Extrinsic Defects Leading to Increased Erythrocyte Destruction—Immune Causes
After completion of this chapter, the reader will be able to:
1. Define immune hemolytic anemia and indicate the types of antibodies involved.
2. Compare and contrast the mechanisms of immune hemolysis mediated by immunoglobulin M (IgM) and IgG antibodies.
3. Describe typical laboratory findings in immune hemolytic anemia and the importance of the direct antiglobulin test (DAT).
4. Compare and contrast four types of autoimmune hemolytic anemia in terms of the immunoglobulin class involved, the temperature for optimal reactivity of the autoantibody, the proteins detected by the DAT on the patient’s red blood cells, the presence of complement activation, the type and site of hemolysis, and the specificity of the autoantibody.
5. Relate results of the DAT to the pathophysiology and clinical findings in autoimmune hemolytic anemia.
6. Describe three mechanisms of drug-induced immune hemolysis.
7. Compare and contrast the pathophysiology of immune hemolysis due to drug-dependent and drug-independent antibodies, including the related laboratory findings.
8. Describe two types of hemolytic transfusion reactions, the usual immunoglobulin class involved, the typical site of hemolysis, and important laboratory findings.
9. Describe the cause, pathophysiology, and laboratory findings in Rh and ABO hemolytic disease of the fetus and newborn.
10. Given a patient history and results of a complete blood count, peripheral blood film examination, pertinent biochemical tests on serum and urine, and the direct and indirect antiglobulin tests, determine the type of immune hemolysis.
Case Study
| Patient Results | Reference Range | |
| WBCs (×109/L) | 5.9 | 4.5-11.5 |
| RBCs (×1012/L) | 1.40 | 4.00-5.40 |
| Hb (g/dL) | 4.2 | 12.0-15.0 |
| Hct (%) | 13 | 35-49 |
The platelet count was within the reference range. Spherocytes and polychromasia were observed on the peripheral blood film (Figure 25-1). The film also revealed a slight increase in neutrophilic bands and 2 nucleated RBCs per 100 WBCs (not shown in figure). An increase in urobilinogen was noted on the routine urinalysis report, but the result for blood was negative. Urine and sputum culture results were negative. Significant findings from the chemistry profile included elevated levels of serum indirect bilirubin and lactate dehydrogenase. Subsequently, a direct antiglobulin test (DAT) and 3 units of red blood cells were ordered. A positive result on the DAT for IgG was reported. The indirect antiglobulin test on the patient’s serum and on an eluate prepared from the patient’s cells yielded negative results for all panel cells tested.
1. Is this type of anemia caused by an intrinsic or extrinsic defect?
2. What does the positive DAT result imply?
3. What mechanisms can lead to the development of drug-induced immune hemolytic anemia?
4. Describe the mechanism that is the most probable cause of this patient’s anemia.
5. In addition to the transfusion, what additional action should be taken?
Overview of Immune Hemolytic Anemias
Immune hemolytic anemia and nonimmune hemolytic anemia are the two broad categories comprising the extrinsic hemolytic anemias, disorders in which red blood cells (RBCs) are structurally and functionally normal, but a condition outside of the RBCs causes premature hemolysis. The nonimmune extrinsic hemolytic anemias are the result of physical or mechanical injury to the RBCs and are covered in Chapter 24. The immune hemolytic anemias are conditions in which RBC survival is shortened due to an antibody-mediated mechanism. The antibody may be an autoantibody (directed against a self RBC antigen), an alloantibody (directed against an RBC antigen of another person), or an antibody directed against a drug taken by the patient (or its metabolite). Some antibodies are able to activate the classical complement pathway, which results in the attachment of activated complement proteins to the RBC membrane. RBCs with bound antibody or complement may be prematurely removed from the circulation extravascularly by macrophages (due to their receptors for complement and the Fc component of antibody), intravascularly by complement-mediated hemolysis, by or a combination of both mechanisms.1 Anemia develops when the amount of hemolysis exceeds the ability of the bone marrow to replace the RBCs lost. The degree of anemia varies from asymptomatic and mild to severe and life-threatening.
The immune hemolytic anemias may be classified into the following groups: autoimmune hemolytic anemia, drug-induced immune hemolytic anemia, and alloimmune hemolytic anemia (Box 25-1).1,2 It is important to determine the cause of an immune hemolytic anemia so that the appropriate therapy can be administered to the patient.
Pathophysiology of Immune Hemolysis
In immune hemolysis an antibody binds to an antigen on the surface of RBCs and that binding results in premature removal of those cells from the circulation through extravascular or intravascular hemolysis (see Chapter 22). The two classes or isotypes of antibodies involved in most immune hemolytic anemias are immunoglobulin G (IgG) and IgM. IgG is a monomer in a Y-like structure with two identical heavy chains (γ H chains) and two identical light chains (either κ or λ) connected by disulfide bonds.3 At the top of the Y-like structure are two antigen binding (Fab) domains, each formed from the N-terminus of the variable domain of one light and one heavy chain. IgG has one Fc domain (the stem of the Y) consisting of the C-terminus of the two heavy chains. IgM is a pentamer consisting of five monomeric units connected by disulfide linkages at the C-termini of their heavy chains (µ H chains).3,4 Because the composition, structure, and size of IgG and IgM are different, their properties and mechanisms in mediating hemolysis are also different.
