[level-membership-for-pulmolory-and-respiratory-category]
Pulmonary Vascular Disease
After reading this chapter you will be able to:
 State how many patients develop venous thromboembolism each year.
State how many patients develop venous thromboembolism each year.
 Describe how and where thromboemboli originate.
Describe how and where thromboemboli originate.
 Describe how pulmonary emboli alter lung and cardiac function.
Describe how pulmonary emboli alter lung and cardiac function.

 Describe how pulmonary embolism is diagnosed and managed.
Describe how pulmonary embolism is diagnosed and managed.
 Describe hemodynamic findings associated with pulmonary hypertension.
Describe hemodynamic findings associated with pulmonary hypertension.

 State patients who are at risk for the development of IPAH.
State patients who are at risk for the development of IPAH.
 Identify the clinical features associated with IPAH.
Identify the clinical features associated with IPAH.


Venous Thromboembolic Disease
Venous thromboembolism is a major health problem in the United States. The prevalence of venous thromboembolism, which includes PE and DVT, has remained relatively constant over time and has been calculated to be 117 cases per 100,000 persons (i.e., DVT at 48 cases per 100,000 and PE at 69 cases per 100,000). It is estimated that 200,000 to 300,000 new cases occur yearly in the United States.1
Venous thromboembolism is treatable but requires prompt diagnosis and therapy to avert serious consequences. One-third of deaths from PE occur within 1 hour of onset of symptoms, and more than 70% of patients who die of PE are not suspected to have PE before death.2 Although mortality from PE has decreased in recent years,3 the death rate for the first episode of PE among hospitalized patients may be 17.4% at 3 months.3,4 Recurrent PE is associated with a much higher mortality rate because only one-quarter of patients survive 3 months.5 The diagnosis is not suspected in approximately two-thirds of patients who die of PE, and the frequency of recognizable emboli in routine autopsies of adult patients ranges from 1.5% to almost 30%.6–8 In a population-based study of PE as a cause of death in New Mexico, only 34% of 812 postmortem documented cases of PE were diagnosed before death.5 Morpurgo and Schmid9 reported their experience with 92 postmortem cases of massive or submassive PE detected during the years 1986-1989. Only 28% of the cases were diagnosed before death, a finding that emphasizes the underdiagnosis of venous thromboembolic disease.
Two-thirds of cases of initial embolus from which patients survive remain undiagnosed. The mortality rate among these patients with undiagnosed embolism is approximately 30%,10 which emphasizes the importance of recognizing PE. If venous thromboembolism is recognized and treated, the mortality rate decreases to less than 8%, and the long-term outcome is generally favorable.11 The long-term survival rates after venous thromboembolism in an inception cohort of 2218 patients were 72% at 1 month and 63% at 1 year.12
Because the accuracy of the clinical impression (i.e., without testing) of venous thromboembolism is less than 50%,13 objective tests are needed to confirm or exclude the diagnosis. Patients with multiple injuries, immobilization, bed rest, or intravascular catheters and elderly patients are at high risk of venous thromboembolic disease (Table 26-1) and should be considered candidates for testing when appropriate symptoms develop.
TABLE 26-1
Frequency of Venous Thrombosis in Various Hospitalized Patient Groups
| Group | Frequency (%) |
| Orthopedic (e.g., fractured hip) | 54-67 |
| Urologic (e.g., prostatectomy) | 25 |
| Surgical patients >40 yr old | 28 |
| Gynecologic surgery | 18 |
| Cardiovascular surgery (e.g., acute myocardial infarction) | 39 |
| Obstetrics | 3 |
From Arroliga AC, Matthay MA, Matthay RA: Pulmonary thromboembolism and other pulmonary vascular diseases. In George RB, et al, editors: Chest medicine: essentials of pulmonary and critical care medicine, ed 3, Baltimore, 1995, Williams & Wilkins.
Pathogenesis
Pulmonary emboli arise from detached portions of venous thrombi that form, in most cases, in deep veins of the lower extremities or pelvis (86%); the point of origin of pulmonary emboli is actually found in only one-half of patients.9 A small percentage of pulmonary emboli arise from the right-sided heart chambers (3.15%) or the superior vena cava (3%).9
Conditions that favor thrombus formation include blood stasis, the presence of hypercoagulable states, and vessel wall abnormalities (factors known as Virchow’s triad). Causes of blood stasis include local pressure, venous obstruction, and immobilization. Other causes of stasis include congestive heart failure, shock and dehydration, varicose veins, and enlargement of the right heart chambers. Several conditions enhance the intravascular coagulability of the blood and predispose to venous thromboembolic disease (Box 26-1).14 The most frequent causes of an inherited hypercoagulable state are the factor V Leiden mutation and the prothrombin gene mutation, which together account for 50% to 60% of cases.15 The major acquired risk factors for venous thromboembolism include recent major surgery, trauma, immobilization, antiphospholipid antibody syndrome, malignancy, pregnancy, oral contraceptives, and myeloproliferative disorders.16 Vessel wall abnormalities are found most often in patients who have sustained trauma or have undergone major surgery.
Pathology
Stasis, an important factor for the formation of DVT, is rarely the only risk factor.17 Deposition of platelets and fibrin in the venous valve cups of the lower extremities occurs as a result of stasis. The combination of diminished blood flow and the presence of trauma and toxins can worsen endothelial damage and promote the release of mediators that encourage adhesion, aggregation, and degranulation of platelets. The result is activation of the coagulation cascade and production of thrombi and fibrin.
PE is a frequent complication of DVT, occurring in more than 50% of cases with DVT confirmed on phlebography.18 PE occurs when a fragment of the thrombus in the venous system travels to the pulmonary circulation. Pulmonary emboli occur more frequently in the lower lobes and are more often found in the right lung than in the left lung, a phenomenon probably related to the flow distribution that favors the right lung and the lower lobes.6 Embolism to the pulmonary circulation produces pulmonary hemorrhage in the ischemic or infarcted lung in less than 10% of cases of PE. Infarction, secondary to thromboembolism, is less common in the lung than in other tissues because the lung has two blood supplies: the pulmonary arterial circulation and the bronchial circulation. At a capillary level, extensive connections exist within the pulmonary and bronchial circulations that prevent serious damage to lung tissue deprived of its pulmonary artery supply.6 Patients with underlying cardiovascular disease may have impairment of the remaining bronchial circulation with resultant lung tissue necrosis when emboli occur.
Pulmonary infarction is associated with thromboembolic obstruction of a medium-sized pulmonary artery. Generally, infarcts occur at the lung bases, are pleural-based, and may be accompanied by pleural effusion. Microscopic examination of the lung in pulmonary infarction shows necrosis of alveolar walls, alveoli filled with red blood cells, and a mild inflammatory response in the periphery.6
 Rule of Thumb
Rule of ThumbPathophysiology
The sudden obstruction of a pulmonary arterial branch causes a decrease in or total cessation of blood flow to the distal area of the lung that leads to respiratory and hemodynamic alterations.19 In the appropriate context, massive PE should be suspected anytime there is unexplained hypotension accompanied by an elevated central venous pressure (jugular vein distention).20 It is a catastrophic entity that frequently results in acute right ventricular failure and death. Death from massive PE is the result of cardiovascular collapse rather than of respiratory failure.
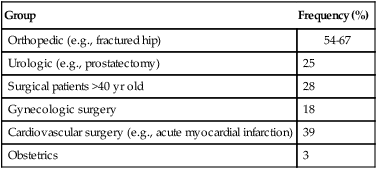
Embolic obstruction of the pulmonary artery increases the alveolar dead space, causes bronchoconstriction, and decreases the production of alveolar surfactant. Wasted or dead space areas occur when areas of the lung parenchyma are ventilated but not perfused. The response is to increase total ventilation (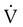 ). The increased
). The increased 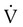 contributes to the sensation of dyspnea that accompanies PE. Bronchoconstriction from diminished carbon dioxide concentration, regional hypoxia, and the production of serotonin and histamine cause further ventilation/perfusion (
contributes to the sensation of dyspnea that accompanies PE. Bronchoconstriction from diminished carbon dioxide concentration, regional hypoxia, and the production of serotonin and histamine cause further ventilation/perfusion (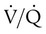 ) mismatching.21
) mismatching.21
Not all patients with PE have significant arterial hypoxemia, but the presence of a widened alveolar-arterial oxygen (O2) tension gradient and reduced PaO2 are common. Hypoxemia develops because of 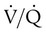 mismatch, intrapulmonary shunt, and cardiogenic shock. Shock is caused by obstruction of the pulmonary vasculature by massive emboli or by numerous small emboli in the presence of cardiopulmonary disease. Cardiac output decreases, and O2 delivery declines. With the decrease in O2 delivery, the peripheral tissues increase O2 extraction causing venous O2 desaturation. In patients with significantly increased right heart pressures, intracardiac right-to-left shunt may develop when blood flows through a patent foramen ovale.19,21 In addition, the depletion of surfactant material as a result of embolic occlusion can lead to atelectasis and intrapulmonary shunt, which can cause hypoxemia.19
mismatch, intrapulmonary shunt, and cardiogenic shock. Shock is caused by obstruction of the pulmonary vasculature by massive emboli or by numerous small emboli in the presence of cardiopulmonary disease. Cardiac output decreases, and O2 delivery declines. With the decrease in O2 delivery, the peripheral tissues increase O2 extraction causing venous O2 desaturation. In patients with significantly increased right heart pressures, intracardiac right-to-left shunt may develop when blood flows through a patent foramen ovale.19,21 In addition, the depletion of surfactant material as a result of embolic occlusion can lead to atelectasis and intrapulmonary shunt, which can cause hypoxemia.19
The main hemodynamic consequence of PE is increased resistance to blood flow caused by obstruction of the pulmonary arterial bed. The hemodynamic consequences are determined by the extent of the cross-sectional area of the pulmonary circulation involved, the underlying cardiopulmonary reserve, and the neurohumoral response to the embolism. Pulmonary hypertension occurs when 50% of the pulmonary vascular bed has been occluded.19,21 To maintain the same flow at a higher pressure, the right ventricle must work harder. The result is an increase in right ventricular work that causes the right ventricle to become dilated and ischemic. The thin-walled right ventricle is not designed to work with acute heavy pressure loads. When the mean pulmonary artery pressure increases to greater than 40 mm Hg during an acute first PE, the right ventricle fails, and hemodynamic collapse and death occur.22 The exact role of vasoconstriction in the pathogenesis of pulmonary hypertension is uncertain, but vasoconstrictors such as serotonin and thromboxane A2 may also play a role in the development of pulmonary hypertension after acute PE.
Although the usual course of PE is to resolve rapidly (because the body lyses the embolism with endogenous fibrinolytic agents), permanent residual emboli do occur.23 Massive emboli are likely to resolve within weeks, particularly in young patients. Overall, less than 10% of patients have perfusion defects after 6 weeks. Vascular patency is restored when the unresolved emboli organize or form scars against the vessel wall.
Clinical Features
A high index of suspicion for venous thromboembolism is crucial to make the diagnosis for patients at risk. No specific signs or symptoms indicate the presence of venous thromboembolic disease, and a significant proportion of patients are asymptomatic (32%).2,24 The physical findings of DVT in the lower extremities include erythema and warm skin in one-third of patients and swelling and tenderness in three-fourths of patients. In patients who have swelling above and below the knee, fever, and a history of immobility and cancer, the likelihood of finding DVT on a venogram is only 42%.25
The most frequent symptoms in patients with confirmed PE are dyspnea, followed by pleuritic chest pain and cough (Table 26-2).26 The onset of dyspnea is usually rapid, within seconds (46%) or minutes (26%).26 Hemoptysis occurs in 13% to 20% of patients. The combination of dyspnea of sudden onset, fainting, and acute chest pain should raise suspicion of PE. In one study, this combination of symptoms was present in 96% of patients with confirmed PE compared with 59% of patients in whom PE was suspected but not confirmed.27 In some patients, dyspnea lasts only a few minutes, and this episode may be wrongly dismissed as being trivial.19,27–29
TABLE 26-2
Clinical Characteristics in Patients With Pulmonary Embolism and No Cardiopulmonary Disease
| Symptoms | Frequency (%) | Signs | Frequency (%) |
| Dyspnea at rest or with exercise | 73 | Tachypnea | 54 |
| Pleuritic pain | 44 | Tachycardia | 24 |
| Calf or thigh pain | 44 | Rales | 18 |
| Cough | 34 | Decreased breath sounds | 17 |
| Orthopnea | 28 | Loud P2 | 15 |
| Wheezing | 21 | Jugular venous distention | 14 |
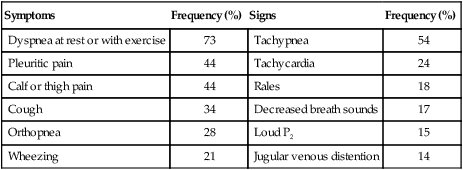
Modified from Stein PD, Beemath A, Matta F, et al: Clinical characteristics of patients with acute pulmonary embolism: data from PIOPED II. Am J Med 120:871–879, 2007.
There are no characteristic physical findings of PE. The most frequent physical findings include tachypnea, rales on chest examination, and tachycardia. Similar to dyspnea, these signs may be short-lived. Other common physical findings include an accentuated pulmonary component of the second heart sound (loud P2) consistent with pulmonary hypertension. Fever may be present in 54% of patients.27–29 Similar to what occurs in the diagnosis of DVT, less than 35% of patients in whom PE is clinically suspected actually have it.2
Chest Radiograph
The chest radiograph cannot confirm the presence of PE but is helpful to rule out other potentially life-threatening conditions, such as pneumothorax or pneumonia, which can manifest in a similar way. In dyspneic patients, a normal chest radiograph may be a clue to the presence of PE; however, chest radiography is abnormal in more than 80% of cases. Abnormalities include enlargement of the right descending pulmonary artery (66%), elevation of the diaphragm (61%), cardiomegaly (55%), and small pleural effusion (50%). Parenchymal densities (patchy infiltrates or round nodular lesions) predominantly appearing next to the pleural surface are present in patients who have infarction or atelectasis. Other, less common findings include the Westermark sign, in which there is pulmonary hyperlucency caused by a marked reduction in blood flow. The so-called Hampton hump, an opacity in the costophrenic angle, is present in 25% to 30% of patients.27
Electrocardiogram
The electrocardiogram (ECG) is helpful to rule out other diagnoses, such as acute myocardial infarction and pericarditis. The ECG is frequently abnormal in patients with PE (87% of the time), but the ECG abnormalities accompanying PE are nonspecific in 70% to 75% of cases; tachycardia and ST segment depression are most common.27 Abnormalities such as T wave inversion in right precordial leads, depression of the ST segment, and T wave inversion in V1 and V2 may be present. A so-called S1Q3T3 pattern is associated with massive PE and is present in 19% of such patients.25
Arterial Blood Gases
Most patients with acute PE have hypoxemia and hypocapnia,27 but a significant percentage of patients (15% to 25%) with or without previous cardiopulmonary disease have a PaO2 exceeding 80 mm Hg.29 Although a widened alveolar-arterial O2 gradient is frequently present, a normal alveolar-arterial O2 gradient may occur in approximately 20% of patients with angiographically documented PE.27,29 Although an arterial blood gas measurement may be helpful in identifying patients with hypoxemia or hypocapnia accompanying PE, arterial blood gas measurements can never secure the diagnosis of PE.
Diagnostic Modalities
By-Products of Thrombin and Plasmin
Clot formation is invariably associated with thrombingeneration. Measurement of cross-linked fibrin split products (D-dimers) has been found to be sensitive for the diagnosis of acute venous thromboembolism. The specificity of D-dimer enzyme-linked immunosorbent assay (ELISA) can exclude all but 5% to 10% of patients with acute PE, so this test has been used as an important tool for early assessment.30 The specificity of the test is only 39%, but a value less than the recommended cutoff for current quantitative ELISA assays has been shown to rule out venous thromboembolic disease in 98% of patients.31–33 In patients in whom DVT is suspected clinically, negative results of the D-dimer assay combined with negative findings on impedance plethysmography have a negative predictive value of 98% for DVT (i.e., if the test is negative, the chance of a DVT is only 2%).31 D-dimer results have been particularly useful in the emergency department and outpatient area for the evaluation of patients with suspected DVT31 and PE.33 In patients with a low pretest probability of DVT or PE and a negative D-dimer result, the negative predictive value for the strategy has been greater than 99%.32,33
Although there are several laboratory methods to measure D-dimer levels, tests using ELISAs are the most widely used and best performing among the D-dimer assays regarding the sensitivity and negative likelihood ratio. For excluding PE or DVT, a negative result on quantitative rapid ELISA is as diagnostically useful as a normal lung scan or negative duplex ultrasonography finding. D-dimer ELISA can be used to exclude PE in outpatients with a low to moderate suspicion without the need for further costly testing. However, inpatients should undergo an imaging study as the initial test for PE because most will already have elevated D-dimer levels because of comorbid conditions.33
Testing for Lower Extremity Deep Venous Thrombosis
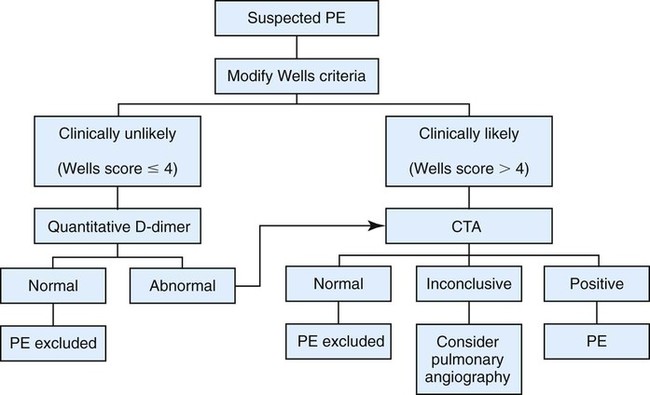
To evaluate the clinical pretest probability of DVT, the Wells criteria are frequently used. These criteria include the following clinical parameters: presence of cancer, immobilization, localized tenderness, swelling, edema, previous DVT, collateral superficial veins, and absence of an alternative diagnosis.34 In cases in which there is a moderate to high pretest probability, several modalities could be used for diagnosing DVT in the extremities, such as compression ultrasonography, impedance plethysmography, and venography. In patients with low pretest probability of DVT and a negative D-dimer, further testing may be unnecessary.31,35–37
Impedance Plethysmography
Impedance plethysmography is a noninvasive method and measures electrical impedance to blood flow, which changes with inflation and deflation of a lower extremity cuff. The quality of the test is operator-dependent and requires that the patient be supine and lay still for at least 2 minutes. The test has a sensitivity and specificity of 91% and 96% for symptomatic proximal venous thrombi. A lower sensitivity of 65% has been reported.38,39
Compression Ultrasonography
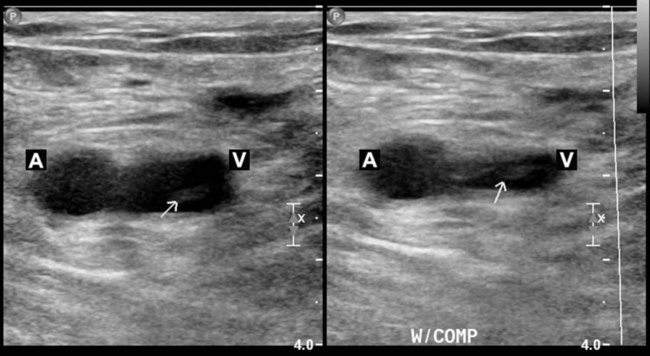
Compression ultrasonography has proved to be sensitive and specific for the diagnosis of symptomatic proximal DVT. This test is noninvasive, portable, and accurate and is the modality of choice for the diagnosis of DVT. Compression ultrasonography combines B-mode scanning with a tightly focused pulse Doppler beam directed at the vessels of interest. DVT is diagnosed with the findings of venous noncompressibility, an echogenic filling defect, absence of Doppler flow, free-floating thrombus in the vein, and venous distention.40 The most reliable sign of DVT is lack of compressibility of the vein, although a free-floating thrombus has the highest embolic potential (Figure 26-1). The sensitivity and specificity of compression ultrasonography in symptomatic patients vary between 95% and 100% for the detection of a proximal lower extremity thrombus.40,41 Areas not well visualized with compression ultrasonography include the iliac veins, the superficial femoral veins in the adductor canal, and the calf veins. However, the accuracy of compression ultrasonography, even with the addition of color Doppler ultrasonography, is moderate to low for the detection of DVT in patients at high risk who do not have symptoms.42 These results suggest that ultrasonography, although sensitive and specific for the diagnosis of DVT, is not a good screening test for patients at high risk who do not have symptoms.
