Chapter 67 Pharmacotherapy of Age-Related Macular Degeneration
Introduction
Age-related macular degeneration (AMD) is a spectrum of related diseases that have in common the progressive decline of vision as a consequence of dysfunction of the central retina and its underlying supporting elements, principally the retinal pigment epithelium (RPE) and choroid in older adults.1 Although the disease is seen in all racial and ethnic groups, it is more commonly encountered in females and light-skinned individuals and typically presents after the seventh decade.2–5 The disease has been traditionally classified into early and late stages.1
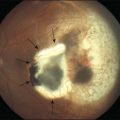
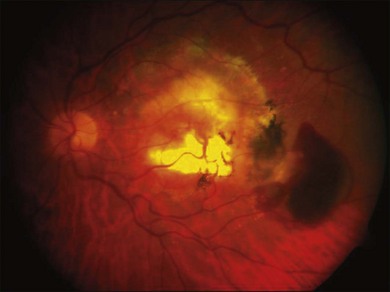
The early stages consist of alterations in the coloration of the macular pigment epithelium, both hypo- and hyperpigmentation, and the presence of drusen of greater than 125 µm in diameter. The early phases have been variously referred to as atrophic, nonexudative, preangiogenic, or dry AMD (Fig. 67.1). In contrast, exudative or neovascular AMD is typically a later-onset phenomenon, occurring in eyes with high-risk characteristics, including the presence of extensive soft drusen, Bruch’s membrane thickening, and focal hyperpigmentation. This phase of the disease is characteristically accompanied by rapid loss of vision over a period of 6–12 months and the development of a central disciform fibrotic scar. Without intervention, the visual acuity generally decreases to the range 20/200 or worse over the 12 months following the onset of this phase.3,5 In light of major improvements in therapy for this condition in the past 5–10 years, previously employed forms of treatment including thermal laser photocoagulation and surgery are now viewed to have limited benefit for this condition, particularly when it presents with involvement of the center of the fovea.6–10 In recent years, AMD therapy has become increasingly targeted toward specific molecular pathways. The discovery of multiple genes and markers for AMD and neovascularization has identified a significant number of new targets for pharmacotherapy.
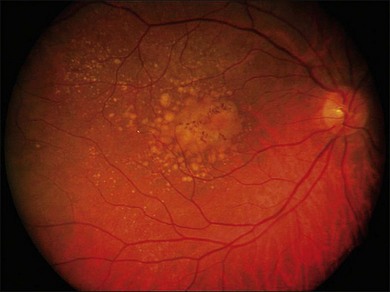
Relevant to the purpose of this chapter, it is important to understand that the mechanisms of vision loss in AMD differ according to the stage, and that different stages may be amenable to different points of attack.2,5 Importantly, at least two separate and distinct mechanisms for vision loss occur in exudative AMD: (1) proliferation of new vessels accompanied by secondary fibrosis and disorganization of the pigment epithelium and outer retina, which is a somewhat gradual process; (2) secondary alterations in the permeability of retinal and choroidal vessels accompanied by retinal pigment epithelial dysfunction leading to the accumulation of serous or serosanguineous fluid beneath the RPE, neurosensory retina, or within the retina itself, and are associated with more acute visual dysfunction. Preventing the vision loss associated with advanced forms of nonexudative disease has proven more challenging. In recent years an increased understanding of the mechanisms underlying drusen formation, pigment epithelial senescence, and loss of photoreceptors and choriocapillaris has made it more feasible to address therapeutically the visual dysfunction arising from the atrophic form of the disease (Fig. 67.2).
Etiologic factors
A variety of factors are thought to play a role in the development of AMD, including genetic susceptibility and a host of potentially modifiable environmental factors, including comorbidities, diet, medications, and light exposure.11
Genetic susceptibility
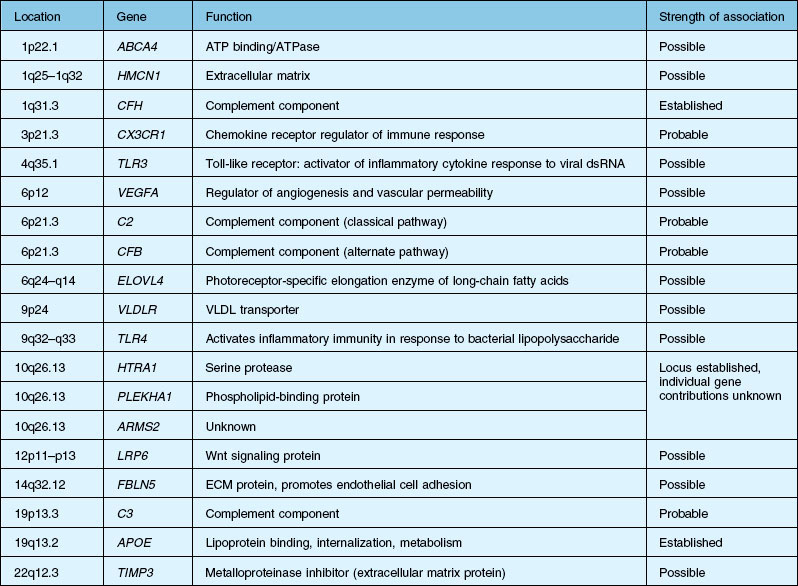
Over the past decade, high resolution genome scans for susceptibility loci in populations enriched for either choroidal neovascularization (CNV), geographic atrophy (GA), or both suggest susceptibility loci on numerous chromosomes. A number of candidate genes and their functions are listed in Table 67.1.2,12–15 An exhaustive discussion of the genetics and postulated mechanisms of ARMD is beyond the scope of this chapter; however, several genes deserve mention.
A significant breakthrough in the study of AMD genetics came in 2005, when it was discovered that single nucleotide polymorphisms (SNPs) in the regulation of the complement activation locus (RCA) of chromosome 1q31.3 increased the risk of AMD. The RCA locus contains the gene encoding complement factor H (CFH). The tyr402his protein polymorphism increased the risk of AMD between 2.7- and 7.4-fold and appeared to account for between 43% and 50% of the attributable risk of AMD. Additionally, it has been found that CFH genotypes may influence an individual’s response to certain treatment modalities and medications, including bevacizumab, photodynamic therapy with verteporfin, as well as AREDS (Age-Related Eye Disease Study) nutritional supplementation.16–20 CFH is responsible for downregulating activation of the complement system, including c5b-9, known as the membrane attack complex (MAC). MAC is thought to be important in the immune-mediated damage to Bruch’s membrane and an important pathogenic component of drusen.21,22
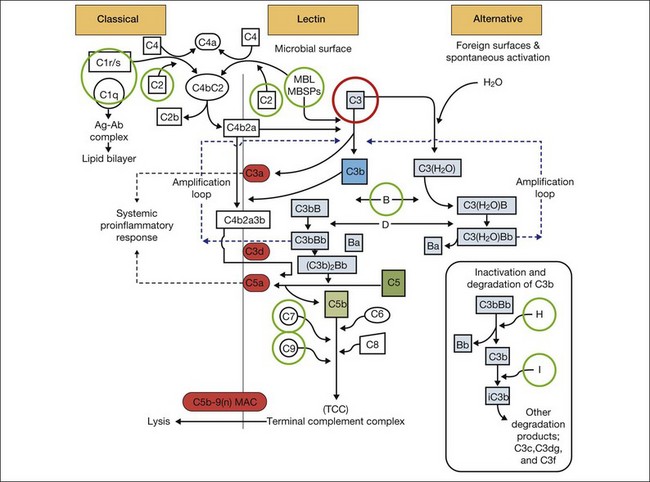
It is clear that both immune dysregulation and inflammation play key roles in the development and progression of AMD. Two recent studies found protective associations against the development of advanced AMD in those with genetic polymorphisms of the complement components C2 (OR 0.35–0.46) and complement factor B (CFB) (OR 0.35–0.44), while C3 (OR 2.66–3.9) was strongly associated with the development of GA.13,23 Figure 67.3 illustrates other complement components implicated in the pathogenesis of AMD. Additionally, elevated levels of the acute phase reactant CRP have been found to correlate with neovascular AMD. It is still unclear how genotypic variations in complement lead to an increased inflammatory state and the AMD phenotype. However, clinicopathologic specimens demonstrate the presence of inflammatory cells in Bruch’s membrane, and of bioactive fragments of C3 (C3a) and C5 (C5a) in drusen of AMD eyes and elevated VEGF expression in RPE cells.24,25 The inflammatory cascade is further characterized by retinal microglial activation and choroidal macrophage infiltration.26,27
The gene products for tissue inhibitor for metalloproteinase 3 (TIMP-3, chromosome 22) and EFEMP1 (chromosome 2, encoding an elastin-related protein, fibulin 3), which act to inhibit the stimulatory effects of VEGF on vascular cells, have been independently associated with advanced AMD in genome scans mentioned previously.28,29 Multiple distinct alleles in exon 5 of the gene encoding TIMP-3 have been causally linked to an autosomal dominant form of exudative macular degeneration with strong phenotypic similarities to exudative AMD,30–32 although it is not thought to account for the majority of patients with exudative AMD.33 TIMP-3 is also found in increasing quantities and is associated with thickening of Bruch’s membrane during aging, including within drusen and the RPE.34–36 An Arg to Trp mutation in EFEMP1 is thought to cause the abnormal accumulation of extensive drusen in malattia leventinese, an inherited macular degenerative disease with phenotypic characteristics closely correlated with advanced nonexudative AMD.34,37 The increased frequency of genome alterations coding for these two proteins (TIMP-3 and fibulin), which are important in the regulation of the extracellular matrix, particularly Bruch’s membrane, provides support for the hypothesis proposed by Blumenkranz that generalized increased susceptibility of elastic fibers to photic degeneration is an important risk factor for choroidal neovascularization.3
Early onset macular retinal degenerations such as Stargardt disease, characterized by the accumulation of lipofuscin within the RPE, have shed light on both the pathophysiology of RPE apoptosis and potential therapeutic avenues.38,39 While initial reports also suggested that the ABCR gene linked to Stargardt disease might also be a dominant susceptibility locus for AMD, subsequent investigators have been unable to confirm this apparent increased frequency.40,41 Nonetheless, increased amounts of lipofuscin are present in both Stargardt disease and in the earlier stages of the nonexudative AMD, and both diseases are characterized by GA in their later stages, suggesting that an understanding of the common pathways expressed by these two different diseases may provide some pharmacologic insights. Biochemical studies on the lipofuscin found in both Stargardt disease and AMD confirm that the principal component is N-retinylidene-N-retinylethanolamine (A2E), which accumulates in cells of the RPE and results in RPE apoptosis, photoreceptor death, and vision loss.22,42,43 This process is thought to be initiated by light activation of rhodopsin in the course of normal visual transduction as the first reactant in A2E biosynthesis. Isotretinoin (Accutane, discussed later), which slows the synthesis of 11-cis retinaldehyde and regeneration of rhodopsin, is able to slow the accumulation of lipofuscin in ABCR knockout mice and also aged wild-type mice.42 This suggests that compounds with mechanisms of action similar to isotretinoin, which is structurally related to vitamin A, may represent another therapeutic avenue for the slowing or prevention of AMD.22,39,41–43
There is additional discussion of AMD genetic susceptibility in Chapter 64 (Pathogenetic mechanisms in age-related macular degeneration.)
Environmental factors
Diet
The principal environmental factors thought to be associated with age-related macular degeneration include diet, history of smoking, light exposure, and use of supplemental medications. Recent AREDS reports have established a relationship between dietary intake of omega-3 long-chain polyunsaturated fatty acids and reduced risk of neovascular AMD (OR 0.61), as well as progression from drusen to GA.44–46 A large case control study suggested that higher dietary intake of carotenoids, particularly lutein and zeaxanthin, was associated with a lower risk of AMD, with the highest quintile having a 43% reduction compared with those in the lowest quintile. In that study no reduction could be found with the use of oral vitamin A, vitamin E or vitamin C.47 Subsequent studies suggest that the use of foods, particularly fruits rich in antioxidants and carotenoids, when consumed at the level of three servings or more per day, result in a pooled multivariate reduced relative risk of 0.64 compared with persons who consumed less than 1.5 servings per day.2 However, there have been a number of studies that have demonstrated little to no benefit with dietary regimens.28,48–50,51
Smoking*
Most studies have consistently implicated smoking as a statistically significant risk factor for the development of late-stage AMD.2,54 In a meta-analysis of three pooled studies from the United States, The Netherlands, and Australia, smoking was one of only two significant associations identified with incident AMD along with total serum cholesterol. Current smoking is associated with a 4.55-fold increased risk of neovascular AMD (vs. “never” smokers) and a 2.54-fold increased risk of atrophic AMD (vs. “never” smokers).55 Based on a pooled analysis of data, the risks of both neovascular and atrophic AMD seem to decrease once one stops smoking.55
Light exposure
Although there is a considerable body of compelling experimental data to suggest that increased retinal irradiance is positively correlated with an increased likelihood of advanced AMD, the epidemiological evidence supporting this hypothesis is modest.3,56,57 In the Beaver Dam eye study, participants exposed to summer sun for more than five hours a day during selected periods of time were at increased risk for increased retinal pigment (relative risk 3.17) and early AMD (relative risk 2.14) after 10 years of follow-up compared with those exposed to less than 2 hours per day.57
The underlying rationale for the hypothesis that increased light stress predisposes to AMD rests on the role of oxidative stress inherent with photo bleaching of the photoreceptors and, to a lesser extent, pigment epithelium. In particular, reactive oxygen intermediates (ROI) including hydrogen peroxide, singlet oxygen, and other short-lived species, which arise as the byproducts of cellular metabolism, are known to have a toxic effect on cellular membranes.48 It is felt that the cumulative effects of chronic low levels of these species result in profound damage to the retina and pigment epithelium through lipid peroxidation, mitochondrial DNA damage and induction of apoptosis.2,48 Experimental studies have confirmed the deleterious effect of chronic light and in particular blue and ultraviolet light on the RPE in part through the creation of A2E oxiranes, a major component of lipofuscin. The presence of natural antioxidants derived from dietary sources including lutein, zeaxanthin, lycopene, and ascorbate are thought to mitigate the effects of photo-oxidation by quenching free radicals and other intermediate species.48 It has been hypothesized that this represents one, but not the only mechanism of action, of the antioxidants in the AREDS study.58
Use of medications
Meta-analyses of pooled prospective studies suggest that certain medications may be associated with an increased or decreased risk of age-related macular degeneration. Antihypertensive medications, particularly beta-blockers, are associated with modest increased risk, whereas hormone replacement therapy in women, and tricyclic antidepressants, confer some relative protection.59 These observations may be able to be further exploited in the development of new classes of drugs in addition to avoiding potentially harmful drug interactions.
The reader is referred to Chapter 63 (Epidemiology and risk factors for age-related macular degeneration).
Systemic risk factors
As previously noted, there appears to be a strong association between levels of ocular inflammation or systemic inflammatory disease and age-related macular degeneration. It has long been known that there is a relationship between VEGF and inflammation.2,60 Inflammatory foci and white cells are commonly seen in the choroid in autopsy specimens of patients with CNV.61–65 To the extent that inflammation does have a causative effect on the development of AMD, therapy aimed at reducing inflammation through complement inhibition or regulation, immunomodulation or stabilization of intracellular organelles such as lysosomes and resultant proteolysis, may be successful pharmacologic strategies. In animal models, intravitreal triamcinolone acetonide has been shown to inhibit preretinal and choroidal neovascularization induced by laser injury.66 There is increasing evidence of the linkage between VEGF, inflammation, and intracellular adhesion molecule 1 (ICAM-1) (Fig. 67.4). Triamcinolone modulates permeability and intracellular adhesion and ICAM-1 expression in cell culture, and it has further been shown that upregulation of ICAM-1 and invasion by CD18 positive leucocytes follows laser injury and precedes neovascularization.67,68 Comparable laser injury in ICAM-1 knockout mice demonstrated considerable reduction in neovascularization, confirming the likely participation of this class of molecules in VEGF-mediated angiogenesis. Other therapies that target CD18-positive leucocytes or ICAM-1 are likely to be beneficial in reducing the angiogenic stimulus associated with inflammation and aging.
The pathophysiology of exudative amd: the crucial role of cytokines
Definition and steps in angiogenesis
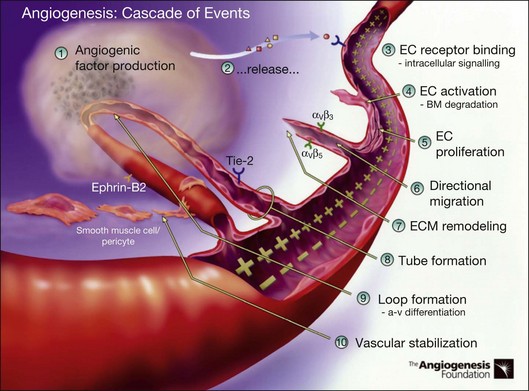
Angiogenesis refers to the creation of new blood vessels from existing blood vessels, and therefore is contrasted from the process of vasculogenesis seen characteristically in utero in which vessels are created de novo. The angiogenesis cascade has been characterized as occurring in multiple sequenced steps (adapted from The Angiogenesis Foundation, www.angio.org/understanding/process.php [current as of February 15, 2012]):
1. Release of angiogenic growth factors by diseased tissue.
2. Binding of the angiogenic growth factors to adjacent existing vascular endothelial cells (EC).
3. Activation of EC to produce molecules, including enzymes.
4. Dissolution of the surrounding basement membrane by activated enzymes.
5. Proliferation and migration of EC.
6. Adhesion molecules (integrins) help to serve as framework for advancing EC.
7. Further dissolution of tissue and remodeling with matrix metalloproteinases.
8. Formation of vascular tubes by EC.
9. Formation of blood vessel loops from EC tubes.
10. Stabilization of new blood vessels by smooth muscle cells and pericytes. Antiangiogenic therapies currently under investigation are thought to inhibit angiogenesis at various steps in this process (Fig. 67.5).
VEGF and other positive and negative modulators of angiogenesis
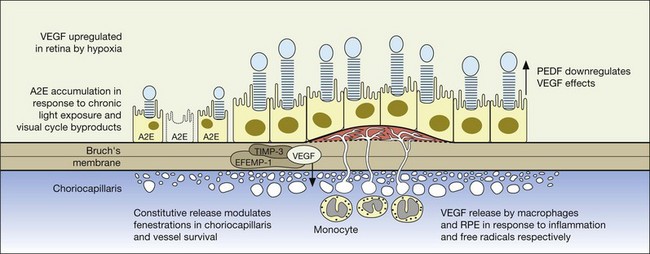
Regardless of the inciting stimulus involved in the development of pathologic neovascularization, it is now well established that VEGF plays a principal role, including not only the neovascular forms of age-related macular degeneration, but also diabetic retinopathy, iris neovascularization, and retinopathy of prematurity. Additionally, other cytokines may play an important role as well, including fibroblast growth factor (FGF), pigment epithelial-derived factor (PEDF), the integrins, angiopoietins, and matrix metalloproteinase inhibitors.2,30,69–72
VEGF undoubtedly represents the putative factor X first postulated by Michaelson associated with pathologic neovascularization.2 First described as a vasopermeability factor associated with tumors, the molecule has since been cloned and well characterized.73,74 VEGF has two important characteristics relevant to its role in the pathogenesis of the neovascular forms of age-related macular degeneration: (1) induction of angiogenesis through endothelial proliferation, migration, and new capillary formation, and (2) enhancement of vascular permeability.
Vascular permeability
VEGF produces important increases in hydraulic conductivity of isolated microvessels that are mediated by increased calcium influx and likely changes in levels of nitric oxide caused by induction of nitric oxide synthetase (NOS). While VEGF is both necessary and sufficient when administered exogenously to induce both these effects in vitro and in vivo, it is also well known that a number of other growth factors participate in this process. Some represent steps in a cascade initiated by VEGF while others act upstream.75,76 The chemical structure of VEGF is that of a heparin-binding homodimeric glycoprotein of 45 kDa.73 VEGF has significant homology to platelet-derived growth factor (PDGF). The human VEGF gene is organized into eight exons separated by seven introns and is localized in chromosome 6p21.3. Although there is only a single gene for VEGF-A, alternate post-translational exon splicing results in the generation of between four and six different isoforms having 121, 145, 165, 183, 189, and 206, amino acids respectively after signal sequence cleavage72,73 (Fig. 67.6). VEGF165 exists in both soluble and bound forms73 (Figs 67.6, 67.7), and is thought to be predominantly responsible for pathologic neovascularization.
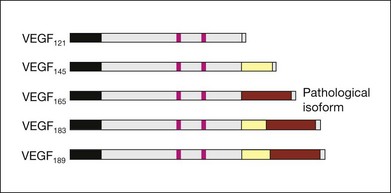
Although VEGF inhibitors share in common an attempt to mitigate the proliferative and permeability effects of VEGF on normal and neovascular tissue, there are likely to be differences in both efficacy and safety related to the choice of agents for several reasons. From a theoretical standpoint, since VEGF165 is the principal isoform involved in pathologic neovascularization, it has been argued that leaving the other three principal isoforms intact (121, which is principally diffusible in soluble form, and 189 and 206 which are principally matrix-bound through high affinity heparin binding sites), avoids the possibility of interference with normal homeostatic mechanisms associated with constitutive VEGF expression. Indeed, previous animal studies suggested that blockade of VEGF164 in the mouse (equivalent to VEGF165 in humans) is as effective as nonselective antibody-mediated VEGF blockade in preventing pathologic neovascularization in a hypoxia-induced angiogenesis model, while not interfering with normal physiologic development of retinal vasculature.60,77 However, it has become apparent with widespread use of bevacizumab and ranibizumab (monoclonal antibodies against multiple VEGF isoforms) that nonselective blockade has few untoward effects on normal retinal vascular physiology.
VEGF receptors
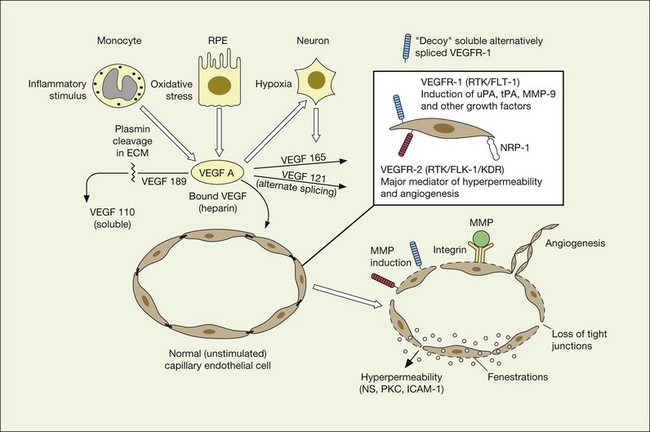
VEGF exerts its effects on cells through two highly related receptor tyrosine kinases (RTKs) VEGFR-1 and VEGFR-2.73 VEGFR-1 also termed FLT-1 (FMS-like tyrosine kinase) was described first, but its role remains open to debate. Like VEGF, VEGFR-1 is upregulated by a hypoxia-inducing factor (HIF)-dependent mechanism. The receptor undergoes weak tyrosine autophosphorylation in response to VEGF. It is thought not to be primarily a mitogenic stimulus but rather a “decoy” receptor, which downregulates the activity of VEGF by sequestering and rendering the factor less available to VEGFR-2 (Fig 67.7, 67.8). This factor may be most important during embryogenesis rather than during pathologic neovascularization as well as in hematopoietic bone marrow-derived cells and neural signaling.73,78
VEGFR-2 (KDR in humans or FLK-1 in mice)
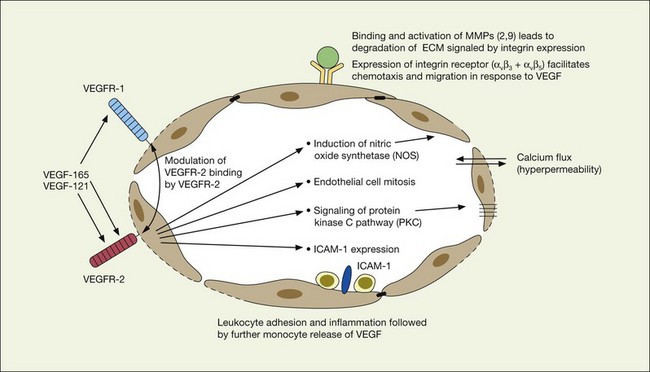
A large body of evidence exists to support the critical and probably rate-limiting role of VEGF in neovascular forms of age-related macular degeneration. High messenger RNA levels and increased VEGF receptor levels are observed in areas of CNV in primates as well as in the extracted neovascular membranes of patients removed at surgery and following autopsy.2,63 Additionally there are strong indications that elevated levels of VEGF are the proximal cause for the hyperpermeability seen not only in diabetic macular edema, but in patients with subsensory and intraretinal fluid associated with CNV as well. The permeability changes resulting from VEGFR-2 are thought to be mediated to a large extent by endothelial nitric oxide synthetase-based generation of increased nitric oxide levels, and associated changes in calcium flux (Fig. 67.8). These alterations can be reversed either by direct blockade of VEGF, the receptor, or NOS using a knockout model.2,74,75 Inactivation of soluble VEGF by monoclonal antibodies directed against it, or inhibition of ICAM also appear to be effective as well. In addition to either the indirect inhibition of VEGF effects through modulation of ICAM, or indirect effects on nitric oxide phosphorylase, nitric oxide synthetase, or protein kinase C, another modulator of permeability, there are several other methods by which VEGF effects can be blocked in the eye and elsewhere. These include the use of a class of molecules termed aptamers, which are chemically synthesized single strand nucleic acids, either RNA or DNA, that bind to target molecules with high selectivity and affinity leaving non-targeted protein functions intact.79,80 Other methods of direct inhibition of VEGF include inhibition of its tyrosine kinase receptors (VEGFR-1 and -2), either by systemic administration or gene transfer. Several laboratories have described the use of small-interfering RNA (siRNAs) in effectively downregulating gene regulation81 (see subsequent sections).
Other cytokines and regulators of angiogenesis
A number of cytokines have been identified that either upregulate VEGF or VEGF-associated effects, or act in an inhibitory capacity. In some instances, the same compounds may perform both functions depending upon the other extracellular signals present in the milieu as well as post-transcriptional or post-translational events, including cleavage by proteases, or binding by soluble receptors such as the family of tyrosine kinases.2 Accordingly, any of this class of normally occurring compounds can be considered either in their native form, through splice or cleavage products, or chemical substitution to be potential therapeutic compounds in the treatment of pathologic neovascularization. Examples of stimulatory cytokines in addition to vascular endothelial growth factor include basic and acidic fibroblast growth factor, the angiopoietins, transforming growth factor (TGF), IL-8, and platelet-derived growth factor (PDGF). Naturally occurring inhibitory factors include interferon alpha, thrombospondin 1, angiostatin, endostatin, hemopexin-like domain of matrix metalloproteinase 2 (PEX), and pigment epithelial-derived factor (PEDF).2,82–90 Additionally, it is likely there are many other naturally occurring compounds as well, as yet not recognized or well characterized biochemically. The relative balance between naturally occurring stimulatory and inhibitory factors is thought to contribute to the tight control of ocular vascular homeostasis, and may explain the differential ability of normal vascular as well as neovascular tissue to respond exquisitely both spatially and temporally to various stimuli.
Naturally occurring upregulators of angiogenesis
Fibroblast growth factor and integrins
Basic fibroblast growth factor (bFGF/FGF2) has been found to be elevated in excised choroidal neovascular membranes from patients undergoing surgery for AMD, and is capable of inducing pathologic neovascularization when implanted within the eye or adjacent to the retina.69,70 Laser models of choroidal neovascularization are accompanied by upregulation of bFGF-associated mRNA, which precedes the development of histologically identifiable new vessels. While bFGF is sufficient when inserted exogenously in creating choroidal neovascularization, it is not necessary; BFGF gene knockout animals are also capable of developing laser-induced CNV bFGF upregulation of angiogenesis is thought to be in part mediated by adhesion-mediated signals including expression of the cell membrane-associated integrins such as the alpha ν beta 3 integrin.2,69,70 Although the alpha ν beta 3 and another integrin, alpha ν beta 5, appear to differentially represent the stimulatory effects of bFGF and VEGF respectively in laboratory studies, there is conflicting evidence as to whether these pathways are distinct. As a result, considered therapeutic approaches to integrin inhibition may be directed at downstream events such as intracellular signaling in addition to targeted inhibition of the individual receptors through monoclonal antibodies or related mechanisms.
Platelet-derived growth factor
Platelet-derived growth factor (PDGF) is a homo- or heterodimeric glycoprotein that stimulates pericyte and vascular smooth muscle cell recruitment in angiogenesis, which are essential in the maintenance and survival of new vessels. The PDGF receptors (PDGFR-A and PDGFR-B) are tyrosine kinases, and are expressed differentially on different cell types, with PDGFR-B being more extensively expressed in endothelial cells, vascular smooth muscle cells, and fibroblasts, among others.91,92 In the absence of PDGFR-B signaling, vascular pericytes detach, and the integrity of vessel is lost. Immature vessels are highly sensitive to the withdrawal of VEGF prior to pericyte recruitment and coverage; conversely, when pericyte coverage is complete, new vessels are resistant to VEGF withdrawal. This is the so-called plasticity window, during which time vessels can regress rapidly with changes in exposure to growth factors.93,94 PDGF blockade in animal models of corneal neovascularization has resulted in prevention and regression of new vessels.95,96 PDGF blockade alone or combined with VEGF inhibition (taking advantage of the plasticity window) may thus serve as a potent inhibitor of neovascular AMD.
Angiopoietins
Angiopoietins, which are also highly specific for endothelial cells, perform a variety of other regulatory activities related to supporting cells and the extracellular matrix. A laboratory study suggests that two different isoforms, Angiopoietin 1 and Angiopoietin 2, appear to have differential and counteracting effects on the vasculature. Angiopoietin 2, which is upregulated by both hypoxia and VEGF, binds to an endothelial cell receptor TIE2 and enhances VEGF-mediated retinal neovascularization, but does not by itself stimulate endothelial cells or proliferation in vitro.2,97 Angiopoietin 1 appears to play a maturation role and is associated with nonleaky behavior and may have potential therapeutic benefit through its inhibition of inflammatory pathways.2
Matrix metalloproteinases and tissue inhibitors of metalloproteinases
Expression of matrix metalloproteinases (MMPs) and their associated RNA, particularly MMP-2 and MMP-9, have been associated with pathologic neovascularization. Both matrix metalloproteinases, which are bound to the integrin receptors alpha ν beta 3 and alpha ν beta 5 on endothelial cells, are activated by VEGF and other regulatory cytokines and act to digest the extracellular matrix and thereby facilitate the spreading and chemotaxis of actively proliferating endothelial cells as they aggregate to form new capillaries.2 Matrix metalloproteinases (MMPs) appear to be modulated and principally downregulated by naturally occurring tissue inhibitors of metalloproteinases (TIMPs) of which TIMP-1, TIMP-2, and TIMP-4 are thought to be soluble, and TIMP-3 principally extracellular matrix-bound. TIMP-3 is thought to play an important and possibly critical role in the natural modulation of matrix metalloproteinase regulation in Bruch’s membrane, and is found in increased amounts in drusen and thickened basement membranes associated with age-related macular degeneration.36,98 Mutations in exon-5 of the TIMP-3 molecule are associated with Sorsby’s fundus dystrophy,31,32,35 and an experimental model that either delivers increased amounts of TIMP-3 or induces overexpression of TIMP-3 by gene therapy demonstrates potent antiangiogenic effects for this molecule.99,100
Naturally occurring downregulators of angiogenesis
Pigment epithelial-derived factor
Pigment epithelial-derived factor (PEDF), which is secreted by the pigment epithelium in vivo as well as in cell culture, has been shown to be biochemically identical to the product of the wild type of retinoblastoma tumor suppressor gene (RB) and is thought to induce differentiation of retinoblastoma cells, inhibit microglial growth, and other important regulatory functions in addition to its effects on angiogenesis.56 Its relevance to AMD is likely related to its role in counterbalancing VEGF. In a study of donor human eyes with and without AMD, relative VEGF and PEDF levels were measured using immunoreactivity assays. Interestingly, VEGF levels were not significantly different between the two groups of eyes, even in the presence of advanced exudative change. In contrast, PEDF levels were much lower in the RPE and choroid of eyes with AMD, suggesting that it is lower levels of PEDF in the ocular milieu that may contribute to the formation of choroidal neovascularization.101 Results from in vitro studies have suggested that the role of PEDF is regulation or inhibition of VEGF receptor signaling, rather than direct VEGF antagonism.102
Other cytokines
Another interesting class of regulatory cytokines that appear to have an inhibitory effect on angiogenesis relate to the class of aminoacyl-tRNA synthetases, which are associated with the first step of protein synthesis. Tryptophanyl-tRNA synthetase (TrpRS, a homolog of TyrRS) has no effect in its native form on angiogenesis or angiogenesis signaling. In contrast, an alternatively spliced fragment reported to be stimulated by interferon-alpha and lacking a portion of the amino terminal fragment, has potent antiangiogenic effects in both in vitro and in vivo.103
Thrombospondin 1 (TSP-1) has been described as both an up- and downregulator of VEGF. Under specified conditions TSP-1, which is produced by platelets and monocytes, enhances simulated VEGF release and is dependent upon binding to alpha ν beta 5 and alpha ν beta 1 integrins. In separate studies, TSP-1 has also been noted to inhibit endothelial cell proliferation, migration, and angiogenesis. VEGF and TSP-1 appear to participate in a feedback loop and its potential role as a therapeutic agent remains uncertain.2 Angiostatin, is a 38 kDa internal fragment of plasminogen that encompasses the first four cringles of the molecule. It shows inhibitory effects on vascular endothelial proliferation in vitro and in vivo, particularly within tumors.90,104 Endostatin, a cleavage product of collagen XVIII, is structurally related to and shares homology with angiostatin, and inhibits tumor-associated angiogenesis.89 Each of these classes is further discussed in the following section as they relate to preclinical or clinical studies.
Agents currently in use or under investigation: non-neovascular AMD
Antioxidants, vitamins, and cofactors
The rationale for the use of antioxidants, multivitamins and other cofactors is based upon several lines of evidence, including the essential role of vitamin A in the visual transduction cycle, the known and predicted effects of oxidative stress resulting in reactive oxygen intermediates, and the requirement of certain critical metal ions including zinc in the functioning of critical proteases and other naturally occurring defense mechanisms against oxidative injury. Prior to the release of the data from the AREDS study, there remained some controversy as to whether antioxidant multivitamin therapy was efficacious, with some studies suggesting a protective effect, and others not.49,50,105
Age-Related Eye Disease Study (AREDS) and related supplements
The AREDS was a large, well-designed multicenter randomized clinical trial, which evaluated the effect of high-dose vitamin C and E, beta-carotene, and zinc supplements on AMD progression and visual acuity. A total 4757 persons were enrolled and stratified into one of four categories of increasing severity of disease. Due to the low rate of progression of category 1, consisting of drusen area of less than five small drusen of 63 µm or less, statistical analysis was only performed on the remaining 3640 patients with categories 2, 3, 4. Only the latter two (categories 3 and 4) were ultimately found to be at more than a trivial risk for progression. Patients in category 2 with at least one intermediate size druse, but not extensive drusen, had only a 1.3% probability of progression to advanced AMD by year 5 and no meaningful inference could be drawn regarding their risk with or without intervention. Patients in category 3 had either extensive intermediate drusen, large drusen, or non-central geographic atrophy, and patients in category 4 had visual acuity of less than 20/32 due to AMD in one eye related either to geographic atrophy involving the center of the macula or choroidal neovascularization in the fellow eye, but not the study eye, which had visual acuity 20/32 or better. The average follow-up of the 3640 enrolled study participants aged 55–80 was 6.3 years, and by comparison with placebo, patients treated with antioxidants plus zinc demonstrated a statistically significant odds reduction for the development of advanced AMD (OR 0.72, 99% CI 0.52–0.98). The odds ratio for zinc alone and antioxidants alone were 0.75 and 0.8, respectively, and the odds reduction estimates increased when only patients with category 3 or category 4 drusen were included, reducing to 0.66, 0.71, and 0.76 for antioxidants plus zinc, zinc alone, and antioxidants alone respectively. These data are further reflected in Fig. 67.9.
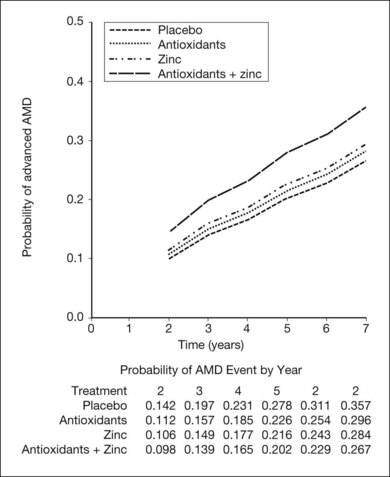
Based upon these findings, and extrapolation of the US population at risk, it has been estimated that of the 8 million persons in the United States at least age 55 or greater with AMD, if those at highest risk including categories 3 and 4 were to receive therapy with the formulation employed in the AREDS trial, as many as 300 000 of the 1.3 million at highest risk for advancement might avoid progression to the severe forms of the disease.106 Additionally, based upon recent epidemiologic data, exclusion of beta-carotene is recommended for persons with a current history of smoking, or a long smoking history based upon a theoretical increased risk of lung cancer.
The macular carotenoids thought to be the principal contributors to the yellow coloration of the macula include lutein and zeaxanthin. One study suggested that a high dietary intake of these carotenoids protected against AMD, resulting in a 43% reduction in risk for AMD when the upper quartile of use was compared with the lowest quartile.47 Recent AREDS reports have supported these findings, showing in cohort studies that those with a high level of dietary intake of lutein and zeaxanthin were less likely to have advanced AMD, as well as large or extensive intermediate drusen.107 High dietary intake of ω-3 long-chain polyunsaturated fatty acids were also found by the AREDS investigators to be inversely related to progression to both GA as well as neovascular AMD.44–46 The critical dependence of the naturally occurring antioxidant enzymes (superoxide dismutase, catalase, and glutathione peroxidase) as well as the matrix metalloproteinases, on copper, zinc and manganese, provides a potential mechanism by which AREDS supplementation of both copper and zinc could be beneficial. The naturally occurring antioxidants such as vitamin E and vitamin C might have a beneficial effect on the progression of AMD as well.48 At present no definitive recommendations can be made regarding dietary supplementation with lutein and zeaxanthin, as well as ω-3 long-chain polyunsaturated fatty acids. However, the AREDS2 randomized multicenter phase III clinical trial (NCT00345176) is currently underway, and is designed to address: (1) the role of lutein (10 mg)/zeaxanthin (2 mg) and ω-3 long-chain polyunsaturated fatty acids (350 mg docosahexaenoic acid (DHA)/650 mg eicosapentaenoic acid (EPA)) in the prevention of development of GA or CNV; and (2) the effect of possible deletion of β-carotene and lowering the daily zinc oxide dose from 80 mg to 25 mg. The AREDS study is discussed also in Chapter 65 (Age-related macular degeneration: Non-neovascular early AMD, intermediate AMD, and geographic atrophy).
Visual cycle inhibitors*
Visual cycle modulators are intended to reduce the accumulation of toxic fluorophores such as A2E in RPE cells. Fenretinide (ReVision Therapeutics, Inc.) treatment causes a dose-dependent, reversible reduction in circulating retinal binding protein (RBP) and retinol by displacing retinol from RBP, as well as interfering with RBP binding to transthyretin (TTR), thus facilitating their elimination via glomerular filtration. In ABCA4−/− mice, fenretinide reduces RPE lipofuscin and A2E accumulation, although it causes modest delays in dark adaptation.108 Notably, however, despite low blood retinol levels, RPB−/− mice acquire normal vision by 5 months of age when given a vitamin A-sufficient diet.109,110 Thus, it is not clear that blockade of vitamin A transport to RPE by inhibition of vitamin A binding to RBP will block vitamin A uptake by the RPE during long-term administration unless dietary vitamin A is restricted also. Patients in a phase IIb clinical trial of fenretinide received placebo, 100 mg, or a 300 mg daily dose for 24 months (NCT00429936). At the conclusion of the two-year study, an exploratory and ad hoc analysis of the data revealed that 15 (18.3%) of 82 patients in the placebo arm progressed to CNV, compared with 15 (9.2%) of 164 patients receiving fenretinide at either dose (P = 0.039). In the 300 mg fenretinide dose cohort, analysis of GA lesion growth by color fundus photography showed a trend for slowing of lesion growth, particularly among patients who had RBP and retinol levels reduced by more than 50%. Although fenretinide can affect dark adaptation111–118 and ERG readings,119–122 and is associated with symptoms of dry eye, 116,117 it was generally well tolerated in this study.
Accutane (13-cis-retinoic acid) inhibits the conversion of all-trans-retinyl esters (in retinosomes) to 11-cis-retinol and the conversion of 11-cis-retinol to 11-cis-retinal by retinol dehydrogenase and also reduces lipofuscin accumulation in ABCA4−/− mice.42,123 This oral agent is associated with a high incidence of nyctalopia.124 Another drug known as ACU-4429 (Acucela) is an orally administered compound that inhibits conversion of all-trans-retinyl ester to 11-cis-retinol via blockade of RPE65. ACU-4429 also reduces lipofuscin and A2E accumulation in the RPE of ABCA4−/− mice. A phase I clinical trial (NCT00942240) in 46 healthy volunteers was completed successfully.125 The most common adverse events were vision-related, occurring in 50% of patients receiving the medication, which included dyschromatopsia (32%), unspecified visual disturbance (29%), night blindness (18%), blurred vision (11%), and photophobia (8%). All adverse events were mild or moderate in intensity and were transient in nature, resolving within a few days after onset. There was dose-dependent suppression of the ERG b-wave as expected. A dose escalation phase II study is underway in patients with GA (NCT01002950).
Complement modulators*
In general, complement modulating agents work by replacing a defective complement component; by inhibiting the activation of convertases; by promoting the decay of convertases; or by blocking effector molecules (Fig. 67.10).126 The following examples illustrate some of the promise as well as the challenges associated with manipulation of the complement pathway.
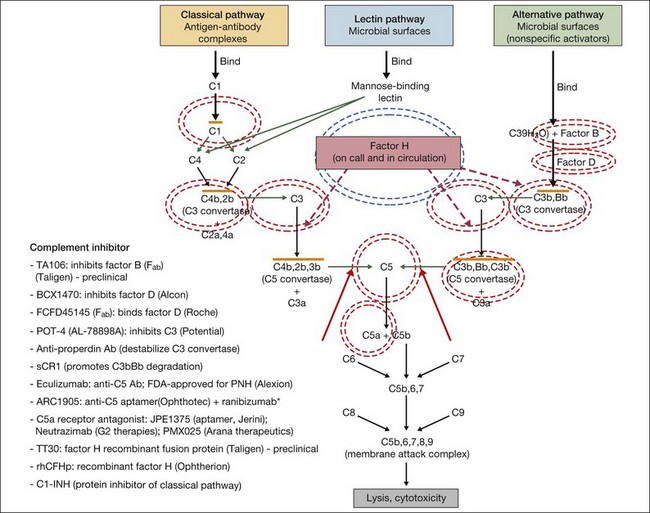
Fig. 67.10 Numerous compounds that modulate the complement pathway are in development for or in clinical trials for AMD treatment. Red circles indicate the parts of the complement pathway that are being modified. The complement pathway illustrated is adapted from Donoso et al.126
(Reproduced from Zarbin MA, Rosenfeld PJ. Pathway-based therapies for age-related macular degeneration: an integrated survey of emerging treatment alternatives. Retina 2010;30:1350–67 with permission of Springer Science+Business Media.)
Inhibition of C5 blocks terminal complement activity, but proximal complement functions remain intact, e.g., C3a anaphylatoxin production, C3b opsonization, and immune complex and apoptotic body clearance. ARC1905 (Ophthotech Corp.) is an anti-C5 aptamer delivered by intravitreal injection. It binds C5 with high affinity (KD = ~700 pM) and prevents cleavage to C5a and C5b. ARC1905 is in a phase I trial (NCT00950638) for patients with GA. Eculizumab (SOLIRIS, Alexion Pharmaceuticals) is a humanized monoclonal antibody that binds to and prevents cleavage of C5 and is administered intravenously. Eculizumab is FDA-approved for the treatment of paroxysmal nocturnal hemoglobinuria and is in phase II trials (NCT00935883) for treatment of nonexudative AMD, including patients with high-risk drusen or GA. C5a receptor blockade – e.g., JPE1375 (Jerini); PMX025 (Arana Therapeutics); Neutrazimab (G2 Therapies) – might have both advantages and disadvantages over direct C5a inhibition. C5a receptor blockade might inhibit some important inflammatory pathways,24 although without disabling membrane attack complex formation.
Factor D is the rate-limiting enzyme in the activation of the complement alternative pathway. It plays an important role in the positive feedback loop that results in the amplification of proinflammatory effectors.127 FCFD45145 (Genentech/Roche) is a monoclonal antibody fragment (Fab) directed against Factor D. A 108-patient placebo-controlled phase I clinical trial (NCT00973011) of intravitreal therapy for GA is complete, and the medication was well tolerated up to a 10 mg dose. Patients were treated monthly or every other month during an 18-month period. A phase II study (NCT01229215) is underway.
Replacement of complement factor H should inhibit inflammation in AMD patients with risk-enhancing mutations in CFH. This approach, which probably would require genetic screening prior to treatment, involves restoration of complement homeostasis so there is no increased risk of infection with therapy. Replacement of defective CFH is being developed by Alexion. TT30 is a recombinant fusion protein comprising complement receptor type 2 and the regulatory domain of factor H. The receptor domain of TT30 binds iC3b/C3d-coated cells, delivering the regulatory domain of CFH to areas where complement regulation is necessary.127 Preclinical testing is underway. Alexion is also exploring factor B inhibition using a humanized antibody fragment (TA106).
The differential expression of complement components in different ocular tissues presents difficulty in determining the optimal route of administration of these agents. For example, factor D is deposited diffusely in the human retina as well as in the choroid,128 while factors B, H, C3, C5, and C5b-9 are found primarily in the choroid, Bruch’s membrane, and/or subjacent to the RPE.128,129 Thus, intravitreal injection may not be the ideal route of delivery for all the complement inhibitors under study.
Another approach to complement modulation is the silencing of genes by preventing mRNA expression, as deletion of genes closely related to CFH (i.e., CFHR1 and CFHR3) appears to be strongly protective against AMD.130 However, short-interfering RNA (siRNA) therapies in the eye may be toxic, and it seems that the deletion of CFHR1 and CFHR3 protect against development of AMD at least in part because the deletion tags a protective haplotype and does not occur in association with the Y402H single nucleotide polymorphism.131 See below for further discussion of siRNAs and other complement inhibitors for exudative AMD.
Agents currently in use or under investigation: neovascular AMD
VEGF inhibitors
A variety of methods are now available to directly inhibit the VEGF165 molecule as well as its various other isoforms. These include the use of monoclonal antibodies directed at one or more isoforms, oligonucleotides with chain-specific sequences corresponding to the VEGF protein, and molecules directed at one or more of the VEGF receptors, including native decoys, tyrosine kinase inhibitors, fusion proteins, and TIE2 receptors. Finally, because VEGF is thought to initiate a cascade of intracellular signals initially, and subsequently extracellular events, it is possible to inhibit VEGF effects either through prevention of secretion of the molecule, direct inhibition of the molecule in the extracellular space, blockade of the receptors, or through interruption in the downstream intracellular signaling pathways leading to both intra- and extracellular events.132 In addition to the sections that follow, the interested reader is referred to Chapter 66 (Neovascular (exudative or “wet”) age-related macular degeneration).
Direct VEGF inhibitors
Monoclonal antibody: bevacizumab (Avastin)
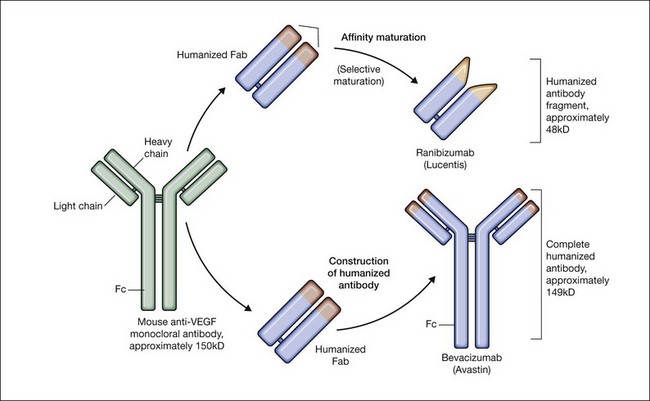
Bevacizumab is a humanized monoclonal antibody (IgG1) against human VEGF-A that selectively inhibits all isoforms and bioactive proteolytic breakdown products of VEGF-A (Fig. 67.11).133 Bevacizumab is an immunoglobulin G molecule that is comprised of amino acid sequences which are about 93% human and 7% murine. Preclinical studies in animal models of various tumor cell lines as well as different forms of ocular neovascularization indicated that the fully sized antibody had excellent efficacy against the primary permeability and proliferative effects of VEGF isoforms. Following extensive clinical testing, it was found to be effective in slowing tumor growth through its effects on angiogenesis.73 Bevacizumab was FDA-approved in 2004 as a treatment for metastatic colorectal cancer when given intravenously at a dose of 5 mg/kg and infused every 2 weeks in combination with 5-fluorouracil. Bevacizumab was shown to increase survival, response rate, and duration of response compared with conventional therapy consisting of irinotecan, fluorouracil, and leukovorin for metastatic colorectal cancer.134 Additional phase III clinical trials with bevacizumab have since resulted in FDA-approval for the treatment of lung, kidney, and brain cancers.
In 2004, a study investigating the use of systemic bevacizumab in patients with exudative AMD (Systemic Avastin for Neovascular AMD – SANA) was initiated at the Bascom Palmer Eye Institute. This study was the first demonstration that bevacizumab was effective for this disease. In this open-label, uncontrolled study, 18 patients underwent two to three intravenous infusions of bevacizumab (5 mg/kg) to evaluate the potential safety and efficacy of systemic bevacizumab over 6 months.135,136 Although clinical trials using intravitreal pegaptanib and ranibizumab for CNV were underway, the prior use of bevacizumab for ocular disease in humans using any route had previously been untested. While no serious adverse events were observed, and only a mild transient increase in blood pressure was identified, the SANA study was too small to establish the safety of bevacizumab. However, significant improvements in visual acuity and OCT central retinal thickness measurements were observed comparable to the changes observed in the early phase Lucentis trials.
While systemic bevacizumab (5 mg/kg) was shown to reduce leakage from CNV, decrease OCT central retinal thickness measurements, and significantly improve vision in exudative AMD, the intravenous use of bevacizumab for exudative AMD was never widely adopted, because the intravitreal approach used up to 500-fold less drug, is much less expensive, and is perceived to be safer due to the smaller dose of drug. In the first-reported case of intravitreal bevacizumab,137 a patient with recurrent CNV secondary to AMD, who had previously been treated with verteporfin photodynamic therapy (PDT) in combination with triamcinolone acetonide followed by treatment with pegaptanib injections, was shown to improve with a decrease in OCT central retinal thickness, subretinal fluid, as well as improvement of subjective visual distortion within 1 week of a single injection of 1.0 mg bevacizumab.
Initially, there was concern that a molecule as large as the 149 kDa bevacizumab may not penetrate the retina and effectively treat CNV compared with ranibizumab, which is one-third the size. However, it became apparent that bevacizumab could penetrate the retina based upon the empiric observations of decreased subretinal fluid on OCT examination and improved visual acuities in treated patients. Subsequently, Han et al. were the first to show that a full-length immunoglobulin the size of bevacizumab was capable of penetrating the rabbit retina after an intravitreal injection.138 Sharar et al. used qualitative immunofluorescence to confirm that intravitreal bevacizumab was able to completely penetrate the retina by 24 hours and was essentially absent at 4 weeks after an injection.139 Moreover, Dib et al. demonstrated bevacizumab molecules in the subretinal space of all six eyes studied two hours after an intravitreal bevacizumab injection of 0.05 ml (1.25 mg), confirming the initial observations that the molecule could rapidly diffuse through the retina.140
The ocular pharmacokinetics of various other monoclonal antibodies injected into the vitreous cavity of monkeys and rabbits demonstrated a half-life of about 5.6 days.141,142 Studies of bevacizumab in rabbits and monkeys suggest a half-life in the range of 4–6 days.143–145 A human study reported that a single dose of intravitreal bevacizumab had a half-life of 3 days and was likely to provide complete intravitreal VEGF blockade for a minimum of 4 weeks.146 One human study suggested a half-life of 6.7 days and yet another study has reported a half-life of as long as 9.8 days.147,148 It is likely that human ocular pharmacokinetics of bevacizumab varies from patient to patient, depending on factors such as the extent of vitreous liquefaction and the phakic status of the eye.
Since the first case of intravitreal bevacizumab for the treatment of exudative AMD was reported, numerous retrospective studies and a few small prospective studies using a dose range between 1.0 and 2.5 mg have been published, all demonstrating clinically significant improvement in mean visual acuity, reduction in fluorescein angiographic leakage, and resolution of OCT-visualized edema in up to 90% of bevacizumab-treated patients. These reports also supported the apparent clinical safety of bevacizumab. As of the time of writing (February 2012), despite the subsequent approval of ranibizumab in 2006, the off-label use of intravitreal bevacizumab has become the most common treatment for exudative AMD in the United State.149,150 In addition, it has also become frequently used for wide range of other forms of macular new vessels including pathologic high myopia, multifocal choroiditis, and ocular histoplasmosis; it has also been used successfully for treatment of clinically significant macular edema from diabetic retinopathy, retinal venous occlusions, and retinopathy of prematurity.151–159 It is being explored for treatment of other macular conditions not associated with new vessels or known elevated VEGF levels, including central serous choroidopathy and uveitides, although its efficacy in those conditions is not yet firmly established. Prior to the release of the Comparison of Age-Related Macular Degeneration Treatments Trial (CATT, discussed below), most of the early pilot studies were small (less than 100 patients) and uncontrolled, with different retreatment criteria and outcome measures; most physicians have frequently extrapolated the results from the phase III ranibizumab trials for these indications to bevacizumab.160 However, two randomized, controlled studies deserve mention.
The ABC trial compared intravitreal bevacizumab (three loading doses every 6 weeks, followed by additional injections at 6-week intervals as needed) to standard therapy, defined at the time of recruitment as verteporfin PDT for predominantly classic CNV, or pegaptanib injection or sham injection for minimally classic or occult CNV. A total of 131 patients were recruited and randomized 1 : 1 to receive either bevacizumab or standard therapy. A significantly higher proportion of the bevacizumab group improved by 15 letters compared with the standard therapy group (32% vs 3%, P < 0.001), irrespective of lesion type. Additionally, more patients lost fewer than 15 letters in the bevacizumab group (91% vs 67%, P < 0.001), and a larger proportion of bevacizumab patients achieved 6/12 or better vision by week 54 of the study. By study end, mean change in vision was +7.0 letters in the bevacizumab group, versus −9.4 letters in the standard care group. Serious ocular and nonocular adverse events were rare in all groups, with no serious intraocular infections in any of the intervention groups. Two myocardial infarctions, one fatal, occurred in the bevacizumab group.161 This study represented the first level 1 evidence of the efficacy of bevacizumab for the treatment of AMD.
A small randomized study conducted at the Veterans Affairs Boston Healthcare System Hospital compared bevacizumab and ranibizumab directly. A total of 28 patients were randomized in a 2 : 1 ratio to receive bevacizumab or ranibizumab, of which 22 patients completed one year of follow-up. Injections were given monthly for 3 months, followed by additional injections given on the basis of OCT findings primarily, or vision or clinical exam. Visual and anatomic results were similar between the two groups: patients receiving bevacizumab improved an average of 7.6 letters, compared with 6.3 letters in the ranibizumab group (P = 0.74). The change in central macular thickness as determined by OCT was −50 µm in the bevacizumab group, versus −91 µm in the ranibizumab group (P = 0.29). The number of ranibizumab injections administered was significantly lower than the number of bevacizumab injections (4 vs 8, P = 0.001). The authors hypothesized two possible reasons: (1) tachyphylaxis to bevacizumab; or (2) a more robust early anatomic response to ranibizumab, leading to a higher number of bevacizumab injections given on an as-needed basis determined primarily by OCT findings.162 This study, although small in size, was the first head-to-head trial of bevacizumab and ranibizumab, and demonstrated the similar efficacy of bevacizumab to ranibizumab.
Antigen binding fragment: ranibizumab (Lucentis)
Ranibizumab is a humanized anti-VEGF-A recombinant Fab fragment that has been affinity matured to increase its binding affinity for VEGF-A (see Fig. 67.11). Ranibizumab binds within the VEGFR-binding domain of all biologically active isoforms of VEGF-A. Two randomized, double-masked, pivotal phase III clinical trials have demonstrated that monthly intravitreal injections of ranibizumab are an effective and safe treatment for subfoveal CNV in AMD patients.163,164
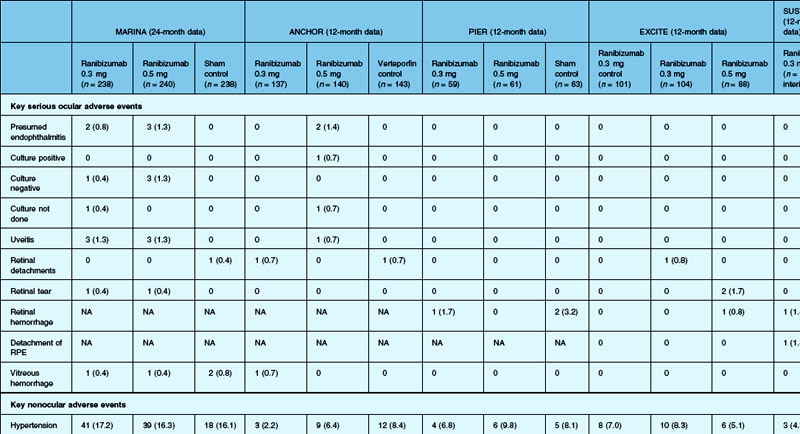
The MARINA study assessed the response of minimally classic or occult CNV to ranibizumab.163 Patients (n = 716) were assigned randomly to receive sham injection (n = 238), 0.3 mg (n = 238), or 0.5 mg (n = 240) ranibizumab. After 4 months of follow-up, approximately 90% of ranibizumab-treated patients had lost less than 15 letters on the Bailey – Lovie (ETDRS) chart as compared to 53% of the sham-injected patients. This treatment response was independent of lesion size, initial visual acuity, or whether the lesion was classified as minimally classic or occult with no classic CNV on fluorescein angiography. Vision improved by at least 15 letters in approximately 33% of patients receiving 0.5 mg ranibizumab at 24 months of follow-up versus 4% in the sham-injected patients (Fig. 67.12).165 Approximately 1% of patients receiving intravitreal ranibizumab developed endophthalmitis. The risk of cataract was approximately 0.2%. There were no cases of retinal detachment among patients receiving intravitreal therapy. Despite the theoretical risk of systemic vascular complications, there was no imbalance among treated and control groups regarding hypertension, and the risk of myocardial infarction among sham-injected, 0.3 mg, and 0.5 mg cohorts was 1.7%, 2.5%, and 1.3%, respectively. The risk of stroke among the three groups was 0.8%, 1.3%, and 2.5%, respectively. The risk of nonocular hemorrhage was 5.5%, 9.2%, and 8.8% in each of the three cohorts, respectively. None of these differences was statistically significant, although the authors recognized that the study was not powered to detect small differences.
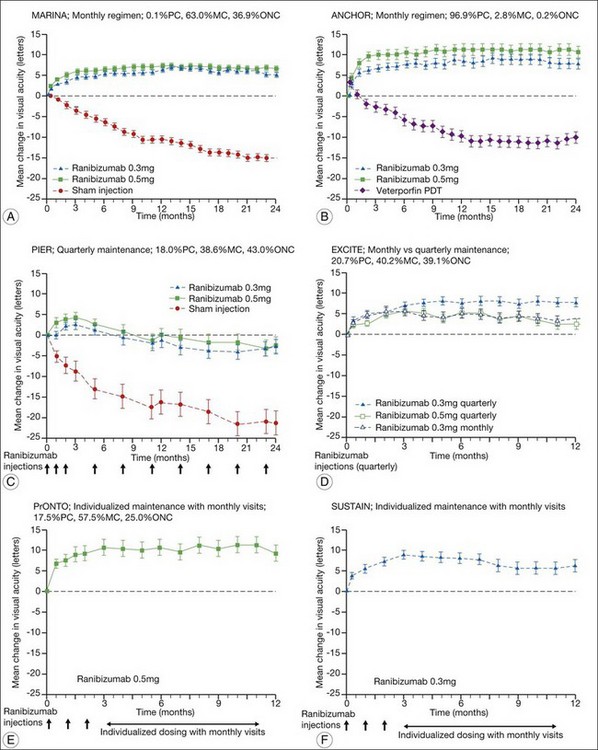
The ANCHOR study assessed the response of patients with predominantly classic CNV as determined by fluorescein angiography.164 Patients (n = 423) were randomly assigned to receive verteporfin-PDT plus sham injection (n = 143) or sham PDT plus injection of either 0.3 mg (n = 140) or 0.5 mg (n = 140) ranibizumab. At 12 months follow-up, approximately 95% of ranibizumab-treated patients lost less than 15 letters of vision versus 64% in the verteporfin active-treatment control group. Forty percent of patients treated with 0.5 mg ranibizumab gained at least 15 letters vision versus 6% in the verteporfin treatment cohort. At month 24, the visual benefit from ranibizumab remained statistically (P < 0.0001 vs PDT) and clinically significant: 89.9–90.0% of ranibizumab-treated patients had lost <15 letters from baseline (vs 65.7% of PDT patients); 34–41.0% had gained ≥15 letters (vs 6.3% of PDT group); and, on average, visual acuity was improved from baseline by 8.1–10.7 letters (versus a mean decline of 9.8 letters in the PDT groups).166 Changes in lesion anatomic characteristics on fluorescein angiography also favored ranibizumab (all comparisons P < 0.0001 vs PDT). Overall, there was no imbalance among groups in rates of serious ocular and nonocular adverse events. In the pooled ranibizumab groups, 3 (1.1%) of 277 patients developed presumed endophthalmitis in the study eye (rate per injection = 3/5921, or 0.05%). Retinal detachment was observed in one patient in both the verteporfin (0.7%) and 0.3 mg ranibizumab (0.7%) cohorts. There were no cases of lens injury. The risk of hypertension was the same in all cohorts. The risk of myocardial infarction among verteporfin, 0.3 mg ranibizumab, and 0.5 mg ranibizumab cohorts was 0.7%, 0.7%, and 2.1%, respectively. The risk of stroke or cerebral infarction was 0.7% in each of the three cohorts. The risk of nonocular hemorrhage was 2.1%, 5.1%, and 6.4% in each of the three cohorts. Immunoreactivity to ranibizumab before treatment was 1.5%, 3.2%, and 0.8% among the verteporfin, 0.3 mg ranibizumab, and 0.5 mg ranibizumab cohorts, respectively. Immunoreactivity was present in 1.6%, 1.6%, and 3.9% of patients in each of these cohorts, respectively, after 12 months of treatment. (The concern regarding immunoreactivity is that patients who develop an immune response might exhibit increased intraocular inflammation following intravitreal injection and consequently might not respond to the medication as well as patients who do not exhibit such a response. In practice, most retina surgeons have not observed this phenomenon and the drug has been minimally immunogenic in the doses tested.167)
In both the MARINA and ANCHOR trials, visual acuity improvement seemed to reach a plateau by the 4-month time point (Fig. 67.12). Because monthly injections were considered inconvenient and daunting for some patients and their family members, and entailed risk as well as considerable expense, studies were undertaken to evaluate alternative treatment regimens. The PIER and EXCITE studies provided useful information in this regard.168,169 In PIER, a phase IIIb multicenter, randomized, double-masked trial, patients with subfoveal CNV were randomly assigned to sham injection (n = 63), 0.3 mg ranibizumab (n = 60), or 0.5 mg ranibizumab (n = 61). Patients received sham or ranibizumab injections every 4 weeks for three doses followed by additional treatment every 3 months. By month 12, mean changes from baseline visual acuity were −16.3, −1.6, and −0.2 letters for the sham, 0.3 mg, and 0.5 mg groups, respectively (P ≤ 0.0001, each ranibizumab dose vs sham). Ranibizumab arrested CNV growth and reduced leakage from CNV. The treatment effect declined, however, in the ranibizumab groups during quarterly dosing. At month 3, for example, the mean changes from baseline vision had been gains of +2.9 and +4.3 letters for the 0.3 mg and 0.5 mg doses, respectively. During study year 2, eligible sham-group patients crossed over to 0.5 mg ranibizumab quarterly. Later in year 2, all eligible randomized patients rolled over to 0.5 mg ranibizumab monthly dosing. The ranibizumab-treated patients showed an improvement in mean visual acuity during the first three months of the study, but this improvement was not sustained. Nonetheless, the 0.5 mg ranibizumab cohort had a mean visual acuity change that was 16 letters better than that of the sham cohort by month 12 (P < 0.0001). By month 24, visual acuity had decreased an average of 21.4, 2.2, and 2.3 letters from baseline in the sham, 0.3 mg, and 0.5 mg groups (P < 0.0001 for each ranibizumab group vs sham). The visual acuity of sham patients who crossed over to ranibizumab decreased over time, with an average loss of 3.5 letters 10 months after crossover. The visual acuity of patients in the 0.3 mg and 0.5 mg cohorts who rolled over to monthly ranibizumab injections increased for an average gain of 2.2 and 4.1 letters, respectively, 4 months after transition. These data indicate that injecting patients with ranibizumab every 3 months (after an induction phase of three monthly injections) does not produce the same chance for visual benefit as monthly injection, at least during the first 12 months of therapy. Ranibizumab appeared to provide additional visual benefit to treated patients who rolled over to monthly dosing, but not to patients who began receiving ranibizumab after >14 months of sham injections.
The EXCITE study, was a 12-month, multicenter, randomized, double-masked, active-controlled, phase IIIb study designed to demonstrate the noninferiority of a quarterly treatment versus monthly intravitreal ranibizumab treatment regimen in patients with AMD-associated CNV.170 Patients with primary or recurrent subfoveal CNV secondary to AMD (353 patients), including those with predominantly classic, minimally classic, or occult (no classic component) lesions, were enrolled. Patients were randomized (1 : 1 : 1) to 0.3 mg quarterly, 0.5 mg quarterly, or 0.3 mg monthly doses of ranibizumab. Treatment comprised three consecutive monthly injections followed by a 9-month maintenance phase (either monthly or quarterly injection). In the per-protocol population (293 patients), visual acuity increased from baseline to month 12 by 4.9, 3.8, and 8.3 letters in the 0.3 mg quarterly (104 patients), 0.5 mg quarterly (88 patients), and 0.3 mg monthly (101 patients) dosing groups, respectively. Similar results were observed in the intent-to-treat (ITT) population (353 patients). The mean decrease in CRT from baseline to month 12 in the ITT population was −96.0 µm in the 0.3 mg quarterly, −105.6 µm in the 0.5 mg quarterly, and −105.3 µm in the 0.3 mg monthly group. At month 12, the visual acuity gain in the monthly treatment cohort was higher than that of the quarterly regimens. The noninferiority of a quarterly regimen was not achieved with reference to 5.0 letters.
The SUSTAIN study is a 12-month, phase III, multicenter, open-label, single-arm study involving 513 ranibizumab-naive patients with AMD-associated CNV.171 In this study, patients received three initial monthly injections of ranibizumab (0.3 mg) and thereafter pro re nata (PRN) retreatment for 9 months based on prespecified retreatment criteria. Patients switched to 0.5 mg ranibizumab after approval in Europe. The average number of retreatments from months 3 to 11 was 2.7. Mean best-corrected visual acuity increased steadily from baseline to month 3 to reach +5.8 letters, decreased slightly from month 3 to 6, and remained stable from month 6 to 12, reaching +3.6 at month 12. The mean change in CRT was −101.1 µm from baseline to month 3 and −91.5 µm from baseline to month 12.
In summary, data from the ANCHOR, MARINA, PIER, EXCITE, and SUSTAIN studies indicated that after a loading dose period of three monthly injections, subsequently monthly ranibizumab injections give superior visual acuity outcomes compared to quarterly or PRN injections (Fig. 67.13).
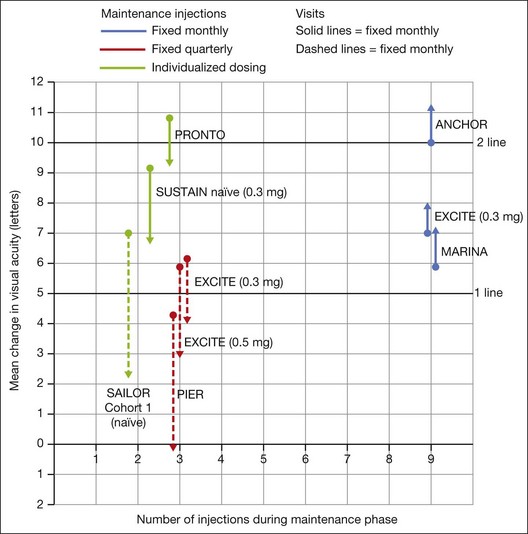
In the PrONTO (Prospective OCT Imaging of Patients with Neovascular AMD Treated with Intraocular Lucentis) study, patients (n = 40) received 0.5 mg ranibizumab at entry, month 1, and month 2.172 Optical coherence tomography (OCT) measurements were obtained at baseline and at least monthly after injection (more frequently during the first two months after entry). Fluorescein angiograms were obtained at baseline and every 3 months thereafter. Retreatment with ranibizumab was done only if one or more of the following conditions was observed: (1) OCT central thickness (CRT) increased 100 µm; (2) ≥5 letter visual loss associated with subretinal fluid (as judged with OCT); (3) new onset classic CNV; (4) new macular hemorrhage; (5) persistent subretinal or intraretinal fluid was present 1 month after the previous injection. One day after the first injection, there was a decrease in the mean OCT thickness of 47 µm. By month 12, the mean visual acuity improved by 9.3 letters (P <0.001), and the mean central thickness decreased by 178 µm compared with baseline (P <0.001). Mean visual acuity improvement was 9.3 letters, and the mean central thickness had decreased by 178 µm. The average number of injections over the first year was 5.6. Visual acuity improved ≥15 letters in 35% of patients, and once macular fluid resorbed completely, the mean interval before another injection was 4.5 months. During the second year, the retreatment criteria were amended to include retreatment if any qualitative increase in the amount of fluid was detected using OCT.173 Forty patients were enrolled and 37 completed the 2-year study. At month 24, the mean visual acuity improved by 11.1 letters (P < .001) and the OCT-CRT decreased by 212 µm (P < 0.001). Visual acuity improved by 15 letters or more in 43% of patients. These visual acuity and OCT outcomes were achieved with an average of 9.9 injections over 24 months. The PrONTO Study data suggested that OCT-guided variable-dosing regimens with intravitreal ranibizumab were capable of achieving visual acuity outcomes comparable to those from the phase III clinical studies, but with fewer intravitreal injections.
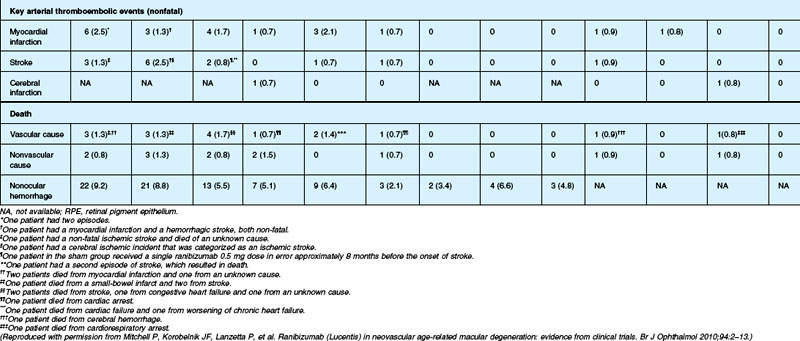
Although the risk of arterial thromboembolic events was not increased to a statistically significant degree among ranibizumab-treated patients in these phase III studies (overall risk ~2.1% in ranibizumab-treated patients during year 1 vs ~1.1% among controls), some concerns were expressed about relative safety compared with alternative therapies, efficacy aside, as the studies were not powered for statistical significance between the two different ranibizumab doses. Year 2 data from the MARINA study indicate the overall rate of antiplatelet trialists’ collaboration (APTC)-defined arterial thromboembolic events, which includes nonfatal myocardial infarction, nonfatal stroke, and death from a vascular or unknown cause,174 was 4.6%, 4.6%, and 3.8% among the 0.5 mg ranibizumab, 0.3 mg ranibizumab, and control cohorts, respectively. Year 2 data from the ANCHOR study indicate the overall rate of APTC arterial thromboembolic events was 5%, 4.4%, and 4.2% in the 0.5 mg, 0.3 mg, and verteporfin-PDT cohorts, respectively. The PIER168,169 and EXCITE170 studies had similar results in this regard.
The SAILOR study was a phase IIIb study whose objectives are to evaluate the safety of 0.3 mg and 0.5 mg Lucentis in patients with AMD-associated subfoveal CNV. In cohort 1 of this study, the dose was randomly assigned and administered once a month for 3 months and thereafter as needed based on retreatment criteria. In SAILOR, there was a numerically higher rate of cerebrovascular stroke with 0.5 mg ranibizumab compared with 0.3 mg ranibizumab (1.2 vs 0.7%), which was not statistically significant in patients with a history of stroke.175 In the SUSTAIN study, a total of 249 patients (48.5%) reported ocular adverse events, 5 patients (1.2%) with ocular serious adverse events involving the study eye (retinal hemorrhage, cataract, retinal pigment epithelial tear, reduced visual acuity, vitreous hemorrhage), 19 patients (3.7%) with arteriothromboembolic events were observed, with 8 deaths (1.5%).171 The most frequent adverse events in the study eye were reduced visual acuity (18.5%), retinal hemorrhage (7.2%), increased intraocular pressure (7.0%), and conjunctival hemorrhage (5.5%). Overall, the ocular and systemic risks associated with ranibizumab use appear to be low and within a reasonable range when taking into account the associated likelihood of visual improvement in properly selected patients (Table 67.2).165
Comparison of Age-related Macular Degeneration Treatments Trial (CATT)
1. Does one treatment offer superior visual outcomes to another?
2. What is the optimal treatment regimen and interval?
3. Is the safety profile of bevacizumab comparable to that of ranibizumab, which has been subjected to rigorous phase I – phase IV evaluation?
The CATT was designed to address these questions.150
1. Bevacizumab monthly injections (8.0 letters gained) and ranibizumab monthly injections (+8.5 letters) yielded equivalent visual outcomes.
2. Bevacizumab given as needed (+5.9 letters) was also equivalent to ranibizumab given as needed (+6.8 letters).
3. Ranibizumab given as needed was equivalent to ranibizumab given monthly.
4. Ranibizumab given as needed was also equivalent to bevacizumab given monthly.
5. Bevacizumab given as needed was neither inferior nor not-inferior (inconclusive) to bevacizumab given monthly.
6. Bevacizumab given as needed was neither inferior nor not-inferior (inconclusive) to ranibizumab given monthly.
Detailed visual and anatomic outcome data are shown in Fig. 67.14 and Table 67.3.
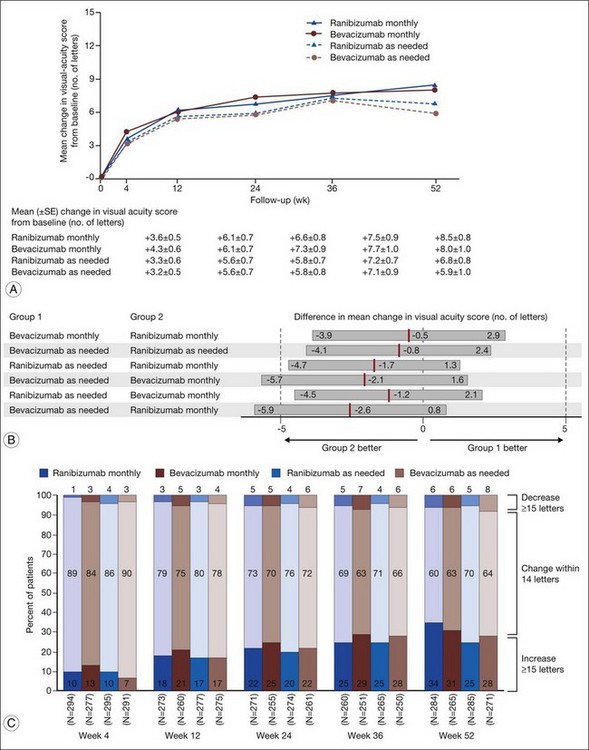
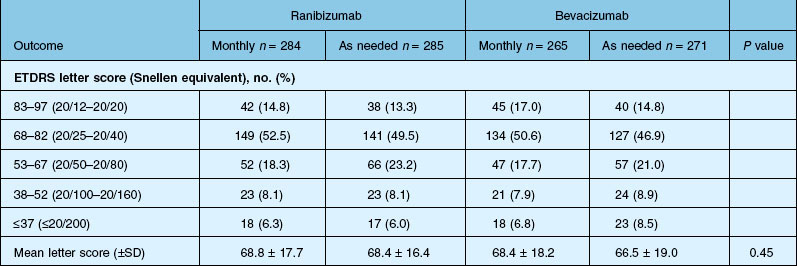
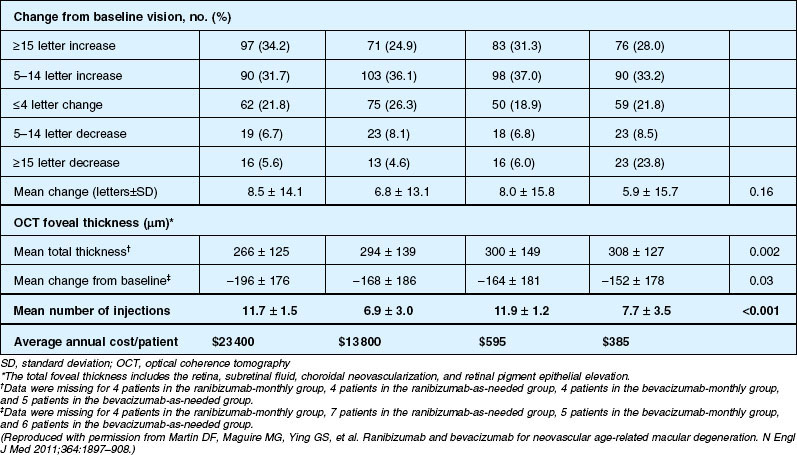
Secondary visual outcomes were similarly equivalent, with 91.5% (bevacizumab as needed) to 95.4% (ranibizumab as needed) of patients not having a decrease in vision of 15 letters or more from baseline, and 24.9% (ranibizumab as needed) to 34.2% (ranibizumab monthly) experiencing a gain of at least 15 letters. Anatomic outcomes showed that all groups had substantial decreases in retinal thickness as measured by OCT at 1 year, although the monthly ranibizumab group experienced a greater decrease in thickness (196 ± 176 µm) than those patients receiving bevacizumab as needed (152 ± 178 µm, P = 0.03). The mean number of injections given in the as-needed groups was 7.7 ± 3.5 (bevacizumab) and 6.9 ± 3.0 (ranibizumab) out of 13 possible, translating to an average yearly cost of $385 vs $13 800, respectively. The average cost to patients given monthly injections was $595 for bevacizumab and $23 400 for ranibizumab (Table 67.3).
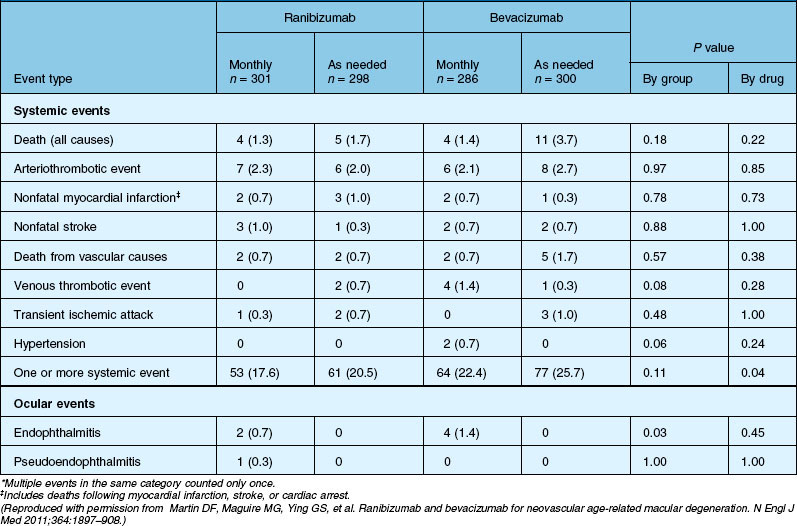
Deaths occurred in 2.0% of the entire study group over the one-year period, with no significant difference among any of the groups. The occurrence of arteriothrombotic events was 2–3% for each group, also without significant differences. Venous thrombotic events occurred in 4 out of 286 patients receiving bevacizumab monthly (P = 0.08 difference between all groups) but occurred infrequently in all groups. When serious systemic adverse events were pooled among drug-specific groups, they occurred in 24.1% of patients receiving bevacizumab, vs 19.0% of those receiving ranibizumab (P = 0.04). Endophthalmitis occurred in 0.05% of the total number of injections, without a significant difference between medication groups. All other serious ocular adverse events occurred with rare frequency (Table 67.4).
The impact of the one-year CATT study results was significant, establishing the equivalent visual outcomes of ranibizumab and bevacizumab when given monthly. Although bevacizumab as needed, when compared to ranibizumab or bevacizumab monthly, yielded inconclusive results, all other groups showed similar efficacy. Rosenfeld proposed in explanation that the treatment effect for bevacizumab may be less durable in some patients; thus, giving more frequent injections might improve visual outcomes in that subgroup.176 Importantly, the first-year study results also appeared to establish that as-needed ranibizumab (as well as monthly bevacizumab) was as effective as monthly ranibizumab, which not only has significant implications for treatment cost, but also for the cumulative risk of injection-associated adverse events as well as patient anxiety surrounding treatment administration. Interestingly, although the effect of the medication on retinal thickness was more prominent for ranibizumab, this did not translate to better vision as compared to bevacizumab. Finally, the study authors recognized that the CATT was insufficiently powered to determine a difference in infrequently occurring systemic and ocular adverse events, and although bevacizumab was associated with a higher overall rate of systemic adverse events, these events involved organ systems not typically associated with systemic anti-VEGF therapy. In particular, the rate of arteriothrombotic and venous thrombotic events was no different between the two medications, and was low in all groups. Additionally, the risk of adverse events was not noted to correlate with increased exposure to medication, with a higher rate of occurrence in patients receiving injections on an as-needed basis.
Interested readers might also refer to the one-year interim report from the IVAN study. The efficacy and safety of ranibizumab and bevacizumab intravitreal injections to treat neovascular age-related macular degeneration were compared, and participants were randomize to four groups: ranibizumab or bevacizumab, given either every month (continuous) or as needed (discontinuous), with monthly review. The authors concluded: “The comparison of visual acuity at 1 year between bevacizumab and ranibizumab was inconclusive. Visual acuities with continuous and discontinuous treatment were equivalent. Other outcomes are consistent with the drugs and treatment regimens having similar efficacy and safety.”176a
The second year results of CATT, published in mid-2012, described the visual impact of switching from monthly to as-needed treatment after the first year, as well as any differences in disease activity or progression, as demonstrated by fluid on OCT, leakage on fluorescein angiography, and lesion size. In addition, the use of high-resolution spectral-domain OCT in year two allowed for comparison between standard and high-resolution imaging in the detection of disease activity.177
Anatomic outcomes showed improved retinal thickness in patients treated monthly as compared to those treated as needed (29 µm difference, P = 0.005). Additionally, the proportion of patients without fluid as detected by OCT was lower in both the ranibizumab group as well as the monthly group (up to 45.5% in the monthly ranibizumab group); similarly, the proportion of patients with no fluorescein leakage was higher in patients treated monthly, and lesion area remained stable in these patients while growing in the as-needed group. However, the study also found that the proportion of monthly patients developing geographic atrophy was significantly higher in patients receiving monthly treatment as compared to the as-needed group, with the highest rate in those receiving ranibizumab monthly. (Vision and anatomic outcomes are detailed in Fig. 67.15 and Table 67.5).
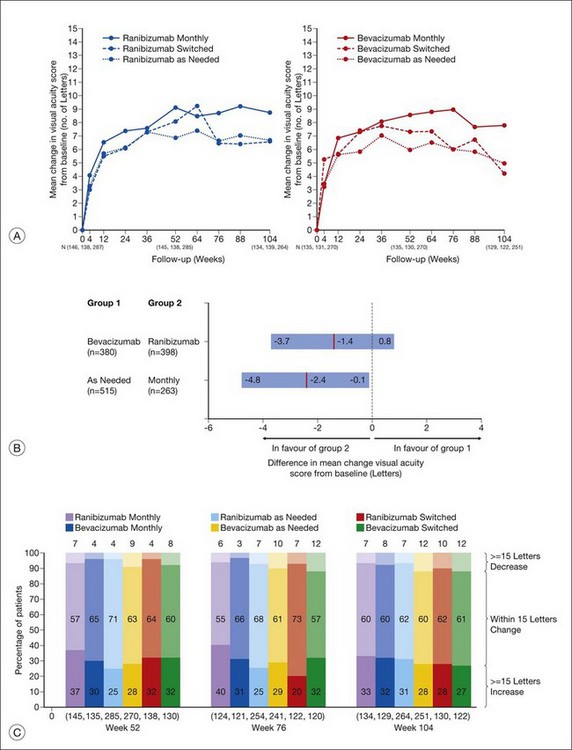
(Reproduced with permission from Martin DF, Maguire MG, Fine SL, et al. Ranibizumab and bevacizumab for treatment of neovascular age-related macular degeneration: two-year results. Ophthalmology 2012. Corrected proof published online 2012 May 2. http//:www.ophsource.org/periodicals/ophtha/article/S0161-6420(12)00321-1/ [cited 2012 June 8].)
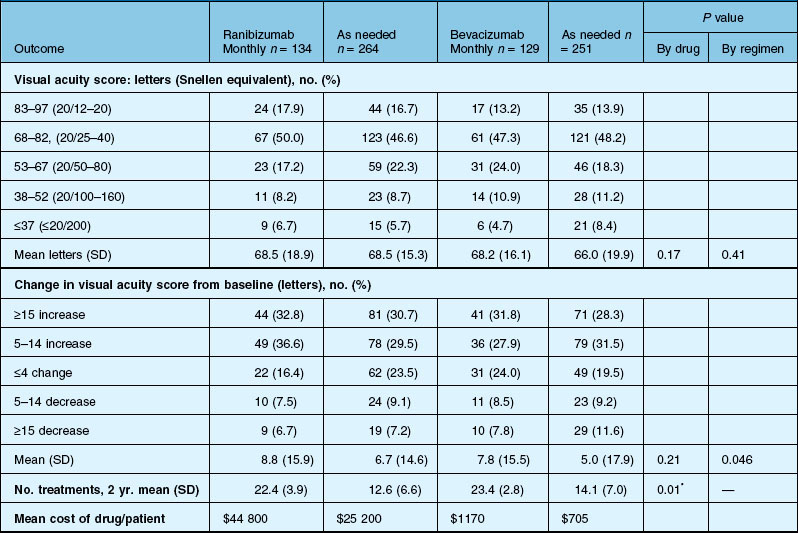
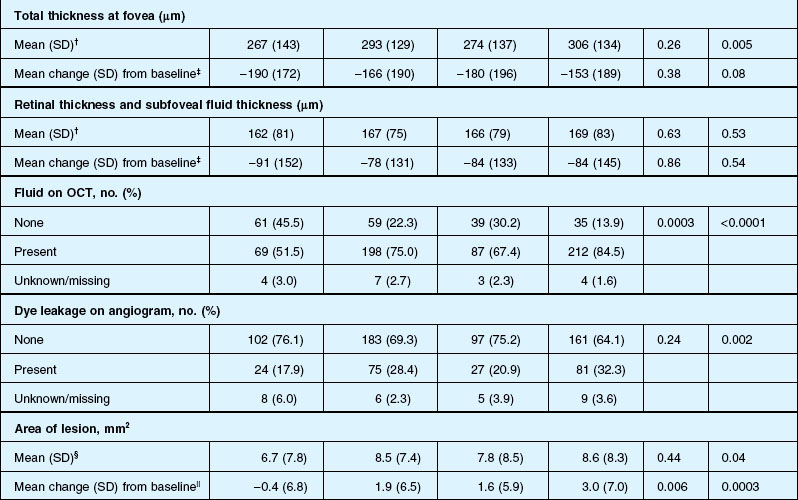
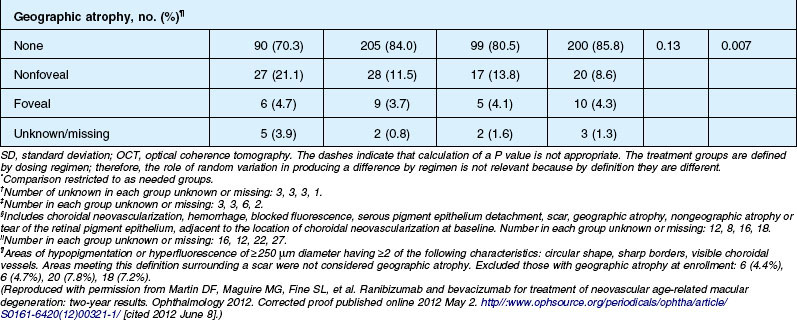
In contrast to patients maintaining monthly therapy through two years, those switching to as-needed therapy at week 52 lost from 1.8 (ranibizumab) to 3.6 (bevacizumab) letters after year one (P = 0.03 when comparing monthly to as-needed therapy), resulting in very similar visual outcomes to patients maintained on as-needed therapy since study enrollment for both medications. Final visual acuity was similar for all groups, without significant differences between bevacizumab and ranibizumab, or between monthly and as-needed therapy. Anatomically, retinal thickness increased significantly, with a range of +19 µm (ranibizumab) to +31 µm (bevacizumab) in patients who were reassigned to as-needed therapy. The proportion of patients without fluid on OCT was significantly higher in patients who switched regimens, as well as in patients receiving bevacizumab. As in patients receiving the same regimen through year two, the proportion demonstrating dye leakage was higher in switched patients. Although the lesion area was not significantly different in either group, the rate of development of geographic atrophy was higher in ranibizumab patients as well as those receiving monthly dosing (Table 67.6).
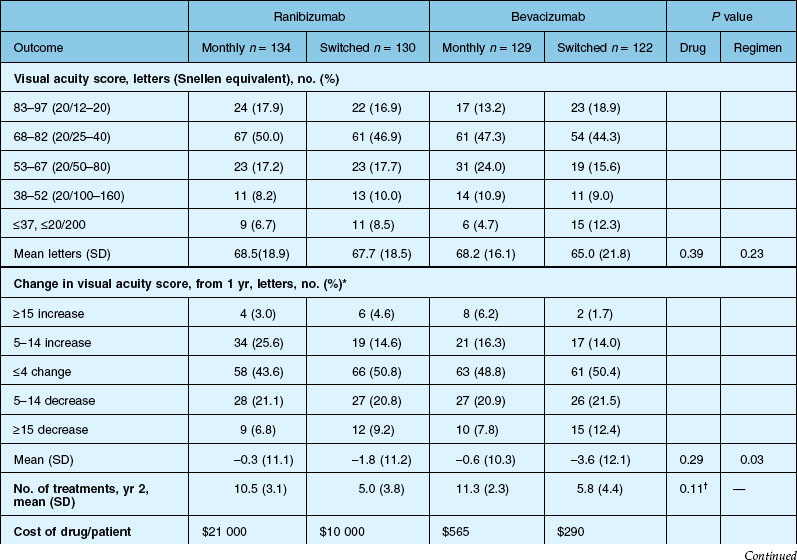
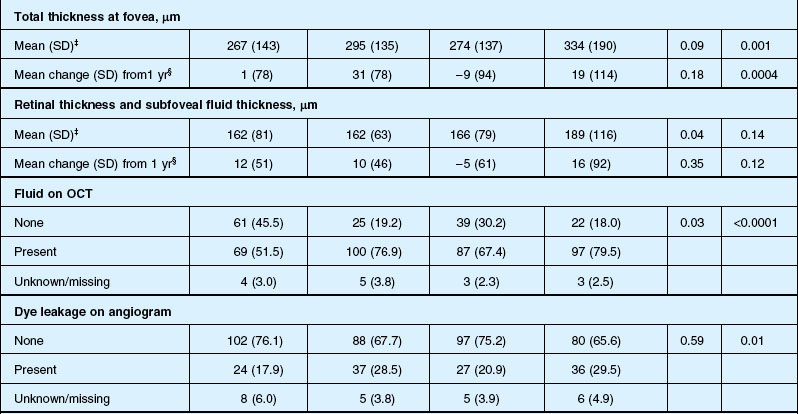
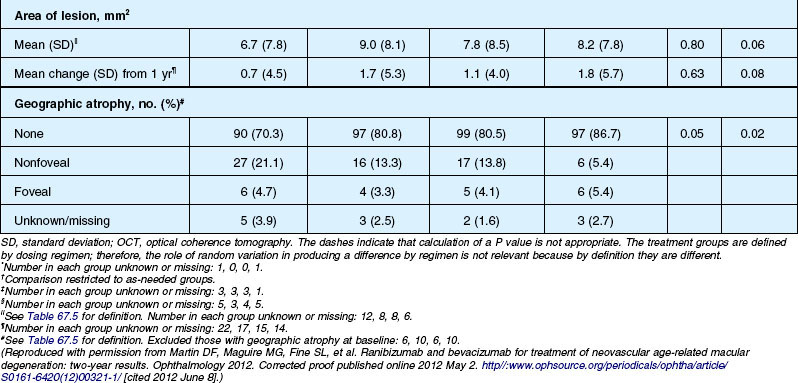
As reported in the first year of results, the rate of systemic adverse events was significantly higher in the bevacizumab group than in the ranibizumab group, at 39.9% as compared to 31.7% overall, and 24.4% vs. 18.0% in the second year alone. Again, this difference persisted after excluding vascular events previously associated with systemic anti-VEGF drugs (P = 0.02), while the difference in events previously associated with anti-VEGF therapy approached statistical significance (P = 0.07). In addition, as noted in the first year, over the course of two years, patients treated as needed had a higher rate of systemic serious adverse events than those treated monthly (risk ratio of 1.30, P = 0.009), although this difference in the second year alone was not significant. There was no difference between drugs in the proportion of patients who died (6.1% vs 5.3%) or in the number of arteriothrombotic (5.0% vs 4.7%) or venous thrombotic events (1.7% vs 0.5%). Endophthalmitis occurred in 7 patients receiving bevacizumab and 4 patients receiving ranibizumab (P = 0.38), with 10 of these 11 cases occurring in patients receiving monthly therapy (Table 67.7).
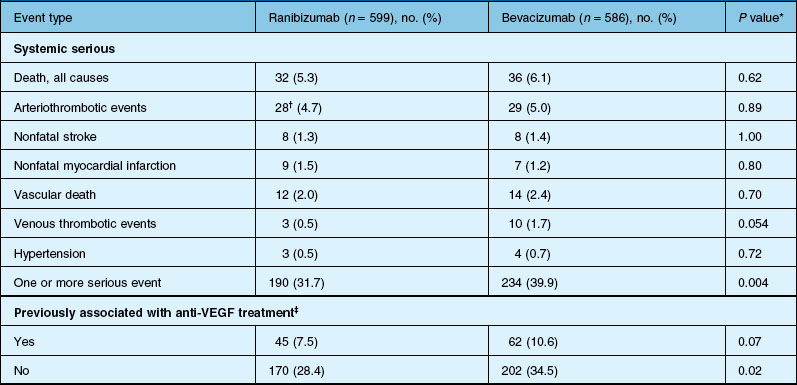
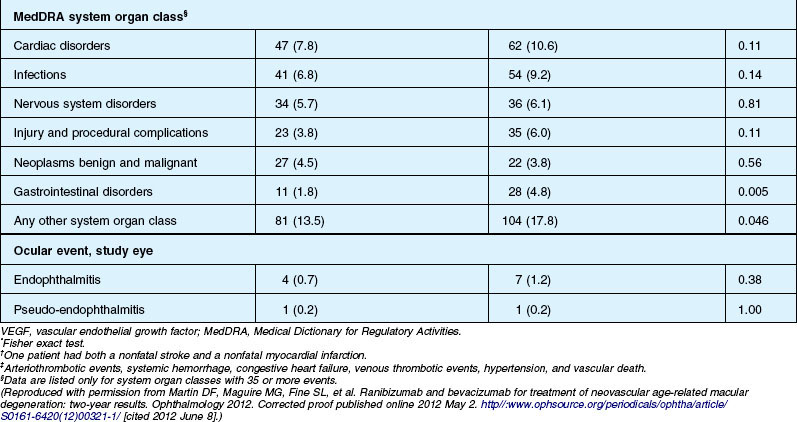
1. In contrast to the first-year results, longer-term follow-up suggests that as-needed therapy is somewhat less effective than monthly therapy overall in terms of visual gain.
2. Visual acuity and visual gain are associated with, but do not closely mirror retinal thickness or the presence of fluid.
3. One year of monthly therapy does not appear to confer lesion stability when subsequently changing to as-needed therapy.
4. When given as needed, bevacizumab is required more frequently, on average 1.5 more injections over the two-year period.
5. Geographic atrophy occurs with greater frequency in patients receiving monthly therapy.
6. Spectral domain OCT does not appear to increase the accuracy of identifying retinal fluid by the treating physician.
7. Endophthalmitis occurs at similar rates in ranibizumab- and bevacizumab-treated patients, although (as one may expect) it is appears to be associated with an increased rate of injections, occurring most often in patients treated monthly.
8. The increased rate of systemic serious adverse events in bevacizumab persisted in the second year. The authors remained unsure of the reason for the increased risk ratio of systemic adverse events. As in the first year, the rate of vascular events previously associated with anti-VEGF therapy was similar in both groups.
Soluble receptor: aflibercept (VEGF-TRAP EYE)
Aflibercept (VEGF-TRAP EYE, EYLEA™) is a product of Regeneron Pharmaceuticals. It is soluble fusion protein which combines ligand-binding elements taken from the extracellular components of VEGF receptors 1 and 2 fused to the Fc portion of IgG1,178 It has at least a 200-fold higher affinity for VEGF than ranibizumab,179 and aflibercept is thought to penetrate all layers of the retina.180 Unlike bevacizumab and ranibizumab that inhibit just VEGF-A, aflibercept also binds VEGF-B and PlGF.179,181 Aflibercept has undergone phase I and II clinical trials in wet AMD, and recently concluded phase III clinical testing. The phase I study, known as CLEAR-IT 1 (CLinical Evaluation of Anti-angiogenesis in the Retina Intravitreal Trial), showed safety and a functional and anatomical improvement with a dose of 0.5 and 2 mg.182 The phase II trial, known as Clear-IT 2, evaluated the biologic effects and safety of aflibercept during a 12-week fixed-dosing period in patients with exudative AMD followed by PRN dosing out to 1 year.183,184 Patients were randomized 1 : 1 : 1 : 1 : 1 to aflibercept during the fixed-dosing phase (day 1 to week 12): 0.5 or 2 mg every 4 weeks on day 1 and at weeks 4, 8, and 12; or 0.5, 2, or 4 mg every 12 weeks on day 1 and at week 12. At week 12, treatment with aflibercept resulted in a significant mean decrease in central retinal thickness from baseline in all groups combined (P < 0.0001). The reduction with the 2 mg every 4 weeks and 0.5 mg every 4 weeks regimens was significantly greater than each of the quarterly dosing regimens. Visual acuity increased significantly by a mean of 5.7 letters at 12 weeks in the combined group (P < 0.0001), with the greatest mean gain of >8 letters in the monthly dosing groups.
After 1 year, there was no difference in the outcomes when the three aflibercept groups were compared with the ranibizumab group.185,186 This was true with respect to visual acuity, OCT central retinal thickness, and safety outcomes. The most important outcome for patients and clinicians was the observation that aflibercept 2 mg dosed every 2 months was equivalent to ranibizumab dosed monthly suggesting that aflibercept could be dosed less frequently. The phase III main results manuscript is expected to be published in 2012.
KH902
KH902 (Chengdu Kanghong Biotechnology) is also a fusion protein that combines ligand-binding elements taken from the extracellular domains of VEGF receptors 1(Flt-1) and 2 (KDR) to the Fc portion of IgG1.187 However, compared with aflibercept (VEGF TRAP-EYE), KH902 also includes the extracellular domain 4 of VEGF receptor 2(KDRd4) which may stabilize the three-dimensional structure and increase the efficiency of dimerization.188 KH902 has a much higher affinity for VEGF due to this additional extracellular domain and may have a longer half-life in the vitreous.187,189 In vivo experiments have shown that intravitreal KH902 was able to inhibit leakage and growth of CNV in rhesus monkeys without signs of toxicity at doses of 300 and 500 µg.187 In a phase I, dose escalation study, intravitreal injections of KH902 up to a dose of 3.0 mg were well tolerated in 28 patients. On day 42 after a single injection, the mean change in visual acuity from baseline was +19.6 letters with no subjects losing 1 letter or more and 57% of patients gaining 15 letters or more from baseline. The mean change in center-point retinal thickness from baseline was −77.2 µm and the mean decrease in CNV area was 12.6%. No safety concerns were detected after a single, intravitreal injection. A phase III trial is currently underway in China.
Adeno-associated viral vector (AAV) gene transduction
As previously discussed, the angiogenic effects of VEGF are mediated through the endothelial receptor tyrosine kinases, VEGFR-1 (also called Flt-1) and VEGFR-2 (Flk-1/KDR),73 with Flt-1 having a tenfold-higher affinity for VEGF.190 A naturally existing soluble form of Flt-1 (sFlt-1) has no transmembrane domains, and thus has no signal transduction properties, instead forming a heterodimer with Flt-1 and Flk-1 extracellular domains.191 This molecule has provided a unique target for gene therapy using an adeno-associated viral (AAV) vector.
AAV vectors are particular suited for gene therapy in part because of their low immunogenicity and pathogenicity, and ability to induce long-term gene expression in the eye. They have been used with success for gene transduction of PEDF for animal models of CNV,192 and have been tested in human trials for RPE65-associated Leber congenital amaurosis.193–195Moreover, for a soluble gene product such as sFlt-1, gene therapy may be especially effective, as transduction need not be cell-specific.188
AAV-mediated sFlt-1 (and related fusion protein) gene therapy has been successful in a number of animal models, including rodent and primate.190,196–199 Lai et al. tested gene transfer using an injected AAV vector with full-length sFlt-1 on both mice and monkeys.190 In the mouse, they found a marked decrease in hyperfluorescence on fluorescein angiography in sFlt-1 transduced eyes when compared to noninjected and control AAV-Green Fluorescent Protein (GFP)-injected eyes. Morphologically, they found that more extensive CNV-induced photoreceptor loss occurred in the noninjected and control eyes when compared to sFlt-1 eyes. Electroretinographic studies showed no significant loss of function as a result of sFlt-1 transduction. Primate studies showed similarly encouraging results, including prevention of CNV induced by laser, without evidence of toxicity on histology. Importantly, there was long-term expression of transduced gene products, with detection of sFlt-1 mRNA at up to 17 months after injection. A similar recent primate study tested an AAV vector tied to a gene for a novel fusion protein of a single domain of sFlt-1 and a human IgG1 heavy chain Fc fragment (termed sFLT01), finding both long-term expression over 5 months, as well as inhibition of CNV in a dose-dependent fashion.198 A phase I clinical trial is currently recruiting for AAV2-sFLT01 gene therapy for neovascular AMD (NCT01024998).
Oligonucleotide aptamer (pegaptanib – Macugen)
The first VEGF inhibitor approved for use in macular degeneration was pegaptanib. Although it has largely been replaced by the monoclonal anti-VEGF antibodies bevacizumab and ranibizumab, it represented the first targeted molecular therapy for macular degeneration, and therefore is of particular historical interest. Pegaptanib is an aptamer, or a short single-stranded nucleic acid sequence of specified strand shape and length, which has a high degree of specificity and affinity for a target polypeptide.200 In the case of pegaptanib, a large library of RNA sequences ranging from 1014 to 1015 sequences are incubated with the target molecule, VEGF isoform 165. The molecule is capable of selectively binding only VEGF165, and not the other isoforms or other receptors and providing sustained inhibition for in excess of 20 days in vitro corresponding to a desired intraocular concentration in excess of the minimal inhibitory concentration necessary to inhibit VEGF165 for a period of approximately 6 weeks.201
Phase II/III double-masked, multicenter clinical trials were conducted in humans in the United States and in Europe concurrently.202 A total of 1186 patients were randomized to one of three treatment doses or sham. Results showed lower risk of moderate and severe vision loss with pegaptanib vs control, with a favorable safety profile. As stated previously, pegaptanib is currently used infrequently for the treatment of exudative AMD, except in the context of combination therapy, or in some cases, maintenance therapy following induction with ranibizumab or bevacizumab.203
Small interfering RNA (siRNA)
RNA interference is the process of sequence-specific post-transcriptional gene silencing in animals and plants, which is initiated by double-stranded RNA (dsRNA) that is homologous in sequence to the silent gene. Confronted with double-stranded RNA eukaryotic cells respond by destroying their own messenger RNA (mRNA) that shares these sequences with the double-strand.204 The process appears to be mediated by 21 and 22 nucleotide siRNAs that are generated by ribonuclease 3 cleavage from longer dsRNAs. siRNA differs from an aptamer in being double- rather than single-stranded RNA; mechanistically, the presence of siRNA results in the inhibition of protein synthesis. In this context, neovascularization is theoretically prevented because VEGF mRNA is destroyed prior to translation, in contrast to the inhibition through the binding of free VEGF165 by an aptamer such as pegaptanib. In testing this hypothesis, siRNA specific for human vascular endothelial growth factor (VEGF) and an enhanced green fluorescent protein (EGFP) were designed and tested as active molecule and negative control in both in vitro cell lines chemically induced to simulate hypoxia as well as in a laser-induced model of CNV in mice. siRNA was delivered by coinjection with recombinant viruses carrying small hVEGF cDNA to induce expression of the appropriate siRNA in the subretinal space. Production of siRNA was confirmed by the expression of EGFP in the subretinal space with positive controls, and as evidence of VEGF transgene expression, which resulted in significant inhibition of CNV compared with negative controls after laser photocoagulation.204 Conventionally injected siRNA at dosages of 70, 150 or 350µg in a 0.05 ml volume with a vehicle serving as a negative control was successful in reducing CNV in a primate model following standardized laser injury between 15 and 36 days after treatment in conjunction with reduced permeability, angiographically and reduced lesion sizes measured by planimetric methods in a masked fashion81 (Fig. 67.16).
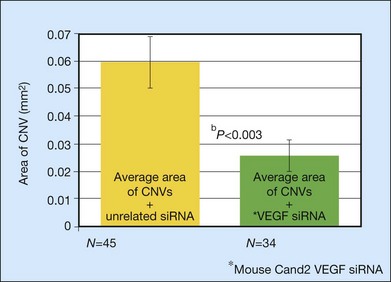
Fig. 67.16 Effects of siRNA treatment in murine model of choroidal neovascularization.
(Reproduced with permission from Reich SJ, Fosnot J, Kuroki A, et al. Small interfering RNA (siRNA) targeting VEGF effectively inhibits ocular neovascularization in a mouse model. Mol Vis 2003;9:210–6.)
A recent phase I study of Sirna-027, a siRNA molecule targeting a conserved region of VEGFR-1, has been conducted. Twenty-six patients were divided into cohorts of 3–6, each receiving sequentially escalating doses of 200–1600 µg in order to determine the maximally tolerated dose. The study authors reported no dose-limiting toxicity, and few medication-specific adverse events. Additionally, visual acuity improved by a mean of +5.5 letters, and stabilized or improved in the vast majority of study participants. Foveal thickness as measured by OCT showed improvement of up to 20.3% in one cohort. The study illustrated the therapeutic principle, as well as showing a favorable safety profile of siRNAs for the treatment of neovascular AMD.205 Two other molecules have also undergone clinical trials. RTP801i-14 (PF-04523655, Quark/Pfizer), a small inhibitory RNA (siRNA) that targets VEGF production via the REDD1/mTOR/HIF1-alpha pathway, is currently in a phase II study (NCT00713518) as adjunctive therapy to ranibizumab for treatment of AMD-associated CNV.
Interestingly, recent evidence suggests that the effect of siRNAs on CNV may in fact be a class effect of the molecule, and not contingent upon sequence specificity of the dsRNA.206–208 The effect is instead postulated to occur through a toll-like receptor, TLR3, expressed on the endothelial cell surface, and mediated by the interferon-gamma and IL-12 cytokines.208 How this may influence further study of siRNA pharmacotherapy is unclear.
Intracellular signaling blockers
PDGF/PDGFR inhibitors*
Currently, an anti-PDGF-B pegylated aptamer, E10030, is under study. A phase I trial in which the aptamer was administered intravitreally in combination with ranibizumab has been completed. Initial results in this safety trial were promising: visual acuity improved by 21–27 letters at 8 weeks, with 59% of patients showing moderate visual improvement at 12 weeks, versus 36% of the ranibizumab cohort.209 A phase II trial is currently underway (NCT01089517).
Another therapeutic avenue currently under examination is the inhibition of the PDGF receptor tyrosine kinase. Pazopanib (GlaxoSmithKline) is a tyrosine kinase inhibitor that blocks the action of PDGFR, as well as VEGFR-1, -2, and -3, kit, and FGFR-1. It has been shown to be effective in preclinical models of CNV.210 It is administered topically and has been tested in a phase I trial (NCT01154062). A phase II study (NCT00612456) demonstrated a mean 4.3-letter increase in visual acuity after treatment with topical pazopanib (5 mg/ml) three times daily.211 Patients with the CFH TT genotype (i.e., the alleles least likely to be associated with AMD) exhibited the best response, both from the standpoint of visual acuity as well as reduction in retinal thickness. PTK787 (Vatalanib, Novartis) is an oral protein kinase inhibitor that targets all known VEGFRs, including VEGFR-1, -2, and -3, PDGFR, and kit and has been tested previously as a treatment for CNV in phase I/II clinical studies (NCT00138632).
Additionally, a tetravalent, bispecific antibody against VEGF-A and PDGFR-B is in development and has shown activity both in vitro as well as in a mouse model of tumorigenesis.212 This may have therapeutic potential for the treatment of AMD as well.
TrpRS
Another class of compounds that appear to play an important role in intracellular signaling following VEGF stimulation are the human aminoacyl-t-RNA synthetases. These molecules, which catalyze the first step of protein synthesis by aminoacylation of transfer RNA appear to have novel cytokine functions in addition to their role in protein synthesis. The catalytic core domain of tryptophanyl-tRNA synthetase (TrpRS) is a close homolog of the catalytic domain of tyrosine RS (TyrRS).103 In normal human cells the TrpRS exists as two forms. The major form is a full-length protein, which is stimulated by interferon gamma and has no cytokine activity. The other is truncated TrpRS (miniTrpRS) in which most of the extra amino terminal domain is deleted through alternate splicing of the mRNA. In addition to its production by alternative splicing, shorter fragment lengths of TrpRS can also be created by PMN Elastase cleavage of full human length TrpRS into two forms of mini TrpRS, both of which show cytokine-related activity. When cells are induced with VEGF165 and then treated with TrpRS, migration is inhibited by the miniTrp fraction, but not the full-length fraction. Following digestion with leukocyte elastase, the smaller of the two mini-TrpRS (T2-TrpRS) in which the entire amino terminal domain has been deleted, appears to be the most potent antiangiogenesis agent with dose-dependent angiostatic activity. The role of TrpRS is complex and incompletely understood. The mechanism of action of this molecule (which is bound to endothelial cells within the retina) appears to be not as an intrinsic inhibitor, but as a downregulator of VEGF-induced pathways, suggesting it represents a portion of the downstream signal cascade following VEGF stimulation, along with NOS, and expression of surface integrins.213 Additionally, in a mouse model of oxygen-induced retinopathy, T2-TrpRS induced nonpathologic revascularization of areas of retina with oxygen-induced vascular obliteration even when expressed at levels insufficient to cause regression of neovascular tufts.214 The study authors hypothesized that T2-TrpRS may act primarily to enhance revascularization of the retina, hence reducing the hypoxic stimulus leading to pathologic neovascularization, although they recognized that this was an unlikely explanation given the apparent potent angiostatic activity of the molecule. This dual nature of T2-TrpRS may be useful especially in eyes prone to hypoxia or vascular insufficiency, including in patients with advanced age. Study of the TrpRS class of molecules is still in preclinical stages.
Protein kinase C inhibitors
Protein kinase C (PKC) isoforms, particularly the beta isoform, are thought to play an important regulatory role in intracellular signaling following VEGF stimulation of endothelial and other cells through phosphorylation mechanisms.215 Most of the work detailing the interaction between VEGF effects on vascular permeability and PKC-β has been performed in experimental diabetic retinopathy and in preliminary human trials, and ongoing clinical studies of the PKC-β inhibitor ruboxistaurin in diabetes have shown promising results. Experience using PKC in experimental CNV is limited; however, one protein kinase C inhibitor, PKC412, has been tested in a porcine model of CNV. PKC412, which blocks several isoforms of PKC, was administered to pigs via a periocular injection within microspheres containing either 25% or 50% drug. Ten to 20 days following laser rupture and injection, experimentally produced CNV at Bruch’s membrane laser rupture sites were smaller in eyes receiving drug than those receiving control microspheres without drugs. Additionally, 20 days following periocular injection, high levels of the inhibitor were measured in the choroid, vitreous, and retina for eyes with 50% loading and to a lesser extent with 25% loading.216
Complement inhibitors*
Complement inhibitors are thought to be a promising class of agents for the treatment of both atrophic and exudative AMD. POT-4 (AL-78898A, Alcon/Potentia Pharmaceuticals), a cyclic peptide derivative of Compstatin 13 amino acids in length, is a C3 inhibitor administered by intravitreal injection. Gel-like deposits form in the vitreous when POT-4 is injected at high concentrations (>0.45 mg dose), which may last as long as 6 months, thus providing a sustained-release delivery system. A phase I study of POT-4 in AMD eyes with CNV was completed successfully without any safety concerns at doses up to 1.05 mg (NCT00473928), although the systemic effects of intravitreal administration are unknown. A phase II study is currently underway (NCT01157065). C3 inhibition should block complement activation arising from many of the currently described complement pathway mutations, as the classical, lectin, fibrinolytic, and alternative pathways all generate the bioactive fragments C3a and C5a and the membrane attack complex (C5b,6,7,8,9) via C3 cleavage (see Fig. 67.3). This targets a relatively large population of AMD patients; however, the broad degree of complement inhibition may theoretically increase risk of intravitreal injection-associated endophthalmitis. In a murine model, it seems that C3 deficiency does not increase the risk of Staphylococcus aureus endophthalmitis.217 In contrast, in a guinea-pig model, complement depletion with cobra venom factor does seem to increase the risk of S. aureus, S. epidermidis, and Pseudomonas aeruginosa endophthalmitis.209,218,219 Other treatments related to complement factors are being examined in the context of nonexudative AMD and have been discussed previously.
Naturally occurring inhibitory polypeptides and inducible cleavage products
While the protein miniTrpRS might also be considered in this category, a broader category is reserved for those agents that are thought to be naturally secreted into the extracellular space rather than acting as intracellular signals and include normal inducible homeostatic regulators. An example is PEDF, which is thought to modulate neuronal differentiation in the outer retina following apical secretion by the pigment epithelium in addition to its downregulation of VEGF effects on endothelial cells. In addition to these compounds, there are pathologically expressed circulating molecules that are formed as a result of cleavage by specific proteases from larger pre-existing polypeptides that have intrinsic antiangiogenic potency, including angiostatin and endostatin.220
Pigment epithelial-derived factor
Pigment epithelial-derived factor (PEDF) was first purified in the conditioned media of human retinal pigment epithelial cells that induced neuronal differentiation as well as inhibiting microglial growth. The protein shares sequence and structural homology with the serum proteinase inhibitor family (Serpen). It acts in counterpoint to VEGF, inhibiting signaling pathways of the VEGF receptors.101,102 One of its most striking abilities is the capacity to inhibit endothelial cell migration in a dose-dependent manner with a greater degree of potency than either angiostatin, thrombospondin-1 or endostatin. It has been possible to use adenovirus vector-mediated gene transfer of cDNA constructs to iris pigment epithelium (IPE) resulting in autologous IPE cells gaining the capacity to support the choriocapillaris and outer retina in vivo.221 Iris pigment epithelial cells overexpressing PEDF have been found to be capable of rescuing photoreceptors in RCS rats, reducing laser-induced CNV, and partially reversing the effects of ischemic retinopathy in various animal models.87 An open-label phase I study of AdPEDF.11 in patients with CNV was designed to test both the safety and feasibility of adenoviral gene transfer for retinal diseases.222 In the 28 patients administered the gene vector, there were no serious adverse events, and only transient intraocular pressure rise and intraocular inflammation without evidence of adenoviral infection. Additionally, although the study was not designed to determine efficacy, there appeared to be a trend toward visual stability in patients receiving a 108 particle units (PU) or higher dose of the vector, as well as stability in lesion size in patients receiving ≥108 PU, compared with increase in size in those receiving 106–107.5 PU. The study authors concluded that gene transformation using an adenoviral vector was both safe and viable for future study.
Pathologically expressed circulating molecules
Angiostatin and endostatin
Angiostatin is an internal proteolytic fragment of a known protein, plasminogen, expressed in association with tumor growth in the serum such that it inhibits primary metastatic tumor growth by blocking tumor angiogenesis. Angiostatin does not appear to have a specific gene locus resulting in transcription, but rather represents a cryptic fragment produced in response to pathologic rather than physiologic conditions, such as primary tumor growth. The molecule has a molecular weight of 38 kDa and inhibits endothelial cell proliferation in vitro and angiogenesis in vivo.104 It appears that tumor cells do not themselves express angiostatin or other fragments of plasminogen. Rather gelatinase A, also known as matrix metalloproteinase-2, which is produced by rapidly growing tumor cells such as experimental Lewis lung carcinoma, performs the proteolysis of plasminogen.90 When administered systemically, angiostatin can cause regression of a wide variety of malignant tumors through its inhibitory effects on angiogenesis. Adenoassociated viral (AAV) vectors causing overexpression of angiostatin in a murine model of CNV were effective in causing regression. Expression of the angiostatin gene in chorioretinal tissue for up to 150 days was possible as was reduction in the average size of CNV lesions in treated eyes as compared with control eyes. There were no significant adverse events reported in the animal model using target gene therapy.86,223
Another compound from this class, endostatin, is thought to represent a proteolytic fragment of 20 kD created by digestion of the C terminal fragment of collagen XVIII.89 Endostatin inhibits both endothelial cell proliferation in vitro and angiogenesis and tumor growth in vivo and is capable of being delivered by a variety of methods including sustained delivery or adenoviral vector.88 Zinc binding of endostatin is thought to be essential for its antiangiogenic activity and metal chelating agents can induce internal degradation of endostatin with associated loss of activity.224 Full-length human type 18 collagen cDNAs that encode 1516 or 336 residue alpha chains exhibit homology with endostatin. Incorporated with adenoviral vectors and administered intravenously, endostatin levels have been demonstrated to rise, with concomitant reduction in CNV size in experimental models. The zinc requirement for endostatin may be one component of the apparent beneficial effect of zinc in the AREDS trial.88 Recently, there has been enthusiasm for lentiviral vectors for the delivery of angiostatin or endostatin genes, and a vector has been developed for the delivery of both simultaneously.224,225 Animal models of CNV have demonstrated significant reduction in area and hyperpermeability, without any significant adverse events. This has led to a dose-escalating phase I clinical trial testing the safety of subretinal administration of Retinostat, a vector that delivers both the endostatin and angiostatin genes (NCT01301443). This study is underway at the time of writing.
Extracellular matrix modulators
A variety of targets exist outside the cell in the extracellular matrix (ECM) for therapeutic intervention. Some, such as the alpha ν beta 3 and alpha ν beta 5 integrins, are expressed on the cell surface in response to VEGF stimulation and participate in the adhesion of regulatory molecules as well as the interaction with metalloproteinases responsible for degradation of the ECM, facilitating migration and sprouting of new vessels.226 Others, such as the naturally occurring tissue inhibitors of metalloproteinase (TIMP) or other synthetic metalloproteinase inhibitors may precede the VEGF activation step and represent both early and late targets for intervention.
Integrin antagonists
Examination of tissues from eyes with pathologic neovascularization indicate that at least two cytokine-dependent pathways of angiogenesis exist and both may be important in pathologic neovascularization. In in vivo models of angiogenesis induced by basic fibroblast growth factor (bFGF) or tumor necrosis factor-alpha (TNF-α), the cell surface-expressed integrin alpha ν beta 3 predominates, whereas in models initiated by VEGF transforming growth factor-alpha (TGF-α), or phorbol ester the pathway is characterized by dependence on and expression of the related cytokine alpha ν beta 5.69,70 These findings are further confirmed by evidence that inhibition of protein kinase C (using the specific inhibitor calphostin C) blocked angiogenesis induced by VEGF and TGF-α, but had only a small effect on bFGF or TNF-α-mediated angiogenesis. This suggests the specificity of the two pathways: (1) that dependent on alpha ν beta 3 and relatively independent of PKC; and (2) the second potentiated by alpha ν beta 5, and dependent upon both VEGF stimulation and PKC activation.69,70
A cyclic peptide antagonist (cyclo RGDv) of alpha ν beta 3 and alpha ν beta 5 was capable of dramatically reducing retinal vessel blood growth compared with a control peptide in a neonatal mouse model of retinopathy of prematurity. Other molecules in preclinical studies, including XJ735 (a selective alpha ν beta 3 antagonist), and XK002, a combined antagonist of alpha ν beta 3 and alpha ν beta 5, also inhibit retinal neovascularization in a murine model.227 Cyclic RGDfV peptide was found to represent an antagonist of vitronectin receptor type integrins by other groups.2
Another approach rather than specific receptor binding with polypeptide molecules is the use of selective monoclonal antibodies directed against cell surface integrins. Specific mouse monoclonal antibodies to integrins alpha ν beta 3 (LM609) and alpha ν beta 5 (P1F6) have been prepared, and in addition to demonstrating the presence of expressed integrins on vascular cell surfaces, have also been used therapeutically to inhibit neovascularization. Antibodies directed against alpha ν beta 3 appeared to selectively block bFGF-induced angiogenesis whereas antibodies directed against antibeta 1 and alpha ν beta 5 had dramatically less effect and appeared to spare pre-existing preformed vessels.69,70 Other approaches have included the conjugation of mitomycin C dextran to a monoclonal anti-integrin alpha ν beta 3 antibody, which was more effective than the use of a mouse monoclonal alone in a rat model of laser-induced CNV.228 Yet another strategy involves the targeting of an proapoptotic peptide domain to a homing target that localizes to the alpha ν beta 3/beta 5 integrins expressed preferentially in neovascular endothelial cells, thus inducing apoptosis in the newly forming vessels. This strategy takes advantage of the expression pattern of the alpha ν beta 3/beta 5 integrins without specifically targeting their function or downstream effects, and has proven effective in a mouse model of retinopathy of prematurity.229
The integrin alpha 5 beta 1 has also been identified as a significant player in choroidal neovascularization.230–232 It has been found to be significantly expressed in laser-induced CNV in animal models, and an inhibitor of alpha 5 beta 1 have been show to reverse corneal neovascularization as well as hypoxia-induced retinal neovascularization.233,234 Currently, antagonists to alpha 5 beta 1 have shown mixed results in the inhibition or reversal of CNV in animal models. There is currently a phase I trial underway evaluating Volociximab, an anti-alpha 5 beta 1 monoclonal antibody already being tested for cancer therapy, for exudative AMD (NCT00782093).235
MMP inhibitors
The naturally occurring class of tissue inhibitors of metalloproteinase (TIMP), particularly TIMP-3, are thought to be critically important in the homeostasis of the extracellular matrix and thereby to factors contributing to the development of CNV. As discussed earlier, large quantities of TIMP-3 have been found with increasing age in Bruch’s membrane where it is thought to be a normal structural component and particularly in areas where there is pathologic thickening of Bruch’s membrane.30,36,98 The carboxy terminus of TIMP-3, unlike the soluble TIMP-1, TIMP-2, and TIMP-4, is exclusively membrane-bound, while the amino terminus appears to inhibit MMPs 2 and 9, which are known to be expressed in choroidal new vessels membranes removed from patients with subfoveal CNV.99 TIMP-3 naturally inhibits VEGF and is capable of reducing angiogenesis in vitro and in vivo including CAM assays and also when overexpressed by selective gene therapy in rat eyes undergoing laser photocoagulation for the induction of CNV.99,100 Pharmacologic therapies directed at replacement or modification of TIMP-3 and potentially other related compounds may have utility as a pharmacologic strategy not only for regulation of the extracellular matrix in response to VEGF stimulation late, but also potentially earlier in regulating the homeostatic mechanisms that lead to Bruch’s membrane thickening and the other inducible factors for CNV preceding VEGF activation (Fig. 67.17).236
Fig. 67.17 Computerized rendering of TIMP-1 interacting as its active NH site with MMP-3 catalytic domain.
(Reproduced with permission from Gomis-Ruth FX, Maskos K, Betz M, et al. Mechanism of inhibition of the human matrix metalloproteinase stromelysin-1 by TIMP-1. Nature 1997;389:77–81.)
Matrix metalloproteinase inhibitors (MMPI) have been tested for their ability to inhibit pathologic ocular neovascularization. AG3340, a synthetic MMP2,9 inhibitor was shown to significantly inhibit neovascularization in a mouse model of retinopathy of prematurity leading to a human phase II/III trial.237 However, the study was unable to confirm any inhibitory effect of the small molecular weight binding inhibitor of MMP-2 MMP-9 in any dose tested either with regards to lesion size, or final visual acuity and efforts were halted. Other MMP inhibitors continue to be tested in preclinical studies.
Other molecules
A variety of molecules with varying mechanisms of action are undergoing preliminary evaluation. Combretastatin A-4a naturally occurring agent from tree bark that binds tubulin and causes necrosis and shrinkage of tumors by inhibition of the blood supply is capable of suppressing experimental CNV in response to photocoagulation.25,238 Preliminary trials of a phosphate prodrug of combretastatin A4 (fosbretabulin, OXiGENE) are underway in humans to test its efficacy in early stage neovascularization AMD.
Squalamine
Squalamine is an antiangiogenic amino sterol derived from shark liver which inhibits iris neovascularization when administered by intravenous injection, although it is ineffective when administered by intravitreal injection.239,240 Squalamine also induces regression of experimental retinopathy of prematurity in a mouse model241 and has been tested in a phase I study of CNV in patients with no safety concerns expressed leading to several phase II trials. Initial visual results were promising, with improvement or stability in those patients receiving higher doses of the drug. Interestingly, the fellow non-study eyes with advanced macular degeneration also showed significant improvement after drug administration. A phase III study was initiated; however, it was terminated.
The role of steroids and other immunomodulators
Steroids
Steroids have long known to be associated with neovascularization reduction by mechanisms that were not clearly understood. Folkman and colleagues were the first to suggest that the antiangiogenic effects of steroids could be separated into two categories: (1) those related to anti-inflammatory effects paralleling convention glucocorticoid and mineralocorticoid activity; and (2) a separate structural configuration of the pregnan nucleus conferring distinct antiangiogenic capability.83,84 Although preclinical studies suggested benefit in terms of VEGF-stimulated vascular permeability and angiogenesis, and while steroids have long been used with success for diabetic macular edema, clinical studies employing conventional steroids alone for AMD have shown relatively unimpressive effects on the natural history of the disease. In the era of anti-VEGF therapy, steroid therapy is largely an adjunct; however, steroid monotherapy is still being examined in continuing clinical trials. At this time, a sustained release fluocinolone implant (Iluvien, Alimera Pharmaceuticals) is in phase II clinical trials for both choroidal neovascularization (NCT00605423) as well as geographic atrophy in AMD (NCT00695318). Additionally, there has been enthusiasm for potential beneficial effects associated with the use of intravitreal triamcinolone combined with conventional photodynamic therapy (PDT) using verteporfin for subfoveal CNV242–252 (discussed below in a separate section on combination therapy).
Rapamycin
Rapamycin (sirolimus) is a macrocyclic, naturally occurring lactone that was initially isolated from the species Streptomyces hygroscopicus and given its name from Rapa Nui (Easter Island) during the course of a search for novel antifungal agents. Rapamycin is structurally related to the immunosuppressive agent FK506 (tacrolimus), which has been used extensively for the prevention of organ transplant rejection. Rapamycin requires direct interaction with at least two intracellular proteins to exert its action of arresting cell phase progression. FK binding protein (FKBP12) forms a rapamycin FK complex, which in turn binds to and inhibits the activity of the mammalian target of rapamycin (mTOR) through which it exerts its intracellular affects. Other FKBPs have been identified and interact with heat shock proteins. When bound to mTOR, phosphorylation does not occur, and the cells’ mitotic ability is blocked in addition to other effects involving protein kinase C delta, and IL-2 dependent functions that control the progression of T cells into the S phase.253
Rapamycin inhibits primary metastatic tumor growth by inhibition with angiogenesis through direct inhibition of vascular endothelial growth factors as well as endothelial cell responses to VEGF.254 Additionally, rapamycin, through its intrinsic immunomodulatory effects, may reduce macrophage chemotaxis and activation with concomitant reduction in release of VEGF and other angiogenic cytokines.
In experimental models, rapamycin has been shown to inhibit the development of ocular neovascularization in rats in response to subretinal matrigel placement orally at a dose of 2.5 mg per kilograms per day, or as an intraocular injection. Inhibition was also seen in a murine model of retinopathy of prematurity and laser-induced neovascularization when administered by an intraperitoneal route in doses of 2–4 mg per kilogram per day.255
A recent phase I/II pilot study has been conducted, comparing the effectiveness of adjunctive systemic administration of one of three immunosuppressive agents (including rapamycin) when combined with anti-VEGF treatment (either ranibizumab or bevacizumab), against anti-VEGF therapy alone.256 The prospective randomized, unmasked trial assigned patients 1 : 1 : 1 : 1 to receive either intravenous daclizumab, intravenous infliximab, oral rapamycin tablets, or observation for a study duration of 6 months. All patients were administered intravitreous bevacizumab (1.25 mg/0.05 mL or 2.5 mg/1.0 mL) or ranibizumab (0.5 mg) at intervals determined by the treating physician based on the presence of intraretinal or subretinal fluid on OCT imaging. The primary study outcome was the number of injections required in each group over the study period, which was also compared to the number of injections required prior to joining the study. There was a decrease in the number of injections in patients receiving daclizumab as well as rapamycin, although the study was small, and differences were not significant. However, the results do suggest that there may be a role for immunomodulators in the treatment of AMD. There is currently a phase I/II study underway examining subconjunctival administration of sirolimus for the treatment of geographic atrophy (NCT00766649).
Photodynamic Therapy
Pharmacology of photodynamic sensitizers
Photodynamic therapy actually represents the combination of a pharmacologic therapy and a laser-based therapy in a two-step process capable of successfully treating subfoveal CNV. Photosensitizers have biophysical properties well suited to the thrombotic closure of abnormal neovascularization while preserving normal physiologic vessels. Most agents possess strong absorption properties in the far-red spectral region (660–780 nm), where light has the greatest penetration through blood and tissue. Photosensitizers also selectively bind to abnormal neovascularization through its expression of increased numbers of lipoprotein receptors contrasted with normal mature vessels, thus achieving the desirable feature of selectivity. Most photosensitizing molecules, including verteporfin, which has been approved for human use, as well as a number of agents under clinical evaluation, are structurally related to porphyrins (Fig. 67.18). Porphyrins are fused tetrapyrrolic macromolecules found in nature as pigments such as protoporphyrin IX, the nonprotein portion of hemoglobin.
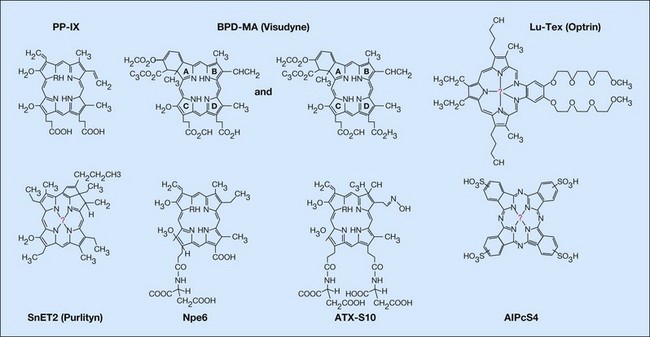
Other molecules undergoing evaluation although not yet approved for use in humans include Tin-ethyl-etiopurpurin (Purlytin), monotexafin lutetium (optrin, Lu-tex), Npe6, and ATX-S10. Their chemical formulas are listed in Fig. 67.18. Purlytin was subjected to a large multicenter phase II/III clinical trial, which demonstrated a trend towards efficacy in selected subgroups, although it failed to meet the preapproved regulatory efficacy endpoint.
Lu-tex underwent a phase I dose escalation trial, in humans, which demonstrated its ability to achieve photodynamic closure, although associated with a greater degree of surrounding choroidal damage than verteporfin and also associated with peripheral paresthesias and occasional periocular pain in selected patients and the trial was discontinued. A phase I dose-ranging study of NPE6 was terminated. ATX-S10 has been the subject of continuing preclinical studies and at the time of this writing no human trials are under way.257
Verteporfin (Visudyne)
Verteporfin photodynamic therapy (PDT) was the first photosensitizer approved for the treatment of exudative AMD. The technique involves infusion of 6 mg/m2 verteporfin over a 10 minute period followed by laser irradiation using a 689 nm diode laser (light dose: 50 J/cm2; power density: 600 mW/cm2; duration: 83 seconds) 15 minutes after the start of the infusion. Verteporfin demonstrated safety and efficacy in selectively localizing to choroidal neovascular membranes in preclinical trials in nonhuman primates. The mechanism of action is postulated to occur by activation of verteporfin to a triplet state following laser irradiation, resulting in the creation of singlet and reactive oxygen species that induces endothelial cellular damage and, ultimately, occlusion of the neovascular membranes through activation of the clotting cascade.258–262 Unfortunately, the generation of free radicals necessary for the photothrombotic effect, may also serve as a proangiogenic stimulus, possibly accounting for the apparent benefit associated with concomitant administration of steroids.
Verteporfin PDT was demonstrated to be safe and effective for treatment of CNV due to AMD in phase I and II testing.263,264 The Treatment of Age-related macular degeneration with Photodynamic Therapy (TAP) Study demonstrated that patients with CNV due to AMD demonstrated a beneficial effect following verteporfin PDT. The greatest benefit was seen among the subset of patients with predominantly classic subfoveal CNV due to AMD, with 67% of verteporfin-treated eyes versus 39% of placebo-treated eyes prevented from progressing to moderate visual loss – loss of 15 or more letters on the Early Treatment Diabetic Retinopathy Study (ETDRS) scale or approximately 3 lines of vision – P < 0.001. Lesions that were less than 50% classic did not demonstrate a treatment benefit. These results were valid through 2- and 3-year follow-up.265,266 Further discussion of the role of verteporfin in AMD is covered in Chapter 66 (Neovascular (exudative or “wet”) age-related macular degeneration). Presently, verteporfin PDT is approved for the treatment of AMD lesions that are predominantly classic subfoveal CNV, or for occult or minimally classic subfoveal CNV less than 4 disc diameters in size.202,265,267
Combination therapies*
The principles of combination therapy have been well established in the treatment of infectious diseases such as HIV,268 or in cancer therapy. The combination of VEGF blockade (e.g., with bevacizumab) with chemotherapy or radiation therapy, for example, results in a greater antitumor effect than with either treatment alone.269,270 Combining AMD treatments with differing mechanisms of action may have synergistic effects that might result in: (1) better visual outcome; (2) reduced frequency of treatment; (3) greater patient convenience (e.g., subconjunctival versus intravitreal injection); (4) lower risk of adverse events such as endophthalmitis; and/or (5) less likelihood of “escape” (a phenomenon in which cells (e.g., infectious agents or tumor cells) develop alternative pathways that allow them to overcome the inhibition of a pathway essential for their survival or growth). A number of variations have been examined, including retrospective and prospective trials of verteporfin photodynamic therapy with anti-VEGF therapy and/or intraocular or periocular triamcinolone or dexamethasone.242–245,247–251,271–277 The efficacy and safety of verteporfin PDT has been demonstrated in multicenter, double-masked, randomized placebo-controlled studies.202,278 However, PDT may affect the choriocapillary bed surrounding the CNV,279 resulting in upregulation of VEGF that might further potentiate CNV growth.280 Thus, adding an anti-VEGF agent to PDT has theoretical merit. Combining anti-VEGF therapy and photodynamic therapy (with or without steroid) is thought to reduce the number of treatments needed to stabilize the CNV, although this benefit has not been observed in all studies.281
Large, multicenter, randomized studies (DENALI (NCT00433017), MONT BLANC (NCT00433017), RADICAL (NCT00492284) have been completed, and 12-month results published in 2012 indicate that the combination of anti-VEGF therapy and PDT has shown no great advantage over anti-VEGF therapy alone. The phase IIIb DENALI study involved the combination of verteporfin PDT (Visudyne, Novartis Pharma AG, QLT, Inc.) with ranibizumab (Lucentis, Genentech/Roche). Three ranibizumab loading doses were followed by additional injections on a monthly as-needed basis in patients (n = 321) with subfoveal CNV of all lesion types secondary to AMD. Verteporfin PDT was administered on day 1 and at 3-month intervals as needed. At month 12, patients in the standard fluence combination group gained on average 5.3 letters from baseline, and patients in the reduced fluence combination group gained on average 4.4 letters. Patients in the ranibizumab monthly monotherapy group gained on average 8.1 letters at month 12. DENALI (n = 255) did not demonstrate noninferiority in terms of visual acuity gain for verteporfin PDT ranibizumab combination therapy compared with ranibizumab monthly monotherapy. On average, patients in the combination groups required 2.2 (standard fluence) or 2.8 (reduced fluence) additional ranibizumab injections after the mandatory three loading doses as compared to an average of 7.6 additional injections in the ranibizumab monthly monotherapy cohort.282
Similarly, in the MONT BLANC study, patients were randomized 1 : 1 to as-needed (PRN) combination (standard-fluence verteporfin 6 mg/m2 PDT and ranibizumab 0.5 mg) or PRN ranibizumab monotherapy (sham infusion PDT with ranibizumab 0.5 mg) following a loading dose of three injections. At month 12, visual improvement was +2.5 letters in the combination therapy group and +4.4 letters in the ranibizumab monotherapy group (P = 0.0048). The noninferiority limit of 7 letters was met; however, other endpoints, including the proportion of patients with a treatment-free interval of >/=3 months (96 vs 92%, P = 0.036) and the number of ranibizumab retreatments (1.9 vs 2.2, P = 0.1373 ) were similar, suggesting that adding verteporfin PDT does not reduce the number of ranibizumab injections required.283
The RADICAL study, a phase II trial, included 162 patients randomized to one of four treatment arms: double therapy of PDT with reduced-fluence verteporfin PDT (25 J/cm2: 300 mW/cm2 for 83 seconds) followed by ranibizumab (n = 43), reduced-fluence verteporfin PDT (25 J/cm2: 300 mW/cm2 for 83 seconds) followed by ranibizumab-dexamethasone triple therapy (n = 39), very low-fluence verteporfin PDT (15 J/cm2: 180 mW/cm2 for 83 seconds) followed by ranibizumab dexamethasone triple therapy (n = 39), or ranibizumab monotherapy (n = 41). At 24 months follow-up, mean visual acuity change from baseline was not statistically different among the treatment groups. Through 24 months, patients in the triple therapy half-fluence group had a mean of 4.2 retreatment visits compared with 8.9 for patients who received ranibizumab monotherapy (P < 0.001). At month 24, mean visual acuity in the triple therapy half-fluence group improved 1.8 letters fewer (95% CI 11.1 letters fewer to 7.6 letters better) compared with the ranibizumab monotherapy group (P = 0.71).284
Anti-VEGF therapy is also being combined with macular radiation therapy. Two different radiation approaches are currently in clinical trials: the internal approach, which requires a pars plana vitrectomy and the introduction of a radioactive (beta-radiation) probe (Epi-Rad90™ Ophthalmic System, NeoVista, Inc., NCT00454389),285,286 and the office-based external approach (X-ray radiation),287 which is delivered through the inferior sclera (IRay, Oraya Therapeutics Inc., NCT01016873). Detailed discussion of radiation therapy for AMD is beyond the scope of this chapter, but the rationale for combining anti-VEGF therapy and irradiation is that the anti-inflammatory, antineovascular, and antifibrotic effects of radiation therapy might logically be combined with the dehydrating effects of anti-VEGF therapy. Combination intraocular epiretinal irradiation and ranibizumab appears to result in a reduced need for intravitreal injection (with comparable visual outcome) compared with intravitreal ranibizumab therapy alone.285,286 Phase III (NCT00454389, CABERNET) and phase IV (NCT01006538, MERLOT) studies are underway.
1 Bird AC, Bressler NM, Bressler SB, et al. An international classification and grading system for age-related maculopathy and age-related macular degeneration. The International ARM Epidemiological Study Group. Surv Ophthalmol. 1995;39:367–374.
2 Ambati J, Ambati BK, Yoo SH, et al. Age-related macular degeneration: etiology, pathogenesis, and therapeutic strategies. Surv Ophthalmol. 2003;48:257–293.
3 Blumenkranz MS, Russell SR, Robey MG, et al. Risk factors in age-related maculopathy complicated by choroidal neovascularization. Ophthalmology. 1986;93:552–558.
4 Ferris FL, 3rd. Senile macular degeneration: review of epidemiologic features. Am J Epidemiol. 1983;118:132–151.
5 Ferris FL, 3rd., Fine SL, Hyman L. Age-related macular degeneration and blindness due to neovascular maculopathy. Arch Ophthalmol. 1984;102:1640–1642.
6 Bressler NM, Hawkins BS, Sternberg P, Jr., et al. Are the submacular surgery trials still relevant in an era of photodynamic therapy? Ophthalmology. 2001;108:435–436.
7 Argon laser photocoagulation for senile macular degeneration. Results of a randomized clinical trial. Arch Ophthalmol. 1982;100:912–918.
8 Argon laser photocoagulation for neovascular maculopathy. Three-year results from randomized clinical trials. Macular Photocoagulation Study Group. Arch Ophthalmol. 1986;104:694–701.
9 Laser photocoagulation of subfoveal neovascular lesions of age-related macular degeneration. Updated findings from two clinical trials. Macular Photocoagulation Study Group. Arch Ophthalmol. 1993;111:1200–1209.
10 Slakter JS, Yannuzzi LA, Sorenson JA, et al. A pilot study of indocyanine green videoangiography-guided laser photocoagulation of occult choroidal neovascularization in age-related macular degeneration. Arch Ophthalmol. 1994;112:465–472.
11 Leibowitz HM, Krueger DE, Maunder LR, et al. The Framingham Eye Study monograph: An ophthalmological and epidemiological study of cataract, glaucoma, diabetic retinopathy, macular degeneration, and visual acuity in a general population of 2631 adults, 1973–1975. Surv Ophthalmol. 1980;24:335–610.
12 Patel N, Adewoyin T, Chong NV. Age-related macular degeneration: a perspective on genetic studies. Eye (Lond). 2008;22:768–776.
13 Francis PJ, Hamon SC, Ott J, et al. Polymorphisms in C2, CFB and C3 are associated with progression to advanced age related macular degeneration associated with visual loss. J Med Genet. 2009;46:300–307.
14 Seddon JM, Gensler G, Rosner B. C-reactive protein and CFH, ARMS2/HTRA1 gene variants are independently associated with risk of macular degeneration. Ophthalmology. 2010;117:1560–1566.
15 Conley YP, Jakobsdottir J, Mah T, et al. CFH, ELOVL4, PLEKHA1 and LOC387715 genes and susceptibility to age-related maculopathy: AREDS and CHS cohorts and meta-analyses. Hum Mol Genet. 2006;15:3206–3218.
16 Brantley MA, Jr., Fang AM, King JM, et al. Association of complement factor H and LOC387715 genotypes with response of exudative age-related macular degeneration to intravitreal bevacizumab. Ophthalmology. 2007;114:2168–2173.
17 Brantley MA, Jr., Edelstein SL, King JM, et al. Association of complement factor H and LOC387715 genotypes with response of exudative age-related macular degeneration to photodynamic therapy. Eye (Lond). 2009;23:626–631.
18 Tsuchihashi T, Mori K, Horie-Inoue K, et al. Complement factor H and high-temperature requirement A-1 genotypes and treatment response of age-related macular degeneration. Ophthalmology. 2011;118:93–100.
19 Feng X, Xiao J, Longville B, et al. Complement factor H Y402H and C-reactive protein polymorphism and photodynamic therapy response in age-related macular degeneration. Ophthalmology. 2009;116:1908–1912. e1
20 Klein ML, Francis PJ, Rosner B, et al. CFH and LOC387715/ARMS2 genotypes and treatment with antioxidants and zinc for age-related macular degeneration. Ophthalmology. 2008;115:1019–1025.
21 Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389.
22 Edwards AO, Ritter R, 3rd., Abel KJ, et al. Complement factor H. polymorphism and age-related macular degeneration. Science. 2005;308:421–424.
23 Chen Y, Zeng J, Zhao C, et al. Assessing susceptibility to age-related macular degeneration with genetic markers and environmental factors. Arch Ophthalmol. 2011;129:344–351.
24 Nozaki M, Raisler BJ, Sakurai E, et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci U S A. 2006;103:2328–2333.
25 Hageman GS, Luthert PJ, Victor Chong NH, et al. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20:705–732.
26 Patel M, Chan CC. Immunopathological aspects of age-related macular degeneration. Semin Immunopathol. 2008;30:97–110.
27 Penfold PL, Madigan MC, Gillies MC, et al. Immunological and aetiological aspects of macular degeneration. Prog Retin Eye Res. 2001;20:385–414.
28 Albig AR, Schiemann WP. Fibulin-5 antagonizes vascular endothelial growth factor (VEGF) signaling and angiogenic sprouting by endothelial cells. DNA Cell Biol. 2004;23:367–379.
29 Klenotic PA, Munier FL, Marmorstein LY, et al. Tissue inhibitor of metalloproteinases-3 (TIMP-3) is a binding partner of epithelial growth factor-containing fibulin-like extracellular matrix protein 1 (EFEMP1). Implications for macular degenerations. J Biol Chem. 2004;279:30469–30473.
30 Fariss RN, Apte SS, Olsen BR, et al. Tissue inhibitor of metalloproteinases-3 is a component of Bruch’s membrane of the eye. Am J Pathol. 1997;150:323–328.
31 Felbor U, Stohr H, Amann T, et al. A second independent Tyr168Cys mutation in the tissue inhibitor of metalloproteinases-3 (TIMP3) in Sorsby’s fundus dystrophy. J Med Genet. 1996;33:233–236.
32 Lin RJ, Blumenkranz MS, Binkley J, et al. A novel His158Arg mutation in TIMP3 causes a late-onset form of Sorsby fundus dystrophy. Am J Ophthalmol. 2006;142:839–848.
33 De La Paz MA, Pericak-Vance MA, Lennon F, et al. Exclusion of TIMP3 as a candidate locus in age-related macular degeneration. Invest Ophthalmol Vis Sci. 1997;38:1060–1065.
34 Marmorstein LY, Munier FL, Arsenijevic Y, et al. Aberrant accumulation of EFEMP1 underlies drusen formation in Malattia Leventinese and age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99:13067–13072.
35 Chong NH, Alexander RA, Gin T, et al. TIMP-3, collagen, and elastin immunohistochemistry and histopathology of Sorsby’s fundus dystrophy. Invest Ophthalmol Vis Sci. 2000;41:898–902.
36 Kamei M, Hollyfield JG. TIMP-3 in Bruch’s membrane: changes during aging and in age-related macular degeneration. Invest Ophthalmol Vis Sci. 1999;40:2367–2375.
37 Stone EM, Lotery AJ, Munier FL, et al. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat Genet. 1999;22:199–202.
38 Mata NL, Weng J, Travis GH. Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc Natl Acad Sci U S A. 2000;97:7154–7159.
39 Zhang K, Garibaldi DC, Kniazeva M, et al. A novel mutation in the ABCR gene in four patients with autosomal recessive Stargardt disease. Am J Ophthalmol. 1999;128:720–724.
40 Allikmets R. Further evidence for an association of ABCR alleles with age-related macular degeneration. The International ABCR Screening Consortium. Am J Hum Genet. 2000;67:487–491.
41 Bernstein PS, Leppert M, Singh N, et al. Genotype-phenotype analysis of ABCR variants in macular degeneration probands and siblings. Invest Ophthalmol Vis Sci. 2002;43:466–473.
42 Radu RA, Mata NL, Nusinowitz S, et al. Treatment with isotretinoin inhibits lipofuscin accumulation in a mouse model of recessive Stargardt’s macular degeneration. Proc Natl Acad Sci U S A. 2003;100:4742–4747.
43 Radu RA, Mata NL, Bagla A, et al. Light exposure stimulates formation of A2E oxiranes in a mouse model of Stargardt’s macular degeneration. Proc Natl Acad Sci U S A. 2004;101:5928–5933.
44 SanGiovanni JP, Chew EY, Clemons TE, et al. The relationship of dietary lipid intake and age-related macular degeneration in a case-control study: AREDS report no. 20. Arch Ophthalmol. 2007;125:671–679.
45 SanGiovanni JP, Chew EY, Agron E, et al. The relationship of dietary omega-3 long-chain polyunsaturated fatty acid intake with incident age-related macular degeneration: AREDS report no. 23. Arch Ophthalmol. 2008;126:1274–1279.
46 Sangiovanni JP, Agron E, Meleth AD, et al. ω-3 Long-chain polyunsaturated fatty acid intake and 12-y incidence of neovascular age-related macular degeneration and central geographic atrophy: AREDS report 30, a prospective cohort study from the Age-Related Eye Disease Study. Am J Clin Nutr. 2009;90:1601–1607.
47 Seddon JM, Ajani UA, Sperduto RD, et al. Dietary carotenoids, vitamins A, C, and E, and advanced age-related macular degeneration. Eye Disease Case-Control Study Group. JAMA. 1994;272:1413–1420.
48 Beatty S, Koh H, Phil M, et al. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2000;45:115–134.
49 Cho E, Stampfer MJ, Seddon JM, et al. Prospective study of zinc intake and the risk of age-related macular degeneration. Ann Epidemiol. 2001;11:328–336.
50 Cho E, Seddon JM, Rosner B, et al. Prospective study of intake of fruits, vegetables, vitamins, and carotenoids and risk of age-related maculopathy. Arch Ophthalmol. 2004;122:883–892.
51 Heuberger RA, Mares-Perlman JA, Klein R, et al. Relationship of dietary fat to age-related maculopathy in the Third National Health and Nutrition Examination Survey. Arch Ophthalmol. 2001;119:1833–1838.
52 Zarbin MA, Rosenfeld PJ. Pathway-based therapies for age-related macular degeneration: an integrated survey of emerging treatment alternatives. Retina. 2010;30:1350–1367.
53 Zarbin M, Rosenfeld PJ. Review of emerging treatments for age-related macular degeneration. In: Stratton RD, Hauswirth WW, Gardner TW. Oxidative stress in applied basic research and clinical practice: Studies in retinal and choroidal disorders. New York: Humana Press, 2012.
54 Tomany SC, Wang JJ, Van Leeuwen R, et al. Risk factors for incident age-related macular degeneration: pooled findings from 3 continents. Ophthalmology. 2004;111:1280–1287.
55 Thornton J, Edwards R, Mitchell P, et al. Smoking and age-related macular degeneration: a review of association. Eye (Lond). 2005;19:935–944.
56 Darzins P, Mitchell P, Heller RF. Sun exposure and age-related macular degeneration. An Australian case–control study. Ophthalmology. 1997;104:770–776.
57 Tomany SC, Cruickshanks KJ, Klein R, et al. Sunlight and the 10-year incidence of age-related maculopathy: the Beaver Dam Eye Study. Arch Ophthalmol. 2004;122:750–757.
58 A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119:1417–1436.
59 van Leeuwen R, Tomany SC, Wang JJ, et al. Is medication use associated with the incidence of early age-related maculopathy? Pooled findings from 3 continents. Ophthalmology. 2004;111:1169–1175.
60 Ishida S, Usui T, Yamashiro K, et al. VEGF164-mediated inflammation is required for pathological, but not physiological, ischemia-induced retinal neovascularization. J Exp Med. 2003;198:483–489.
61 Arroyo JG, Michaud N, Jakobiec FA. Choroidal neovascular membranes treated with photodynamic therapy. Arch Ophthalmol. 2003;121:898–903.
62 Ghazi NG, Jabbour NM, De La Cruz ZC, et al. Clinicopathologic studies of age-related macular degeneration with classic subfoveal choroidal neovascularization treated with photodynamic therapy. Retina. 2001;21:478–486.
63 Grossniklaus HE, Ling JX, Wallace TM, et al. Macrophage and retinal pigment epithelium expression of angiogenic cytokines in choroidal neovascularization. Mol Vis. 2002;8:119–126.
64 Schnurrbusch UE, Welt K, Horn LC, et al. Histological findings of surgically excised choroidal neovascular membranes after photodynamic therapy. Br J Ophthalmol. 2001;85:1086–1091.
65 Moshfeghi DM, Kaiser PK, Grossniklaus HE, et al. Clinicopathologic study after submacular removal of choroidal neovascular membranes treated with verteporfin ocular photodynamic therapy. Am J Ophthalmol. 2003;135:343–350.
66 Ciulla TA, Criswell MH, Danis RP, et al. Intravitreal triamcinolone acetonide inhibits choroidal neovascularization in a laser-treated rat model. Arch Ophthalmol. 2001;119:399–404.
67 Penfold PL, Wen L, Madigan MC, et al. Triamcinolone acetonide modulates permeability and intercellular adhesion molecule-1 (ICAM-1) expression of the ECV304 cell line: implications for macular degeneration. Clin Exp Immunol. 2000;121:458–465.
68 Sakurai E, Taguchi H, Anand A, et al. Targeted disruption of the CD18 or ICAM-1 gene inhibits choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44:2743–2749.
69 Friedlander M, Theesfeld CL, Sugita M, et al. Involvement of integrins alpha v beta 3 and alpha v beta 5 in ocular neovascular diseases. Proc Natl Acad Sci U S A. 1996;93:9764–9769.
70 Friedlander M, Brooks PC, Shaffer RW, et al. Definition of two angiogenic pathways by distinct alpha v integrins. Science. 1995;270:1500–1502.
71 Miller JW. Vascular endothelial growth factor and ocular neovascularization. Am J Pathol. 1997;151:13–23.
72 Robinson CJ, Stringer SE. The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J Cell Sci. 2001;114:853–865.
73 Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004;25:581–611.
74 Senger DR, Galli SJ, Dvorak AM, et al. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983–985.
75 Fukumura D, Gohongi T, Kadambi A, et al. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc Natl Acad Sci U S A. 2001;98:2604–2609.
76 Sennlaub F, Courtois Y, Goureau O. Inducible nitric oxide synthase mediates retinal apoptosis in ischemic proliferative retinopathy. J Neurosci. 2002;22:3987–3993.
77 Usui T, Ishida S, Yamashiro K, et al. VEGF164(165) as the pathological isoform: differential leukocyte and endothelial responses through VEGFR1 and VEGFR2. Invest Ophthalmol Vis Sci. 2004;45:368–374.
78 Mayerhofer M, Valent P, Sperr WR, et al. BCR/ABL induces expression of vascular endothelial growth factor and its transcriptional activator, hypoxia inducible factor-1alpha, through a pathway involving phosphoinositide 3-kinase and the mammalian target of rapamycin. Blood. 2002;100:3767–3775.
79 Elbashir SM, Harborth J, Lendeckel W, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498.
80 El-Hashemite N, Walker V, Zhang H, et al. Loss of Tsc1 or Tsc2 induces vascular endothelial growth factor production through mammalian target of rapamycin. Cancer Res. 2003;63:5173–5177.
81 Tolentino MJ, Brucker AJ, Fosnot J, et al. Intravitreal injection of vascular endothelial growth factor small interfering RNA inhibits growth and leakage in a nonhuman primate, laser-induced model of choroidal neovascularization. Retina. 2004;24:660.
82 Dawson DW, Volpert OV, Gillis P, et al. Pigment epithelium-derived factor: a potent inhibitor of angiogenesis. Science. 1999;285:245–248.
83 Folkman J, Ingber DE. Angiostatic steroids. Method of discovery and mechanism of action. Ann Surg. 1987;206:374–383.
84 Folkman J, Weisz PB, Joullie MM, et al. Control of angiogenesis with synthetic heparin substitutes. Science. 1989;243:1490–1493.
85 Fung WE. Interferon alpha 2a for treatment of age-related macular degeneration. Am J Ophthalmol. 1991;112:349–350.
86 Lai CC, Wu WC, Chen SL, et al. Suppression of choroidal neovascularization by adeno-associated virus vector expressing angiostatin. Invest Ophthalmol Vis Sci. 2001;42:2401–2407.
87 Mori K, Duh E, Gehlbach P, et al. Pigment epithelium-derived factor inhibits retinal and choroidal neovascularization. J Cell Physiol. 2001;188:253–263.
88 Mori K, Ando A, Gehlbach P, et al. Inhibition of choroidal neovascularization by intravenous injection of adenoviral vectors expressing secretable endostatin. Am J Pathol. 2001;159:313–320.
89 O’Reilly MS, Boehm T, Shing Y, et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–285.
90 O’Reilly MS, Wiederschain D, Stetler-Stevenson WG, et al. Regulation of angiostatin production by matrix metalloproteinase-2 in a model of concomitant resistance. J Biol Chem. 1999;274:29568–29571.
91 Hellstrom M, Kalen M, Lindahl P, et al. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126:3047–3055.
92 Tolentino MJ. Current molecular understanding and future treatment strategies for pathologic ocular neovascularization. Curr Mol Med. 2009;9:973–981.
93 Benjamin LE, Golijanin D, Itin A, et al. Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J Clin Invest. 1999;103:159–165.
94 Benjamin LE, Hemo I, Keshet E. A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Development. 1998;125:1591–1598.
95 Dell S, Peters S, Muther P, et al. The role of PDGF receptor inhibitors and PI3-kinase signaling in the pathogenesis of corneal neovascularization. Invest Ophthalmol Vis Sci. 2006;47:1928–1937.
96 Perez-Santonja JJ, Campos-Mollo E, Lledo-Riquelme M, et al. Inhibition of corneal neovascularization by topical bevacizumab (Anti-VEGF) and Sunitinib (anti-VEGF and anti-PDGF) in an animal model. Am J Ophthalmol. 2010;150:519–528. e1
97 Oshima Y, Deering T, Oshima S, et al. Angiopoietin-2 enhances retinal vessel sensitivity to vascular endothelial growth factor. J Cell Physiol. 2004;199:412–417.
98 Crabb JW, Miyagi M, Gu X, et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99:14682–14687.
99 Anand-Apte B, Pepper MS, Voest E, et al. Inhibition of angiogenesis by tissue inhibitor of metalloproteinase-3. Invest Ophthalmol Vis Sci. 1997;38:817–823.
100 Takahashi T, Nakamura T, Hayashi A, et al. Inhibition of experimental choroidal neovascularization by overexpression of tissue inhibitor of metalloproteinases-3 in retinal pigment epithelium cells. Am J Ophthalmol. 2000;130:774–781.
101 Bhutto IA, McLeod DS, Hasegawa T, et al. Pigment epithelium-derived factor (PEDF) and vascular endothelial growth factor (VEGF) in aged human choroid and eyes with age-related macular degeneration. Exp Eye Res. 2006;82:99–110.
102 Amaral J, Becerra SP. Effects of human recombinant PEDF protein and PEDF-derived peptide 34-mer on choroidal neovascularization. Invest Ophthalmol Vis Sci. 2010;51:1318–1326.
103 Otani A, Slike BM, Dorrell MI, et al. A fragment of human TrpRS as a potent antagonist of ocular angiogenesis. Proc Natl Acad Sci U S A. 2002;99:178–183.
104 Cao Y, Xue L. Angiostatin. Semin Thromb Hemost. 2004;30:83–93.
105 Newsome DA, Swartz M, Leone NC, et al. Oral zinc in macular degeneration. Arch Ophthalmol. 1988;106:192–198.
106 Bressler NM, Bressler SB, Congdon NG, et al. Potential public health impact of Age-Related Eye Disease Study results: AREDS report no. 11. Arch Ophthalmol. 2003;121:1621–1624.
107 SanGiovanni JP, Chew EY, Clemons TE, et al. The relationship of dietary carotenoid and vitamin A, E, and C intake with age-related macular degeneration in a case-control study: AREDS report no. 22. Arch Ophthalmol. 2007;125:1225–1232.
108 Radu RA, Han Y, Bui TV, et al. Reductions in serum vitamin A arrest accumulation of toxic retinal fluorophores: a potential therapy for treatment of lipofuscin-based retinal diseases. Invest Ophthalmol Vis Sci. 2005;46:4393–4401.
109 Quadro L, Blaner WS, Salchow DJ, et al. Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO J. 1999;18:4633–4644.
110 Vogel S, Piantedosi R, O’Byrne SM, et al. Retinol-binding protein-deficient mice: biochemical basis for impaired vision. Biochemistry. 2002;41:15360–15368.
111 Decensi A, Torrisi R, Bruno S, et al. Randomized trial of fenretinide in superficial bladder cancer using DNA flow cytometry as an intermediate end point. Cancer Epidemiol Biomarkers Prev. 2000;9:1071–1078.
112 Follen M, Atkinson EN, Schottenfeld D, et al. A randomized clinical trial of 4-hydroxyphenylretinamide for high-grade squamous intraepithelial lesions of the cervix. Clin Cancer Res. 2001;7:3356–3365.
113 Camerini T, Mariani L, De Palo G, et al. Safety of the synthetic retinoid fenretinide: long-term results from a controlled clinical trial for the prevention of contralateral breast cancer. J Clin Oncol. 2001;19:1664–1670.
114 Garaventa A, Luksch R, Lo Piccolo MS, et al. Phase I trial and pharmacokinetics of fenretinide in children with neuroblastoma. Clin Cancer Res. 2003;9:2032–2039.
115 Puduvalli VK, Yung WK, Hess KR, et al. Phase II study of fenretinide (NSC 374551) in adults with recurrent malignant gliomas: A North American Brain Tumor Consortium study. J Clin Oncol. 2004;22:4282–4289.
116 Mariani L, Formelli F, De Palo G, et al. Chemoprevention of breast cancer with fenretinide (4-HPR): study of long-term visual and ophthalmologic tolerability. Tumori. 1996;82:444–449.
117 Conley B, O’Shaughnessy J, Prindiville S, et al. Pilot trial of the safety, tolerability, and retinoid levels of N-(4-hydroxyphenyl) retinamide in combination with tamoxifen in patients at high risk for developing invasive breast cancer. J Clin Oncol. 2000;18:275–283.
118 Caruso RC, Zujewski J, Iwata F, et al. Effects of fenretinide (4-HPR) on dark adaptation. Arch Ophthalmol. 1998;116:759–763.
119 Kaiser-Kupfer MI, Peck GL, Caruso RC, et al. Abnormal retinal function associated with fenretinide, a synthetic retinoid. Arch Ophthalmol. 1986;104:69–70.
120 Decensi A, Torrisi R, Polizzi A, et al. Effect of the synthetic retinoid fenretinide on dark adaptation and the ocular surface. J Natl Cancer Inst. 1994;86:105–110.
121 Decensi A, Fontana V, Fioretto M, et al. Long-term effects of fenretinide on retinal function. Eur J Cancer. 1997;33:80–84.
122 Marmor MF, Jain A, Moshfeghi D. Total rod ERG suppression with high dose compassionate Fenretinide usage. Doc Ophthalmol. 2008;117:257–261.
123 Radu RA, Mata NL, Nusinowitz S, et al. Isotretinoin treatment inhibits lipofuscin accumulation in a mouse model of recessive Stargardt’s macular degeneration. Novartis Found Symp. 2004;255:51–63. discussion 7, 177–8
124 Sieving PA, Chaudhry P, Kondo M, et al. Inhibition of the visual cycle in vivo by 13-cis retinoic acid protects from light damage and provides a mechanism for night blindness in isotretinoin therapy. Proc Natl Acad Sci U S A. 2001;98:1835–1840.
125 Kubota R, Boman NL, David R, et al. Safety and effect on rod function of Acu-4429, a novel small-molecule visual cycle modulator. Retina. 2012;32:183–188.
126 Donoso LA, Kim D, Frost A, et al. The role of inflammation in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2006;51:137–152.
127 Emlen W, Li W, Kirschfink M. Therapeutic complement inhibition: new developments. Semin Thromb Hemost. 2010;36:660–668.
128 Anderson DH, Radeke MJ, Gallo NB, et al. The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog Retin Eye Res. 2010;29:95–112.
129 Anderson DH, Mullins RF, Hageman GS, et al. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. 2002;134:411–431.
130 Hughes AE, Orr N, Esfandiary H, et al. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat Genet. 2006;38:1173–1177.
131 Schmid-Kubista KE, Tosakulwong N, Wu Y, et al. Contribution of copy number variation in the regulation of complement activation locus to development of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2009;50:5070–5079.
132 Campochiaro PA. Ocular neovascularisation and excessive vascular permeability. Expert Opin Biol Ther. 2004;4:1395–1402.
133 Steinbrook R. The price of sight–ranibizumab, bevacizumab, and the treatment of macular degeneration. N Engl J Med. 2006;355:1409–1412.
134 Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–2342.
135 Michels S, Rosenfeld PJ, Puliafito CA, et al. Systemic bevacizumab (Avastin) therapy for neovascular age-related macular degeneration twelve-week results of an uncontrolled open-label clinical study. Ophthalmology. 2005;112:1035–1047.
136 Moshfeghi AA, Rosenfeld PJ, Puliafito CA, et al. Systemic bevacizumab (Avastin) therapy for neovascular age-related macular degeneration: twenty-four-week results of an uncontrolled open-label clinical study. Ophthalmology. 2006;113:2002e1–12.
137 Rosenfeld PJ, Moshfeghi AA, Puliafito CA. Optical coherence tomography findings after an intravitreal injection of bevacizumab (avastin) for neovascular age-related macular degeneration. Ophthalmic Surg Lasers Imaging. 2005;36:331–335.
138 Han DP. Intravitreal human immune globulin in a rabbit model of Staphylococcus aureus toxin-mediated endophthalmitis: a potential adjunct in the treatment of endophthalmitis. Trans Am Ophthalmol Soc. 2004;102:305–320.
139 Shahar J, Avery RL, Heilweil G, et al. Electrophysiologic and retinal penetration studies following intravitreal injection of bevacizumab (Avastin). Retina. 2006;26:262–269.
140 Dib E, Maia M, Longo-Maugeri IM, et al. Subretinal bevacizumab detection after intravitreous injection in rabbits. Invest Ophthalmol Vis Sci. 2008;49:1097–1100.
141 Mordenti J, Thomsen K, Licko V, et al. Intraocular pharmacokinetics and safety of a humanized monoclonal antibody in rabbits after intravitreal administration of a solution or a PLGA microsphere formulation. Toxicol Sci. 1999;52:101–106.
142 Mordenti J, Cuthbertson RA, Ferrara N, et al. Comparisons of the intraocular tissue distribution, pharmacokinetics, and safety of 125I-labeled full-length and Fab antibodies in rhesus monkeys following intravitreal administration. Toxicol Pathol. 1999;27:536–544.
143 Bakri SJ, Kitzmann AS. Retinal pigment epithelial tear after intravitreal ranibizumab. Am J Ophthalmol. 2007;143:505–507.
144 Miyake T, Sawada O, Kakinoki M, et al. Pharmacokinetics of bevacizumab and its effect on vascular endothelial growth factor after intravitreal injection of bevacizumab in macaque eyes. Invest Ophthalmol Vis Sci. 2010;51:1606–1608.
145 Nomoto H, Shiraga F, Kuno N, et al. Pharmacokinetics of bevacizumab after topical, subconjunctival, and intravitreal administration in rabbits. Invest Ophthalmol Vis Sci. 2009;50:4807–4813.
146 Beer PM, Wong SJ, Hammad AM, et al. Vitreous levels of unbound bevacizumab and unbound vascular endothelial growth factor in two patients. Retina. 2006;26:871–876.
147 Krohne TU, Eter N, Holz FG, et al. Intraocular pharmacokinetics of bevacizumab after a single intravitreal injection in humans. Am J Ophthalmol. 2008;146:508–512.
148 Zhu Q, Ziemssen F, Henke-Fahle S, et al. Vitreous levels of bevacizumab and vascular endothelial growth factor-A in patients with choroidal neovascularization. Ophthalmology. 2008;115:1750–1755. 5 e1
149 Brechner RJ, Rosenfeld PJ, Babish JD, et al. Pharmacotherapy for neovascular age-related macular degeneration: an analysis of the 100% 2008 medicare fee-for-service part B claims file. Am J Ophthalmol. 2011;151:887–895. e1
150 Martin DF, Maguire MG, Ying GS, et al. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N Engl J Med. 2011;364:1897–1908.
151 Arevalo JF, Maia M, Garcia-Amaris RA, et al. Intravitreal bevacizumab for refractory pseudophakic cystoid macular edema: the Pan-American Collaborative Retina Study Group results. Ophthalmology. 2009;116:1481–1487. 7 e1
152 Caccavale A, Romanazzi F, Imparato M, et al. Central serous chorioretinopathy: a pathogenetic model. Clin Ophthalmol. 2011;5:239–243.
153 Cervantes-Castaneda RA, Giuliari GP, Gallagher MJ, et al. Intravitreal bevacizumab in refractory uveitic macular edema: one-year follow-up. Eur J Ophthalmol. 2009;19:622–629.
154 Ehrlich R, Ciulla TA, Maturi R, et al. Intravitreal bevacizumab for choroidal neovascularization secondary to presumed ocular histoplasmosis syndrome. Retina. 2009;29:1418–1423.
155 El Matri L, Kort F, Bouraoui R, et al. Intravitreal bevacizumab for the treatment of choroidal neovascularization secondary to angioid streaks: one year of follow-up. Acta Ophthalmol. 2010;89:641–646.
156 Gregori NZ, Rattan GH, Rosenfeld PJ, et al. Safety and efficacy of intravitreal bevacizumab (avastin) for the management of branch and hemiretinal vein occlusion. Retina. 2009;29:913–925.
157 Mintz-Hittner HA, Kennedy KA, Chuang AZ. Efficacy of intravitreal bevacizumab for stage 3+ retinopathy of prematurity. N Engl J Med. 2011;364:603–615.
158 Nakanishi H, Tsujikawa A, Yodoi Y, et al. Prognostic factors for visual outcomes 2-years after intravitreal bevacizumab for myopic choroidal neovascularization. Eye (Lond). 2011;25:375–381.
159 Wu L, Arevalo JF, Berrocal MH, et al. Comparison of two doses of intravitreal bevacizumab as primary treatment for macular edema secondary to branch retinal vein occlusions: results of the Pan American Collaborative Retina Study Group at 24 months. Retina. 2009;29:1396–1403.
160 Chiang A, Regillo CD. Preferred therapies for neovascular age-related macular degeneration. Curr Opin Ophthalmol. 2011;22:199–204.
161 Tufail A, Patel PJ, Egan C, et al. Bevacizumab for neovascular age related macular degeneration (ABC Trial): multicentre randomised double masked study. BMJ. 2010;340:c2459.
162 Subramanian ML, Ness S, Abedi G, et al. Bevacizumab vs ranibizumab for age-related macular degeneration: early results of a prospective double-masked, randomized clinical trial. Am J Ophthalmol. 2009;148:875–882.
163 Rosenfeld PJ, Brown DM, Heier JS, et al. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. 2006;355:1419–1431.
164 Brown DM, Kaiser PK, Michels M, et al. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med. 2006;355:1432–1444.
165 Mitchell P, Korobelnik JF, Lanzetta P, et al. Ranibizumab (Lucentis) in neovascular age-related macular degeneration: evidence from clinical trials. Br J Ophthalmol. 2010;94:2–13.
166 Brown DM, Michels M, Kaiser PK, et al. Ranibizumab versus verteporfin photodynamic therapy for neovascular age-related macular degeneration: Two-year results of the ANCHOR study. Ophthalmology. 2009;116:57–65.
167 Rosenfeld PJ, Schwartz SD, Blumenkranz MS, et al. Maximum tolerated dose of a humanized anti-vascular endothelial growth factor antibody fragment for treating neovascular age-related macular degeneration. Ophthalmology. 2005;112:1048–1053.
168 Regillo CD, Brown DM, Abraham P, et al. Randomized, double-masked, sham-controlled trial of ranibizumab for neovascular age-related macular degeneration: PIER Study year 1. Am J Ophthalmol. 2008;145:239–248.
169 Abraham P, Yue H, Wilson L. Randomized, double-masked, sham-controlled trial of ranibizumab for neovascular age-related macular degeneration: PIER study year 2. Am J Ophthalmol. 2010;150:315–324.
170 Schmidt-Erfurth U, Eldem B, Guymer R, et al. Efficacy and safety of monthly versus quarterly ranibizumab treatment in neovascular age-related macular degeneration: the EXCITE study. Ophthalmology. 2011;118:831–839.
171 Holz FG, Amoaku W, Donate J, et al. Safety and efficacy of a flexible dosing regimen of ranibizumab in neovascular age-related macular degeneration: the SUSTAIN study. Ophthalmology. 2011;118:663–671.
172 Fung AE, Lalwani GA, Rosenfeld PJ, et al. An optical coherence tomography-guided, variable dosing regimen with intravitreal ranibizumab (Lucentis) for neovascular age-related macular degeneration. Am J Ophthalmol. 2007;143:566–583.
173 Lalwani GA, Rosenfeld PJ, Fung AE, et al. A variable-dosing regimen with intravitreal ranibizumab for neovascular age-related macular degeneration: year 2 of the PrONTO Study. Am J Ophthalmol. 2009;148:43–58.
174 Collaborative overview of randomised trials of antiplatelet therapy – I: Prevention of death, myocardial infarction, and stroke by prolonged antiplatelet therapy in various categories of patients. Antiplatelet Trialists’ Collaboration. BMJ. 1994;308:81–106.
175 Schmidt-Erfurth U. Clinical safety of ranibizumab in age-related macular degeneration. Expert Opin Drug Saf. 2010;9:149–165.
176 Rosenfeld PJ. Bevacizumab versus ranibizumab for AMD. N Engl J Med. 2011;364:1966–1967.
176a IVAN Study Investigators. Ranibizumab versus bevacizumab to treat neovascular age-related macular degeneration: one-year findings from the IVAN randomized trial. Ophthalmology. 2012. Corrected proof published online 2012 May 11 http//:www.ophsource.org/periodicals/ophtha/article/S0161-6420(12)00358-2/ [cited 2012 June 8 ]
177 Martin DF, Maguire MG, Fine SL, et al. Ranibizumab and bevacizumab for treatment of neovascular age-related macular degeneration: two-year results. Ophthalmology. 2012. Corrected proof published online 2012 May 2 http//:www.ophsource.org/periodicals/ophtha/article/S0161-6420(12)00321-1/ [cited 2012 June 8]
178 Holash J, Davis S, Papadopoulos N, et al. VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci U S A. 2002;99:11393–11398.
179 Chappelow AV, Kaiser PK. Neovascular age-related macular degeneration: potential therapies. Drugs. 2008;68:1029–1036.
180 Cao J, Song H, Renard RA, et al. Systemic or intravitreal administration of VEGF Trap suppresses vascular leak and leukostasis in the retinas of diabetic rats. Invest Ophthalmol Vis Sci. 47, 2006.
181 Rudge JS, Thurston G, Davis S, et al. VEGF trap as a novel antiangiogenic treatment currently in clinical trials for cancer and eye diseases, and VelociGene- based discovery of the next generation of angiogenesis targets. Cold Spring Harb Symp Quant Biol. 2005;70:411–418.
182 Nguyen QD, Shah SM, Browning DJ, et al. A phase I study of intravitreal vascular endothelial growth factor trap-eye in patients with neovascular age-related macular degeneration. Ophthalmology. 2009;116:2141–2148.
183 Brown DM, Heier JS, Ciulla T, et al. Primary endpoint results of a phase II study of vascular endothelial growth factor trap-eye in wet age-related macular degeneration. Ophthalmology. 2011;118:1089–1097.
184 Heier JS, Boyer D, Nguyen QD, et al. The 1-year results of CLEAR-IT 2, a phase 2 study of vascular endothelial growth factor trap-eye dosed as-needed after 12-week fixed dosing. Ophthalmology. 2011;118:1098–1106.
185 http://investor.regeneron.com/releasedetail.cfm?ReleaseID=532099. v. 2012 [cited 2012 Feb 15]
186 http://files.shareholder.com/downloads/REGN/1026966568x0x440864/8BEB55ED-1AA6-43DB-8783-EA5C0C4C78DE/REGN_Angiogenesis_Final_021311.pdf. v. 2012 [cited 2012 Feb 15]
187 Zhang M, Zhang J, Yan M, et al. Recombinant anti-vascular endothelial growth factor fusion protein efficiently suppresses choridal neovasularization in monkeys. Mol Vis. 2008;14:37–49.
188 Suto K, Yamazaki Y, Morita T, et al. Crystal structures of novel vascular endothelial growth factors (VEGF) from snake venoms: insight into selective VEGF binding to kinase insert domain-containing receptor but not to fms-like tyrosine kinase-1. J Biol Chem. 2005;280:2126–2131.
189 Zhang M, Yu D, Yang C, et al. The pharmacology study of a new recombinant human VEGF receptor-fc fusion protein on experimental choroidal neovascularization. Pharm Res. 2009;26:204–210.
190 Lai CM, Shen WY, Brankov M, et al. Long-term evaluation of AAV-mediated sFlt-1 gene therapy for ocular neovascularization in mice and monkeys. Mol Ther. 2005;12:659–668.
191 Kendall RL, Wang G, Thomas KA. Identification of a natural soluble form of the vascular endothelial growth factor receptor, FLT-1, and its heterodimerization with KDR. Biochem Biophys Res Commun. 1996;226:324–328.
192 Mori K, Gehlbach P, Yamamoto S, et al. AAV-mediated gene transfer of pigment epithelium-derived factor inhibits choroidal neovascularization. Invest Ophthalmol Vis Sci. 2002;43:1994–2000.
193 Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med. 2008;358:2240–2248.
194 Bainbridge JW, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. 2008;358:2231–2239.
195 Cideciyan AV, Hauswirth WW, Aleman TS, et al. Human RPE65 gene therapy for Leber congenital amaurosis: persistence of early visual improvements and safety at 1 year. Hum Gene Ther. 2009;20:999–1004.
196 Bainbridge JW, Mistry A, De Alwis M, et al. Inhibition of retinal neovascularisation by gene transfer of soluble VEGF receptor sFlt-1. Gene Ther. 2002;9:320–326.
197 Gehlbach P, Demetriades AM, Yamamoto S, et al. Periocular gene transfer of sFlt-1 suppresses ocular neovascularization and vascular endothelial growth factor-induced breakdown of the blood-retinal barrier. Hum Gene Ther. 2003;14:129–141.
198 Lukason M, DuFresne E, Rubin H, et al. Inhibition of choroidal neovascularization in a nonhuman primate model by intravitreal administration of an AAV2 vector expressing a novel anti-VEGF molecule. Mol Ther. 2011;19:260–265.
199 Igarashi T, Miyake K, Masuda I, et al. Adeno-associated vector (type 8)-mediated expression of soluble Flt-1 efficiently inhibits neovascularization in a murine choroidal neovascularization model. Hum Gene Ther. 2010;21:631–637.
200 Jellinek D, Green LS, Bell C, et al. Inhibition of receptor binding by high-affinity RNA ligands to vascular endothelial growth factor. Biochemistry. 1994;33:10450–10456.
201 Gragoudas ES, Adamis AP, Cunningham ET, et al. Pegaptanib for neovascular age-related macular degeneration. N Engl J Med. 2004;351:2805–2816.
202 Verteporfin therapy of subfoveal choroidal neovascularization in age-related macular degeneration: two-year results of a randomized clinical trial including lesions with occult with no classic choroidal neovascularization – verteporfin in photodynamic therapy report 2. Am J Ophthalmol. 2001;131:541–560.
203 Friberg TR, Tolentino M, Weber P, et al. Pegaptanib sodium as maintenance therapy in neovascular age-related macular degeneration: the LEVEL study. Br J Ophthalmol. 2010;94:1611–1617.
204 Reich SJ, Fosnot J, Kuroki A, et al. Small interfering RNA (siRNA) targeting VEGF effectively inhibits ocular neovascularization in a mouse model. Mol Vis. 2003;9:210–216.
205 Kaiser PK, Symons RC, Shah SM, et al. RNAi-based treatment for neovascular age-related macular degeneration by Sirna-027. Am J Ophthalmol. 2010;150:33–39.
206 Ashikari M, Tokoro M, Itaya M, et al. Suppression of laser-induced choroidal neovascularization by nontargeted siRNA. Invest Ophthalmol Vis Sci. 2010;51:3820–3824.
207 Gu L, Chen H, Tuo J, et al. Inhibition of experimental choroidal neovascularization in mice by anti-VEGFA/VEGFR2 or non-specific siRNA. Exp Eye Res. 2010;91:433–439.
208 Kleinman ME, Yamada K, Takeda A, et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature. 2008;452:591–597.
209 Ni Z, Hui P. Emerging pharmacologic therapies for wet age-related macular degeneration. Ophthalmologica. 2009;223:401–410.
210 Takahashi K, Saishin Y, King AG, et al. Suppression and regression of choroidal neovascularization by the multitargeted kinase inhibitor pazopanib. Arch Ophthalmol. 2009;127:494–499.
211 Charters L. Pazopanib explored for neovascular age-related macular degeneration. Ophthalmology Times. 2010;1:34. Mar
212 Mabry R, Gilbertson DG, Frank A, et al. A dual-targeting PDGFRbeta/VEGF-A molecule assembled from stable antibody fragments demonstrates anti-angiogenic activity in vitro and in vivo. MAbs. 2010;2:20–34.
213 Wakasugi K, Slike BM, Hood J, et al. A human aminoacyl-tRNA synthetase as a regulator of angiogenesis. Proc Natl Acad Sci U S A. 2002;99:173–177.
214 Banin E, Dorrell MI, Aguilar E, et al. T2-TrpRS inhibits preretinal neovascularization and enhances physiological vascular regrowth in OIR as assessed by a new method of quantification. Invest Ophthalmol Vis Sci. 2006;47:2125–2134.
215 Aiello LP. The potential role of PKC beta in diabetic retinopathy and macular edema. Surv Ophthalmol. 2002;47(Suppl 2):S263–S269.
216 Saishin Y, Silva RL, Callahan K, et al. Periocular injection of microspheres containing PKC412 inhibits choroidal neovascularization in a porcine model. Invest Ophthalmol Vis Sci. 2003;44:4989–4993.
217 Engelbert M, Gilmore MS. Fas ligand but not complement is critical for control of experimental Staphylococcus aureus endophthalmitis. Invest Ophthalmol Vis Sci. 2005;46:2479–2486.
218 Giese MJ, Mondino BJ, Glasgow BJ, et al. Complement system and host defense against staphylococcal endophthalmitis. Invest Ophthalmol Vis Sci. 1994;35:1026–1032.
219 Aizuss DH, Mondino BJ, Sumner HL, et al. The complement system and host defense against Pseudomonas endophthalmitis. Invest Ophthalmol Vis Sci. 1985;26:1262–1266.
220 Brooks SE, Gu X, Samuel S, et al. Reduced severity of oxygen-induced retinopathy in eNOS-deficient mice. Invest Ophthalmol Vis Sci. 2001;42:222–228.
221 Semkova I, Kreppel F, Welsandt G, et al. Autologous transplantation of genetically modified iris pigment epithelial cells: a promising concept for the treatment of age-related macular degeneration and other disorders of the eye. Proc Natl Acad Sci U S A. 2002;99:13090–13095.
222 Campochiaro PA, Nguyen QD, Shah SM, et al. Adenoviral vector-delivered pigment epithelium-derived factor for neovascular age-related macular degeneration: results of a phase I clinical trial. Hum Gene Ther. 2006;17:167–176.
223 Lai YK, Shen WY, Brankov M, et al. Potential long-term inhibition of ocular neovascularisation by recombinant adeno-associated virus-mediated secretion gene therapy. Gene Ther. 2002;9:804–813.
224 Kachi S, Binley K, Yokoi K, et al. Equine infectious anemia viral vector-mediated codelivery of endostatin and angiostatin driven by retinal pigmented epithelium-specific VMD2 promoter inhibits choroidal neovascularization. Hum Gene Ther. 2009;20:31–39.
225 Balaggan KS, Binley K, Esapa M, et al. EIAV vector-mediated delivery of endostatin or angiostatin inhibits angiogenesis and vascular hyperpermeability in experimental CNV. Gene Ther. 2006;13:1153–1165.
226 Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science. 1994;264:569–571.
227 Luna J, Tobe T, Mousa SA, et al. Antagonists of integrin alpha v beta 3 inhibit retinal neovascularization in a murine model. Lab Invest. 1996;75:563–573.
228 Kamizuru H, Kimura H, Yasukawa T, et al. Monoclonal antibody-mediated drug targeting to choroidal neovascularization in the rat. Invest Ophthalmol Vis Sci. 2001;42:2664–2672.
229 Lahdenranta J, Sidman RL, Pasqualini R, et al. Treatment of hypoxia-induced retinopathy with targeted proapoptotic peptidomimetic in a mouse model of disease. FASEB J. 2007;21:3272–3278.
230 Umeda N, Kachi S, Akiyama H, et al. Suppression and regression of choroidal neovascularization by systemic administration of an alpha5beta1 integrin antagonist. Mol Pharmacol. 2006;69:1820–1828.
231 Zahn G, Vossmeyer D, Stragies R, et al. Preclinical evaluation of the novel small-molecule integrin alpha5beta1 inhibitor JSM6427 in monkey and rabbit models of choroidal neovascularization. Arch Ophthalmol. 2009;127:1329–1335.
232 Wang W, Wang F, Lu F, et al. The anti-angiogenic effects of integrin α5β1 inhibitor (ATN-161) in vitro and in vivo. Invest Ophthalmol Vis Sci. 2011;52:7213–7220.
233 Maier AK, Kociok N, Zahn G, et al. Modulation of hypoxia-induced neovascularization by JSM6427, an integrin alpha5beta1 inhibiting molecule. Curr Eye Res. 2007;32:801–812.
234 Muether PS, Dell S, Kociok N, et al. The role of integrin alpha5beta1 in the regulation of corneal neovascularization. Exp Eye Res. 2007;85:356–365.
235 Kuwada SK. Drug evaluation: Volociximab, an angiogenesis-inhibiting chimeric monoclonal antibody. Curr Opin Mol Ther. 2007;9:92–98.
236 Gomis-Ruth FX, Maskos K, Betz M, et al. Mechanism of inhibition of the human matrix metalloproteinase stromelysin-1 by TIMP-1. Nature. 1997;389:77–81.
237 Garcia C, Bartsch DU, Rivero ME, et al. Efficacy of Prinomastat (AG3340), a matrix metalloprotease inhibitor, in treatment of retinal neovascularization. Curr Eye Res. 2002;24:33–38.
238 Nambu H, Nambu R, Melia M, et al. Combretastatin A-4 phosphate suppresses development and induces regression of choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44:3650–3655.
239 Genaidy M, Kazi AA, Peyman GA, et al. Effect of squalamine on iris neovascularization in monkeys. Retina. 2002;22:772–778.
240 Jones SR, Kinney WA, Zhang X, et al. The synthesis and characterization of analogs of the antimicrobial compound squalamine: 6 beta-hydroxy-3-aminosterols synthesized from hyodeoxycholic acid. Steroids. 1996;61:565–571.
241 Higgins RD, Yan Y, Geng Y, et al. Regression of retinopathy by squalamine in a mouse model. Pediatr Res. 2004;56:144–149.
242 Spaide RF, Sorenson J, Maranan L. Combined photodynamic therapy with verteporfin and intravitreal triamcinolone acetonide for choroidal neovascularization. Ophthalmology. 2003;110:1517–1525.
243 Spaide RF, Sorenson J, Maranan L. Photodynamic therapy with verteporfin combined with intravitreal injection of triamcinolone acetonide for choroidal neovascularization. Ophthalmology. 2005;112:301–304.
244 Chan A, Blumenkranz MS, Wu KH, et al. Photodynamic therapy with and without adjunctive intravitreal triamcinolone acetonide: a retrospective comparative study. Ophthalmic Surg Lasers Imaging. 2009;40:561–569.
245 Chaudhary V, Mao A, Hooper PL, et al. Triamcinolone acetonide as adjunctive treatment to verteporfin in neovascular age-related macular degeneration: a prospective randomized trial. Ophthalmology. 2007;114:2183–2189.
246 Luttrull JK, Spink CJ. Prolongation of choroidal hypofluorescence following combined verteporfin photodynamic therapy and intravitreal triamcinolone acetonide injection. Retina. 2007;27:688–692.
247 Maberley D. Photodynamic therapy and intravitreal triamcinolone for neovascular age-related macular degeneration: a randomized clinical trial. Ophthalmology. 2009;116:2149–2157.
248 Hatta Y, Ishikawa K, Nishihara H, et al. Effect of photodynamic therapy alone or combined with posterior subtenon triamcinolone acetonide or intravitreal bevacizumab on choroidal hypofluorescence by indocyanine green angiography. Retina. 2010;30:495–502.
249 Kovacs KD, Quirk MT, Kinoshita T, et al. A retrospective analysis of triple combination therapy with intravitreal bevacizumab, posterior sub-tenon’s triamcinolone acetonide, and low-fluence verteporfin photodynamic therapy in patients with neovascular age-related macular degeneration. Retina. 2011;31:446–452.
250 Liggett PE, Colina J, Chaudhry NA, et al. Triple therapy of intravitreal triamcinolone, photodynamic therapy, and pegaptanib sodium for choroidal neovascularization. Am J Ophthalmol. 2006;142:1072–1074.
251 Arias L, Garcia-Arumi J, Ramon JM, et al. Photodynamic therapy with intravitreal triamcinolone in predominantly classic choroidal neovascularization: one-year results of a randomized study. Ophthalmology. 2006;113:2243–2250.
252 Arias L, Garcia-Arumi J, Ramon JM, et al. Optical coherence tomography analysis of a randomized study combining photodynamic therapy with intravitreal triamcinolone. Graefes Arch Clin Exp Ophthalmol. 2008;246:245–254.
253 Napoli KL, Taylor PJ. From beach to bedside: history of the development of sirolimus. Ther Drug Monit. 2001;23:559–586.
254 Guba M, von Breitenbuch P, Steinbauer M, et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat Med. 2002;8:128–135.
255 Dejneka NS, Kuroki AM, Fosnot J, et al. Systemic rapamycin inhibits retinal and choroidal neovascularization in mice. Mol Vis. 2004;10:964–972.
256 Nussenblatt RB, Byrnes G, Sen HN, et al. A randomized pilot study of systemic immunosuppression in the treatment of age-related macular degeneration with choroidal neovascularization. Retina. 2010;30:1579–1587.
257 Woodburn KW, Engelman CJ, Blumenkranz MS. Photodynamic therapy for choroidal neovascularization: a review. Retina. 2002;22:391–405. quiz 527–8
258 Flower RW, von Kerczek C, Zhu L, et al. Theoretical investigation of the role of choriocapillaris blood flow in treatment of subfoveal choroidal neovascularization associated with age-related macular degeneration. Am J Ophthalmol. 2001;132:85–93.
259 Husain D, Kramer M, Kenny AG, et al. Effects of photodynamic therapy using verteporfin on experimental choroidal neovascularization and normal retina and choroid up to 7 weeks after treatment. Invest Ophthalmol Vis Sci. 1999;40:2322–2331.
260 Husain D, Miller JW, Michaud N, et al. Intravenous infusion of liposomal benzoporphyrin derivative for photodynamic therapy of experimental choroidal neovascularization. Arch Ophthalmol. 1996;114:978–985.
261 Kramer M, Miller JW, Michaud N, et al. Liposomal benzoporphyrin derivative verteporfin photodynamic therapy. Selective treatment of choroidal neovascularization in monkeys. Ophthalmology. 1996;103:427–438.
262 Miller JW, Walsh AW, Kramer M, et al. Photodynamic therapy of experimental choroidal neovascularization using lipoprotein-delivered benzoporphyrin. Arch Ophthalmol. 1995;113:810–818.
263 Miller JW, Schmidt-Erfurth U, Sickenberg M, et al. Photodynamic therapy with verteporfin for choroidal neovascularization caused by age-related macular degeneration: results of a single treatment in a phase 1 and 2 study. Arch Ophthalmol. 1999;117:1161–1173.
264 Schmidt-Erfurth U, Miller JW, Sickenberg M, et al. Photodynamic therapy with verteporfin for choroidal neovascularization caused by age-related macular degeneration: results of retreatments in a phase 1 and 2 study. Arch Ophthalmol. 1999;117:1177–1187.
265 Photodynamic therapy of subfoveal choroidal neovascularization in pathologic myopia with verteporfin. 1-year results of a randomized clinical trial – VIP report no. 1. Ophthalmology. 2001;108:841–852.
266 Blumenkranz MS, Bressler NM, Bressler SB, et al. Verteporfin therapy for subfoveal choroidal neovascularization in age-related macular degeneration: three-year results of an open-label extension of 2 randomized clinical trials – TAP Report no. 5. Arch Ophthalmol. 2002;120:1307–1314.
267 Blinder KJ, Bradley S, Bressler NM, et al. Effect of lesion size, visual acuity, and lesion composition on visual acuity change with and without verteporfin therapy for choroidal neovascularization secondary to age-related macular degeneration: TAP and VIP report no. 1. Am J Ophthalmol. 2003;136:407–418.
268 Mathis S, Khanlari B, Pulido F, et al. Effectiveness of protease inhibitor monotherapy versus combination antiretroviral maintenance therapy: a meta-analysis. PLoS One. 2011;6:e22003.
269 Klement G, Baruchel S, Rak J, et al. Continuous low-dose therapy with vinblastine and VEGF receptor-2 antibody induces sustained tumor regression without overt toxicity. J Clin Invest. 2000;105:R15–R24.
270 Lee CG, Heijn M, di Tomaso E, et al. Anti-vascular endothelial growth factor treatment augments tumor radiation response under normoxic or hypoxic conditions. Cancer Res. 2000;60:5565–5570.
271 Piermarocchi S, Sartore M, Lo Giudice G, et al. Combination of photodynamic therapy and intraocular triamcinolone for exudative age-related macular degeneration and long-term chorioretinal macular atrophy. Arch Ophthalmol. 2008;126:1367–1374.
272 Roth DB, Kulkarni KM, Walsman S, et al. Intravitreal triamcinolone acetonide preceding photodynamic therapy for exudative age-related macular degeneration. Ophthalmic Surg Lasers Imaging. 2009;40:467–471.
273 Sacu S, Michels S, Prager F, et al. Randomised clinical trial of intravitreal Avastin vs photodynamic therapy and intravitreal triamcinolone: long-term results. Eye (Lond). 2009;23:2223–2227.
274 Augustin AJ, Puls S, Offermann I. Triple therapy for choroidal neovascularization due to age-related macular degeneration: verteporfin PDT, bevacizumab, and dexamethasone. Retina. 2007;27:133–140.
275 Bakri SJ, Couch SM, McCannel CA, et al. Same-day triple therapy with photodynamic therapy, intravitreal dexamethasone, and bevacizumab in wet age-related macular degeneration. Retina. 2009;29:573–578.
276 Kaiser PK, Boyer DS, Garcia R, et al. Verteporfin photodynamic therapy combined with intravitreal bevacizumab for neovascular age-related macular degeneration. Ophthalmology. 2009;116:747–755.
277 Forte R, Bonavolonta P, Benayoun Y, et al. Intravitreal ranibizumab and bevacizumab in combination with full-fluence verteporfin therapy and dexamethasone for exudative age-related macular degeneration. Ophthalmic Res. 2011;45:129–134.
278 Azab M, Boyer DS, Bressler NM, et al. Verteporfin therapy of subfoveal minimally classic choroidal neovascularization in age-related macular degeneration: 2-year results of a randomized clinical trial. Arch Ophthalmol. 2005;123:448–457.
279 Schmidt-Erfurth U, Michels S, Barbazetto I, et al. Photodynamic effects on choroidal neovascularization and physiological choroid. Invest Ophthalmol Vis Sci. 2002;43:830–841.
280 Schmidt-Erfurth U, Schlotzer-Schrehard U, Cursiefen C, et al. Influence of photodynamic therapy on expression of vascular endothelial growth factor (VEGF), VEGF receptor 3, and pigment epithelium-derived factor. Invest Ophthalmol Vis Sci. 2003;44:4473–4480.
281 Lim JY, Lee SY, Kim JG, et al. Intravitreal bevacizumab alone versus in combination with photodynamic therapy for the treatment of neovascular maculopathy in patients aged 50 years or older: 1-year results of a prospective clinical study. Acta Ophthalmol. 2012;90:61–67.
282 Kaiser PK, Boyer DS, Cruess AF, et al. Verteporfin plus ranibizumab for choroidal neovascularization in age-related macular degeneration: Twelve-month results of the DENALI Study. Ophthalmology. 2012;119:1001–1010.
283 Larsen M, Schmidt-Erfurth U, Lanzetta P, et al. Verteporfin plus ranibizumab for choroidal neovascularization in age-related macular degeneration: Twelve-month MONT BLANC Study Results. Ophthalmology. 2012;119:992–1000.
284 Retina Today: Bryn Mawr Communications LLC. RADICAL demonstrated fewer retreatment visits for AMD combination therapy. 2010.
285 Avila MP, Farah ME, Santos A, et al. Twelve-month short-term safety and visual-acuity results from a multicentre prospective study of epiretinal strontium-90 brachytherapy with bevacizumab for the treatment of subfoveal choroidal neovascularisation secondary to age-related macular degeneration. Br J Ophthalmol. 2009;93:305–309.
286 Avila MP, Farah ME, Santos A, et al. Twelve-month safety and visual acuity results from a feasibility study of intraocular, epiretinal radiation therapy for the treatment of subfoveal CNV secondary to AMD. Retina. 2009;29:157–169.
287 Moshfeghi DM, Kaiser PK, Gertner M. Stereotactic low-voltage x-ray irradiation for age-related macular degeneration. Br J Ophthalmol. 2011;95:185–188.
* Selected text adapted with kind permission of Springer Science+Business Media.52,53
* Selected text adapted with kind permission of Springer Science+Business Media.52,53
* Selected text adapted with kind permission of Springer Science+Business Media.52,53
* Selected text adapted with kind permission of Springer Science+Business Media.52,53
* Selected text adapted with kind permission of Springer Science+Business Media.52,53
* Selected text adapted with kind permission of Springer Science+Business Media.52,53