The classical complement pathway is an important mediator of immune hemolysis. The major proteins of the classical complement pathway are designated C1 through C9, and their components or fragments are designated with lowercase suffixes. The first protein, C1, has three components: C1q, C1r, C1s. After an antibody binds antigen on the RBC surface, C1q must bind to two adjacent Fc domains to activate the pathway.4 Theoretically only one IgM molecule is needed for activation due to its pentameric structure with five Fc domains; however, with the monomeric IgG, at least two molecules in close proximity are required.4 Therefore IgM antibodies are highly effective in activating complement, whereas IgG antibodies are unable to activate the pathway unless there is a sufficient number of IgG molecules on the RBC surface.4,5 In addition, subclasses IgG3 and IgG1 have high binding affinity for C1q, whereas subclasses IgG2 and IgG4 have minimal ability to bind complement.4,5
The binding of C1q to adjacent Fc domains activates C1r, which subsequently activates C1s. C1s activates C4 and then C2, which results in the binding of a small number of C4bC2a complexes to the RBC membrane. The C4bC2a complex is an active C3 convertase enzyme that cleaves C3 in plasma; this results in the binding of many C3b molecules on the RBC surface. C3b binds to C4bC2a to activate C5 to C5b. Next, C5b, C6, C7, C8, and C9 form the membrane attack complex (MAC). The MAC inserts into the lipid bilayer, forming a pore that allows water and small ions to enter the cell, which eventually causes intravascular lysis.3 Negative regulators inhibit various complement proteins and complexes in the pathway to prevent uncontrolled activation and excessive hemolysis.1,4
Hemolysis mediated by IgM antibodies requires complement and can result in both extravascular and intravascular hemolysis.5 When IgM molecules attach to the RBC surface in relatively low density, complement activation results in C3b binding to the membrane, but complement inhibitors prevent full activation of the pathway to the terminal membrane attack complex.1,5 C3b-sensitized RBCs are destroyed by extravascular hemolysis, predominantly by macrophages (Kupffer cells) in the liver, which have C3b receptors. Some of the C3b on the RBCs can be cleaved, however, which leaves the C3d fragment on the cell. RBCs sensitized with only C3d are not prematurely removed from circulation, because macrophages lack a C3d receptor.5 In severe cases of immune hemolysis involving heavy sensitization of RBCs with IgM antibody, significantly more complement is activated, which overwhelms the complement inhibitors. In these cases, complement activation proceeds from C1 to C9 and results in rapid intravascular hemolysis.5
Hemolysis mediated by IgG antibodies occurs with or without complement and predominantly by extravascular mechanisms.5 RBCs sensitized with IgG are removed from circulation by macrophages in the spleen, which have receptors for the Fc component of IgG1 and IgG3.1,5 IgG antibodies are not efficient in activating complement; therefore, intravascular hemolysis by full activation of complement from C1 to C9 is rare (except with anti-P in paroxysmal cold hemoglobinuria).5 However, if there is a high density of IgG3 or IgG1 bound to antigens on the RBCs, some complement is activated and C3b binds to the membrane. If both IgG and C3b are on the RBC membrane, there is faster clearance from the circulation by macrophages in both the spleen and liver.1,5 Often, IgG-sensitized RBCs are only partially phagocytized by macrophages, which results in the removal of some membrane. Spherocytes are the result of this process, and they are the characteristic cell of IgG-mediated hemolysis.5 The spherocytes are eventually removed from circulation by entrapment in the red pulp of the spleen (splenic cords), where they are rapidly phagocytized by macrophages (see Chapter 8).5 The mechanisms of immune hemolysis are summarized in Table 25-1.
TABLE 25-1
Major Mechanisms of Immune Hemolysis
| IgM Mediated | IgG Mediated | |
| Extravascular hemolysis | IgM activation of classical complement pathway from C1 to C3b only; clearance of C3b-sensitized RBCs by macrophages mainly in liver | Clearance of IgG-sensitized RBCs by macrophages mainly in spleen Formation of spherocytes by partial phagocytosis of IgG-sensitized RBCs; spherocytes cleared by macrophages after entrapment in spleen IgG* activation of classical complement pathway from C1 to C3b only; requires high-density IgG on RBC surface; clearance of IgG- and C3b-sensitized RBCs by macrophages in spleen and liver |
| Intravascular hemolysis | Full IgM activation of classical complement pathway from C1 to C9 and direct RBC lysis; requires high-density IgM on RBCs to overcome complement inhibitors | Full IgG activation of classical complement pathway from C1 to C9 and direct RBC lysis; requires very-high-density IgG on RBCs for activation and to overcome complement inhibitors; uncommon |
Laboratory Findings in Immune Hemolytic Anemia
Laboratory findings in immune hemolytic anemia are similar to the findings in other hemolytic anemias and include decreased hemoglobin (Hb), increased reticulocyte count, increased levels of indirect serum bilirubin and lactate dehydrogenase, and decreased serum haptoglobin level. If the hemolysis is predominantly intravascular, the haptoglobin level will be moderately to severely decreased, plasma hemoglobin will be increased, and the patient may have hemoglobinuria or even hemosiderinuria (in cases of chronic hemolysis) (see Chapter 22). The mean cell volume may be increased due to the reticulocytosis, and leukocytosis and thrombocytosis may be present due to the increased erythroid proliferation in the bone marrow.1,6 Findings on the peripheral blood film include polychromasia (due to the reticulocytosis), spherocytes (due to IgG-mediated membrane damage by macrophages), and RBC agglutination (in some cases of IgM autoantibodies).5 Nucleated RBCs, occasional schistocytes, and erythrophagocytosis may also be observed on the peripheral blood film.1
To determine if the hemolysis is due to an immune mechanism, a direct antiglobulin test (DAT) is performed. The DAT detects in vivo sensitization of the RBC surface by IgG, C3d, or both.2 In the DAT procedure, polyspecific antihuman globulin (AHG) is added to saline-washed patient RBCs. Polyspecific AHG has specificity for the Fc portion of human IgG and complement component C3d and will agglutinate the RBCs if a critical number of one or both molecules is present on the RBC surface.2 If the DAT result is positive with polyspecific AHG, then the cells are tested with monospecific anti-IgG and anti-C3d to identify the type of sensitization. If IgG is detected on the RBCs, elution procedures are used to remove the antibody. The specificity of the antibody may be determined by assessing the reaction of the eluate with screening and panel reagent RBCs (RBCs genotyped for the major RBC antigens) using the indirect antiglobulin (AHG) test. Identification of any circulating alloantibodies or autoantibodies by the indirect antiglobulin test is also important in the investigation.2 The DAT result may be negative in patients with some immune hemolytic anemias.2 A positive DAT result has been reported in apparently healthy individuals and in blood donors with no evidence of hemolysis.7,8 In addition, other disorders beside immune hemolytic anemia can cause a positive DAT finding.2 Therefore diagnosis of immune hemolytic anemia cannot rely solely on the DAT and must take into account the patient history, symptoms, recent medications, previous transfusions, coexisting conditions including pregnancy, as well as the results of the applicable hematologic, biochemical, and serologic tests.2,5
Autoimmune Hemolytic Anemia
Autoimmune hemolytic anemia (AIHA) is a rare disorder characterized by premature RBC destruction and anemia caused by autoantibodies that bind the RBC surface with or without complement activation. Autoantibodies may arise as a result of immune system dysregulation and loss of immune tolerance, exposure to an antigen similar to an autoantigen, B lymphocyte neoplasm, or other unknown reason.1,6 The type, amount, and duration of antigen exposure and genetic and environmental factors may also contribute to the development of autoantibodies.5,6 The anemia can be mild or severe, and the onset acute or gradual. The severity of the anemia depends on the autoantibody characteristics (titer, ability to react at 37° C, ability to activate complement, and specificity and affinity for the autoantigen), antigen characteristics (density on RBCs, immunogenicity), as well as patient factors (age; ability of the bone marrow to compensate for the hemolysis; function of macrophages, complement proteins and regulators; and underlying conditions).1,6
The autoimmune hemolytic anemias may be divided into four major categories based on the characteristics of the autoantibody and the mechanism of hemolysis: warm autoimmune hemolytic anemia, cold agglutinin disease, paroxysmal cold hemoglobinuria, and mixed-type autoimmune hemolytic anemia (Table 25-2).2,5,6
TABLE 25-2
Characteristics of Autoimmune Hemolytic Anemias
| Warm Autoimmune Hemolytic Anemia | Cold Agglutinin Disease | Paroxysmal Cold Hemoglobinuria | Mixed-Type Autoimmune Hemolytic Anemia | |
| Immunoglobulin class | IgG (rarely IgM, IgA) | IgM | IgG | IgG, IgM |
| Optimum reactivity temperature of autoantibody | 37° C | 4° C; reactivity extends to >30° C | 4° C | 4°-37° C |
| Sensitization detected by direct antiglobulin test | IgG or IgG + C3d; only C3d uncommon | C3d | C3d | IgG and C3d |
| Complement activation | Variable | Yes | Yes | Yes |
| Hemolysis | Extravascular primarily | Extravascular; rarely intravascular | Intravascular | Extravascular and intravascular |
| Autoantibody specificity | Panreactive or Rh complex; rarely specific Rh or other antigen | I (most), i (some), Pr (rare) | P | Panreactive; unclear specificity |
Warm Autoimmune Hemolytic Anemia
Warm autoimmune hemolytic anemia (WAIHA) is the most commonly encountered autoimmune hemolytic anemia comprising up to 70% of cases.5 The autoantibodies causing WAIHA react optimally at 37° C, and the vast majority of them are IgG.1 WAIHA is most commonly found in adults older than 40 years of age and in children younger than 4 years of age.6 WAIHA may be classified as idiopathic or secondary. In patients with idiopathic WAIHA, the etiology is unknown. Secondary WAIHA may be found in many conditions such as lymphoproliferative diseases (chronic lymphocytic leukemia, B-lymphocyte lymphomas, Waldenström macroglobulinemia), nonlymphoid neoplasms (thymoma and cancers of the colon, kidney, lung, and ovary), autoimmune disorders (rheumatoid arthritis, scleroderma, polyarteritis nodosa, Sjögren syndrome, systemic lupus erythematosus), immunodeficiency disorders, and viral infections.1,6
The onset of WAIHA is usually insidious with symptoms of anemia (fatigue, dizziness, dyspnea), but some cases can be acute and life-threatening with fever, jaundice, splenomegaly, and hepatomegaly, especially in children with WAIHA secondary to viral infections.1,6 An underlying lymphoproliferative disorder is suggested in adults with massive splenomegaly, lymphadenopathy, fever, petechiae, ecchymosis, or renal failure.1,6
Although most autoantibodies that cause WAIHA are IgG, rare cases involving IgA autoantibodies as well as cases with fatal outcomes caused by warm-reacting IgM antibodies have been reported.1,2,9 Hemolysis is predominantly extravascular in WAIHA, and cases of fulminant intravascular hemolysis are rare.5
The result of the DAT is positive in over 95% of patients, with approximately 85% of patients having IgG alone or both IgG and C3d on their RBCs, and 10% to 14% having C3d only. Between 1% and 4% of patients have a negative DAT result.1,2,5 A negative DAT result may be caused by IgA or IgM autoantibodies that are not detected by the polyspecific AHG, by the presence of IgG or C3d in an amount below the reagent detection limit, by the dissociation of IgG antibodies with low avidity during the washing phase of the DAT, or by various technical errors.1,2 Therefore a negative DAT result does not rule out autoimmune hemolytic anemia.
Warm autoantibodies are usually panreactive; that is, they will agglutinate all screening and panel cells, donor RBCs, and the patient’s own RBCs, so the specificity of the autoantibody is not apparent.2,5 In some cases Rh complex specificity can be demonstrated.2 Rarely, a specific autoantibody to an antigen in the Rh blood group system is identified. Autoantibodies to other antigens (such as LW, Jk, K, Di, Ge, Lu, M, N, S, U, Ena, and Wrb) are rarely encountered.2,5,6 For most patients (approximately 80%) the autoantibody can be detected in the serum. Because the autoantibody is panreactive, it may mask reactions of alloantibodies with RBC panel cells. If an RBC transfusion is necessary, it is crucial to perform tests to determine if clinically significant alloantibodies are also present. Donor cells that lack the antigens corresponding to all clinically significant alloantibodies identified are selected for transfusion.1,2,5
Anemia in WAIHA can be mild or severe, with RBC life span sometimes reduced to 5 days or less.1 Laboratory findings for serum and urine reflect the predominantly extravascular hemolysis that occurs in IgG-mediated immune hemolysis. Polychromasia and spherocytes are the typical findings on the peripheral blood film (see Figure 25-1). Occasionally the WAIHA is accompanied by immune thrombocytopenic purpura and a decreased platelet count; that condition is known as Evans syndrome.10
In symptomatic but non–life-threatening WAIHA, a corticosteroid such as prednisone is the initial treatment of choice.1,2 Approximately 70% to 80% of patients show improvement with prednisone, but most adult patients need to be on a long-term maintenance dosage to remain asymptomatic.1,6 Children generally respond very well to corticosteroids and experience a complete remission.6 Splenectomy is an option in patients with chronic WAIHA that is refractive to prednisone therapy or requires long-term, high-dose prednisone therapy, and a favorable response is achieved in 50% to 75% of patients.1,6 Immunosuppressive drugs, such as cyclophosphamide or azathioprine, are used for refractory WAIHA, but the side effects may be severe.1 Rituximab (monoclonal anti-CD20) has also been used. It causes minimal side effects and produces a response rate of 17% to 100% according to various published case reports.1 Intravenous gamma globulin or plasma exchange is also used in severe disease, but is not recommended for routine or long-term use.1,6 In secondary WAIHA, successful management of the underlying condition often controls the hemolysis and anemia. Life-threatening WAIHA requires RBC transfusion. If the autoantibody has broad specificity, all RBC units may be incompatible with the autoantibody.1,2 In such cases a minimum volume of RBCs is used, and the patient is carefully monitored during the transfusion.
Cold Agglutinin Disease
Cold agglutinins are autoantibodies of the IgM class that react optimally at 4° C. Cold agglutinins are commonly found in healthy individuals. These nonpathologic cold agglutinins are polyclonal, occur in low titers (less than 1 : 64 at 4° C), and have no reactivity above 30° C.1,5 Most pathologic cold agglutinins are monoclonal, occur at high titers (greater than 1 : 1000 at 4° C), and are capable of reacting at temperatures greater than 30° C.2,6 Because pathologic cold agglutinins can react at body temperature, they may induce cold agglutinin disease (CAD). Cold agglutinins that are able to bind RBC antigens near or at 37° C (high thermal amplitude) cause more severe symptoms.1,11 CAD comprises approximately one fourth of the cases of autoimmune hemolytic anemia.6
Chronic CAD is a rare hemolytic anemia that typically occurs in middle-aged and elderly individuals.12 In one recent study, the median age at onset was 67 years, with a range of 30 to 92 years.13 Chronic CAD can be idiopathic, with no known cause, or secondary due to lymphoproliferative neoplasms such as B-lymphocyte lymphomas, Waldenström macroglobulinemia, or chronic lymphocytic leukemia.12 In chronic CAD, the autoantibody is usually monoclonal IgM with κ light chains.2,5,11
Acute CAD occurs secondary to Mycoplasma pneumoniae infection, infectious mononucleosis, and other viral infections. These cold agglutinins are polyclonal IgM with a normal distribution of κ and λ light chains.2,5
In CAD, the IgM autoantibody binds to RBCs after exposure to the cold, particularly in the peripheral circulation and the vessels of the skin where temperatures can drop to 30° C.6,11 During the brief transit through these colder areas, IgM autoantibodies activate the classical complement pathway. When the RBCs return to the central circulation, the IgM antibody dissociates, but C3b components remain on the cell.2,11 Hemolysis is predominantly extravascular by hepatic macrophages, which have receptors for C3b.1,11 However, if the autoantibody has a high thermal amplitude, or there is a deficiency in complement regulatory proteins, full complement activation and intravascular hemolysis can occur.1
In chronic CAD, the clinical manifestations are variable. Most patients have a mild anemia with hemoglobin ranging from 9 to 12 g/dL,12 but others can develop life-threatening anemia with hemoglobin levels falling below 5 g/dL, especially after exposure to cold temperatures.5,13 Individuals often experience fluctuation between mild and severe symptoms, and approximately half require transfusions over the course of the disease.11 Symptoms include fatigue, weakness, dyspnea, pallor (due to the anemia), and acrocyanosis.12 Acrocyanosis is a bluish discoloration of the extremities (fingers, toes, feet, ear lobes, nose) due to RBC autoagglutination, which causes local capillary stasis.1,2,12 Some patients also have episodes of hemoglobinuria, especially after exposure to cold temperatures.2,12 In contrast, patients with acute CAD may have mild to severe hemolysis that appears abruptly within 2 to 3 weeks after the onset of infectious mononucleosis, another viral infection, or M. pneumoniae infection, but it resolves spontaneously within days to a few weeks.12
The DAT result is positive because of the presence of C3d on the RBC surface. The specificity of the cold agglutinin is most often anti-I, but can be anti-i or, very rarely, anti-Pr.2 Virtually all adult RBCs are positive for the I antigen, so anti-I will agglutinate all screening and panel cells, donor RBCs, and the patient’s own RBCs at room temperature and higher, depending on the thermal amplitude of the autoantibody. Anti-I will show weaker or negative reactions with cord RBCs (cord cells are negative for I antigen, but positive for i antigen).2
A cold agglutinin method is used to determine the titer of the antibody at 4° C. Pathologic cold agglutinins can reach titers of 1 : 10,000 to 1 : 1,000,000 at 4° C.1,5 Blood specimens to determine the titer of cold agglutinins are maintained at 37° C after collection to prevent the binding of the autoantibody to the patient’s own RBCs, which can falsely decrease the antibody titer in the serum. Alternatively, a sample anticoagulated with ethylenediaminetetraacetic acid (EDTA) can be warmed for 10 to 15 minutes at 37° C to dissociate autoabsorbed antibody prior to determining the titer.
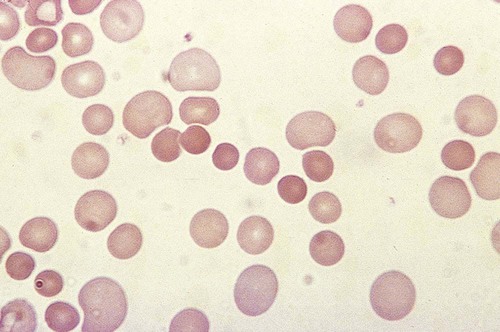
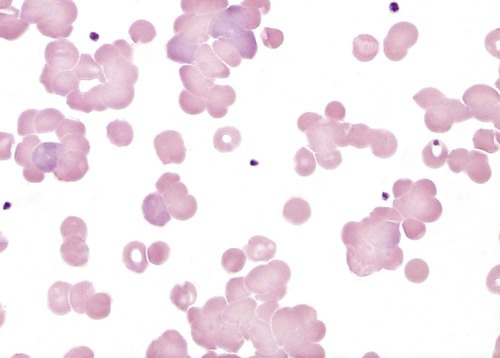
When a high-titer cold agglutinin is present, an EDTA-anticoagulated blood specimen can show visible agglutinates in the tube at room temperature or below.1 The agglutination can also be observed on a peripheral blood film (Figure 25-2). Blood specimens from patients with cold agglutinins must be warmed to 37° C for 15 minutes before analysis by automated hematology analyzers. RBC agglutination grossly elevates the mean cell volume, reduces the RBC count, and has unpredictable effects on other indices (see Chapter 39). When the sample is warmed at 37° C, the antibody dissociates from the RBCs, and agglutination usually disappears. If not, a new specimen is collected and maintained at 37° C for the entire time before testing. To avoid agglutination on a peripheral blood film, the slide can also be warmed to 37° C prior to the application of blood. Cold agglutinins can also interfere with ABO typing.2
Acute CAD associated with infections is self-limited and the cold agglutinin titers are usually less than 1 : 4000.12 If the hemolysis is mild, no treatment is required; however, patients with severe hemolysis require transfusion and supportive care.12
Patients with chronic CAD and mild anemia are regularly monitored and advised to avoid cold temperatures.1 In chronic CAD with moderate to severe symptoms, rituximab (anti-CD20) produces partial remission in about half of patients, but median remission is less than a year.14,15 In a recent study, a combination of rituximab with fludarabine resulted in a better response rate (76%) and median duration of remission (66 months); however, hematologic toxicity was reported in almost half of the patients.16 Plasmapheresis may be used in severe cases but provides only temporary benefit.1 Corticosteroid therapy and immunosuppressive therapy with cyclophosphamide and chlorambucil are not effective for most patients.12 Splenectomy is also not effective, because C3b-sensitized RBCs in IgM-mediated autoimmune hemolysis are cleared primarily by the liver.1,5,12
RBC transfusion is reserved for patients with life-threatening anemia or cardiovascular or cerebrovascular symptoms.1 If transfusion is needed, the presence of clinically significant alloantibodies must be ruled out.2 In CAD cases involving an autoantibody with wide thermal amplitude, detection of coexisting warm-reactive alloantibodies can be time consuming and difficult. During transfusion, the patient is kept warm, and a blood warmer is used to minimize the in vivo reactivity of the cold autoantibody.1
Paroxysmal Cold Hemoglobinuria
Paroxysmal cold hemoglobinuria (PCH) is an acute form of cold-reactive hemolytic anemia. PCH can be idiopathic or secondary. Historically, secondary PCH was associated with late-stage syphilis, but now it is most commonly seen in young children after a respiratory viral infection.1,2,12 PCH is rare in adults. The incidence of PCH in children has been reported to be as high as 32% to 40% of children with autoimmune hemolytic anemia, with a median age at presentation of 5 years.17–19
The anti-P autoantibody, also called the Donath-Landsteiner antibody, is a complement-binding IgG hemolysin with specificity for the P antigen on RBCs. The anti-P autoantibody is biphasic in that at cold temperatures it binds to the P antigen on RBCs and partially activates complement (C1 to C4), but full complement activation (C3 through C9) and hemolysis occur only upon warming to 37° C.19 Exposure to cold temperatures is not required for the hemolytic manifestations in vivo, however, and the reasons for this have yet to be explained.2,6 The anti-P autoantibody binds antigen optimally at 4° C and has a thermal amplitude of less than 20° C. At warmer temperatures, the anti-P autoantibody dissociates from the RBCs. The titer is usually less than 1 : 64.6,12
Children typically present with acute fever, malaise, and back, leg, and/or abdominal pain 1 to 2 weeks after an upper respiratory tract infection.6,12 Pallor, jaundice, and dark urine due to hemoglobinuria are frequently present.12 The abrupt onset of hemolysis causes a rapidly progressing and severe anemia, with hemoglobin levels often dropping below 5 gm/dL.12
Reticulocytosis is typical, but can be preceded by reticulocytopenia.6,12 The peripheral blood film shows polychromasia and spherocytes, but schistocytes, nucleated RBCs, anisocytosis, poikilocytosis, and erythrophagocytosis can also be observed.6,12 At first, leukopenia may be present; later, leukocytosis occurs.6,12 In addition, laboratory findings typical for intravascular hemolysis are found. Because the anti-P autoantibody is dissociated from the RBCs at body temperature, the DAT result is usually positive for C3d only.2
The classic Donath-Landsteiner test for anti-P is done by collecting blood samples in two tubes, one for the patient test and the other for the patient control.19 The patient test sample is incubated first at 4° C for 30 minutes (to allow anti-P binding to the P antigen and partial complement activation on the RBCs), then at 37° C for 30 minutes (to allow full activation of the complement pathway to lysis). The patient control tube is kept at 37° C for both incubations (or a total of 60 minutes). After centrifugation, the supernatant is examined for hemolysis. A positive test result for anti-P is indicated by hemolysis in the patient test sample incubated first at 4° C and then at 37° C, and no hemolysis in the patient control sample kept at 37° C. In the control tube, the anti-P is not able to bind to antigen at 37° C, so complement is not activated and hemolysis does not occur. Initial test results may be falsely negative due to low complement and/or anti-P levels in the patient sample because of the brisk hemolysis in vivo.19 Incubating patient serum with complement and papain-treated compatible group O RBCs increases the sensitivity of the test in detecting anti-P. The enzyme treatment provides greater exposure of the P antigen on the RBC surface for antibody binding.19
PCH is severe but self-limited and resolves in several days to a few weeks with an excellent prognosis.1,12 In most patients, the anemia is severe and can be life-threatening, so transfusion is usually needed until the symptoms resolve. Because the anti-P autoantibody reacts only at lower temperatures and P antigen–negative blood is very rare, P-positive blood can be transfused.2
Mixed-Type Autoimmune Hemolytic Anemia
Mixed-type autoimmune hemolytic anemia occurs very infrequently.20 In this condition, the patient simultaneously develops an IgG autoantibody with optimum reactivity at 37° C (WAIHA) and a pathologic IgM autoantibody that reacts optimally at 0° C to 10° C but has a thermal amplitude of greater than 30° C (CAD).20,21 Patients with WAIHA and a nonpathogenic cold agglutinin (i.e., an agglutinin that does not react at a temperature greater than 20° C) should not be classified as having a mixed-type autoimmune hemolytic anemia, because the cold agglutinin is not clinically significant.12,20,21
The hemolysis results from a combination of extravascular and intravascular mechanisms. The disease course appears to be chronic with intermittent episodes of severe anemia.5,6,12,21 The DAT results can be positive with IgG only, C3d only, or IgG and C3d.2 The warm autoantibody is typically panreactive with unclear specificity, whereas the cold-reacting antibody usually has anti-I specificity.5 Treatment is the same as that described for WAIHA.5
Drug-Induced Immune Hemolytic Anemia
Drug-induced immune hemolytic anemia (DIIHA) is very rare, with an estimated annual incidence of about 1 per million persons.22 This condition is suspected when there is a sudden decrease in hemoglobin after administration of a drug, clinical and biochemical evidence of extravascular or intravascular hemolysis, and a positive DAT result.23 Over 125 drugs have been reported to cause DIIHA, with the most common drug categories being antimicrobial, antiinflammatory, and antineoplastic drugs.24,25 The most common drugs implicated in DIIHA in the last 10 years are cefotetan, ceftriaxone, and piperacillin.22 Severe, even fatal, cases have been reported.
Mechanisms of Drug-Induced Immune Hemolysis
Various theories have been proposed to explain the mechanisms of DIIHA.22 Three generally accepted mechanisms involve an antibody produced by the patient as a result of exposure to the drug and include (1) drug adsorption, (2) drug–RBC membrane protein immunogenic complex, and (3) RBC autoantibody induction. A fourth mechanism, drug-induced nonimmunologic protein adsorption, can result in a positive DAT result, but no drug or RBC antibody is produced by the patient. This mechanism is discussed at the end of this section.
1. Drug adsorption: The patient produces an IgG antibody to a drug. When the drug is taken by the patient, the drug binds strongly to the patient’s RBCs. The IgG drug antibody binds to the drug attached to the RBCs, usually without complement activation. Because the offending antibody is IgG and is strongly attached to the RBCs via the drug, hemolysis is extravascular by splenic macrophages, which remove the antibody- and drug-coated RBCs from the circulation.
2. Drug–RBC membrane protein immunogenic complex: A drug binds loosely to an RBC membrane protein to form a drug–RBC protein immunogenic complex or epitope. The patient produces an IgM and/or IgG antibody that binds to the complex on the RBCs and complement is fully activated, which causes acute intravascular hemolysis.
3. RBC autoantibody induction: A drug induces the patient to produce IgG warm-reactive autoantibodies against RBC self-antigens. These autoantibodies react at 37° C, and the laboratory findings are indistinguishable from those in WAIHA. Hemolysis is extravascular and is mediated by macrophages predominantly in the spleen.
Several authors have suggested that all drug-induced immune hemolysis is explained by a single mechanism, known as the unifying theory. This theory proposes that a drug interacts with the RBC membrane and generates multiple immunogenic epitopes that can elicit an immune response to (1) the drug alone, (2) the drug–RBC membrane protein combination, or (3) an RBC membrane protein alone.24,26
Antibody Characteristics
Antibodies implicated in DIIHA can be divided into two general types: drug-dependent (most common) and drug-independent antibodies.2,24 Some drugs are able to induce a combination of both type of antibodies.22,27,28
Drug-Dependent Antibodies
Drug-dependent antibodies only react in vitro when the suspected drug or its metabolite is present.2,24 There are two types of drug-dependent antibodies:
1. Antibodies that react only with drug-treated cells2: These are IgG drug antibodies that bind to the drug when it is strongly associated with the RBC surface (drug adsorption mechanism). Because they have bound IgG, the RBCs are cleared from the circulation extravascularly by macrophages in the spleen, and a hemolytic anemia gradually develops. Complement is not usually activated. If the DIIHA is not recognized, the patient may continue to take the drug to a point in which life-threatening anemia develops.2,22 Examples of drugs that elicit antibodies in this category are penicillin and cefotetan.2,24 Laboratory features include a positive DAT reaction with anti-IgG, whereas the reaction with anti-C3d is usually negative. In the indirect AHG test, the patient’s serum and an eluate of the patient’s cells react only with drug-treated RBCs and not with untreated RBCs.2,6
2. Antibodies that react only in the presence of the drug2: These IgG and/or IgM antibodies bind to the drug or its metabolite only when it is weakly associated in a drug–RBC membrane protein complex (drug–RBC membrane protein immunogenic complex mechanism). The antibodies activate complement and trigger acute intravascular hemolysis that may progress to renal failure.2 Hemolysis occurs abruptly after short periods of drug exposure or upon readministration of the drug.22 Examples of drugs that elicit antibodies in this category are piperacillin, quinidine, and ceftriaxone.22 Laboratory features include a positive DAT reaction with anti-C3d and occasionally with anti-IgG. In the indirect AHG test, the patient’s serum reacts with untreated, normal RBCs only in the presence of the drug. Because C3d is the predominant protein on the RBCs, the result of the indirect AHG test on an eluate of the patient’s RBC is usually negative.2,6
Drug-Independent Antibodies
Drug-independent antibodies are IgG, warm-reactive, RBC autoantibodies induced by the drug (RBC autoantibody induction mechanism). These autoantibodies have the same serologic reactivity as those causing WAIHA, and they do not require the presence of the drug for in vitro reactivity. Hemolysis is extravascular, mediated by macrophages predominantly in the spleen, usually with a gradual onset of anemia. Examples of drugs that elicit antibodies in this category are fludarabine, methyldopa, and procainamide.22,24 Laboratory features include a positive DAT reaction with anti-IgG. In the indirect AHG test, the patient’s serum and an eluate of the patient’s cells generally react at 37° C with all screening and panel RBCs and with the patient’s own RBCs.2,6
Nonimmune Drug-Induced Hemolysis
In drug-induced nonimmunologic protein adsorption, the patient does not produce an antibody to the drug or to RBCs. The mechanism is also called the membrane modification method, because certain drugs such as high-dose cephalosporins and cisplatin can alter the RBC membrane so that numerous proteins, including IgG and complement, adsorb on the RBC surface.24,27 This phenomenon results in a positive DAT finding, but only rarely has hemolysis been reported. The indirect AHG test on the patient’s serum and an eluate of the patient’s RBCs yields negative results.2
Treatment
After a DIIHA is recognized and confirmed, the first treatment is to discontinue the drug. Most patients will gradually show improvement within a few days to several weeks.6,22 In cases in which a warm-reacting autoimmune antibody is present, the positive DAT result may persist for months after a hematologic recovery.22 If the anemia is severe, the patient may require RBC transfusion or plasma exchange.22 Regardless of mechanism, future episodes of DIIHA are prevented by avoidance of the drug.
