[level-membership-for-pediatrics-category]
Chapter 120 Primary Combined Antibody and Cellular Immunodeficiencies
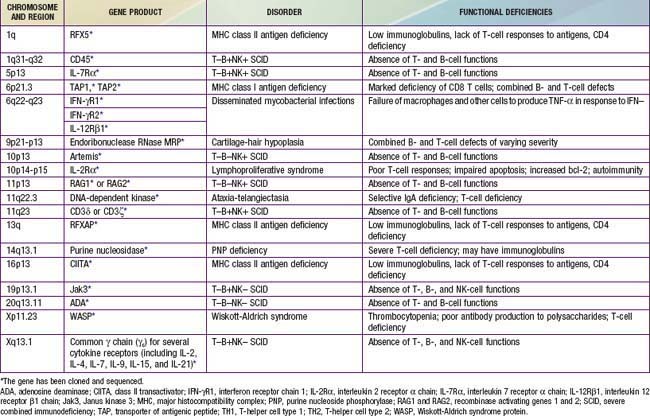
Patients with combined antibody and cellular defects have severe, frequently opportunistic infections that lead to death in infancy or childhood unless they are provided hematopoietic stem cell transplantation early in life. These are thought to be rare defects, although the true incidences are unknown because there has been no newborn screening for any of these defects. It is possible that many affected children die of infection during infancy without being diagnosed. The defective gene products for many combined immunodeficiencies are identified (Table 120-1). Because life threatening infection may occur in infancy, screening for SCID has been recommended by the U.S. Secretary of Health and Human Services to be included in the state newborn screening programs. Live, vaccine-derived infections have occurred during this time of life and knowledge of SCID status could prevent these infections. In addition, early identification and subsequent bone marrow transplantation before life-threatening infections and end organ injury is the best approach to therapy.
120.1 Severe Combined Immunodeficiency (SCID)
Pathogenesis
SCID results from mutations in any 1 of 13 known genes that encode components of the immune system crucial for lymphoid cell development (Table 120-2). All patients with SCID have very small thymuses (<1 g) that usually fail to descend from the neck, contain no thymocytes, and lack corticomedullary distinction or Hassall corpuscles. The thymic epithelium appears histologically normal. Both the follicular and paracortical areas of the spleen are depleted of lymphocytes. Lymph nodes, tonsils, adenoids, and Peyer patches are absent or extremely underdeveloped.
Table 120-2 PATHOPHYSIOLOGY MECHANISMS THAT ACCOUNT FOR SEVERE COMBINED IMMUNE DEFICIENCY (SCID)
| DISEASE MECHANISM | GENE DEFECTS |
|---|---|
| Increased apoptosis | |
| • Due to mitochondrial energy failure | AK2 |
| • Due to accumulation of toxic metabolites | ADA |
| • Due to abnormal actin polymerization | CORO1A |
| Impaired cytokine-mediated signaling | |
| • Due to defects of the common γ chain | IG2RG (X-linked SCID) |
| • Due to defects of the IL-7R α chain | IL7R |
| • Due to defects of Jak3 | JAK3 |
| Impaired signaling through the pre–T cell receptor | |
| • Due to defective V(D)J recombination | RAG1, RAG2, DCLRE1C, LIG4,* PRKDC |
| • Due to impaired expression of CD3 subunits | CD3D, CD3E, CD3Z |
| Impaired signaling in the periphery | ORA1 |
| Unknown mechanism | RMRP* |
* These gene defects are most often associated with a milder clinical phenotype than SCID.
From Pessach I, Walter J, Notarangelo LD: Recent advances in primary immunodeficiencies: identification of novel genetic defects and unanticipated phenotypes, Pediatr Res 65:3R–12R, 2009.
Clinical Manifestations
Because all molecular types of SCID lack T cells, infants with SCID have lymphopenia (<2,500/mm3) that is present at birth, indicating that the condition could be diagnosed in all affected infants if white blood cell counts with manual differential counts were routinely performed on all cord bloods and the absolute lymphocyte count calculated. These infants also have an absence of lymphocyte proliferative responses to mitogens, antigens, and allogeneic cells in vitro. Patients with adenosine deaminase (ADA) deficiency have the lowest absolute lymphocyte counts, usually <500/mm3. Serum immunoglobulin concentrations are low or absent, and no antibodies are formed after immunizations. Analyses of lymphocyte populations and subpopulations demonstrate distinctive phenotypes for the various genetic forms of SCID (see Table 120-2). T cells are extremely low or absent in all types; when detected, in most cases they are transplacentally derived maternal T cells.
X-Linked Severe Combined Immunodeficiency (SCIDX1) Due To Mutations in the Gene Encoding the Common Cytokine Receptor γ Chain (γC)
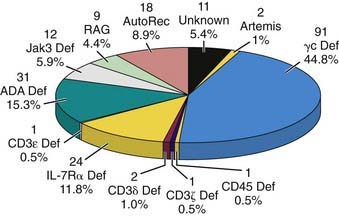
X-linked SCID (X-SCD) is the most common form of SCID in the USA, accounting for 47% of cases (Fig. 120-1). Clinically, immunologically, and histopathologically, affected individuals appear similar to those with other forms of SCID except for having uniformly low percentages of T and NK cells and an elevated percentage of B cells (T−, B+, NK−), a characteristic feature shared only with Janus kinase 3 (Jak3)–deficient SCID. The abnormal gene in X-SCD was mapped to Xq13, cloned, and found to encode the common γ chain (γc) for several cytokine receptors, including IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21. The shared γc functions both to increase the affinity of the receptor for the respective cytokine and to enable the receptors to mediate intracellular signaling. Incapacitation of the receptors for all of these developmentally crucial cytokines by genetic mutations in γc provides an explanation for the severity of the immunodeficiency in SCIDX1. In the 1st 136 patients studied, 95 distinct mutations spanning all 8 IL2RG exons were identified, most of them consisting of small changes at the level of 1 to a few nucleotides. These mutations resulted in abnormal γc chains in two thirds of the cases and absent γc protein in the remainder. Carriers can be detected by demonstrating nonrandom X-chromosome inactivation or the deleterious mutation in their T, B, or NK lymphocytes. Unless donor B or NK cells develop, patients with X-SCID lack B- and NK-cell function after bone marrow transplantation because the abnormal γc persists in those host cells, despite excellent reconstitution of T-cell function by donor-derived T cells.
Autosomal Recessive Severe Combined Immunodeficiency
This pattern of inheritance of SCID is less common in the USA than in Europe. Mutated genes on autosomal chromosomes have been identified in 12 forms of SCID: ADA deficiency; Jak3 deficiency; IL-7 receptor α chain (IL-7Rα) deficiency; RAG1 or RAG2 deficiency; Artemis deficiency; ligase 4 deficiency; DNA–protein kinase catalytic subunit (DNA-PKcs) deficiency; CD3δ, CD3ε, CD3ζ deficiency; and CD45 deficiency (see Fig. 120-1).
IL-7Rα Deficiency
Patients with IL-7Rα-deficient SCID have a distinctive lymphocyte phenotype in that, though lacking T cells, they have normal or elevated numbers of both B and NK cells (T−, B+, NK+). This is the third most common form of SCID, accounting for 12% of cases in the USA (see Fig. 120-1). In contrast to patients with γc– and Jak3-deficient SCID, the immunologic defect in these patients is completely correctable by bone marrow stem cell transplantation, as the host B and NK cells appear to be normal.
120.2 Combined Immunodeficiency (CID)
Cartilage Hair Hypoplasia
Genetics and Pathogenesis
CHH is an autosomal recessive condition. Numerous mutations that co-segregate with the CHH phenotype have been identified in the untranslated RNase MRP gene, which has been mapped to chromosome 9p21-p13 in Amish and Finnish families (see Table 120-1). The RNase MRP endoribonuclease consists of an RNA molecule bound to several proteins and has at least two functions: cleavage of RNA in mitochondrial DNA synthesis and nucleolar cleaving of pre-RNA. Mutations in RMRP cause CHH by disrupting a function of RNase MRP RNA that affects multiple organ systems. In vitro studies show decreased numbers of T cells and defective T-cell proliferation due to an intrinsic defect related to the G1 phase, resulting in a longer cell cycle for individual cells. NK cells are increased in number and function.
