Physiology of neuromuscular transmission
The neuromuscular junction
The neuromuscular junction is a synapse between the tightly apposed presynaptic motor neuron terminal and the postsynaptic muscle fiber; this is where a chemical process (release of acetylcholine [ACh] from the nerve ending) leads to an electrical event (muscle membrane depolarization) resulting in a mechanical effect (muscle contraction) (Figure 44-1). Large motor nerve axons branch as they course distally within skeletal muscle. Ultimately, the axons divide into 10 to 100 smaller terminal nerve fibers, lose their myelin sheath, and innervate a single muscle fiber. The combination of the terminal neural fibers that originate from 1 axon and the muscle fibers they innervate form a motor unit. The average number of muscle fibers innervated by a single motor neuron defines the innervation ratio, which, in humans, varies between 1:5 and 1:2000. For smaller muscles that are specialized in fine and precise movement (such as the hand and ocular muscles), the innervation ratio is low (1:5, or 5 muscle fibers per neuron), whereas large antigravity muscles have very high innervation ratios (1:2000). Transmission from nerve to muscle is mediated by ACh, which is synthesized in the nerve terminal and stored in specialized vesicles. Each nerve terminal contains approximately 500,000 vesicles (also called quanta, and each containing 6-10,000 ACh molecules) arranged in a specialized region of the membrane where the synaptic vesicles gather, the active zone. ACh is released by exocytosis into the junctional cleft after an appropriate nerve impulse reaches the nerve terminal and diffuses across the 50- to 70-nm cleft to bind the nicotinic cholinoceptors on the postjunctional muscle membrane and initiate muscle contraction.
Function of the neuromuscular junction
Acetylcholine synthesis
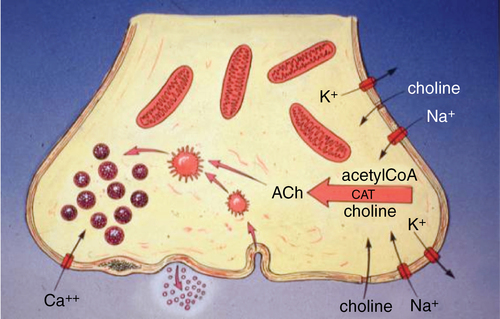
ACh is synthesized from acetyl coenzyme A and choline under the catalytic influence of choline O-acetyltransferase (CAT) enzyme in the axoplasm (Figure 44-2). The ACh is transported into vesicles by a specific carrier-mediated system. Approximately 80% of the ACh present in the nerve terminal is located in the vesicles, with the remainder dissolved in the axoplasm.
Transmitter mobilization
Postsynaptic receptors
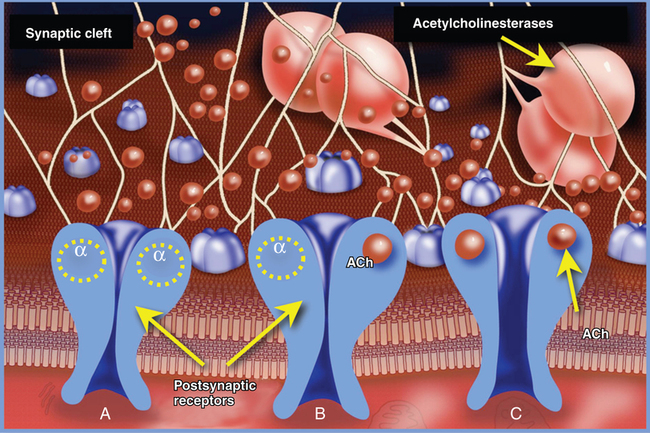
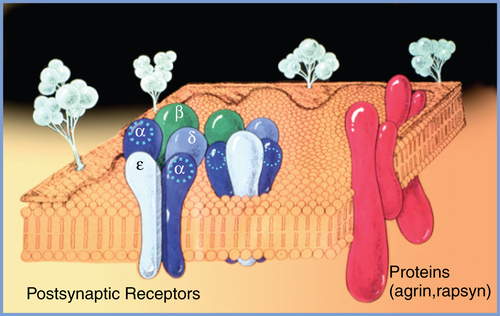
The postsynaptic muscle membrane at the cleft contains multiple invaginations that markedly increase the membrane surface area. At the top of these folds are high concentrations (up to 10,000-20,000 receptors/μm2) of nicotinic acetylcholine receptors (nAChRs). Outside the synaptic cleft area, the concentration of receptors is at least 1000 times lower. The nAChRs are pentameric proteins consisting of two α subunits (protomers), and one β, δ, and ε subunit each; the receptors are anchored to the postsynaptic muscle membrane by proteins such as agrin and rapsyn. In the adult mammal, the receptors are designated as α2 β, δ and ε (Figure 44-3). Stereochemically, they are arranged in a counterclockwise order as α, ε, α, δ, β. The fetal nAChR is similar to that of the adult, except that the fetal nAChR has a γ protomer that is replaced in the adult by the ε protomer. The five subunits of the nAChR form a rosette surrounding a central transmembrane pore with a diameter of approximately 0.7 nm. Each α subunit possesses a recognition site for ACh at the αε and αδ subunit interfaces. When ACh binds to the two α recognition sites, the receptor undergoes a conformational change and the central pore opens, allowing sodium flux that produces a brief (6.5-ms) current.
Upregulation and downregulation
Upregulation
An increase in the number of nAChRs develops on the postjunctional membrane in conditions involving decreased stimulation of the neuromuscular junction over time (Box 44-1). Upregulation leads to hypersensitivity to the agonists ACh and succinylcholine (SCh) and decreased sensitivity to antagonists such as nondepolarizing NMBAs. Upregulation can lead to lethal potassium release from cells after SCh administration in patients with motor neuron lesions, burns, muscle atrophy from disuse, and severe trauma and infections, as well as in those who have received NMBAs over a prolonged period in the intensive care unit. The phenomenon can develop in 3 to 5 days when there is total loss of ACh activity at the end plate. Pretreatment with NMBAs does not predictably prevent SCh-induced hyperkalemia. In other conditions for which chronic anticonvulsant therapy is prescribed, such as cerebral palsy and epilepsy, resistance to NMBAs is seen without potassium release after SCh use.