Principles Governing Antimicrobial Therapy in the Intensive Care Unit
ADEQUACY OF INITIAL EMPIRIC ANTIBIOTIC THERAPY
Penetration at the Site of Infection: Tissue-Targeted Therapy
UNINTENDED CONSEQUENCES OF ANTIBIOTIC THERAPY
CLINICIAN RESPONSES TO MULTIDRUG RESISTANCE
Mechanisms of Action and of Resistance
The Clinical Relevance for Understanding Antibiotic Resistance
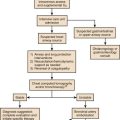
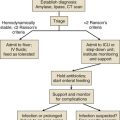
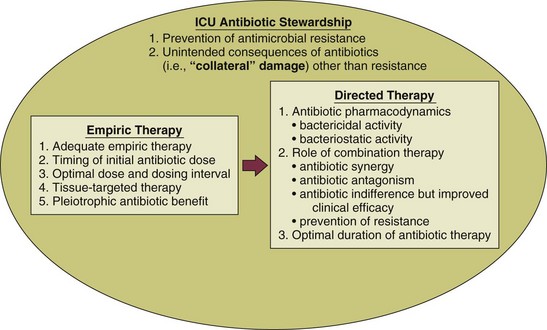
In the critical care setting, the selection of optimal antibiotic therapy often entails a two-stage process: empiric therapy, followed by directed therapy once the pathogen and type of infection are clearly identified. Figure 51.1 incorporates this progression in antibiotic therapy and summarizes some principles that contribute to the goal of preserving the antibiotic armamentarium while attempting to achieve optimal clinical efficacy—the components of antibiotic stewardship. A challenge for the critical care physician is to recognize that antibiotic therapy for critically ill patients has potential ramifications for other patients in that unit over the following weeks. If antibiotic-resistant organisms or organisms with certain virulence factors are selected by a pattern of antibiotic use, those pathogens can become part of the ecology of intensive care units (ICUs) and can then be transmitted to other patients. See Figure 51.2 for clinical examples of the principles of empiric therapy. In a joint 2007 guideline paper for the development of an institutional program to enhance antimicrobial stewardship, the Infectious Diseases Society of America (IDSA) and the Society for Healthcare Epidemiology of America (SHEA) wrote on the importance of education as an essential element in any program designed to influence prescribing behavior.1 This chapter focuses on the elements of antibiotic stewardship that play a clinically relevant role in the use of antibiotics prescribed to patients in ICUs. Knowledge of these variables is essential in clinical practice and can serve as the basis for the insights necessary for an enhanced level of antibiotic prescribing.
Adequacy of Initial Empiric Antibiotic Therapy
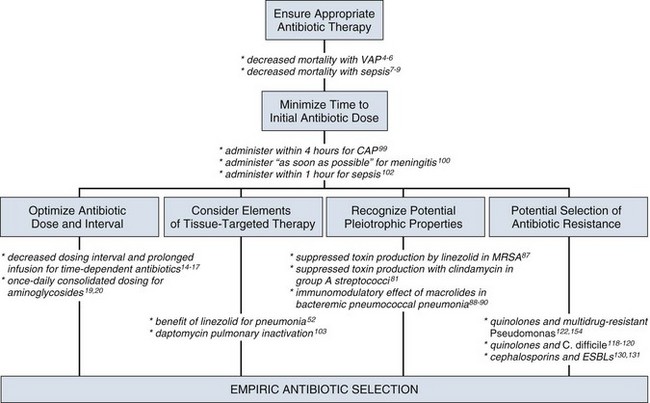
For many years clinicians felt that antibiotic therapy could be adjusted on day 2 or 3 into a clinical course, once either bacterial susceptibility was known or the clinical course of the patient had been defined, with no negative aspects of such changes. Beginning in the 1990s, several reports challenging this tenet were published regarding such infectious disease processes as sepsis and ventilator-associated pneumonia (VAP).2–8 In these reports investigators used the term inadequate to describe situations in which the organism causing an infection was not covered with antibiotic therapy as indicated by in vitro susceptibility. The published studies exhibited variability in sample size, inconclusiveness regarding whether isolated organisms were pathogens or colonizers, and lack of consistent identification of confounders that may contribute to mortality rates. On the basis of such variables, it is not possible to definitively prove that such inadequate therapy increased mortality rates. Nevertheless, the consensus of multiple studies concerned with adequate versus inadequate initial therapy in the critically ill led to the interpretation that initial inadequate therapy contributed to mortality rates.9
Shortly after the reports emphasizing the importance of initial, adequate therapy, a growing awareness of other important variables that influenced clinical outcomes in ICU patients began to emerge. In a 2005 joint guideline for management of patients with hospital-acquired pneumonia (HAP), VAP, and health care–associated pneumonia (HCAP), the American Thoracic Society (ATS) and the IDSA modified definitions for the terms used in the selection of antibiotic therapy.10 Inappropriate replaced the term inadequate; furthermore, adequate was adopted to refer to therapy that included not only the correct antibiotic based on the susceptibility of the organism but also optimal dose, correct route of administration, and use, if necessary, of combination therapy.10
Optimal Dose
As the literature regarding the treatment of infections in the ICU has evolved over the years, studies have identified several elements that influence the dose of an antibiotic that is most likely to result in the best clinical efficacy. The manner in which antibiotics kill bacteria varies among different classes of drugs, but the two pharmacodynamic categories of killing that have been best categorized are time-dependent killing and concentration-dependent killing.11 In time-dependent killing, also referred to as concentration-independent killing, maximum bacterial killing occurs when the drug concentration remains above the minimal inhibitory concentration (MIC). Examples of antibiotics that demonstrate time-dependent killing include β-lactam antibiotics (i.e., penicillins, cephalosporins, carbapenems, and monobactams) and vancomycin. Conversely, in concentration-dependent killing, maximum bacterial killing occurs when the peak drug concentration is approximately 10 times the MIC. Examples of agents with concentration-dependent killing are fluoroquinolones and aminoglycosides. The relevance of such antibiotic properties in the management of patients with serious infections has been well demonstrated.
The pharmacologic properties of vancomycin have been nicely summarized, emphasizing the fact that vancomycin exhibits time-dependent killing.12 Accordingly, the length of time that concentrations of vancomycin are maintained above the pathogen’s MIC is critical to bacterial eradication, with a key variable in the treatment of pneumonia being the percentage of time that drug levels in the alveolar space exceed the MIC. As drug levels decline, organisms have the potential to regrow, increasing the chance of clinical failure. The clinical relevance of this pharmacologic principle may support the development of the practice habit of more frequent (every 6 hours) or continuous-infusion vancomycin dosing for infections like pneumonia in which vancomycin penetration to the target site may not be optimal.
Because a goal with time-dependent antibiotics is to maintain levels above the MIC of the organism for as long as possible during the dosing cycle, research has explored extended-infusion dosing of several time-dependent agents, including β-lactam antibiotics and vancomycin. In a study of 194 patients with infection caused by Pseudomonas aeruginosa, piperacillin-tazobactam was administered intravenously either every 4 to 6 hours over 30 minutes or every 8 hours over 4 hours.13 The 14-day mortality rate was significantly lower among patients who received extended-infusion therapy than among patients who received intermittent-infusion therapy (12.2% versus 31.6%, respectively; P = 0.04). In another study designed to assess blood levels of antibiotic based on both dose and pattern of administration, meropenem was administered to two study groups, each with eight healthy volunteers.14 One group received 500 mg as an intravenous infusion over 30 minutes three times a day or a 250-mg loading dose followed by a 1500-mg continuous infusion over 24 hours; the second group received 1000 mg as an intravenous infusion over 30 minutes three times a day or a 500-mg loading dose followed by a 3000-mg continuous infusion over 24 hours. Investigators performed pharmacokinetic calculations and used Monte Carlo simulations for 10,000 simulated subjects. The results of the analyses of the probability of MIC attainment with the high dose were 4 mg/L with continuous infusion and 0.5 mg/L with intermittent infusion. With the low dose, results were 2 mg/L with continuous infusion and 0.25 mg/L with intermittent infusion. Such data emphasize that intermittent infusion of a low dose of a time-dependent drug may result in MICs adequate to treat relatively sensitive organisms such as Klebsiella pneumoniae but may result in less-than-optimal killing of organisms that have intrinsically higher MICs (for example, P. aeruginosa). Other reports have explored the efficacy of continuous-infusion vancomycin.15,16 A systematic review and meta-analysis suggests that extended or continuous infusion of carbapenems or piperacillin/tazobactam may be associated with lower mortality.17
The postantibiotic effect (PAE), in which microbial killing persists despite loss of detectable serum levels, complements the concentration-dependent killing of gram-negative bacilli exhibited by aminoglycosides, and these two properties may serve as the basis for once-daily aminoglycoside therapy.18 It has been suggested that giving the same total dose in larger concentration less often will result in better killing, longer PAE period, and reduced aminoglycoside toxicity that has been associated with elevated trough levels. Such pharmacologic and clinical data were the foundation for the move toward once-daily aminoglycoside dosing.
In such a dosing schedule, the single doses of gentamicin or tobramycin that have been used once daily have included 5 mg/kg and 7 mg/kg of body weight. The experience with once-daily therapy using 7 mg/kg in 2184 adult patients has been reported.19 Excluded from such therapy were patients with ascites, burns involving greater than 20% total body surface area, pregnancy, end-stage renal disease requiring dialysis, and enterococcal endocarditis. The review stated that it was unnecessary to draw standard peak and trough samples and that monitoring could be completed by obtaining a single random blood sample between 6 and 14 hours after the start of an aminoglycoside infusion. The treating clinician could subsequently adjust the dosing interval in accordance with a provided nomogram. Several important observations were made in this large group of patients: (1) Despite the prolonged drug-free period, bacterial regrowth was not clinically evident; (2) no increase in either ototoxicity or nephrotoxicity was found; and (3) efficacy was promoted in a cost-effective manner.19 A meta-analysis evaluating the safety and efficacy of once-daily aminoglycosides in 1200 patients from 16 trials found no difference concerning efficacy and safety between single-dose and multiple-dose regimens.20
To achieve optimal clinical benefits while minimizing the unintended consequences of selecting resistant bacteria, clinicians must simultaneously consider multiple variables when using concentration-dependent drugs in the treatment of serious infections. In a study of lower respiratory tract infections caused by P. aeruginosa, ciprofloxacin was administered intravenously as a dose of 200 to 300 mg every 12 hours.21 Resistance emerged at a rate greater than 70% during therapy. This was similar to the 75% rate predicted by a pharmacodynamic study.22 By contrast, a randomized comparison of imipenem and ciprofloxacin for treatment of nosocomial pneumonia used a ciprofloxacin regimen of 400 mg given intravenously every 8 hours and noted emergence of resistance during therapy in 33% of cases in which Pseudomonas was the causative pathogen.23 This rate was similar to the 38% predicted by the pharmacodynamic study previously cited.22 These data emphasize the importance of considering not only the dosing interval but also the dose of antibiotic administered when using concentration-dependent antibiotics to treat infections in the critically ill.
In addition to dosing interval and initial dose, antibiotic clearance plays an integral role in adequacy of antibiotic therapy. Although it is routinely accepted that reductions in antibiotic dosing are appropriate in advancing acute kidney injury,24 clinicians may fail to escalate drug administration in the increasingly reported clinical phenomenon of augmented renal clearance (ARC).25 ARC is defined as the enhanced renal elimination of circulating solutes due to an increased creatinine clearance (CrCl > 130 mL/minute/1.73 m2). Prevalence of ARC is higher in burn,26 major trauma,27 traumatic brain injury,28 and febrile neutropenic29 ICU patients; however, it is difficult to traditionally recognize as derived estimates of glomerular filtration, such as the modification of diet in renal disease (MDRD) equation and Cockcroft-Gault (CG) formula are less accurate than direct measurement.30 In 48 ICU patients with measured CrCl, Udy and colleagues demonstrated that CrCl values 130 mL/minute/1.73 m2 or greater were associated with initial trough concentrations of β-lactams less than MIC in 82% (P < 0.001). Moreover, CrCl remained a significant predictor of subtherapeutic concentrations after multivariate analysis.31 ARC has importance in vancomycin dosing as well. Baptista and associates performed a prospective, single-center, observational cohort study of 93 consecutive septic patients in the ICU treated with vancomycin by continuous infusion after a weight-based loading dose. Investigators compared patients with and without ARC. A statistically significant correlation between subtherapeutic vancomycin serum concentration and ARC was observed in the first 3 days of treatment.32
An interaction between renal clearance and hypoalbuminemia also plays a prominent role in pharmacokinetics. In fact, Ulldemolins and colleagues demonstrated that in critically ill patients with hypoalbuminemia, unbound flucloxacillin concentrations fell below concentrations necessary for treatment of methicillin-susceptible Staphylococcus aureus (MSSA) 4 hours after standard bolus infusions.33 The combination of increased volume of distribution due to hypoalbuminemia combined with ARC presents a daily situation in which lower achieved antibacterial exposures could result in subtherapeutic dosing, particularly for time-dependent antibiotics.34 Because accurate and timely drug exposure is necessary for optimal clinical benefit,3 ARC and associated factors like hypoalbuminemia must be identified to prevent subtherapeutic pharmacologic treatment.
