Pharmacology and Nonanesthetic Drugs During Pregnancy and Lactation
Tony Gin MBChB, MD, FANZCA, FHKAM, Jerome Yankowitz MD
Chapter Outline
Drug therapy during pregnancy can be complex because the physiologic changes of pregnancy may alter drug disposition and effect. Maternal medications may have direct effects on the fetus after placental transfer or indirect effects through changes in placental and uterine function. Even after delivery, drug transfer to breast milk may be a concern. Nevertheless, pregnant women still require medications to treat many acute and chronic conditions. The challenge is finding the balance between the benefits and risks of therapy.
It would seem prudent to use only drugs considered safe in pregnancy. Unfortunately, the potential adverse effects of many drugs remain unclear. Pregnant women are not usually included in early clinical trials, and because of the low incidence of some complications, the first suggestion of adverse effects may be revealed only from post-marketing surveillance and registries of complications. This uncertainty about safety, and the difficulties in determining what public information is reliable, may convince mothers to refuse appropriate drug treatment. New improved drugs are available in many areas of therapeutics, but many prescribers prefer to use older drugs that have a longer empirical history of safety.
Maternal variations in drug effect are not usually difficult to manage, and a change in dosing regimen or the choice of drug may be all that is required. The greatest fears and concerns are for potential fetal effects that may manifest as teratogenicity with fetal loss or congenital malformations, fetal growth restriction (also known as intrauterine growth restriction), preterm labor, and other complications. Impaired development and behavioral problems may manifest after delivery.
The anesthesia provider should understand the implications of pregnancy on drug disposition and effect. Although not usually responsible for primary maternal drug therapy, he or she will encounter women taking many different medications and may sometimes need to administer a variety of nonanesthetic drugs during the peripartum period, either for maintenance of chronic therapy or for acute indications, especially when managing critically ill patients.
In this chapter, how maternal physiology affects pharmacology is summarized, teratology and fetal effects of the main classes of drugs the anesthesia provider is likely to encounter are addressed, and drug transfer to breast milk is reviewed. The more common perioperative drugs are used as examples. Specific drugs used in the management of individual obstetric conditions are discussed in other chapters.
Changes in Drug Disposition and Effect
Pharmacogenetics
Genetic differences are responsible for some of the variation in drug response among individuals. Pregnancy does not obviously modify these pharmacogenetic differences, although some obstetric conditions such as preeclampsia are related to complex genetic factors. There are, however, some examples in which underlying genetic differences do affect obstetric management.1
The metabolism of codeine to morphine is greatly affected by polymorphisms of the cytochrome P450 (CYP) isoenzyme CYP2D6. It has been recognized only recently that mothers who are ultrarapid metabolizers may produce and transfer sufficient morphine through breast milk to cause neonatal central nervous system (CNS) depression and even death.2
Two of the possible changes at the β2-adrenergic receptor are an arginine-to-glycine substitution at codon 16 (Arg16Gly) and a glutamine-to-glutamate substitution at codon 27 (Glu27Gln). When β2-receptor agonists were used for tocolysis, Arg16 homozygotes had longer gestation and better neonatal outcome.3 In the management of hypotension during spinal anesthesia for cesarean delivery, Gly16 homozygotes and Glu27 homozygotes required less ephedrine.4
The µ-opioid receptor gene may have an adenine-to-guanine substitution at nucleotide position 118 (A118G). There have been many studies of the effects of this polymorphism on opioid dose requirements and response, although the studies in obstetric patients have had conflicting results. For example, laboring women who were AA homozygous had an increased intrathecal fentanyl requirement for analgesia.5 In contrast, AA homozygous women receiving intrathecal morphine for postcesarean analgesia reported less pain and required less patient-controlled morphine but had a higher incidence of nausea and vomiting.6 Wong7 summarized the conflicting evidence and noted that the true effect of this polymorphism is probably small. Many other genetic factors influence opioid disposition and response, and even more factors influence pain perception. Indeed, a meta-analysis found that A118G polymorphism only explained 7% of the variability in opioid requirements.8 Thus, at present there are no indications for pharmacogenetic testing in routine obstetric practice.9
Pharmacokinetic Changes
The major physiologic changes during pregnancy would be expected to alter drug disposition.10,11 However, the magnitude and time course of these changes vary throughout pregnancy and among individuals. The results of many older studies are unreliable because the studies were often of low quality. Thus, making generalizations about the effects of pregnancy on drug disposition can be difficult, and individualized dosing is important.
Maternal Pharmacokinetics
Absorption and Uptake.
Oral absorption and bioavailability are not usually affected by pregnancy, although nausea and vomiting may limit oral intake. Intestinal motility is decreased during pregnancy, but gastric emptying is only delayed during labor or after opioid administration. Cardiac output is increased by 30% to 50% during pregnancy, and the increased blood flow to skin and mucous membranes will enhance absorption from these sites. Reduced functional residual capacity and increased minute ventilation lead to increased pulmonary uptake of inhalational anesthetic agents.
Distribution.
The increased cardiac output during pregnancy increases distribution of drug to all tissues. Drugs acting peripherally (e.g., neuromuscular blockers) will be delivered to their site of action more quickly. However, the onset of intravenous and inhalational anesthetics is dependent on the time course of their cerebral drug concentrations. A delay in the increase in arterial and brain anesthetic concentrations will result from increased peripheral perfusion. Increased peripheral perfusion will, however, increase the return of drug during the elimination phase. Total body water increases on average by 8 L, and intravascular plasma volume is increased by 40%, whereas extravascular volume increases by a variable amount, depending on weight gain and edema. Thus, hydrophilic drugs, such as neuromuscular blockers, will have a small increase in the volume of distribution. Body fat is increased on average by 4 kg, but this is unimportant given the large volume of distribution of lipophilic drugs.
Changes in protein binding are more important clinically. Plasma albumin concentration is reduced to about 70% of normal, whereas α1-acid glycoprotein concentration is largely unchanged. Protein binding of drugs may be reduced by increased concentrations of free fatty acids and other endogenous displacing substances. This leads to increased concentrations of free drug, but with chronic drug administration this is offset by increased clearance of that free drug. The total (free + bound) concentration of drug will decrease, and it may be necessary to reset the therapeutic target range lower to compensate. Thus, it is important to know whether monitored concentrations are for free or total drug. Only a few drugs (e.g., theophylline, phenytoin) require monitoring and modification of dose because of changes in protein binding.
Metabolism.
Most drugs are metabolized in the liver, and the rate of metabolism may depend on hepatic blood flow or intrinsic enzyme activity. Although cardiac output is increased in pregnancy, it is not clear whether blood flow to the liver is significantly increased. Two studies using clearance of markers concluded that hepatic blood flow was unchanged,12,13 whereas another using Doppler ultrasonography reported unchanged hepatic arterial flow during pregnancy but increased portal venous flow after 28 weeks’ gestation.14 More importantly, some cytochrome P450 isoenzymes (CYP3A4, CYP2D6, and CYP2C9) and uridine diphosphate glucuronosyltransferase (UGT) isoenzymes (UGT1A4 and UGT2B7) have increased activity during pregnancy,10,15 which increases the metabolism of drugs such as phenytoin (CYP2C9), midazolam (CYP3A4), and morphine (UGT2B7). Other isoenzymes (CYP1A2 and CYP2C19) have decreased activity, which reduces the metabolism of drugs such as caffeine and theophylline (CYP1A2).
Elimination.
Renal blood flow is increased by 60% to 80% and glomerular filtration rate is increased by 50% in pregnancy; thus, the renal excretion of unchanged drugs such as cephalosporin antibiotics is increased. There is also increased activity of transporter proteins such as renal P-glycoprotein, which may contribute to the increased clearance of digoxin in pregnancy.16 Increased minute ventilation enhances elimination of inhalational anesthetic agents.
The physiologic changes of pregnancy will affect individual drugs depending on their physicochemical characteristics and metabolic pathways. Bioavailability is not usually changed significantly. Changes in volume of distribution as a result of changes in protein binding may affect drugs such as phenytoin, but monitoring and modification of therapy is usually straightforward. Drugs metabolized by the liver may require increases or decreases in dose, depending on the metabolic pathway involved. Drugs excreted unchanged by the kidneys often require an increased dose.
Placental Transfer and Metabolism
Our understanding of placental transfer and metabolism is rapidly improving (see Chapter 4). Early research was often limited to measuring drug concentrations in the umbilical vessels and maternal vein at delivery. Results were variable and difficult to interpret, especially for drugs such as anesthetic agents that are administered shortly before delivery. Umbilical blood samples are obtained at variable times after drug exposure, well before steady-state conditions are achieved. The theory of a placental barrier was proposed because maternal and fetal concentrations were often different. However, differences in concentrations of binding proteins are mainly responsible for the fetal-maternal distribution of drugs at steady state.17 The fetal concentration of albumin is slightly greater than in the mother, but α1-acid glycoprotein concentration is only a third of the maternal value at term. Umbilical-to-maternal blood ratios of total drug may be misleading because it is the free drug that equilibrates across the placenta. Maternal-to-fetal ratios of drugs do not provide information on the rate of drug transfer or the amount of drug that has already been transferred to the fetus.
Drug transfer across the placenta was previously thought to occur mainly by diffusion. This would favor the movement of lipophilic drugs, and placental perfusion would be an important factor affecting transfer. Fetal pH is lower than maternal pH, so that weak bases become more ionized in the fetus, thus limiting their transfer back across the placenta. Normally, the difference in pH is only 0.1 and this “ion trapping” is irrelevant, but fetal acidosis can significantly increase the fetal concentration of drugs such as local anesthetics.
It is now known that the placenta contains many drug transporters that can modify fetal drug exposure, and these are particularly relevant when trying to use transplacental pharmacotherapy to deliver drugs to the fetus.18,19 For example, in the treatment of sustained fetal tachyarrhythmia, placental P-glycoprotein, an adenosine triphosphate–dependent drug efflux pump, will reduce net transfer of substrates such as digoxin and verapamil from the mother. With maternal human immunodeficiency virus (HIV) infection, treatment of the fetus is also required, but drug transporters limit the transfer of some antiviral drugs, such as protease inhibitors.
The placenta also contains many enzymes, especially those such as UGT that catalyze phase II conjugation reactions.19,20 Clearance of substrates by UGT in full-term placentas may be sufficient to contribute to overall maternal metabolism.
Fetal and Neonatal Elimination.
The fetus and neonate metabolize drugs, but at a reduced rate compared with adults.21,22 The fetal circulation guides drug transferred across the placenta to undergo first-pass hepatic metabolism, but some drug will bypass the liver. Renal blood flow is minimal until near term, and any excreted products would just pass into the amniotic fluid to be swallowed. Elimination of drugs by the fetus is thus mainly reliant on placental transfer. It would seem prudent to minimize the amount of drug transferred to the neonate and choose drugs that are eliminated rapidly. Relatively large minute ventilation promotes neonatal elimination of inhalational anesthetic agents, and this may be further increased by assisted ventilation.
Pharmacodynamic Changes
Changes in the concentrations of various hormones may alter the response to other substances. In particular, progesterone and endorphins may enhance sedation and anti-nociception, respectively. A pharmacodynamic difference specifically refers to a change in response to a given effect-site concentration, but it is difficult during pregnancy to carry out the high-fidelity studies necessary for accurate pharmacokinetic-pharmacodynamic modeling. Thus, the demonstrations of pharmacodynamic changes in pregnancy have been limited to specific experimental designs where there are large differences in effect.
General Anesthesia
Early animal studies showed that maternal anesthetic requirements were reduced during pregnancy. Minimum alveolar concentration (MAC) values for inhalational agents were reduced by 25% to 40% in pregnant ewes23 and by 16% to 19% in pregnant rats.24 Ethical and practical difficulties with research in pregnant women delayed confirmation of this finding in humans. Isoflurane MAC (determined using transcutaneous electrical stimulation instead of the classic skin incision) was decreased by 28% in women undergoing termination of pregnancy at 8 to 12 weeks’ gestation.25 Similar reductions in MAC were found for enflurane (30%) and halothane (27%).26 MAC was reduced by 30% in the immediate postpartum period, with a return to nonpregnant values by 12 to 72 hours after delivery (Figure 14-1).27,28
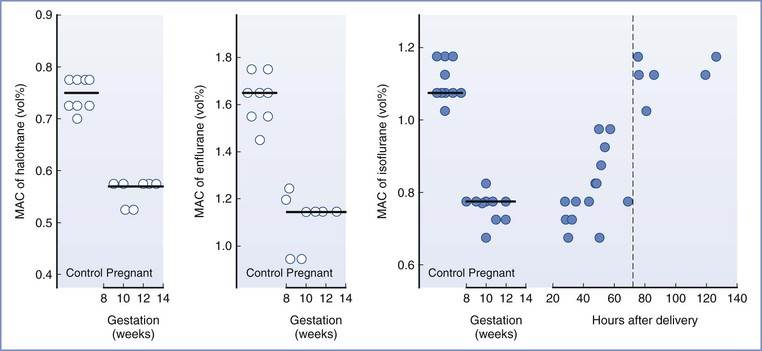
FIGURE 14-1 Changes in minimum alveolar concentration (MAC), determined by response to transcutaneous electrical stimulation, for halothane, enflurane, and isoflurane in early pregnancy and for isoflurane in the early postpartum period. (Reproduced with permission from Gin T. Obstetric pharmacology. In Evers AS, Maze M, Kharasch ED, editors. Anesthetic Pharmacology: Basic Principles and Clinical Practice, 2nd edition. Cambridge, Cambridge University Press, 2011:948-62. Original data from references 25, 26, and 27.)
Progesterone is probably the cause of the reduced anesthetic requirements during pregnancy; chronic progesterone administration reduced MAC in rabbits, dogs, and sheep.29–31 Although human studies have not found a good correlation between progesterone concentrations and the reduction in anesthetic requirement, a poor correlation may be expected if the effect of progesterone is not dose-dependent; it is possible that progesterone concentrations only need to exceed a low threshold to decrease anesthetic requirements. Lower concentrations of sevoflurane were required to maintain anesthesia in nonpregnant women during the luteal phase of the menstrual cycle, when progesterone concentrations are elevated, compared with during the follicular phase.32 The progesterone concentrations during pregnancy are much greater than those seen during the luteal phase of the menstrual cycle. The reduced MAC during pregnancy may also be a result of the increased endogenous endorphins that mediate the increase in nociceptive threshold during pregnancy; it is well known that opioids reduce MAC.
Pregnancy also alters other measures of anesthetic effect. In early pregnancy, the isoflurane concentration required for hypnosis was reduced by 31% and the bispectral index (BIS) was decreased at isoflurane concentrations over the range 0.1% to 2.0%.33 During the second trimester, the sevoflurane concentration required to achieve a targeted BIS of 50 was reduced by 31%.34 Both in early and term pregnancy, the median concentration of nitrous oxide required for loss of consciousness (MACawake) was reduced by 25% to 27%.35 One study did not show any difference in electroencephalographic (EEG) measures between women having cesarean delivery or gynecologic surgery, but there were many confounding factors such as the study being conducted partly during and partly after surgery, the concurrent use of significant doses of fentanyl, large variations in EEG measures, and small sample size.36
The data for intravenous anesthetic agents are more variable, partly because of methodologic challenges. It is difficult to produce a stable effect-site concentration of intravenous drugs to allow accurate measurement of drug effect. Increased cardiac output usually results in an increase in intravenous anesthetic dose requirements to produce central effects, and this change would counter any decrease in requirements with pregnancy. The bolus dose of thiopental for hypnosis (failure to open eyes to command) was 17% lower, and that for anesthesia (no purposeful movement to a transcutaneous electrical stimulus) was 18% lower in early pregnancy compared with nonpregnant women (Figure 14-2).37 A similar reduction was found in the early postpartum period, less than 60 hours after delivery.38
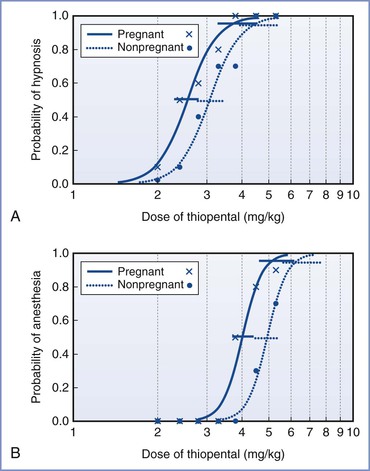
FIGURE 14-2 Calculated dose-response curves (log[dose] scale) for thiopental for hypnosis (A) and anesthesia (B) in pregnant and nonpregnant women. The 95% confidence intervals for the values of ED50 and ED95 are also displayed, slightly offset for clarity. Raw data are shown for pregnant women (x) and nonpregnant women (•). (Modified with permission from Gin T, Mainland P, Chan MTV, Short TG. Decreased thiopental requirements in early pregnancy. Anesthesiology 1997; 86:73-8.)
Studies using target-controlled infusions may not be reliable because the pharmacokinetic models may not be accurate in pregnancy, and they are known to predict concentrations poorly at induction of anesthesia. These methodologic problems may be the reason one study found no differences in the concentration of propofol required for loss of consciousness in early pregnancy.39 Another study used a slow infusion of propofol for induction of anesthesia and found that the dose and calculated effect-site concentrations at loss of consciousness were 8% lower than in nonpregnant women.40 The reduction in anesthetic requirement for intravenous agents appears to be less (8% to 18%) than that for inhalational agents (approximately 30%). It is not known whether this reflects real differences between the drugs or the methodologic problems just outlined.
Local Anesthesia
The spread of neuraxial block is increased in pregnant women (see Chapter 2). This has been shown as early as the first trimester for epidural anesthesia41 and the second trimester for spinal anesthesia.42 One small study suggested that although the spread of epidural block was increased, the latency and density of sensory and motor block were not.43 However, two more recent studies showed that the median effective dose of intrathecal bupivacaine for motor block was decreased by 13% to 35% in pregnant women at term.44,45 Magnetic resonance imaging has confirmed that pregnant women have increased epidural blood volume, decreasing the capacity of the epidural space and decreasing the volume of lumbar cerebrospinal fluid.46,47 These mechanical factors would explain the increased spread of local anesthetic. However, several studies have also shown that there is increased sensitivity to local anesthetics during pregnancy. The onset of conduction block in the vagus nerve with bupivacaine was faster in pregnant versus nonpregnant rabbits.48,49 Sciatic nerve block was of longer duration and the lidocaine content in the nerves was lower at the time of return of deep pain in pregnant versus nonpregnant rats.50 Sensory nerve action potentials were inhibited to a greater extent during median nerve block at the wrist with lidocaine in pregnant versus nonpregnant women.51 The increased sensitivity may be caused by progesterone because exogenously administered progesterone increased the susceptibility of rabbit vagus nerves to bupivacaine.52 One study found no changes in conduction block in pregnant rats and suggested that enhanced block may be due to pregnancy-induced changes that facilitate diffusion of local anesthetic or an interaction with endogenous analgesic systems.53
Analgesia
Pregnancy is associated with increases in nociceptive response thresholds that are mediated by endogenous opioid systems.54,55 The changes in threshold can be reproduced using exogenous progesterone and estrogen and appear to involve spinal cord kappa (κ) and delta (δ) opioid receptors and descending spinal α2-noradrenergic pathways.56 Early human studies produced mixed results, probably because of methodologic problems. Recent controlled studies showed that heat pain threshold was increased in term pregnant women, and this persisted during the first 24 to 48 hours after delivery.57,58 Given the many different factors that influence pain behavior, especially those unique to pregnancy and delivery, it is difficult to determine how this change in pain threshold influences perioperative analgesic requirements.
Drug Use during Pregnancy
General Teratology
Teratology is the study of abnormal development or birth defects. Teratogens are substances that act to irreversibly alter growth, structure, or function of the developing embryo.59 Ideally, preclinical studies would identify teratogens, but drug teratogenicity unfortunately can be markedly species-specific. For example, thalidomide produces phocomelia in primates but not in rodents.
In the United States, major malformations affect 2% to 3% of neonates.60 A major malformation is defined as one that is incompatible with survival (e.g., anencephaly), one that requires major surgery for correction (e.g., cleft palate, congenital heart disease), or one that causes mental retardation. If all minor malformations (e.g., ear tags, extra digits) are included, the incidence of congenital anomalies may be as high as 7% to 10%. Exogenous causes of birth defects (e.g., radiation, infections, maternal metabolic disorders, drugs, environmental chemicals) account for almost 10% of all major birth defects and therefore affect only 0.2% to 0.3% of all births. Drug exposure explains only 2% to 3% of birth defects, and the majority of birth defects are of unknown etiology.
To avoid unnecessary and potentially teratogenic exposures, nonpharmacologic techniques should be used when possible and drugs should be used only when necessary. The risk-to-benefit ratio should justify the use of a drug given to a pregnant woman, and the minimum effective dose should be employed. Long-term effects of fetal drug exposure may not become apparent for many years. Therefore, physicians and patients should exercise caution in the use of any drug during pregnancy. On the other hand, the physician should ask the following question: what would be the appropriate treatment in the nonpregnant patient with the same condition? In most cases, the answer is the same as that for women who are pregnant.61
Sensitive serum pregnancy tests can diagnose pregnancy as early as 1 week after conception. Before drug therapy is started, a sensitive test should be used if there is any question about drug safety during a potential pregnancy.
It is also important to remember that the male partner may be taking teratogenic drugs and the drug may be present in semen at low concentrations. Although the magnitude of fetal risk is unclear, men are advised to avoid drugs such as thalidomide, ribavirin, and isotretinoin if their partners could become pregnant.
The maternal and fetal genotype and phenotype can affect individual susceptibility to an agent. For example, fetuses with low levels of the enzyme epoxide hydrolase are more likely to manifest fetal hydantoin syndrome than those with normal levels of this enzyme.62
Drug teratogenicity is affected by the timing of exposure. Teratogen exposure in the first 2 weeks after conception is generally thought to be an all-or-nothing phenomenon (i.e., having either no effect or resulting in spontaneous fetal loss). Among women with a 28-day menstrual cycle, the classic period of susceptibility to teratogenic agents is during the period of organogenesis, which occurs primarily at  to 8 weeks after conception (31 to 71 days, or 4 to 10 weeks, after the first day of the last menstrual period) (Figure 14-3). During organogenesis, each organ system has different critical periods of sensitivity and there may be striking differences in effect. When administered between 35 and 37 days’ gestation, thalidomide produces ear malformations; when administered between 41 and 44 days’ gestation, it produces amelia or phocomelia. After this period, embryonic development is characterized primarily by increasing organ size; thus, the principal effect of exposure consists of growth restriction and/or effects on the nervous system and gonadal tissue. For example, diethylstilbestrol exposure during the second trimester results in uterine anomalies that do not become apparent until after puberty. Fetal alcohol syndrome may occur with chronic exposure to alcohol during pregnancy.
to 8 weeks after conception (31 to 71 days, or 4 to 10 weeks, after the first day of the last menstrual period) (Figure 14-3). During organogenesis, each organ system has different critical periods of sensitivity and there may be striking differences in effect. When administered between 35 and 37 days’ gestation, thalidomide produces ear malformations; when administered between 41 and 44 days’ gestation, it produces amelia or phocomelia. After this period, embryonic development is characterized primarily by increasing organ size; thus, the principal effect of exposure consists of growth restriction and/or effects on the nervous system and gonadal tissue. For example, diethylstilbestrol exposure during the second trimester results in uterine anomalies that do not become apparent until after puberty. Fetal alcohol syndrome may occur with chronic exposure to alcohol during pregnancy.
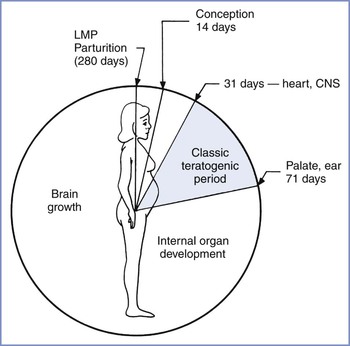
FIGURE 14-3 Gestational clock showing the classic teratogenic period. CNS, central nervous system; LMP, day of last menstrual period. (From Niebyl JR. Drug Use in Pregnancy. 2nd edition. Philadelphia, Lea & Febiger, 1988:2.)
The drug dosing regimen can influence teratogenicity. In most cases, administration of a low dose has no effect whereas malformations may occur at intermediate doses and death may occur at higher doses. Fetal death may allow organ-specific teratogenic activity to go unnoticed. Small doses administered over several days may have an effect different from that observed with the same total dose administered at one time. Sequential drug administration may induce the production of an enzyme that metabolizes the drug and thus results in less exposure. Constant exposure may destroy cells that would have catabolized the drug if it had been administered in periodic doses. Combinations of agents may produce different degrees of malformation and growth restriction from those that occur with drugs administered individually. For example, fetuses whose mothers receive combination anticonvulsant therapy are at the highest risk for malformations, including neural tube defects and facial dysmorphic features.
U.S. Food and Drug Administration Categories
In 1979, the U.S. Food and Drug Administration (FDA) introduced a drug classification system to discourage nonessential use of medications during pregnancy (Box 14-1).
Unfortunately, maternal anxiety related to medication use can lead to unnecessary pregnancy terminations. Several characteristics of the FDA drug classification system contribute to public perception—and misperception—of the dangers of using medication during pregnancy. Although only 20 to 30 commonly used drugs are known teratogens, 7% of all the medications that are listed in the Physicians’ Desk Reference are classified as Category X.63,64 All new medications are classified as Category C, leading to an exaggerated impression of the danger of many medications. In addition, the manufacturer’s prescribing information for many drugs may state that the drug is not approved for use in pregnancy despite a long history of uncomplicated unlicensed or “off-label” use.65
The FDA categories imply a progressive fetal risk from Category A to X; however, the drugs in different categories may pose similar risks but may be listed in different categories on the basis of risk-to-benefit considerations. In addition, the categories create the impression that drugs within a category present similar risks whereas the category definition permits inclusion (in the same category) of drugs that vary in type, degree, and extent of risk.
Use of potentially teratogenic drugs in pregnancy is surprisingly commonplace. Over 63% of pregnant patients in Canada filled a prescription for at least one drug, with almost 8% filling a prescription for a Category D or X medication.66
When counseling patients or responding to queries from physicians, we prefer to avoid referring to the Physicians’ Desk Reference. Rather, we use specific descriptions in teratogen databases to provide the best information available. Many resources are freely available online, in addition to the commercially available databases (Table 14-1).
TABLE 14-1
Internet Resources for Additional Drug and Teratogen Information
| American Academy of Pediatrics: The Transfer of Drugs and Other Chemicals into Human Milk | http://pediatrics.aappublications.org/content/108/3/776.full.html |
| The American Botanical Council | http://www.herbalgram.org |
| The American College of Obstetricians and Gynecologists | http://www.acog.org/About_ACOG/ACOG_Departments/Resource_Center/WEBTREATS_Teratology_Toxicology |
| Motherisk | http://www.motherisk.org |
| Organization of Teratology Information Specialists: Fact sheets on exposure during pregnancy to a variety of diseases, medications, and herbal remedies | http://otispregnancy.org/otis_fact_sheets.asp |
| The National Library of Medicine PubMed | http://www.ncbi.nlm.nih.gov/sites/entrez?db=pubmed |
| The National Institutes of Health National Center for Complementary and Alternative Medicine | http://nccam.nih.gov |
| The National Institutes of Health Office of Dietary Supplements | http://dietary-supplements.info.nih.gov |
| Perinatology.com: Drugs in Pregnancy and Breastfeeding | http://www.perinatology.com/exposures/druglist.htm |
| The Reproductive Toxicology Center* | http://www.reprotox.org |
| RxList: The Internet Drug Index | http://www.rxlist.com |
| SafeFetus.com | http://www.safefetus.com |
| University of Washington Clinical Teratology Web* | http://depts.washington.edu/~terisweb |
| U.S. Food and Drug Administration Office of Women’s Health | http://www.fda.gov/AboutFDA/CentersOffices/OC/OfficeofWomensHealth/ |
| Drugs and Lactation Database (LactMed) | http://toxnet.nlm.nih.gov/cgi-bin/sis/htmlgen?LACT |
* Databases, including Reprotox, Reprotext, Teris, and Shepard’s Catalog of Teratogenic Agents, can be purchased from these websites.
The Teratology Society has suggested abandonment of the FDA classification scheme.63 In 1997, the FDA held a public meeting to discuss labeling of drugs. There was consensus that the current classification scheme is probably oversimplified and confusing, does not address the range of clinical situations or the range of possible effects, and should be replaced with narrative labeling. Subsequently, a concept paper was presented that outlined a new model for labeling and included sections such as “clinical management statement,” “summary risk assessment,” and “discussion of data” for both pregnant and breast-feeding women.67 This proposal has not yet been implemented. The FDA Office of Women’s Health has created a pregnancy registry website, which lists a variety of registries that women who have used specific medications during pregnancy can consult (see Table 14-1). In 2008, the FDA stated they will eliminate the A, B, C, D, X classification system.68 As of 2013, the new system has yet to be implemented despite completion of the 90-day comment period some time earlier.69
Analgesics
There is no known teratogenic risk associated with the use of acetaminophen (paracetamol),70 which is the preferred mild analgesic or antipyretic during pregnancy.
Nonsteroidal anti-inflammatory drugs (NSAIDs) have not been associated with an increased risk for birth defects overall, but the occasional review has suggested an association with specific defects.71 For example, aspirin is not associated with an overall increase in rates of congenital malformations, but one review suggested a higher risk for gastroschisis.72 Aspirin is not usually the first choice of NSAID, but low-dose aspirin may still have a role in some situations, such as preventing fetal loss associated with antiphospholipid antibody syndrome.73 Studies of the use of NSAIDs and aspirin in the first trimester have reported increased risk for pregnancy loss (adjusted odds ratio [OR], 1.8 to 8.1).71 In the second trimester, their use has been associated with fetal cryptorchidism. In the third trimester, NSAIDs and aspirin are usually avoided because of significant fetal risks, such as renal injury, oligohydramnios, and intrauterine constriction of the ductus arteriosus, an effect that increases with advancing gestational age.74 Renal injury, necrotizing enterocolitis, and intracranial hemorrhage are other potential complications.
Opioids such as propoxyphene and codeine have no known teratogenic risk,75 but they have well-known potential for addiction. Excessive antepartum use can also lead to neonatal opioid-withdrawal symptoms.76 Surprisingly, a recent case-control study found that maternal opioid treatment (mostly codeine and hydrocodone) between 1 month before pregnancy and the first trimester was associated with an increased OR of 1.8 to 2.7 for various cardiac birth defects, spina bifida, and gastroschisis.77
Tramadol has analgesic effects from weak opioid activity and inhibition of serotonin and norepinephrine uptake. Despite availability in some countries for more than 30 years, few data are available regarding potential adverse effects in pregnancy. Tramadol exposure in early pregnancy was associated with a higher number of spontaneous abortions, and it should be avoided in the first trimester.78 Chronic tramadol use in later pregnancy may result in neonatal withdrawal syndrome.
Sedatives
Human epidemiologic studies of the possible teratogenic effects of various tranquilizers are inconsistent. One report of nearly 400 patients found a 12% incidence of birth defects in the offspring of meprobamate users.79 Another study of similar size did not identify any higher risk for malformations.80 These latter two articles found similar results for chlordiazepoxide.79,80 A recent study evaluating meprobamate used in high doses to attempt suicide did not show any teratogenic or fetotoxic effects.81 A sample of 35 pregnant women who self-poisoned with chlordiazepoxide showed no association with congenital abnormalities but did show dose-related fetal growth restriction.82
Some studies have suggested that first-trimester exposure to diazepam increases the risk for cleft lip with or without cleft palate, neural tube defects, intestinal atresia, and limb defects.83 Other reports have not suggested an increase in rate of congenital abnormalities after fetal exposure to benzodiazepines. In a case-control study of 611 infants with cleft lip or cleft palate and 2498 controls with other birth defects, after adjustment of the data for potential confounders, no association between diazepam and cleft palate was found (OR, 0.8 for cleft lip with or without cleft palate, with 95% confidence interval [CI] of 0.4 to 1.7, and OR, 0.8 for cleft palate alone, with 95% CI of 0.2 to 2.5).84 Reanalysis of the Hungarian Case-Control Surveillance of Congenital Abnormality data also showed a weak relationship between exposure to diazepam in early pregnancy and neural tube defects, limb deficiency defects, and possibly intestinal atresia or stenosis.83 One study found no difference in the incidence of congenital anomalies between 460 women exposed to benzodiazepines during pregnancy and 424 control women without such exposure (i.e., 3.1% versus 2.6%, respectively).84 Perinatal use of diazepam has been associated with hypotonia, hypothermia, and respiratory depression.85 Overall, it appears that the teratogenic risk of benzodiazepines is small at most,86,87 but there may be a small risk for preterm birth and low birth weight.88 Benzodiazepines should only be used in the first trimester if the perceived benefit offsets the possible teratogenic risks and later neonatal effects of continued use. Some benzodiazepines such as temazepam are classified as FDA Category X.
Anticonvulsants
Epilepsy is the most common serious neurologic problem during pregnancy.89 It has been estimated that 3 to 5 births per thousand will be to women with epilepsy.90 All anticonvulsants cross the placenta. The fetal congenital anomaly rate in pregnant women with epilepsy who ingest anticonvulsant drugs is 4% to 8%, compared with a background incidence in the general population of 2% to 3%.91,92 A twofold higher risk for minor malformations also exists in this population.92 Cleft lip, with or without cleft palate, and congenital heart disease are especially common. Administration of valproic acid or carbamazepine entails a 1% risk for neural tube defects and other malformations; thus, alpha-fetoprotein screening and targeted ultrasonography are appropriate for patients taking these agents. In addition, the offspring of epileptic women have a 2% to 3% incidence of epilepsy, which is five times that of the general population.
Holmes et al.93 attempted to refute the unproven theory that women with epilepsy have a genetic propensity to have children with a higher risk for birth defects that is separate from the greater risk associated with the use of anticonvulsants. These investigators studied children of women who had a history of seizures but took no medications during the pregnancy. There was no difference in physical features or cognitive function between these children and a group of matched controls. In a second study, Holmes et al.94 screened 128,049 pregnant women at delivery to identify the following three groups of infants: (1) those exposed to anticonvulsant drugs, (2) those unexposed to anticonvulsant drugs but with a maternal history of seizures, and (3) those unexposed to anticonvulsant drugs with no maternal history of seizures (control group). The frequency of anticonvulsant embryopathy was higher in the 223 infants exposed to one anticonvulsant drug than in the 508 control infants (20.6% versus 8.5%, respectively; OR, 2.8; 95% CI, 1.1 to 9.7). The frequency was also higher in 93 infants exposed to two or more anticonvulsant drugs than in the controls (28.0% versus 8.5%; OR, 4.2; 95% CI, 1.1 to 5.1). The 98 infants whose mothers had a history of epilepsy but took no anticonvulsant drugs during pregnancy did not have a higher frequency of abnormalities than the control infants. The investigators concluded that “a distinctive pattern of physical abnormalities in infants of mothers with epilepsy is associated with the use of anticonvulsant drugs during pregnancy, rather than with epilepsy itself.” A more recent meta-analysis also did not support the view that epilepsy per se represents a teratogenic risk.95
Possible causes of these congenital malformations include genetic differences in drug metabolism, the specific drugs themselves, and deficiency states (e.g., decreased folate levels) induced by the drugs. No congenital malformations appear to be unique to any one anticonvulsant. The characteristics of these syndromes are so similar that the broad term fetal anticonvulsant syndrome, consisting primarily of orofacial, cardiovascular, and digital malformations, has been applied to almost every anticonvulsant drug.96
Among women taking phenytoin, there is a 2% to 5% risk for major congenital anomalies, primarily midline heart defects, orofacial clefts, and urogenital defects.91 Fetal hydantoin syndrome is a constellation of minor anomalies, such as craniofacial abnormalities (short nose, flat nasal bridge, wide lips, hypertelorism, ptosis, epicanthal folds, low-set ears, and low hairline) and limb anomalies (distal digital hypoplasia, absent nails, and altered palmar crease). In addition, neonatal growth and performance delays have been documented. The risk for fetal hydantoin syndrome for the child of a woman taking phenytoin is approximately 10%.94 Phenytoin may act as a competitive inhibitor of the placental transport of vitamin K. This results in a decrease in fetal coagulation factors II, VII, IX, and X. In addition, phenytoin may induce fetal hepatic metabolism of the coagulation factors. The resulting reduction in fetal coagulation factors is associated with a higher risk for hemorrhagic disease of the newborn.97 To help prevent this coagulopathy, some physicians advocate oral vitamin K supplementation (10 mg daily) for pregnant epileptic patients during the last month of pregnancy in addition to the parenteral administration of vitamin K to the neonate at birth.98 Several anticonvulsant medications have metabolites that typically are eliminated by the enzyme epoxide hydrolase. In one study, 19 women taking phenytoin underwent amniocentesis. All 4 of the women with low enzyme activity in amniocytes had affected fetuses. The 15 fetuses with normal amniocyte epoxide hydrolase activity did not have the characteristics of fetal hydantoin syndrome.62
Carbamazepine is used to treat all types of seizure disorders, with the exception of petit mal epilepsy. It is most commonly used in the treatment of psychomotor (temporal lobe) epilepsy and grand mal epilepsy. In a prospective study involving 72 women with epilepsy who were taking carbamazepine, the incidence of congenital anomalies was higher in the 35 fetuses exposed only to this drug. There was an 11% incidence of craniofacial defects, a 26% incidence of fingernail hypoplasia, and a 20% incidence of developmental delay.99 This constellation of fetal effects, named fetal carbamazepine syndrome, closely resembles the malformations seen in cases of fetal hydantoin syndrome. In addition, maternal carbamazepine exposure has been specifically associated with spina bifida. An analysis of all available data involving cohorts of pregnant women ingesting carbamazepine supports the conclusion that fetal exposure to this drug carries a 0.5% to 1% risk for spina bifida.100 Although it is generally agreed that the use of carbamazepine in pregnancy is associated with a risk for neural tube defects and other anomalies, the exact magnitude of the risk from use of carbamazepine alone is unclear.101–104
Phenobarbital is used in the treatment of partial and generalized tonic-clonic seizures and status epilepticus.105 Fetal exposure to phenobarbital has been associated with major malformations, such as congenital heart defects and orofacial clefting. Fetal phenobarbital syndrome is characterized by minor dysmorphic features similar to those seen with fetal hydantoin syndrome.91 Fetal exposure to phenobarbital has also been associated with decreased intellectual and cognitive development in neonates and children. Maternal phenobarbital use during pregnancy can result in hemorrhagic disease of the newborn and neonatal withdrawal symptoms after delivery. The withdrawal symptoms consist mostly of irritability, begin at about 7 days of life, and usually last for 2 to 6 weeks.105
Valproic acid is used to treat absence and generalized tonic-clonic seizures. Infants exposed to valproic acid have a 1% to 2% risk for spina bifida. The neural tube defect tends to be lumbosacral. Fetal valproic acid exposure has also been associated with cardiac defects, orofacial clefting, and genitourinary anomalies. Fetal valproate syndrome has been described; it is characterized by dysmorphic features, including epicanthal folds, shallow orbits, hypertelorism, low-set ears, flat nasal bridge, upturned nasal tip, microcephaly, thin vermillion borders, downturned mouth, thin overlapping fingers and toes, and hyperconvex fingernails.91 Jentink et al.106 found a markedly increased risk for spina bifida (OR, 12.7) craniosynostosis, cleft palate, atrial septal defect, and hypospadias. They concluded that valproic acid monotherapy should be avoided during pregnancy. A report of the Quality Standards Subcommittee and Therapeutics and Technology Subcommittee of the American Academy of Neurology and the American Epilepsy Society concluded that valproic acid and polytherapy should be avoided if possible during the first trimester to decrease the risk for major congenital malformations.107 They also concluded that, if possible, avoidance of valproic acid and polytherapy throughout pregnancy should be considered, and avoidance of phenytoin and phenobarbital throughout pregnancy may be considered, to prevent poor cognitive outcomes.107 Other reviews have also confirmed that valproic acid and carbamazepine are associated with neural tube defects and that valproic acid is associated with hypospadias, thus lending further support to limiting its use in women of reproductive age.
There are insufficient data to establish risks for birth defects with the newer anticonvulsants.108,109 Felbamate was approved by the FDA for monotherapy, but later its use was severely restricted because of its association with aplastic anemia and hepatic failure. Gabapentin was initially released in the United States as an adjunctive treatment for partial seizures and secondarily generated tonic-clonic seizures. This agent inhibits dopamine release in the CNS. Lamotrigine appears to have efficacy comparable to that of carbamazepine in the monotherapy for partial epilepsy. An inhibitor of dihydrofolate reductase, lamotrigine decreases embryonic folate levels in experimental animals. This finding raises the concern that human use of lamotrigine may result in developmental toxicity. The manufacturer has established a registry to evaluate this possibility. A preliminary report from this registry has described a 6% rate of congenital malformations in fetuses exposed to this drug; this rate does not represent a clear increase in the baseline rate of malformations. However, there have not been sufficient numbers of fetal exposures to support any definitive conclusions.110–112
Many patients who present to prenatal clinics are already taking these newer anticonvulsants. Patients should be counseled that even though little information is available, no clear evidence of their teratogenicity exists. Some investigators have suggested avoiding the newer anticonvulsants until evidence of their safety is accumulated.105 Even in the two decades since newer agents such as lamotrigine were introduced, definitive evaluation of their teratogenic potential has not been published.
Some women may be taking anticonvulsant drugs without having been reevaluated recently for their need to continue drug therapy. If a patient with idiopathic epilepsy has been seizure-free for 2 years and has normal EEG findings, the neurologist may try to withdraw the drug before pregnancy.113
If a patient first presents for care during pregnancy, most authorities agree that the benefits of anticonvulsant therapy during pregnancy outweigh the risks of discontinuing the drug. The blood level of the drug should be monitored to minimize the dose needed to ensure a therapeutic level of drug.
Antidepressants
A summation of 14 studies assessing the effect of fetal exposure to tricyclic antidepressants evaluated 414 cases of first-trimester exposure114; when the study data were pooled or viewed individually, no significant association between fetal exposure to tricyclic antidepressants and congenital malformations was found.114 In a surveillance study of Michigan Medicaid recipients,115 467 neonates had been exposed to amitriptyline and 75 neonates had been exposed to imipramine during the first trimester; there was no association between tricyclic antidepressant use and congenital anomalies. However, a large review of data from the Swedish Medical Birth Register reported that use of tricyclic antidepressants was associated with a greater risk for congenital malformations compared with other antidepressants.116 Results of the latter study are making their way into reviews that state a concern about tricyclic antidepressants.117 Overall, tricyclic antidepressants have been replaced by selective serotonin reuptake inhibitors (SSRIs) as first-line therapy for depression (see Chapter 51).118
The SSRIs include sertraline, paroxetine, fluoxetine, and citalopram. No higher risk for major malformations or developmental (language and behavior) abnormalities was identified with their use in earlier studies.119–121 In a multicenter evaluation of birth defects, use of SSRIs was not associated with a higher risk for cardiac defects.122 However, in analyses of the individual medications, sertraline was associated with an increased risk for septal defects (OR, 2.0; 95% CI, 1.2 to 4.0), and paroxetine was associated with a higher risk for right ventricular outflow tract obstruction (OR, 3.3; 95% CI, 1.3 to 8.8). Sertraline was also associated with an increased risk for omphalocele, but this association was based on only three subjects. Another study found significantly higher risks for craniosynostosis, omphalocele, and anencephaly in association with exposure to SSRIs as a group; this study also found an association between paroxetine and right ventricular outflow tract lesions.123 The association of septal heart defects with sertraline124,125 and citalopram has been seen in other studies.124
A cohort study compared postnatal outcome for infants exposed to fluoxetine in late gestation (up to the time of delivery) with that for infants whose exposure was limited to the first trimester.126 Infants exposed in the third trimester had a greater incidence of perinatal complications, including preterm delivery, admission to the special care nursery, poor neonatal adaptation, lower mean birth weight, and shorter body length.126 A subsequent study suggested an association between the maternal use of SSRIs in late pregnancy and a higher risk for persistent pulmonary hypertension of the newborn (PPNH).127 The association between SSRIs and PPHN has been confirmed in other studies, which showed an increase from the background rate of 1.2/1000 neonates to about 3/1000, and appears to be a class effect.128
The American College of Obstetricians and Gynecologists (ACOG) has recommended that use of paroxetine in pregnant women (and in women planning pregnancy) be avoided, if possible.129 In addition, it was suggested that fetal echocardiography should be considered in women who have used paroxetine during early pregnancy. Further, the ACOG has stated that treatment with all SSRIs should be individualized.129 A report from the American Psychiatric Association and the ACOG concluded that although antidepressant use in pregnancy is well studied, available research has not yet adequately controlled for other factors such as maternal illness and behaviors that can adversely affect pregnancy.130
Lithium
In the International Registry of Lithium Babies, 25 of 217 (11.5%) infants exposed to lithium during the first trimester of pregnancy were malformed.131 Eighteen infants had cardiovascular anomalies, and six had the rare Ebstein anomaly, which occurs only once in 20,000 nonexposed pregnancies. Subsequent studies have suggested that ascertainment bias may have flawed the findings; the reported risk for anomalies after lithium exposure is much less than that reported by the registry.
In a cohort study linking the Swedish Birth Registry with the records of women with bipolar disorder, 59 infants were identified whose mothers had been treated with lithium early in pregnancy.132 Four (6.8%) of the 59 infants exposed to lithium had congenital heart disease, compared with 2 (0.9%) of 228 infants not exposed to lithium (relative risk [RR], 7.7; 95% CI, 1.5 to 41.2). None of the infants had Ebstein anomaly. In a prospective cohort study of 148 women treated with lithium during the first trimester and 148 controls not exposed to any known teratogen, there was no significant difference between groups in the incidence of major congenital anomalies or cardiac malformations,132 although one lithium-exposed infant did have Ebstein anomaly. The investigators concluded that lithium is not a major human teratogen, but they recommended that women exposed to lithium be offered ultrasonography and fetal echocardiography.133 If the data are pooled, these two cohort studies do not suggest a statistically significant increase in risk for congenital malformations or cardiac malformations in women exposed to lithium during pregnancy. Although the risk for congenital malformations associated with intrauterine lithium exposure is likely to be lower than previously reported, an absence of risk cannot be assumed from the available data. Recent reviews of available data have come to the same conclusion that the risk for anomalies is small, but it is prudent to perform fetal echocardiography after first-trimester exposure.134
Two published cases associated polyhydramnios with maternal lithium treatment.135,136 Nephrogenic diabetes insipidus has been reported in adults taking lithium; thus, the presumed mechanism of polyhydramnios is fetal diabetes insipidus. Polyhydramnios may signal fetal lithium toxicity.
Pregnancy accelerates the excretion of lithium, so serum lithium levels should be monitored in pregnant women.137 Perinatal effects of lithium include hypotonia, lethargy, and poor feeding in the infant. In addition, complications (e.g., goiter, hypothyroidism) similar to those seen in adults taking lithium have been noted in neonates.
Some authorities recommend discontinuation of lithium and substitution with another medication during pregnancy. However, the discontinuation of lithium is associated with a 70% chance of relapse of the affective disorder in 1 year, as opposed to a 20% risk for relapse with continuation of lithium therapy.131
Cardiovascular Drugs
There are no reports of teratogenicity related to the use of inotropic agents such as dopamine, dobutamine, or digoxin. Physicians should monitor the maternal digoxin level to ensure a therapeutic level of drug during pregnancy.
Methyldopa and labetalol are often used to treat mild chronic hypertension during pregnancy. There is no evidence of teratogenicity or other adverse fetal effects with either of these drugs. In one randomized double-blind trial of 152 women with hypertension, there were no malformations in either the labetalol group or the placebo group, although the exposure to labetalol occurred in the second and third trimesters.138
In a large analysis of published trials involving beta-adrenergic receptor antagonist therapy, there was little or no information on teratogenicity for the multiple agents studied, including atenolol, labetalol, metoprolol, oxprenolol, pindolol, and propranolol.139 Maternal administration of propranolol may result in modest fetal growth restriction.140 It seems prudent to use ultrasonography to assess intrauterine fetal growth in women receiving propranolol. Maternal administration of propranolol within 2 hours of delivery may result in neonatal bradycardia.75 Atenolol was associated with lower birth weight and a trend toward more frequent preterm delivery than with other antihypertensive drugs and with no therapy. These effects were more pronounced when the drug was administered earlier in pregnancy and for a long duration.141 In one study, treatment of hypertension (mostly with atenolol) reduced the risk for severe hypertension and preterm labor.141 In a randomized clinical trial, the same group observed that atenolol prevented preeclampsia but resulted in the birth of infants who weighed 440 g less than infants in the placebo group.142
The angiotensin-converting enzyme (ACE) inhibitors initially did not appear to be teratogenic when administered during the first trimester.143 However, a 2007 study suggested an increase in the incidence of congenital anomalies, particularly cardiac and CNS defects, after first-trimester exposure to ACE inhibitors.144 Some authorities contend that this study had major limitations.145 A very large epidemiologic database study of 465,754 mother-infant pairs showed no difference in abnormality rate between women who used ACE inhibitors during the first trimester, those who used other antihypertensive agents, and those who had hypertension but used no medications.146 The authors postulated that any increased risk for birth defects may be due to the hypertension. A different set of investigators made the same conclusion.147 Later in pregnancy, ACE inhibitors can cause fetal renal failure and oligohydramnios, which may result in fetal limb contractures, craniofacial deformities, and pulmonary hypoplasia148; therefore, use of ACE inhibitors should be avoided if possible during this period.
Amiodarone is structurally similar to thyroxine and contains 37% iodine by weight. In a review of 64 reported pregnancies in which amiodarone was administered to the mothers, there was no clear increase in the incidence of malformations. However, 11 (17%) infants had evidence of hypothyroidism, and two (3%) neonates had goiter.149,150 The use of amiodarone in pregnancy was associated with mild abnormalities of neurodevelopment in some of the hypothyroid infants, although this was also observed in some euthyroid infants.150 This agent is most often used during pregnancy to treat fetal arrhythmias, and first-trimester exposure is rare.
Respiratory Drugs
Maternal asthma is relatively common in pregnancy, affecting 4% to 8% of pregnant women. It is important to maintain appropriate treatment because asthma can increase the risk for adverse fetal and maternal outcomes.151
Inhaled β2-adrenergic receptor agonists, cromolyn sodium, and corticosteroids have not been associated with congenital malformations. Currently, albuterol is the preferred short-acting β2-receptor agonist, and salmeterol is the preferred long-acting β2-receptor agonist. Budesonide is the recommended inhaled corticosteroid because it has a long history of safe usage, but there is no evidence against the other options, which include beclomethasone, ciclesonide, fluticasone, flunisolide, mometasone, and triamcinolone.
Severe persistent asthma may require systemic oral corticosteroid therapy, and this has been associated with low birth weight and a three- to five-fold increase in the relative risk for cleft lip and palate.152,153 However, other studies have found no adverse effects and, given the risks from severe asthma, oral corticosteroids should be used if required.
The 5-lipoxygenase inhibitors and leukotriene receptor antagonists are newer agents with no evidence of teratogenicity.154 Women using montelukast enrolled in several teratogen information services around the world showed no increased risk for birth defects in 160 live births.155
Methylxanthines such as theophylline and aminophylline have no adverse fetal effects, but the protein binding and metabolism of theophylline are both reduced during pregnancy, making it necessary to monitor drug concentrations and adjust maintenance doses.
Anticoagulants
Warfarin use in early pregnancy can result in an embryopathy similar to the X-linked chondrodysplasia punctata. The embryopathy can occur with fetal exposure between 6 and 12 weeks’ gestation. Because deficiency of arylsulfatase E is responsible for the chondrodysplasia, the embryopathy may result from inhibition of arylsulfatase E by warfarin. The period between 6 and 9 weeks’ gestation is especially critical. Fetal warfarin syndrome consists of nasal hypoplasia, depressed nasal bridge (often with a deep groove between the alae and nasal tip), stippled epiphyses, nail hypoplasia, mental retardation, and growth restriction. Second- and third-trimester exposures can result in other adverse fetal effects, including microcephaly, blindness, deafness, and growth restriction. Three prospective studies of women exposed only in the second and third trimesters found that the incidence of malformations must be exceedingly low because there was no evidence of fetal or neonatal CNS or eye abnormalities.156 Some evidence suggests that a lower dose (< 5 mg/day) has less teratogenic potential.157
Heparin is a large, water-soluble molecule that does not cross the placenta. Maternal administration does not have an adverse effect on the fetus, and heparin is the drug of choice for most pregnant women who require anticoagulation daily. Daily administration of 20,000 units of standard unfractionated heparin for more than 20 weeks may be associated with maternal bone demineralization.158 Unfractionated heparin should be used for prolonged periods only when it is clearly necessary.
Low-molecular-weight heparin (LMWH) has some advantages over standard unfractionated heparin.159 LMWH does not cross the placenta and has a longer half-life than standard unfractionated heparin, which typically allows once-daily dosing in nonpregnant patients. In addition, LMWH has a more predictable dose-response relationship in nonpregnant patients, which obviates monitoring. However, pregnancy is associated with a higher volume of distribution and accelerated clearance of LMWH. Use of LMWH thromboprophylaxis during pregnancy requires adjustments in the dose of LMWH to accommodate for the changes in the pharmacokinetics of this drug that occur during pregnancy. For example, pregnant women may require twice-daily doses.160
Full anticoagulation is necessary in pregnant women with cardiac valve prostheses. In a systematic review of anticoagulation in pregnant women with mechanical heart valves, Chan et al.161 evaluated outcomes with the following three anticoagulation regimens: (1) oral anticoagulants (most commonly warfarin) given throughout pregnancy, (2) heparin administered during the first trimester and then warfarin for the duration of pregnancy, and (3) heparin administered throughout pregnancy. The data demonstrated progressively higher rates of maternal death with regimens 1, 2, and 3 (1.8%, 4.2%, and 15.0%, respectively). The use of warfarin throughout pregnancy was associated with warfarin embryopathy in 6.4% of live-born infants. The substitution of heparin at or before 6 weeks’ gestation eliminated that risk.161
For women with prosthetic heart valves, the American College of Chest Physicians (ACCP) has recommended use of one of the following three regimens: (1) adjusted-dose LMWH twice-daily throughout pregnancy, with doses adjusted to achieve the manufacturer’s peak anti–factor Xa LMWH levels 4 hours after subcutaneous injection; (2) adjusted-dose unfractionated heparin administered subcutaneously every 12 hours throughout pregnancy, with doses adjusted to keep the mid-interval activated partial thromboplastin time at least twice control or to attain an anti–factor Xa heparin level of 0.35 to 0.70 units/mL; or (3) unfractionated heparin or LMWH as just described until the 13th week of pregnancy, with substitution by vitamin K antagonists until close to delivery, when unfractionated heparin or LMWH is resumed.160 In women at very high risk for thromboembolism in whom there is concern about the efficacy and safety of unfractionated heparin or LMWH, it is suggested to administer vitamin K antagonists throughout pregnancy with replacement by unfractionated heparin or LMWH close to delivery.160 In high-risk women with prosthetic heart valves, it is suggested to add low-dose aspirin 75 to 100 mg/day.160
Antiemetics
Most women in early pregnancy have nausea, with or without vomiting (see Chapter 2 and Chapter 16).162 Complementary therapies such as acupressure and ginger may be effective, but approximately 10% of women will require drug therapy. Although many different drugs can be effective, vitamin B6 should be the drug of choice.163 A combination of vitamin B6 with the antihistamine doxylamine was previously available as Bendectin, but this was withdrawn by the manufacturer in 1983 because of allegations of teratogenicity that were later found to be false. Other formulations are now available, and there is no evidence of teratogenicity with them or other antihistamines. Trimethobenzamide is structurally related to antihistamines such as diphenhydramine, but it has only weak antihistamine activity. The mechanism of action is unclear, but it probably acts at the chemoreceptor trigger zone. It has few side effects but also variable efficacy. Nausea and vomiting can also be treated with phenothiazines, but they may cause sedation.
The prokinetic agent and dopamine antagonist metoclopramide is not sedating, but it has a “black box” warning because chronic usage has been associated with rare cases of tardive dyskinesia. There are no specific safety concerns during pregnancy. A multicenter study did not observe an increased incidence of anomalies,164 and a large trial of 3458 women exposed to metoclopramide in the first trimester found no association with increased risk for birth defects, low birth weight, preterm delivery, or perinatal death.165
The 5-hydroxytryptamine receptor antagonists are widely used and very effective antiemetics with few side effects, so they are also increasingly being used in pregnancy. Ondansetron was similar in efficacy to promethazine for the treatment of hyperemesis gravidarum but less sedating.166 There is no evidence that it has a teratogenic effect.167
Corticosteroids such as dexamethasone are now commonly used for postoperative and chemotherapy-induced nausea and vomiting. Methylprednisolone has been used for refractory nausea and vomiting during pregnancy. However, a meta-analysis concluded that the use of glucocorticoids before 10 weeks’ gestation was associated with a threefold to fivefold increased risk for cleft lip with or without cleft palate152; and thus, they should not be used during this period.
Antihistamines
Some pregnant patients may be treated with antihistamines for allergies or upper respiratory tract infections. These drugs provide symptomatic therapy with no influence on the course of the disease. If considered necessary, combinations of drugs should be avoided if possible. Topical nasal sprays result in less fetal exposure than systemic medication. One study suggested an association between pseudoephedrine and defects attributable to vascular disruption, including gastroschisis, small intestinal atresia, and hemifacial microsomia.168 Physicians should discourage the use of nonprescription drugs for trivial indications because the long-term fetal effects of the chronic maternal use of these drugs are unknown.
Sedating or first-generation antihistamines are available in over-the-counter medications and have not been reported to increase fetal risk.169 Examples include chlorpheniramine, diphenhydramine, methapyrilene, thonzylamine, pyrilamine, tripelennamine, phenyltoloxamine, and buclizine. Despite conflicting reports, a meta-analysis found no evidence to implicate brompheniramine as a teratogen.170 In the Boston Collaborative Program,171 none of the sedating antihistamines was associated with malformations. Two combination products—triprolidine with pseudoephedrine (Actifed) and phenylpropanolamine with chlorpheniramine (Ornade)—were not associated with malformations. In a cohort of 1502 women, antihistamines were not associated with congenital malformations.172 Azatadine was not found to be teratogenic among 127 Michigan Medicaid recipients.115
Limited safety information is available for the nonsedating antihistamines. In a cohort study, 114 women exposed to astemizole were matched with 114 women exposed to known nonteratogens.173 There were two major malformations in the astemizole group and two in the control group. In a study of 39 women exposed to cetirizine, the rate of malformations was no higher than in a control group.174 This finding was replicated in a study performed through a teratogen information service.175 There are no controlled human studies for loratadine or fexofenadine. Desloratadine is the major metabolite of loratadine; most data do not point to a risk for congenital abnormalities.169
Several antihistamines have primary indications not directly related to upper respiratory complaints. Hydroxyzine is used for treatment of pruritus, meclizine for dizziness, diphenhydramine for sleep and pruritus, and doxylamine (a component of the former Bendectin) for treatment of nausea and vomiting of pregnancy. A meta-analysis of antihistamines used mostly for morning sickness in early pregnancy found a protective effect against malformations (OR, 0.76; 95% CI, 0.60 to 0.94).176 This apparent benefit may have resulted from an association between maternal nausea and good fetal outcomes rather than from a direct effect of antihistamines.
Anti-infective Drugs
Sepsis is a leading cause of maternal mortality. When administering perioperative antibiotics, anesthesia providers should be aware of the considerations regarding their use during pregnancy.177 Although penicillins and cephalosporins are considered first-line treatment because of their long safety record, therapy is ultimately guided by local microbiology policies and bacterial sensitivity.
Tetracyclines should not be administered after the fifth week of pregnancy. They bind to developing enamel and cause discoloration of the teeth. They affect deciduous teeth when administered between approximately 26 weeks’ gestation and 6 months of age in the infant, and they affect permanent teeth only if administered to children between approximately 6 months and 5 years of age. In addition, tetracyclines deposit in developing osseous sites and inhibit bone growth beginning in the second trimester.177
Quinolones (e.g., ciprofloxacin, norfloxacin) should not be used in pregnancy or in children. They have a high affinity for bone tissue and cartilage and may cause arthropathies in children. However, no malformations or musculoskeletal problems were noted in 38 infants exposed during the first trimester.177
Malaria is a significant cause of maternal and fetal death. Pregnant women should avoid traveling to malaria-endemic areas, but chloroquine or mefloquine can be used for malaria chemoprophylaxis. They are not associated with an increase in spontaneous abortions or congenital malformations. Doxycycline, primaquine, and atovaquone-proguanil are not to be used in pregnancy.
Treatment of herpes simplex and herpes zoster infections in the first trimester with acyclovir or valacyclovir has not been associated with an increased risk for birth defects.178
Zidovudine has been studied because of its role in the treatment of acquired immunodeficiency syndrome (AIDS). In a prospective cohort study, children exposed to it during the perinatal period in the Pediatric AIDS Clinical Trials Group Protocol 076 (PACTG 076) were studied to a median age of 4.2 years; no adverse effects were observed.179 Combination antiretroviral therapy has not been associated with major infant toxicity, even when the therapy was initiated in the first trimester of pregnancy.180
The Antiretroviral Pregnancy Registry was established in 1989 to detect any major teratogenic effect of the antiretroviral drugs. Each year it enrolls approximately 15% of all HIV-positive women who give birth to live infants in the United States plus a small number from other countries. It depends on voluntary reporting of prenatal exposure; therefore, drug-associated adverse events may not necessarily reflect true rates. Results of retrospective and clinical studies are also reviewed. Through January 2013, no apparent increase in the frequency of birth defects after first-trimester exposure to antiretroviral drugs compared with population-based comparators was reported.181 A modest but statistically significant elevation of defect rates with didanosine and nelfinavir was reported, the clinical relevance of which is unclear.
It is generally appropriate to administer vaccinations during pregnancy because the risks of the disease may outweigh the risk of the vaccine. No evidence exists of risk to the fetus from vaccinating pregnant women with inactivated virus or bacterial vaccines or toxoids. However, live vaccines pose a theoretical risk to the fetus, so vaccination with live attenuated virus (e.g., varicella, measles-mumps-rubella [MMR]) or tuberculosis (e.g., bacillus Calmette Guérin [BCG]) is considered contraindicated.182 Influenza vaccination is recommended with the inactivated virus preparation.183 Prevention and treatment of influenza with either zanamivir or oseltamivir is possible during pregnancy184; no adverse fetal effects have been reported.185
Caffeine
No evidence suggests that caffeine has teratogenic effects in humans. Early uncontrolled studies suggested that heavy ingestion of caffeine was associated with an increased incidence of spontaneous abortion, low birth weight, preterm delivery, and stillbirth. However, these studies did not control for the use of tobacco and alcohol. A contemporary meta-analysis showed no association with caffeine consumption and preterm birth.186 In 2010, the ACOG published a committee opinion based on studies of caffeine use in pregnancy.187 They concluded that (1) daily intake of less than 200 mg of caffeine was not associated with an increased risk for miscarriage, but contradictory data did not allow for a recommendation regarding daily intake above this level; (2) moderate caffeine intake does not appear to contribute to preterm birth; and (3) the association between caffeine intake and fetal growth restriction was equivocal. A subsequent review concluded with more certainty that moderate or even high amounts of caffeine-containing foods and beverages did not increase the risk for congenital malformations, miscarriage, or growth restriction.188
Smoking Cessation Therapies
Smoking during pregnancy is clearly linked to multiple adverse fetal, neonatal, and childhood effects. Drug interventions to stop smoking include nicotine replacement therapy, antidepressants, bupropion (dopamine and norepinephrine reuptake inhibitor and nicotine antagonist), bromocriptine (dopamine agonist), varenicline (nicotine partial agonist), and cytisine (plant alkaloid). Bupropion and bromocriptine therapy were not associated with congenital malformations or other adverse outcomes.189 Animal data show no increase in malformations with varenicline, but there are no human data.
Specific Highly Teratogenic Drugs
Some drugs are so highly teratogenic that two simultaneous forms of reliable contraception are recommended or required during treatment of either partner, sometimes to be continued for months or years after stopping the drug. Some examples are (1) thalidomide, which is still used for erythema nodosum leprosum and multiple myeloma; (2) the antiviral ribavirin, which is used for hepatitis C and viral hemorrhagic fevers; (3) isotretinoin, which is used for cystic acne; and (4) acitretin, which is used for severe psoriasis.
Drug Use during Lactation
General Principles
Nursing mothers are understandably concerned about the transfer of drugs and chemicals to breast milk. Correct advice is important to prevent them from unnecessarily stopping breast-feeding or discontinuing appropriate drug treatment. Unfortunately, it can be difficult to decide which of the many, sometimes conflicting, sources of public information to trust. Pharmaceutical information leaflets from the manufacturers often discourage the use of drugs during breast-feeding simply as a general precaution. The most comprehensive up-to-date information is found in the Drugs and Lactation database (LactMed) of the National Library of Medicine’s Toxicology Data Network (TOXNET).190 The American Academy of Pediatrics (AAP) has published policy statements on the benefits of breast-feeding191 and the use of drugs during lactation.192 The Centers for Disease Control and Prevention (CDC) provides online information regarding breast-feeding and various toxins and infectious diseases.193
Only a few types of drugs such as cytotoxic and immunosuppressive drugs (e.g., cyclophosphamide, methotrexate) and radioactive compounds are strongly contraindicated during breast-feeding.59 Mothers breast-feeding infants with glucose-6-phosphate-dehydrogenase (G6PD) deficiency should avoid drugs such as sulfonamides, including the combination of sulfamethoxazole and trimethoprim, nitrofurantoin, and primaquine.
Maternal drugs may also affect lactation, and some are used therapeutically for this purpose. Drugs that increase the secretion of prolactin can stimulate milk production; these include dopamine antagonists such as phenothiazines, haloperidol, metoclopramide, and domperidone, as well as sulpiride, risperidone, and methyldopa. Drugs that decrease the production of milk include diuretics, estrogen, and dopamine agonists such as bromocriptine, cabergoline, lisuride, and quinagolide. Bromocriptine is no longer approved for postpartum lactation suppression because of its association with puerperal seizures, stroke, and myocardial infarction. Women who smoke also have lower milk production.
Drug transfer to breast milk occurs by passive diffusion, but transporter systems are also present.194 The rate of passive transfer into breast milk depends on the lipid solubility, molecular weight, degree of ionization, and protein binding of the drug. Nonionized molecules of small molecular weight (e.g., ethanol) are readily transferred into breast milk. Drugs that have more than 85% maternal protein binding are often not detectable in the infant.195
The amount of a drug in breast milk is a variable fraction of the maternal blood concentration, which is proportional to the maternal dose. Quoted maternal milk-to-plasma ratios can vary because drug transfer is a time-dependent process. Even when calculated under steady-state conditions, there can be large individual variation. Absolute infant dose (µg/kg/day) can be calculated as the product of the average concentration in milk and the estimated daily volume of milk intake, and relative infant dose can be estimated by dividing absolute infant dose by the maternal dose.196 It has been suggested that a relative infant dose less than 10% is generally safe,196 but this will also depend on the oral bioavailability of the drug in the infant and the relative toxicity of individual agents. However, physicians and patients should be aware of the following disclaimers. First, in the case of toxic drugs, any exposure may be inappropriate. Second, the infant may be allergic to a drug consumed by the mother. Third, there may be unknown, long-term effects of even small doses of drugs. Fourth, individual variability in drug disposition may lead to unexpectedly high maternal blood and breast milk concentrations. Finally, infants have immature enzyme systems and metabolic pathways and some drugs are eliminated more slowly. The benefits of breast-feeding are well known, and the risk of drug exposure must be weighed against these benefits.
Lactation is not fully established during the first several days postpartum. The neonate receives only a small volume of colostrum, and little drug is excreted through milk at this time. Thus, only very small amounts of drugs administered after vaginal or cesarean delivery would reach the neonate and significant effects should be unlikely. However, neonatal metabolism and elimination are also poorly developed, and several days of maternal opioid analgesia with drugs such as meperidine (pethidine) and codeine may result in neonatal accumulation and side effects.
When a mother requires a daily dose of a drug during lactation, the minimum effective dose should be used. Some mothers requiring long-term therapy would already have been taking drugs during pregnancy, and the fetus would have been exposed to concentrations much greater than those achieved in the infant through breast-feeding. Thus, if a drug has been acceptable during pregnancy, it is often reasonable to continue it during breast-feeding unless there are drug-specific factors to the contrary.197 For example, poor neonatal elimination of lamotrigine may eventually lead to drug accumulation in the infant and adverse effects. In general, medications should be taken after breast-feeding, and long-acting preparations should be avoided. If the infant nurses less frequently overnight, ingestion of a drug dose at night after nursing will decrease the infant’s exposure.
Anesthetic Drugs
There are generally no concerns regarding anesthetic drugs and perioperative medicines in the breast milk of women who require an anesthetic.198,199 However, the prolonged use of postoperative medications such as analgesics will obviously increase the infant dose, the potential effects of which are discussed next.
Analgesics
No harmful effects of acetaminophen or NSAIDs have been noted except for aspirin.71 Some NSAIDs specifically recommended as compatible with breast-feeding are ibuprofen, flurbiprofen, naproxen, and celecoxib. Other NSAIDs may be discouraged simply because of limited information, although there are some concerns about diflunisal and carisoprodol based on animal data.192 Surprisingly, the FDA has singled out ketorolac with a “black box” warning that it is “contraindicated in nursing mothers because of the potential adverse effects of prostaglandin-inhibiting drugs on neonates”; the basis for this decision is unclear. Theoretically, NSAIDs with antiplatelet effects should be avoided by mothers who are breast-feeding neonates with platelet dysfunction. With aspirin, there is limited transfer of salicylic acid into breast milk because it exists mostly in the ionized form. After single oral doses, peak milk levels occur at approximately 3 hours with milk-to-plasma concentration ratios between 0.03 and 0.05.200 However, maternal ingestion of high doses (e.g., more than 16 tablets per day) may result in maternal and breast milk concentrations sufficiently high to affect platelet aggregation in the infant. Reduced neonatal clearance of salicylic acid may lead to drug accumulation and toxic effects, even when repeated exposures are small.201 Because of these concerns, the World Health Organization (WHO) Working Group on Human Lactation has classified salicylates as unsafe for use by nursing women.202 Low doses of aspirin prescribed as anti-platelet therapy may be acceptable.192
It was thought previously that opioids used by nursing mothers were highly unlikely to have adverse effects on breast-fed infants. Normal maternal doses of codeine, morphine, tramadol, and meperidine do not have obvious adverse effects on most nursing infants.78,192 The dose detectable in breast milk is 1% to 2% of the mother’s dose and is unlikely to have significant pharmacologic activity. However, in one patient who took propoxyphene in a suicide attempt, the breast milk concentration of propoxyphene was half that of the maternal serum level.203 Theoretically, a breast-feeding infant could receive up to 1 mg of propoxyphene per day if the mother were to consume the maximum dose.
Neonates are particularly vulnerable because their drug metabolism and elimination are poorly developed. Neonates of mothers receiving meperidine by intravenous patient-controlled analgesia after cesarean delivery had significant neurobehavioral depression by the third day.204 The cumulative maternal meperidine dose at 48 hours postpartum was 14 mg/kg. No neonatal depression was seen in a morphine group in whom the cumulative maternal dose at 48 hours was 2.1 mg/kg. Both opioids and their major metabolites accumulated in colostrum. In a subsequent study,205 the cumulative opioid doses at 48 hours were lower (meperidine 4.7 mg/kg and morphine 0.54 mg/kg), but infants in the morphine group were still more alert and oriented. With lower maternal morphine doses, concentrations in colostrum may even be undetectable.206
Recently, it has been recognized that infants of breast-feeding mothers taking codeine may have CNS depression. In 2006, the full-term 13-day-old infant of a mother taking a modest dose of codeine for episiotomy pain died of an apparent morphine overdose. It was discovered that the mother was heterozygous for a CYP2D6*2A allele with CYP2D6*2×2 gene duplication, making her an ultra-rapid metabolizer of codeine. This condition resulted in a breast milk morphine concentration of 87 ng/mL, compared with the expected typical range of 1.9 to 20.5 ng/mL.207 This case prompted the FDA to release a Public Health Advisory advising caution in the use of codeine-containing medications by breast-feeding women.208 The FDA recommended that the lowest effective dose for the shortest necessary duration should be used and that women should be taught how to recognize signs of high morphine levels in their infants. The number of ultra-rapid metabolizers is estimated to vary between 1 and 28 per 100 people.208 In addition, genetic variability in UGT2B7 may increase the formation of active morphine-6-glucuronide. It is estimated that 1.4% of Western European women would have both the CYP2D6 and UGT2B7 variants promoting neonatal depression.209
Ultra-rapid metabolism has been reported as a problem for breast-feeding only with codeine, although the FDA suggested that “it has the potential to affect other opioids.”208 Maternal oxycodone for postpartum analgesia has been associated with neonatal depression.210 However, polymorphisms have less effect on the metabolism and analgesic effects of oxycodone, hydrocodone, and tramadol than codeine.211
Sedatives
Long-acting drugs such as diazepam are metabolized slowly in the infant, and accumulation can lead to sedation and poor feeding. Perinatal use of diazepam has been associated with hypotonia, hypothermia, and respiratory depression.85 If a sedative must be used in a lactating woman, a relatively short-acting agent with inactive metabolites, such as oxazepam, lorazepam, alprazolam, or midazolam, is recommended.212 The infant should be monitored for sedation during maternal drug use and for withdrawal symptoms after the medication is stopped or after discontinuation of breast-feeding.213
Anticonvulsants
Carbamazepine and valproic acid have low excretion into breast milk and are considered the safest choices.214 Studies have detected only small amounts of phenytoin, phenobarbital, and diazepam in breast milk.215,216 However, infants eliminate phenobarbital and diazepam slowly, so these agents may accumulate. Women taking a barbiturate or a benzodiazepine should observe their infants for evidence of sedation and withdrawal.92,192 There are conflicting recommendations for the newer anticonvulsants. Lamotrigine concentrations in breast-fed infants can be very high, and some recommend that the drug should be avoided.214 However, other authors note that the mother has usually been taking this drug throughout pregnancy already and that there is no evidence to recommend stopping the drug.217
Cruikshank et al.218 measured breast milk magnesium concentrations in 10 preeclamptic women who were receiving magnesium sulfate 1 g/h intravenously for 24 hours after delivery. The mean breast milk magnesium concentration was 6.4 ± 0.4 mg/dL, compared with 4.8 ± 0.5 mg/dL in controls. Breast milk calcium concentrations were not affected by magnesium sulfate therapy.
Antidepressants and Lithium
Psychotropic agents may be of concern because many of these medications have long half-lives and the effect of even small doses on the developing nervous system is not known. Nevertheless, the many medications available to treat depression or postpartum depression appear to have little if any immediate effect on the infant.219 Many antidepressants have low milk-to-plasma ratios. There are few reports of neonatal problems with the use of SSRIs and related drugs.220 Fluoxetine and its metabolite have relatively long elimination half-lives, so other drugs such as sertraline and paroxetine are preferred.221
Breast milk concentrations of lithium are one third to one half of maternal serum concentrations,222,223 and infant serum concentrations during breast-feeding are much lower than fetal serum concentrations that occur when mothers take lithium during pregnancy. Neonatal clearance of lithium is reduced, and it may be useful to monitor neonatal lithium concentrations.
Cardiovascular Drugs
Amiodarone concentrations in the infant are unpredictable but may be sufficiently high to cause cardiac effects. Iodine released during its metabolism may cause thyroid dysfunction. Amiodarone has a very long half-life and is still excreted in breast milk weeks after stopping the drug.
Beta-adrenergic receptor antagonists with low protein binding have relatively higher transfer to breast milk, and those excreted renally are more likely to accumulate in neonates. Thus, atenolol with 10% protein binding and 85% renal excretion would not be the best choice. Breast milk concentrations of atenolol are approximately three times maternal plasma concentrations.224 Even though the total infant dose is only 1% of the maternal therapeutic dose, and the infant plasma concentration would not normally cause side effects in the infant, atenolol has been associated with neonatal cyanosis and bradycardia.
Other common drugs such as ACE inhibitors and antihypertensives are considered safe. However, diuretics may reduce milk production. Maternal protein binding of digoxin limits infant drug exposure; after a maternal dose of 0.25 mg, a peak breast milk concentration of 0.6 to 1.0 ng/mL occurs and the milk-to-plasma concentration ratio at the 4-hour peak is between 0.8 and 0.9 ng/mL. In 24 hours an infant might receive approximately 1% of the maternal digoxin dose,225 and no adverse effects have been reported in nursing infants of mothers taking this drug. Breast milk clonidine concentrations are almost twice maternal serum concentrations. However, this exposure does not seem to have any adverse effects on the infant.226
There are no safety data for statins, but they are not recommended during breast-feeding because of concerns that they may disrupt infant lipid metabolism.
Respiratory Drugs and Corticosteroids
Salbutamol, terbutaline, and salmeterol inhalers are considered compatible with breast-feeding. Maximum milk concentrations of theophylline are achieved between 1 and 3 hours after an oral dose. It has been calculated that the nursing infant receives less than 1% of the maternal dose. Such exposure appears to have no adverse effects.
Inhaled and oral corticosteroids are also considered safe. Katz and Duncan227 obtained breast milk 2 hours after an oral dose of 10 mg of prednisone in one nursing mother. They detected breast milk concentrations of prednisone and prednisolone that would be unlikely to result in any deleterious effect on the infant. McKenzie et al.228 administered 5 mg of radioactive prednisolone to seven patients and found that 0.14% (a negligible quantity) of the radioactive label was secreted in the milk in the subsequent 60 hours. Thus, breast-feeding is not contraindicated in mothers taking corticosteroids. Even at a maternal dosage of 80 mg/day, the nursing infant would ingest a dose equivalent to less than 10% of its endogenous cortisol production.229
Anticoagulants
Most mothers who require anticoagulation may continue to nurse their infants with no problems. Warfarin is 98% protein bound. Orme et al.230 reported no warfarin in breast milk or infant plasma in seven women taking warfarin 5 to 12 mg/day. Similarly, de Swiet and Lewis231 found that warfarin appears in breast milk in insignificant quantities. Heparin does not cross significantly into breast milk and is not active when administered orally.
Antihistamines
No harmful effects have been noted with maternal use of antihistamines.232 These drugs do not appear to affect the milk supply. Little antihistamine is excreted into breast milk, further confirming safety of use during lactation.169
In theory, histamine type 2 (H2)-receptor antagonists might suppress gastric acidity or cause CNS stimulation in the infant, but these effects have not been confirmed in published studies. Famotidine, nizatidine, and roxatidine are less concentrated in breast milk and may be preferable to cimetidine.233
Anti-infective Drugs
Penicillin and its derivatives are safe in nursing mothers. With the usual therapeutic doses of ampicillin, the milk-to-plasma concentration ratio is 0.2 or less and no adverse effects are noted in nursing infants.234 Theoretically, infant diarrhea or candidiasis might occur with prolonged therapy. Cephalosporins appear in trace amounts in breast milk and are also safe.
Sulfonamides displace bilirubin from binding sites on albumin, so these drugs are best avoided during the first 5 days of life or in mothers of preterm infants with hyperbilirubinemia. Sulfonamides appear in breast milk in small amounts. Sulfasalazine has been associated with diarrhea and bloody diarrhea in an infant.192 Sulfonamides should be avoided in infants with G6PD deficiency.
Tetracyclines are normally avoided during breast-feeding because of the potential for tooth staining and delayed bone growth. The breast milk concentration of tetracycline is about half the maternal plasma concentration, but tetracycline has a high affinity for both calcium and protein, and the amount of free tetracycline available for systemic absorption is very small. Thus, some references consider a short course of tetracyclines to be compatible with breast-feeding. Similarly, quinolones are usually avoided in pregnancy, but there is disagreement over their safety during breast-feeding.
Maternal administration of acyclovir does not contraindicate breast-feeding. If a mother takes 1 g/day, the infant probably receives less than 1 mg/day, a very low dose.235
Most vaccines, except for yellow-fever and smallpox, can be administered during breast-feeding.192
Caffeine
Moderate maternal intake of caffeine does not adversely affect the breast-fed infant. One study noted that breast milk contains only 1% of the total maternal dose of caffeine.236 If a mother drinks excessive amounts of coffee, caffeine might accumulate in the infant and the infant might show signs of caffeine stimulation (e.g., irritability, poor sleeping pattern). Nursing mothers should limit their intake to a moderate level of caffeinated beverages (e.g., 2 to 3 cups per day).192
References
1. Landau R, Kraft JC. Pharmacogenetics in obstetric anesthesia. Curr Opin Anaesthesiol. 2010;23:323–329.
2. Lam J, Matlow JN, Ross CJ, et al. Postpartum maternal codeine therapy and the risk of adverse neonatal outcomes: the devil is in the details. Ther Drug Monit. 2012;34:378–380.
3. Landau R, Morales MA, Antonarakis SE, et al. Arg16 homozygosity of the β2-adrenergic receptor improves the outcome after β2-agonist tocolysis for preterm labor. Clin Pharmacol Ther. 2005;78:656–663.
4. Smiley RM, Blouin JL, Negron M, Landau R. β2-adrenoceptor genotype affects vasopressor requirements during spinal anesthesia for cesarean delivery. Anesthesiology. 2006;104:644–650.
5. Landau R, Kern C, Columb MO, et al. Genetic variability of the μ-opioid receptor influences intrathecal fentanyl analgesia requirements in laboring women. Pain. 2008;139:5–14.
6. Sia AT, Lim Y, Lim EC, et al. A118G single nucleotide polymorphism of human μ-opioid receptor gene influences pain perception and patient-controlled intravenous morphine consumption after intrathecal morphine for postcesarean analgesia. Anesthesiology. 2008;109:520–526.
7. Wong CA. The promise of pharmacogenetics in labor analgesia … tantalizing, but not there yet. Int J Obstet Anesth. 2012;21:105–108.
8. Walter C, Lotsch J. Meta-analysis of the relevance of the OPRM1 118A>G genetic variant for pain treatment. Pain. 2009;146:270–275.
9. American College of Obstetricians and Gynecologists Committee on Genetics. Pharmacogenetics. ACOG Committee Opinion No. 488. [Washington, DC] Obstet Gynecol. 2011;117:1240–1241.
10. Hodge LS, Tracy TS. Alterations in drug disposition during pregnancy: implications for drug therapy. Expert Opin Drug Metab Toxicol. 2007;3:557–571.
11. Feghali MN, Mattison DR. Clinical therapeutics in pregnancy. J Biomed Biotechnol. 2011;2011:783528.
12. Munnell EW, Taylor HCJ. Liver blood flow in pregnancy—hepatic vein catheterization. J Clin Invest. 1947;26:952–956.
13. Robson SC, Mutch E, Boys RJ, Woodhouse KW. Apparent liver blood flow during pregnancy: a serial study using indocyanine green clearance. Br J Obstet Gynaecol. 1990;97:720–724.
14. Nakai A, Sekiya I, Oya A, et al. Assessment of the hepatic arterial and portal venous blood flows during pregnancy with Doppler ultrasonography. Arch Gynecol Obstet. 2002;266:25–29.
15. Isoherranen N, Thummel KE. Drug metabolism and transport during pregnancy: how does drug disposition change during pregnancy and what are the mechanisms that cause such changes? Drug Metab Dispos. 2013;41:256–262.
17. Hill MD, Abramson FP. The significance of plasma protein binding on the fetal/maternal distribution of drugs at steady-state. Clin Pharmacokinet. 1988;14:156–170.
18. Staud F, Cerveny L, Ceckova M. Pharmacotherapy in pregnancy; effect of ABC and SLC transporters on drug transport across the placenta and fetal drug exposure. J Drug Target. 2012;20:736–763.
19. Rubinchik-Stern M, Eyal S. Drug interactions at the human placenta: what is the evidence? Front Pharmacol. 2012;3:126.
20. Syme MR, Paxton JW, Keelan JA. Drug transfer and metabolism by the human placenta. Clin Pharmacokinet. 2004;43:487–514.
21. Anderson BJ, Allegaert K. The pharmacology of anaesthetics in the neonate. Best Pract Res Clin Anaesthesiol. 2010;24:419–431.
22. de Wildt SN. Profound changes in drug metabolism enzymes and possible effects on drug therapy in neonates and children. Expert Opin Drug Metab Toxicol. 2011;7:935–948.
23. Palahniuk RJ, Shnider SM, Eger EI 2nd. Pregnancy decreases the requirement for inhaled anesthetic agents. Anesthesiology. 1974;41:82–83.
24. Strout CD, Nahrwold ML. Halothane requirement during pregnancy and lactation in rats. Anesthesiology. 1981;55:322–323.
25. Gin T, Chan MT. Decreased minimum alveolar concentration of isoflurane in pregnant humans. Anesthesiology. 1994;81:829–832.
26. Chan MT, Mainland P, Gin T. Minimum alveolar concentration of halothane and enflurane are decreased in early pregnancy. Anesthesiology. 1996;85:782–786.
27. Chan MT, Gin T. Postpartum changes in the minimum alveolar concentration of isoflurane. Anesthesiology. 1995;82:1360–1363.
28. Zhou HH, Norman P, DeLima LG, et al. The minimum alveolar concentration of isoflurane in patients undergoing bilateral tubal ligation in the postpartum period. Anesthesiology. 1995;82:1364–1368.
29. Datta S, Migliozzi RP, Flanagan HL, Krieger NR. Chronically administered progesterone decreases halothane requirements in rabbits. Anesth Analg. 1989;68:46–50.
30. Tanifuji Y, Yasuda N, Kobayashi K, Eger E II. Effect of progesterone and estrogen on halothane MAC in dogs. Anesthesiology. 1986;65:A351.
31. Thomas BA, Anzalone TA, Rosinia FA. Progesterone decreases the MAC of desflurane in the nonpregnant ewe. Anesthesiology. 1995;83:A952.
32. Erden V, Yangin Z, Erkalp K, et al. Increased progesterone production during the luteal phase of menstruation may decrease anesthetic requirement. Anesth Analg. 2005;101:1007–1011.
33. Gin T, Chan MTV. Pregnancy reduces the bispectral index during isoflurane anesthesia. Anesthesiology. 1997;87:A305.
34. Chan MT, Gin T. Pregnancy potentiates the sedative effects of sevoflurane. Anesthesiology. 2008;109:A616.
35. Gin T, Chan MTV, Lau TK, Lam KK. Pregnancy potentiates the sedative effects of nitrous oxide. Anesthesiology. 1998;89:A1042.
36. Ueyama H, Hagihira S, Takashina M, et al. Pregnancy does not enhance volatile anesthetic sensitivity on the brain: an electroencephalographic analysis study. Anesthesiology. 2010;113:577–584.
37. Gin T, Mainland P, Chan MT, Short TG. Decreased thiopental requirements in early pregnancy. Anesthesiology. 1997;86:73–78.
38. Mainland P, Chan MTV, Gin T. Reduced thiopental requirement in the postpartum period. Anesthesiology. 1997;87:A358.
39. Higuchi H, Adachi Y, Arimura S, et al. Early pregnancy does not reduce the C50 of propofol for loss of consciousness. Anesth Analg. 2001;93:1565–1569.
40. Mongardon N, Servin F, Perrin M, et al. Predicted propofol effect-site concentration for induction and emergence of anesthesia during early pregnancy. Anesth Analg. 2009;109:90–95.
41. Fagraeus L, Urban BJ, Bromage PR. Spread of epidural analgesia in early pregnancy. Anesthesiology. 1983;58:184–187.
42. Hirabayashi Y, Shimizu R, Saitoh K, Fukuda H. Spread of subarachnoid hyperbaric amethocaine in pregnant women. Br J Anaesth. 1995;74:384–386.
43. Arakawa M. Does pregnancy increase the efficacy of lumbar epidural anesthesia? Int J Obstet Anesth. 2004;13:86–90.
44. Camorcia M, Capogna G, Columb MO. Effect of sex and pregnancy on the potency of intrathecal bupivacaine: determination of ED50 for motor block with the up-down sequential allocation method. Eur J Anaesthesiol. 2011;28:240–244.
45. Zhan Q, Huang S, Geng G, Xie Y. Comparison of relative potency of intrathecal bupivacaine for motor block in pregnant versus non-pregnant women. Int J Obstet Anesth. 2011;20:219–223.
46. Hirabayashi Y, Shimizu R, Fukuda H, et al. Soft tissue anatomy within the vertebral canal in pregnant women. Br J Anaesth. 1996;77:153–156.
47. Onuki E, Higuchi H, Takagi S, et al. Gestation-related reduction in lumbar cerebrospinal fluid volume and dural sac surface area. Anesth Analg. 2010;110:148–153.
48. Datta S, Lambert DH, Gregus J, et al. Differential sensitivities of mammalian nerve fibers during pregnancy. Anesth Analg. 1983;62:1070–1072.
49. Flanagan HL, Datta S, Lambert DH, et al. Effect of pregnancy on bupivacaine-induced conduction blockade in the isolated rabbit vagus nerve. Anesth Analg. 1987;66:123–126.
50. Popitz-Bergez FA, Leeson S, Thalhammer JG, Strichartz GR. Intraneural lidocaine uptake compared with analgesic differences between pregnant and nonpregnant rats. Reg Anesth. 1997;22:363–371.
51. Butterworth JF 4th, Walker FO, Lysak SZ. Pregnancy increases median nerve susceptibility to lidocaine. Anesthesiology. 1990;72:962–965.
52. Flanagan HL, Datta S, Moller RA, Covino BG. Effect of exogenously administered progesterone on susceptibility of rabbit vagus nerves to bupivacaine. Anesthesiology. 1988;69:A676.
53. Dietz FB, Jaffe RA. Pregnancy does not increase susceptibility to bupivacaine in spinal root axons. Anesthesiology. 1997;87:610–616.
54. Gintzler AR. Endorphin-mediated increases in pain threshold during pregnancy. Science. 1980;210:193–195.
55. Jayaram A, Singh P, Carp H. SCH 32615, an enkephalinase inhibitor, enhances pregnancy-induced analgesia in mice. Anesth Analg. 1995;80:944–948.
56. Gintzler AR, Liu NJ. The maternal spinal cord: biochemical and physiological correlates of steroid-activated antinociceptive processes. Prog Brain Res. 2001;133:83–97.
57. Draisci G, Catarci S, Vollono C, et al. Pregnancy-induced analgesia: a combined psychophysical and neurophysiological study. Eur J Pain. 2012;16:1389–1397.
58. Carvalho B, Angst MS, Fuller AJ, et al. Experimental heat pain for detecting pregnancy-induced analgesia in humans. Anesth Analg. 2006;103:1283–1287.
59. Buhimschi CS, Weiner CP. Medications in pregnancy and lactation. Part 1. Teratology. Obstet Gynecol. 2009;113:166–188.
60. Niebyl JR, Simpson JL. Drugs and environmental agents in pregnancy and lactation: embryology, teratology, epidemiology. Gabbe SG, Niebyl JR, Simpson JL. Obstetrics: Normal and Problem Pregnancies. 5th edition. Churchill Livingstone: Philadelphia; 2007.
61. Yankowitz J. Use of medications in pregnancy: General principles, teratology, and current developments. Yankowitz J, Niebyl JR. Drug Therapy in Pregnancy. 3rd edition. Lippincott Williams & Wilkins: Philadelphia; 2001.
62. Buehler BA, Delimont D, van Waes M, Finnell RH. Prenatal prediction of risk of the fetal hydantoin syndrome. N Engl J Med. 1990;322:1567–1572.
63. Teratology Society Public Affairs Committee. FDA classification of drugs for teratogenic risk. Teratology. 1994;49:446–447.
64. Friedman JM. Report of the Teratology Society Public Affairs Committee symposium on FDA classification of drugs. Teratology. 1993;48:5–6.
65. Howell PR, Madej TH. Administration of drugs outside of Product License: awareness and current practice. Int J Obstet Anesth. 1999;8:30–36.
66. Daw JR, Mintzes B, Law MR, et al. Prescription drug use in pregnancy: a retrospective, population-based study in British Columbia, Canada (2001-2006). Clin Ther. 2012;34:239–249.e2.
67. Doering PL, Boothby LA, Cheok M. Review of pregnancy labeling of prescription drugs: is the current system adequate to inform of risks? Am J Obstet Gynecol. 2002;187:333–339.
68. Law R, Bozzo P, Koren G, Einarson A. FDA pregnancy risk categories and the CPS: do they help or are they a hindrance? Can Fam Physician. 2010;56:239–241.
69. U S Food and Drug Administration. Pregnancy and Lactation Labeling. [Available at] http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/Labeling/ucm093307.htm [Accessed July 1, 2013] .
70. Feldkamp ML, Meyer RE, Krikov S, Botto LD. Acetaminophen use in pregnancy and risk of birth defects: findings from the National Birth Defects Prevention Study. Obstet Gynecol. 2010;115:109–115.
71. Bloor M, Paech M. Nonsteroidal anti-inflammatory drugs during pregnancy and the initiation of lactation. Anesth Analg. 2013;116:1063–1075.
72. Kozer E, Nikfar S, Costei A, et al. Aspirin consumption during the first trimester of pregnancy and congenital anomalies: a meta-analysis. Am J Obstet Gynecol. 2002;187:1623–1630.
73. Farquharson RG, Quenby S, Greaves M. Antiphospholipid syndrome in pregnancy: a randomized, controlled trial of treatment. Obstet Gynecol. 2002;100:408–413.
74. Moise KJ Jr. Effect of advancing gestational age on the frequency of fetal ductal constriction in association with maternal indomethacin use. Am J Obstet Gynecol. 1993;168:1350–1353.
75. Heinonen OP, Slone D, Shapiro S. Birth Defects and Drugs in Pregnancy. Publishing Sciences Group: Littleton, MA; 1977.
76. Tyson HK. Neonatal withdrawal symptoms associated with maternal use of propoxyphene hydrochloride (Darvon). J Pediatr. 1974;85:684–685.
77. Broussard CS, Rasmussen SA, Reefhuis J, et al. Maternal treatment with opioid analgesics and risk for birth defects. Am J Obstet Gynecol. 2011;204:314 e1–11.
78. Bloor M, Paech MJ, Kaye R. Tramadol in pregnancy and lactation. Int J Obstet Anesth. 2012;21:163–167.
79. Milkovich L, van den Berg BJ. Effects of prenatal meprobamate and chlordiazepoxide hydrochloride on human embryonic and fetal development. N Engl J Med. 1974;291:1268–1271.
80. Hartz SC, Heinonen OP, Shapiro S, et al. Antenatal exposure to meprobamate and chlordiazepoxide in relation to malformations, mental development, and childhood mortality. N Engl J Med. 1975;292:726–728.
81. Timmermann G, Acs N, Banhidy F, Czeizel AE. A study of teratogenic and fetotoxic effects of large doses of meprobamate used for a suicide attempt by 42 pregnant women. Toxicol Ind Health. 2008;24:97–107.
82. Gidai J, Acs N, Banhidy F, Czeizel AE. A study of the teratogenic and fetotoxic effects of large doses of chlordiazepoxide used for self-poisoning by 35 pregnant women. Toxicol Ind Health. 2008;24:41–51.
83. Kjaer D, Horvath-Puho E, Christensen J, et al. Use of phenytoin, phenobarbital, or diazepam during pregnancy and risk of congenital abnormalities: a case-time-control study. Pharmacoepidemiol Drug Saf. 2007;16:181–188.
84. Ornoy A, Arnon J, Shechtman S, et al. Is benzodiazepine use during pregnancy really teratogenic? Reprod Toxicol. 1998;12:511–515.
85. Gillberg C. “Floppy infant syndrome” and maternal diazepam. Lancet. 1977;2:244.
86. Wikner BN, Kallen B. Are hypnotic benzodiazepine receptor agonists teratogenic in humans? J Clin Psychopharmacol. 2011;31:356–359.
87. Bellantuono C, Tofani S, Di Sciascio G, Santone G. Benzodiazepine exposure in pregnancy and risk of major malformations: a critical overview. Gen Hosp Psychiatry. 2013;35:3–8.
88. Wikner BN, Stiller CO, Bergman U, et al. Use of benzodiazepines and benzodiazepine receptor agonists during pregnancy: neonatal outcome and congenital malformations. Pharmacoepidemiol Drug Saf. 2007;16:1203–1210.
89. Walker SP, Permezel M, Berkovic SF. The management of epilepsy in pregnancy. BJOG. 2009;116:758–767.
90. Harden CL, Hopp J, Ting TY, et al. Management issues for women with epilepsy—Focus on pregnancy (an evidence-based review). I. Obstetrical complications and change in seizure frequency: report of the Quality Standards Subcommittee and Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Epilepsia. 2009;50:1229–1236.
91. Malone FD, D’Alton ME. Drugs in pregnancy: anticonvulsants. Semin Perinatol. 1997;21:114–123.
92. Morrell MJ. Guidelines for the care of women with epilepsy. Neurology. 1998;51:S21–S27.
93. Holmes LB, Rosenberger PB, Harvey EA, et al. Intelligence and physical features of children of women with epilepsy. Teratology. 2000;61:196–202.
94. Holmes LB, Harvey EA, Coull BA, et al. The teratogenicity of anticonvulsant drugs. N Engl J Med. 2001;344:1132–1138.
95. Fried S, Kozer E, Nulman I, et al. Malformation rates in children of women with untreated epilepsy: a meta-analysis. Drug Saf. 2004;27:197–202.
96. Morrell MJ. The new antiepileptic drugs and women: efficacy, reproductive health, pregnancy, and fetal outcome. Epilepsia. 1996;37(Suppl 6):S34–S44.
97. Cornelissen M, Steegers-Theunissen R, Kollee L, et al. Increased incidence of neonatal vitamin K deficiency resulting from maternal anticonvulsant therapy. Am J Obstet Gynecol. 1993;168:923–928.
98. Cornelissen M, Steegers-Theunissen R, Kollee L, et al. Supplementation of vitamin K in pregnant women receiving anticonvulsant therapy prevents neonatal vitamin K deficiency. Am J Obstet Gynecol. 1993;168:884–888.
99. Jones KL, Lacro RV, Johnson KA, Adams J. Pattern of malformations in the children of women treated with carbamazepine during pregnancy. N Engl J Med. 1989;320:1661–1666.
100. Rosa FW. Spina bifida in infants of women treated with carbamazepine during pregnancy. N Engl J Med. 1991;324:674–677.
101. Morrow J, Russell A, Guthrie E, et al. Malformation risks of antiepileptic drugs in pregnancy: a prospective study from the UK Epilepsy and Pregnancy Register. J Neurol Neurosurg Psychiatry. 2006;77:193–198.
102. Meador K, Reynolds MW, Crean S, et al. Pregnancy outcomes in women with epilepsy: a systematic review and meta-analysis of published pregnancy registries and cohorts. Epilepsy Res. 2008;81:1–13.
103. Wide K, Winbladh B, Kallen B. Major malformations in infants exposed to antiepileptic drugs in utero, with emphasis on carbamazepine and valproic acid: a nation-wide, population-based register study. Acta Paediatr. 2004;93:174–176.
104. Ornoy A. Neuroteratogens in man: an overview with special emphasis on the teratogenicity of antiepileptic drugs in pregnancy. Reprod Toxicol. 2006;22:214–226.
105. Levy RH, Yerby MS. Effects of pregnancy on antiepileptic drug utilization. Epilepsia. 1985;26(Suppl 1):S52–S57.
106. Jentink J, Loane MA, Dolk H, et al. Valproic acid monotherapy in pregnancy and major congenital malformations. N Engl J Med. 2010;362:2185–2193.
107. Harden CL, Meador KJ, Pennell PB, et al. Management issues for women with epilepsy—Focus on pregnancy (an evidence-based review). II. Teratogenesis and perinatal outcomes: Report of the Quality Standards Subcommittee and Therapeutics and Technology Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Epilepsia. 2009;50:1237–1246.
108. Werler MM, Ahrens KA, Bosco JL, et al. Use of antiepileptic medications in pregnancy in relation to risks of birth defects. Ann Epidemiol. 2011;21:842–850.
109. Wlodarczyk BJ, Palacios AM, George TM, Finnell RH. Antiepileptic drugs and pregnancy outcomes. Am J Med Genet A. 2012;158A:2071–2090.
110. Tennis P, Eldridge RR. Preliminary results on pregnancy outcomes in women using lamotrigine. Epilepsia. 2002;43:1161–1167.
111. Vajda FJ, Hitchcock A, Graham J, et al. Foetal malformations and seizure control: 52 months data of the Australian Pregnancy Registry. Eur J Neurol. 2006;13:645–654.
112. Shor S, Koren G, Nulman I. Teratogenicity of lamotrigine. Can Fam Physician. 2007;53:1007–1009.
113. Kalviainen R, Tomson T. Optimizing treatment of epilepsy during pregnancy. Neurology. 2006;67:S59–S63.
114. Altshuler LL, Cohen L, Szuba MP, et al. Pharmacologic management of psychiatric illness during pregnancy: dilemmas and guidelines. Am J Psychiatry. 1996;153:592–606.
115. Briggs GG, Freeman RK, Yaffe SJ. Drugs in Pregnancy and Lactation. 8th edition. Lippincott Williams & Wilkins: Philadelphia; 2008.
116. Reis M, Kallen B. Delivery outcome after maternal use of antidepressant drugs in pregnancy: an update using Swedish data. Psychol Med. 2010;40:1723–1733.
117. Stewart DE. Clinical practice. Depression during pregnancy. N Engl J Med. 2011;365:1605–1611.
118. Patil AS, Kuller JA, Rhee EH. Antidepressants in pregnancy: a review of commonly prescribed medications. Obstet Gynecol Surv. 2011;66:777–787.
119. Goldstein DJ, Corbin LA, Sundell KL. Effects of first-trimester fluoxetine exposure on the newborn. Obstet Gynecol. 1997;89:713–718.
120. Nulman I, Rovet J, Stewart DE, et al. Child development following exposure to tricyclic antidepressants or fluoxetine throughout fetal life: a prospective, controlled study. Am J Psychiatry. 2002;159:1889–1895.
121. Hendrick V, Smith LM, Suri R, et al. Birth outcomes after prenatal exposure to antidepressant medication. Am J Obstet Gynecol. 2003;188:812–815.
122. Louik C, Lin AE, Werler MM, et al. First-trimester use of selective serotonin-reuptake inhibitors and the risk of birth defects. N Engl J Med. 2007;356:2675–2683.
123. Alwan S, Reefhuis J, Rasmussen SA, et al. Use of selective serotonin-reuptake inhibitors in pregnancy and the risk of birth defects. N Engl J Med. 2007;356:2684–2692.
124. Pedersen LH, Henriksen TB, Vestergaard M, et al. Selective serotonin reuptake inhibitors in pregnancy and congenital malformations: population based cohort study. BMJ. 2009;339:b3569.
125. Kornum JB, Nielsen RB, Pedersen L, et al. Use of selective serotonin-reuptake inhibitors during early pregnancy and risk of congenital malformations: updated analysis. Clin Epidemiol. 2010;2:29–36.
126. Chambers CD, Johnson KA, Dick LM, et al. Birth outcomes in pregnant women taking fluoxetine. N Engl J Med. 1996;335:1010–1015.
127. Chambers CD, Hernandez-Diaz S, Van Marter LJ, et al. Selective serotonin-reuptake inhibitors and risk of persistent pulmonary hypertension of the newborn. N Engl J Med. 2006;354:579–587.
128. Kieler H, Artama M, Engeland A, et al. Selective serotonin reuptake inhibitors during pregnancy and risk of persistent pulmonary hypertension in the newborn: population based cohort study from the five Nordic countries. BMJ. 2012;344:d8012.
129. American College of Obstetricians and Gynecologists Committee on Practice Bulletins—Obstetrics. Use of psychiatric medications during pregnancy and lactation. ACOG Practice Bulletin No. 92. [Washington, DC, April 2008 (Reaffirmed 2012)] Obstet Gynecol. 2008;111:1001–1020.
130. Yonkers KA, Wisner KL, Stewart DE, et al. The management of depression during pregnancy: a report from the American Psychiatric Association and the American College of Obstetricians and Gynecologists. Obstet Gynecol. 2009;114:703–713.
131. Linden S, Rich CL. The use of lithium during pregnancy and lactation. J Clin Psychiatry. 1983;44:358–361.
132. Kallen B, Tandberg A. Lithium and pregnancy: a cohort study on manic-depressive women. Acta Psychiatr Scand. 1983;68:134–139.
133. Jacobson SJ, Jones K, Johnson K, et al. Prospective multicentre study of pregnancy outcome after lithium exposure during first trimester. Lancet. 1992;339:530–533.
134. Yacobi S, Ornoy A. Is lithium a real teratogen? What can we conclude from the prospective versus retrospective studies? A review. Isr J Psychiatry Relat Sci. 2008;45:95–106.
135. Krause S, Ebbesen F, Lange AP. Polyhydramnios with maternal lithium treatment. Obstet Gynecol. 1990;75:504–506.
136. Ang MS, Thorp JA, Parisi VM. Maternal lithium therapy and polyhydramnios. Obstet Gynecol. 1990;76:517–519.
137. Schou M, Amdisen A, Steenstrup OR. Lithium and pregnancy. II. Hazards to women given lithium during pregnancy and delivery. Br Med J. 1973;2:137–138.
138. Pickles CJ, Symonds EM, Broughton Pipkin F. The fetal outcome in a randomized double-blind controlled trial of labetalol versus placebo in pregnancy-induced hypertension. Br J Obstet Gynaecol. 1989;96:38–43.
139. Magee LA, Bull SB, Koren G, Logan A. The generalizability of trial data; a comparison of beta-blocker trial participants with a prospective cohort of women taking beta-blockers in pregnancy. Eur J Obstet Gynecol Reprod Biol. 2001;94:205–210.
140. Redmond GP. Propranolol and fetal growth retardation. Semin Perinatol. 1982;6:142–147.
141. Easterling TR, Carr DB, Brateng D, et al. Treatment of hypertension in pregnancy: effect of atenolol on maternal disease, preterm delivery, and fetal growth. Obstet Gynecol. 2001;98:427–433.
142. Easterling TR, Brateng D, Schmucker B, et al. Prevention of preeclampsia: a randomized trial of atenolol in hyperdynamic patients before onset of hypertension. Obstet Gynecol. 1999;93:725–733.
143. Burrows RF, Burrows EA. Assessing the teratogenic potential of angiotensin-converting enzyme inhibitors in pregnancy. Aust N Z J Obstet Gynaecol. 1998;38:306–311.
144. Cooper WO, Hernandez-Diaz S, Arbogast PG, et al. Major congenital malformations after first-trimester exposure to ACE inhibitors. N Engl J Med. 2006;354:2443–2451.
145. Ray JG, Vermeulen MJ, Koren G. Taking ACE inhibitors during early pregnancy: is it safe? Can Fam Physician. 2007;53:1439–1440.
146. Li DK, Yang C, Andrade S, et al. Maternal exposure to angiotensin converting enzyme inhibitors in the first trimester and risk of malformations in offspring: a retrospective cohort study. BMJ. 2011;343:d5931.
147. Walfisch A, Al-maawali A, Moretti ME, et al. Teratogenicity of angiotensin converting enzyme inhibitors or receptor blockers. J Obstet Gynaecol. 2011;31:465–472.
148. Piper JM, Ray WA, Rosa FW. Pregnancy outcome following exposure to angiotensin-converting enzyme inhibitors. Obstet Gynecol. 1992;80:429–432.
149. Tan HL, Lie KI. Treatment of tachyarrhythmias during pregnancy and lactation. Eur Heart J. 2001;22:458–464.
150. Bartalena L, Bogazzi F, Braverman LE, Martino E. Effects of amiodarone administration during pregnancy on neonatal thyroid function and subsequent neurodevelopment. J Endocrinol Invest. 2001;24:116–130.
151. Rocklin RE. Asthma, asthma medications and their effects on maternal/fetal outcomes during pregnancy. Reprod Toxicol. 2011;32:189–197.
152. Park-Wyllie L, Mazzotta P, Pastuszak A, et al. Birth defects after maternal exposure to corticosteroids: prospective cohort study and meta-analysis of epidemiological studies. Teratology. 2000;62:385–392.
153. Carmichael SL, Shaw GM. Maternal corticosteroid use and risk of selected congenital anomalies. Am J Med Genet. 1999;86:242–244.
154. Bakhireva LN, Jones KL, Schatz M, et al. Safety of leukotriene receptor antagonists in pregnancy. J Allergy Clin Immunol. 2007;119:618–625.
155. Sarkar M, Koren G, Kalra S, et al. Montelukast use during pregnancy: a multicentre, prospective, comparative study of infant outcomes. Eur J Clin Pharmacol. 2009;65:1259–1264.
156. Jones KL, Smith DW. Smith’s Recognizable Patterns of Human Malformation. 5th edition. WB Saunders: Philadelphia; 1997.
157. Vitale N, De Feo M, De Santo LS, et al. Dose-dependent fetal complications of warfarin in pregnant women with mechanical heart valves. J Am Coll Cardiol. 1999;33:1637–1641.
158. Dahlman T, Lindvall N, Hellgren M. Osteopenia in pregnancy during long-term heparin treatment: a radiological study post partum. Br J Obstet Gynaecol. 1990;97:221–228.
159. James A. Practice bulletin no. 123: thromboembolism in pregnancy. [Committee on Practice Bulletins—Obstetrics] Obstet Gynecol. 2011;118:718–729.
160. Bates SM, Greer IA, Middeldorp S, et al. VTE, thrombophilia, antithrombotic therapy, and pregnancy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141:e691S–736S.
161. Chan WS, Anand S, Ginsberg JS. Anticoagulation of pregnant women with mechanical heart valves: a systematic review of the literature. Arch Intern Med. 2000;160:191–196.
162. Niebyl JR. Clinical practice. Nausea and vomiting in pregnancy. N Engl J Med. 2010;363:1544–1550.
163. American College of Obstetricians and Gynecologists. Nausea and vomiting of pregnancy. ACOG Practice Bulletin No. 52. [Washington, DC] 2004.
164. Berkovitch M, Mazzota P, Greenberg R, et al. Metoclopramide for nausea and vomiting of pregnancy: a prospective multicenter international study. Am J Perinatol. 2002;19:311–316.
166. Sullivan CA, Johnson CA, Roach H, et al. A pilot study of intravenous ondansetron for hyperemesis gravidarum. Am J Obstet Gynecol. 1996;174:1565–1568.
167. Pasternak B, Svanstrom H, Hviid A. Ondansetron in pregnancy and risk of adverse fetal outcomes. N Engl J Med. 2013;368:814–823.
168. Werler MM. Teratogen update: pseudoephedrine. Birth Defects Res A Clin Mol Teratol. 2006;76:445–452.
169. So M, Bozzo P, Inoue M, Einarson A. Safety of antihistamines during pregnancy and lactation. Can Fam Physician. 2010;56:427–429.
170. Seto A, Einarson T, Koren G. Evaluation of brompheniramine safety in pregnancy. Reprod Toxicol. 1993;7:393–395.
171. Aselton P, Jick H, Milunsky A, et al. First-trimester drug use and congenital disorders. Obstet Gynecol. 1985;65:451–455.
172. Schatz M, Zeiger RS, Harden K, et al. The safety of asthma and allergy medications during pregnancy. J Allergy Clin Immunol. 1997;100:301–306.
173. Pastuszak A, Schick B, D’Alimonte D, et al. The safety of astemizole in pregnancy. J Allergy Clin Immunol. 1996;98:748–750.
174. Einarson A, Bailey B, Jung G, et al. Prospective controlled study of hydroxyzine and cetirizine in pregnancy. Ann Allergy Asthma Immunol. 1997;78:183–186.
175. Paulus W, Schloemp S, Sterzik K, Stoz F. Pregnancy outcome after exposure to cetirizine/levocetirizine in the first trimester—a prospective controlled study. Reprod Toxicol. 2004;19:258.
176. Seto A, Einarson T, Koren G. Pregnancy outcome following first trimester exposure to antihistamines: meta-analysis. Am J Perinatol. 1997;14:119–124.
177. Mylonas I. Antibiotic chemotherapy during pregnancy and lactation period: aspects for consideration. Arch Gynecol Obstet. 2011;283:7–18.
178. Pasternak B, Hviid A. Use of acyclovir, valacyclovir, and famciclovir in the first trimester of pregnancy and the risk of birth defects. JAMA. 2010;304:859–866.
179. Culnane M, Fowler M, Lee SS, et al. Lack of long-term effects of in utero exposure to zidovudine among uninfected children born to HIV-infected women. Pediatric AIDS Clinical Trials Group Protocol 219/076 Teams. JAMA. 1999;281:151–157.
180. McGowan JP, Crane M, Wiznia AA, Blum S. Combination antiretroviral therapy in human immunodeficiency virus–infected pregnant women. Obstet Gynecol. 1999;94:641–646.
181. The Antiretroviral Pregnancy Registry Interim Report. [Available at] http://www.apregistry.com/forms/interim_report.pdf [Accessed June 29, 2013] .
182. CDC. Guidelines for vaccinating pregnant women. April 2013. [Available at] http://www.cdc.gov/vaccines/pubs/preg-guide.htm [Accessed June 29, 2013] .
183. American College of Obstetricians and Gynecologists Committee on Obstetric Practice. Influenza vaccination during pregnancy. ACOG Committee Opinion No. 468. [Washington, DC] Obstet Gynecol. 2010;116:1006–1007.
184. Beigi RH. Influenza during pregnancy: a cause of serious infection in obstetrics. Clin Obstet Gynecol. 2012;55:914–926.
185. Xie HY, Yasseen AS 3rd, Xie RH, et al. Infant outcomes among pregnant women who used oseltamivir for treatment of influenza during the H1N1 epidemic. Am J Obstet Gynecol. 2013;208:293 e1–7.
186. Maslova E, Bhattacharya S, Lin SW, Michels KB. Caffeine consumption during pregnancy and risk of preterm birth: a meta-analysis. Am J Clin Nutr. 2010;92:1120–1132.
187. American College of Obstetricians and Gynecologists. Moderate caffeine consumption during pregnancy. ACOG Committee Opinion No. 462. [Washington, DC] Obstet Gynecol. 2010;116:467–468.
188. Brent RL, Christian MS, Diener RM. Evaluation of the reproductive and developmental risks of caffeine. Birth Defects Res B Dev Reprod Toxicol. 2011;92:152–187.
189. Einarson A, Riordan S. Smoking in pregnancy and lactation: a review of risks and cessation strategies. Eur J Clin Pharmacol. 2009;65:325–330.
190. Drugs and Lactation Database (LactMed). [Available at] http://toxnet.nlm.nih.gov/cgi-bin/sis/htmlgen?LACT [Accessed July 10, 2013] .
191. Breastfeeding and the use of human milk. Pediatrics. 2012;129:e827–e841.
192. Sachs HC, Committee On Drugs. The transfer of drugs and therapeutics into human breast milk: an update on selected topics. Pediatrics. 2013;132:e796–e809.
193. Breastfeeding. Diseases and Conditions. [Available at] http://www.cdc.gov/breastfeeding/disease/index.htm [Accessed July 10, 2013] .
194. Ito S, Alcorn J. Xenobiotic transporter expression and function in the human mammary gland. Adv Drug Deliv Rev. 2003;55:653–665.
195. Anderson GD. Using pharmacokinetics to predict the effects of pregnancy and maternal-infant transfer of drugs during lactation. Expert Opin Drug Metab Toxicol. 2006;2:947–960.
196. Bennett PN, Jensen AA. Drugs and Human Lactation: A Comprehensive Guide to the Content and Consequences of Drugs, Micronutrients, Radiopharmaceuticals, and Environmental and Occupational Chemicals in Human Milk. Elsevier Science Limited: Amsterdam; 1996.
197. Henderson E, Mackillop L. Prescribing in pregnancy and during breast feeding: using principles in clinical practice. Postgrad Med J. 2011;87:349–354.
198. Montgomery A, Hale TW. ABM clinical protocol #15: analgesia and anesthesia for the breastfeeding mother, revised 2012. Breastfeed Med. 2012;7:547–553.
199. Chu TC, McCallum J, Yii MF. Breastfeeding after anaesthesia: a review of the pharmacological impact on children. Anaesth Intensive Care. 2013;41:35–40.
200. Findlay JW, DeAngelis RL, Kearney MF, et al. Analgesic drugs in breast milk and plasma. Clin Pharmacol Ther. 1981;29:625–633.
201. McNamara PJ, Burgio D, Yoo SD. Pharmacokinetics of acetaminophen, antipyrine, and salicylic acid in the lactating and nursing rabbit, with model predictions of milk to serum concentration ratios and neonatal dose. Toxicol Appl Pharmacol. 1991;109:149–160.
202. Bennett PN. World Health Organization. Regional Office for Europe. Drugs and Human Lactation: a Guide to the Content and Consequences of Drugs, Micronutrients, Radiopharmaceuticals, and Environmental and Occupational Chemicals in Human Milk. Elsevier: Amsterdam; 1988.
203. Catz CS, Giacoia GP. Drugs and breast milk. Pediatr Clin North Am. 1972;19:151–166.
204. Wittels B, Scott DT, Sinatra RS. Exogenous opioids in human breast milk and acute neonatal neurobehavior: a preliminary study. Anesthesiology. 1990;73:864–869.
205. Wittels B, Glosten B, Faure EA, et al. Postcesarean analgesia with both epidural morphine and intravenous patient-controlled analgesia: neurobehavioral outcomes among nursing neonates. Anesth Analg. 1997;85:600–606.
206. Baka NE, Bayoumeu F, Boutroy MJ, Laxenaire MC. Colostrum morphine concentrations during postcesarean intravenous patient-controlled analgesia. Anesth Analg. 2002;94:184–187.
207. Koren G, Cairns J, Chitayat D, et al. Pharmacogenetics of morphine poisoning in a breastfed neonate of a codeine-prescribed mother. Lancet. 2006;368:704.
208. Public Health Advisory. Use of codeine by some breastfeeding mothers may lead to life-threatening side effects in nursing babies 8/17/2007. [Available at] http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/PublicHealthAdvisories/ucm054717.htm [Accessed June 29, 2013] .
209. Madadi P, Ross CJ, Hayden MR, et al. Pharmacogenetics of neonatal opioid toxicity following maternal use of codeine during breastfeeding: a case-control study. Clin Pharmacol Ther. 2009;85:31–35.
210. Lam J, Kelly L, Ciszkowski C, et al. Central nervous system depression of neonates breastfed by mothers receiving oxycodone for postpartum analgesia. J Pediatr. 2012;160:33–37.
211. Madadi P, Avard D, Koren G. Pharmacogenetics of opioids for the treatment of acute maternal pain during pregnancy and lactation. Curr Drug Metab. 2012;13:721–727.
212. Chisholm CA, Kuller JA. A guide to the safety of CNS-active agents during breastfeeding. Drug Saf. 1997;17:127–142.
214. Fortinguerra F, Clavenna A, Bonati M. Psychotropic drug use during breastfeeding: a review of the evidence. Pediatrics. 2009;124:e547–e556.
215. Cole AP, Hailey DM. Diazepam and active metabolite in breast milk and their transfer to the neonate. Arch Dis Child. 1975;50:741–742.
216. Nau H, Rating D, Hauser I, et al. Placental transfer and pharmacokinetics of primidone and its metabolites phenobarbital, PEMA and hydroxyphenobarbital in neonates and infants of epileptic mothers. Eur J Clin Pharmacol. 1980;18:31–42.
217. Madadi P, Ito S. Perinatal exposure to maternal lamotrigine: clinical considerations for the mother and child. Can Fam Physician. 2010;56:1132–1134.
218. Cruikshank DP, Varner MW, Pitkin RM. Breast milk magnesium and calcium concentrations following magnesium sulfate treatment. Am J Obstet Gynecol. 1982;143:685–688.
219. Pearlstein T, Howard M, Salisbury A, Zlotnick C. Postpartum depression. Am J Obstet Gynecol. 2009;200:357–364.
220. Rowe H, Baker T, Hale TW. Maternal medication, drug use, and breastfeeding. Pediatr Clin North Am. 2013;60:275–294.
221. Sie SD, Wennink JM, van Driel JJ, et al. Maternal use of SSRIs, SNRIs and NaSSAs: practical recommendations during pregnancy and lactation. Arch Dis Child Fetal Neonatal Ed. 2012;97:F472–F476.
222. Sykes PA, Quarrie J, Alexander FW. Lithium carbonate and breast-feeding. Br Med J. 1976;2:1299.
223. Schou M, Amdisen A. Lithium and pregnancy. 3. Lithium ingestion by children breast-fed by women on lithium treatment. Br Med J. 1973;2:138.
224. White WB, Andreoli JW, Wong SH, Cohn RD. Atenolol in human plasma and breast milk. Obstet Gynecol. 1984;63:42S–44S.
225. Loughnan PM. Digoxin excretion in human breast milk. J Pediatr. 1978;92:1019–1020.
226. Hartikainen-Sorri AL, Heikkinen JE, Koivisto M. Pharmacokinetics of clonidine during pregnancy and nursing. Obstet Gynecol. 1987;69:598–600.
227. Katz FH, Duncan BR. Entry of prednisone into human milk. N Engl J Med. 1975;293:1154.
228. McKenzie SA, Selley JA, Agnew JE. Secretion of prednisolone into breast milk. Arch Dis Child. 1975;50:894–896.
229. Ost L, Wettrell G, Bjorkhem I, Rane A. Prednisolone excretion in human milk. J Pediatr. 1985;106:1008–1011.
230. Orme ML, Lewis PJ, de Swiet M, et al. May mothers given warfarin breast-feed their infants? Br Med J. 1977;1:1564–1565.
231. De Swiet M, Lewis PJ. Excretion of anticoagulants in human milk. N Engl J Med. 1977;297:1471.
232. Lione A, Scialli AR. The developmental toxicity of the H1 histamine antagonists. Reprod Toxicol. 1996;10:247–255.
233. Anderson PO. Drug use during breast-feeding. Clin Pharm. 1991;10:594–624.
234. Wilson JT, Brown RD, Cherek DR, et al. Drug excretion in human breast milk: principles, pharmacokinetics and projected consequences. Clin Pharmacokinet. 1980;5:1–66.
235. Meyer LJ, de Miranda P, Sheth N, Spruance S. Acyclovir in human breast milk. Am J Obstet Gynecol. 1988;158:586–588.
236. Berlin CM Jr, Denson HM, Daniel CH, Ward RM. Disposition of dietary caffeine in milk, saliva, and plasma of lactating women. Pediatrics. 1984;73:59–63.