Testing for Pulmonary Embolism
Noninvasive tests for the diagnosis of PE include 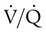 scan and helical or spiral computed tomography (CT) angiography scan of the chest. Either of these tests, depending on the resources available, may be the initial diagnostic examination if the presence of acute PE is clinically suspected.43 Echocardiography can suggest the diagnosis (right ventricular dilation, dysfunction, or thrombus) and provide prognostic information.44 In certain cases when these noninvasive tests are nondiagnostic, pulmonary angiography may be needed to confirm or exclude the diagnosis of PE.
scan and helical or spiral computed tomography (CT) angiography scan of the chest. Either of these tests, depending on the resources available, may be the initial diagnostic examination if the presence of acute PE is clinically suspected.43 Echocardiography can suggest the diagnosis (right ventricular dilation, dysfunction, or thrombus) and provide prognostic information.44 In certain cases when these noninvasive tests are nondiagnostic, pulmonary angiography may be needed to confirm or exclude the diagnosis of PE.
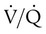 scanning involves the inhalation of a radiolabeled gas (usually xenon-133, xenon-127, krypton-181m, or technetium-99m) and the intravenous injection of macroaggregated albumin tagged with a gamma-emitting radioisotope. The distribution of lung ventilation (
scanning involves the inhalation of a radiolabeled gas (usually xenon-133, xenon-127, krypton-181m, or technetium-99m) and the intravenous injection of macroaggregated albumin tagged with a gamma-emitting radioisotope. The distribution of lung ventilation (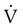 ) and lung perfusion (
) and lung perfusion (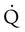 ) is studied, and areas of mismatch where
) is studied, and areas of mismatch where 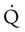 is less than
is less than 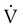 are sought. The presence of mismatches most often indicates embolic occlusion of the blood vessel, although other rare causes of mismatches exist, such as extrinsic compression of the vessel by a mass, intraluminal obstruction by angiosarcoma, or obliteration of a vessel by vasculitis. The addition of
are sought. The presence of mismatches most often indicates embolic occlusion of the blood vessel, although other rare causes of mismatches exist, such as extrinsic compression of the vessel by a mass, intraluminal obstruction by angiosarcoma, or obliteration of a vessel by vasculitis. The addition of 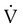 scan increases the specificity of
scan increases the specificity of 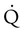 scan.45 Generally, with the presence of a parenchymal abnormality, the
scan.45 Generally, with the presence of a parenchymal abnormality, the 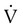 defect coincides with the
defect coincides with the 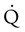 defect, and matched abnormalities are found. Normal results of a
defect, and matched abnormalities are found. Normal results of a 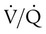 scan exclude the presence of a clinically significant PE in the context of a low clinical probability of PE. In these cases, anticoagulant therapy can be safely withheld.46 Abnormal
scan exclude the presence of a clinically significant PE in the context of a low clinical probability of PE. In these cases, anticoagulant therapy can be safely withheld.46 Abnormal 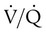 scan results can be classified as high probability, intermediate (or indeterminate) probability, and low probability for PE, according to the size of the defect and the degree of mismatch between the
scan results can be classified as high probability, intermediate (or indeterminate) probability, and low probability for PE, according to the size of the defect and the degree of mismatch between the 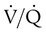 scan and chest radiographic abnormalities.45 Diagnostic accuracy is greatest when
scan and chest radiographic abnormalities.45 Diagnostic accuracy is greatest when 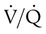 scan results are combined with clinical probability (Table 26-3).47 The presence of concomitant cardiopulmonary disease (e.g., COPD), even if severe, does not diminish the diagnostic usefulness of
scan results are combined with clinical probability (Table 26-3).47 The presence of concomitant cardiopulmonary disease (e.g., COPD), even if severe, does not diminish the diagnostic usefulness of 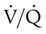 scans in the diagnosis of acute PE.48–50
scans in the diagnosis of acute PE.48–50
TABLE 26-3
| High probability | Two or more large (>75% of a segment) segmental 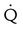 defects without corresponding defects without corresponding 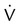 or abnormalities on chest radiograph or abnormalities on chest radiograph |
One large segment 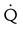 defect and two or more moderate (25%-75% of a segment) segmental defect and two or more moderate (25%-75% of a segment) segmental 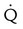 defects without corresponding defects without corresponding 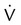 or abnormalities on chest radiograph or abnormalities on chest radiograph |
|
Four or more moderate segmental 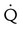 defects without corresponding defects without corresponding 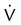 or abnormalities on chest radiograph or abnormalities on chest radiograph |
|
| Intermediate probability | One moderate or up to two large segment 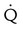 defects without corresponding defects without corresponding 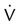 defect or abnormalities on chest radiograph defect or abnormalities on chest radiograph |
Corresponding 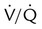 defects and parenchymal opacity in lower lung zone on chest radiograph defects and parenchymal opacity in lower lung zone on chest radiograph |
|
Corresponding 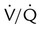 defects and small pleural effusion defects and small pleural effusion |
|
Single moderate matched 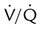 defects with normal findings on chest radiograph defects with normal findings on chest radiograph |
|
| Findings difficult to categorize as normal, low, or high probability | |
| Low probability | Multiple matched 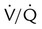 defects, regardless of size, with normal findings on chest radiograph defects, regardless of size, with normal findings on chest radiograph |
Corresponding 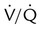 defects and parenchymal opacity in upper or middle lung zone on chest radiograph defects and parenchymal opacity in upper or middle lung zone on chest radiograph |
|
Corresponding 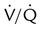 defects and large pleural effusion defects and large pleural effusion |
|
Any 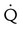 defects with substantially larger abnormality on chest radiograph defects with substantially larger abnormality on chest radiograph |
|
| Defects surrounded by normally perfused lung (stripe sign) | |
Single or multiple small (<25% of a segment) segmental 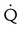 defects with a normal chest radiograph defects with a normal chest radiograph |
|
Nonsegmental 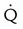 defects (cardiomegaly, aortic impression, enlarged hila) defects (cardiomegaly, aortic impression, enlarged hila) |
|
| Normal | No 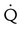 defects; defects; 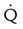 outlines the shape of the lung on chest radiograph outlines the shape of the lung on chest radiograph |
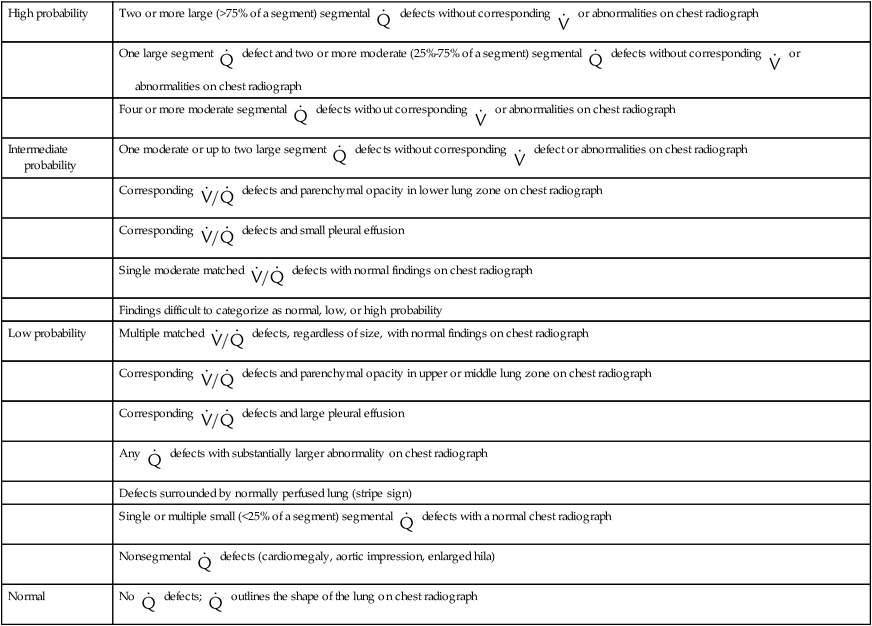
Modified from Worsley DF, Alavi A, Palevsky JH: Role of radionuclide imaging in patients with suspected pulmonary embolism. Radiol Clin North Am 31:849, 1993.
For the one-third of patients who do not receive a definitive diagnosis on the basis of the results of noninvasive studies, pulmonary angiography is the test of choice. The mortality rate for pulmonary angiography is 0.5%, and the prevalence of major nonfatal complications is 1%. Nevertheless, patients in a medical intensive care unit are at higher risk of morbidity and mortality (approximately 4%), including respiratory failure, renal failure, and hematoma necessitating transfusion, than other patients.47 Pulmonary angiography signs of acute PE include filling defects and cutoff of the pulmonary arteries. Other signs on angiography include absent, decreased, or delayed filling of pulmonary arteries; delayed venous emptying; pruning; and abnormal tapering. None of these findings is as specific as filling defects, in particular, in the presence of other cardiopulmonary diseases. Table 26-4 shows the probability of finding PE with angiography on the basis of results of  scan and clinical probability.47 A definite diagnosis can be established with noninvasive diagnostic tools in two-thirds of cases.51
scan and clinical probability.47 A definite diagnosis can be established with noninvasive diagnostic tools in two-thirds of cases.51
TABLE 26-4
| Scan Interpretation | High Clinical Probability (%) | Intermediate Clinical Probability (%) | Low Clinical Probability (%) |
| High probability | 96 | 88 | 56 |
| Intermediate probability | 66 | 28 | 16 |
| Low probability | 40 | 16 | 4 |
| Near-normal/normal | 0 | 6 | 2 |
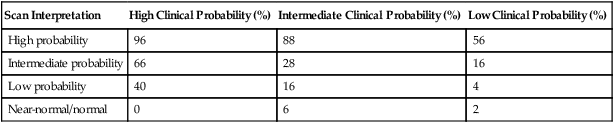
From Arroliga AC, Matthay MA, Matthay RA: Pulmonary thromboembolism and other pulmonary vascular diseases. In George RB, et al, editors: Chest medicine: essentials of pulmonary and critical care medicine, ed 3, Baltimore, 1995, Williams & Wilkins.