Alloimmune Hemolytic Anemias
Hemolytic Transfusion Reaction
Acute Hemolytic Transfusion Reaction
Acute hemolytic transfusion reactions (AHTRs) occur within minutes to hours of the initiation of a transfusion.29 The most common cause of AHTR is the accidental transfusion of ABO-incompatible donor red cells into a recipient. An example is the transfusion of group A red cells into a group O recipient. The recipient has preformed, naturally occurring anti-A (IgM) that is capable of fully activating complement to C9 upon binding to the A antigen on donor red cells. There is rapid, complement-mediated intravascular hemolysis and activation of the coagulation system. ABO-incompatible transfusions are usually due to clerical error and have been estimated to occur in approximately 1 in 38,000 to 1 in 70,000 RBC transfusions.29 The severity of AHTR is variable and is affected by the infusion rate and volume of blood transfused.30 AHTR carries an estimated mortality rate of 2%.29 AHTRs can occur due to incompatibilities involving other blood group systems, but these are rare.29
The immediate investigation of a suspected HTR includes (1) a clerical check for errors, (2) examination of a posttransfusion specimen for hemolysis, and (3) performance of the DAT on the posttransfusion specimen.29 If an AHTR occurred, hemoglobinemia and hemoglobinuria are detectable, and the DAT result is positive. DAT findings may be negative, however, if all the donor cells are lysed.29 The hemoglobin and serum haptoglobin levels decrease, but the serum indirect bilirubin will not begin to rise until 2 to 3 days after the episode. The ABO and Rh typing, antibody screen, and cross-matching are repeated on the recipient and the donor blood to identify the blood group incompatibility. Coagulation tests reveal and assess the risk of DIC.
Delayed Hemolytic Transfusion Reaction
A delayed hemolytic transfusion reaction (DHTR) may occur days to weeks after transfusion as the titer of alloantibodies increases.31 Often, the patient has been alloimmunized by a pregnancy or previous transfusion, but the antibody titer was below the level of serologic detection at the time of transfusion. The second exposure to the antigen results in an increase in titer (anamnestic response). The antibody is usually IgG, is reactive at 37° C, and may or may not be able to partially or fully activate complement. The antibodies most often implicated in DHTRs are directed against the following antigens: Jka, Jkb, E, C, c, K, Fya, S, and s.31 The patient’s antibody binds to the transfused RBCs, which leads to extravascular hemolysis, with or without complement activation. Inadequate posttransfusion hemoglobin response, positive DAT results for IgG and/or C3d, morphologic evidence of hemolysis, and indirect bilirubinemia are the principal signs.31
Hemolytic Disease of the Fetus and Newborn
Hemolytic disease of the fetus and newborn (HDFN) occurs when an IgG alloantibody produced by the mother crosses the placenta into the fetal circulation and binds to fetal RBCs that are positive for the corresponding antigen. The IgG-sensitized fetal RBCs are cleared from the circulation by macrophages in the fetal spleen, and an anemia gradually develops. There is erythroid hyperplasia in the fetal bone marrow and extramedullary erythropoiesis in the fetal spleen, liver, kidneys, and adrenal glands.32 Many nucleated RBCs are released into the fetal circulation. If the anemia is severe in utero, it can lead to generalized edema, ascites, and a condition called hydrops fetalis, which is fatal if untreated.32,33
Laboratory Findings
During the first trimester of pregnancy, ABO and Rh typing is performed on the mother’s blood along with an indirect AHG test for antibodies in her serum.33 An unimmunized D-negative mother receives Rh immune globulin at 28 weeks’ gestation and again within 72 hours of delivery of a D-positive infant to prevent alloimmunization to the D antigen.33 Rh-negative women who experience spontaneous or induced abortion also receive Rh immune globulin. If the mother is alloimmunized to the D antigen, the titer of anti-D is determined by the indirect AHG test. Antibody titers are repeated at 18 weeks’ gestation and every 2 to 4 weeks thereafter until delivery. Titration of the antibody does not predict the severity of HDFN; rather, it helps determine when to monitor for HDFN by additional methods, such as spectrophotometric analysis of amniotic fluid bilirubin.34 Amniocentesis is accurate at predicting severe fetal anemia, but it is an invasive procedure and carries some risk of fetal loss. If severe fetal anemia and HDFN due to anti-D is suspected, a percutaneous umbilical fetal blood sample can be obtained and tested for the hemoglobin level to determine the severity of the anemia.32
At delivery, testing for the newborn is performed on umbilical cord blood.33 Neonates with Rh HDFN have a decreased hemoglobin level, increased reticulocyte count, and increased level of serum indirect bilirubin. The peripheral blood film shows polychromasia and many nucleated RBCs. ABO (only forward grouping) and Rh typing as well as the DAT are also performed. The DAT result shows positivity for IgG, and anti-D can be demonstrated in an eluate of the infant’s RBCs.33
Treatment for the Affected Infant
Treatment for a fetus affected by HDFN may include intrauterine transfusion, which can be used to correct fetal anemia and prevent hydrops fetalis.32,33 After delivery, the neonate may need exchange transfusions and phototherapy to reduce the level of serum indirect bilirubin and prevent kernicterus.