Penetration at the Site of Infection: Tissue-Targeted Therapy
The importance of antibiotic pharmacokinetic properties has recently received heightened attention for the treatment of pneumonia caused by multidrug-resistant gram-positive cocci. Indeed, pulmonary pharmacokinetics underlie the findings that the use of linezolid for pneumonia caused by methicillin-resistant S. aureus (MRSA), when compared with vancomycin in traditional regimens, results in improved clinical outcomes.35–38 Understanding the relevance of pharmacokinetic properties will help the critical care physician navigate through the recent published literature for these infections.
Pulmonary pharmacokinetics specifically address the tissue penetration and distribution of antibiotics within the lung.39 Early studies investigating antibiotic penetration in pulmonary infections occurring in the ICU were related to aminoglycosides and fluoroquinolones. Although aminoglycoside levels in the interstitium of the lung are acceptable, levels in pulmonary secretions reach a mean of only about 20% of the concomitant serum level.40 By contrast, the concentrations of quinolones in lung tissue significantly exceed the concomitant serum concentrations, and levels in bronchial secretions also have been reported to exceed those in serum.41 Despite the fact that quinolones have better penetration into the lung and less potential for nephrotoxicity than aminoglycosides, available data show a trend toward improved survival in patients with VAP treated with an aminoglycoside-containing, but not with a quinolone-containing, combination.42 A concern with fluoroquinolones in combination therapy directed against gram-negative organisms is the selection of resistance, particularly in organisms such as P. aeruginosa, in which the resultant resistance may be to multiple classes of antibiotics.43 Because of the coexistent potential for clinical efficacy and nephrotoxicity with aminoglycosides, some investigators have suggested, on the basis of clinical trials, discontinuation of the aminoglycoside after 5 days if the patient is improving.44
Existing pharmacokinetic evidence reveals the extremely poor lung tissue penetration of vancomycin. Cruciani and associates investigated vancomycin pharmacokinetics in 30 human lung tissue sections after administration of a 1-g dose over 1 hour.45 A comparison of serum-to-tissue concentration over the dosing interval was used to generate a graph allowing determination of a concentration ratio. Overall, the serum-to-lung tissue concentration ratio was determined to be 21%. Not surprisingly, investigation has confirmed even poorer penetration into epithelial lining fluid.46 These data raise concern that the traditional dosing regimens of vancomycin (1 g given intravenously every 12 hours) and low target serum trough concentrations (5 to 10 µg/mL) will generate lung tissue concentrations below the MIC for S. aureus. The issues surrounding suboptimal vancomycin dosing are reflected in the 2005 published ATS/IDSA guidelines for treatment for adults with HAP, VAP, and HCAP, in which it was noted that retrospective pharmacokinetic modeling suggested that the vancomycin failures may be related to inadequate dosing. Because of these concerns the authors recommend that trough levels for vancomycin should be 15 to 20 µg/mL.10 This recommendation of intensified vancomycin dosing is supported in a joint vancomycin clinical guideline for treating all complicated infectious caused by MRSA.47
In contrast with the poor pulmonary pharmacokinetic properties of vancomycin, several studies of linezolid confirm excellent lung penetration in healthy volunteer subjects and with in vitro modeling.48–50 Boselli and coworkers investigated the steady-state plasma pharmacokinetic variables and epithelial lining fluid concentrations of linezolid administered to critically ill patients with VAP.51 Epithelial lining fluid concentrations of linezolid approximated 100% of corresponding plasma values, with drug concentrations that exceeded the susceptibility breakpoint (4 mg/mL) for S. aureus throughout the greater part of the dosing interval. The principle of antibiotic pharmacokinetics has supported the conclusion that linezolid is superior to vancomycin in traditional dosing regimens for MRSA pneumonia based on retrospective analysis of the two multinational, double-blind, randomized studies published to date.38 A subsequent randomized, controlled clinical trial of linezolid versus vancomycin in the treatment of nosocomial pneumonia favored linezolid; furthermore, a subgroup analysis of this investigation did not suggest a benefit in those patients with vancomycin troughs of 15 mg/L or more on day 3.52
In light of the difficulty in achieving adequate drug levels in pulmonary tissue, many clinicians consider the use of topical, aerosolized antibiotics to deliver drug directly to the site of infection. The agents most commonly employed in this manner are tobramycin and colistin.53 Most of our experience with, and evidence supporting, nebulized tobramycin originates from small studies and meta-analyses assessing the utility of these agents in the management of chronic respiratory disease such as cystic fibrosis, not in the treatment of acute infection.54 Although the risk of toxicity appears lower with the administration of nebulized tobramycin, certain populations, such as those with renal insufficiency, may be at risk.55 Because of the increasing prevalence of multidrug-resistant gram-negative organisms such as Acinetobacter baumannii and P. aeruginosa, clinicians have employed colistimethate via intravenous infusion56 or aerosolization57,58 as therapy for VAP and HAP. Intravenous colistimethate appears to be safe (with a risk of nephrotoxicity similar to that for intravenous aminoglycosides) and effective, though there are few randomized, controlled trials from which to draw firm conclusions; furthermore, the optimal dosing strategy is unclear.59 Although the aerosolized route of colistimethate administration appears to be safe,60 its efficacy has not been established.
Role of Combination Therapy
Synergy
One of the most frequently cited justifications for combination therapy is for the achievement of synergy, in which antimicrobial combinations are more effective than single agents. The best-recognized example of synergistic antimicrobial therapy is in treatment of enterococcal endocarditis, in which treatment with penicillin or ampicillin alone has been associated with a high rate of relapse when compared with therapy with penicillin or ampicillin in combination with streptomycin or gentamicin.61,62 Discussions of combination therapy have raised the question of whether the use of multiple drugs in the treatment of an infection may result in improved clinical outcomes. For example, in the treatment of bacteremia with S. aureus, some investigators have used a semisynthetic penicillinase-resistant penicillin (e.g., nafcillin or oxacillin) in combination with a brief course (3 to 5 days) of an aminoglycoside, based on data showing more rapid clearing of bacteremia.63 Data from this trial did not show a decrease in mortality rates in the study population of nonaddicts with primarily left-sided endocarditis caused by S. aureus when compared with those patients who received nafcillin alone. For P. aeruginosa the mechanism of synergy between antipseudomonal penicillins and aminoglycosides is similar to that of enterococci.64 Despite this microbiologic observation, the presence or absence of synergy seemed less important in a different trial assessing outcomes in patients with P. aeruginosa bacteremia; rather, this investigation explored combination therapy given in an attempt to prevent the emergence of resistance.65
In contrast with the beneficial effect of synergy, a combination regimen may result in the detrimental effect of antagonism. The classic example of such an effect was with the treatment of pneumococcal meningitis in the 1950s, in which the fatality rate among patients who received penicillin alone was 21%, in contrast with 79% among those who received both penicillin (a bactericidal agent) plus chlortetracycline (a bacteriostatic agent).66
The clinical importance of antagonism has increased in the era of community-associated methicillin-resistant S. aureus (CA-MRSA). Published evidence supports the fact that MRSA is an independent predictor of mortality rate, ICU length of stay, and overall cost of care.67–70 Because a limited number of therapeutic options exist for the treatment of severe, invasive MRSA infections, effort has been made to identify the in vitro activity of antibiotic combinations that may have clinical applicability. Although the intent of using antibiotic combinations is to achieve additive or synergistic effects, the use of combinations may have the unintended effect of antagonism.
A study using 10 different strains of S. aureus found an overall pattern of antibiotic indifference (no combination effect) in combinations of linezolid with fusidic acid, rifampin, or gentamicin. On the other hand, the combination of linezolid with ciprofloxacin or vancomycin resulted in slight antagonism and reduced bactericidal effect when linezolid was combined with ciprofloxacin and vancomycin against the same strains of staphylococci.71 A subsequent study using the checkerboard broth microdilution method tested linezolid in combination with 28 different antimicrobial agents—including vancomycin and several fluoroquinolones—and demonstrated no antagonistic effect.72 Sahuquillo and colleagues reproduced the effect of antibiotic indifference with vancomycin, but they noted antagonism with levofloxacin in two of the five S. aureus isolates tested.73
Another investigation used the rabbit model of aortic valve endocarditis to evaluate 5-day treatment regimens of linezolid alone, vancomycin alone, and linezolid in combination with vancomycin in 40 rabbits infected with an MRSA strain.74 Those treated with vancomycin alone demonstrated greater mean reductions in valvular vegetation bacterial counts than those in the other treatment groups (P = 0.05). Vancomycin also sterilized aortic valve vegetations in three of eight rabbits; by contrast, none of the rabbits treated with linezolid had sterile aortic valve vegetations. A noteworthy finding in this study was that the treatment regimen of linezolid plus vancomycin lowered the peak linezolid levels in serum to below those obtained with regimens with linezolid alone. Even though in vitro synergy testing revealed additive or indifferent activity between the two drugs, the rabbit model revealed in vivo antagonism. A potential explanation offered by the investigators for the observed antagonism between vancomycin and linezolid was the effect of combining a bacteriostatic (linezolid) with a bactericidal drug (vancomycin). The observed reduction in peak linezolid levels in serum with the combination of the two drugs suggests a role for additional mechanisms in the interaction between the two antibiotics. Unfortunately, the clinical significance of these findings is not yet known.
In the absence of definitive data on optimal management of S. aureus infections in seriously ill patients, a pattern has emerged in some health care systems to prescribe combination therapy for this pathogen. The overall assessment of the data is that antibiotic indifference appears to best characterize the drug interaction profile of linezolid with vancomycin. Because some data, albeit unsubstantiated in controlled clinical trials, seem to cast doubt on the advisability of the combination of linezolid with vancomycin71 and the fluoroquinolones,73 it is important for the intensivist to be aware of the potential consequences of doing so. In short, the role of combination therapy in the treatment of serious MRSA infection, except in a few specific instances, is unclear.75
Enhanced Efficacy Against a Pathogen
A common question in clinical medicine is whether combination therapy will result in increased efficacy against a pathogen via a mechanism other than synergy. In an attempt to find a more definitive answer to this question, a meta-analysis of 64 trials with 7586 patients comparing β-lactam monotherapy versus β-lactam plus an aminoglycoside in immunocompetent patients with sepsis was conducted.76 This report did not identify a statistically significant advantage of combination therapy among the 1835 patients with gram-negative infections for whom the data were analyzed. In contrast with the results in the previously cited study,65 no improved survival was observed for the 426 patients who had infection caused by P. aeruginosa. An additional finding was that the rates of development of resistance did not differ in the two treatment groups. Nephrotoxicity, however, developed significantly more often in those patients who received combination therapy.
Prevention of the Emergence of Resistance
Clinicians frequently use combination therapy for P. aeruginosa in an attempt to prevent the emergence of resistance. Despite the importance of this subject, no definitive data are available to prove that combination therapy will prevent the emergence of Pseudomonas resistance;77 however, results of clinical trials23 and concern about this possibility based on limited data have been the basis for such use of combination therapy. A meta-analysis of eight randomized controlled clinical trials compared β-lactam monotherapy and β-lactam plus aminoglycoside combination therapy to assess if combination therapy may decrease the risk of the emergence of resistance.78 Among initially antimicrobial-susceptible isolates in this analysis, combination therapy did not impact the development of antimicrobial resistance. Furthermore, in the meta-analysis of 64 trials comparing β-lactam monotherapy and β-lactam plus aminoglycoside combination therapy in immunocompetent patients with sepsis, there was no difference in the rate of development of resistance.76
Increased Opportunity for Achieving Appropriate Therapy
Even though issues of synergy and reduction in the emergence of resistance frequently are invoked in discussions of combination therapy, the relevant data do not prove consistent benefits. In patients in the intensive care setting, an important advantage of combination therapy is that it provides the clinician with broader antibacterial coverage for potentially multidrug-resistant microorganisms.79 Additionally, because inappropriate initial therapy may result in increased mortality rate, a combination of antibiotics has the potential benefit of providing coverage against a pathogen that may not be the most likely on a statistical basis but is a reasonable consideration in life-threatening clinical settings confronting the critical care physician. The use of antibiotic combinations for empiric therapy increases the probability that at least one of the agents will have activity against the causative organism.
Immunomodulating Effect of Antibiotics
Streptococcal toxic shock syndrome is a clinical infection in which bacteria produce exotoxins that act as host superantigens, precipitating shock, multiple organ failure, and death. Although Streptococcus pyogenes demonstrates exquisite in vitro sensitivity to penicillin, experimental studies of infection by this pathogen have demonstrated reduced efficacy against organisms in the stationary phase of bacterial growth. This phenomenon has been termed the Eagle effect, whereby high organism population density and slow organism division make treatment with an antibiotic dependent on cell wall synthesis ineffective.80 Some cite the Eagle effect as a justification for use of the bacteriostatic antibiotic clindamycin in the treatment of toxic shock syndrome. In addition, clindamycin is an antibiotic that inhibits bacterial protein synthesis, and this pharmacodynamic property is independent of the stage of bacterial growth.81 Clindamycin inhibits bacterial exotoxin production, facilitates phagocytosis of S. pyogenes by inhibiting M protein synthesis, and suppresses the production of penicillin-binding proteins (PBPs). Furthermore, evidence exists that demonstrates clindamycin has immunomodulatory effects, suppressing monocyte synthesis of tumor necrosis factor-α (TNF-α).82,83 All of these pleiotropic qualities have resulted in the recommendation for clindamycin use in necrotizing skin or soft tissue infections and toxic shock syndrome caused by S. pyogenes.81
The dramatic increase in the worldwide incidence of highly virulent, community-acquired infection with CA-MRSA has resulted in increasing reports of necrotizing skin and soft tissue infections as well as necrotizing pneumonia confronting the critical care physician.84–86 CA-MRSA virulence has been attributed to expression of several virulence factors: α-hemolysins (AH), toxic shock syndrome toxin-1 (TSST-1), staphylococcal enterotoxin B, and Panton-Valentine leukocidin (PVL). The association of staphylococcal virulence with the current CA-MRSA epidemic prompted Stevens and colleagues to investigate the impact of antibiotics on the expression of virulence-associated exotoxin genes.87 These investigators were able to demonstrate markedly suppressed in vitro production of staphylococcal toxin genes by clindamycin and linezolid such that no PVL production was noted up to 12 hours after antibiotic administration. Of interest, subinhibitory concentrations of the cell wall–active agent nafcillin were found to increase toxin production. These findings led the investigators to conclude that the inhibition of protein synthesis is an important consideration in the selection of antimicrobial agents for treatment of serious infections caused by toxin-producing gram-positive cocci.87
A growing body of evidence exists to support the benefit of macrolide therapy for bacteremic pneumonia caused by Streptococcus pneumoniae.88–96 Although multiple explanations have been proposed, efficacy appears to extend beyond the drug’s spectrum of antimicrobial activity. The macrolide class of antibiotics exerts a broad range of immunomodulatory effects, including suppression of harmful interleukin host responses and inhibition of neutrophil oxidant burst and degranulation.88–90 These pleiotropic effects have received an increasing focus of attention and further illustrate how immune modulation influences recommendations for therapy in the intensive care setting.
Timing
The impact of the timing of antibiotic therapy has been addressed in several ways with regard to patients in the ICU. In an analysis based on 107 consecutive patients receiving mechanical ventilation and antibiotic treatment for VAP, Iregui and colleagues noted that 30.8% (33 of 107) received antibiotic treatment that was delayed for 24 hours or more after initially meeting diagnostic criteria for VAP; these patients were classified as having initially delayed appropriate antibiotic therapy (IDAAT).97 Two major variables were identified in these patients with IDAAT: (1) a delay in writing an antibiotic order (in 75.8% of the cases); and (2) the presence of a bacterial species resistant to the initially prescribed antibiotic regimen (in 18.2%). The investigators found that hospital mortality rate was 69.7% for the patients with IDAAT, in contrast with only 28.4% for the patients without IDAAT (P < 0.01). An earlier study noted that, in patients with VAP, modification of the antibiotic regimen to cover pathogens based on the susceptibility report did not eliminate the increased mortality rate associated with inadequate empiric therapy.5 Acknowledgment of this finding was the basis for the statement that secondary modifications of an initially failing antibiotic regimen do not substantially improve the outcome for critically ill patients.98 These results challenge the clinician to promptly order antibiotics that cover the involved pathogens even before culture results are available.
The importance of antibiotic timing in patients with community-acquired pneumonia was assessed in a retrospective cohort study of pneumonia in 18,209 Medicare patients.99 In this trial, conducted in a random sample of inpatients 65 years of age or older with community-acquired pneumonia who had not received antibiotics as outpatients, the influence on clinical outcome was assessed for use of antibiotics prescribed according to standard guidelines published at the time of the analysis and not identification of pathogens isolated from the patients. Of the patients who received antibiotics within 4 hours of hospital arrival, 83.2% were prescribed a guideline-recommended regimen, in contrast with 71.8% of the patients who received antibiotics after 4 hours of arrival. The results of this analysis found an association between the administration of antibiotics within 4 hours of hospital arrival and a decrease in mortality rate and length of stay. The investigators postulated that antibiotics may interrupt or minimize the effects of the acute lung injury process that occurs as part of the systemic inflammatory response in patients with bacterial pneumonia.
The IDSA practice guidelines for management of adult patients with meningitis note the lack of prospective clinical data on the relationship of the timing of antimicrobial administration of antimicrobial agents to clinical outcome in patients with bacterial meningitis.100 Data from a retrospective cohort study of 269 adult patients with community-acquired bacterial meningitis provide some insights into the timing of antibiotic therapy in the absence of definitive recommendations.101 Using these and other data, the IDSA acknowledged that evidence for definitive recommendations is inadequate and concluded that a reasonable assumption is to administer treatment for bacterial meningitis before the infection advances to a high level of clinical severity.100 Referring to meningitis as a “neurologic emergency,” the guideline recommended that appropriate therapy for meningitis be initiated as soon as possible after the diagnosis is considered to be likely.100 In support of the importance of prompt timing, this document also noted the potential in certain patients for administration of antibiotics before hospital admission if the patient initially presents outside the hospital.
For the critical care clinician, sepsis is a clinical entity in which adequate antibiotic therapy has been associated with improved clinical outcomes.2,3,7,8,79 In recognition of the importance of prompt therapy in influencing clinical outcomes in patients with sepsis, the 2008 Surviving Sepsis Campaign guidelines offered a specific recommendation regarding the timing of antimicrobial therapy for the septic patient: “Intravenous antibiotic therapy should be started as early as possible and within the first hour of recognition of septic shock and severe sepsis without septic shock. Appropriate cultures should be obtained before initiating antibiotic therapy but should not prevent prompt administration of antimicrobial therapy.”102
Special Pharmacologic Properties
As the focus of antibiotic research has expanded beyond the characterization of in vitro properties for a particular agent, the importance of antibiotic performance at the in vivo target tissue level is becoming increasingly recognized. It was with the 2003 introduction of a novel antibiotic, daptomycin, for treatment of infections caused by resistant gram-positive cocci that the relevance of “organ-specific deactivation” initially was described.103 Daptomycin is an intravenous cyclic lipopeptide with rapid, concentration-dependent killing and bactericidal activity against a broad spectrum of gram-positive cocci.104–106 It demonstrates a unique mechanism of action, with calcium-dependent insertion into the phospholipid bacterial cell membrane. This results in cell depolarization via potassium efflux, causing disruption of DNA, RNA, and protein synthesis.107
As a result of two multicenter, randomized controlled trials comparing it with penicillinase-resistant penicillin or vancomycin,108 daptomycin, at a daily dose of 4 mg/kg (for patients with CrCl greater than 30 mL/minute), received U.S. Food and Drug Administration (FDA) approval for the treatment of complicated skin and soft tissue infections. A subsequent randomized, open-label trial investigated use of daptomycin in S. aureus bacteremia and endocarditis.109 This trial prompted FDA approval of daptomycin for treatment of S. aureus bacteremia, including right-sided endocarditis, at a daily intravenous dose of 6 mg/kg.
Phase 3 clinical trials also were conducted for the treatment of community-acquired pneumonia in hospitalized patients. Despite daptomycin’s potent in vitro bactericidal activity against S. pneumoniae, clinical outcomes were disappointing and inferior to those with the comparator, ceftriaxone. Although daptomycin is known to exhibit poor penetration into epithelial lining fluid, the reason for treatment failure was not fully elucidated until Silverman and colleagues described a unique organ-specific inactivation process.103 Daptomycin’s inactivation was linked to its mechanism of bactericidal action: calcium-dependent membrane lipid binding.103,107,110,111 Using a mouse model, investigators demonstrated drug sequestration and inactivation by binding to phospholipid vesicles that are found in pulmonary surfactant.103 For this reason daptomycin is not considered an appropriate therapeutic agent for treatment of pneumonia, largely because of the presence of surfactant at the target tissue. This phenomenon appears to be unique to daptomycin103 but raises the question of unrecognized organ-specific inactivation for other classes of antibiotics and other target organs.
Unintended Consequences of Antibiotic Therapy
Traditional teaching about antibiotics focused on three classic parameters: efficacy, safety, and cost-effectiveness. With respect to safety, the major considerations were allergic reactions and adverse effects. Within the pandemic of antibiotic resistance, an important new safety issue should be added: unintended consequences of antibiotic therapy. An insightful report termed such unintended consequences the “collateral damage” of antibiotics.112 In this report, collateral damage referred to the ecologic adverse effect of selecting drug-resistant organisms and the unwanted development of colonization or infection with multidrug-resistant organisms. Some important new considerations recently described in the literature, however, may influence initial antibiotic selection for serious infections in the ICU.
The increasing prevalence of MRSA infections in patients in the critical care setting has required an ongoing evaluation of factors that propagate and sustain such a pattern of resistance. The role of inadequate handwashing in the spread of nosocomial MRSA in hospitals has been well known for many years.113 As rates of MRSA have increased in infections such as HAP,114 increased emphasis has been placed on other variables that contribute to the role that MRSA now plays as the etiologic agent of HAP and VAP. In recognition of the infectious disease concept that colonization is an antecedent event to infection, attention has been directed to factors that may increase colonization by MRSA. Bisognano and colleagues provided preliminary evidence by evaluating the occurrence and frequency of increased adhesion in clinical isolates of fluoroquinolone-resistant MRSA and MSSA. This increased adhesion was mediated by fluoroquinolone-induced increases in fibronectin-binding proteins (FnBPs).115 Although this report does not prove that suboptimal levels of fluoroquinolones contribute in a clinically significant way to increased production of FnBPs and higher levels of bacterial attachment, the data do challenge the clinician to consider unintended antibiotic consequences as an explanation for increasing rates of infection with MRSA.
In addition to infection with MRSA, a problem that has gained increasing attention in recent years has been infection by strains of Clostridium difficile with markedly increased levels of toxin production.116 A postulated cause for this increase in toxin production is the phenomenon of hypermutation. Spontaneous mutations that lead to bacterial resistance occur with a frequency that generally is in the range from 10−6 to 10−8. Hypermutation has been used to refer to a situation in which the mutation rate exceeds that recognized for spontaneous mutations.117 Certain factors may lead to genetic damage in bacteria with a resultant increase in the potential for a mutation that can lead to resistance, and antibiotics that have an effect on DNA have been shown with in vitro experiments to potentially contribute to this predisposition to resistance.117
The data regarding the role of antibiotics in causing DNA damage that leads to increased resistance via hypermutation become relevant when one considers potential mechanisms for the increasing toxin production in C. difficile that has led to such problems as toxic megacolon, bowel perforation, and death. In an attempt to better understand outbreaks of clinical disease caused by toxin-producing strains of C. difficile, the genetics of epidemic strains were studied.118,119 In these studies, an 18-base-pair deletion in the tcdC gene, which normally downregulates production of toxins A and B, was noted. An additional finding was the presence of binary toxin genes. These factors were thought to contribute to the hyperproduction of toxin expressed in clinical outbreaks. The epidemiologic analysis of these outbreaks identified fluoroquinolone therapy as a risk factor in recent C. difficile outbreaks.118–120 These observations challenge the critical care clinician to again consider the potential of unintended antibiotic consequences in the selection of empiric antibiotic therapy in seriously ill patients.
In addition to C. difficile, the effect of antibiotics on toxin production has been studied in gram-positive organisms. In a study investigating the effects that cell wall–active antibiotics and protein synthesis inhibitors have on transcription and translation of genes for PVL, AH, and TSST-1, Miller and associates demonstrated that subinhibitory concentrations of nafcillin induced and prolonged messenger RNA (mRNA) expression for PVL, AH, and TSST-1.86 A clinical interpretation of these data suggested by the investigators is that inadvertent use of β-lactam antibiotics to treat MRSA infections may contribute to worse outcomes.
Clinician Responses to Multidrug Resistance
In the studies addressing increased mortality rates associated with inadequate initial antimicrobial therapy for serious infections,2–7 unanticipated bacterial resistance was a frequent reason for not initially selecting a drug to which the organism was susceptible. An important insight is provided in the study by Trouillet and colleagues, who evaluated the risk factors for resistance in patients with VAP.121 The conclusions of that study were that use of antibiotics within the past 15 days and mechanical ventilation of at least 7 days’ duration were the most important factors. When these two parameters were used in subcategories to evaluate the predisposition for selecting resistant organisms, antibiotic use was a more influential factor than was mechanical ventilation. This fact highlights the dual role that antibiotics play in the ICU: (1) treatment of infection and (2) potential selection of the resistant organisms that lead to the next episode of infection. The challenge for the clinician is to achieve the appropriate degree of balance between these two opposing effects.
Mechanisms of Action and of Resistance
Penetration Through the Bacterial Cell Wall
For an antibiotic to reach its target binding site, it must penetrate the bacterial cell wall. One route that can be used by certain antibiotics is through trimers of proteins referred to as porins, which are located in the bacterial cell wall. Originally intended as a point of access for nutrients into the bacteria and excretory products out of the bacteria, porin channels have been used as the route of entry for certain antibiotics, most notably, the carbapenems. When an organism becomes deficient in these porins, then carbapenems are not able to enter the bacteria, which ultimately leads to a lack of binding to PBPs and resultant clinical resistance. The most important example of resistance mediated by porin channel closure is the resistance of P. aeruginosa to carbapenems. For the clinician in the ICU, it is important to know that P. aeruginosa possesses three mechanisms of resistance regulated by genetic operons on the chromosome: (1) AmpC β-lactamases; (2) efflux pumps; and (3) outer membrane porin alterations.122,123 Because the clinician will not have information at the time of empiric therapy regarding which, if any, of these mechanisms might be in play in the P. aeruginosa being targeted, regimens for infections like hospital-acquired pneumonia often target more than one of these mechanisms of pseudomonal resistance.10
Enzymatic Degradation
Type 1 β-lactamase
Type 1 β-lactamases (AmpCs) are chromosomally mediated, with the product of the ampC gene (hence the term AmpC β-lactamases) being the archetype. See Table 51.1 for organisms commonly felt to produce type 1 β-lactamase, their substrate, and antibiotics felt to be stable in the presence of the enzyme. According to data from the Centers for Disease Control and Prevention’s National Nosocomial Infections Surveillance (NNIS) system, these organisms account for 10% to 33% of nosocomial infections involving blood, surgical site, urine, and lung.124 Even though these pathogens do not always produce type 1 enzymes, this potential does exist and must be taken into consideration if initial antibiotic therapy is to be appropriate. Because type 1 β-lactamases have an affinity for cephalosporins (and have therefore been referred to by some authors as cephalosporinases), third-generation cephalosporins are not predictably stable in the presence of type 1 enzymes.125 Also lacking stability are the β-lactamase inhibitors, of which tazobactam is the most likely to resist destruction by these enzymes.
Table 51.1
Characteristics of Type 1 β-Lactamase–Expressing Organisms
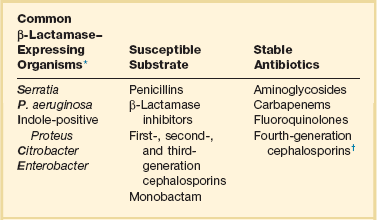
*“SPICE” (or “SPACE”) Bugs: If Acinetobacter is substituted for indole-positive Proteus, then the mnemonic SPACE applies.
Exposure to specific antibiotics may influence a microorganism’s ability to produce type 1 β-lactamases. In a classic paper from the infectious diseases literature, two mechanisms have been described by which this occurs: (1) induction and (2) the selection of spontaneous mutant strains (previously referred to as stable de-repression).126 Sanders and Sanders126 explored the property of induction when they performed an investigation in which they incubated an organism with the potential to produce type 1 β-lactamases overnight in the presence of antibiotic. After this incubation, they performed an assay for type 1 β-lactamase. If they subsequently detected the enzyme, they described the process as induction. Strong inducing antibiotics identified in this report were cefoxitin, imipenem, and clavulanic acid. Of note, upon removal of the inducing antibiotic, the β-lactamase production ceased before the next dose of drug was due to be given. The investigators thus described induction as a reversible in vitro phenomenon.126
What has been proved to occur in patients is the selection of spontaneous mutant strains of bacteria.126 As stated earlier, organisms that possess the ampC gene typically possess complex regulatory mechanisms that prevent overexpression of the gene; however, a certain number of bacteria within clinical isolates (often in the 10−6 to 10−7 range) will have spontaneous mutation(s) that allows them to overproduce type 1 β-lactamase.126 When certain broad-spectrum antibiotics are given, the sensitive nonmutated organisms are killed; however, the genetic mutant strains proliferate and become the predominant organisms. Once this occurs, the clinical isolate will now exhibit resistance to a broad range of β-lactam antibiotics. Most notable of the antibiotics that have been described in the literature to select these “stably de-repressed” mutants are the third-generation cephalosporins.127,128
Extended-Spectrum β-Lactamases
An increasingly common problem facing clinicians and clinical microbiologists is the presence of ESBLs, which are broad-spectrum β-lactamase enzymes produced by such pathogens as Klebsiella, Escherichia coli, Enterobacter, and Proteus. First described in the 1980s, these enzymes may occur on the basis of a change of only one amino acid in the β-lactamases normally produced by these pathogens.129 Despite the minimal structural change, the enzymes have the capacity to inactivate many broad-spectrum β-lactam drugs. Of note, use of any of several classes of antibiotics, notably third-generation cephalosporins and fluoroquinolones, has been identified as a risk factor for selecting ESBLs.130,131
The resistance that occurs in such ESBL producers is of importance for the clinician in initial antibiotic selection in two regards. First, with β-lactam antibiotic therapy, even antibiotics that generally are stable in the presence of β-lactamases may not have predictable activity. Inferior outcomes associated with extended-spectrum cephalosporins such as cefepime and with β-lactam/β-lactamase inhibitor combinations, such as piperacillin-tazobactam, have been described;132 one proposed mechanism is inoculum effect, with diminished susceptibility as the size of the inoculum is increased from 105 to 107 organisms.132,133 An alternative explanation for the lack of activity by certain β-lactam antibiotics has been proposed by Craig and Bhavnani.134 As the number of bacterial colony-forming units (CFUs) increases from 5 × 105 to 5 × 107, approximately 100 times more β-lactamase is released from bacteria lysed after antibiotic exposure. Variations in antibiotic efficacy may rest on the increased release of enzyme in infections with larger inoculums of organisms (e.g., pneumonia and intra-abdominal abscess). Thus, in vitro susceptibility may not predict in vivo outcome for treatment of infectious diseases.
Unlike the type 1 β-lactamase, the ESBL is not chromosomal; rather, it is carried on a large plasmid, which may contain genetic material encoding the machinery that conveys resistance to many classes of antibiotics. For example, the resistance genes for aminoglycosides may be located on the same plasmid as those encoding an ESBL. As a result, gentamicin and tobramycin resistance often occurs in ESBL producers.133 With fluoroquinolones, multiple mechanisms may contribute to quinolone resistance in ESBL producers. Topoisomerase mutations (i.e., target modification) may be associated with decreased binding. The qnr gene codes for a protein that wraps around DNA gyrase, thereby preventing quinolones from attaching to target binding sites.135,136 In addition, ESBL-producing organisms may possess efflux pumps, and because fluoroquinolones are subject to extrusion from bacteria by means of these pumps, efflux is a potential mechanism of quinolone resistance in ESBL producers.137
During the early years of infection with ESBL-producing organisms, infections occurred almost exclusively in either hospitalized patients or in those who had been exposed to antibiotics. In the early 2000s, ESBL infections that occurred in the community setting in patients who had never been hospitalized and who had not been previously exposed to antibiotic were reported.138 Risk factors for ESBL infections included rates in the 35% range of infections caused by ESBL-producing organisms in patients who had no previous health care contact.139 With the awareness of community ESBL infections came the identification of large increases within relatively short periods of time of specific types of ESBLs (e.g., the enzyme CTX-M, which refers to a cefotaximase that was initially identified in Munich, Germany) occurring predominately in urinary tract isolates of E. coli in community settings.140 Perhaps the best explanation for why infection with ESBL-producing pathogens might originate in the community setting in patients without the risk factors for either selective antibiotic pressure or person-to-person spread of the organism was suggested in the review by Jacoby and Munoz-Price.133 Genes for CTX-M–type enzymes are found on the chromosome of Kluyvera, a genus of rarely pathogenic commensal organisms found in the gastrointestinal tract. The postulation was that pathogenic organisms such as E. coli producing CTX-M–type enzymes appear to have emerged by plasmid acquisition of β-lactamase genes from commensal organisms with which they have close proximity in an environmental reservoir (e.g., the gastrointestinal tract). It is awareness of such a trend for community-acquired resistant pathogens that may influence the initial empiric therapy of patients with either a gastrointestinal or urinary tract source of infection who present ill enough to require ICU admission.
Although infection with ESBL-producing organisms has been associated with significant clinical consequences, no definitive recommendations exist for how to optimally treat such infections, and no antibiotic presently on the market has an FDA-approved indication for treating infections caused by ESBL-producing pathogens. In the absence of such data but with the important ramifications of such infections, recommendations must be extracted from the medical literature. The reviews by Jacoby and Munoz-Price133 and by Paterson and Bonomo129 have listed carbapenems as a drug of choice for infections caused by ESBL-producing pathogens. Influencing this recommendation are the facts that (1) ESBL-producing pathogens are often resistant to fluoroquinolones and aminoglycosides because resistance mechanisms for these classes of antibiotics are often carried on the same large plasmid containing the genetic elements for ESBL production and (2) carbapenems (although β-lactam agents) are paradoxically stable in the presence of ESBLs.
It is important for the clinician to consider whether one carbapenem might be more favorable than another for the treatment of infections caused by ESBL-producing organisms. In the era of antimicrobial stewardship, a principle of importance in such considerations is whether the collateral damage of antibiotics that may result in selecting resistant strains of P. aeruginosa can be prevented. Although not a universally accepted tenet, the concept of pseudomonal sparing is of potential importance. The clinical approach to carbapenems that is useful in such a consideration is to divide this class of antibiotics into those agents with activity against P. aeruginosa (i.e., imipenem, meropenem, doripenem) versus the carbapenems without significant activity against P. aeruginosa (i.e., ertapenem). Incorporating the data from Sanders and Sanders,126 selection of strains of resistant P. aeruginosa occurs when a spontaneous mutant with the genetics for drug resistance is allowed to grow because sensitive nonmutant strains of the organism have been killed by antibiotic therapy. Although numerous references regarding antibiotic selective pressure leading to drug-resistant P. aeruginosa have been published, two publications are representative of how a process might be favored by pseudomonal-active antibiotics.
In a 2-year case-control study of 2613 patients admitted to three ICUs in a large teaching hospital in Paris, France, prolonged receipt of antibiotics with specific antipseudomonal activity, most notably ciprofloxacin, was associated with the emergence of multidrug-resistant P. aeruginosa.141 The authors concluded that if treatment with an antibiotic active against gram-negative bacteria is needed, agents with little antipseudomonal activity should be preferred over those with specific antipseudomonal activity to limit the emergence of multidrug-resistant P. aeruginosa. Using data from 779 isolates of carbapenem-resistant P. aeruginosa, Carmeli and colleagues showed by univariate analysis that the risk factors for isolation of carbapenem-resistant P. aeruginosa were the administration of group 2 carbapenems (imipenem, meropenem) (P < 0.0001), aminoglycosides (P = 0.034), or penicillins (P = 0.05).142 The group 1 carbapenem ertapenem was not associated with imipenem-resistant P. aeruginosa (P = 0.2).
Since the introduction of ertapenem onto the market, there have been theoretical concerns that use of this nonpseudomonal carbapenem might select for strains of carbapenem-resistant P. aeruginosa. Ecology studies conducted over the first 9 years that ertapenem was on the market specifically addressed this concern, and those studies have been published in a review article on the topic.143 Results from the 10 clinical studies evaluating the effect of ertapenem use on the susceptibility of Pseudomonas to carbapenems uniformly showed that ertapenem use did not result in decreased Pseudomonas susceptibility to the antipseudomonal carbapenems.
With the increasing prevalence of ESBL infections, including those in the community setting that might be severe enough to require ICU admission, an unanswered question is whether noncarbapenem therapy might be appropriate for some patients. This question was addressed in a post-hoc analysis from Spain of patients from six published prospective cohorts who had bloodstream infections due to ESBL-producing strains of E. coli.144 Most of these patients had the urinary tract as the site of origin of the bloodstream infection. These results suggested that amoxicillin/clavulanic acid and piperacillin/tazobactam might be suitable alternatives to carbapenems for treating patients with bloodstream infections due to ESBL-producing E. coli if the isolate was susceptible in vitro to these agents. Unanswered by the review is the optimal management of ESBL-producing E. coli bloodstream infections originating outside the urinary tract and of infections caused by pathogens other than E. coli that produce ESBLs.
Carbapenemases
The name carbapenemase has the potential to be misleading. In an insightful discussion of carbapenemases written at the time when these enzymes were just beginning to be commonly recognized as causes of clinical infections, it was acknowledged that KPC carbapenemases in actuality hydrolyze β-lactams of all classes, with the most efficient hydrolysis observed for nitrocefin, cephalothin, cephaloridine, benzylpenicillin, ampicillin, and piperacillin.145 That review went on to state that the carbapenems imipenem and meropenem, as well as the β-lactam antibiotics cefotaxime and aztreonam, were hydrolyzed with one tenth the efficiency of the penicillins and early cephalosporins. The microbiologically important message was that the KPC family of carbapenemases has a broad hydrolysis spectrum that includes most β-lactam antibiotics. It is with such a basic science foundation that one can begin to approach the role of antibiotics in selecting organisms that produce carbapenemases.
There has been an inclination by some clinicians to assume based on the name carbapenemase that carbapenem antibiotics are the most likely to select such a pattern of resistance. Although a definitive answer for such a question cannot be found from prospective clinical trials, multiple reports in the literature support the conclusion that several classes of antibiotics have been linked to subsequent infection with carbapenemase-producing bacteria. As a basic piece of information, one can assume that patients become colonized with such resistant pathogens and that any broad-spectrum antibiotic has the ability to kill the normal flora and thereby allow the resistant strains to grow and to cause clinical disease. In the absence of identifying a specific class of antibiotics responsible for selecting carbapenemase-producing bacteria, there has been a focus in the medical literature to identify other variables that contribute to the problem. Two representative papers provide an important insight related to the association between prior antimicrobial therapy and the subsequent identification of carbapenemase-producing bacteria. In a 4-year case-control study (n = 102), the only covariate independently associated with carbapenem-resistant Enterobacteriaceae in all multivariate analyses was the cumulative number of prior antibiotic exposures.146 A 26-month double case-control study (n = 96 ESBL-CRKP and 55 ESBL-CSKP) from Greece identified both prior cumulative exposure to antibiotics and increasing duration of prior treatment as risk factors.147 Shown in this study to be associated with the isolation of carbapenemases was therapy with β-lactam/β-lactamase inhibitor or with the combination of fluoroquinolone and carbapenem. These data are consistent with previous reports that no particular class of antibiotic is the predominant predisposing factor for selection of carbapenemase production. Knowledge of this information by the ICU physician is important because it (1) debunks the myth that carbapenemases are selected only by one class of antibiotics (and, therefore, that restriction of only one class of antibiotics can manage such a problem) and (2) supports the intensity and duration of antibiotic therapy as the most important variables in creating the milieu in which carbapenemase-producing bacteria are selected.
Therapy for infections caused by carbapenemase-producing bacteria has been extremely challenging, and at the present time there are neither definitive nor predictable recommendations that guarantee clinical success. In the absence of such data, the clinician is challenged to make clinically relevant deductions from the published literature. The findings from 15 studies and reports in treating infections caused by KPCs showed success rates of 75% with aminoglycosides, 73% with polymyxin combinations, 71% with tigecycline, 40% with carbapenems, and 14% with polymyxin monotherapy.148 At the time of writing of this chapter, the antimicrobial agents accepted as having the most potential activity based on in vitro susceptibility are tigecycline, polymyxins, and aztreonam.149 There are evolving data suggesting a potential role for carbapenems as a therapeutic option if (1) the MIC of the infecting organism is 4 mg/L or less and (2) the carbapenem is given in combination with another active agent.150
The examples of carbapenem resistance (whether on the basis of carbapenemase or other mechanism of resistance) in Pseudomonas, Acinetobacter, Stenotrophomonas, and Klebsiella underscore an important concept in antimicrobial therapy for the critical care clinician, for whom such patterns of resistance are becoming increasingly prevalent in the ICU. When multidrug resistance occurs, it cannot be predictably assumed that such resistance is on the basis of exposure to the broadest class of antibiotic to which the organism is resistant. To accept such a flawed assessment could result in limited use of that class of antibiotic, which could then shift to increased use of the actual class of antibiotic that led to the pattern of resistance. The other clinically relevant observation in many of the recently described outbreaks is that inadequate infection control contributed to the spread of resistant strains that had been selected by antibiotics.132,151,152
Efflux Pumps
Efflux pumps are three-component systems, contained within the bacterial cell wall, that allow bacteria to eliminate antibiotics that have entered. Initially described in 1980 as a mechanism of resistance in tetracyclines, efflux was recognized in 1988 as a contributor to fluoroquinolone resistance.153 In recent years, the contribution of efflux to clinical resistance has broadened, with important implications for treatment of serious infections. The composition of this system has been nicely detailed.154,155 The pump itself (also referred to as the transporter) lies in the cytoplasmic membrane and is designated MexB, MexD, or MexF. It is attached via a linker lipoprotein (MexA, MexC, or MexE) in the periplasm to the third component, the exit portal (OprM, OprJ, or OprN), which lies in the outer membrane. These three components of the efflux pump normally are under repressor gene control and therefore are not clinically active.
P. aeruginosa has several efflux systems, with MexAB-OprM and MexEF-OprN having particular clinical significance: The MexAB-OprM system contributes to both intrinsic and acquired resistance; and the MexEF-OprN system contributes only to acquired resistance.156 The MexAB-OprM system is expressed constitutively in cells grown in standard laboratory media, where it contributes to intrinsic resistance to a number of antimicrobials, including fluoroquinolones, tetracyclines, piperacillin, cefepime, aztreonam, and certain carbapenems.123 Of the β-lactams, only carbapenems appear to be poor substrates for MexAB-OprM; however, different carbapenems vary with regard to their susceptibility to efflux. Meropenem and doripenem are subject to efflux, and expression of the MexAB-OprM efflux system has been correlated with resistance to meropenem.157 By contrast, imipenem is not subject to efflux.156 It has been suggested that this may be due to the need for efflux systems with MexAB-OprM to access their substrates within the cytoplasmic membrane. Meropenem is much more amphiphilic than imipenem, which may explain why imipenem does not act as a substrate for MexAB-OprM whereas meropenem does.157 Such a microbiologic property may explain why a gram-negative isolate may be susceptible to imipenem but resistant to meropenem.
The ability of fluoroquinolones to select certain nfxc (mexT) mutants has been discussed.122,154 In addition to resistance to fluoroquinolones, these mutants may have decreased susceptibility and even clinical resistance to carbapenems (e.g., imipenem, meropenem) that occurs on the basis of either closure of porin channels in the outer membrane of the bacterial cell wall (with resultant impermeability) or upregulation of an efflux pump (which allows the bacteria to eliminate drug that has penetrated the organism’s cell wall). Such newly recognized information may help explain patterns of increasing carbapenem resistance in health care institutions in which carbapenem use has not recently increased.
The Clinical Relevance for Understanding Antibiotic Resistance
A classic example of multidrug resistance in gram-negative organisms is the resistance in P. aeruginosa. As previously noted, multiple resistance mechanisms regulated by genetic operons on the chromosomes of P. aeruginosa have been described, including efflux pumps, AmpC β-lactamases, and outer membrane porin closure.122,158 For infections such as HAP and VAP, in which P. aeruginosa is a pathogen targeted with empiric therapy and for which fluoroquinolones are offered as an option in the initial regimen,10,15,159 data on the risk of fluoroquinolones leading to multidrug resistance via efflux mechanisms become especially noteworthy. Additionally, because of the ability of certain organisms such as P. aeruginosa to express multiple mechanisms of resistance, many experts recommend avoiding the use of traditional antipseudomonal agents (i.e., a pseudomonal-sparing regimen) when treating infections in which Pseudomonas is not a suspected pathogen, as the use of overuse these agents may result in the development of multidrug resistance in Pseudomonas isolates.141
The experience with carbapenem-resistant A. baumannii in Brooklyn, New York, provides an important insight into the problem of selection of resistance by one class of antibiotic to an entirely different class. In a study to evaluate the endemicity of A. baumannii, all unique patient isolates of this pathogen were collected from 15 Brooklyn hospitals over a 3-month period.152 Antibiotic susceptibilities, ribotyping, and the relationship between antibiotic use and resistance rates were determined. Among the 224 carbapenem-resistant strains of A. baumannii, ribotyping demonstrated that one strain accounted for two thirds of the isolates and was present in all of the 15 participating hospitals. The strongest predisposition to selection for this pathogen was cephalosporin use. Known A. baumannii resistance mechanisms include chromosomally associated β-lactamases and porin protein mutations,160 so it can be assumed that this represents selection of carbapenem-resistant mutant strains by the cephalosporins.
A retrospective analysis of critically ill trauma patients with late-onset gram-negative pneumonia showed that the antibiotic most associated with pneumonia due to Stenotrophomonas maltophilia was cefepime.161 These data on cephalosporins as a risk factor for S. maltophilia infection are similar to those in an earlier report that identified use of ceftazidime and imipenem as associated with similar rates of S. maltophilia acquisition in hospitalized patients.162 The suggestion by these reports is that broad-spectrum agents, more so than one specific agent, may kill sensitive bacteria and allow pathogens such as Stenotrophomonas to become clinically expressed.
De-Escalation
Decreasing Broadness or Number of Agents
Classically, there have been three ways that de-escalation can occur. The most intuitively understood is changing from a broad-spectrum agent to a narrower spectrum agent based on culture and sensitivity data. As is well known to those who practice in the intensive care setting, culture data are often negative in the sickest patients, and the option of de-escalating based on culture data is often not available. The de-escalating process becomes less clear when diagnostic studies do not yield an obvious pathogen. Perhaps an appropriate manner of employing the information gained by a negative culture is to allow the nonrecovery of certain organisms to influence the alteration of an existing regimen. HAP, including VAP, provides a clinically applicable example of this concept. In an attempt to minimize mortality rates, patients with HAP typically receive as empiric therapy a three-drug regimen that includes coverage against MRSA and P. aeruginosa. Guidelines of the ATS/IDSA for HAP have suggested that clinically improving patients not on antibiotics when cultures are obtained may have therapy against MRSA and P. aeruginosa stopped if sputum cultures are negative for these pathogens.10
A third technique for de-escalating is to shorten the duration of therapy.
Duration of Therapy
Because antibiotics may be a risk factor for resistance,121 it is important in the process of antibiotic stewardship to identify opportunities to minimize exposure to these therapeutic agents. One possibility is by decreasing the duration of therapy to the shortest duration necessary to achieve an optimal clinical outcome.163 Often the decisions regarding traditional durations of therapy find their basis not in controlled trials or prospective studies, but rather in expert opinion. Although certain infections, such as S. aureus bacteremia, require well-defined and often prolonged durations of intravenous therapy,164,165 trials in VAP have challenged some of the classic tenets and have resulted in recommendations for shorter durations of therapy when compared to the traditional regimens.
Dennesen and coworkers166 evaluated the response to antimicrobial therapy administered according to ATS guidelines in 27 patients diagnosed with VAP in a study that initially used a bronchoalveolar lavage (BAL) along with clinical parameters to confirm the diagnosis of VAP but subsequently used semiquantitative tracheal aspirates for microbiologic surveillance. All patients in this study received appropriate antibiotic therapy. After initiation of antibiotic therapy, Tmax (maximal temperature over a 24-hour period), PaO2/FIO2 (ratio used to quantify impairment of oxygen gas exchange in the lung), white blood cell count, and semiquantitative cultures of endotracheal aspirate were monitored. Resolution of clinical parameters occurred primarily within the first 6 days of therapy. Using cultures of endotracheal aspirates, the investigators found that colonization with P. aeruginosa persisted throughout the duration of treatment, whereas colonization with S. aureus, H. influenzae, and S. pneumoniae resolved shortly after initiation of therapy. Acquired colonization, predominantly with resistant pathogens such as P. aeruginosa or members of Enterobacteriaceae, usually occurred in week 2 and frequently preceded a recurrent episode of lung infection. On the basis of these data, it was suggested that 7 days may be an appropriate duration of therapy for patients with VAP.
Ibrahim and colleagues167 evaluated a clinical guideline for the treatment of VAP using 7 days of therapy. A total of 102 patients were prospectively evaluated, 50 before institution of the guidelines and 52 after institution. In addition to more frequent choice of adequate initial antibiotic treatment after implementation of the clinical guideline, patients also had shorter antibiotic courses when treated under the guideline. No mortality rate difference was noted between the two groups. As was the case with the study by Dennesen and coworkers,166 a second episode of VAP was more likely to occur in the patients receiving the longer, traditional duration of therapy.
Chastre and associates168 conducted a prospective, multicenter, randomized, double-blind study of 401 patients with VAP confirmed by quantitative cultures obtained by bronchoscopic protected specimen brush (PSB) or BAL, or both. Only patients who had received initial appropriate antibiotic therapy were included. Therapy was divided into two categories: short course, which was given for 8 days in 197 patients, versus long course, which was given for 15 days in 204 patients. Clinical efficacy was similar between the short-course and long-course groups. Because slightly more patients with nonfermenting gram-negative bacilli (e.g., P. aeruginosa and A. baumannii) assigned to the 8-day regimen had pulmonary infection recurrences, the investigators were unable to demonstrate the noninferiority of the 8-day regimen for infection by such pathogens compared with the 15-day course of therapy. Multiresistant pathogens more frequently caused recurrences in patients who received the 15-day regimen. Even though the findings from this study do not definitively prove that therapy for HAP or VAP can be limited to 7 days, they lend support to the findings by Dennesen166 and Ibrahim167 and their colleagues.
Minimizing Clinical Resistance
For many years, the standard approach to antibiotic prescribing occurred in a homogeneous manner in which a single antibiotic or limited numbers of antibiotics were used as the “workhorse” agents in empiric therapy.169 Such an approach to antibiotic prescribing often was associated with restricted formularies, the thought being that limiting broad-spectrum agents might prevent the emergence of resistance. Unfortunately, this approach did not take into consideration the possibility that the inadequate therapy in seriously ill patients might not be reversible when the initial antibiotics were changed to the correct agents once susceptibilities were known. In addition, it was during this era of restricted formularies that the proliferation of bacterial resistance began to occur. Because an inherent risk with homogeneous antibiotic use is the selective pressure that when applied can lead to resistance, other approaches must be considered.
Heterogeneous antibiotic use, in which antibiotic selection is based not on hospital mandates but rather on issues related both to the patient and the pathogens involved in the infectious process, may decrease selective pressure.169 Heterogeneity can be achieved in any of several ways. A strategy by which multiple or all classes of antibiotics are available for use (i.e., antibiotic heterogeneity) has been suggested as part of a broader effort aimed at curtailing antibiotic resistance within ICUs. As a potential alternative to cycling, a mathematical model was developed to compare antibiotic cycling with mixing, in which antibiotic variation is random as opposed to the regulation that occurs with a process like antibiotic cycling.170 The premise for such a comparison is that mixing imposes greater fluctuation in selective conditions, thereby yielding greater heterogeneity than occurs with cycling. In this study, the results were underlaid by a simple ecologic explanation that led to the conclusion that cycling is unlikely to be effective and may even hinder resistance control and that mixing may yield more favorable results in terms of preserving antibiotic susceptibility.
In contrast with homogeneous antibiotic use, which was developed to control resistance, heterogeneous use is aimed at managing resistance.169 Because resistance at some level is an inevitable part of medical practice, the latter approach seems more insightful.
Conclusions
At a time when bacterial resistance is becoming more prevalent but during which antibiotic development is relatively stagnant, the critical care clinician is faced with the somewhat daunting challenge of achieving clinical efficacy without compromising the antibiotic armamentarium that exists at present. For this to be accomplished, the interplay of several important concepts will drive the decision process. As depicted in Figure 51.3, these considerations include the organism, the host, and the targeted tissue, as well as specific issues related to the ICU itself.
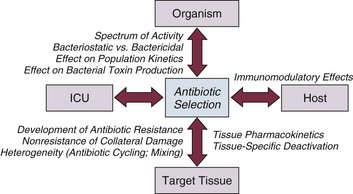
References
1. Dellit, TH, Owens, RC, McGowan, JE, et al. Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America guidelines for developing an institutional program to enhance antimicrobial stewardship. Clin Infect Dis. 2007; 44(2):159–177.
2. Harbarth, S, Garbino, J, Pugin, J, et al. Inappropriate initial antimicrobial therapy and its effect on survival in a clinical trial of immunomodulating therapy for severe sepsis. Am J Med. 2003; 115(7):529–535.
3. Ibrahim, EH, Sherman, G, Ward, S, et al. The influence of inadequate antimicrobial treatment of bloodstream infections on patient outcomes in the ICU setting. Chest. 2000; 118(1):146–155.
4. Kollef, MH, Ward, S. The influence of mini-BAL cultures on patient outcomes: Implications for the antibiotic management of ventilator-associated pneumonia. Chest. 1998; 113(2):412–420.
5. Luna, CM, Vujacich, P, Niederman, MS, et al. Impact of BAL data on the therapy and outcome of ventilator-associated pneumonia. Chest. 1997; 111(3):676–685.
6. Rello, J, Gallego, M, Mariscal, D, et al. The value of routine microbial investigation in ventilator-associated pneumonia. Am J Respir Crit Care Med. 1997; 156(1):196–200.
7. Vallés, J, Rello, J, Ochagavía, A, et al. Community-acquired bloodstream infection in critically ill adult patients: Impact of shock and inappropriate antibiotic therapy on survival. Chest. 2003; 123(5):1615–1624.
8. Kumar, A, Ellis, P, Arabi, Y, et al. Initiation of inappropriate antimicrobial therapy results in a fivefold reduction of survival in human septic shock. Chest. 2009; 136(5):1237–1248.
9. Kollef, MH. Inadequate antimicrobial treatment: An important determinant of outcome for hospitalized patients. Clin Infect Dis. 2000; 31(Suppl 4):S131–S138.
10. American Thoracic Society/Infectious Diseases Society of America. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and health care-associated pneumonia. Am J Respir Crit Care Med. 2005; 171(4):388–416.
11. Ebert, SC, Craig, WA. Pharmacodynamic properties of antibiotics: Application to drug monitoring and dosage regimen design. Infect Control Hosp Epidemiol. 1990; 11(6):319–326.
12. Bodi, M, Ardanuy, C, Rello, J. Impact of gram-positive resistance on outcome of nosocomial pneumonia. Crit Care Med. 2001; 29(4 Suppl):N82–86.
13. Lodise, TP, Jr., Lomaestro, B, Drusano, GL. Piperacillin-tazobactam for Pseudomonas aeruginosa infection: Clinical implications of an extended-infusion dosing strategy. Clin Infect Dis. 2007; 44(3):357–363.
14. Krueger, WA, Bulitta, J, Kinzig-Schippers, M, et al. Evaluation by Monte Carlo simulation of the pharmacokinetics of two doses of meropenem administered intermittently or as a continuous infusion in healthy volunteers. Antimicrob Agents Chemother. 2005; 49(5):1881–1889.
15. Rello, J, Paiva, JA, Baraibar, J, et al. International conference for the development of consensus on the diagnosis and treatment of ventilator-associated pneumonia. Chest. 2001; 120(3):955–970.
16. Wysocki, M, Delatour, F, Faurisson, F, et al. Continuous versus intermittent infusion of vancomycin in severe staphylococcal infections: Prospective multicenter randomized study. Antimicrob Agents Chemother. 2001; 45(9):2460–2467.
17. Falagas, ME, Tansarli, GS, Ikawa, K, Vardakas, KZ. Clinical outcomes with extended or continuous versus short-term intravenous infusion of carbapenems and piperacillin/tazobactam: A systematic review and meta-analysis. Clin Infect Dis. 2013; 56(2):272–282.
18. Vogelman, B, Craig, WA. Kinetics of antimicrobial activity. J Pediatr. 1986; 108(5 Pt 2):835–840.
19. Nicolau, DP, Freman, CD, Belliveau, PP, et al. Experience with a once-daily aminoglycoside program administered to 2,184 adult patients. Antimicrob Agents Chemother. 1995; 39(3):650–655.
20. Galloe, AM, Graudal, N, Christensen, HR, Kampmann, JP. Aminoglycosides: Single or multiple daily dosing? A meta-analysis on efficacy and safety. Eur J Clin Pharmacol. 1995; 48(1):39–43.
21. Peloquin, CA, Cumbo, TJ, Nix, DE, et al. Evaluation of intravenous ciprofloxacin in patients with nosocomial lower respiratory tract infections. Impact of plasma concentrations, organism, minimum inhibitory concentration, and clinical condition on bacterial eradication. Arch Intern Med. 1989; 149(10):2269–2273.
22. Drusano, GL, Louis, A, Deziel, M, et al. The crisis of resistance: Identifying drug exposures to suppress amplification of resistant mutant subpopulations. Clin Infect Dis. 2006; 42(4):525–532.
23. Fink, MP, Snydman, DR, Niederman, MS, et al. Treatment of severe pneumonia in hospitalized patients: Results of a multicenter, randomized, double-blind trial comparing intravenous ciprofloxacin with imipenem-cilastatin. The Severe Pneumonia Study Group. Antimicrob Agents Chemother. 1994; 38(3):547–557.
24. Gilbert, B, Robbins, P, Livornese, LL, Jr. Use of antibacterial agents in renal failure. Infect Dis Clin North Am. 2009; 23(4):899–924.
25. Udy, AA, Roberts, JA, Boots, RJ, et al. Augmented renal clearance: Implications for antibacterial dosing in the critically ill. Clin Pharmacokinet. 2010; 49(1):1–16.
26. Conil, JM, Georges, B, Fourcade, O, et al. Assessment of renal function in clinical practice at the bedside of burn patients. Br J Clin Pharmacol. 2007; 63(5):583–594.
27. Cherry, RA, Eachempati, SR, Hydo, L, Barie, PS. Accuracy of short-duration creatinine clearance determinations in predicting 24-hour creatinine clearance in critically ill and injured patients. J Trauma. 2002; 53(2):267–271.
28. Udy, A, Boots, R, Senthuran, S, et al. Augmented creatinine clearance in traumatic brain injury. Anesth Analg. 2010; 111(6):1505–1510.
29. Lortholary, O, Lefort, A, Tod, M, et al. Pharmacodynamics and pharmacokinetics of antibacterial drugs in the management of febrile neutropenia. Lancet Infect Dis. 2008; 8(10):612–620.
30. Baptista, JP, Udy, AA, Sousa, E, et al. A comparison of estimates of glomerular filtration in critically ill patients with augmented renal clearance. Crit Care. 2011; 15(3):R139.
31. Udy, AA, Varghese, JM, Altukroni, M, et al. Subtherapeutic initial β-lactam concentrations in select critically ill patients: Association between augmented renal clearance and low trough drug concentrations. Chest. 2012; 142(1):30–39.
32. Baptista, JP, Sousa, E, Martins, PJ, Pimentel, JM. Augmented renal clearance in septic patients and implications for vancomycin optimisation. Int J Antimicrob Agents. 2011; 39(5):420–423.
33. Ulldemolins, M, Roberts, JA, Wallis, SC, et al. Flucloxacillin dosing in critically ill patients with hypoalbuminaemia: special emphasis on unbound pharmacokinetics. J Antimicrob Chemother. 2010; 65(8):1771–1778.
34. Ulldemolins, M, Roberts, JA, Rello, J, et al. The effects of hypoalbuminaemia on optimizing antibacterial dosing in critically ill patients. Clin Pharmacokinet. 2011; 50(2):99–110.
35. Kollef, MH, Rello, J, Cammarata, SK, et al. Clinical cure and survival in gram-positive ventilator-associated pneumonia: Retrospective analysis of two double-blind studies comparing linezolid with vancomycin. Intensive Care Med. 2004; 30(3):388–394.
36. Rubinstein, E, Cammarata, S, Oliphant, T, et al. Linezolid (PNU-100766) versus vancomycin in the treatment of hospitalized patients with nosocomial pneumonia: A randomized, double-blind, multicenter study. Clin Infect Dis. 2001; 32(3):402–412.
37. Wunderink, RG, Cammarata, SK, Oliphant, TH, et al. Continuation of a randomized, double-blind, multicenter study of linezolid versus vancomycin in the treatment of patients with nosocomial pneumonia. Clin Ther. 2003; 25(3):980–992.
38. Wunderink, RG, Rello, J, Cammarata, SK, et al. Linezolid vs. vancomycin: Analysis of two double-blind studies of patients with methicillin-resistant Staphylococcus aureus nosocomial pneumonia. Chest. 2003; 124(5):1789–1797.
39. Honeybourne, D. Antibiotic penetration into lung tissues. Thorax. 1994; 49(2):104–106.
40. Thys, JP, Klastersky, J, Mombelli, G. Peak or sustained antibiotic serum levels for optimal tissue penetration. J Antimicrob Chemother. 1981; 8(Suppl C):29–36.
41. Berre, J, Thys, JP, Husson, M, et al. Penetration of ciprofloxacin in bronchial secretions after intravenous administration. J Antimicrob Chemother. 1988; 22(4):499–504.
42. Fowler, RA, Flavin, KE, Barr, J, et al. Variability in antibiotic prescribing patterns and outcomes in patients with clinically suspected ventilator-associated pneumonia. Chest. 2003; 123(3):835–844.
43. Niederman, MS. Reexamining quinolone use in the intensive care unit: Use them right or lose the fight against resistant bacteria. Crit Care Med. 2005; 33(2):443–444.
44. Gruson, D, Hilbert, G, Vargas, F, et al. Rotation and restricted use of antibiotics in a medical intensive care unit. Impact on the incidence of ventilator-associated pneumonia caused by antibiotic-resistant gram-negative bacteria. Am J Respir Crit Care Med. 2000; 162(3 Pt 1):837–843.
45. Cruciani, M, Gatti, G, Lazzarini, L, et al. Penetration of vancomycin into human lung tissue. J Antimicrob Chemother. 1996; 38(5):865–869.
46. Lamer, C, de Beco, V, Soler, P, et al. Analysis of vancomycin entry into pulmonary lining fluid by bronchoalveolar lavage in critically ill patients. Antimicrob Agents Chemother. 1993; 37(2):281–286.
47. Rybak, M, Lomaestro, B, Rotschafer, JC, et al. Therapeutic monitoring of vancomycin in adult patients: A consensus review of the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists. Am J Health Syst Pharm. 2009; 66(1):82–98.
48. Conte, JE, Jr., Golden, JA, Kipps, J, Zurlinden, E. Intrapulmonary pharmacokinetics of linezolid. Antimicrob Agents Chemother. 2002; 46(5):1475–1480.
49. Honeybourne, D, Tobin, C, Jevons, G, et al. Intrapulmonary penetration of linezolid. J Antimicrob Chemother. 2003; 51(6):1431–1434.
50. MacGowan, AP, Alasdair, P. Pharmacokinetic and pharmacodynamic profile of linezolid in healthy volunteers and patients with gram-positive infections. J Antimicrob Chemother. 2003; 51(Suppl 2):17–25.
51. Boselli, E, Breilh, D, Rimmele, T, et al. Pharmacokinetics and intrapulmonary concentrations of linezolid administered to critically ill patients with ventilator-associated pneumonia. Crit Care Med. 2005; 33(7):1529–1533.
52. Wunderink, RG, Niederman, MS, Kollef, MH, et al. Linezolid in methicillin-resistant Staphylococcus aureus nosocomial pneumonia: A randomized, controlled study. Clin Infect Dis. 2012; 54(5):621–629.
53. Wood, GC. Aerosolized antibiotics for treating hospital-acquired and ventilator-associated pneumonia. Expert Rev Anti Infect Ther. 2011; 9(11):993–1000.
54. Conway, SP. Nebulized antibiotic therapy: The evidence. Chron Respir Dis. 2005; 2(1):35–41.
55. Kahler, DA, Schowengerdt, KO, Fricker, FJ, et al. Toxic serum trough concentrations after administration of nebulized tobramycin. Pharmacotherapy. 2003; 23(4):543–545.
56. Montero, M, Horcajada, JP, Sorlí, L, et al. Effectiveness and safety of colistin for the treatment of multidrug-resistant Pseudomonas aeruginosa infections. Infection. 2009; 37(5):461–465.
57. Sabuda, DM, Laupland, K, Pitout, J, et al. Utilization of colistin for treatment of multidrug-resistant Pseudomonas aeruginosa. Can J Infect Dis Med Microbiol. 2008; 19(6):413–418.
58. Kofteridis, DP, Alexopoulou, C, Valachis, A, et al. Aerosolized plus intravenous colistin versus intravenous colistin alone for the treatment of ventilator-associated pneumonia: A matched case-control study. Clin Infect Dis. 2010; 51(11):1238–1244.
59. Imberti, R, Cusato, M, Villani, P, et al. Steady-state pharmacokinetics and BAL concentration of colistin in critically ill patients after IV colistin methanesulfonate administration. Chest. 2010; 138(6):1333–1339.
60. Rattanaumpawan, P, Lorsutthitham, J, Ungprasert, P, et al. Randomized controlled trial of nebulized colistimethate sodium as adjunctive therapy of ventilator-associated pneumonia caused by gram-negative bacteria. J Antimicrob Chemother. 2010; 65(12):2645–2649.
61. Moellering, RC, Jr., Wennersten, C, Weinberg, AN. Studies on antibiotic synergism against enterococci. I. Bacteriologic studies. J Lab Clin Med. 1971; 77(5):821–828.
62. Rahal, JJ, Jr. Antibiotic combinations: The clinical relevance of synergy and antagonism. Medicine. 1978; 57(2):179–195.
63. Korzeniowski, O, Sande, MA. Combination antimicrobial therapy for Staphylococcus aureus endocarditis in patients addicted to parenteral drugs and in nonaddicts: A prospective study. Ann Intern Med. 1982; 97(4):496–503.
64. Moellering, RC, Jr., Eliopoulos, GM. Principles of anti-infective therapy. In: Mandel GL, Bennett JE, Dolin R, eds. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 6th ed. Philadelphia: Churchill Livingstone; 2005:242–253.
65. Hilf, M, Yu, VL, Sharp, JA, et al. Antibiotic therapy for Pseudomonas aeruginosa bacteremia: Outcome correlations in a prospective study of 200 patients. Am J Med. 1989; 87(5):540–546.
66. Lepper, MH, Dowling, HF. Treatment of pneumococcic meningitis with penicillin compared with penicillin plus aureomycin: Studies including observations on an apparent antagonism between penicillin and aureomycin. Arch Intern Med. 1951; 88(4):489–494.
67. Blot, SI, Vandewoude, KH, Hoste, EA, Colardyn, FA. Outcome and attributable mortality in critically ill patients with bacteremia involving methicillin-susceptible and methicillin-resistant Staphylococcus aureus. Arch Intern Med. 2002; 162(19):2229–2235.
68. Cosgrove, SE, Sakoulas, G, Perencevich, EN, et al. Comparison of mortality associated with methicillin-resistant and methicillin-susceptible Staphylococcus aureus bacteremia: A meta-analysis. Clin Infect Dis. 2003; 36(1):53–59.
69. Shorr, AF, Combes, A, Kollef, MH, Chastre, J. Methicillin-resistant Staphylococcus aureus prolongs intensive care unit stay in ventilator-associated pneumonia, despite initially appropriate antibiotic therapy. Crit Care Med. 2006; 34(3):700–706.
70. Shorr, AF, Tabak, YP, Gupta, V, et al. Morbidity and cost burden of methicillin-resistant Staphylococcus aureus in early onset ventilator-associated pneumonia. Crit Care. 2006; 10(3):R97.
71. Grohs, P, Kitzis, MD, Gutmann, L. In vitro bactericidal activities of linezolid in combination with vancomycin, gentamicin, ciprofloxacin, fusidic acid, and rifampin against Staphylococcus aureus. Antimicrob Agents Chemother. 2003; 47(1):418–420.
72. Sweeney, MT, Zurenko, GE. In vitro activities of linezolid combined with other antimicrobial agents against staphylococci, enterococci, pneumococci, and selected gram-negative organisms. Antimicrob Agents Chemother. 2003; 47(6):1902–1906.
73. Sahuquillo, AJM, Colombo, GM, Gil, BA, et al. In vitro activity of linezolid in combination with doxycycline, fosfomycin, levofloxacin, rifampicin, and vancomycin against methicillin-susceptible Staphylococcus aureus. Rev Esp Quimioter. 2006; 19(3):252–257.
74. Chiang, FY, Climo, M. Efficacy of linezolid alone or in combination with vancomycin for treatment of experimental endocarditis due to methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2003; 47(9):3002–3004.
75. Liu, C, Bayer, A, Cosgrove, SE, et al. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. 2011; 52(3):e18–55.
76. Paul, M, Benuri-Silbiger, I, Soares-Weiser, K, Leibovici, L. Beta lactam monotherapy versus beta lactam-aminoglycoside combination therapy for sepsis in immunocompetent patients: Systematic review and meta-analysis of randomised trials. BMJ. 2004; 328(7441):668–681.
77. Allan, JD, Jr. Antibiotic combinations. Med Clin North Am. 1987; 71(6):1079–1091.
78. Bliziotis, IA, Samonis, G, Vardakas, KZ, et al. Effect of aminoglycoside and beta-lactam combination therapy versus beta-lactam monotherapy on the emergence of antimicrobial resistance: A meta-analysis of randomized, controlled trials. Clin Infect Dis. 2005; 41(2):149–158.
79. Harbarth, S, Nobre, V, Pittet, D. Does antibiotic selection impact patient outcome? Clin Infect Dis. 2007; 44(1):87–93.
80. Eagle, H. Experimental approach to the problem of treatment failure with penicillin. I. Group A streptococcal infection in mice. Am J Med. 1952; 13(4):389–399.
81. Bisno, AL, Stevens, DL. Streptococcal infections of skin and soft tissues. N Engl J Med. 1996; 334(4):240–245.
82. Gemmell, CG, Peterson, PK, Schmeling, D, et al. Potentiation of opsonization and phagocytosis of Streptococcus pyogenes following growth in the presence of clindamycin. J Clin Invest. 1981; 67(5):1249–1256.
83. Stevens, DL, Bryant, AE, Hackett, SP. Antibiotic effects on bacterial viability, toxin production, and host response. Clin Infect Dis. 1995; 20(Suppl 2):S154–S157.
84. Francis, JS, Doherty, MC, Lopatin, U, et al. Severe community-onset pneumonia in healthy adults caused by methicillin-resistant Staphylococcus aureus carrying the Panton-Valentine leukocidin genes. Clin Infect Dis. 2005; 40(1):100–107.
85. Fridkin, SK, Hagerman, JC, Morrison, M, et al. Methicillin-resistant Staphylococcus aureus disease in three communities. N Engl J Med. 2005; 352(14):1436–1444.
86. Miller, LG, Perdreau-Remington, F, Rieg, G, et al. Necrotizing fasciitis caused by community-associated methicillin-resistant Staphylococcus aureus in Los Angeles. N Engl J Med. 2005; 352(14):1445–1453.
87. Stevens, DL, Ma, Y, Salmi, DB, et al. Impact of antibiotics on expression of virulence-associated exotoxin genes in methicillin-sensitive and methicillin-resistant Staphylococcus aureus. J Infect Dis. 2007; 195(2):202–211.
88. Baddour, LM, Yu, VL, Klugman, KP, et al. Combination antibiotic therapy lowers mortality among severely ill patients with pneumococcal bacteremia. Am J Respir Crit Care Med. 2004; 170(4):440–444.
89. Garcia Vazquez, E, Mensa, J, Martinez, JA, et al. Lower mortality among patients with community-acquired pneumonia treated with a macrolide plus a beta-lactam agent versus a beta-lactam agent alone. Eur J Clin Microbiol Infect Dis. 2005; 24(3):190–195.
90. Martinez, FJ. Monotherapy versus dual therapy for community-acquired pneumonia in hospitalized patients. Clin Infect Dis. 2004; 38(Suppl 4):S328–S340.
91. Martinez, JA, Horcajada, JP, Almela, M, et al. Addition of a macrolide to a beta-lactam-based empirical antibiotic regimen is associated with lower in-hospital mortality for patients with bacteremic pneumococcal pneumonia. Clin Infect Dis. 2003; 36(4):389–395.
92. Mufson, MA, Stanek, RJ. Bacteremic pneumococcal pneumonia in one American city: A 20-year longitudinal study, 1978-1997. Am J Med. 1999; 107(1A):34S–43S.
93. Mufson, MA, Stanek, RJ. Revisiting combination antibiotic therapy for community-acquired invasive Streptococcus pneumoniae pneumonia. Clin Infect Dis. 2006; 42(2):304–306.
94. Waterer, GW. Monotherapy versus combination antimicrobial therapy for pneumococcal pneumonia. Curr Opin Infect Dis. 2005; 18(2):157–163.
95. Waterer, GW, Somes, GW, Wunderink, RG. Monotherapy may be suboptimal for severe bacteremic pneumococcal pneumonia. Arch Intern Med. 2001; 161(15):1837–1842.
96. Weiss, K, Low, DE, Cortes, L, et al. Clinical characteristics at initial presentation and impact of dual therapy on the outcome of bacteremic Streptococcus pneumoniae pneumonia in adults. Can Respir J. 2004; 11(8):589–593.
97. Iregui, M, Ward, S, Sherman, G, et al. Clinical importance of delays in the initiation of appropriate antibiotic treatment for ventilator-associated pneumonia. Chest. 2002; 122(1):262–268.
98. Höffken, G, Niederman, MS. Nosocomial pneumonia: The importance of a de-escalating strategy for antibiotic treatment of pneumonia in the ICU. Chest. 2002; 122(6):2183–2196.
99. Houck, PM, Bratzler, DW, Nsa, W, et al. Timing of antibiotic administration and outcomes for Medicare patients hospitalized with community-acquired pneumonia. Arch Intern Med. 2004; 164(6):637–644.
100. Tunkel, AR, Hartman, BJ, Kaplan, SL, et al. Practice guidelines for the management of bacterial meningitis. Clin Infect Dis. 2004; 39(9):1267–1284.
101. Aronin, SI, Peduzzi, P, Quagliarello, VJ. Community-acquired bacterial meningitis: Risk stratification for adverse clinical outcome and effect of antibiotic timing. Ann Intern Med. 1998; 129(11):862–869.
102. Dellinger, RP, Levy, MM, Carlett, JM, et al. Surviving sepsis campaign: International guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med. 2008; 36(1):296–327.
103. Silverman, JA, Mortin, LI, VanPraagh, AD, et al. Inhibition of daptomycin by pulmonary surfactant: In vitro modeling and clinical impact. J Infect Dis. 2005; 191(12):2149–2152.
104. Fuchs, PC, Barry, AL, Brown, SD. In vitro bactericidal activity of daptomycin against staphylococci. J Antimicrob Chemother. 2002; 49(3):467–470.
105. King, A, Phillips, I. The in vitro activity of daptomycin against 514 gram-positive aerobic clinical isolates. J Antimicrob Chemother. 2001; 48(2):219–223.
106. Wise, R, Andrews, JM, Ashby, JP. Activity of daptomycin against gram-positive pathogens: A comparison with other agents and the determination of a tentative breakpoint. J Antimicrob Chemother. 2001; 48(4):563–567.
107. Silverman, JA, Perlmutter, NG, Shapiro, HM. Correlation of daptomycin bactericidal activity and membrane depolarization in Staphylococcus aureus. Antimicrob Agents Chemother. 2003; 47(8):2538–2544.
108. Arbeit, RD, Maki, D, Tally, FP, et al. The safety and efficacy of daptomycin for the treatment of complicated skin and skin-structure infections. Clin Infect Dis. 2004; 38(12):1673–1681.
109. Fowler, VG, Jr., Boucher, HW, Corey, GR, et al. Daptomycin versus standard therapy for bacteremia and endocarditis caused by Staphylococcus aureus. N Engl J Med. 2006; 355(7):653–665.
110. Jung, D, Rozek, A, Okon, M, Hancock, REW, et al. Structural transitions as determinants of the action of the calcium-dependent antibiotic daptomycin. Chem Biol. 2004; 11(7):949–957.
111. Lakey, JH, Ptak, M. Fluorescence indicates a calcium-dependent interaction between the lipopeptide antibiotic LY146032 and phospholipid membranes. Biochemistry. 1988; 27(13):4639–4645.
112. Paterson, DL. “Collateral damage” from cephalosporin or quinolone antibiotic therapy. Clin Infect Dis. 2004; 38(Suppl 4):S341–S345.
113. Hota, B. Infection control or formulary control: What is the best tool to reduce nosocomial infections due to methicillin-resistant Staphylococcus aureus? Clin Infect Dis. 2006; 42(6):785–787.
114. National Nosocomial Infections Surveillance System. National Nosocomial Infections Surveillance (NNIS) System Report, data summary from January 1992 through June 2004, issued October 2004. Am J Infect Control. 2004; 32(8):470–485.
115. Bisognano, C, Vaudaux, P, Rohner, P, et al. Induction of fibronectin-binding proteins and increased adhesion of quinolone-resistant Staphylococcus aureus by subinhibitory levels of ciprofloxacin. Antimicrob Agents Chemother. 2000; 44(6):1428–1437.
116. Cohen, SH, Gerding, DN, Johnson, S, et al. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol. 2010; 31(5):431–455.
117. Blazquez, J. Hypermutation as a factor contributing to the acquisition of antimicrobial resistance. Clin Infect Dis. 2003; 37(9):1201–1209.
118. Loo, VG, Poirier, L, Miller, LA, et al. A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality. N Engl J Med. 2005; 353(23):2442–2449.
119. McDonald, LC, Killgore, GE, Thompson, A, et al. An epidemic, toxin gene-variant strain of Clostridium difficile. N Engl J Med. 2005; 353(23):2433–2441.
120. Pépin, J, Saheb, N, Coulombe, MA, et al. Emergence of fluoroquinolones as the predominant risk factor for Clostridium difficile-associated diarrhea: A cohort study during an epidemic in Quebec. Clin Infect Dis. 2005; 41(9):1254–1260.
121. Trouillet, JL, Chastre, J, Vuagnat, A, et al. Ventilator-associated pneumonia caused by potentially drug-resistant bacteria. Am J Respir Crit Care Med. 1998; 157(2):531–539.
122. Lister, PD, Wolter, DJ. Levofloxacin-imipenem combination prevents the emergence of resistance among clinical isolates of Pseudomonas aeruginosa. Clin Infect Dis. 2005; 40(Suppl 2):S105–S114.
123. Quale, J, Bratu, S, Gupta, J, Landman, D, et al. Interplay of efflux system, ampC, and oprD expression in carbapenem resistance of Pseudomonas aeruginosa clinical isolates. Antimicrob Agents Chemother. 2006; 50(5):1633–1641.
124. Gaynes, R, Edwards, JR. The National Nosocomial Infections Surveillance System, overview of nosocomial infections caused by gram-negative bacilli. Clin Infect Dis. 2005; 41:848–854.
125. Sanders, WE, Jr., Sanders, CC. Enterobacter spp. : Pathogens poised to flourish at the turn of the century. Clin Microbiol Rev. 1997; 10(2):220–241.
126. Sanders, CC, Sanders, WE, Jr. Type I beta-lactamases of gram-negative bacteria: Interactions with beta-lactam antibiotics. J Infect Dis. 1986; 154(5):792–800.
127. Chow, JW, Fine, MJ, Shlaes, DM, et al. Enterobacter bacteremia: Clinical features and emergence of antibiotic resistance during therapy. Ann Intern Med. 1991; 115(8):585–590.
128. Jacobson, KL, Cohen, SH, Inciardi, JF, et al. The relationship between antecedent antibiotic use and resistance to extended-spectrum cephalosporins in group I beta-lactamase-producing organisms. Clin Infect Dis. 1995; 21(5):1107–1113.
129. Paterson, DL, Bonomo, RA. Extended-spectrum beta-lactamases: A clinical update. Clin Microbiol Rev. 2005; 18(4):657–686.
130. Paterson, DL, Ko, W-C, Von Gottberg, A, et al. International prospective study of Klebsiella pneumoniae bacteremia: Implications of extended-spectrum beta-lactamase production in nosocomial infections. Ann Intern Med. 2004; 140(1):26–32.
131. Rodríguez-Baño, J, Navarro, MD, Romero, L, et al. Clinical and molecular epidemiology of extended-spectrum beta-lactamase-producing Escherichia coli as a cause of nosocomial infection or colonization: Implications for control. Clin Infect Dis. 2006; 42(1):37–45.
132. Paterson, DL, Ko, W-C, Von Gottberg, A, et al. Antibiotic therapy for Klebsiella pneumoniae bacteremia: Implications of production of extended-spectrum beta-lactamases. Clin Infect Dis. 2004; 39(1):31–37.
133. Jacoby, GA, Munoz-Price, LS. The new beta-lactamases. N Engl J Med. 2005; 352(4):380–391.
134. Craig, WA, Bhavnani, SM, Ambrose, PG. The inoculum effect: Fact or artifact? Diagn Microbiol Infect Dis. 2004; 50(4):229–230.
135. Wang, M, Sahm, DF, Jacoby, GA, et al. Emerging plasmid-mediated quinolone resistance associated with the qnr gene in Klebsiella pneumoniae clinical isolates in the United States. Antimicrob Agents Chemother. 2004; 48(4):1295–1299.
136. Wang, CX, Cai, PQ, Guo, Y, et al. Emerging plasmid-mediated quinolone resistance associated with the qnrA gene in Enterobacter cloacae clinical isolates in China. J Hosp Infect. 2006; 63(3):349–350.
137. Gruteke, P, Goessens, W, Van Gils, J, et al. Patterns of resistance associated with integrons, the extended-spectrum beta-lactamase SHV-5 gene, and a multidrug efflux pump of Klebsiella pneumoniae causing a nosocomial outbreak. J Clin Microbiol. 2003; 41(3):1161–1166.
138. Pitout, JD, Nordmann, P, Laupland, KB, Poirel, L. Emergence of Enterobacteriaceae producing extended-spectrum beta-lactamases (ESBLs) in the community. J Antimicrob Chemother. 2005; 56(1):52–59.
139. Ben-Ami, R, Rodríguez-Baño, J, Arslan, H, et al. A multinational survey of risk factors for infection with extended-spectrum beta-lactamase-producing Enterobacteriaceae in nonhospitalized patients. Clin Infect Dis. 2009; 49(5):682–690.
140. Qi, C, Pilla, V, Yu, JH, Reed, K. Changing prevalence of Escherichia coli with CTX-M-type extended-spectrum beta-lactamases in outpatient urinary E. coli between 2003 and 2008. Diagn Microbiol Infect Dis. 2010; 67(1):87–91.
141. Paramythiotou, E, Lucet, JC, Timsit, JF, et al. Acquisition of multidrug-resistant Pseudomonas aeruginosa in patients in intensive care units: Role of antibiotics with antipseudomonal activity. Clin Infect Dis. 2004; 38(5):670–677.
142. Carmeli, Y, Lidji, SK, Shabtai, E, et al. The effects of group 1 versus group 2 carbapenems on imipenem-resistant Pseudomonas aeruginosa: An ecological study. Diagn Microbiol Infect Dis. 2011; 70(3):367–372.
143. Nicolau, DP, Carmeli, Y, Crank, CW, et al. Carbapenem stewardship: Does ertapenem affect Pseudomonas susceptibility to other carbapenems? A review of the evidence. Int J Antimicrob Agents. 2012; 39(1):11–15.
144. Rodriguez-Bano, J, Navarro, MD, Retamar, P, et al. Beta-Lactam/beta-lactam inhibitor combinations for the treatment of bacteremia due to extended-spectrum beta-lactamase-producing Escherichia coli: A post hoc analysis of prospective cohorts. Clin Infect Dis. 2012; 54(2):167–174.
145. Queenan, AM, Bush, K. Carbapenemases: The versatile beta-lactamases. Clin Microbiol Rev. 2007; 20(3):440–458.
146. Patel, N, Harrington, S, Dihmess, A, et al. Clinical epidemiology of carbapenem-intermediate or -resistant Enterobacteriaceae. J Antimicrob Chemother. 2011; 66(7):1600–1608.
147. Kritsotakis, EI, Tsioutis, C, Roumbelaki, M, et al. Antibiotic use and the risk of carbapenem-resistant extended-spectrum-β-lactamase-producing Klebsiella pneumoniae infection in hospitalized patients: Results of a double case-control study. J Antimicrob Chemother. 2011; 66(6):1383–1391.
148. Hirsch, EB, Tam, VH. Detection and treatment options for Klebsiella pneumoniae carbapenemases (KPCs): An emerging cause of multidrug-resistant infection. J Antimicrob Chemother. 2010; 65(6):1119–1125.
149. Quale, J, Spelman, D. Carbapenemases. In: Basow D, ed. UpToDate. Waltham, MA: UpToDate, 2012.
150. Daikos, GL, Markogiannakis, A. Carbapenemase-producing Klebsiella pneumoniae: When might we still consider treating with carbapenems? Clin Microbiol Infect. 2010; 17(8):1135–1141.
151. Bratu, S, Landman, D, Haag, R, et al. Rapid spread of carbapenem-resistant Klebsiella pneumoniae in New York City: A new threat to our antibiotic armamentarium. Arch Intern Med. 2005; 165(12):1430–1435.
152. Landman, D, Quale, JM, Mayorga, D, et al. Citywide clonal outbreak of multiresistant Acinetobacter baumannii and Pseudomonas aeruginosa in Brooklyn, NY: The preantibiotic era has returned. Arch Intern Med. 2002; 162(13):1515–1520.
153. Levy, SB. Active efflux mechanisms for antimicrobial resistance. Antimicrob Agents Chemother. 1992; 36(4):695–703.
154. Livermore, DM. Multiple mechanisms of antimicrobial resistance in Pseudomonas aeruginosa: Our worst nightmare? Clin Infect Dis. 2002; 34(5):634–640.
155. Nikaido, H. Antibiotic resistance caused by gram-negative multidrug efflux pumps. Clin Infect Dis. 1998; 27(Suppl 1):S32–S41.
156. Masuda, N, Sakagawa, E, Ohya, S, et al. Substrate specificities of MexAB-OprM, MexCD-OprJ, and MexXY-oprM efflux pumps in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2000; 44(12):3322–3327.
157. Poole, K. Multidrug efflux pumps and antimicrobial resistance in Pseudomonas aeruginosa and related organisms. J Mol Microbiol Biotechnol. 2001; 3(2):255–264.
158. Livermore, DM. Of Pseudomonas, porins, pumps and carbapenems. J Antimicrob Chemother. 2001; 47(3):247–250.
159. Chastre, J, Fagon, JY. Ventilator-associated pneumonia. Am J Respir Crit Care Med. 2002; 165(7):867–903.
160. Urban, C, Segal-Maurer, S, Rahal, JJ. Considerations in control and treatment of nosocomial infections due to multidrug-resistant Acinetobacter baumannii. Clin Infect Dis. 2003; 36(10):1268–1274.
161. Hanes, SD, Demirkan, K, Tolley, E, et al. Risk factors for late-onset nosocomial pneumonia caused by Stenotrophomonas maltophilia in critically ill trauma patients. Clin Infect Dis. 2002; 35(3):228–235.
162. Carmeli, Y, Samore, MH. Comparison of treatment with imipenem vs. ceftazidime as a predisposing factor for nosocomial acquisition of Stenotrophomonas maltophilia: A historical cohort study. Clin Infect Dis. 1997; 24(6):1131–1134.
163. Hayashi, Y, Paterson, DL. Strategies for reduction in duration of antibiotic use in hospitalized patients. Clin Infect Dis. 2011; 52(10):1232–1240.
164. Fowler, VG, Jr., Olsen, MK, Corey, GR, et al. Clinical identifiers of complicated Staphylococcus aureus bacteremia. Arch Intern Med. 2003; 163(17):2066–2072.
165. Mermel, LA, Allon, M, Bouza, E, et al. Clinical practice guidelines for the diagnosis and management of intravascular catheter-related infection: 2009 Update by the Infectious Diseases Society of America. Clin Infect Dis. 2009; 49(1):1–45.
166. Dennesen, PJW, van der Ven, AJ, Kessels, AGH, et al. Resolution of infectious parameters after antimicrobial therapy in patients with ventilator-associated pneumonia. Am J Respir Crit Care Med. 2001; 163(6):1371–1375.
167. Ibrahim, EH, Ward, S, Sherman, G, et al. Experience with a clinical guideline for the treatment of ventilator-associated pneumonia. Crit Care Med. 2001; 29(6):1109–1115.
168. Chastre, J, Wolff, M, Fagon, JY, et al. Comparison of 8 vs. 15 days of antibiotic therapy for ventilator-associated pneumonia in adults: A randomized trial. JAMA. 2003; 290(19):2588–2598.
169. Burke, JP, Pestotnik, SL. Antibiotic use and microbial resistance in intensive care units: Impact of computer-assisted decision support. J Chemother. 1999; 11(6):530–535.
170. Bergstrom, CT, Lo, M, Lipsitch, M. Ecological theory suggests that antimicrobial cycling will not reduce antimicrobial resistance in hospitals. Proc Natl Acad Sci U S A. 2004; 101(36):13285–13290.