Helical CT angiography has been used extensively in the diagnostic evaluation of PE and has become the principal diagnostic imaging method to evaluate suspected PE (Figure 26-2).52,53 The reported sensitivity of helical CT angiography ranges from 53% to 100%, and the specificity ranges from 81% to 100%.53 The variability is probably due to the experience of the radiologists and image quality.54 Studies indicate that helical CT scanning detects large pulmonary emboli involving main and lobar emboli. However, this test is generally unable to detect smaller pulmonary emboli. One potential advantage of helical CT angiography is its ability to identify alternative diagnoses in cases in which PE is not present (e.g., pneumonia, pleural disease). Helical CT angiography scanning can be as cost-effective as 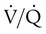 scanning and duplex ultrasound examination of the lower extremity but only when combined with D-dimer testing.
scanning and duplex ultrasound examination of the lower extremity but only when combined with D-dimer testing.
Multicenter trials suggest that helical CT scanning is safe to use for ruling out PE, at least in patients with a low or intermediate clinical probability of embolism. The PIOPED (Prospective Investigation of Pulmonary Embolism Diagnosis) II trial evaluated the accuracy of multidetector CT angiography alone and combined with venous phase imaging (CT angiography–CT venography) for the diagnosis of acute PE. Among 824 patients with a reference diagnosis and a completed CT study, CT angiography was inconclusive in 51 because of poor image quality. Excluding such inconclusive studies, the sensitivity of CT angiography was 83%, and the specificity was 96%. CT angiography–CT venography was inconclusive in 87 of 824 patients because the image quality of either CT angiography or CT venography was poor. The sensitivity of CT angiography–CT venography for PE was 90%, and specificity was 95%. The predictive value of either CT angiography or CT angiography–CT venography is high with a concordant clinical assessment, but additional testing is necessary when the clinical probability is inconsistent with the imaging results.53,55 Several algorithms for the diagnosis of PE are available, but no approach has proven superior to others.56–58 Figure 26-3 summarizes the diagnostic approach to PE using CT angiography.
Mini Clini
Respiratory Distress After Hip Replacement
 Problem
ProblemBecause of the history of surgery on the right hip, DVT and PE are the most likely diagnoses. The next examinations are duplex ultrasonography of the lower extremities followed by a 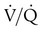 radionuclide study or a CT angiography scan of the chest. DVT should be sought in patients diagnosed with PE to investigate the origin of the thrombus and because patients with PE found to have a coexisting DVT are at increased risk for mortality.59 The presence of a “normal” PaO2 of 85 mm Hg in this patient may be misleading. The wide alveolar-arterial gradient probably is caused by the presence of a pulmonary embolus. The patient needs to be anticoagulated (e.g., with heparin, continuous intravenous drip) followed by warfarin.
radionuclide study or a CT angiography scan of the chest. DVT should be sought in patients diagnosed with PE to investigate the origin of the thrombus and because patients with PE found to have a coexisting DVT are at increased risk for mortality.59 The presence of a “normal” PaO2 of 85 mm Hg in this patient may be misleading. The wide alveolar-arterial gradient probably is caused by the presence of a pulmonary embolus. The patient needs to be anticoagulated (e.g., with heparin, continuous intravenous drip) followed by warfarin.
Other diagnostic modalities have been used to make the diagnosis of PE. Magnetic resonance imaging (MRI) has been suggested as an alternative noninvasive method for confirming the presence or absence of DVT. The sensitivity, specificity, and accuracy of MRI all are approximately 97%.60 MRI with radial pulse acquisition seems accurate in the diagnosis of acute DVT. Because of respiratory and cardiac motion artifacts and suboptimal resolution from the adjacent air-containing lung, MRI may not be useful in the acute setting.61
Treatment
Prophylaxis
Prophylactic therapy reduces the risk of venous thromboembolism in patients at risk. The frequency of proximal DVT ranges from 2% to 4% among general surgical patients undergoing minor surgery to 40% to 80% among patients at the highest risk, such as patients who have undergone hip or knee surgery.62 Patients at moderate to high risk include patients with acute spinal cord injury, myocardial infarction, ischemic stroke, or other medical conditions such as heart failure and pneumonia.62 Patients admitted to medical intensive care units are another group at risk for DVT; 33% of these patients have been found to have DVT.63 Compliance in the use of prophylaxis varies between 28% and 100%.62
Pharmacologic choices for prophylaxis include low-dose subcutaneous heparin, low-molecular-weight heparin, and fondaparinux.64–66 Mechanical measures to reduce venous stasis include early ambulation, wearing elastic stockings, pneumatic calf compression, and electrical stimulation of calf muscles. Mechanical methods are reserved for patients with contraindications to anticoagulant thromboprophylaxis.67 Current prophylactic strategies for DVT and PE are summarized in Table 26-5. Most hospitalized patients who are immobile need prophylaxis for venous thromboembolism.
TABLE 26-5
Thromboembolism Risk and Recommended Thromboprophylaxis in Hospitalized Patients
| Risk | DVT Risk Without Prophylaxis | Suggested Option | |
| Low | (a) Minor surgery in mobile patient | <10% | (a) No specific prophylaxis |
| (b) Medical patients fully mobile | (b) Early and aggressive ambulation | ||
| Moderate | (a) Most general, open gynecologic or urologic surgery | 10%-40% | (a) LMWH, UF heparin, or fondaparinux |
| (b) Medical patients, bed rest or sick | (b) LMWH, UF heparin, or fondaparinux | ||
| (c) High bleeding risk | (c) Mechanical prophylaxis | ||
| High | (a) Hip or knee arthroplasty, major trauma, hip fracture, and spinal cord injury | 40%-80% | (a) LMWH, fondaparinux, warfarin (INR 2-3) |
| (b) High bleeding risk | (b) Mechanical prophylaxis | ||
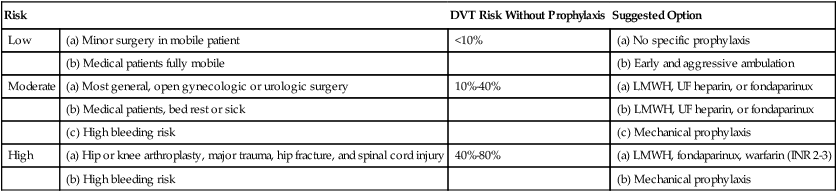
INR, International normalized ratio; LMWH, low molecular weight heparin; UF, unfractionated.
Modified from Geerts WH, Bergqvist D, Pineo GF, et al: Prevention of venous thromboembolism. American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 133:381S–453S, 2008.
 Rule of Thumb
Rule of Thumb
Most hospitalized patients who are immobile need prophylaxis for venous thromboembolism.
Management of Venous Thromboembolism
Anticoagulation
Heparin is the standard therapy for venous thromboembolic disease. Unfractionated heparin is the time-honored drug treatment, but low-molecular-weight heparin (e.g., enoxaparin) is widely used and has been endorsed in some guidelines as first-line therapy.68 Heparin has an immediate action and is relatively safe. It potentiates the action of antithrombin and heparin cofactor 2 and in this way inactivates thrombin, factor IXa, and factor Xa. Heparin does not lyse existing clots but prevents formation and propagation of new clots. Unfractionated heparin should be administered as a bolus followed by a continuous infusion.62 Low-molecular-weight heparin is administered subcutaneously, once or twice a day, and does not characteristically require blood test monitoring to ensure therapeutic benefit.68
The heparin regimen should be selected to maximize its antithrombotic effect without increasing the risk of bleeding. It is very important to achieve a therapeutic effect in the first 24 to 48 hours of starting therapy. The goal of unfractionated heparin therapy is to maintain an activated partial thromboplastin time greater than 1.5 times the control value.69 Clinical recurrence of DVT and PE is rare when heparin is infused at doses of at least 1250 units/hr.69 The fastest way to achieve a therapeutic heparin effect is to follow an established nomogram. Several nomograms are available, and one is shown in Table 26-6.69–71 These nomograms have been well accepted by clinicians and have led to aggressive heparin dosing and improvement in intermediate outcome. The use of nomograms has been associated with decreasing time to achieve therapeutic activated partial thromboplastin time (85% to 90% of patients achieve a therapeutic level within 24 hours) and a decrease in the variance of these parameters without any changes in bleeding rate.71 The complications of intravenous heparin administration include major bleeding (3.8%) and thrombocytopenia caused by IgG antiheparin antibodies (2.5% to 3% of patients to whom heparin is given therapeutically, <0.5% of patients to whom it is given prophylactically). If thrombocytopenia or bleeding occurs, heparin should be discontinued promptly.
TABLE 26-6
Weight-Based Nomogram for Administration of Heparin
| Initial dose | 80 U/kg bolus, then 18 U/kg/hr |
| aPTT < 35 sec (<1.2× control value) | 80 U/kg bolus, then 4 U/kg/hr |
| aPTT 35-45 sec (1.2-1.5× control value) | 40 U/kg bolus, then 2 U/kg/hr |
| aPTT 46-70 sec (1.5-2.3× control value) | No change |
| aPTT 71-90 sec (2.3-3× control value) | Decrease infusion rate by 2 U/kg/hr |
| aPTT > 90 sec (>3× control value) | Hold infusion 1 hr, then decrease infusion rate by 3 U/kg/hr |
aPTT, Activated partial thromboplastin time.
Note: Doses are calculated on actual body weight.
Modified from Raschke RA, Gollihake B, Pierce JC: The effectiveness of implementing the weight-based heparin nomogram as a practice guideline. Arch Intern Med 156:1645, 1996.
Heparin should be given for a minimum of 5 to 7 days.69 Oral anticoagulation can be started at the same time, not before, as the initiation of heparin.72 Coumarin derivatives are drugs that inhibit the formation of vitamin K–dependent factors II, VII, IX, and X. The coumarin derivative warfarin sodium is the most commonly used oral anticoagulant. Warfarin should be overlapped with heparin for a minimum of 5 days and until the international normalized ratio has been therapeutic for at least 24 hours to achieve a full antithrombotic effect.72 Warfarin should not be used solely for the initial management of venous thromboembolism because the peak effect is delayed for at least 72 to 96 hours. Because warfarin also decreases production of proteins C and S, a relative hypercoagulable state may occur in the first 24 hours as a result of the depletion of these proteins. The loading dose of warfarin varies between 5 mg/day and 10 mg/day. The 5-mg/day dose produces a lesser degree of anticoagulation and avoids the development of a potential hypercoagulable state caused by the decrease in the level of protein C during the first 36 hours of therapy.73
Management of Deep Venous Thrombosis
Patients generally need treatment with oral anticoagulants for 6 months, although in patients with transient risk of DVT (postoperative period), a 4- to 6-week course of anticoagulation may be adequate.69,74 Patients who need therapy for more than 6 months include patients with idiopathic venous thromboembolism. Patients who need therapy for more than 1 year or for life are patients with a history of cancer, anticardiolipin antibody, or antithrombin deficiency.69
Low-molecular-weight heparin, as a single dose or twice a day given via the subcutaneous route, is the suggested therapy for proximal DVT if no contraindication exists.72 This agent has been shown to be as effective and as safe as, and less expensive than, intravenous heparin therapy. In selected patients, low-molecular-weight heparin can be administered at home in an efficacious and safe way that can potentially decrease the number of days of hospital admission for acute DVT.69 It has been suggested that $250 million could be saved annually in the United States if patients were treated in the outpatient setting. Patients chosen for outpatient therapy should be in stable condition, should have a low risk of bleeding, and should not have renal insufficiency. At-home administration of low-molecular-weight heparin should be closely supervised.
The role of thrombolytic therapy with streptokinase, urokinase, or tissue plasminogen activator is not well defined in the management of acute DVT. Administration of early thrombolytic therapy decreases the pain and the incidence of postphlebitic syndrome, but the risks and benefits of this therapy are not well established.69 A systematic review of the efficacy and the safety of the use of recombinant tissue plasminogen activator in the management of lower extremity DVT did not support the routine use of this medication.75 It may be indicated in patients with massive proximal DVT and high risk of limb gangrene.72 Knowledge of the patient’s values and preferences must be used to guide the best decision.76
Management of Pulmonary Embolism
The management of PE depends on the extent of PE and the status of the cardiopulmonary system. Therapy with unfractionated or low-molecular-weight heparin followed by oral coumarin in a regimen similar to that for acute DVT is the treatment of choice. When the heparin effect is therapeutic within the first 24 hours, the risk of recurrent PE, which is associated with higher mortality, is decreased.77
Patients with an acute pulmonary embolus need additional supportive measures. Supplemental O2 should be administered to patients who have hypoxemia, and adequate analgesia should be prescribed for patients who have pain and anxiety. Resuscitation with fluids and vasopressor agents is necessary for patients who develop hypotension and shock. The vasopressors of choice include agents that may reduce pulmonary vascular resistance and increase cardiac output, such as norepinephrine and dopamine.78,79 Anticoagulation prevents further clot formation but does not lyse existing thromboemboli or decrease thrombus size. In the care of patients with severe hypoxemia, acute right heart failure, or shock, thrombolytic therapy may be administered for lysis of the emboli. Persistent hypotension secondary to massive PE is the most commonly accepted indication for thrombolytic therapy; however, no major trial has conclusively shown a mortality benefit of this intervention.72 When thrombolytic therapy with streptokinase and urokinase is used, heparin should not be infused concurrently. However, the use of heparin is optional in the treatment of patients receiving tissue plasminogen activator or reteplase.69
Other options in the care of a patient with confirmed massive PE, in whom thrombolysis is either contraindicated or unsuccessful, include pulmonary embolectomy, catheter tip embolectomy (physical removal of the embolism), and catheter tip fragmentation. Because of associated risks, these techniques should be used in centers with appropriate experience.43 Patients who have undergone attempts at embolectomy and catheter extraction have had massive embolism and shock.69
For patients in whom anticoagulation is contraindicated (e.g., because of bleeding risk), placement of a filter into the inferior vena cava to prevent movement of clot from the lower extremities to the pulmonary arteries is a treatment option. Another reason for placing an inferior vena cava filter is that a recurrent embolism has occurred despite adequate anticoagulation or that the patient has experienced multiple past emboli and is considered to be unable to tolerate another pulmonary embolus. Filter placement reduces the risk of PE in the period immediately after insertion but is associated over the longer term with a higher incidence of recurrent DVT.69,80
Pulmonary Hypertension
Pulmonary hypertension is defined by an elevation in mean pulmonary artery pressure greater than 25 mm Hg at rest.81 Pulmonary hypertension is grouped in five categories; this classification that was updated in 2008 by the Fourth World Symposium on Pulmonary Hypertension in Dana Point, California (Box 26-2).82 The importance of the clinical system, outside of allowing a better understanding of pathophysiology, is to give a framework for understanding important branch-points in the management and treatment of different conditions known to cause pulmonary hypertension. The first category, pulmonary artery hypertension (PAH), is characterized by an elevation in pulmonary artery pressure associated with high pulmonary vascular resistance (≥3 Wood units) and normal left ventricular filling pressures (pulmonary artery occlusion pressure ≤15 mm Hg). PAH may be associated with several conditions, including congenital heart disease, collagen vascular disease, cirrhosis of the liver, viral infections (e.g., HIV), and drugs and toxins (diet pills or anorexiants).82 In patients in whom no underlying etiology of pulmonary hypertension can be identified, the disease is referred to as idiopathic pulmonary arterial hypertension (IPAH), previously known as primary pulmonary hypertension.83–86 Pulmonary hypertension can also develop as a consequence of PE, and this entity is known as chronic thromboembolic pulmonary hypertension.87
Pathogenesis
The initial event of IPAH is probably an insult to the pulmonary endothelium (the cells that line the blood vessel). A genetic predisposition is probably necessary. The damage to the endothelium alters the balance between vasoconstrictive mediators such as thromboxane and endothelin I and vasodilators such as nitric oxide and prostacyclin, and vasoconstriction results. Vasoconstriction may not be the primary event, but it is an important component in the pathogenesis of IPAH.88–90 In addition to vasoconstriction, inflammation, thrombosis, cell proliferation, apoptosis, and fibrosis lead to pulmonary vascular remodeling and irreversible PAH.91 More recent research suggests the presence of other potential pathways involved in the pathogenesis of PAH, including downregulation of potassium channels,92 increased matrix metalloproteinases,93 decreased vasoactive intestinal peptide,94 elevated serotonin,95 transforming growth factor beta,96 and others.97 Potential new biomarkers and lines of therapies could result from these discoveries.83–86,88–90,98
Epidemiology and Clinical Findings
IPAH is more common among women than among men, with a ratio of 3 : 1. Approximately 7% of all cases are heritable. IPAH can occur at any age, although it is more common from ages 20 to 50 years. On average, the diagnosis of IPAH is delayed for 2 years after the onset of IPAH. The condition frequently is misdiagnosed as anxiety or depression because it is characterized by onset of vague symptoms and hyperventilation. The most common initial symptom is dyspnea (60% of patients). Angina, probably caused by underperfusion of the right ventricle or stretching of the large pulmonary arteries, is present in approximately 50% of patients. Syncope (passing out) is present in 8% of patients as an early symptom. Other symptoms include cough, hemoptysis, hoarseness, and Raynaud phenomenon (blanching of the fingers on exposure to cold) in approximately 10% of patients. Physical findings associated with IPAH include a loud second heart sound and a right-sided third or fourth heart sound. Other common signs are a palpable right ventricular heave and impulse of the pulmonary artery and both pulmonary ejection and pulmonary tricuspid regurgitation murmurs. Signs of right ventricular failure are common. Cyanosis often is present as a result of low cardiac output or the presence of an intracardiac right-to-left shunt that occurs as cor pulmonale develops. Clubbing does not occur in IPAH. Findings on chest radiograph include enlargement of the main and hilar pulmonary arteries, “pruning” (or narrowing) of the peripheral arteries, enlargement of the right ventricle and atrium, and pleural effusion, although the chest radiograph may remain normal in 6% of patients. In pulmonary venoocclusive disease, a histopathologic type of IPAH, there is an increase in vascular markings, and so-called Kerley B lines may be present on the chest radiograph (thin lines that represent congested pulmonary lymphatics that extend from the pleural surface into the lung).83–86
Diagnosis
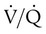 scan, CT angiography, and pulmonary artery catheterization.
scan, CT angiography, and pulmonary artery catheterization.Laboratory tests include complete blood count, HIV, rheumatologic panel, and liver function testing. These tests help identify conditions associated with PAH. ECG findings usually include right-axis deviation, right ventricular hypertrophy, and strain. Pulmonary function tests are useful to rule out the presence of significant restrictive or obstructive airway disease. The most common abnormality on pulmonary function testing of patients with IPAH is a low carbon monoxide diffusing capacity (DLCO), associated with normal pulmonary mechanics. The echocardiogram may show dilation of the right ventricle and right atrium and tricuspid regurgitation (Figure 26-4).
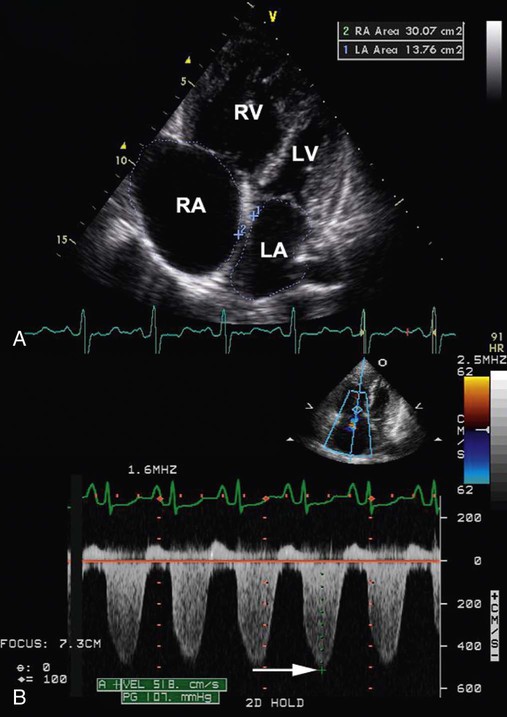
 lung scan, which helps to rule out the possibility of chronic thromboembolic pulmonary hypertension, a mimic of IPAH that has different treatment. In patients with IPAH, the
lung scan, which helps to rule out the possibility of chronic thromboembolic pulmonary hypertension, a mimic of IPAH that has different treatment. In patients with IPAH, the  scan may be normal or show only patchy subsegmental defects. In patients with chronic thromboembolic pulmonary hypertension, the
scan may be normal or show only patchy subsegmental defects. In patients with chronic thromboembolic pulmonary hypertension, the  scan shows segmental defects; in these cases, confirmation of chronic thromboembolic pulmonary hypertension requires pulmonary angiography. High-resolution CT is helpful to rule out associated etiologies and may be useful in evaluating the small group of patients with chronic interstitial disease and normal chest radiographs.
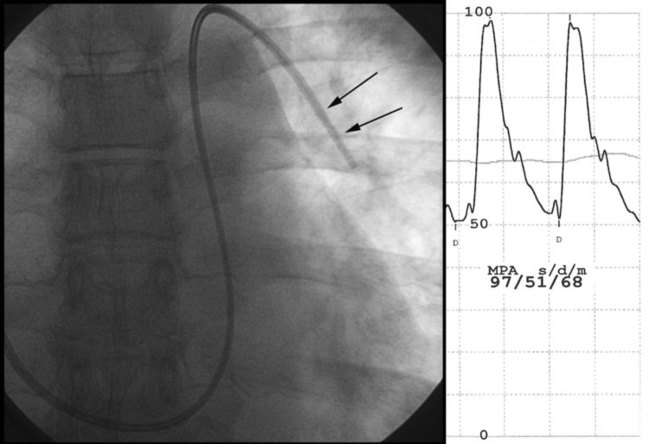
scan shows segmental defects; in these cases, confirmation of chronic thromboembolic pulmonary hypertension requires pulmonary angiography. High-resolution CT is helpful to rule out associated etiologies and may be useful in evaluating the small group of patients with chronic interstitial disease and normal chest radiographs.Right heart catheterization is required to confirm the diagnosis and determine the degree of hemodynamic impairment, the presence of vasoreactivity, and the prognosis of patients with PAH (Figure 26-5). Patients with severe degrees of pulmonary hypertension, high right atrial pressure, and low cardiac output have a very poor prognosis.14,83–86
Management of Pulmonary Hypertension
IPAH can be life-threatening and is associated with a poor prognosis. Without therapy, only 33% of patients are alive 5 years after the onset of the disorder. During the past decade, treatment has improved considerably.99–104 Current treatment options include using calcium channel blockers, prostanoids, endothelin receptor antagonists, and phosphodiesterase 5 (PDE5) inhibitors (discussed later).
General Measures
Oral anticoagulation improves survival in IPAH and is recommended in all patients with IPAH unless there is a contraindication to anticoagulation.105 The recommended target international normalized ratio is approximately 2. The role of anticoagulation in other forms of PAH is less clear. Supplemental O2 should be used to maintain O2 saturation greater than 90%, especially because hypoxemia is a major cause of pulmonary vasoconstriction. Diuretics are indicated for right ventricular volume overload, and digoxin is reserved for patients with refractory right ventricular failure and for rate control in atrial flutter or fibrillation.103,104
Calcium Channel Blockers
Patients with IPAH who respond to vasodilators in the short-term have improved survival with long-term use of calcium channel blockers. These agents should be considered in all patients who have significant and definite response to a short-acting vasodilator such as nitric oxide. Nitric oxide is the preferred agent for pulmonary vasodilator testing because its half-life is very short, it does not affect cardiac output, and it enhances 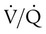 matching.106 It is usually administered by mask at 10 to 40 parts per million for 2 to 5 minutes.106 Only a small fraction of patients with IPAH qualify for and benefit from long-term therapy with oral calcium channel blockers.103,104
matching.106 It is usually administered by mask at 10 to 40 parts per million for 2 to 5 minutes.106 Only a small fraction of patients with IPAH qualify for and benefit from long-term therapy with oral calcium channel blockers.103,104
Prostanoids
Prostanoids available for treating patients with pulmonary hypertension, especially IPAH, include epoprostenol, treprostinil, and iloprost. Epoprostenol, delivered via continuous intravenous infusion, improves exercise capacity, hemodynamic variables, and survival in patients with IPAH and is the treatment of choice for severely ill patients.107 Epoprostenol therapy is complicated, however, by the instability of the drug at room temperature and the need for continuous intravenous infusion because of the short half-life of the drug. Common side effects include headache, flushing, jaw pain, diarrhea, nausea, skin rash, and musculoskeletal pain. Catheter-related complications include infection, sometimes serious (e.g., bacteremia), and thrombosis. By changing the buffer, a thermostable epoprostenol was developed and has been approved for clinical use by the U.S. Food and Drug Administration (FDA).
Another prostanoid, treprostinil, is a stable prostacyclin analogue with a longer half-life, allowing for subcutaneous,108 intravenous,109 or inhaled delivery.110 In addition to side effects seen with epoprostenol, patients receiving treprostinil subcutaneously may experience pain at the infusion site. Inhaled treprostinil is administered by using the Tyvaso Inhalation System (ultrasonic, pulsed-delivery device). It is initially dosed at 3 inhalations four times a day. If this dose is tolerated, it may be increased up to 9 inhalations four times a day.
Iloprost is a stable prostacyclin analogue that can be delivered via inhalation and is an effective therapy for PAH.111 Because of the relatively short duration of action of inhaled iloprost, it needs to be administered as 1 to 2 inhalations six to nine times a day. For its administration, the I-neb adaptive aerosol delivery (AAD) or Prodose AAD system should be used. Common side effects include cough, flushing, and headache. Inhaled iloprost may be useful as an adjunct to oral therapy. The main impediment to the use of prostanoids has been the route of delivery. The development of orally administered prostanoids is under way.99,100,103,104
Endothelin Receptor Antagonists
Endothelin antagonists represent another class of medications available for treating pulmonary hypertension. Bosentan, an orally administered nonselective endothelin-1 receptor antagonist, improves walking distance, hemodynamic variables, and functional class in patients with PAH.112 The main side effect is the asymptomatic increase in hepatic aminotransferase levels, which necessitates monitoring liver function at least monthly in all patients receiving the medication. Ambrisentan is a selective type A endothelin-1 receptor antagonist that is also beneficial in patients with PAH. Its main side effect is peripheral edema.113,114 All endothelin receptor antagonists are potent teratogens, and meticulous contraception must be observed by patients receiving these medications.
Phosphodiesterase 5 Inhibitors
Sildenafil, a PDE5 inhibitor, reduces pulmonary artery pressure and is effective in the treatment of pulmonary hypertension.101 By inhibiting PDE5, sildenafil stabilizes cyclic guanosine monophosphate (cGMP), the second messenger of nitric oxide, allowing a more sustained effect of endogenous nitric oxide, an indirect but effective and practical way of using the nitric oxide–cGMP pathway. Tadalafil, a long-acting PDE5 inhibitor, also improves outcomes in PAH and has some differences in the acute effects compared with sildenafil.115,116 These medications are usually well-tolerated; rarely, patients can have vision or hearing loss, priapism, and hypotension.
Surgical Therapy
Atrial Septostomy
The role of balloon atrial septostomy in the treatment of patients with PAH is uncertain. Septostomy might be beneficial in the setting of severe disease with recurrent syncope or right heart failure despite maximal medical therapy. The procedure can also be used as a palliative bridge to lung transplantation. The rationale for its use is that the controlled creation of an atrial septal defect would allow right-to-left shunting, leading to increased systemic output and systemic O2 transport despite the accompanying decrease in systemic arterial O2 saturation. The shunt at the atrial level would also allow decompression of the right atrium and right ventricle, alleviating signs and symptoms of right heart failure. Balloon atrial septostomy is a high-risk procedure and should be performed only in experienced centers to reduce the procedural risks.103
Lung Transplantation
Single-lung or double-lung transplantation has been used successfully in the treatment of patients with IPAH. Patients who undergo lung transplantation have an immediate decrease in pulmonary artery pressure at the time of surgery and rapid improvement in right heart function despite severe preoperative cor pulmonale.103 This option is reserved for special cases of patients not responsive to medical treatment who have an indicator of poor prognosis (syncope, refractory right heart failure, function class III/IV, or severe hypoxemia).117 Perioperative mortality for transplantation is higher in PAH, but after this period some patients have an excellent response with dramatic improvements in symptoms and quality of life.118 Although lung transplantation is an alternative in the treatment of patients with IPAH, the disadvantages of transplantation are the need for lifelong immunosuppression and the morbidity and mortality of lung transplantation, which increase over time. The survival rate 3 years after lung transplantation is approximately 60%. By the time patients with PAH and CHD are considered for transplantation, they are usually poor candidates because of multiple organ system failures.
 Rule of Thumb
Rule of ThumbPulmonary Hypertension in Chronic Lung Disease
Pulmonary hypertension is a frequent complication of chronic pulmonary disease (see Chapter 23). Approximately 50% of elderly patients with COPD have pulmonary hypertension with significant reduction in survival and quality of life. The pulmonary hypertension associated with COPD is multifactorial. Loss of vascular surface caused by destruction of lung parenchyma, compression of the vascular bed as a result of hyperinflation, hyperviscosity of the blood as a result of polycythemia, and left ventricular dysfunction are important contributory factors. Alveolar hypoxia, because of its potent pulmonary vasoconstrictive effect, is probably the most important factor contributing to pulmonary hypertension in patients with COPD. Sustained alveolar hypoxia causes pulmonary vasoconstriction and eventually medial hypertrophy, fibrosis of the intima, and narrowing of the lumen of the pulmonary blood vessels. The increases in pulmonary artery pressure and vascular resistance lead to an increase in the afterload of the right ventricle with dilation and hypertrophy in an effort to maintain the cardiac output.
Patients with COPD may have worsening of dyspnea and a decrease in exercise tolerance without a change in the degree of airway obstruction. The presence of pulmonary hypertension in patients with COPD correlates with the severity of the disease. Patients with severe hypoxemia (PaO2 < 55 mm Hg) may have more elevated pulmonary artery pressures, although the mean pulmonary artery pressure owing to COPD alone rarely exceeds 35 to 40 mm Hg.119–122 Patients with mean pulmonary artery pressure higher than this have a poor prognosis.123
 Problem
Problem scan or pulmonary angiogram can exclude chronic thromboembolic disease, a CT scan of the chest and pulmonary function tests can help determine the presence of parenchymal lung disease, and blood serologic tests can be used to evaluate for connective tissue disease. A 6-minute walk test may help in determining the functional capacity of the patient and assess the patient’s response to treatment. Several treatment options are currently available for patients with pulmonary hypertension. The best option depends on the underlying diagnosis (if any) and the severity of the disease.
scan or pulmonary angiogram can exclude chronic thromboembolic disease, a CT scan of the chest and pulmonary function tests can help determine the presence of parenchymal lung disease, and blood serologic tests can be used to evaluate for connective tissue disease. A 6-minute walk test may help in determining the functional capacity of the patient and assess the patient’s response to treatment. Several treatment options are currently available for patients with pulmonary hypertension. The best option depends on the underlying diagnosis (if any) and the severity of the disease.O2 therapy is the main treatment that improves survival among patients with COPD and pulmonary hypertension, although smoking cessation and lung volume reduction (in selected individuals) may also confer survival benefits in patients with COPD. Vasodilator agents used for IPAH could potentially be used in these patients, but the results of large clinical trials are not yet available.121,123
[/level-membership-for-pulmolory-and-respiratory-category][not-level-membership-for-pulmolory-and-respiratory-category]
Pulmonary Vascular Disease
After reading this chapter you will be able to:
State how many patients develop venous thromboembolism each year.
Describe how and where thromboemboli originate.
Describe how pulmonary emboli alter lung and cardiac function.
Describe how pulmonary embolism is diagnosed and managed.
Describe hemodynamic findings associated with pulmonary hypertension.
State patients who are at risk for the development of IPAH.
Identify the clinical features associated with IPAH.
Venous Thromboembolic Disease
Venous thromboembolism is a major health problem in the United States. The prevalence of venous thromboembolism, which includes PE and DVT, has remained relatively constant over time and has been calculated to be 117 cases per 100,000 persons (i.e., DVT at 48 cases per 100,000 and PE at 69 cases per 100,000). It is estimated that 200,000 to 300,000 new cases occur yearly in the United States.1
Venous thromboembolism is treatable but requires prompt diagnosis and therapy to avert serious consequences. One-third of deaths from PE occur within 1 hour of onset of symptoms, and more than 70% of patients who die of PE are not suspected to have PE before death.2 Although mortality from PE has decreased in recent years,3 the death rate for the first episode of PE among hospitalized patients may be 17.4% at 3 months.3,4 Recurrent PE is associated with a much higher mortality rate because only one-quarter of patients survive 3 months.5 The diagnosis is not suspected in approximately two-thirds of patients who die of PE, and the frequency of recognizable emboli in routine autopsies of adult patients ranges from 1.5% to almost 30%.6–8 In a population-based study of PE as a cause of death in New Mexico, only 34% of 812 postmortem documented cases of PE were diagnosed before death.5 Morpurgo and Schmid9 reported their experience with 92 postmortem cases of massive or submassive PE detected during the years 1986-1989. Only 28% of the cases were diagnosed before death, a finding that emphasizes the underdiagnosis of venous thromboembolic disease.
Two-thirds of cases of initial embolus from which patients survive remain undiagnosed. The mortality rate among these patients with undiagnosed embolism is approximately 30%,10 which emphasizes the importance of recognizing PE. If venous thromboembolism is recognized and treated, the mortality rate decreases to less than 8%, and the long-term outcome is generally favorable.11 The long-term survival rates after venous thromboembolism in an inception cohort of 2218 patients were 72% at 1 month and 63% at 1 year.12
Because the accuracy of the clinical impression (i.e., without testing) of venous thromboembolism is less than 50%,13 objective tests are needed to confirm or exclude the diagnosis. Patients with multiple injuries, immobilization, bed rest, or intravascular catheters and elderly patients are at high risk of venous thromboembolic disease (Table 26-1) and should be considered candidates for testing when appropriate symptoms develop.
TABLE 26-1
Frequency of Venous Thrombosis in Various Hospitalized Patient Groups
| Group | Frequency (%) |
| Orthopedic (e.g., fractured hip) | 54-67 |
| Urologic (e.g., prostatectomy) | 25 |
| Surgical patients >40 yr old | 28 |
| Gynecologic surgery | 18 |
| Cardiovascular surgery (e.g., acute myocardial infarction) | 39 |
| Obstetrics | 3 |
From Arroliga AC, Matthay MA, Matthay RA: Pulmonary thromboembolism and other pulmonary vascular diseases. In George RB, et al, editors: Chest medicine: essentials of pulmonary and critical care medicine, ed 3, Baltimore, 1995, Williams & Wilkins.
Pathogenesis
Pulmonary emboli arise from detached portions of venous thrombi that form, in most cases, in deep veins of the lower extremities or pelvis (86%); the point of origin of pulmonary emboli is actually found in only one-half of patients.9 A small percentage of pulmonary emboli arise from the right-sided heart chambers (3.15%) or the superior vena cava (3%).9
Conditions that favor thrombus formation include blood stasis, the presence of hypercoagulable states, and vessel wall abnormalities (factors known as Virchow’s triad). Causes of blood stasis include local pressure, venous obstruction, and immobilization. Other causes of stasis include congestive heart failure, shock and dehydration, varicose veins, and enlargement of the right heart chambers. Several conditions enhance the intravascular coagulability of the blood and predispose to venous thromboembolic disease (Box 26-1).14 The most frequent causes of an inherited hypercoagulable state are the factor V Leiden mutation and the prothrombin gene mutation, which together account for 50% to 60% of cases.15 The major acquired risk factors for venous thromboembolism include recent major surgery, trauma, immobilization, antiphospholipid antibody syndrome, malignancy, pregnancy, oral contraceptives, and myeloproliferative disorders.16 Vessel wall abnormalities are found most often in patients who have sustained trauma or have undergone major surgery.
Pathology
Stasis, an important factor for the formation of DVT, is rarely the only risk factor.17 Deposition of platelets and fibrin in the venous valve cups of the lower extremities occurs as a result of stasis. The combination of diminished blood flow and the presence of trauma and toxins can worsen endothelial damage and promote the release of mediators that encourage adhesion, aggregation, and degranulation of platelets. The result is activation of the coagulation cascade and production of thrombi and fibrin.
PE is a frequent complication of DVT, occurring in more than 50% of cases with DVT confirmed on phlebography.18 PE occurs when a fragment of the thrombus in the venous system travels to the pulmonary circulation. Pulmonary emboli occur more frequently in the lower lobes and are more often found in the right lung than in the left lung, a phenomenon probably related to the flow distribution that favors the right lung and the lower lobes.6 Embolism to the pulmonary circulation produces pulmonary hemorrhage in the ischemic or infarcted lung in less than 10% of cases of PE. Infarction, secondary to thromboembolism, is less common in the lung than in other tissues because the lung has two blood supplies: the pulmonary arterial circulation and the bronchial circulation. At a capillary level, extensive connections exist within the pulmonary and bronchial circulations that prevent serious damage to lung tissue deprived of its pulmonary artery supply.6 Patients with underlying cardiovascular disease may have impairment of the remaining bronchial circulation with resultant lung tissue necrosis when emboli occur.
Pulmonary infarction is associated with thromboembolic obstruction of a medium-sized pulmonary artery. Generally, infarcts occur at the lung bases, are pleural-based, and may be accompanied by pleural effusion. Microscopic examination of the lung in pulmonary infarction shows necrosis of alveolar walls, alveoli filled with red blood cells, and a mild inflammatory response in the periphery.6
Pathophysiology
The sudden obstruction of a pulmonary arterial branch causes a decrease in or total cessation of blood flow to the distal area of the lung that leads to respiratory and hemodynamic alterations.19 In the appropriate context, massive PE should be suspected anytime there is unexplained hypotension accompanied by an elevated central venous pressure (jugular vein distention).20 It is a catastrophic entity that frequently results in acute right ventricular failure and death. Death from massive PE is the result of cardiovascular collapse rather than of respiratory failure.
Embolic obstruction of the pulmonary artery increases the alveolar dead space, causes bronchoconstriction, and decreases the production of alveolar surfactant. Wasted or dead space areas occur when areas of the lung parenchyma are ventilated but not perfused. The response is to increase total ventilation (). The increased contributes to the sensation of dyspnea that accompanies PE. Bronchoconstriction from diminished carbon dioxide concentration, regional hypoxia, and the production of serotonin and histamine cause further ventilation/perfusion () mismatching.21
Not all patients with PE have significant arterial hypoxemia, but the presence of a widened alveolar-arterial oxygen (O2) tension gradient and reduced PaO2 are common. Hypoxemia develops because of mismatch, intrapulmonary shunt, and cardiogenic shock. Shock is caused by obstruction of the pulmonary vasculature by massive emboli or by numerous small emboli in the presence of cardiopulmonary disease. Cardiac output decreases, and O2 delivery declines. With the decrease in O2 delivery, the peripheral tissues increase O2 extraction causing venous O2 desaturation. In patients with significantly increased right heart pressures, intracardiac right-to-left shunt may develop when blood flows through a patent foramen ovale.19,21 In addition, the depletion of surfactant material as a result of embolic occlusion can lead to atelectasis and intrapulmonary shunt, which can cause hypoxemia.19
The main hemodynamic consequence of PE is increased resistance to blood flow caused by obstruction of the pulmonary arterial bed. The hemodynamic consequences are determined by the extent of the cross-sectional area of the pulmonary circulation involved, the underlying cardiopulmonary reserve, and the neurohumoral response to the embolism. Pulmonary hypertension occurs when 50% of the pulmonary vascular bed has been occluded.19,21 To maintain the same flow at a higher pressure, the right ventricle must work harder. The result is an increase in right ventricular work that causes the right ventricle to become dilated and ischemic. The thin-walled right ventricle is not designed to work with acute heavy pressure loads. When the mean pulmonary artery pressure increases to greater than 40 mm Hg during an acute first PE, the right ventricle fails, and hemodynamic collapse and death occur.22 The exact role of vasoconstriction in the pathogenesis of pulmonary hypertension is uncertain, but vasoconstrictors such as serotonin and thromboxane A2 may also play a role in the development of pulmonary hypertension after acute PE.
Although the usual course of PE is to resolve rapidly (because the body lyses the embolism with endogenous fibrinolytic agents), permanent residual emboli do occur.23 Massive emboli are likely to resolve within weeks, particularly in young patients. Overall, less than 10% of patients have perfusion defects after 6 weeks. Vascular patency is restored when the unresolved emboli organize or form scars against the vessel wall.
Clinical Features
A high index of suspicion for venous thromboembolism is crucial to make the diagnosis for patients at risk. No specific signs or symptoms indicate the presence of venous thromboembolic disease, and a significant proportion of patients are asymptomatic (32%).2,24 The physical findings of DVT in the lower extremities include erythema and warm skin in one-third of patients and swelling and tenderness in three-fourths of patients. In patients who have swelling above and below the knee, fever, and a history of immobility and cancer, the likelihood of finding DVT on a venogram is only 42%.25
The most frequent symptoms in patients with confirmed PE are dyspnea, followed by pleuritic chest pain and cough (Table 26-2).26 The onset of dyspnea is usually rapid, within seconds (46%) or minutes (26%).26 Hemoptysis occurs in 13% to 20% of patients. The combination of dyspnea of sudden onset, fainting, and acute chest pain should raise suspicion of PE. In one study, this combination of symptoms was present in 96% of patients with confirmed PE compared with 59% of patients in whom PE was suspected but not confirmed.27 In some patients, dyspnea lasts only a few minutes, and this episode may be wrongly dismissed as being trivial.19,27–29
TABLE 26-2
Clinical Characteristics in Patients With Pulmonary Embolism and No Cardiopulmonary Disease
| Symptoms | Frequency (%) | Signs | Frequency (%) |
| Dyspnea at rest or with exercise | 73 | Tachypnea | 54 |
| Pleuritic pain | 44 | Tachycardia | 24 |
| Calf or thigh pain | 44 | Rales | 18 |
| Cough | 34 | Decreased breath sounds | 17 |
| Orthopnea | 28 | Loud P2 | 15 |
| Wheezing | 21 | Jugular venous distention | 14 |
Modified from Stein PD, Beemath A, Matta F, et al: Clinical characteristics of patients with acute pulmonary embolism: data from PIOPED II. Am J Med 120:871–879, 2007.
There are no characteristic physical findings of PE. The most frequent physical findings include tachypnea, rales on chest examination, and tachycardia. Similar to dyspnea, these signs may be short-lived. Other common physical findings include an accentuated pulmonary component of the second heart sound (loud P2) consistent with pulmonary hypertension. Fever may be present in 54% of patients.27–29 Similar to what occurs in the diagnosis of DVT, less than 35% of patients in whom PE is clinically suspected actually have it.2
Chest Radiograph
The chest radiograph cannot confirm the presence of PE but is helpful to rule out other potentially life-threatening conditions, such as pneumothorax or pneumonia, which can manifest in a similar way. In dyspneic patients, a normal chest radiograph may be a clue to the presence of PE; however, chest radiography is abnormal in more than 80% of cases. Abnormalities include enlargement of the right descending pulmonary artery (66%), elevation of the diaphragm (61%), cardiomegaly (55%), and small pleural effusion (50%). Parenchymal densities (patchy infiltrates or round nodular lesions) predominantly appearing next to the pleural surface are present in patients who have infarction or atelectasis. Other, less common findings include the Westermark sign, in which there is pulmonary hyperlucency caused by a marked reduction in blood flow. The so-called Hampton hump, an opacity in the costophrenic angle, is present in 25% to 30% of patients.27
Electrocardiogram
The electrocardiogram (ECG) is helpful to rule out other diagnoses, such as acute myocardial infarction and pericarditis. The ECG is frequently abnormal in patients with PE (87% of the time), but the ECG abnormalities accompanying PE are nonspecific in 70% to 75% of cases; tachycardia and ST segment depression are most common.27 Abnormalities such as T wave inversion in right precordial leads, depression of the ST segment, and T wave inversion in V1 and V2 may be present. A so-called S1Q3T3 pattern is associated with massive PE and is present in 19% of such patients.25
Arterial Blood Gases
Most patients with acute PE have hypoxemia and hypocapnia,27 but a significant percentage of patients (15% to 25%) with or without previous cardiopulmonary disease have a PaO2 exceeding 80 mm Hg.29 Although a widened alveolar-arterial O2 gradient is frequently present, a normal alveolar-arterial O2 gradient may occur in approximately 20% of patients with angiographically documented PE.27,29 Although an arterial blood gas measurement may be helpful in identifying patients with hypoxemia or hypocapnia accompanying PE, arterial blood gas measurements can never secure the diagnosis of PE.
Diagnostic Modalities
By-Products of Thrombin and Plasmin
Clot formation is invariably associated with thrombingeneration. Measurement of cross-linked fibrin split products (D-dimers) has been found to be sensitive for the diagnosis of acute venous thromboembolism. The specificity of D-dimer enzyme-linked immunosorbent assay (ELISA) can exclude all but 5% to 10% of patients with acute PE, so this test has been used as an important tool for early assessment.30 The specificity of the test is only 39%, but a value less than the recommended cutoff for current quantitative ELISA assays has been shown to rule out venous thromboembolic disease in 98% of patients.31–33 In patients in whom DVT is suspected clinically, negative results of the D-dimer assay combined with negative findings on impedance plethysmography have a negative predictive value of 98% for DVT (i.e., if the test is negative, the chance of a DVT is only 2%).31 D-dimer results have been particularly useful in the emergency department and outpatient area for the evaluation of patients with suspected DVT31 and PE.33 In patients with a low pretest probability of DVT or PE and a negative D-dimer result, the negative predictive value for the strategy has been greater than 99%.32,33
Although there are several laboratory methods to measure D-dimer levels, tests using ELISAs are the most widely used and best performing among the D-dimer assays regarding the sensitivity and negative likelihood ratio. For excluding PE or DVT, a negative result on quantitative rapid ELISA is as diagnostically useful as a normal lung scan or negative duplex ultrasonography finding. D-dimer ELISA can be used to exclude PE in outpatients with a low to moderate suspicion without the need for further costly testing. However, inpatients should undergo an imaging study as the initial test for PE because most will already have elevated D-dimer levels because of comorbid conditions.33
Testing for Lower Extremity Deep Venous Thrombosis
To evaluate the clinical pretest probability of DVT, the Wells criteria are frequently used. These criteria include the following clinical parameters: presence of cancer, immobilization, localized tenderness, swelling, edema, previous DVT, collateral superficial veins, and absence of an alternative diagnosis.34 In cases in which there is a moderate to high pretest probability, several modalities could be used for diagnosing DVT in the extremities, such as compression ultrasonography, impedance plethysmography, and venography. In patients with low pretest probability of DVT and a negative D-dimer, further testing may be unnecessary.31,35–37
Impedance Plethysmography
Impedance plethysmography is a noninvasive method and measures electrical impedance to blood flow, which changes with inflation and deflation of a lower extremity cuff. The quality of the test is operator-dependent and requires that the patient be supine and lay still for at least 2 minutes. The test has a sensitivity and specificity of 91% and 96% for symptomatic proximal venous thrombi. A lower sensitivity of 65% has been reported.38,39
Compression Ultrasonography
Compression ultrasonography has proved to be sensitive and specific for the diagnosis of symptomatic proximal DVT. This test is noninvasive, portable, and accurate and is the modality of choice for the diagnosis of DVT. Compression ultrasonography combines B-mode scanning with a tightly focused pulse Doppler beam directed at the vessels of interest. DVT is diagnosed with the findings of venous noncompressibility, an echogenic filling defect, absence of Doppler flow, free-floating thrombus in the vein, and venous distention.40 The most reliable sign of DVT is lack of compressibility of the vein, although a free-floating thrombus has the highest embolic potential (Figure 26-1). The sensitivity and specificity of compression ultrasonography in symptomatic patients vary between 95% and 100% for the detection of a proximal lower extremity thrombus.40,41 Areas not well visualized with compression ultrasonography include the iliac veins, the superficial femoral veins in the adductor canal, and the calf veins. However, the accuracy of compression ultrasonography, even with the addition of color Doppler ultrasonography, is moderate to low for the detection of DVT in patients at high risk who do not have symptoms.42 These results suggest that ultrasonography, although sensitive and specific for the diagnosis of DVT, is not a good screening test for patients at high risk who do not have symptoms.
Testing for Pulmonary Embolism
Noninvasive tests for the diagnosis of PE include scan and helical or spiral computed tomography (CT) angiography scan of the chest. Either of these tests, depending on the resources available, may be the initial diagnostic examination if the presence of acute PE is clinically suspected.43 Echocardiography can suggest the diagnosis (right ventricular dilation, dysfunction, or thrombus) and provide prognostic information.44 In certain cases when these noninvasive tests are nondiagnostic, pulmonary angiography may be needed to confirm or exclude the diagnosis of PE.
scanning involves the inhalation of a radiolabeled gas (usually xenon-133, xenon-127, krypton-181m, or technetium-99m) and the intravenous injection of macroaggregated albumin tagged with a gamma-emitting radioisotope. The distribution of lung ventilation () and lung perfusion () is studied, and areas of mismatch where is less than are sought. The presence of mismatches most often indicates embolic occlusion of the blood vessel, although other rare causes of mismatches exist, such as extrinsic compression of the vessel by a mass, intraluminal obstruction by angiosarcoma, or obliteration of a vessel by vasculitis. The addition of scan increases the specificity of scan.45 Generally, with the presence of a parenchymal abnormality, the defect coincides with the defect, and matched abnormalities are found. Normal results of a scan exclude the presence of a clinically significant PE in the context of a low clinical probability of PE. In these cases, anticoagulant therapy can be safely withheld.46 Abnormal scan results can be classified as high probability, intermediate (or indeterminate) probability, and low probability for PE, according to the size of the defect and the degree of mismatch between the scan and chest radiographic abnormalities.45 Diagnostic accuracy is greatest when scan results are combined with clinical probability (Table 26-3).47 The presence of concomitant cardiopulmonary disease (e.g., COPD), even if severe, does not diminish the diagnostic usefulness of scans in the diagnosis of acute PE.48–50
TABLE 26-3
| High probability | Two or more large (>75% of a segment) segmental defects without corresponding or abnormalities on chest radiograph |
| One large segment defect and two or more moderate (25%-75% of a segment) segmental defects without corresponding or abnormalities on chest radiograph |
|
| Four or more moderate segmental defects without corresponding or abnormalities on chest radiograph |
|
| Intermediate probability | One moderate or up to two large segment defects without corresponding defect or abnormalities on chest radiograph |
| Corresponding defects and parenchymal opacity in lower lung zone on chest radiograph |
|
| Corresponding defects and small pleural effusion |
|
| Single moderate matched defects with normal findings on chest radiograph |
|
| Findings difficult to categorize as normal, low, or high probability | |
| Low probability | Multiple matched defects, regardless of size, with normal findings on chest radiograph |