Defective Expression of Major Histocompatibility Complex Antigens
MHC Class I Antigen Deficiency
Isolated deficiency of MHC class I (HLA-A, -B, and -C) antigens, the bare lymphocyte syndrome, is rare. The resulting immunodeficiency is much milder than in SCID, contributing to a later age of presentation. Sera from affected children contain normal quantities of MHC class I antigens and β2-microglobulin, but MHC class I antigens are not detected on any cells in the body. There is a deficiency of CD8 but not CD4 T cells. Mutations have been found in 2 genes within the MHC locus on chromosome 6 that encode the peptide transporter proteins TAP1 and TAP2 (see Fig. 118-1). TAP functions to transport antigenic peptides from the cytoplasm across the Golgi apparatus membrane to join the α chain of MHC class I antigens and β2-microglobulin. All these are then assembled into a MHC class I complex that can then move to the cell surface. If the assembly of the complex cannot be completed because there is no antigenic peptide, the MHC class I complex is destroyed in the cytoplasm.
MHC Class II Antigen Deficiency
Four different molecular defects resulting in impaired expression of MHC class II antigens have been identified (see Table 120-1 and Fig. 118-1). One form is a mutation in the gene on chromosome 1q that encodes a protein called RFX5, a subunit of RFX, which is a multiprotein complex that binds the X box motif of MHC-II promoters. A second form is caused by mutations in a gene on chromosome 13q that encodes a second 36-kD subunit of the RFX complex, called RFX-associated protein (RFXAP). The most common cause of MHC class II defects is mutation in RFXANK, the gene encoding a 3rd subunit of RFX. In a 4th type, there is a mutation in the gene on chromosome 16p13 that encodes a novel MHC class II transactivator (CIITA), a non–DNA-binding co-activator that controls the cell-type specificity and inducibility of MHC-II expression. All 4 of these defects cause impairment in the coordinate expression of MHC class II molecules on the surface of B cells and macrophages.
Immunodeficiency with Thrombocytopenia and Eczema (Wiskott-Aldrich Syndrome)
Genetics and Pathogenesis
The abnormal gene, on the proximal arm of the X chromosome at Xp11.22-11.23 near the centromere, encodes a 501–amino acid proline-rich cytoplasmic protein restricted in its expression to hematopoietic cell lineages. The Wiskott-Aldrich syndrome protein (WASP) (see Fig. 119-1) binds CDC42H2 and rac, members of the Rho family of guanosine triphosphatases. WASP appears to control the assembly of actin filaments required for microvesicle formation downstream of protein kinase C and tyrosine kinase signaling. Carriers can be detected by nonrandom X-chromosome inactivation in several hematopoietic cell lineages or by demonstration of the deleterious mutation.
Ataxia-Telangiectasia
Genetics and Pathogenesis
The mutated gene responsible for this defect, ataxia telangiectasia mutation (ATM), was mapped to the long arm of chromosome 11 (11q22-23) and has been cloned (see Fig. 119-1). The gene product is a DNA-dependent protein kinase localized predominantly to the nucleus and involved in mitogenic signal transduction, meiotic recombination, and cell cycle control. Cells from patients as well as those of heterozygous carriers have increased sensitivity to ionizing radiation, defective DNA repair, and frequent chromosomal abnormalities.
120.3 Defects of Innate Immunity
Hyper-IgE Syndrome
The hyper-IgE syndrome is a relatively rare primary immunodeficiency syndrome characterized by recurrent severe staphylococcal abscesses of the skin, lungs, and other viscera as well as sinusitis, mastoiditis, and markedly elevated levels of serum IgE (Table 120-3). C. albicans is the second most common pathogen. More than 200 patients with hyper-IgE syndrome have been reported. The most common form of this condition (autosomal dominant) is now known to be caused by mutations in the gene encoding STAT-3. These mutations result in a dominant negative effect on the expression of STAT-3 by the other nonmutated gene. Rarely, autosomal recessive forms of the hyper-IgE syndrome have been reported, mainly in Turkey, and a mutation in the gene encoding Tyk2 was found in one such patient but not in the others.
Table 120-3 CLINICAL FEATURES OF AUTOSOMAL DOMINANT HYPER IgE SYNDROME (AD-HIES)
IMMUNOLOGIC (APPROXIMATE % FREQUENCY)
SOMATIC (APPROXIMATE % FREQUENCY)
From Freeman AF, Holland SM: Clinical manifestations, etiology, and pathogenesis of the hyper-IgE syndromes, Pediatr Res 65:32R–37R, 2009.
120.4 Treatment of Cellular or Combined Immunodeficiency
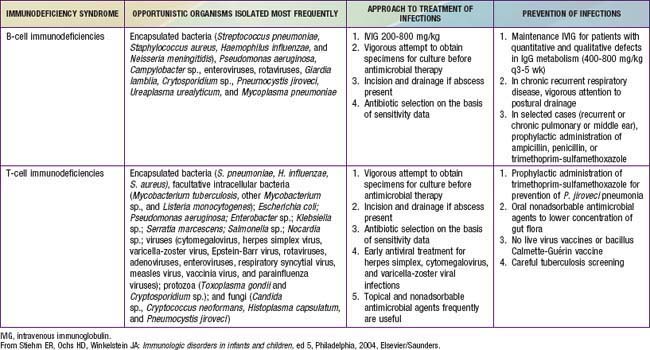
Good supportive care including prevention and treatment of infections is critical while patients await more definitive therapy (Table 120-4). Having knowledge of the pathogens causing disease with specific immune defects is also useful (see Table 120-4).
120.5 Immune Dysregulation with Autoimmunity or Lymphoproliferation
Autoimmune Lymphoproliferative Syndrome (ALPS)
ALPS, also known as Canale-Smith syndrome, is a disorder of abnormal lymphocyte apoptosis leading to polyclonal populations of T cells (double-negative T cells), which express CD3 and α/β antigen receptors but do not have CD4 or CD8 co-receptors (CD3 + T cell receptor α/β+ CD4− CD8−). These T cells respond poorly to antigens or mitogens and do not produce growth or survival factors (interleukin 2). The genetic deficit in most patients is a germ line or somatic mutation in the Fas gene, which produces a cell surface receptor of the tumor necrosis factor receptor superfamily (TNFRSF6), which, when stimulated by its ligand, will produce programmed cell death (Table 120-5). Persistent survival of these lymphocytes leads to immune dysregulation and autoimmunity.
Table 120-5 ALPS CASE CRITERIA AND ALPS CLASSIFICATION
REQUIRED
SUPPORTING
ALPS Ia = due to mutation in TNFRSF6
ALPS Ib = due to mutation in the gene for Fas ligand
ALPS II = due to mutation in the gene for caspase 10
ALPS III = ALPS without defined genetic cause
From Straus SE, Sneller M, Lenardo MJ, et al: An inherited disorder of lymphocyte apoptosis: the autoimmune lymphoproliferative syndrome, Ann Intern Med 130:591–601, 1999; Bleesing JJH, Straus SE, Fleisher TA: Autoimmune lymphoproliferative syndrome: a human disorder of abnormal lymphocyte survival, Pediatr Clin North Am 47:1291–1310, 2000.
Clinical Manifestations
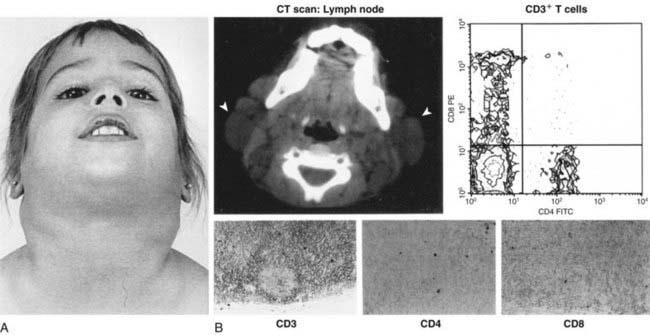
ALPS is characterized by autoimmunity, chronic persistent or recurrent lymphadenopathy, splenomegaly, hepatomegaly (in 50%), and hypergammaglobulinemia (IgG, IgA). Many patients present in the 1st yr of life, and most are symptomatic by yr 5. Lymphadenopathy can be striking (Fig. 120-2). Splenomegaly may produce hypersplenism with cytopenias. Autoimmunity also produces anemia (Coombs positive hemolytic anemia) or thrombocytopenia or a mild neutropenia. Lymphoproliferative process (lymphadenopathy, splenomegaly) may regress over time, but autoimmunity does not and is characterized by frequent exacerbations and recurrences. Other autoimmune features include urticaria, uveitis, glomerulonephritis, hepatitis, vasculitis, glomulonephritis, vasculitis, panniculitis, arthritis, and central nervous system involvement (seizures, headaches, encephalopathy).
Diagnosis
Laboratory abnormalities depend on the lymphoproliferative organ response (hypersplenism) or the degree of autoimmunity (anemia, thrombocytopenia). There may be lymphocytosis or lymphopenia. Criteria for the diagnosis are noted in Table 120-5. Flow cytometry helps identify the lymphocyte type (see Fig. 120-2). Functional genetic analysis for the TNFRSF6 gene often reveals a heterozygous mutation.
Adeli MM, Buckley RH. Why newborn screening for severe combined immunodeficiency is essential: a case report. Pediatrics. 2010;126(2):e465-e469.
Aiuti A, Cattaneo F, Galimberti S, et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. 2009;360:447-458.
Bouma G, Burns SO, Thrasher AJ. Wiskott-Aldrich Syndrome: immunodeficiency resulting from defective cell migration and impaired immunostimulatory activation. Immunobiology. 2009;214:778-790.
Buckley RH. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu Rev Immunol. 2004;22:625-655.
Casrouge A, Zhang SY, Eidenschenk C, et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science. 2006;314:308-312.
Freeman AF, Holland SM. Clinical manifestations, etiology, and pathogenesis of the hyper-IgE syndromes. Pediatr Res. 2009;6:32R-37R.
Geha RS, Notarangelo LD, Casanova JL, et al. Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. J Allergy Clin Immunol. 2007;120:776-794.
Hacein-Bey-Abina S, Hauer J, Lim A, et al. Efficacy of gene therapy for x-linked severe combined immunodeficiency. N Engl J Med. 2010;363:355-364.
Hacein-Bey-Abina S, Garrigue A, Wang GP, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118:3132-3142.
Holland SM, Deleo FR, Elloumi HZ, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;357:1608-1619.
Howe SJ, Mansour MR, Schwarzwaelder K, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118:3143-3150.
Ku CL, Yang K, Bustamante J, et al. Inherited disorders of human Toll-like receptor signaling: immunological implications. Immunol Rev. 2005;203:10-20.
Lagresle-Peyrou C, Six EM, Picard C, et al. Human adenylate kinase 2 deficiency causes a profound hematopoietic defect associated with sensorineural deafness. Nat Genet. 2009;41:106-111.
Ma CS, Chew GY, Simpson N, et al. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med. 2008;205:1551-1557.
Meyer-Bahlburg A, Becker-Herman S, Humblet-Baron S, et al. Wiskott-Aldrich syndrome protein deficiency in B cells results in impaired peripheral homeostasis. Blood. 2008;112:4158-4169.
Minegishi Y, Saito M, Tsuchiya S, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448:1058-1062.
Morinishi Y, Imai K, Nakagawa N, et al. Identification of severe combined immunodeficiency by T-cell receptor excision circles quantification using neonatal Guthrie cards. J Pediatr. 2009;155:829-833.
Pannicke U, Honig M, Hess I, et al. Reticular dysgenesis (aleukocytosis) is caused by mutations in the gene encoding mitochondrial adenylate kinase 2. Nat Genet. 2009;41:101-105.
Patel NC, Hertel PM, Estes MK, et al. Vaccine-acquired rotavirus in infants with severe combined immunodeficiency. N Engl J Med. 2010;362:314-319.
Pessach I, Walter J, Notarangelo LD. Recent advances in primary immunodeficiencies: identification of novel genetic defects and unanticipated phenotypes. Pediatr Res. 2009;65:3R-12R.
Railey MD, Lokhnygina Y, Buckley RH. Long-term clinical outcome of patients with severe combined immunodeficiency who received related donor bone marrow transplants without pretransplant chemotherapy post-transplant GVHD prophylaxis. J Pediatr. 2009;155:834-840.
Renner ED, Puck JM, Holland SM, et al. Autosomal recessive hyperimmunoglobulin E syndrome: a distinct disease entity. J Pediatr. 2004;144:93-99.
Roberts JL, Lauritsen JHP, Cooney M, et al. T-B+NK+ severe combined immunodeficiency caused by complete deficiency of the CD3 zeta subunit of the T cell antigen receptor complex. Blood. 2007;109:3198-3206.
Rosenzweig SD, Holland SM. Defects in the interferon-gamma and interleukin-12 pathways. Immunol Rev. 2005;203:38-47.
Routes JM, Grossman WJ, Verbsky J, et al. Statewide newborn screening for severe T-cell lymphopenia. JAMA. 2009;302:2465-2470.
Sarzotti-Kelsoe M, Win CM, Parrott RE, et al. Thymic output, T-cell diversity, and T-cell function in long-term human SCID chimeras. Blood. 2009;114:1445-1453.
Su MA, Anderson MS. Monogenic autoimmune diseases: insights into self-tolerance. Pediatr Res. 2009;65:20R-25R.
Tangye SG, Cook MC, Fulcher DA. Insights into the role of STAT3 in human lymphocyte differentiation as revealed by the hyper-IgE syndrome. J Immunol. 2009;182:21-28.
van der Burg M., Ijspeert H, Verkaik NS, et al. A DNA-PKcs mutation in a radiosensitive T-B- SCID patient inhibits Artemis activation and nonhomologous end-joining. J Clin Invest. 2009;119:91-98.
von Bernuth H, Picard C, Jin Z, et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science. 2008;321:691-696.
Zhang Q, Davis JC, Lamborn IT, et al. Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med. 2009;361:2046-2054.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 120 Primary Combined Antibody and Cellular Immunodeficiencies
Patients with combined antibody and cellular defects have severe, frequently opportunistic infections that lead to death in infancy or childhood unless they are provided hematopoietic stem cell transplantation early in life. These are thought to be rare defects, although the true incidences are unknown because there has been no newborn screening for any of these defects. It is possible that many affected children die of infection during infancy without being diagnosed. The defective gene products for many combined immunodeficiencies are identified (Table 120-1). Because life threatening infection may occur in infancy, screening for SCID has been recommended by the U.S. Secretary of Health and Human Services to be included in the state newborn screening programs. Live, vaccine-derived infections have occurred during this time of life and knowledge of SCID status could prevent these infections. In addition, early identification and subsequent bone marrow transplantation before life-threatening infections and end organ injury is the best approach to therapy.
120.1 Severe Combined Immunodeficiency (SCID)
Pathogenesis
SCID results from mutations in any 1 of 13 known genes that encode components of the immune system crucial for lymphoid cell development (Table 120-2). All patients with SCID have very small thymuses (<1 g) that usually fail to descend from the neck, contain no thymocytes, and lack corticomedullary distinction or Hassall corpuscles. The thymic epithelium appears histologically normal. Both the follicular and paracortical areas of the spleen are depleted of lymphocytes. Lymph nodes, tonsils, adenoids, and Peyer patches are absent or extremely underdeveloped.
Table 120-2 PATHOPHYSIOLOGY MECHANISMS THAT ACCOUNT FOR SEVERE COMBINED IMMUNE DEFICIENCY (SCID)
| DISEASE MECHANISM | GENE DEFECTS |
|---|---|
| Increased apoptosis | |
| • Due to mitochondrial energy failure | AK2 |
| • Due to accumulation of toxic metabolites | ADA |
| • Due to abnormal actin polymerization | CORO1A |
| Impaired cytokine-mediated signaling | |
| • Due to defects of the common γ chain | IG2RG (X-linked SCID) |
| • Due to defects of the IL-7R α chain | IL7R |
| • Due to defects of Jak3 | JAK3 |
| Impaired signaling through the pre–T cell receptor | |
| • Due to defective V(D)J recombination | RAG1, RAG2, DCLRE1C, LIG4,* PRKDC |
| • Due to impaired expression of CD3 subunits | CD3D, CD3E, CD3Z |
| Impaired signaling in the periphery | ORA1 |
| Unknown mechanism | RMRP* |
* These gene defects are most often associated with a milder clinical phenotype than SCID.
From Pessach I, Walter J, Notarangelo LD: Recent advances in primary immunodeficiencies: identification of novel genetic defects and unanticipated phenotypes, Pediatr Res 65:3R–12R, 2009.
Clinical Manifestations
Because all molecular types of SCID lack T cells, infants with SCID have lymphopenia (<2,500/mm3) that is present at birth, indicating that the condition could be diagnosed in all affected infants if white blood cell counts with manual differential counts were routinely performed on all cord bloods and the absolute lymphocyte count calculated. These infants also have an absence of lymphocyte proliferative responses to mitogens, antigens, and allogeneic cells in vitro. Patients with adenosine deaminase (ADA) deficiency have the lowest absolute lymphocyte counts, usually <500/mm3. Serum immunoglobulin concentrations are low or absent, and no antibodies are formed after immunizations. Analyses of lymphocyte populations and subpopulations demonstrate distinctive phenotypes for the various genetic forms of SCID (see Table 120-2). T cells are extremely low or absent in all types; when detected, in most cases they are transplacentally derived maternal T cells.
X-Linked Severe Combined Immunodeficiency (SCIDX1) Due To Mutations in the Gene Encoding the Common Cytokine Receptor γ Chain (γC)
X-linked SCID (X-SCD) is the most common form of SCID in the USA, accounting for 47% of cases (Fig. 120-1). Clinically, immunologically, and histopathologically, affected individuals appear similar to those with other forms of SCID except for having uniformly low percentages of T and NK cells and an elevated percentage of B cells (T−, B+, NK−), a characteristic feature shared only with Janus kinase 3 (Jak3)–deficient SCID. The abnormal gene in X-SCD was mapped to Xq13, cloned, and found to encode the common γ chain (γc) for several cytokine receptors, including IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21. The shared γc functions both to increase the affinity of the receptor for the respective cytokine and to enable the receptors to mediate intracellular signaling. Incapacitation of the receptors for all of these developmentally crucial cytokines by genetic mutations in γc provides an explanation for the severity of the immunodeficiency in SCIDX1. In the 1st 136 patients studied, 95 distinct mutations spanning all 8 IL2RG exons were identified, most of them consisting of small changes at the level of 1 to a few nucleotides. These mutations resulted in abnormal γc chains in two thirds of the cases and absent γc protein in the remainder. Carriers can be detected by demonstrating nonrandom X-chromosome inactivation or the deleterious mutation in their T, B, or NK lymphocytes. Unless donor B or NK cells develop, patients with X-SCID lack B- and NK-cell function after bone marrow transplantation because the abnormal γc persists in those host cells, despite excellent reconstitution of T-cell function by donor-derived T cells.
Autosomal Recessive Severe Combined Immunodeficiency
This pattern of inheritance of SCID is less common in the USA than in Europe. Mutated genes on autosomal chromosomes have been identified in 12 forms of SCID: ADA deficiency; Jak3 deficiency; IL-7 receptor α chain (IL-7Rα) deficiency; RAG1 or RAG2 deficiency; Artemis deficiency; ligase 4 deficiency; DNA–protein kinase catalytic subunit (DNA-PKcs) deficiency; CD3δ, CD3ε, CD3ζ deficiency; and CD45 deficiency (see Fig. 120-1).
IL-7Rα Deficiency
Patients with IL-7Rα-deficient SCID have a distinctive lymphocyte phenotype in that, though lacking T cells, they have normal or elevated numbers of both B and NK cells (T−, B+, NK+). This is the third most common form of SCID, accounting for 12% of cases in the USA (see Fig. 120-1). In contrast to patients with γc– and Jak3-deficient SCID, the immunologic defect in these patients is completely correctable by bone marrow stem cell transplantation, as the host B and NK cells appear to be normal.