Hemolytic Disease of the Fetus and Newborn Caused by Other Blood Group Antigens
ABO HDFN is more common than Rh disease and may occur during the first pregnancy. Unlike Rh disease, ABO disease is asymptomatic or produces mild hyperbilirubinemia and anemia. ABO HDFN is seen in some infants with group A or B blood born to group O mothers who, in addition to the usual IgM ABO antibodies, produce IgG anti-A and anti-B, which are capable of crossing the placenta. The disease is milder than Rh HDFN, because most of the maternal anti-A and anti-B antibodies are the IgM isotype and do not cross the placenta.32
The DAT result for the newborn with ABO HDFN is only weakly positive and may be negative. Spherocytes and polychromasia on the peripheral blood film are typical.32 Table 25-3 presents a comparison of HDFN caused by ABO and Rh incompatibility.
TABLE 25-3
Characteristics of Rh and ABO Hemolytic Disease of the Fetus and Newborn
| Rh | ABO | |
| Blood groups | ||
| Mother | Rh (D) negative | O |
| Child | Rh (D) positive | A or B |
| Severity of disease | Severe | Mild |
| Jaundice | Severe | Mild |
| Spherocytes on peripheral blood film | Rare | Usually present |
| Anemia | Severe | If present, mild |
| Direct antiglobulin test result | Positive | Negative or weakly positive |
HDFN can be caused by other IgG antibodies, particularly antibodies to the K, c, and Fya antigens.33 HDFN due to other blood group antibodies is rare.33 Varying degrees of anemia, jaundice, and kernicterus are the adverse clinical outcomes in all forms of HDFN.
Summary
• The immune hemolytic anemias are classified into autoimmune hemolytic anemia, drug-induced immune hemolytic anemia, and alloimmune hemolytic anemia.
• Hemolysis mediated by IgM requires complement; hemolysis may be extravascular (mainly in the liver) if complement is partially activated to C3b, or intravascular if complement is fully activated to C9.
• Hemolysis mediated by IgG occurs with or without complement activation; IgG-sensitized RBCs are removed from the circulation by macrophages in the spleen; partial phagocytosis produces spherocytes, which are trapped in the spleen and phagocytized; IgG- and C3b-sensitized RBCs are removed by macrophages in the spleen and liver.
• Laboratory findings in immune hemolytic anemia include decreased hemoglobin level, increased reticulocyte count, increased levels of serum indirect bilirubin and lactate dehydrogenase, and decreased serum haptoglobin level. The peripheral blood film may show polychromasia, spherocytes (IgG-mediated hemolysis), or RBC agglutination (cold agglutinins). The DAT detects in vivo sensitization of RBCs by IgG and/or C3d.
• The classification of autoimmune hemolytic anemia includes WAIHA, CAD, PCH, and mixed-type autoimmune hemolytic anemia.
• WAIHA is the most common form of autoimmune hemolytic anemia and involves IgG autoantibodies with optimum reactivity at 37° C. The anemia varies from mild to severe, and characteristic morphologic features on the peripheral blood film are polychromasia and spherocytes.
• CAD is caused by an IgM autoantibody with optimum reactivity at 4° C and a thermal amplitude of greater than 30° C. RBC agglutination may be observed on a peripheral blood film, and agglutinates may cause interference with the CBC analysis on automated instruments.
• PCH is due to a biphasic IgG autoantibody with anti-P specificity. The antibody binds to the P antigen on the RBCs and partially activates complement at 4° C; complete complement activation and hemolysis occurs upon warming the sample to 37° C.
• In DIIHA, the patient produces antibodies to (1) a drug only, (2) a complex of a drug loosely bound to an RBC membrane protein, or (3) an RBC membrane protein only. In vitro reactions of antibodies in DIIHA may be drug dependent or drug independent.
• AHTRs occur within minutes to hours after the start of an RBC transfusion and most often involve transfusion of ABO-incompatible blood; the hemolysis is predominantly intravascular. DHTRs may occur days or weeks after the transfusion and represent an anamnestic response to a donor antigen; the hemolysis is extravascular.
• HDFN occurs when an IgG alloantibody produced by the mother crosses the placenta into the fetal circulation and binds to fetal RBCs that are positive for the corresponding antigen. The IgG-sensitized fetal RBCs are cleared from the circulation by macrophages in the fetal spleen, and an anemia gradually develops; the usual laboratory findings in the neonate are anemia, hyperbilirubinemia, and a positive DAT result.
• ABO HDFN is more common than Rh HDFN and produces no symptoms or mild anemia. Rh HDFN due to anti-D results in severe anemia; administration of Rh immune globulin at 28 weeks’ gestation and after delivery of a D-positive infant prevents immunization to the D antigen in D-negative pregnant women.
Review Questions
1. The pathophysiology of immune hemolysis with IgM antibodies always involves:
2. In hemolysis mediated by IgG antibodies, which abnormal RBC morphology is typically observed on the peripheral blood film?
3. The most important finding in the diagnostic investigation of a suspected autoimmune hemolytic anemia is:
a. Detection of a low hemoglobin and hematocrit
b. Observation of hemoglobinemia in a specimen
c. Recognition of a low reticulocyte count
5. The hemolysis in PCH is usually:
a. Extravascular with complement activation to C3b
b. Extravascular with no complement activation
6. The difference between nonpathologic and pathologic cold agglutinins is that the pathologic types:
a. Are IgG and react optimally at 37° C
b. Have a thermal amplitude greater than 30° C
7. Which one of the following statements is true about DHTR:
a. It is usually due to an ABO incompatibility
b. Hemoglobinemia and hemoglobinuria frequently occur
c. It is due to an anamnestic response after repeat exposure to a blood group antigen
8. Chronic secondary CAD is most often associated with:
9. A 63-year-old man is being evaluated because of a decrease in hemoglobin of 5 gm/dL after a second cycle of fludarabine for treatment of chronic lymphocytic leukemia. The patient’s DAT result is strongly positive for IgG only, and antibody testing on his serum and an eluate of his RBCs yields positive results with all panel cells and the patient’s own cells. This suggests which mechanism of immune hemolysis for this patient?
10. A Group A Rh-negative mother gave birth to a Group O Rh-positive baby. The baby is at risk for HDFN if: