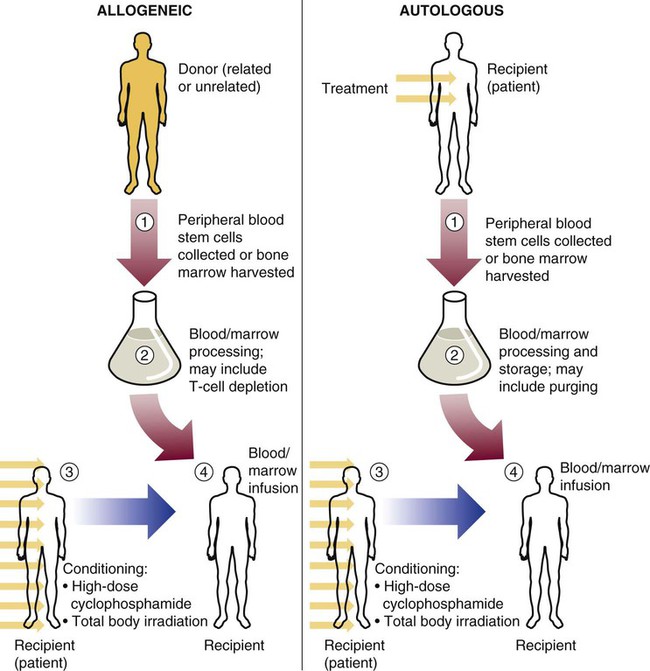
Stem cell transplants for treatment of malignant disease have come from donors of three general types: (1) an identical twin donor (syngeneic transplant), (2) a donor genetically different from the recipient (allogeneic transplant), or (3) the patient’s own marrow or peripheral blood stem cells (autologous transplant) (Figure 29-2). Syngeneic transplants are most desirable because of the perfect match of cells. For obvious reasons, however, they are rare, and they are not discussed further in this chapter.
Summary
• Most malignancies of the hematopoietic system are acquired genetic diseases.
• Most leukocyte malignancies are not localized, but rather are systemic at the initiation of the malignant process.
• For most leukocyte neoplasms, causes directly related to the development of the malignancy are unknown, but a few exceptions exist. Some known causes include environmental toxins, certain viruses, previous chemotherapy, and familial predisposition.
• There are several classification schemes for leukocyte neoplasia, including the FAB system, based primarily on morphology and cytochemical staining, and the WHO system, which retains some elements of the FAB scheme but emphasizes molecular and cytogenetic changes.
• Chromosomal translocations in hematologic malignancies illustrate that a single mutation, or a series of mutations in stepwise fashion, can lead to malignant transformation by disrupting the molecular machinery of the cell.
• Most chromosomal translocations in leukemias involve oncogenes. The “dominant” transforming oncogene is able to alter the gene product and transform the cell into a malignant phenotype, even in the presence of a residual normal allele.
• In contrast to oncogenes, tumor suppressor genes contribute to the malignant process only if both alleles have been lost or otherwise inactivated.
• The formation of oncogenes or the loss of tumor suppressor genes has molecular effects on the proliferation and differentiation of hematologic tissues.
• Current treatments for leukocyte neoplasms can be roughly divided into the following categories: chemotherapy, radiation therapy, supportive therapy, targeted therapy, and stem cell transplantation.
Review Questions
1. Which of the following viruses is known to cause lymphoid malignancies in humans?
2. Tumor suppressor genes cause cancers such as leukemia when mutations result in:
3. Oncogenes are said to act in a dominant fashion because:
a. Cancer is a dominating disease that overtakes the individual’s health
b. The mutation product affects (i.e., dominates) normal cells and mutated cells
c. A mutation in a single allele is sufficient for malignancy to develop
4. All of the following are among the cellular abnormalities produced by oncogenes except:
5. Chemotherapeutic agents are divided into which two major subgroups?
6. G-CSF is provided as supportive treatment during leukemia treatment regimens to:
7. Imatinib is an example of what type of leukemia treatment?
8. Monoclonal antibodies may kill cancer cells by all of the following mechanisms except:
a. Binding to the cancer cells and causing the immune system to destroy them
b. Stimulating the production of anticancer cell antibodies by the patient’s own immune system
9. Hematopoietic stem cells for transplantation are harvested from all of the following tissues except:
10. Compared with autologous bone marrow transplantation, allogeneic transplantation has: