Chapter 40 Phakomatoses and Allied Conditions
The disorders referred to as phakomatoses are notable for their dysplastic nature and tendency to form tumors in various organs, particularly the nervous system. Bielschowsky observed these characteristic features in neurofibromatosis and tuberous sclerosis complex [Bielschowsky, 1914, 1919], and van der Hoeve called particular attention to these disorders and named the disease category the “phakomatoses” (Greek phakos, “mole” or “birthmark”) [Van der Hoeve, 1923]. Von Hippel–Lindau disease and Sturge–Weber syndrome were thought to belong to this category of diseases [Van der Hoeve, 1923, 1932] and Louis-Bar identified a syndrome, ataxia-telangiectasia, that she believed also had the typical clinical characteristics of the phakomatoses [Louis-Bar, 1941]. Some of these conditions have been referred to as “neurocutaneous disorders” because of the frequent involvement of the skin in addition to the nervous system. Cutaneous features are not present in all phakomatoses, however (e.g., von Hippel–Lindau syndrome), and many include features outside the skin and nervous system, so the term can be misleading. Box 40-1 lists disorders associated with specific cutaneous findings; Table 40-1 summarizes the phakomatoses.
Box 40-1 Neurocutaneous Disorders: Specific Cutaneous Abnormalities and Associated Disorders


















Table 40-1 Phakomatoses and their Clinical Features
| Syndrome | Common Non-neurologic Features | Common Neurologic Features |
|---|---|---|
| Neurofibromatosis type 1 | Café-au-lait spots, malignant peripheral nerve sheath tumor, skeletal dysplasia | Neurofibromas, optic glioma, learning disabilities |
| Neurofibromatosis type 2 | Posterior subcapsular cataract | Vestibular schwannomas, other cranial and peripheral nerve schwannomas, meningiomas, ependymomas |
| Schwannomatosis | Schwannomas | |
| Tuberous sclerosis complex | Hypopigmented macules, collagenous plaques, angiofibroma, renal angiomyolipoma, pulmonary lymphangiomyomatosis, periungual fibromas | Cortical dysplasias, subependymal nodules, subependymal giant cell astrocytomas, seizures |
| Sturge–Weber syndrome | Port-wine stain | Leptomeningeal angiomatosis, seizures |
| von Hippel–Lindau disease | Ocular hemangioblastoma, renal cell carcinoma, pheochromocytoma, endolymphatic sac tumor | Hemangioblastomas |
| Maffucci’s syndrome | Multiple endochondromas | |
| Epidermal nevus syndrome | Epidermal nevus, skeletal dysplasia | Seizures, developmental impairment |
| Parry–Romberg syndrome | Facial hemiatrophy | |
| Neurocutaneous melanosis | Multiple melanocytic nevi, melanoma | Leptomeningeal melanosis, seizures, hydrocephalus, Dandy–Walker malformation |
| Klippel–Trenaunay–Weber syndrome | Hemihypertrophy, angioma | Macrocephaly, hydrocephalus |
| Incontinentia pigmenti | Females only; hyperpigmented skin lesions, abnormalities of teeth, hair, and bone | Seizures, developmental impairment |
| Hypomelanosis of Ito | Hypopigmented skin lesions | Seizures |
| Wyburn–Mason syndrome | Retinal arteriovenous malformations | Cerebral arteriovenous malformations |
This chapter surveys the clinical features of the three major phakomatoses: neurofibromatosis, tuberous sclerosis complex, and von Hippel–Lindau syndrome. Also included in this chapter are a number of additional disorders that are commonly considered along with the phakomatoses, including vascular malformation syndromes, pigmentary disorders, and several other rare disorders. Ataxia-telangiectasia is considered in detail in Chapter 67 on the hereditary ataxias.
The Neurofibromatoses
It was only with the report of Young et al. [1970] that the distinction between a peripheral and a central form of neurofibromatosis came to be recognized. The classification was formalized by the National Institutes of Health (NIH) Consensus Development Conference in 1987 [Stumpf, 1988], with the peripheral form of neurofibromatosis referred to as NF1, and the central form, the hallmark of which is bilateral vestibular schwannomas, called NF2. The distinction between the disorders was subsequently verified when the two genes were found to be distinct, first by mapping and later by cloning. More recently, a third disorder, referred to as schwannomatosis, has been split out [MacCollin et al., 2005]. This disorder is characterized by the development of multiple schwannomas of cranial nerves except the vestibular nerve, and of spinal and peripheral nerves.
Neurofibromatosis Type 1
Neurofibromatosis type 1 (NF1) is transmitted as an autosomal-dominant trait and is notable for its great variability of expression. It can involve not only the peripheral and central nervous systems, but also many other systems, including the skin, bone, endocrine, gastrointestinal, and vascular systems. Although von Recklinghausen is credited with the initial clinical and pathologic account of this disease in 1882 [von Recklinghausen, 1882], he cited Tilesius for the first description of a patient with multiple fibrous skin tumors. Wishart [Wishart, 1822] and Smith [Smith, 1849] also provided clinical accounts of the disorder before the report of von Recklinghausen, although early reports failed to recognize the distinction between the disorders known as NF1 and NF2. Diagnostic criteria for NF1 are presented in Box 40-2.
Box 40-2 Diagnostic Criteria for Neurofibromatosis 1
 Six or more café-au-lait macules more than 5 mm in greatest diameter in prepubertal children, and more than 15 mm in greatest diameter in postpubertal children
Six or more café-au-lait macules more than 5 mm in greatest diameter in prepubertal children, and more than 15 mm in greatest diameter in postpubertal children





(Modified from Stumpf D. Consensus development conference of neurofibromatosis. Arch Neurol 1988;45:575–578; Gutmann DH et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 1997;278: 51–57.)
Clinical Characteristics
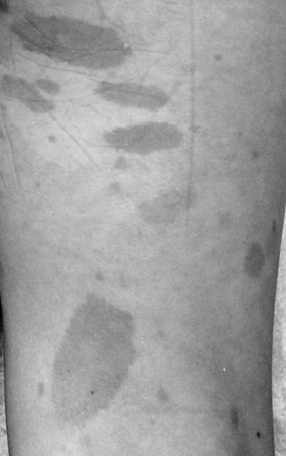
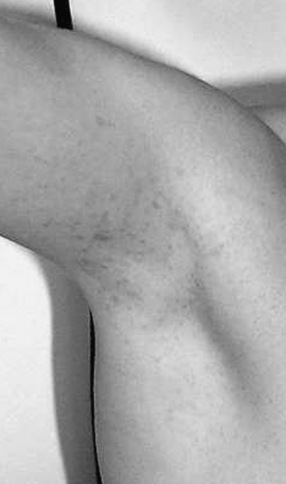
In NF1, the usual presenting signs are cutaneous manifestations. These skin changes include café-au-lait macules, cutaneous neurofibromas, hypopigmented macules, patchy and diffuse areas of hyperpigmentation, juvenile xanthogranulomas, and angiomas. Café-au-lait macules usually are present at birth and range in size from a few millimeters to centimeters [Crowe et al., 1956] (Figure 40-1). They do not significantly increase in number after the first 2 years of life. Six or more café-au-lait macules measuring at least 5 mm across before puberty or 15 mm after puberty constitute one diagnostic criterion for NF1. Rare persons with variant forms of neurofibromatosis, such as spinal neurofibromatosis [Messiaen et al., 2003; Korf et al., 2005], may lack café-au-lait macules, and one rare NF1 gene mutation is associated with café-au-lait macules and no neurofibromas in affected individuals [Upadhyaya et al., 2007]. Individuals with mutation in the SPRED1 gene may also present with multiple café-au-lait macules, skinfold freckles, and macrocephaly, but do not appear to develop neurofibromas or other tumor-related NF1 complications [Brems et al., 2007; Messiaen et al., 2009]. Nevertheless, in a majority of children who present with six or more café-au-lait macules and do not have an alternative diagnosis, signs of NF1 eventually develop [Korf, 1992]. Usually the second sign to appear is skinfold freckling [Crowe, 1964] (Figure 40-2). Freckles begin in the inguinal region in children at 3–4 years of age, and eventually appear in the axillae, at the base of the neck, and in the inframammary region in females. Areas of freckling also can be found over the trunk and extremities. Diffuse, patchy areas of hyperpigmentation also may appear, sometimes overlying plexiform neuromas [Riccardi, 1980]. Areas of hypopigmentation or hypovascularity also may occur. Juvenile xanthogranulomas appear as firm yellowish papules in infants and young children, and eventually regress. Although an association with leukemia has been suggested [Zvulunov, 1996], this has not been verified.
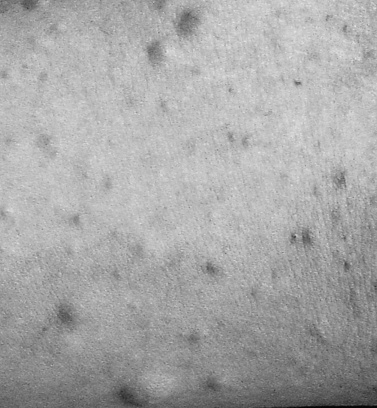
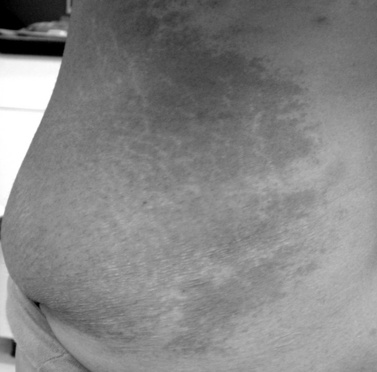
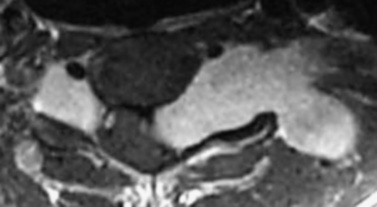
Cutaneous neurofibromas are a prominent finding in NF1 and are located in the dermis or adjacent to it (Figure 40-3). They are discrete, soft or firm papules, ranging in size from a few millimeters to several centimeters, can be flat, sessile, or pedunculated, and can be readily impressed into the skin below. Neurofibromas can develop at any time and in any location, and may affect any component of the peripheral nervous system, from the dorsal root ganglion to the terminal nerve twigs. Plexiform neuromas represent tumors that involve a longitudinal section of nerve and can involve multiple branches of a major nerve [Korf, 1999]. Near the surface of the body they can cause thickening and hypertrophy of the skin and soft tissues (Figure 40-4). They may occur deeper in the body and be detected only by imaging [Tonsgard et al., 1998]. Tumors of the orbit or limbs can cause major physical deformity. Plexiform neurofibromas can be congenital lesions, often growing rapidly in the early months of life; they then may remain quiescent for long periods of time or grow unpredictably. The tumors are easily visualized by magnetic resonance imaging (MRI), and display a characteristic “target sign.” Volumetric MRI may be helpful in following their growth [Dombi et al., 2007; Cai et al., 2009]. Neurofibromas originating at the dorsal roots may grow in a dumbbell shape and invade the spinal canal (Figure 40-5], sometimes causing spinal cord compression. The gastrointestinal tract can also be affected by growth of neurofibromas or ganglioneuromas. These tumors can cause intestinal obstruction or bleeding.
Ophthalmologic features of NF1 include Lisch nodules, glaucoma, and optic glioma. Iris Lisch nodules are melanocytic hamartomas that are highly specific to NF1 [Lisch, 1937; Lewis and Riccardi, 1981]. Their appearance is age-dependent, usually beginning at the age of approximately 6 years. Lisch nodules occur in approximately 95 percent of adults with the disorder [Lubs et al., 1991] and are therefore helpful in diagnosis. Glaucoma usually occurs when a plexiform neurofibroma involves the upper eyelid (Figure 40-6) [Morales et al., 2009]. Orbital plexiform neurofibroma, arising from the trigeminal nerve, often is associated with sphenoid dysplasia, and can present with pulsating exophthalmos or enophthalmos [Jacquemin et al., 2002].
Optic pathway gliomas are found in approximately 15 percent of patients [Lewis et al., 1984]. Most are asymptomatic, but these tumors can manifest with symptoms of decreased visual acuity, visual field defects, or precocious puberty [Listernick et al., 2007]. The glioma can involve the optic nerves, chiasm, optic radiations, and hypothalamus (Figure 40-7); it rarely manifests as the diencephalic syndrome of infancy or precocious puberty. Optic gliomas are pilocytic astrocytomas, but often are slow-growing.
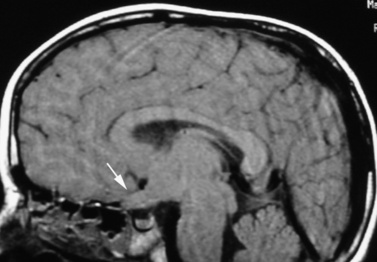
Fig. 40-7 Sagittal magnetic resonance imaging scan of brain showing a glioma of the optic chiasm (arrow).
Aside from optic gliomas, astrocytomas of the cerebrum, brainstem, and cerebellum are the most common intracranial tumors encountered in NF1 [Albers and Gutmann, 2009]. Malignant peripheral nerve sheath tumor occurs in upwards of 8–13 percent of affected persons [Evans et al., 2002]. These manifest with pain or sudden growth, usually within a pre-existing plexiform neurofibroma [D’Agostino, Soule and Miller, 1963]. Various other neoplastic disorders occur more frequently in patients with NF1 than in the general population, including leukemia, especially juvenile myelomonocytic leukemia [Stiller, Chessells and Fitchett, 1994] and pheochromocytoma [Walther et al., 1999a].
Macrocephaly and short stature are common in NF1 and scoliosis has been reported to occur in 10–40 percent of patients [Crawford and Herrera-Soto, 2007]. Scoliosis usually does not develop before the age of 6 years and most commonly involves the thoracic spine. Bowing of the tibia, fibula, and other long bones can be present in early life, with occurrence of spontaneous fractures at the junction of the middle and distal thirds of the bone shaft, resulting in pseudarthrosis [Elefteriou et al., 2009; Stevenson et al., 1999]. Non-ossifying fibromas may occur and can present with pain or fracture [Howlett et al., 1998]. There is also evidence for decreased bone mineral density in children and adults with NF1, which may contribute to an increased risk of fracture [Stevenson et al., 2008; Kuorilehto et al., 2005].
Approximately 50 percent of patients have learning disabilities, with no specific pattern unique to those with NF1 [Hyman, Arthur Shores and North, 2006]. Both verbal and nonverbal disabilities occur, as well as attention-deficit disorder [Mautner et al., 2002] and hypotonia [Souza et al., 2009]. Fewer than 10 percent have mental retardation, and most of these patients have large deletions of the NF1 gene [Kayes et al., 1994]. Seizures occur in approximately 6–10 percent of patients [Korf, Carrazana and Holmes, 1993]. The pathogenesis of the cognitive phenotype is not known. Abnormal cortical architecture and heterotopias have been reported in the brains of some patients with severe cognitive deficits [Rosman and Pearce, 1967]. It has been suggested that the areas of enhanced T2 signal intensity characteristically seen in the brains of children with NF1 (Figure 40-8) may be associated with learning disabilities [Hyman et al., 2007]. These lesions occur in the basal ganglia, brainstem, cerebellum, and internal capsule; tend to disappear with age [Hyman et al., 2003; Ferner et al., 1993]; and are characterized by increased myelin water content and gliosis [DiPaolo et al., 1995].
Vascular anomalies in NF1 include regions of intimal proliferation and fibromuscular changes in small arteries [Friedman et al., 2002]. Renal artery stenosis can lead to hypertension in children [Fossali et al., 2000], and involvement of other vessels can cause vascular insufficiency or hemorrhage as a result of arterial wall dissection [Hinsch et al., 2008]. Stenosis of the internal carotid artery can lead to moyamoya disease and stroke [Cairns and North, 2008], although lesions often are asymptomatic. The stenotic changes can arise spontaneously in children with NF1, but frequently develop after radiation therapy for brain tumors in young children [Kestle, Hoffman and Mock, 1993].
Pathology
Neurofibromas consist of a mixture of cell types, including Schwann cells, fibroblasts, perineurial cells, and mast cells [Pineda, 1965]. They are polyclonal [Fialkow et al., 1971], but genetic studies have confirmed that the “tumor cell” is the Schwann cell [Sherman et al., 2000; Zhu et al., 2002], or in the case of dermal neurofibromas, stem cells referred to as skin-derived precursors [Le et al., 2009]. The other cell types present in the lesion apparently proliferate as a secondary phenomenon, perhaps in response to stimulation by cytokines [Yang et al., 2003]. Mast cells are present in large numbers and have been suspected of being a source of cytokines [Yang et al., 2008]. In plexiform neurofibromas, the pathologic process extends across multiple nerve fascicles instead of occurring at a focal site in a nerve, and may extend across branches of a larger nerve. Malignant peripheral nerve sheath tumor manifests as a malignant tumor of Schwann cell origin, although sometimes rhabdoid elements are present in such tumors [Buck, Mahboubi and Raney, 1977]. Most, if not all, of these neoplasms arise from pre-existing tumors, usually plexiform neurofibromas. The pathology of other NF1-associated lesions is less well understood than that of the neurofibroma.
Genetics
NF1, inherited as an autosomal-dominant trait, has an estimated prevalence of 1 in 3000; about half of cases are new mutations [Crowe, Schull and Neel, 1956; Huson, Harper and Compston, 1988]. The NF1 gene is located at 17q11.2 and encodes a 3818-amino-acid protein referred to as neurofibromin [Cawthon et al., 1990a, b; Viskochil et al., 1990; Wallace et al., 1990]. Neurofibromin is expressed in multiple cell types but is highly expressed in Schwann cells, oligodendrocytes, and neurons [Daston et al., 1992]. The protein includes a functional GTPase-activating protein (GAP) domain that regulates conversion of Ras-guanosine triphosphate to Ras-guanosine diphosphate [Ballester et al., 1990; Xu et al., 1990a, b]. Ras is a membrane-bound intracellular signaling molecule that is activated by complexing with guanosine triphosphate (GTP) on ligand binding to membrane receptor tyrosine kinases. GAP proteins regulate this process by stimulating a GTPase activity that is intrinsic to Ras [Bernards, 2003]. Neurofibromin functions as a tumor suppressor gene with respect to neurofibroma formation [Sherman et al., 2000; Cichowski and Jacks, 2001; Zhu et al., 2002]. The germline mutation of one NF1 allele constitutes the first “hit”; the second hit is a somatic mutation [Upadhyaya et al., 2004] in a Schwann cell that causes that cell to proliferate and attract other cells, such as mast cells, fibroblasts, and perineurial cells, which also proliferate. These cells, being heterozygous for the NF1 mutation, may be hypersensitive to cytokine stimulation [Vogel et al., 1995]. Transformation to malignancy requires additional genetic changes, such as mutation of p53 [Legius et al., 1994]. Biallelic NF1 gene mutation also occurs in melanocytes within café-au-lait macules [Maertens et al., 2007] and in dysplastic bone tissue [Stevenson et al., 2006]. It is unclear whether other dysplastic lesions also occur as a result of a tumor suppressor mechanism, or whether haploinsufficiency of neurofibromin expression itself causes these lesions. Mice rendered heterozygous for an NF1 mutation do display cognitive deficits [Costa et al., 2002], suggesting that haploinsufficiency might account for some of the NF1 phenotype.
NF1 exhibits a wide range of variability of expression and complete penetrance. Mutations are widely scattered across the gene and include a wide variety of mutational mechanisms [Messiaen et al., 2000]. Most of the mutations lead to decreased level of expression of neurofibromin or complete lack of expression. Few genotype–phenotype correlations have been established. Large deletions that include the NF1 gene and multiple contiguous genes over a 1.5-Mb region tend to lead to a particularly severe form of NF1, with mental retardation, early onset of large numbers of neurofibromas, facial dysmorphism, and increased risk of cancer [De Raedt et al., 2003; Kayes et al., 1994; Wu et al., 1995]. Another variant form of neurofibromatosis, familial spinal neurofibromatosis, may represent an additional genotype–phenotype correlation. Persons with this disorder have multiple spinal neurofibromas and subcutaneous tumors but lack skinfold freckling and dermal tumors, and tend to have missense mutations or splicing mutations [Korf, Henson and Stemmer-Rachamimov, 2005]. Individuals with a three-base deletion in exon 17 have only café-au-lait spots and do not develop neurofibromas [Upadhyaya et al., 2007]. Approximately 50 percent of cases of NF1 occur sporadically, as a result of a new mutation of the NF1 gene. Because of the high penetrance of the disorder, unaffected parents of a sporadically affected child have a low risk of recurrence, barring the rare instance of germline mosaicism [Lazaro et al., 1995]. Somatic mosaicism for NF1 may manifest with segmental distribution of features [Tinschert et al., 2000; Vandenbroucke et al., 2004]. Genetic testing for diagnosis of NF1 is available on a clinical basis. It is used to confirm a diagnosis in patients who fulfill only a single diagnostic criterion, to characterize patients with unusual clinical presentations, and to enable prenatal testing. The discovery of mutation in the SPRED1 gene accounting for patients with multiple café-au-lait spots but lacking other features of NF1 [Brems et al., 2007] (now referred to as “Legius syndrome”), provides additional rationale for genetic testing in young children with multiple café-au-lait spots. The majority of mutations are found in the NF1 gene, making it cost-effective to begin with NF1 testing, followed by SPRED1 testing if no NF1 mutation is found [Messiaen et al., 2009].
Management
Treatment of patients with neurofibromatosis is symptomatic. Affected persons should be followed on a regular basis by a physician who is familiar with the disorder to recognize treatable complications early and to provide anticipatory guidance and counseling. Genetic counseling should be provided. Controversy surrounds the use of imaging, especially MRI, in screening patients with NF1. Most of the lesions that will be identified are not amenable to treatment, so such testing may create needless anxiety, and in children the procedure carries the risks associated with sedation. The value of the “baseline” examination is questionable because most of the lesions of NF1 are slow-growing and will be followed both clinically and by imaging once they come to attention [Listernick et al., 1994]. Current consensus guidelines do not recommend routine imaging [Gutmann et al., 1997], although care should be individualized for specific clinical needs.
Neurofibromas of the peripheral nerves need not be removed unless they are subject to repeated irritation and trauma or develop malignant change. Some plexiform neuromas can be removed for cosmetic reasons, although complete resection is difficult and regrowth is common [Needle et al., 1997]. Malignant tumors are managed with appropriate neurosurgical measures, radiation therapy, and chemotherapy. Optic gliomas tend to behave in an indolent manner and therefore are followed clinically without treatment in asymptomatic children [Listernick et al., 2007]. Symptomatic tumors most often are treated with chemotherapy [Packer et al., 1997]; radiation therapy may be associated with second malignant tumors [Sharif et al., 2006] or moyamoya disease [Desai et al., 2006; Kestle, Hoffman and Mock, 1993]. Malignant peripheral nerve sheath tumors tend to be highly malignant, so early diagnosis is essential [Evans et al., 2002]. Patients with unexplained pain or growth of a neurofibroma should be evaluated, with consideration of biopsy. Positron emission tomography (PET) scanning may be helpful in distinguishing a malignant peripheral nerve sheath tumor from plexiform neurofibroma [Ferner et al., 2000; Karabatsou et al., 2009; Warbey et al., 2009].
Clinical trials of drugs to treat specific complications are on-going. These include the use of statins to treat learning disabilities [Krab et al., 2008] and several experimental treatments for neurofibromas [Babovic-Vuksanovic et al., 2007; Gupta et al., 2003; Packer and Rosser, 2002; Packer et al., 2002].
Neurofibromatosis Type 2
Clinical Characteristics and Pathology
Diagnostic criteria for NF2 are presented in Box 40-3 [Gutmann et al., 1997; Stumpf, 1988; Baser et al., 2002]. The defining feature of NF2 is the occurrence of bilateral vestibular schwannomas. Age at onset is highly variable, ranging from early childhood to the seventh decade and beyond [Evans 1999; Mautner et al., 1993]. In view of this variability in age at onset of vestibular tumors, NF2 also should be considered in patients with early onset of associated tumors or those with combinations of associated tumors.
Box 40-3 Diagnostic Criteria for Neurofibromatosis 2




Presumptive NF2







* Gutmann DH et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 1997;278: 51–57.
† Baser ME et al. Evaluation of clinical diagnostic criteria for neurofibromatosis 2. Neurology 2002;59(11):1759–1765.)
(Based on
Vestibular schwannomas commonly manifest with tinnitus and/or hearing loss, and may cause problems with balance [Evans, 1999]. Audiology and auditory brainstem-evoked response testing can be helpful, but definitive diagnosis is based on MRI findings (Figure 40-9). Early tumors may be confined to the internal auditory canal and require careful search with thin MRI slices to be detected. Schwannomas can occur along any other cranial nerve, the fifth being most common after the eighth. Schwannomas also may occur along spinal nerves, with the potential for causing radiculopathy or cord compression, or along peripheral nerves. In some patients, a polyneuropathy develops as a result of Schwann cell proliferation around peripheral nerves [Gijtenbeek et al., 2001; Hagel et al., 2002]. Dermal schwannomas appear as plaquelike lesions, often with associated hair growth. Café-au-lait macules are not a reliable indicator of NF2, unlike in NF1 [Mautner et al., 1997]. Other major central nervous system tumors associated with NF2 are meningiomas and ependymomas. Multiple meningiomas may not be surgically resectable and can be responsible for significant morbidity. Virtually the entire NF2 phenotype is characterized by proliferative lesions; the one exception is the occurrence of posterior subcapsular cataracts or cortical wedge opacities [Pearson-Webb et al., 1986].
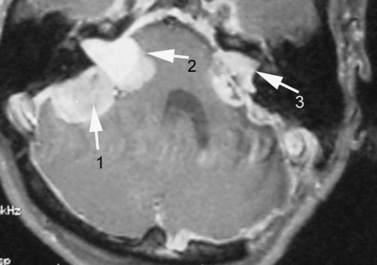
Genetics
NF2 is transmitted as an autosomal-dominant trait with complete penetrance and variable expression. Prevalence is estimated at approximately 1 in 60,000, and birth incidence at 1 in 30,000 [Evans, 2009; Evans et al., 2010]. Approximately half of cases occur sporadically as a result of new mutation. The NF2 gene was mapped to chromosome 22 [Rouleau et al., 1987], and the responsible gene was identified by two groups in 1993 [Rouleau et al., 1993; Trofatter et al., 1993]. The protein is variously referred to as schwannomin or merlin (the latter an acronym for moesin, ezrin, and radixin-like protein]. Merlin is a cytoskeletal protein that appears to play a role in the control of cell growth in tissues [Xiao, Chernoff and Testa, 2003]. Schwannomas are clonal tumors, and the NF2 gene acts as a tumor suppressor in formation of these tumors, as well as other NF2-associated tumors [Seizinger et al., 1987]. Genetic testing for NF2 is available for diagnostic purposes. Some genotype–phenotype correlations have been identified; specifically, missense or splicing mutations tend to predict milder disease than do mutations that lead to protein truncation [Parry et al., 1996; Ruttledge et al., 1996]. Somatic mosaicism for NF2 mutation may produce localized disease or ameliorate disease severity [Baser et al., 2000a].
Management
Patients benefit from multidisciplinary care at a center with experience in dealing with the varied manifestations of the disorder [Evans et al., 1993]. Management of tumors associated with NF2 is primarily surgical [Evans et al., 2005]. Timing of surgery and the decision to treat one or both vestibular tumors depends on tumor size, degree of hearing loss, and involvement of other cranial nerves or compression of the brainstem. Stereotactic radiosurgery is also used for the treatment of vestibular schwannomas [Battista, 2009], though there may be an increased risk of malignancy in residual tumor [Baser et al., 2000b]. Recent trials with the vascular endothelial growth factor (VEGF) inhibitor bevacizumab have shown promising results in reduction in size of vestibular schwannomas [Plotkin et al., 2009; Mautner et al., 2010].
Schwannomatosis
Schwannomatosis is a more recently recognized entity [MacCollin et al., 1996; Evans et al., 1997], characterized only by the occurrence of schwannomas on cranial and spinal nerves other than the vestibular nerve. It often manifests with pain or nerve compression. Diagnostic criteria are provided in Box 40-4. Schwannomatosis is most commonly sporadic, but familial cases have been observed, in which case inheritance is autosomal-dominant with incomplete penetrance. The gene responsible for the disorder is designated INI1 (also designated SMARCB1; Hulsebos et al., 2007; Boyd et al., 2008], and encodes a protein component of a chromatin remodeling complex. It is located on chromosome 22 near the NF2 locus but is distinct from that locus. Symptomatic tumors are treated surgically. Many patients require management for chronic pain.
Box 40-4 Diagnostic Criteria for Schwannomatosis
 Patient does not fulfill diagnostic criteria for NF2, and has no vestibular schwannoma on high-resolution MRI and no NF2 mutation
Patient does not fulfill diagnostic criteria for NF2, and has no vestibular schwannoma on high-resolution MRI and no NF2 mutationDefinite Diagnosis


Possible Diagnosis
 Age under 30 years AND two or more nonintradermal schwannomas, at least one with histologic confirmation
Age under 30 years AND two or more nonintradermal schwannomas, at least one with histologic confirmation


(From MacCollin M et al. Diagnostic criteria for schwannomatosis. Neurology 2005;64(11):1838–1845; Baser ME et al. Increasing the specificity of diagnostic criteria for schwannomatosis. Neurology 2006;66(5):730–732.)
Tuberous Sclerosis Complex
Tuberous sclerosis complex is a disorder of autosomal-dominant inheritance that affects multiple organ systems, resulting in manifold clinical expressions. Tuberous sclerosis complex is currently recognized as one of the most common single-gene disorders seen in children and adults, with an estimated incidence of 1 in 5800 live births [Osborne et al., 1991] The first description of tuberous sclerosis complex was by von Recklinghausen, who described a newborn who had died of respiratory distress and was found at postmortem examination to have multiple cardiac tumors and a “great number of cerebral scleroses [von Recklinghausen, 1862].” Bourneville usually is credited with the first detailed description of the cerebral manifestations of the disease, describing “sclérose tubéreuse,” indicating the superficial resemblance of the lesions of a potato [Bourneville and Brissard, 1880]. He attached no significance to the facial skin rash of his first patient, calling it acne rosacea, but he and Brissard believed that the renal tumors and cerebral scleroses were associated findings [Bourneville and Brissard, 1900]. Facial angiofibromas, previously referred to as adenoma sebaceum, were independently described in several reports, but Vogt emphasized the association of adenoma sebaceum and the cerebral scleroses described by Bourneville [Vogt, 1908]. He also described a “classic” triad of clinical features comprising mental retardation, intractable epilepsy, and adenoma sebaceum, which is now known to be present in less than one-third of patients with tuberous sclerosis complex.
Clinical Characteristics
Diagnostic criteria are provided in Box 40-5. The clinical presentation of tuberous sclerosis complex depends on the age of the patient, the organs involved, and the severity of involvement. Of importance, both the brain and the skin have more than one major criterion for diagnosis; therefore, a diagnosis of definite tuberous sclerosis complex can be based on skin findings alone, or on neuroimaging findings alone.
Box 40-5 Diagnostic Criteria for Tuberous Sclerosis Complex

























(From Roach ES et al. Tuberous sclerosis complex consensus conference: Revised clinical diagnostic criteria. J Child Neurol 1998;13(12):624–8.)
Epilepsy is the most common presenting symptom in tuberous sclerosis complex and also is the most common medical disorder. In up to 80–90 percent of persons with tuberous sclerosis complex, seizures will develop during their lifetime, with the onset most frequently in childhood [Gomez, 1999; Thiele, 2004; Chu-Shore et al., 2009]. A majority of children with tuberous sclerosis complex have the onset of seizures during the first year of life, and approximately one-third develop infantile spasms. Almost all seizure types can be seen in persons with tuberous sclerosis complex, including tonic, clonic, tonic-clonic, atonic, myoclonic, atypical absence, partial, and complex partial. Only “pure” absence seizures are not observed.
Infantile spasms will develop in approximately one-third of children with tuberous sclerosis complex, although some reports suggest an incidence as high as 75 percent [Riikonen and Simell, 1990; Fukushima et al., 1998; Hamano et al., 2003; Husain et al., 2000]. Tuberous sclerosis complex is thought to be the most common single cause of infantile spasms, and in some series, 25 percent of symptomatic infantile spasms are secondary to tuberous sclerosis complex. Partial complex seizures precede infantile spasms in approximately one-third of patients with tuberous sclerosis complex in whom infantile spasms develop [Curatolo et al., 2001, 2002]. A strong association between the presence of infantile spasms in tuberous sclerosis complex and subsequent developmental impairment has been noted, although children with tuberous sclerosis complex and infantile spasms can have a normal cognitive outcome [Goh et al., 2005; Yamamoto et al., 1987; Muzykewicz et al., 2009].
The electroencephalogram (EEG) in infantile spasms associated with tuberous sclerosis complex often demonstrates hypsarrhythmia or modified hypsarrhythmia. It is important to realize, however, that the EEG, although usually abnormal, frequently does not have the features of hypsarrhythmia; in some series, up to 70 percent of children with tuberous sclerosis complex and infantile spasms did not have the characteristics of hypsarrhythmia [Curatolo et al., 2001]. Several reports have characterized the EEG patterns of persons with tuberous sclerosis complex and have found a high incidence of abnormalities, including diffuse slowing and epileptiform features [Ganji and Hellman, 1985; Westmoreland, 1999; Muzykewicz et al., 2009].
Tuberous sclerosis complex is associated with a wide range of cognitive and behavioral manifestations. Approximately one-half of persons with tuberous sclerosis complex have normal intelligence, whereas the other half have some degree of cognitive impairment, ranging from mild learning disabilities to severe mental retardation. A bimodal distribution of cognitive abilities is evident, with affected persons falling into a severely cognitively impaired group or a group with normal intelligence [Gillberg et al., 1994; Winterkorn et al., 2007]. Risk factors for cognitive impairment include a history of infantile spasms, intractable epilepsy, and a mutation in the TSC2 gene. Persons with tuberous sclerosis complex, particularly those with cognitive impairment, also are at high risk for developmental disorders. Autistic spectrum disorders affect up to 50 percent of persons with tuberous sclerosis complex [Wiznitzer, 2004; Curatolo et al., 2004; Smalley, 1998], and attention-deficit hyperactivity and related disorders also are common, affecting approximately 50 percent of the patients [de Vries and Watson, 2008]. During adolescence and adulthood, anxiety disorders, depression, or mood disorders develop in a majority of patients with tuberous sclerosis complex [Muzykewicz et al., 2007; Raznahan et al., 2006; Pulsifer et al., 2007].
Cutaneous manifestations are found in up to 96 percent of patients with tuberous sclerosis complex [Gomez et al., 1987; Webb et al., 1996]. Angiofibroma, the skin manifestation initially described in the disorder as adenoma sebaceum, typically appears between the ages of 1 and 4 years and can progress through childhood and adolescence [Gomez et al., 1987; Webb et al., 1996; Pampiglione and Moynahan, 1976]. These lesions typically are pink or red papules that appear in patches or in a butterfly distribution on or about the nose, cheeks, and chin (Figure 40-10).
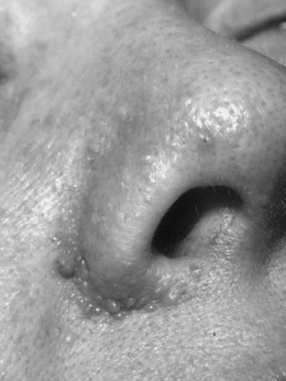
Fig. 40-10 Typical angiofibroma in an adult with tuberous sclerosis complex.
(Courtesy of Dr. TN Darling, Uniformed Services University of Health Sciences, Bethesda, MD.)
Hypopigmented, oval, or leaf-shaped macules, ranging from a few millimeters to several centimeters in length and scattered over the trunk and limbs, are commonly seen [Fitzpatrick, 1991]. The lesions often are apparent at birth and can appear more prominent during the first several years of life as the child’s body size and surface area increase. In fair-skinned persons, visualization of these hypopigmented spots is facilitated by using a Wood’s light, an ultraviolet light that accentuates the hypopigmented spots [Fitzpatrick et al., 1968; Roth and Epstein, 1971]. At least three types of hypopigmented macules occur: polygonal (similar to a thumbprint) is the most frequent shape (0.5–2 cm); an ash leaf-shaped hypopigmented macule is characteristic but is not the most common shape (1–12 cm); and the third common type is a confetti-shaped arrangement of multiple, tiny white macules (1–3 mm) [Fitzpatrick, 1991] (Figure 40-11). Histologic assessment of the hypopigmented spots usually demonstrates a normal number of melanocytes, and on electron microscopy a reduction in the number, diameter, and melanization of melanosomes in the melanocytes from the white macule is seen [Jozwiak and Schwartz, 2003]. If hypopigmented macules occur on the scalp, the affected person will have poliosis, or a patch of gray or white hair [McWilliam and Stephenson, 1978].
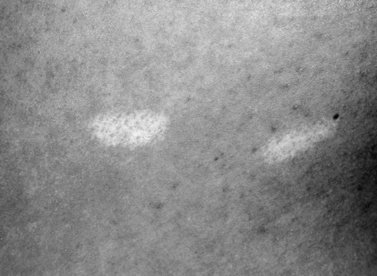
Fig. 40-11 Hypopigmented macule in a child with tuberous sclerosis complex.
(Courtesy of Dr. TN Darling, Uniformed Services University of Health Sciences, Bethesda, MD.)
Another skin manifestation currently considered a major criterion for clinical diagnosis of tuberous sclerosis complex is the shagreen patch, a connective tissue hamartoma that is distributed asymmetrically on the dorsal body surfaces, particularly on the lumbosacral skin (Figure 40-12). In a majority of the cases, the shagreen patch is characterized by multiple and small areas of connective tissue hamartoma, ranging in size from a few millimeters to 1 cm. Present from birth, the shagreen patch is more easily identified as the child grows and body surface area increases. Subungual or periungual fibromas (Koenen’s tumors) are present in at least 20 percent of patients and usually first appear during adolescence, although they can be seen earlier. These typically involve the toes more often than the fingers [Barroeta and Grinspan Bozza, 1962] (Figure 40-13). Oral fibromas or papillomas occur in about 10 percent of patients and usually are found on the anterior aspect of the gingiva [Papanyothou and Verzirtzi, 1975]. Dental enamel pits have been found in all adult patients with tuberous sclerosis complex, compared with 7 percent of controls [Hoff et al., 1975; Mlynarczyk, 1991; Weits-Binnerts et al., 1982].
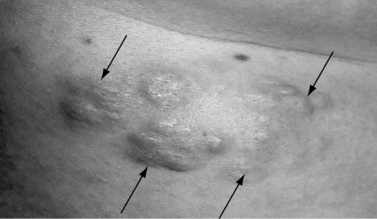
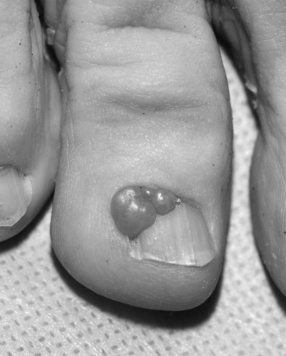
Fig. 40-13 Periungual fibroma on finger of a patient with tuberous sclerosis complex.
(Courtesy of Dr. TN Darling, Uniformed Services University of Health Sciences, Bethesda, MD.)
The kidneys are frequently affected in persons with tuberous sclerosis complex, and after neurologic manifestations, renal involvement is the most common cause of morbidity and mortality [Franz, 2004]. The two main types of renal lesions are angiomyolipoma and renal cysts. Angiomyolipoma are present in up to 80 percent of patients with tuberous sclerosis complex and can develop in either childhood or adulthood [Rakowski et al., 2006]. Persons with tuberous sclerosis complex can have multiple small angiomyolipomas on the surface of the kidneys, throughout the kidney, or one or more larger lesions. The larger lesions are considered to be at greater risk of becoming symptomatic, particularly when they reach 4–6 cm in size. They can produce nonspecific complaints such as flank pain, but they also carry a risk of potentially life-threatening hemorrhage from rupture of dysplastic, aneurysmal blood vessels in the angiomyolipoma. Renal cysts are seen in fewer than 20 percent of persons with tuberous sclerosis complex and are rarely, if ever, symptomatic. Polycystic kidney disease occurs in 3–5 percent of patients with tuberous sclerosis complex and, when present, usually reflects a contiguous gene syndrome, because the polycystic kidney disease gene is adjacent to the TSC2-tuberin gene on chromosome 16 [Brook-Carter et al., 1994].
The cardiac manifestation, rhabdomyoma, is seen in 50–60 percent of persons with tuberous sclerosis complex [Jozwiak et al., 1994]. Typically, rhabdomyomas, which can frequently be detected prenatally, are maximal at birth and early childhood, and undergo spontaneous regression during the first few years of life. If symptomatic, they result in outflow tract obstruction or valve dysfunction. If the lesions involve the cardiac conduction system, they can predispose the patient to dysrhythmias not only in infancy and childhood, but also throughout life.
Pulmonary involvement in tuberous sclerosis complex includes lymphangioleiomyomatosis, multifocal micronodular pneumocyte hyperplasia, and pulmonary cysts. While multifocal micronodular pneumocyte hyperplasia is seen fairly commonly in both men and women with tuberous sclerosis complex, lymphangioleiomyomatosis is thought to occur almost exclusively in women. Although lymphangioleiomyomatosis was once thought to be quite rare, affecting less than 1 percent of women, recent studies have found such abnormalities in up to 40 percent of women with tuberous sclerosis complex, many of whom are asymptomatic [Moss et al., 2001].
Retinal hamartomas are relatively common, affecting at least 50 percent of patients, although typically they are not clinically significant [Rowley et al., 2001]. A nodular (mulberry) tumor can be seen on or about the optic nerve head, and round or oval gray–yellow glial patches can be central or peripheral. The large retinal tumors can be cystic [Walsh and Hoyt, 1969; Messinger and Clarke, 1937]. Papilledema is not present, except in those patients with an intracranial mass lesion that obstructs the normal circulation of the cerebrospinal fluid, resulting in increased intracranial pressure [Kapp et al., 1967].
Hamartomas also can be found in other organ systems, including stomach, intestine, colon, pancreas, and liver. Hepatic angiomyolipoma and cysts have been reported in up to 24 percent of persons with tuberous sclerosis complex and are thought to be asymptomatic and nonprogressive [Fricke et al., 2004]. Sclerotic and hypertrophic lesions of bone often can be seen, although these typically are not symptomatic.
Clinical Laboratory Testing
As a result of the multi-organ involvement in tuberous sclerosis complex, a variety of clinical testing is recommended both at time of diagnosis and subsequently, to monitor for involvement and allow appropriate intervention (Table 40-2).
Table 40-2 Diagnostic and Follow-Up Management in Tuberous Sclerosis Complex
| Evaluation | Initial Testing | Follow-up Testing |
|---|---|---|
| Neuroimaging | At diagnosis | Every 1–3 years until age 20 |
| Neuropsychologic testing | At diagnosis | At school entry and as indicated |
| Electroencephalogram | If seizures occur | As indicated |
| Opthalmologic examination | At diagnosis | As indicated |
| Echocardiogram, electrocardiogram | At diagnosis | As indicated |
| Renal ultrasound examination | At diagnosis | Every 1–3 years, more frequently as indicated |
| Chest computed tomography | At onset of adulthood (women only) | As indicated |
(From Roach ES et al. Tuberous sclerosis consensus conference: Recommendations for diagnostic evaluation. National Tuberous Sclerosis Association. J Child Neurol 1999;14(6):401–407.)
Neuroimaging studies, particularly MRI and also computed tomography (CT), are important in confirming the diagnosis of tuberous sclerosis complex, demonstrating cortical tubers, subependymal nodules (Figure 40-14), and subependymal giant cell tumors (Figure 40-15). Brain MRI is the preferred imaging modality, because it allows better delineation of cortical tubers and other cortical abnormalities, such as radial migration lines. The imaging characteristics of tubers change with the age of the patient, which is thought to be related to myelination. In neonates, tubers appear hyperintense on T1 sequences and hypointense on T2. With increasing age, tubers appear isointense on T1 and hyperintense on T2. In addition to T1- and T2-weighted MRI sequences, fluid-attenuated inversion recovery (FLAIR) sequences appear most useful for identifying tubers and other cortical and subcortical abnormalities. Both CT and MRI can identify subependymal nodules; calcification of the nodules is readily apparent on CT scan. In view of the risk of development of subependymal nodules into subependymal giant cell tumors, which occurs in 5–10 percent of persons with tuberous sclerosis complex, follow-up neuroimaging is recommended annually if possible until the age of 20 years [Goh et al., 2004; Roach et al., 1999]. For unclear reasons, subependymal giant cell tumors lose the propensity to grow after early adulthood.
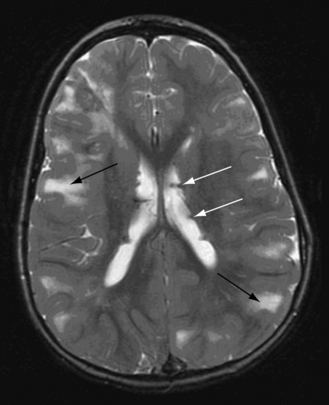
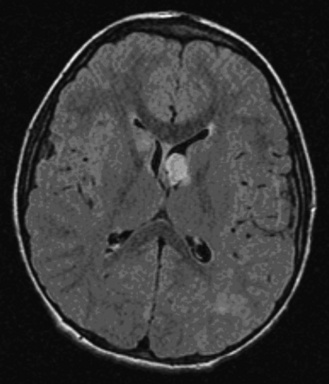
Genetics
Tuberous sclerosis complex is transmitted as an autosomal-dominant trait with variable penetrance and an estimated incidence of 1 in 5800 live births worldwide [Gomez, 1999]. Wide phenotypic variability of clinical manifestations and severity has been noted, even within families having the same mutation [Lyczkowski et al., 2007]. Currently, no known effect of paternal or maternal age or of birth order on disease phenotype has been recognized. Approximately two-thirds of cases are sporadic and the result of apparent spontaneous mutations. Both somatic and germline mosaicism have been described in many patients [Kwiatkowska et al., 1999; Verhoef et al., 1999].
Two genes, TSC1 and TSC2, have been identified for tuberous sclerosis complex. A disease causing mutation in one of these two genes can be identified in approximately 85 percent of persons with Definite tuberous sclerosis complex according to current criteria [Kwiatkowski et al., 2003]. TSC1 located at 9q34; it was cloned in 1997, and the protein product, hamartin, was identified and characterized [Sampson et al., 1989; Smith et al., 1990; Connor and Sampson, 1991; Haines et al., 1991; Hornigold et al., 1997; van Slegtenhorst et al., 1997]. The TSC2 gene is located on 16p13 [Kandt et al., 1992; Consortium, 1993] and encodes a protein referred to as tuberin.
Tuberin and hamartin interact with one another and function as tumor suppressor molecules. Loss of heterozygosity has been identified in hamartomas from persons with TSC1 and TSC2 mutations, particularly in kidney and lung tissue, but less commonly in cortical tubers or subependymal giant cell astrocytomas [Henske et al., 1996]. Tuberin has GTPase-activating properties, similar to the NF1 protein product. Hamartin and tuberin are components of the mammalian target of rapamycin (mTOR) pathway, which is involved in many functions, including regulation of cell size [Gao and Pan, 2001; Potter et al., 2001; Tapon et al., 2001; Crino et al., 2006; Huang and Manning, 2008]. In vivo, it appears that tuberin can be phosphorylated by Akt, at least in part regulating its activity. In normal cells, the tuberin/hamartin complex acts as an inhibitor of mTOR activity. On growth factor stimulation or other stimuli, however, tuberin is phosphorylated by Akt, which leads to the inactivation of inhibitory activity of TSC1/TSC2 and resultant cell growth. In cells containing mutations affecting the function of hamartin or tuberin, mTOR and S6 kinase activities are significantly increased, and cell growth is no longer regulated by the PI3-kinase-TSC1/TSC2 signaling pathway, which is thought to lead to the development of hamartoma.
Management
Tuberous sclerosis complex affects most organ systems, and management and treatment recommendations vary according to organ manifestations (see Table 40-2). Affected persons, both children and adults, should be managed with regular follow-up evaluations by a physician who is familiar with tuberous sclerosis complex, to recognize treatable manifestations early and to provide anticipatory guidance and counseling.
With regard to neurologic manifestations, management focuses on treatment of epilepsy and behavioral disorders and on identification of learning disabilities. Treatment of epilepsy in tuberous sclerosis complex is similar to that for partial epilepsies resulting from other causes, and includes antiepileptic medications, the vagus nerve stimulator, and the ketogenic diet [Thiele, 2004]. Vigabatrin is particularly effective in treating infantile spasms in patients with tuberous sclerosis complex [Curatolo et al., 2008]. Epilepsy surgery has a very important role in the management of patients who have pharmacoresistant epilepsy [Weiner et al., 2004; Jansen et al., 2007].
Rapamycin, an mTOR antagonist, has been shown to reduce the size of subependymal giant cell tumors and renal angiomyolipoma in tuberous sclerosis complex, and may also reduce the progression of pulmonary lymphangioleiomyomatosis [Bissler et al., 2008; Franz et al., 2006]. In animal models of tuberous sclerosis complex, rapamycin has also been shown to prevent epilepsy if given prenatally, and to improve cognitive deficits [Zeng et al., 2008; Ehninger et al., 2008]. On-going multicenter trials are evaluating the role of rapamycin and other mTOR antagonists in the management of tuberous sclerosis complex.
Von Hippel–Lindau Disease
Von Hippel–Lindau disease is inherited as an autosomal-dominant trait and is characterized by retinal, cerebellar, and spinal hemangioblastomas, cystic tumors of the pancreas, kidney, and epididymis, renal cell carcinoma, endolymphatic sac tumors, and, in some families, pheochromocytoma. The basic pathologic lesion is a capillary hemangioblastoma. The retinal lesions, originally described by Panas and Remy, were not recognized as hemangioblastomas [Panas and Remy, 1879]. Fuchs posited that the retinal lesions were arteriovenous malformations [Fuchs, 1882], and Collins believed that the retinal lesions were hemangioblastomas [Collins, 1894]. Von Hippel believed that the retinal lesions were hemangioblastomas but labeled them as angiomatosis retinae [von Hippel, 1911]. The postmortem examination in one patient with retinal lesions reported by von Hippel revealed cerebellar tumor, hypernephroma, and cystic lesions of the pancreas, kidney, and epididymis. Lindau recognized the similarity of the tissue type of the retinal lesions and some cerebellar tumors, and also observed that the same type of tumor occasionally was found in the medulla and spinal cord [Lindau, 1926].
Clinical Characteristics
Retinal hemangioblastoma is one of the earliest manifestations of the disease, and although it has been reported in early childhood [Ridley et al., 1986; Webster et al., 1999], it usually is first observed during the third decade of life (Figure 40-16A). The early retinal lesion has the appearance of an aneurysmal dilatation of a peripheral retinal vessel; typically, tortuous vessels later manifest, with an arteriovenous pair leading to small, elevated retinal lesions [Macmichael, 1970]. These lesions commonly are located in the retinal periphery and can easily be overlooked unless careful ophthalmoscopy is performed. Fluorescein angiography is helpful in demonstrating the lesion [Atuk et al., 1979; Augsburger et al., 1981; Greenwald and Weiss, 1984]. An accumulation of fluid beneath the retina can occur, and retinal detachment with progressive visual loss commonly is observed as the first manifestation of retinal abnormality [Goldberg and Duke, 1968; Kupersmith and Berenstein, 1981; Hardwig and Robertson, 1984; Webster et al., 1999]. Wong et al. studied 335 Von Hippel–Lindau patients with hemangioblastomas in at least one eye and found unilateral tumors in 42.1 percent and bilateral tumors in 57.9 percent [Wong et al., 2008a]. Vascular proliferation leading to visual loss has been reported in rare patients [Wong et al., 2008b].
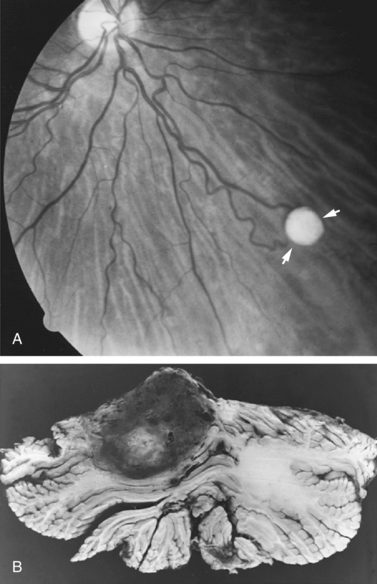
Fig. 40-16 Von Hippel–Lindau disease.
(A, Courtesy of Dr. Creig Hoyt, University of California Medical Center, San Francisco, CA.)
Although they usually affect the cerebellum [Slater et al., 2003], central nervous system hemangioblastomas sometimes are found in the medulla [Pavesi et al., 2010] and spinal cord [Kanno et al., 2009], and rarely occur in the cerebral hemispheres. The tumor usually is found in patients after the third decade of life, but has been reported to occur rarely in children; initial symptoms and signs are those of a space-occupying lesion of the posterior fossa. Symptoms and signs tend to be associated with tumors that display a cystic component [Slater et al., 2003]. Hemangioblastomas of the medulla or spinal cord are associated with syringomyelia in approximately 80 percent of patients [Salazar and Lamiell, 1980]. The tumor rarely is found in the supratentorial region, and can occur in the pituitary gland, third ventricle, or cerebral hemispheres (i.e., frontal, temporal, parietal, and parieto-occipital lobes].
A variety of renal lesions has been found in von Hippel–Lindau disease, including benign cysts, hemangiomas, adenomas, and malignant hypernephromas. Cystic lesions vary in size, ranging from a few millimeters to several centimeters across, and, although they can occur unilaterally, these lesions are more often bilateral and multiple [Melmon and Rosen, 1964]. The cystic lesions can be so extensive that they mimic polycystic kidney disease. A prominent cause of morbidity and mortality is renal cell carcinoma, occurring with a frequency next to that of the retinal and cerebellar hemangioblastoma [Neumann et al., 1998].
Cystic lesions can also occur in the pancreas, adrenal gland, and epididymis. Other organs less commonly affected with cystic changes include the liver, spleen, and lung. Endolymphatic sac tumors associated with von Hippel–Lindau disease can cause hearing loss [Manski et al., 1997]. Pheochromocytomas occur more often in patients with von Hippel–Lindau disease than in the general population and tend to cluster in certain families. Walther et al. diagnosed pheochromocytoma in 64 of 246 patients with von Hippel–Lindau and found an association with missense mutations in the VHL gene [Walther et al., 1999b].
Pathology
The tumors usually are well circumscribed, can be solid or cystic, and usually are found in the paramedial aspect of the cerebellar cortex (see Figure 40-16B). Characteristic microscopic features include large numbers of thin-walled, closely packed blood vessels lined by plump endothelial cells; the cells are separated by large, pale cells and incorporated in the elaborate network of reticulin fibers.
Genetics
Von Hippel–Lindau disease is inherited as an autosomal-dominant trait. It is estimated to affect about 1 in 36,000 persons [Maher et al., 1991]. Penetrance is found to be nearly complete on careful evaluation. The gene is located on 3p25–26 [Latif et al., 1993], and encodes a protein that regulates a cellular system that senses and responds to hypoxia [Kaelin, 2005]. The VHL gene functions as a tumor suppressor; hence, homozygous mutation occurs in tumors, leading to loss of function and constitutive activation of the hypoxia-sensing pathway. Genetic testing is available and has revealed that specific mutations tend to be found in families with von Hippel–Lindau disease associated with pheochromocytoma [Nordstrom-O’Brien et al., 2010]. Von Hippel–Lindau disease has been subdivided into type 1, in which all of the manifestations may be present except for pheochromocytoma, and type 2, which includes the full set of features. Type 2 is further divided into 2A (pheochromocytoma and other manifestations, but not renal cell carcinoma), 2B (all features), and 2C (isolated pheochromocytoma). [Nordstrom-O’Brien et al., 2010] categorized mutations in 945 Von Hippel–Lindau families. A wide variety of mutations were found in type 1 families, whereas 83 percent of type 2 families had missense mutations. Nonsense and frameshift mutations were more frequent in type 1 families.
Management
Affected individuals should be provided a program of surveillance to insure early recognition of treatable complications. Recommendations of the VHL Family Alliance are provided in Table 40-3. Poulsen et al. reviewed records for 59 Danish Von Hippel–Lindau patients, noting that semi-annual MRIs were not sufficient to prevent neurological complications, and instead recommended annual evaluations [Poulsen et al., 2010]. They also noted CNS hemangioblastomas in 18 percent of children younger than 15 years and in one 8-year-old. Retinal hemangioblastomas should be carefully followed by serial ophthalmologic evaluations when the lesions are small. If, however, visual loss or retinal detachment occurs, the lesions can be treated by either laser photocoagulation or cryocoagulation [Webster et al., 1999]. Central nervous systems lesions are usually treated surgically [Pavesi et al., 2008] or with stereotactic radiation therapy [Kano et al., 2008; Karabagli et al., 2010]. Medications that inhibit angiogenesis are being explored as possible nonsurgical therapies [Sardi et al., 2009].
Sturge–Weber Syndrome (Encephalofacial Angiomatosis)
Sturge–Weber syndrome is characterized by presence of a facial angioma (port-wine stain, or nevus flammeus) and an ipsilateral leptomeningeal angioma; it has an incidence currently estimated at 1 case in 20,000–50,000 persons [Comi, 2007]. Schirmer initially described a patient with a facial vascular nevus who had associated buphthalmos, but he did not mention the central nervous system lesion [Schirmer, 1860]. Sturge initially described this syndrome by providing the clinical findings of a 6-year-old girl with a facial nevus who also had angiomas of the lips, gingiva, palate, floor of the mouth, uvula, and pharynx [Sturge, 1879]. The child had buphthalmos and was hemiparetic, and Sturge suggested that she had a similar vascular nevus of the underlying brain. Not until 1897, however, did Kalischer perform the first neuropathologic study of a patient with similar findings, demonstrating that Sturge’s initial contention of cerebral involvement by vascular nevus was correct [Kalischer, 1897]. Associated intracranial calcification was later described by Weber [Weber, 1929].
Clinical Characteristics
Sturge–Weber syndrome, which occurs sporadically, is characterized by angiomas involving the leptomeninges and ipsilateral skin of the face, typically in the ophthalmic (V1) and maxillary (V2) distributions of the trigeminal nerve. It can extend to other facial areas, including the lips, gingiva, palate, tongue, pharynx, and larynx. The neck, trunk, and extremities also can be involved, either ipsilaterally or contralaterally to the facial angioma. The angioma also can involve the nasopharynx, mucous membrane, and ocular choroidal membrane, resulting in glaucoma in approximately 25 percent of patients (Figure 40-17). Additional ocular findings include iridic heterochromia, strabismus, optic atrophy, and dilated retinal veins. In the brain, the associated ipsilateral leptomeningeal angioma most commonly involves the parietal and occipital regions, but also may involve the temporal region and, on occasion, can affect both hemispheres. Dimitri reported that these patients had intracranial calcifications observed on the skull radiographs and described the typical serpentine “tram-track sign” of calcific intracranial densities [Dimitri, 1923].
Neurologic manifestations vary and depend on location and extent of the leptomeningeal angioma. Seizures occur in 75–90 percent of patients with Sturge–Weber syndrome and may be refractory to treatment [Bebin and Gomez, 1988; Takeoka and Riviello, 2010]. It is hypothesized that the seizure activity results from cortical irritability caused by the leptomeningeal angioma, resulting in regional hypoxia, ischemia, and gliosis, although associated cortical dysgenesis also may be involved. Seizure manifestations are primarily partial motor (40 percent), although some patients can have primary or secondary generalized tonic-clonic (20 percent) and both partial and generalized seizures (40 percent) [Chao, 1959; Bebin and Gomez, 1988]. Other types of seizure activity occur less frequently. Unfortunately, refractory epilepsy develops in a significant number of patients with Sturge–Weber syndrome, ranging in series from 11 to 83 percent [Takeoka, 2010]. Surgical procedures, including focal cortical resection, hemispherectomy, and corpus callosotomy, should be considered if seizure activity proves medically intractable.
Persons with Sturge–Weber syndrome also are at risk for hemiparesis contralateral to the leptomeningeal angioma, which is seen in approximately 33 percent of the patients. The hemiparesis can result from ischemia with venous occlusion and thrombosis due in part to venous congestion resulting from failure of cortical vein development [Hebold, 1913; Chao, 1959; Alexander, 1972; Chamberlain et al., 1989]. Transient weakness also may result from seizure activity, and may become more severe and less transient with recurrent seizure activity. Leptomeningeal venous angiomas can arise in the absence of any facial angioma; although secondary cerebral signs and symptoms similar to those of Sturge–Weber syndrome can occur, these patients are more appropriately considered to have leptomeningeal angiomatosis.
Persons with Sturge–Weber syndrome also are at risk for developmental delay and mental retardation, which occur in 50–60 percent of the patients and are more likely in those with bilateral leptomeningeal involvement [Bebin and Gomez, 1988] and in those with a history of seizures [Sujansky and Conradi, 1995; Comi, 2007]. Headaches also are common, occurring in up to 60 percent of affected persons, and are thought to be secondary to the vascular abnormalities, giving symptoms consistent with migraine [Maria et al., 1999]. EEG studies document decreased amplitude and frequency of electrocerebral activity over the affected hemisphere. Diffuse, multiple, and independent spike foci commonly are present [Aminoff, 1992; Sassower et al., 1994].
Intracranial calcification is evident on radiographs in 90 percent of adult patients. Calcifications uncommonly are present at birth but are manifest in virtually all patients by the end of the second decade of life [Nellhaus et al., 1967]. The intracranial calcifications typically assume a linear, parallel configuration (“tram-track sign”) or a convolutional pattern most commonly seen in the parietal or parieto-occipital regions [Thomas-Sohl et al., 2004] (Figure 40-18). Cranial CT and MRI scans are complementary in evaluating the cerebral changes of Sturge–Weber syndrome, in that the MRI demonstrates thickened cortex, decreased convolutions, and abnormal white matter, whereas cranial CT scans demonstrate more definitively the characteristic calcification. Cranial MRI scans (T2-weighted images) reveal smaller, nonspecific foci of hypointense signal [Chamberlain et al., 1989; Thomas-Sohl et al., 2004]. Gadolinium enhancement may reveal pial angioma, thereby allowing early diagnosis of Sturge–Weber syndrome before calcification [Sugama et al., 1997].
Cerebral angiography discloses decreased cerebral venous drainage with dilatation of the deep cerebral veins. Various other vascular abnormalities have been demonstrated in approximately one-third of patients and include thrombotic lesions, dural venous sinus abnormalities, and arteriovenous malformations [Bentson et al., 1957]. PET provides a sensitive measure of the extent of cerebral metabolic impairment. Serial PET scans in children with Sturge–Weber syndrome can be useful and, when used with other neuroimaging studies, document the progression of the disease [Chugani et al., 1989].
Pathology
Sturge–Weber syndrome is thought to be caused by the presence of residual embryonal blood vessels and their secondary effects on surrounding tissues. During development, a vascular plexus develops around the cephalic portion of the neural tube, under the ectoderm that subsequently becomes facial skin. This plexus forms during the sixth week of gestation and regresses at approximately the ninth week. It is thought that failure of this regression results in residual vascular tissue, subsequently forming the angiomata of the leptomeninges, face, and ipsilateral eye. Neuropathologic studies have demonstrated thickened, hypervascularized leptomeninges that involve the occipital, parietal, or temporoparietal region primarily (Figure 40-19). These meningeal vessels generally are small and tortuous, and rarely enter the underlying brain substance. Calcific deposits are present in the walls of some small cerebral vessels but more commonly are found in the outer pyramidal and molecular cortical layers. Biochemical assays have demonstrated increased calcium content of the gray and white matter, with normal iron content. The pathophysiology of the deposition of intracerebral calcium is not well understood [Weber, 1929; Tingey, 1956; Wachswulth and Lowenthal, 1979; Thomas-Sohl et al., 2004].
Management
Treatment for the neurologic manifestations of Sturge–Weber syndrome includes management of seizure activity and headaches. Approximately 50 percent of children with seizures achieve control with administration of appropriate antiepileptic drugs. Those patients with seizure disorders refractory to medical treatment should be carefully considered for epilepsy surgery with resection of the affected lobe(s) or hemispherectomy. Rochkind and colleagues reported that seizure control after surgery was better in those patients who received antiepileptic drugs [Rochkind et al., 1990]. Aspirin therapy may reduce the incidence of strokelike episodes, and is typically used in individuals with either recurrent vascular events or progressive neurologic deficits [Roach et al., 1985; Comi, 2007]. Treatment options for the facial angioma include laser therapy using various pulsed-dye lasers, as well as pulsed-light sources, and other laser therapies. The current recommendation is to begin treatment as early as possible; infants have received treatment during the first week of life. Treatment of glaucoma, if present, consists of control of intraocular pressure, to prevent optic nerve injury, by medical or surgical intervention. The management of patients with Sturge–Weber syndrome requires the skill of an attentive physician, psychologist, and social worker.
Maffucci’s Syndrome
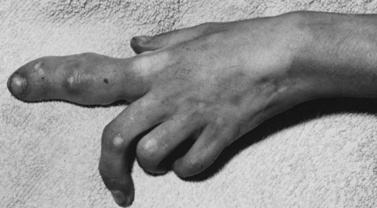
Maffucci’s syndrome is a rare congenital disease characterized by multiple enchondromas with secondary hemangiomas, phlebolithiasis, and malformations of bone. Occasionally, associated skin changes, including patches of vitiligo, café-au-lait spots, and other hyperpigmented patches and nevi, are seen. The reported cases are sporadic, and no gender predilection exists. A related disorder, Ollier’s disease, is characterized by multiple endochondromas without hemangiomas. The initial report of this syndrome by Maffucci described a 40-year-old woman who had multiple enchondromas, some of which had undergone sarcomatous changes [Maffucci, 1881]. Her four children had no stigmata of the disease. Maffucci syndrome’s generally is a sporadic occurrence. Hopyan et al. have identified mutations in the gene encoding the parathyroid hormone/parathyroid hormone-related protein type 1 receptor (PTHR1) in persons with endochondromatosis, suggesting the possibility that the disorder is inherited as autosomal-dominant, with most cases due to new mutation [Hopyan et al., 2002]. Couvineau et al. identified germline PTHR1 mutations in 1 of 61 patients with Ollier’s disease, but in none of 23 patients with Maffucci’s syndrome [Couvineau et al., 2008].
The enchondromas affect the small bones of the hands and feet, or any bone preformed in cartilage; they are apparent during the first few years of life. Initially, a small, firm nodule 1–2 cm in diameter usually is found on digital bone; the nodule is readily palpable. Additional, similar nodules appear shortly thereafter. They are unilateral in about 40 percent of patients and, if bilateral, are strikingly asymmetric [Bean, 1955, 1958]. During childhood, the enchondromas increase in size, ultimately resulting in malformations of the limbs and trunk (Figure 40-20). Patients generally are of small stature and have a distinctive appearance resulting from their deformities. About 20–30 percent of affected individuals have enchondromas that undergo sarcomatous changes [Albregts and Rapini, 1995; Ranger et al., 2009; Ramina et al., 1997]. Neurologic symptoms may result from skull-base chondrosarcomas or from rare gliomas. Cutaneous hemangiomas become apparent during the first decade of life and are not necessarily found in the areas of enchondromas. They can be superficial or deep, and they may affect the lips, tongue, palate, or cheeks. Visceral hemangiomas involving the esophagus, ileum, and anal mucosa have been reported. Patients have cavernous hemangiomas and lymphangiomas. Associated angiofibromas, hemangiomas, hemangioendotheliomas, teratomas, thecomas, and pancreatic adenocarcinomas have been reported [McDermott et al., 2001].
Epidermal Nevus Syndrome
Epidermal nevus syndrome is a heterogeneous group of disorders characterized by patchy cutaneous hamartomatous lesions, central nervous system abnormalities, and various other manifestations (Table 40-4). Most of these disorders occur sporadically and are highly variable in their presentation [Vidaurri-de la Cruz et al., 2004; Happle, 1991]. Nevi can take various forms, including verrucous, sebaceous, and lentiginous, and may evolve over the years. They tend to follow the lines of Blaschko and manifest early in life. Central nervous system manifestations include unilateral lissencephaly, a paucity of white matter, excessive and heterotopic gray matter, apparent schizencephaly, unilateral colpocephaly, and hemimegalencephaly [Hager et al., 1991; Zhang et al., 2003]. Associated neurologic abnormalities include mental retardation and convulsive disorders, occurring in approximately 60 percent of patients; ocular abnormalities in up to 50 percent of patients; and corticospinal tract dysfunction in a smaller number of patients [Holden and Dekaban, 1972; Kurokawa et al., 1981]. Cortical resection has been helpful in some instances [Maher et al., 2003], but no other definitive treatment exists.
| Disorder | Features | Genetics |
|---|---|---|
| Sebaceous nevus syndrome | Congenital sebaceous nevus, neurologic dysfunction, skeletal, ocular anomalies | Sporadic |
| Nevus comedonicus syndrome | Congenital hyperkeratotic papules of follicular origin on face, neck, chest, abdomen, arms; ipsilateral eye and skeletal defects; seizures, mental retardation | Sporadic |
| Becker nevus syndrome | Hyperpigmented plaque associated with hair growth; smooth muscle hyperplasia, rib defects, breast hypoplasia | Sporadic (more common in males) |
| Phakomatosis pigmentokeratotica | Sebaceous and speckled lentiginous nevus; ipsilateral weakness, hypohydrosis; seizures, mental retardation | Sporadic |
| Proteus syndrome | Epidermal nevi; asymmetric overgrowth of soft tissues and bone | Sporadic; some with mosaic PTEN mutations |
| Congenital hemidysplasia with ichthyosiform nevus and limb defects (CHILD) | Ichthiosiform nevus; skeletal aplasia or hypoplasia; abnormalities in brain, lung, heart, kidneys | X-linked dominant; mutations in 3β-hydroxysteroid dehydrogenase gene |
For additional information, see Happle R. How many epidermal nevus syndromes exist? A clinicogenetic classification. J Am Acad Dermatol 1991;25:550–556; Vidaurri-de la Cruz H. Epidermal nevus syndromes: Clinical findings in 35 patients. Pediatr Dermatol 2004;21(4): 432–439.
The patchy manifestations of these disorders have suggested the possibility that they result from somatic mosaicism; this has been demonstrated in some instances of Proteus syndrome, where mosaicism for PTEN has been found [Hobert and Eng, 2009; Orloff and Eng, 2008].
Parry–Romberg Syndrome (Facial Hemiatrophy)
Parry–Romberg syndrome, which typically has onset between 5 and 15 years of age, is characterized by a progressive ipsilateral loss of facial soft tissue, cartilage, and bone. This tissue loss usually involves the tissues between the nose and nasolabial fold or above the maxilla, but progresses to affect most of the ipsilateral face during the ensuing years. The tongue, the gums, and the soft palate may also become involved. The eyelashes, eyebrows, and hair on the involved side can be affected, and ipsilateral blanching of the hair or alopecia can occur (Figure 40-21). Progression of this atrophic process generally lasts between 2 and 10 years, and is believed to cease by the end of the second or beginning of the third decade of life [Wartenberg, 1945; Wolf and Verity, 1974; Rischbieth, 1976; Stone, 2003].
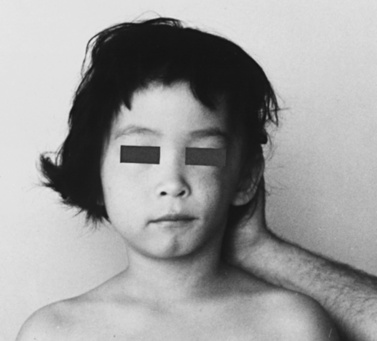
In addition to atrophy of the facial tissues, various other neurologic deficits have been reported, including recurrent headaches, trigeminal neuralgia, ipsilateral Horner’s syndrome, contralateral partial seizures, and hemiparesis [Stone, 2003]. Moreover, an unusual association of the syndrome with multiple benign tumors has been described. Scleroderma and lipodystrophy must be clinically differentiated from this disease. Cranial CT can be normal or document cerebral atrophy; contralateral intracerebral calcification has been described [Asher and Berg, 1982]. No typical or consistent neuropathologic findings have been reported. No specific treatment for the syndrome exists; however, various reconstructive surgical procedures, often using grafts of autogenous fat after disease stabilizes, can result in reasonably good cosmetic results [Hintringer et al., 1991; Mayro et al., 1991].
Neurocutaneous Melanosis
Neurocutaneous melanosis is a rare, nonfamilial, embryonic, neuroectodermal dysplasia characterized by abnormally pigmented cutaneous areas (e.g., giant hairy pigmented nevi, multiple hyperpigmented cutaneous nevi, large congenital melanocytic nevi) and leptomeningeal melanosis [DiRocco et al., 2004]. Virchow [Virchow, 1859] first reported the topographic distribution of melanotic cells in the arachnoid, but Rokitansky provided the initial clinical description of this condition with his report of a 14-year-old, mentally retarded, hydrocephalic girl who had a giant pigmented nevus over her back and multiple hyperpigmented skin lesions [Rokitansky, 1861]. At postmortem examination, a brown–black pigmentation of the pia overlying the cerebral cortex was noted. Van Bogaert named this condition neurocutaneous melanosis [Van Bogaert, 1948].
Diagnosis is usually made in infants and children younger than 2 years of age; however, the condition is present at birth. No gender predilection exists. All patients have areas of abnormal skin hyperpigmentation, the most common pattern of which is multiple giant hairy pigmented nevi. Giant hairy nevi usually have a “bathing suit” or cape-shaped distribution. In one series of 289 patients with large congenital melanocytic nevi, 33 had manifest central nervous system melanosis, wherein the nevi were present in the posterior axial location on the head, neck, back, and/or buttocks. “Satellite” nevi were present in 31 of the 33 patients [DeDavid et al., 1996]. Lovett et al. [Lovett et al., 2009] reviewed a series of 26 patients with large congenital melanocytic nevi who underwent MRI, and found six with neurocutaneous melanosis. The presence of multiple satellite lesions and midline location for pigmented lesions were predictive of neurological involvement.
The clinical presentation of neurocutaneous melanosis depends on the location and extent of involvement of this leptomeningeal lesion. Hydrocephalus is commonly encountered because of cerebrospinal fluid pathway obstruction in the basilar cisterns, the arachnoid villi over the cerebral hemispheric convexities, or both, and intraspinal melanotic arachnoid cyst, lipoma, and intraspinal lipoma have been described as obstructed [Kasantikul et al., 1989; van Heuzen et al., 1989]. Association with Dandy–Walker malformation has been reported [Schreml et al., 2008]. Behavioral abnormalities and recurring seizures can occur, as well as cranial nerve dysfunction and signs of spinal cord and root involvement [Pavlidou et al., 2008]. Findings on cerebrospinal fluid examination can be normal; however, the protein level usually is elevated, and glucose concentration is decreased. Cytologic examination usually reveals abnormal melanin-containing cells [Fox, 1972]. In one study of MRI scans of seven patients with neurocutaneous melanosis, five had regions of T1 shortening in the cerebellum, three of whom also had T2 shortening. Five patients had regions of T1 shortening in the anterior temporal lobes. Other areas of involvement included the pia mater over the cerebellum, pons, medulla, and left parietal lobe. Only two lesions demonstrated enhancement, edema, or necrosis, and both lesions were found to be malignant melanomas at biopsy [Barkovich et al., 1994].
Attempts to treat hydrocephalus by a shunting procedure are palliative. There is a risk of melanoma. The prognosis for neurocutaneous melanosis generally is poor, regardless of the variety of treatment methods used, which include radiation therapy, chemotherapy, or both, and patients rarely live beyond the age of 20 years, though one report suggests a role for aggressive chemotherapy in associated melanoma [Subbiah and Wolff, 2010].
Klippel–Trénaunay–Weber Syndrome
Klippel–Trénaunay–Weber syndrome initially was believed to be characterized only by cutaneous and/or subcutaneous hemangiomas, varicosities, and hypertrophy of the soft tissues and bone of a limb. Additional associated anomalies later recognized included macrocephaly; hydrocephalus; lymphangiomas; hemangiomas of the trunk, intestine, and bladder; and abnormalities of the digits [Klippel and Trénaunay, 1900; Lian and Alhomme, 1945; Meine et al., 1997; Parkes Weber, 1907]. Klippel–Trénaunay–Weber syndrome usually occurs sporadically but has been found to be associated, at least in some patients, with translocations that disrupt the AGGF1 gene, which encodes a protein involved in angiogenesis [Tian et al., 2004]. Hu et al. [2008] also demonstrated association of single nucleotide polymorphisms in this gene with Klippel-Trénaunay-Weber syndrome.
Jacob et al. [1998] reviewed findings in 252 patients, and found port-wine capillary malformations in 98 percent, venous malformations in 72 percent, and limb hypertrophy in 67 percent. Typical skin lesions usually are present at birth but may not be recognized until several years later. They can be single or multiple, and affect the face, trunk, and limbs. The vascular lesions are variable and include capillary hemangiomas, nevus flammeus, telangiectasias, varicosities, phlebectasias, and arteriovenous fistulae; lymphangiectasis also can be present. Vascular lesions usually are found in areas of limb hypertrophy. Rarely, patients with large vascular lesions have a bleeding diathesis; thrombophlebitis can occur, as well as thromboembolic phenomena. Varicosities of various sizes commonly are present and, although usually found in the lower limbs, also can be found on the scalp, chest, and torso [Inceman and Tangun, 1969].
Limb hypertrophy usually is apparent at birth. Prenatal diagnosis has been made by ultrasonography [Hatjis et al., 1981]. Lymphedema can be present. An inordinately progressive growth of the affected body part eventually occurs, leading to the development of various other abnormalities. Abnormal growth of one leg, for example, can result in a pelvic tilt and scoliosis, but the patient’s overall height is not significantly changed [Lindenauer, 1965]. The legs are affected more commonly than the arms.
Additional abnormalities include megalocornea, glaucoma, iridic heterochromia, syndactyly, polydactyly, macrodactyly, and clinodactyly. Hemangiomas of the tongue, pharynx, larynx, and bladder have been described, and labial and scrotal lesions are common. Macrocephaly often is present, and seizures and mental retardation have been reported. Less frequently, associated abnormalities include congenital heart malformations and imperforate anus [Banhayan, 1971; Lindenauer, 1965; Hall, 1971].
Any treatment plan for these patients must be individualized and requires a multidisciplinary team. Nonsurgical management involves compression of the involved limb. MRI or CT venography [Mavili et al., 2009; Bastarrika et al., 2007] can be performed if any vascular surgical procedure is contemplated. Some vascular lesions can be treated with cryotherapy, laser therapy, or sclerotherapy, whereas others can be surgically removed [Redondo et al., 2009; Gloviczki and Driscoll, 2007]. An osteotomy or epiphyseal stapling procedure can occasionally benefit patients with limb hypertrophy, but limb amputations may be required in others.
Incontinentia Pigmenti (Bloch–Sulzberger Syndrome)
Incontinentia pigmenti is transmitted as an X-linked dominant trait, predominantly affecting females [Lenz, 1961]. Most hemizygous males apparently die in utero; rare affected males usually are mosaics or have a 47,XXY karyotype [Kenwrick et al., 2001]. The gene has been identified and is referred to as NEMO; it encodes a protein that participates in the nuclear factor kappa B (NF-κB) signaling pathway [Smahi et al., 2000]. The syndrome is characterized by various hyperpigmented skin lesions that can be apparent at birth and by commonly associated abnormalities involving the central nervous system, eyes, hair, teeth, and bone.
Garrod [1906] likely provided the first description of this syndrome with his report of the case of a 2-year-old, mentally retarded, spastic diplegic girl who had a whorled pattern of brown skin pigmentation on her trunk and limbs. Bardach [1925], however, is most often credited with the first clinical description of the syndrome in twin sisters; Bloch [1926], Sulzberger [1938], and Sieman [1929] each later defined the syndrome more clearly. Bloch introduced the term incontinentia pigmenti, which is still used.
The skin manifestations have been described as having three stages: The first stage typically is characterized by vesiculobullous lesions present at birth or during the first several weeks of life. These lesions appear in groups or in a linear distribution over the trunk and limbs, following the lines of Blaschko. A preponderance of eosinophils is found in the vesicular fluid, and the peripheral blood also can exhibit an eosinophilia. The lesions rupture, resulting in oozing and crusting, and can persist for months. The second stage is characterized by evolution into verrucous lesions beginning after the sixth week of life. The third stage typically is characterized by hyperpigmented brown or gray–brown macular lesions that follow the lines of Blaschko. These pigmented skin lesions usually become more prominent during the first few years of life and then gradually fade. The decrease of abnormal pigmentation may continue throughout adolescence, and in some patients the pigmentation can completely disappear [Carney, 1976].
One-third to one-half of patients have symptoms and signs of neurologic abnormalities manifested by developmental retardation, corticospinal tract dysfunction, and seizures [Carney, 1976]. Microcephaly and hydrocephaly may occur. Approximately one-third of patients have ocular abnormalities, including optic atrophy, papillitis, abnormal retinal pigmentation, nystagmus, strabismus, and cataracts. Visual loss occurs in about 8 percent of patients. The most common ocular abnormalities are retinal detachment and a fibrovascular retrolental membrane [Heathcote et al., 1991].
There are often associated ectodermal and skeletal anomalies [Carney, 1976]. Skin changes include atrophic scarring and alopecia, and nails can be flat and thin, commonly with transverse ridges. Skeletal abnormalities include spina bifida, hemivertebrae, accessory ribs, and syndactyly. Delayed dentition, pegged teeth, and abnormal crown formation are also seen.
Few neuropathologic studies are available. Some reports have described micropolygyria, cortical atrophy, and small areas of leukomalacia; however, these findings can be nonspecific and unrelated to the disease process [Hauw et al., 1977]. MRI studies suggest that the underlying cerebral pathology may be microvascular infarcts associated with the occlusion of small vessels [Hennel et al., 2003; Lee et al., 1995].
Treatment remains symptomatic and supportive. Recent case reports indicate some success in treating early skin lesions with corticosteroids [Kaya et al., 2009] or topical tacrolimus [Jessup et al., 2009].
Incontinentia Pigmenti Achromians (Hypomelanosis of Ito)
Incontinentia pigmenti achromians initially was described by Ito [Ito, 1951] and has been known as systemic achromic nevus and hypomelanosis of Ito. Typical skin changes occur as hypopigmented lesions on any part of the head, trunk, or limbs, either unilaterally or bilaterally. The configuration of the hypopigmented lesions may manifest as linear streaks or whorls of hypopigmentation that follow the lines of Blaschko. The skin lesions are congenital. Multiple associated anomalies are common and can involve the central nervous system or the peripheral nervous system, eyes, and bone [Takematsu et al., 1983; Hamada et al., 1967, 1979]. Common central nervous system abnormalities include mental retardation, language disabilities, seizures, and motor system dysfunction. Ocular abnormalities include strabismus, epicanthic folds, myopia, optic nerve hypoplasia, and hypopigmentation of the fundus; rarely, corneal asymmetry, pannus, and atrophic irides with irregular pupillary margins have been reported. Cataracts and retinal detachments also have been reported. Visual loss has not been reported in patients with incontinentia pigmenti achromians [Takematsu et al., 1983; Weaver et al., 1991].
Characteristic histologic features of skin biopsy specimens include dyskeratosis, increased dermal mastocytes, and pilosebaceous abnormalities. Electron microscopy reveals few melanosomes, sparsely dendritic melanocytes, and a reduction in the number of melanosomes in keratinocytes [Schwartz et al., 1977].
In one study [Ruggieri et al., 1996], MRI was performed in 13 affected patients, and anomalies of the white matter were observed in 7. Abnormal signals in white matter were located primarily in the parietal, periventricular, and subcortical regions of both hemispheres, and asymmetry of the cerebral and cerebellar hemispheres was observed in each of two patients. A correlation between the white matter changes and neurologic symptoms and signs was noted.
Hypomelanosis of Ito occurs sporadically and in many cases has been found to be associated with mosaicism for chromosomal abnormalities. The abnormal cells may be confined to the skin lesions and therefore are detected only by cytogenetic analysis of cultured fibroblasts obtained by skin biopsy. No single chromosome abnormality accounts for all cases; rather, it appears that the streaky hypopigmentation associated with the disorder, or sometimes patches of hyperpigmentation following Blaschko’s lines, are the cutaneous manifestation of mosaicism for various genes or chromosomal regions [Ritter et al., 1990; Taibjee et al., 2004; Zajac et al., 1997; Flannery, 1990].
Wyburn–Mason Syndrome (Retinocephalic Angiomatosis)
One of the first descriptions of a retinal vascular malformation was by Magnus [1874], who viewed it as a medical curiosity. Yates and Paine [1930] reported an extensive retinal and ipsilateral cerebral arteriovenous malformation in a patient who died from cerebral hemorrhage, but the association of a unilateral arteriovenous malformation of the retina, brain, and parts of the face and head initially was described in a postmortem examination report by Brock and Dyke [1932]. Bonnet et al. [1937] emphasized the importance of a coexisting tortuous angioma of the fundus in patients with intracranial arteriovenous malformations. In 1943, Wyburn–Mason described the clinical syndrome in detail [1943]. Although known as Bonnet–Dechaum–Blanc syndrome in Europe and as Wyburn–Mason syndrome in the United Kingdom and the United States, the condition is more appropriately called a retinocephalic vascular malformation. The syndrome is thought to result from an embryonic abnormality in the development of the optic nerve pathway and related vessels from its origin in the mesencephalon all the way to the projection to the retina.
Wyburn–Mason [1943] reviewed reports of 27 patients with retinal angiomas and found signs of intracranial arteriovenous malformations in 22 patients. He later surveyed reports of 20 other patients with midbrain arteriovenous malformations and found 14 of those with associated retinal arteriovenous malformations. He then suggested that the syndrome was more common than had been suspected and that patients with vascular malformations of the retina or brain were likely to have other organs similarly affected. Because many of his patients had not been studied with cerebral angiography and only two patients had neuropathologic confirmation of the lesion, his suggestions were criticized, and the syndrome has been considered rare [Bech and Jensen, 1958]. Schmidt et al. [2008] reviewed the 121 patients with retinal arteriovenous malformations in the literature, and found that 27 had typical retinocephalic angiomatosis, 25 lacked the facial skin lesions, 57 had apparent isolated retinal arteriovenous malformations, and 12 had retinal arteriovenous communications and neurologic signs, but no neuroradiologic evidence of cerebral arteriovenous malformations. The retinal lesions are unilateral and readily observed by ophthalmoscopy. The vascular malformation affects one or all retinal vessels, which are notably dilated and tortuous. Ocular enlargement, conjunctival hyperemia, and proptosis usually occur; a cranial bruit is commonly present. Visual function of the involved eye is rarely normal, and the visual loss varies according to the severity of the vascular malformation. The retrobulbar soft tissue and optic nerve commonly are affected by the malformed vessels [Hoyt and Cameron, 1968; Reck et al., 2005].
The intracranial vascular malformation usually is deep within the brain substance and can involve the mesencephalon, diencephalon, and basal ganglia, extending to the visual pathways and chiasm. Variable involvement of the cranial nerves occurs, including the third, sixth, seventh, and eighth nerves; nystagmus and Parinaud’s syndrome have been reported. Corticospinal tract dysfunction can be unilateral or bilateral, and some patients are ataxic [Hoyt and Cameron, 1968; Theron et al., 1974]. Approximately one-half of patients have vascular malformations that affect the palate, oral mucosa, maxilla, and mandible. Cutaneous lesions also can occur, manifesting as angiomas or punctate erythematous lesions.
The diagnosis of this syndrome is initially considered when the retinal vascular lesion is observed. Cranial CT and MRI scans clearly demonstrate arteriovenous malformation, but only cerebral angiography reliably delineates the extent of the lesion [Fujita et al., 1989; Hopen et al., 1983]. No beneficial treatment method is currently available for this syndrome. The surgical removal of part or all of the extensive vascular malformation cannot be performed with any practical success, and the use of rigorous radiologic interventional techniques has been unsuccessful.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Albers A.C., Gutmann D.H. Gliomas in patients with neurofibromatosis type 1. Expert Rev Neurother. 2009;9(4):535-539.
Albregts A.E., Rapini R.P. Malignancy in maffucci’s syndrome. Dermatol Clin. 1995;13:73-78.
Alexander G.L. Sturge-Weber syndrome. In: Bruyn G.W., Vinken P.J., editors. Handbook of Clinical Neurology. New York: American Elsevier, 1972.
Aminoff M.J. Electrodiagnosis in Clinical Neurology. New York: Churchill Livingstone; 1992.
Asher S.W., Berg B.O. Progressive hemifacial atrophy: Report of three cases, including one observed over 43 years, and computed tomographic findings. Arch Neurol. 1982;39:44-46.
Atuk N.O., McDonald T., Wood T., et al. Familial pheochromocytoma, hypercalcemia, and von hippel-lindau disease. A ten year study of a large family. Medicine (Baltimore). 1979;58:209-218.
Augsburger J.J., Shields J.A., Goldberg R.E. Classification and management of hereditary retinal angiomas. Int Ophthalmol. 1981;4(1–2):93-106.
Babovic-Vuksanovic D., Widemann B., Dombi E., et al. Phase I trial of pirfenidone in children with neurofibromatosis 1 and plexiform neurofibromas. Pediatr Neurol. 2007;36(5):293-300.
Ballester R., Marchuk D., Boguski M., et al. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell. 1990;63:851-859.
Banhayan G.A. Lipomatosis, angiomatosis, and macrocephaly. Arch Pathol. 1971;92:1.
Bardach M. Systematisierte naeuvusbildungen bei einen eineiigen zwillingspan. Z Kinderheilkd. 1925;39:542.
Barkovich A.J., Frieden I.J., Williams M.L. MR of neurocutaneous melanosis. Am J Neuroradiol. 1994;15(5):859-867.
Barroeta S., Grinspan Bozza N. Koenen’s tumor without other clinical manifestations of epiloia. Arch Argent Dermatol. 1962;12:290-292.
Baser M.E., Wallace A.J., Strachan T., et al. Clinical and molecular correlates of somatic mosaicism in neurofibromatosis 2. J Med Genet. 2000;37(7):542-543.
Baser M.E., Evans D.G., Jackler R.K., et al. Neurofibromatosis 2, radiosurgery and malignant nervous system tumours. Br J Cancer. 2000;82(4):998.
Baser M.E., Friedman J.M., Evans D.G.R. Increasing the specificity of diagnostic criteria for schwannomatosis. Neurology. 2006;66(5):730-732.
Baser M.E., Friedman J.M., Wallace A.J., et al. Evaluation of clinical diagnostic criteria for neurofibromatosis 2. Neurology. 2002;59(11):1759-1765.
Bastarrika G., Redondo P., Sierra A., et al. New techniques for the evaluation and therapeutic planning of patients with Klippel-Trenaunay syndrome. J Am Acad Dermatol. 2007;56(2):242-249.
Battista R.A. Gamma knife radiosurgery for vestibular schwannoma. Otolaryngol Clin North Am. 2009;42(4):635-654.
Bean W.B. Dyschondroplasia and hemangiomata (maffucci’s syndrome). Arch Intern Med. 1955;95:767.
Bean W.B. Dyschondroplasia and hemangiomata (maffucci’s syndrome). II. Arch Intern Med. 1958;102:544.
Bebin E.M., Gomez M.R. Prognosis in sturge-weber disease: Comparison of unihemispheric and bihemispheric involvement. J Child Neurol. 1988;3(3209844):181-184.
Bech K., Jensen O.A. Racemose haemangioma of the retina; two additional cases, including one with defects of the visual fields as a complication of arteriography. Acta Ophthalmol (Copenh). 1958;36(4):769-781.
Bentson J.R., Wilson G.H., Newton T.H. Cerebral venous drainage pattern in sturge-weber syndrome. Radiology. 1957;68:327.
Bernards A. Gaps galore! A survey of putative ras superfamily gtpase activating proteins in man and drosophila. Biochim Biophys Acta. 2003;1603:47-82.
Bielschowsky M. Uber tuberose sklerose und ihre beziehungen nur recklinghausenchan krankheit. Z Neurol Psychiatr. 1914;26:133-155.
Bielschowsky M. Entwurf eines systems der heredodegenerationen des zentralnervensystems einschliesslich der zugehoren straitum-krankungen. J Psychol Neurol. 1919;24:48.
Bissler J.J., McCormack F.X., Young L.R., et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358(2):140-151.
Bloch B. Eigentumliche, bisher nicht beschreibene pigmentaffektion (incontinentia pigmenti). Schweiz Med Wochenschr. 1926;7:404.
Bonnet P., Dechaume J., Blanc E. L’aneurysme circsoide de la retine (aneurysme race meux) ses relations avec l’aneurysme circoide de la face et avec “l’aneurysme cirsoide du cerveau”. J Med Lyon. 1937;18:165.
Bourneville D.M., Brissard E. Encephalite ou sclerose tubereuse des circonvolutions cerebrales. Arch Neurol. 1880;1:390-410.
Bourneville D.M., Brissard E. Idiotie et epilepsie symptomatiques de sclerose tubereuse ou hypertrophique. Arch Neurol. 1900;10:29.
Boyd C., Smith M., Kluwe L., et al. Alterations in the SMARCB1 (INI1) tumor suppressor gene in familial schwannomatosis. Clin Genet. 2008;74(4):358-1356.
Brems H., Chmara M., Sahbatou M., et al. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nat Genet. 2007;39(9):1120-1126.
Brock S., Dyke C.G. Venous and arteriovenous angiomas of the brain: Clinical and roentgenographic study of 8 cases. Bull Neurol Inst N Y. 1932;2:247.
Brook-Carter P.T., Peral B., Ward C.J., et al. Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kidney disease–a contiguous gene syndrome. Nature Genet. 1994;8:328.
Buck B.E., Mahboubi S., Raney R.B.Jr. Congenital neurogenous sarcoma with rhabdomyosarcomatous differentiation. J Pediatr Surg. 1977;12(4):581-582.
Cai W., Kassarjian A., Bredella M.A., et al. Tumor burden in patients with neurofibromatosis types 1 and 2 and schwannomatosis: Determination on whole-body MR images. Radiology. 2009;250(3):665-673.
Cairns A., North K. Cerebrovascular dysplasia in neurofibromatosis type 1. J Neurol Neurosurg Psychiatry. 2008;79(10):1165-1170.
Carney R.G. Incontinentia pigmenti. A world statistical analysis. Arch Dermatol. 1976;112(4):535-542.
Cawthon R.M., O’Connell P., Buchberg A.M., et al. Identification and characterization of transcripts from the neurofibromatosis 1 region: The sequence and genomic structure of EVI2 and mapping of other transcripts. Genomics. 1990;7:555-565.
Cawthon R.M., Weiss R., Xu G.F., et al. A major segment of the neurofibromatosis type 1 gene: Cdna sequence, genomic structure, and point mutations. Cell. 1990;62:193-201.
Chamberlain M.C., Press G.A., Hesselink J.R. MR imaging and CT in three cases of sturge-weber syndrome: Prospective comparison. Am J Neuroradiol. 1989;10(3):491-496.
Chao D. Congenital neurocutaneous syndromes in childhood. Pediatrics. 1959;54:189-199.
Chu-Shore C.J., Major P., Camposano S., et al. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia. 2009. doi: EPI2474 [pii) 10.1111/j.1528-1167.2009.02474.x
Chugani H.T., Mazziotta J.C., Phelps M.E. Sturge-Weber syndrome: A study of cerebral glucose utilization with positron emission tomography. J Pediatr. 1989;114(2):244-253.
Cichowski K., Jacks T. NF1 tumor suppressor gene function: Narrowing the GAP. Cell. 2001;104:593-604.
Collins E.T. Intra-Ocular growths. I. Two cases, brother and sister, with peculiar vascular new growth, probably primary retinal, affected both eyes. Trans Ophthalmol Soc UK. 1894;14:141.
Comi A.M. Update on sturge-weber syndrome: Diagnosis, treatment, quantitative measures, and controversies. Lymphat Res Biol. 2007;5(4):257-264.
Connor J.M., Sampson J. Recent linkage studies in tuberous sclerosis. Chromosome 9 markers. Ann N Y Acad Sci. 1991;615:265-273.
Consortium, The European Chromosome 16 Tuberous Sclerosis. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. 1993;75(7):1305.
Costa R.M., Federov N.B., Kogan J.H., et al. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature. 2002;415(6871):526-530.
Couvineau A., Wouters V., Bertrand G., et al. PTHR1 mutations associated with ollier disease result in receptor loss of function. Hum Mol Genet. 2008;17(18):2766-2775.
Crawford A.H., Herrera-Soto J. Scoliosis associated with neurofibromatosis. Orthop Clin North Am. 2007;38(4):553-562.
Crino P.B., Nathanson K.L., Henske E.P. The tuberous sclerosis complex. N Engl J Med. 2006;355(13):1345-1356.
Crowe F.W. Axillary freckling as a diagnostic aid in neurofibromatosis. Ann Intern Med. 1964;61:1142-1143.
Crowe F.W., Schull W.J., Neel J.V. A Clinical, Pathological, and Genetic Study of Multiple Neurofibromatosis. Springfield: Charles C. Thomas; 1956.
Curatolo P., D’Argenzio L., Cerminara C., et al. Management of epilepsy in tuberous sclerosis complex. Expert Rev Neurother. 2008;8(3):457-467.
Curatolo P., Porfirio M.C., Manzi B., et al. Autism in tuberous sclerosis. Eur J Paediatr Neurol. 2004;8(6):327-332.
Curatolo P., Seri S., Verdecchia M., et al. Infantile spasms in tuberous sclerosis complex. Brain Dev. 2001;23:502-507.
Curatolo P., Verdecchia M., Bombardieri R. Tuberous sclerosis complex: A review of neurological aspects. Eur J Paediatr Neurol. 2002;6(1):15-23.
D’Agostino A.N., Soule E.H., Miller R.H. Sarcomas of the peripheral nerves and somatic soft tissues associated with multiple neurofibromatosis (von recklinghausen’s disease). Cancer. 1963;16:1015-1027.
Daston M.M., Scrable H., Nordlund M., et al. The protein product of the neurofibromatosis type 1 gene is expressed at highest abundance in neurons, schwann cells, and oligodendrocytes. Neuron. 1992;8(3):415-428.
DeDavid M., Orlow S.J., Provost N., et al. Neurocutaneous melanosis: Clinical features of large congenital melanocytic nevi in patients with manifest central nervous system melanosis. J Am Acad Dermatol. 1996;35(4):529-538.
Desai S.S., Paulino A.C., Mai W.Y., et al. Radiation-induced moyamoya syndrome. Int J Radiat Oncol Biol Phys. 2006;65(4):1222-1227.
De Raedt T., Brems H., Wolkenstein P., et al. Elevated risk for MPNST in NF1 microdeletion patients. Am J Hum Genet. 2003;72(5):1288-1292.
de Vries P.J., Watson P. Attention deficits in tuberous sclerosis complex (TSC): Rethinking the pathways to the endstate. J Intellect Disabil Res. 2008;52(Pt 4):348-357.
Dimitri V. Tumor cerebral congenito (angioma cavernoso). Rev Assoc Med Argent. 1923;36:1029.
DiPaolo D.P., Zimmerman R.A., Rorke L.B., et al. Neurofibromatosis type 1: Pathologic substrate of high-signal-intensity foci in the brain. Radiology. 1995;195(7754001):721-724.
DiRocco F., Sabatino G., Koutzoglou M. Neurocutaneous melanosis. Childs Nerv Syst. 2004;20:23.
Dombi E., Solomon J., Gillespie A.J., et al. NF1 plexiform neurofibroma growth rate by volumetric MRI: Relationship to age and body weight. Neurology. 2007;68(9):643-647.
Ehninger D., Han S., Shilyansky C., et al. Reversal of learning deficits in a tsc2(+/−) mouse model of tuberous sclerosis. Nat Med. 2008;14(8):843-848.
Elefteriou F., Kolanczyk M., Schindeler A., et al. Skeletal abnormalities in neurofibromatosis type 1: Approaches to therapeutic options. Am J Med Genet A. 2009;149A(10):2327-2328.
Evans D.G. Neurofibromatosis type 2: Genetic and clinical features. Ear Nose Throat J. 1999;78(2):97-100.
Evans D.G. Neurofibromatosis type 2 (NF2): A clinical and molecular review. Orphanet J Rare Dis. 2009;4(1):16.
Evans D.G., Baser M.E., McGaughran J., et al. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39(5):311.
Evans D.G., Baser M.E., O’Reilly B., et al. Management of the patient and family with neurofibromatosis 2: A consensus conference statement. Br J Neurosurg. 2005;19(1):5-12.
Evans D.G., Howard E., Giblin C., et al. Birth incidence and prevalence of tumor-prone syndromes: Estimates from a UK family genetic register service. Am J Med Genet A. 2010;152A(2):327-332.
Evans D.G., Mason S., Huson S.M., et al. Spinal and cutaneous schwannomatosis is a variant form of type 2 neurofibromatosis: A clinical and molecular study. J Neurol Neurosurg Psychiatry. 1997;62(4):361-366.
Evans D.G., Ramsden R., Huson S.M., et al. Type 2 neurofibromatosis: The need for supraregional care? J Laryngol Otol. 1993;107:401-406.
Ferner R.E., Chaudhuri R., Bingham J., et al. MRI in neurofibromatosis 1. The nature and evolution of increased intensity T2 weighted lesions and their relationship to intellectual impairment. J Neurol Neurosurg Psychiatry. 1993;56:492-495.
Ferner R.E., Lucas J.D., O’Doherty M.J., et al. Evaluation of (18)fluorodeoxyglucose positron emission tomography ((18)FDG PET) in the detection of malignant peripheral nerve sheath tumours arising from within plexiform neurofibromas in neurofibromatosis 1. J Neurol Neurosurg Psychiatry. 2000;68(3):353-357.
Fialkow P.J., Sagebiel R.W., Gartler S.M., et al. Multiple cell origin of hereditary neurofibromas. N Engl J Med. 1971;284(6):298-300.
Fitzpatrick T.B. History and significance of white macules, earliest visible sign of tuberous sclerosis. Ann N Y Acad Sci. 1991;615:26-35.
Fitzpatrick T.B., Szabo G., Hori Y., et al. White leaf-shaped macules. Earliest visible sign of tuberous sclerosis. Arch Dermatol. 1968;98(1):1-6.
Flannery D.B. Pigmentary dysplasias, hypomelanosis of ito, and genetic mosaicism. Am J Med Genet. 1990;35(1):18-21.
Fossali E., Signorini E., Intermite R.C., et al. Renovascular disease and hypertension in children with neurofibromatosis. Pediatr Nephrol. 2000;14(8–9):806.
Fox H. Neurocutaneous melanosis. In: Bruyn P.J., Vinken G.W., editors. Handbook of Clinical Neurology. Amsterdam: North Holland, 1972.
Franz D.N. Non-neurologic manifestations of tuberous sclerosis complex. J Child Neurol. 2004;19(9):690-698.
Franz D.N., Leonard J., Tudor C., et al. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59(3):490-498.
Fricke B.L., Donnelly L.F., Casper K.A., et al. Frequency and imaging appearance of hepatic angiomyolipomas in pediatric and adult patients with tuberous sclerosis. Am J Roentgenol. 2004;182(4):1027-1030.
Friedman J.M., Arbiser J., Epstein J.A., et al. Cardiovascular disease in neurofibromatosis 1: Report of the NF1 cardiovascular task force. Genet Med. 2002;4(3):105-111.
Fuchs E. Aneurysma arteri-venosum retinae. Arch Augenheilkd. 1882;11:440.
Fujita H., Nakano K., Kumon Y., et al. (A case of wyburn-mason syndrome). Rinsho Shinkeigaku. 1989;29(8):1039-1044.
Fukushima K., Inoue Y., Fujiwara T., et al. Long-term course of west syndrome associated with tuberous sclerosis. Epilepsia. 1998;39(Suppl 5):50-54.
Ganji S., Hellman C.D. Tuberous sclerosis: Long-term follow-up and longitudinal electroencephalographic study. Clin Electroencephalogr. 1985;16(4):219-224.
Gao X., Pan D. TSC1 and TSC2 tumor suppressors antagonize insulin signaling in cell growth. Genes Dev. 2001;15(11):1383-1392.
Garrod A.E. Peculiar pigmentation of the skin in an infant. Trans Clin Soc Lond. 1906;39:216.
Gijtenbeek J.M., Gabreels-Festen A.A., Lammens M., et al. Mononeuropathy multiplex as the initial manifestation of neurofibromatosis type 2. Neurology. 2001;56(12):1766-1768.
Gillberg I.C., Gillberg C., Ahlsen G. Autistic behaviour and attention deficits in tuberous sclerosis: A population-based study. Dev Med Child Neurol. 1994;36(1):50-56.
Gloviczki P., Driscoll D.J. Klippel-Trenaunay syndrome: Current management. Phlebology. 2007;22(6):291-298.
Goh S., Butler W., Thiele E.A. Subependymal giant cell tumors in tuberous sclerosis complex. Neurology. 2004;63(8):1457-1461.
Goh S., Kwiatkowski D.J., Dorer D.J., et al. Infantile spasms and intellectual outcomes in children with tuberous sclerosis complex. Neurology. 2005;65(2):235-238.
Goldberg M.F., Duke J.R. Von Hippel-Lindau disease. Histopathologic findings in a treated and an untreated eye. Am J Ophthalmol. 1968;66:693-705.
Gomez M.R. Natural history of cerebral tuberous sclerosis. In: Sampson J.R., Whittemore V.H., Gomez M.R., editors. Tuberous Sclerosis Complex: Developmental Perspectives in Psychiatry. New York: Oxford University Press, 1999.
Developmental Perspectives in Psychiatry (Series),. Gomez M.R., Sampson J.R., Whittemore V.H., editors. Tuberous Sclerosis Complex, ed 3, New York: Oxford University Press, 1987. 1999
Greenwald M.J., Weiss A. Ocular manifestations of the neurocutaneous syndromes. Pediatr Dermatol. 1984;2(2):98-117.
Gupta A., Cohen B.H., Ruggieri P., et al. Phase I study of thalidomide for the treatment of plexiform neurofibroma in neurofibromatosis 1. Neurology. 2003;60(1):130-132.
Gutmann D.H., Aylsworth A., Carey J.C., et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA. 1997;278:51-57.
Hagel C., Lindenau M., Lamszus K., et al. Polyneuropathy in neurofibromatosis 2: Clinical findings, molecular genetics and neuropathological alterations in sural nerve biopsy specimens. Acta Neuropathol. 2002;104(2):179-187.
Hager B.C., Dyme I.Z., Guertin S.R., et al. Linear nevus sebaceous syndrome: Megalencephaly and heterotopic gray matter. Pediatr Neurol. 1991;7:45-49.
Haines J.L., Short M.P., Kwiatkowski D.J., et al. Localization of one gene for tuberous sclerosis within 9q32-9q34, and further evidence for heterogeneity. Am J Hum Genet. 1991;49(4):764-772.
Hall B.D. Bladder hemangiomas in Klippel-Trenaunay-Weber syndrome. N Engl J Med. 1971;285(18):1032-1033.
Hamada K., Tanaka T., Ohdo S., et al. Incontinentia pigmenti achromians as part of a neurocutaneous syndrome: A case report. Brain Dev. 1979;1:313-317.
Hamada T., Saito T., Sugai T., et al. Incontinentia pigmenti achromians (ito). Arch Dermatol. 1967;96:673-676.
Hamano S., Tanaka M., Mochizuki M., et al. Long-term follow-up study of west syndrome: Differences of outcome among symptomatic etiologies. J Pediatr. 2003;143(2):231-235.
Happle R. How many epidermal nevus syndromes exist? A clinicogenetic classification. J Am Acad Dermatol. 1991;25:550-556.
Hardwig P., Robertson D.M. Von Hippel-Lindau disease: A familial, often lethal, multi-system phakomatosis. Ophthalmology. 1984;91:263-270.
Hatjis C.G., Philip A.G., Anderson G.G., et al. The in utero ultrasonographic appearance of Klippel-Trenaunay-Weber syndrome. Am J Obstet Gynecol. 1981;139:972-974.
Hauw J.J., Perie G., Bonnette J., et al. (Neuropathological study of incontinentia pigmenti. Anatomical case report [author’s transl]). Acta Neuropathol. 1977;38(2):159-162.
Heathcote J.G., Schoales B.A., Willis N.R. Incontinentia pigmenti (bloch-sulzberger syndrome): A case report and review of the ocular pathological features. Can J Ophthalmol. 1991;26:229-237.
Hebold O. Haemangiom der weichen hirnbaut bei naevus vasculosus des gesichts. Arch Psychiatr Nervenkr. 1913;51:445.
Hennel S.J., Ekert P.G., Volpe J.J., et al. Insights into the pathogenesis of cerebral lesions in incontinentia pigmenti. Pediatr Neurol. 2003;29(2):148-150.
Henske E.P., Scheithauer B.W., Short M.P., et al. Allelic loss is frequent in tuberous sclerosis kidney lesions but rare in brain lesions. Am J Hum Genet. 1996;59(2):400-406.
Hinsch N., Kriener S., Ritter R.G., et al. Fatal haemorrhage due to extensive fragility of medium- and large-sized arteries and veins in a young patient with neurofibromatosis 1. Cardiovasc Pathol. 2008;17(2):108-112.
Hintringer T., Furnschlief E., Bauer M. Reconstruction of facial contour in hemifacial atrophy (romberg disease) using a free revascularized scapula-fat-fascia flap. Wien Med Wochenschr. 1991;141:118-120.
Hobert J.A., Eng C. PTEN hamartoma tumor syndrome: An overview. Genet Med. 2009;11(10):687-694.
Hoff M., van Grunsven M.F., Jongebloed W.L., et al. Enamel defects associated with tuberous sclerosis. A clinical and scanning-electron-microscope study. Oral Surg Oral Med Oral Pathol. 1975;40(2):261-269.
Holden K.R., Dekaban A.S. Neurological involvement in nevus unis lateris and nevus linearis sebaceus. Neurology. 1972;22:879-887.
Hopen G., Smith J.L., Hoff J.T., et al. The wyburn-mason syndrome. Concomitant chiasmal and fundus vascular malformations. J Clin Neuroophthalmol. 1983;3(1):53-62.
Hopyan S., Gokgoz N., Poon R., et al. A mutant PTH/pthrp type I receptor in enchondromatosis. Nat Genet. 2002;30(3):306-310.
Hornigold N., van Slegtenhorst M., Nahmias J., et al. A 1.7-megabase sequence-ready cosmid contig covering the TSC1 candidate region in 9q34. Genomics. 1997;41(3):385-389.
Howlett D.C., Farrugia M.M., Ferner R.E., et al. Multiple lower limb non-ossifying fibromas in siblings with neurofibromatosis. Eur J Radiol. 1998;26(3):280-283.
Hoyt W.F., Cameron R.B. Racemose angioma of the mandible, face, retina, and brain: Report of case. J Oral Surg. 1968;26:596-601.
Hu Y., Li L., Seidelmann S.B., et al. Identification of association of common AGGF1 variants with susceptibility for Klippel-Trenaunay syndrome using the structure association program. Ann Hum Genet. 2008;72(Pt 5):636-643.
Huang J., Manning B.D. The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem J. 2008;412(2):179-190.
Hulsebos T.J.M., Plomp A.S., Wolterman R.A., et al. Germline mutation of INI1/SMARCB1 in familial schwannomatosis. Am J Hum Genet. 2007;80(4):805-810.
Husain A.M., Foley C.M., Legido A., et al. West syndrome in tuberous sclerosis complex. Pediatr Neurol. 2000;23(3):233-235.
Huson S.M., Harper P.S., Compston D.A. Von Recklinghausen neurofibromatosis. A clinical and population study in south-east Wales. Brain. 1988;111(Pt 6):1355-1381.
Hyman S.L., Gill D.S., Shores E.A., et al. Natural history of cognitive deficits and their relationship to MRI t2-hyperintensities in NF1. Neurology. 2003;60(7):1139-1145.
Hyman S.L., Gill D.S., Shores E.A., et al. T2 hyperintensities in children with neurofibromatosis type 1 and their relationship to cognitive functioning. J Neurol Neurosurg Psychiatr. 2007;78(10):1088-1091.
Hyman S.L., Arthur Shores E., North K.N. Learning disabilities in children with neurofibromatosis type 1: Subtypes, cognitive profile, and attention-deficit-hyperactivity disorder. Dev Med Child Neurol. 2006;48(12):973-977.
Inceman S., Tangun Y. Chronic defibrination syndrome due to a giant hemangioma associated with microangiopathic hemolytic anemia. Am J Med. 1969;46(6):997-1002.
Ito M. A singular case of naevus depigmentosus systematicus bilateralis. Jpn J Dermatol. 1951;61:31.
Jacob A.G., Driscoll D.J., Shaughnessy W.J., et al. Klippel-Trenaunay syndrome: Spectrum and management. Mayo Clin Proc. 1998;73(1):28-36.
Jacquemin C., Bosley T.M., Liu D., et al. Reassessment of sphenoid dysplasia associated with neurofibromatosis type 1. Am J Neuroradiol. 2002;23(4):644-648.
Jansen F.E., van Huffelen A.C., Algra A., et al. Epilepsy surgery in tuberous sclerosis: A systematic review. Epilepsia. 2007;48(8):1477-1484.
Jessup C.J., Morgan S.C., Cohen L.M., et al. Incontinentia pigmenti: Treatment of IP with topical tacrolimus. J Drugs Dermatol. 2009;8(10):944-946.
Jozwiak S., Schwartz R.A. Dermatological and Stomatological Manifestations. In: Curatolo P., editor. Tuberous sclerosis complex: From basic science to clinical phenotypes. Cambridge: MacKeith Press, 2003.
Jozwiak S., Kawalec W., Dluzewska J., et al. Cardiac tumours in tuberous sclerosis: Their incidence and course. Eur J Pediatr. 1994;153(3):155-157.
Kaelin W.G.Jr. The Von Hippel-Lindau protein, HIF hydroxylation, and oxygen sensing. Biochem Biophys Res Commun. 2005;9(338):627-638.
Kalischer S. Demonstration des gehirns eines kindes mit teleangiektasie der linksseitigen gesichts-kopfhaut und hirnoberflache. Berliner Klinische Wochenschrift. 1897;34:1059-1067.
Kandt R.S., Haines J.L., Smith M., et al. Linkage of an important gene locus for tuberous sclerosis to a chromosome 16 marker for polycystic kidney disease. Nat Genet. 1992;2(1):37-41.
Kanno H., Yamamoto I., Nishikawa R., et al. Spinal cord hemangioblastomas in Von Hippel-Lindau disease. Spinal Cord. 2009;47(6):447-452.
Kano H., Niranjan A., Mongia S., et al. The role of stereotactic radiosurgery for intracranial hemangioblastomas. Neurosurgery. 2008;63(3):443-450. discussion 450–1
Kapp J.P., Paulson G.W., Odom G.L. Brain tumors with tuberous sclerosis. J Neurosurg. 1967;26(2):191-202.
Karabagli H., Genc A., Karabagli P., et al. Outcomes of gamma knife treatment for solid intracranial hemangioblastomas. J Clin Neurosci. 2010. doi: S0967-5868(09)00706-1 [pii) 10.1016/j.jocn.2009.09.028
Karabatsou K., Kiehl T.R., Wilson D.M., et al. Potential role of 18fluorodeoxyglucose-positron emission tomography/computed tomography in differentiating benign neurofibroma from malignant peripheral nerve sheath tumor associated with neurofibromatosis 1. Neurosurgery. 2009;65(4 Suppl):160-170.
Kasantikul V., Shuangshoti S., Pattanaruenglai A., et al. Intraspinal melanotic arachnoid cyst and lipoma in neurocutaneous melanosis. Surg Neurol. 1989;31:138-141.
Kaya T.I., Tursen U., Ikizoglu G. Therapeutic use of topical corticosteroids in the vesiculobullous lesions of incontinentia pigmenti. Clin Exp Dermatol. 2009;34(8):e611-e613.
Kayes L.M., Burke W., Riccardi V.M., et al. Deletions spanning the neurofibromatosis 1 gene: Identification and phenotype of five patients. Am J Hum Genet. 1994;54(3):424-436.
Kenwrick S., Woffendin H., Jakins T., et al. Survival of male patients with incontinentia pigmenti carrying a lethal mutation can be explained by somatic mosaicism or klinefelter syndrome. Am J Hum Genet. 2001;69(6):1210-1217.
Kestle J.R., Hoffman H.J., Mock A.R. Moyamoya phenomenon after radiation for optic glioma. J Neurosurg. 1993;79(1):32-35.
Klippel M., Trenaunay P. Du naevus varigues osteohypertrophique. Arch Gen Med. 1900;185:641.
Korf B.R. Diagnostic outcome in children with multiple cafe au lait spots. Pediatrics. 1992;90:924-927.
Korf B.R. Neurofibromas and malignant tumors of the peripheral nervous system. In: Friedman J.M., Gutmann D.H., MacCollin M., et al, editors. Neurofibromas and Malignant Tumors of the Peripheral Nervous System. Baltimore: Johns Hopkins, 1999.
Korf B.R., Carrazana E., Holmes G.L. Patterns of seizures observed in association with neurofibromatosis 1. Epilepsia. 1993;34(4):616-620.
Korf B.R., Henson J.W., Stemmer-Rachamimov A. Case records of the Massachusetts General Hospital. Case 13–2005. A 48-year-old man with weakness of the limbs and multiple tumors of spinal nerves. N Engl J Med. 2005;352(17):1800-1808.
Krab L.C., de Goede-Bolder A., Aarsen F.K., et al. Effect of simvastatin on cognitive functioning in children with neurofibromatosis type 1: A randomized controlled trial. JAMA. 2008;300(3):287-294.
Kuorilehto T., Pöyhönen M., Bloigu R., et al. Decreased bone mineral density and content in neurofibromatosis type 1: Lowest local values are located in the load-carrying parts of the body. Osteoporos Int. 2005;16(8):928-936.
Kupersmith M.J., Berenstein A. Visual disturbances in Von Hippel-Lindau disease. Ann Ophthalmol. 1981;13:195-197.
Kurokawa T., Sasaki K., Hanai T., et al. Linear nevus sebaceus syndrome. Report of a case with lennox-gastaut syndrome following infantile spasms. Arch Neurol. 1981;38:375-377.
Kwiatkowska J., Wigowska-Sowinska J., Napierala D., et al. Mosaicism in tuberous sclerosis as a potential cause of the failure of molecular diagnosis. N Engl J Med. 1999;340(9):703-707.
Kwiatkowski D.J., Reeve M.P., Cheadle J.P., et al. Molecular genetics. In: Curatolo P., editor. Tuberous Sclerosis Complex: From Basic Science to Clinical Phenotypes. London: MacKeith Press, 2003.
Latif F., Tory K., Gnarra J., et al. Identification of the Von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317-1320.
Lazaro C., Gaona A., Lynch M., et al. Molecular characterization of the breakpoints of a 12-kb deletion in the NF1 gene in a family showing germ-line mosaicism. Am J Hum Genet. 1995;57(5):1044-1049.
Le L.Q., Shipman T., Burns D.K., et al. Cell of origin and microenvironment contribution for nf1-associated dermal neurofibromas. Cell Stem Cell. 2009;4(5):453-463.
Lee A.G., Goldberg M.F., Gillard J.H., et al. Intracranial assessment of incontinentia pigmenti using magnetic resonance imaging, angiography, and spectroscopic imaging. Arch Pediatr Adolesc Med. 1995;149:573-580.
Legius E., Dierick H., Wu R., et al. TP53 mutations are frequent in malignant NF1 tumors. Genes Chromosomes Cancer. 1994;10:250-255.
Lenz W. On the genetics of incontinentia pigmenti. Ann Paediatr. 1961;196:149-165.
Lewis R.A., Riccardi V.M. Von Recklinghausen neurofibromatosis. Incidence of iris hamartomata. Ophthalmology. 1981;88(4):348-354.
Lewis R.A., Gerson L.P., Axelson K.A., et al. Von Recklinghausen neurofibromatosis. II. Incidence of optic gliomata. Ophthalmology. 1984;91(8):929-935.
Lian C., Alhomme P. Les varices congenitales par dysembryoplasia (le syndrome de Klippel-Trénaunay). Arch Mal Coeur Vaiss. 1945;38:176.
Lindau A. Studien uber kleinhirncysten: Bau, pathogenese und beziehungen zur angiomatosis retinae. Acta Pathol Microbiol Scand. 1926;1(Suppl):1.
Lindenauer J.M. The Klippel-Trénaunay syndrome: Varicosity, hypertrophy and hemangioma with no arteriovenous fistula. Am Surg. 1965;162:203-214.
Lisch K. Uber beteilung der augen, insobesondere das vorkommen von irisknotchun bei der neurofibromatose (Recklinghausen). Z Augenheilk. 1937;93:137-143.
Listernick R., Charrow J., Greenwald M., et al. Natural history of optic pathway tumors in children with neurofibromatosis type 1: A longitudinal study. J Pediatr. 1994;125:63-66.
Listernick R., Ferner R.E., Liu G.T., et al. Optic pathway gliomas in neurofibromatosis-1: Controversies and recommendations. Ann Neurol. 2007;61(3):189-198.
Louis-Bar D. Sur un syndrome professif comprenant des telangiectasis capillaries cutanees et conjunctivales symetriques a disposition naevoide et des troubles cerebelleux. Confin Neurol. 1941;4:32-42.
Lovett A., Maari C., Decarie J.C., et al. Large congenital melanocytic nevi and neurocutaneous melanocytosis: One pediatric center’s experience. J Am Acad Dermatol. 2009;61(5):766-774.
Lubs M.L., Bauer M.S., Formas M.E., et al. Lisch nodules in neurofibromatosis type 1. N Engl J Med. 1991;324(18):1264-1266.
Lyczkowski D.A., Conant K.D., Pulsifer M.B., et al. Intrafamilial phenotypic variability in tuberous sclerosis complex. J Child Neurol. 2007;22(12):1348-1355.
MacCollin M., Chiocca E.A., Evans D.G., et al. Diagnostic criteria for schwannomatosis. Neurology. 2005;64(11):1838-1845.
MacCollin M., Woodfin W., Kronn D., et al. Schwannomatosis: A clinical and pathologic study. Neurology. 1996;46(4):1072-1079.
Macmichael I.M. Von Hippel Lindau’s disease of the optic disc. Trans Ophthalmol Soc UK. 1970;90:877-885.
Maertens O., De Schepper S., Vandesompele Jo, et al. Molecular dissection of isolated disease features in mosaic neurofibromatosis type 1. Am J Hum Genet. 2007;81(2):243-251.
Maffucci A. Di uno case encondroma ed angioma multiplo. Movemento Med-Chir. 1881;3:399.
Magnus H. Aneurysma arteriovenosum retinale. Arch Pathol Anat Physiol. 1874;60:38.
Maher C.O., Cohen-Gadol A.A., Raffel C. Cortical resection for epilepsy in children with linear sebaceous nevus syndrome. Pediatr Neurosurg. 2003;39(3):129-135.
Maher E.R., Iselius L., Yates J.R., et al. Von Hippel-Lindau disease: A genetic study. J Med Genet. 1991;28:443-447.
Manski T.J., Heffner D.K., Glenn G.M., et al. Endolymphatic sac tumors. A source of morbid hearing loss in Von Hippel-Lindau disease. J Am Med Assoc. 1997;277(18):1461-1466.
Maria B.L., Hoang K.B.N., Robertson R.L., et al. Imaging brain structure and function in Sturge Weber syndrome. In: Roach E.S., Bodensteiner J.B., editors. Sturge-Weber Syndrome. Mt. Freedom, NJ: Sturge Weber Foundation, 1999.
Mautner V.F., Kluwe L., Thakker S.D., et al. Treatment of ADHD in neurofibromatosis type 1. Dev Med Child Neurol. 2002;44(3):164.
Mautner V.F., Lindenau M., Baser M.E., et al. Skin abnormalities in neurofibromatosis 2. Arch Dermatol. 1997;133(12):1539-1543.
Mautner V.F., Nguyen R., Kutta H., et al. Bevacizumab induces regression of vestibular schwannomas in patients with\ neurofibromatosis type 2. Neuro Oncol. 12, 2010. 14-8Epub
Mautner V.F., Tatagiba M., Guthoff R., et al. Neurofibromatosis 2 in the pediatric age group. Neurosurgery. 1993;33(1):92-96.
Mavili E., Ozturk M., Akcali Y., et al. Direct CT venography for evaluation of the lower extremity venous anomalies of Klippel-Trenaunay syndrome. Am J Roentgenol. 2009;192(6):W311-W316.
Mayro R.F., DeLozier J.B., Whitaker L.A. Facial reconstruction consideration in rheumatic diseases. Rheum Dis Clin North Am. 1991;17:943-969.
McDermott A.L., Dutt S.N., Chavda S.V., et al. Maffucci’s syndrome: Clinical and radiological features of a rare condition. J Laryngol Otol. 2001;115:845-847.
McWilliam R.C., Stephenson J.B. Depigmented hair. The earliest sign of tuberous sclerosis. Arch Dis Child. 1978;53(12):961-963.
Meine J.G., Schwartz R.A., Janniger C.K. Klippel-Trenaunay-Weber syndrome. Cutis. 1997;60(3):127-132.
Melmon K.L., Rosen S.W. Lindau’s disease. Review of the literature and study of a large kindred. Am J Med. 1964;36:595-617.
Messiaen L., Yao S., Brems H., et al. Clinical and mutational spectrum of neurofibromatosis type 1-like syndrome. JAMA. 2009;302(19):2111-2118.
Messiaen L., Riccardi V., Peltonen J., et al. Independent NF1 mutations in two large families with spinal neurofibromatosis. J Med Genet. 2003;40(2):122-126.
Messiaen L.M., Callens T., Mortier G., et al. Exhaustive mutation analysis of the NF1 gene allows identification of 95 percent of mutations and reveals a high frequency of unusual splicing defects. Hum Mutat. 2000;15(6):541-555.
Messinger H.S., Clarke B.W. Retinal tumors in tuberous sclerosis: Review of the literature and report of a case with special attention to microscopic structure. Arch Ophthalmol. 1937;18:1.
Mlynarczyk G. Enamel pitting. A common sign of tuberous sclerosis. Ann N Y Acad Sci. 1991;615:367-369.
Morales J., Chaudhry I.A., Bosley T.M. Glaucoma and globe enlargement associated with neurofibromatosis type 1. Ophthalmology. 2009;116(9):1725-1730.
Moss J., Avila N.A., Barnes P.M., et al. Prevalence and clinical characteristics of lymphangioleiomyomatosis (LAM) in patients with tuberous sclerosis complex. Am J Respir Crit Care Med. 2001;164(4):669-671.
Muzykewicz D.A., Costello D.J., Halpern E.F., et al. Infantile spasms in tuberous sclerosis complex: Prognostic utility of EEG. Epilepsia. 2009;50(2):290-296.
Muzykewicz D.A., Newberry P., Danforth N., et al. Psychiatric comorbid conditions in a clinic population of 241 patients with tuberous sclerosis complex. Epilepsy Behav. 2007;11(4):506-513.
Needle M.N., Cnaan A., Dattilo J., et al. Prognostic signs in the surgical management of plexiform neurofibroma: The Children’s Hospital of Philadelphia experience, 1974-1994. J Pediatr. 1997;131:678-682.
Nellhaus G., Haberland C., Hill B.J. Sturge-Weber disease with bilateral intracranial calcifications at birth and unusual pathologic findings. Acta Neurol Scand. 1967;43:314-347.
Neumann H.P., Schwarzkopf G., Henske E.P. Renal angiomyolipomas, cysts, and cancer in tuberous sclerosis complex. Semin Pediatr Neurol. 1998;5(4):269-275.
Nordstrom-O’Brien M., van der Luijt R.B., van Rooijen E., et al. Genetic analysis of von Hippel-Lindau disease. Hum Mutat. 2010. doi: 10.1002/humu.21219
Orloff M.S., Eng C. Genetic and phenotypic heterogeneity in the PTEN hamartoma tumour syndrome. Oncogene. 2008;27(41):5387-5397.
Osborne J.P., Fryer A., Webb D. Epidemiology of tuberous sclerosis. Ann N Y Acad Sci. 1991;615:125-127.
Packer R.J., Ater J., Allen J., et al. Carboplatin and vincristine chemotherapy for children with newly diagnosed progressive low-grade gliomas. J Neurosurg. 1997;86(5):747-754.
Packer R.J., Gutmann D.H., Rubenstein A., et al. Plexiform neurofibromas in NF1: Toward biologic-based therapy. Neurology. 2002;58(10):1461-1470.
Packer R.J., Rosser T. Therapy for plexiform neurofibromas in children with neurofibromatosis 1: An overview. J Child Neurol. 2002;17(8):638-641.
Pampiglione G., Moynahan E.J. The tuberous sclerosis syndrome: Clinical and EEG studies in 100 children. J Neurol Neurosurg Psychiatry. 1976;39(7):666-673.
Panas F., Remy D.A. Anatomie Pathologique De L’oeil. Paris: Delahaye; 1879.
Papanyothou P., Verzirtzi E. Tuberous sclerosis with gingival lesions: Report of a case. Oral Surgery. 1975;39:578.
Parkes Weber F. Angioma formation in connection with hypertrophy of limbs and hemihypertrophy. Br J Dermatol. 1907;19:231.
Parry D.M., MacCollin M.M., Kaiser-Kupfer M.I., et al. Germ-line mutations in the neurofibromatosis 2 gene: Correlations with disease severity and retinal abnormalities. Am J Hum Genet. 1996;59:529-539.
Pavesi G., Feletti A., Berlucchi S., et al. Neurosurgical treatment of Von Hippel-Lindau-associated hemangioblastomas: Benefits, risks and outcome. J Neurosurg Sci. 2008;52(2):29-36.
Pavesi G., Berlucchi S., Munari M., et al. Clinical and surgical features of lower brain stem hemangioblastomas in Von Hippel-Lindau disease. Acta Neurochir (Wien). 2010;152(2):287-292.
Pavlidou E., Hagel C., Papavasilliou A., et al. Neurocutaneous melanosis: Report of three cases and up-to-date review. J Child Neurol. 2008;23(12):1382-1391.
Pearson-Webb M.A., Kaiser-Kupfer M.I., Eldridge R. Eye findings in bilateral acoustic (central) neurofibromatosis: Association with presenile lens opacities and cataracts but absence of lisch nodules. N Engl J Med. 1986;315:1553-1554.
Pineda A. Mast cells–their presence and ultrastructural characteristics in peripheral nerve tumors. Arch Neurol. 1965;13(4):372-382.
Plotkin S., Stemmer-Rachamimov A., Barker F., et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med. 2009. doi: NEJMoa0902579 [pii) 10.1056/NEJMoa0902579 [doi)
Potter C.J., Huang H., Xu T. Drosophila tsc1 functions with tsc2 to antagonize insulin signaling in regulating cell growth, cell proliferation, and organ size. Cell. 2001;105(3):357-368.
Poulsen M.L., Budtz-Jorgensen E., Bisgaard M.L. Surveillance in Von Hippel-Lindau disease (vHL). Clin Genet. 2010;77(1):49-59.
Pulsifer M.B., Winterkorn E.B., Thiele E.A. Psychological profile of adults with tuberous sclerosis complex. Epilepsy Behav. 2007;10(3):402-406.
Rakowski S., Winterkorn E., Paul E., et al. Renal manifestations of tuberous sclerosis complex: Incidence, prognosis, and predictive factors. Kidney Int. 2006;70(10):1777-1782.
Ramina R., Coelho Neto M., Meneses M.S., et al. Maffucci’s syndrome associated with a cranial base chondrosarcoma: Case report and literature review. Neurosurgery. 1997;41(1):269-272.
Ranger A., Szymczak A., Hammond R.R., et al. Pediatric thalamic glioblastoma associated with ollier disease (multiple enchondromatosis): A rare case of concurrence. J Neurosurg Pediatr. 2009;4(4):363-367.
Raznahan A., Joinson C., O’Callaghan F., et al. Psychopathology in tuberous sclerosis: An overview and findings in a population-based sample of adults with tuberous sclerosis. J Intellect Disabil Res. 2006;50(Pt 8):561-569.
Reck S.D., Zacks D.N., Eibschitz-Tsimhoni M. Retinal and intracranial arteriovenous malformations: Wyburn-Mason syndrome. J Neuroophthalmol. 2005;25:205-208.
Redondo P., Bastarrika G., Sierra A., et al. Efficacy and safety of microfoam sclerotherapy in a patient with Klippel-Trenaunay syndrome and a patent foramen ovale. Arch Dermatol. 2009;145(10):1147-1151.
Riccardi V.M. Pathophysiology of neurofibromatosis. IV. Dermatologic insights into heterogeneity and pathogenesis. J Am Acad Dermatol. 1980;3(2):157-166.
Ridley M., Green J., Johnson G. Retinal angiomatosis: The ocular manifestations of Von Hippel-Lindau disease. Can J Ophthalmol. 1986;21(7):276-283.
Riikonen R., Simell O. Tuberous sclerosis and infantile spasms. Dev Med Child Neurol. 1990;32(3):203-209.
Rischbieth R.H.C. Progressive facial hemiatrophy (Parry-Romberg syndrome). Proc Aust Assoc Neurol. 1976;13:109.
Ritter C.L., Steele M.W., Wenger S.L., et al. Chromosome mosaicism in hypomelanosis of ito. Am J Med Genet. 1990;35(2301465):14-17.
Roach E.S., DiMario F.J., Kandt R.S., et al. Tuberous sclerosis consensus conference: Recommendations for diagnostic evaluation. National tuberous sclerosis association. J Child Neurol. 1999;14(6):401-407.
Roach E.S., Gomez M.R., Northrup H. Tuberous sclerosis complex consensus conference: Revised clinical diagnostic criteria. J Child Neurol. 1998;13(12):624-628.
Roach E.S., Riela A.R., McLean W.T. Asprin therapy for Sturge-Weber syndrome. Ann Neurol. 1985;18:387.
Rochkind S., Hoffman H.J., Hendrick E.B. Sturge-Weber syndrome: Natural history and prognosis. J Epilepsy. 1990;3(Suppl):293.
Rokitansky C. Eim ausgezeichneter fall von pigment-mal ausgebreiter pigmentierung der inner hirnhaute und ruchemmarkshaute. Allg Wien Med Ztg. 1861;6:113.
Rosman N.P., Pearce J. The brain in multiple neurofibromatosis (Von Recklinghausen’s disease): A suggested neuropathological basis for the associated mental defect. Brain. 1967;90(4):829-838.
Roth J.C., Epstein C.J. Infantile spasms and hypopigmented macules. Early manifestations of tuberous sclerosis. Arch Neurol. 1971;25(6):547-551.
Rouleau G.A., Merel P., Lutchman M., et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature. 1993;363:515-521.
Rouleau G.A., Wertelecki W., Haines J.L., et al. Genetic linkage of bilateral acoustic neurofibromatosis to a DNA marker on chromosome 22. Nature. 1987;329:246-248.
Rowley S.A., O’Callaghan F.J., Osborne J.P. Ophthalmic manifestations of tuberous sclerosis: A population based study. Br J Ophthalmol. 2001;85(4):420-423.
Ruggieri M., Tigano G., Mazzone D., et al. Involvement of the white matter in hypomelanosis of ito (incontinentia pigmenti achromiens). Neurology. 1996;46:485-492.
Ruttledge M.H., Andermann A.A., Phelan C.M., et al. Type of mutation in the neurofibromatosis type 2 gene (NF2) frequently determines severity of disease. Am J Hum Genet. 1996;59(2):331-342.
Salazar F.G., Lamiell J.M. Early identification of retinal angiomas in a large kindred Von Hippel-Lindau disease. Am J Ophthalmol. 1980;89:540-545.
Sampson J.R., Yates J.R., Pirrit L.A., et al. Evidence for genetic heterogeneity in tuberous sclerosis. J Med Genet. 1989;26(8):511-516.
Sardi I., Sanzo M., Giordano F., et al. Monotherapy with thalidomide for treatment of spinal cord hemangioblastomas in a patient with Von Hippel-Lindau disease. Pediatr Blood Cancer. 2009;53(3):464-467.
Sassower K., Duchowny M., Jayakar P. EEG evaluation of children with Sturge-Weber syndrome. J Epilepsy. 1994;7:285.
Schirmer R. Ein fall von teleangiektasie. Graefes Arch Ophthalmol. 1860;7:119.
Schmidt D., Pache M., Schumacher M. The congenital unilateral retinocephalic vascular malformation syndrome (bonnet-dechaume-blanc syndrome or wyburn-mason syndrome): Review of the literature. Surv Ophthalmol. 2008;53(3):227-249.
Schreml S., Gruendobler B., Schreml J., et al. Neurocutaneous melanosis in association with dandy-walker malformation: Case report and literature review. Clin Exp Dermatol. 2008;33(5):611-614.
Schwartz M.F., Esterly N.B., Fretzin D.F., et al. Hypomelanosis of ito (incontinentia pigmenti achromians): A neurocutaneous syndrome. J Pediatr. 1977;90:236-240.
Seizinger B.R., Rouleau G., Ozelius L.J., et al. Common pathogenetic mechanism for three tumor types in bilateral acoustic neurofibromatosis. Science. 1987;236(4799):317-319.
Sharif S., Ferner R., Birch J.M., et al. Second primary tumors in neurofibromatosis 1 patients treated for optic glioma: Substantial risks after radiotherapy. J Clin Oncol. 2006;24(16):2570-2575.
Sherman L.S., Atit R., Rosenbaum T., et al. Single cell ras-gtp analysis reveals altered ras activity in a subpopulation of neurofibroma schwann cells but not fibroblasts. J Biol Chem. 2000;275:30740-30745.
Sieman H.W. Die melanosis corii degenerativa, eine neue pigmentdermatose. Arch Dermatol Syphilol. 1929;157:382.
Slater A., Moore N.R., Huson S.M. The natural history of cerebellar hemangioblastomas in von hippel-lindau disease. Am J Neuroradiol. 2003;24:1570-1574.
Smahi A., Courtois G., Vabres P., et al. Genomic rearrangement in NEMO impairs nf-kappab activation and is a cause of incontinentia pigmenti. The international incontinentia pigmenti (IP) consortium. Nature. 2000;405(6785):466-472.
Smalley S.L. Autism and tuberous sclerosis. J Autism Dev Disord. 1998;28(5):407-414.
Smith M., Smalley S., Cantor R., et al. Mapping of a gene determining tuberous sclerosis to human chromosome 11q14-11q23. Genomics. 1990;6(1):105-114.
Smith R.W. A Treatise on the Pathology, Diagnosis and Treatment of Neuroma. Dublin: Hodges & Smith; 1849.
Souza J., Passos R., Guedes A., et al. Muscular force is reduced in neurofibromatosis type 1. J Musculoskelet Neuronal Interact. 2009;9(1):15-17.
Stevenson D.A., Birch P.H., Friedman J.M., et al. Descriptive analysis of tibial pseudarthrosis in patients with neurofibromatosis 1. Am J Med Genet. 1999;84:413-419.
Stevenson D.A., Murray M., Viskochil D.H., et al. Bone mineral density in children with neurofibromatosis type 1. J Pediatr Orthop. 2008;28(7):791. author reply 791–2
Stevenson D.A., Zhou H., Ashrafi S., et al. Double inactivation of NF1 in tibial pseudarthrosis. Am J Hum Genet. 2006;79(1):143-148.
Stiller C.A., Chessells J.M., Fitchett M. Neurofibromatosis and childhood leukaemia/lymphoma: A population-based UKCCSG study. Br J Cancer. 1994;70(5):969-972.
Stone J. Parry-Romberg syndrome: A global survey of 205 patients using the internet. Neurology. 2003;61(5):674-676.
Stumpf D. Consensus development conference of neurofibromatosis. Arch Neurol. 1988;45:575-578.
Sturge W.A. Case of rare vaso-motor disturbance in the leg. Trans Clin Soc Lond. 12(156), 1879.
Subbiah V., Wolff J.E. Rapid response to therapy of neurocutaneous melanosis with leptomeningeal melanoma. Pediatr Blood Cancer. 2010;54(1):180-181.
Sugama S., Yoshimura H., Ashimine K., et al. Enhanced magnetic resonance imaging of leptomeningeal angiomatosis. Pediatr Neurol. 1997;17:262-265.
Sujansky E., Conradi S. Outcome of Sturge-Weber syndrome in 52 adults. Am J Med Genet. 1995;57(1):35-45.
Sulzberger M.B. Incontinentia-Pigmenti (Bloch-Sulzberger): Report of an additional case with comment on possible relation to a new syndrome of familial and congenital anomalies. Arch Dermatol Syphilol. 1938;38:57.
Taibjee S.M., Bennett D.C., Moss C. Abnormal pigmentation in hypomelanosis of ito and pigmentary mosaicism: The role of pigmentary genes. Br J Dermatol. 2004;151:269-282.
Takematsu H., Sato S., Igarashi M., et al. Incontinentia pigmenti achromians (ito). Arch Dermatol. 1983;119:391-395.
Takeoka, M, Riviello, JJ: Sturge Weber syndrome: http://emedicine.com
Tapon N., Ito N., Dickson B.J., et al. The drosophila tuberous sclerosis complex gene homologs restrict cell growth and cell proliferation. Cell. 2001;105(3):345-355.
Theron J., Newton T.H., Hoyt W.F. Unilateral retinocephalic vascular malformations. Neuroradiology. 1974;7:185-196.
Thiele E.A. Managing epilepsy in tuberous sclerosis complex. J Child Neurol. 2004;19(9):680-686.
Thomas-Sohl K.A., Vaslow D.F., Maria B.L. Sturge-Weber syndrome: A review. Pediatr Neurol. 2004;30(5):303-310.
Tian X.-L., Kadaba R., You S.-A., et al. Identification of an angiogenic factor that when mutated causes susceptibility to Klippel-Trenaunay syndrome. Nature. 2004;427(6975):640-645.
Tingey A.H. Iron and calcium in Sturge-Weber disease. J Ment Sci. 1956;102:178.
Tinschert S., Naumann I., Stegmann E., et al. Segmental neurofibromatosis is caused by somatic mutation of the neurofibromatosis type 1 (NF1) gene. Eur J Hum Genet. 2000;8(6):455-459.
Tonsgard J.H., Kwak S.M., Short M.P., et al. CT imaging in adults with neurofibromatosis-1: Frequent asymptomatic plexiform lesions. Neurology. 1998;50:1755-1760.
Trofatter J.A., MacCollin M.M., Rutter J.L., et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell. 1993;72(5):791-800.
Upadhyaya M., Han S., Consoli C., et al. Characterization of the somatic mutational spectrum of the neurofibromatosis type 1 (NF1) gene in neurofibromatosis patients with benign and malignant tumors. Hum Mutat. 2004;23(2):134-146.
Upadhyaya M., Huson S.M., Davies M., et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delaat): Evidence of a clinically significant NF1 genotype-phenotype correlation. Am J Hum Genet. 2007;80(1):140-151.
Van Bogaert L. La melanose neurocutanee diffuse heredofamiliae. Bull Acad R Med. 1948;13:391.
Van der Hoeve J. Eye symptoms in tuberous sclerosis of the brain and in Recklinghausen disease. Trans Ophthalmol Soc UK. 1923;45:534.
Van der Hoeve J.. The Doyne Memorial Lecture: Eye Symptoms in Phakomatoses. 1932.
van Heuzen E.P., Kaiser M.C., de Slegte R.G. Neurocutaneous melanosis associated with intraspinal lipoma. Neuroradiology. 1989;31:349-351.
van Slegtenhorst M., de Hoogt R., Hermans C., et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997;277(5327):805-808.
Vandenbroucke I., van Doorn R., Callens T., et al. Genetic and clinical mosaicism in a patient with neurofibromatosis type 1. Hum Genet. 2004;114(3):284-290.
Verhoef S., Bakker L., Tempelaars A.M., et al. High rate of mosaicism in tuberous sclerosis complex. Am J Hum Genet. 1999;64(6):1632-1637.
Vidaurri-de la Cruz H., Tamayo-Sánchez L., Durán-McKinster C., et al. Epidermal nevus syndromes: Clinical findings in 35 patients. Pediatr Dermatol. 2004;21(4):432-439.
Virchow R. Pigment and diffuse melanose der arachnoides. Arch Pathol Anat Physiol. 1859;16:180.
Viskochil D., Buchberg A.M., Xu G., et al. Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell. 1990;62(1):187-192.
Vogel K.S., Brannan C.I., Jenkins N.A., et al. Loss of neurofibromin results in neurotrophin-independent survival of embryonic sensory and sympathetic neurons. Cell. 1995;82(5):733-742.
Vogt H. Zur diagnostic der tuberosen sklerose. Z Erforsh Behandl Jugendl Schwachsinns. 1908;2:1-12.
von Hippel E. Die anatomische grundlage der von mir beschriebenen “sehr seltenen erkrankung der netzhaut”. Albrech Graefes Arch Ophthalmol. 1911;79:350.
von Recklinghausen F.D. Ein herz von einen neugeborenene welches mehrere theils nach aussen, theils nach den hohlen prominirende tumoren (myomen) trug: Verh ges geburtsch. Verh Ges Geburtsch. 1862;20:1.
von Recklinghausen F.D.. Über die multiplen fibrome der haut und ihre beziehung zu den multiplen neuromen. In Festshrift Fur Rudolf Virchow. 1882. Berlin
Wachswulth N., Lowenthal A. Determination chemique d’elements mineraux dans les calcifications intracerebrales de la maladie Sturge-Weber syndrome. Childs Brain. 1979;5:233.
Wallace M.R., Marchuk D.A., Andersen L.B., et al. Type 1 neurofibromatosis gene: Identification of a large transcript disrupted in three NF1 patients. Science. 1990;249:181-186.
Walsh F.B., Hoyt W.F. Clinical Neuro-Ophthalmology, ed 3. Baltimore: Williams & Wilkins; 1969.
Walther M.M., Herring J., Enquist E., et al. Von Recklinghausen’s disease and pheochromocytomas. J Urol. 1999;162(5):1582-1586.
Walther M.M., Reiter R., Keiser H.R., et al. Clinical and genetic characterization of pheochromocytoma in Von Hippel-Lindau families: Comparison with sporadic pheochromocytoma gives insight into natural history of pheochromocytoma. J Urol. 1999;162(3 Pt 1):659-664.
Warbey V., Ferner R., Dunn J., et al. [(18)F)FDG PET/CT in the diagnosis of malignant peripheral nerve sheath tumours in neurofibromatosis type-1. Eur J Nucl Med Mol Imaging. 2009. doi: 10.1007/s00259-008-1038-0 [doi)
Wartenberg R. Progressive facial hemiatrophy. Arch Neurol Psychiatr. 1945;54:75.
Weaver R.G., Martin T., Zanolli M.D. The ocular changes of incontinentia pigmenti achromians (hypomelanosis of ito). J Pediatr Ophthalmol Strabismus. 1991;28:160-163.
Webb D.W., Clarke A., Fryer A., et al. The cutaneous features of tuberous sclerosis: A population study. Br J Dermatol. 1996;135(1):1-5.
Weber F.B. A note on the association of extensive haemangiomatous naevus of the skin with cerebral meningeal hemangioma, especially cases of facial vascular naevus with contralateral hemiplegia. Proc R Soc Med. 1929;22:431.
Webster A.R., Maher E.R., Moore A.T. Clinical characteristics of ocular angiomatosis in Von Hippel-Lindau disease and correlation with germline mutation. Arch Ophthalmol. 1999;117(3):371-378.
Weiner H.L., Ferraris N., LaJoie J., et al. Epilepsy surgery for children with tuberous sclerosis complex. J Child Neurol. 2004;19(9):687-689.
Weits-Binnerts J.J., Hoff M., van Grunsven M.F. Dental pits in deciduous teeth, an early sign in tuberous sclerosis. Lancet. 1982;2(8311):1344.
Westmoreland B. The electroencephalogram in tuberous sclerosis. In: Sampson J.R., Whittemore V.H., Gomez M.R., editors. Tuberous Sclerosis Complex. New York: Oxford University Press, 1999.
Winterkorn E.B., Pulsifer M.B., Thiele E.A. Cognitive prognosis of patients with tuberous sclerosis complex. Neurology. 2007;68(1):62-64.
Wishart J. Cases of tumors of the skull, dura mater, and brain. Edinburgh Med J. 1822;18:393.
Wiznitzer M. Autism and tuberous sclerosis. J Child Neurol. 2004;19(9):675-679.
Wolf S.M., Verity M.A. Neurological complications of progressive facial hemiatrophy. J Neurol Neurosurg Psychiatr. 1974;37:997.
Wong W.T., Agron E., Coleman H.R., et al. Clinical characterization of retinal capillary hemangioblastomas in a large population of patients with Von Hippel-Lindau disease. Ophthalmology. 2008;115(1):181-188.
Wong W.T., Yeh S., Chan C.C., et al. Retinal vascular proliferation as an ocular manifestation of Von Hippel-Lindau disease. Arch Ophthalmol. 2008;126(5):637-643.
Wu B.L., Austin M.A., Schneider G.H., et al. Deletion of the entire NF1 gene detected by the FISH: Four deletion patients associated with severe manifestations. Am J Med Genet. 1995;59(8585580):528-535.
Wyburn-Mason R. Arteriovenous aneurysm of mid-brain and retina, facial naevi and mental changes. Brain. 1943;66:163.
Xiao G.H., Chernoff J., Testa J.R. NF2: The wizardry of merlin. Genes Chromosomes Cancer. 2003;38(4):389-399.
Xu G.F., O’Connell P., Viskochil D., et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell. 1990;62:599-608.
Xu G., Lin B., Tanaka K., et al. The catalytic domain of the neurofibromatosis type 1 gene product stimulates ras gtpase and complements ira mutants of S. Cerevisiae. Cell. 1990;63:835.
Yamamoto N., Watanabe K., Negoro T., et al. Long-term prognosis of tuberous sclerosis with epilepsy in children. Brain Dev. 1987;9(3):292-295.
Yang F., Ingram D., Chen S., et al. Nf1-dependent tumors require a microenvironment containing nf1+/− and c-kit-dependent bone marrow. Cell. 2008;135(3):437-448.
Yang F.C., Ingram D.A., Chen S., et al. Neurofibromin-deficient schwann cells secrete a potent migratory stimulus for nf1+/− mast cells. J Clin Invest. 2003;112(12):1851-1861.
Yates A.G., Paine C.G. A case of arteriovenous aneurysm within the brain. Brain. 1930;53:38.
Young D.F., Eldridge R., Gardner W.J. Bilateral acoustic neuroma in a large kindred. J Am Med Assoc. 1970;214(2):347-353.
Zajac V., Kirchhoff T., Levy E.R., et al. Characterisation of X;17(q12;p13) translocation breakpoints in a female patient with hypomelanosis of ito and choroid plexus papilloma. Eur J Hum Genet. 1997;5:61-68.
Zeng L.H., Xu L., Gutmann D.H., et al. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol. 2008;63(4):444-453.
Zhang W., Simos P.G., Ishibashi H., et al. Neuroimaging features of epidermal nevus syndrome. Am J Neuroradiol. 2003;24(7):1468-1470.
Zhu Y., Ghosh P., Charnay P., et al. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science. 2002;296(5569):920-922.
Zvulunov A. Juvenile xanthogranuloma, neurofibromatosis, and juvenile chronic myelogenous leukemia. Arch Dermatol. 1996;132(6):712-713.
