Published on 19/03/2015 by admin
Filed under Dermatology
Last modified 22/04/2025
This article have been viewed 2508 times
Chinmoy Bhate and Robert A. Schwartz
Evidence Levels: A Double-blind study B Clinical trial ≥ 20 subjects C Clinical trial < 20 subjects D Series ≥ 5 subjects E Anecdotal case reports
Peutz–Jeghers syndrome (PJS) is a rare hereditary disorder of polyposis. It is characterized by gastrointestinal polyps, mucocutaneous pigmentation, recurrent abdominal pain from intussusceptions, and an increased risk of intestinal and other malignancies.
The pigmented macules of PJS are typically found around the mouth, eyes, nostrils, and anus; they may also be seen on the hands and feet. These brown to black macules are often round or oval, 1–5 mm in diameter, and irregular in shape. They may occasionally manifest with a blue-gray hue. Clinically and histologically, these are simple lentigines.
Most patients with this autosomal dominant disorder have a germ-line mutation of the STK11/LKB1 (serine/threonine kinase 11) tumor suppressor gene, located at chromosome 19p13.3. A more recently identified second PJS disease locus exists at 19q13.4. The exact mechanism of cancer and hamartoma development remains unclear. Genetic testing may be useful in equivocal cases as well as in counseling of at-risk families; however, it is not required for diagnosis and is not 100% sensitive.
Management is predicated upon the potential for visceral complications and familial inheritance. This includes an evaluation for associated findings, including recurrent intussusceptions, gastrointestinal bleeding, and a variety of malignancies. Disease is most often discovered between the ages of 10 and 30 years. Any child with recurrent, unexplained abdominal pain should raise concern for intussusception, a medical emergency associated with PJS. Gastrointestinal polyps may undergo malignant degeneration. Ovarian neoplasms, especially granulosa cell tumors, may be seen. Both in men and women, sex cord tumors with annular tubules, and sex cord stromal tumors with sexual precocity, may develop. Cancers of the pancreas, stomach, esophagus, lung, uterus, and testes (Sertoli cell) all have an elevated relative risk in patients with PJS. Additionally, women with this disorder appear to carry an increased risk of bilateral breast cancer.
Genetic counseling is indicated. Laboratory studies investigating anemia, iron deficiency, or fecal occult blood are necessary in suspicious cases. Some utilize serum tumor markers, including carcinoembryonic antigen and cancer antigen 19-9 or 125, in addition to endoscopy and ultrasonography, for screening and monitoring. Families should be reassured that the mucocutaneous macules are benign and may improve after puberty. Several therapies have been used with varying responses; however, removal of these mucocutaneous markers may potentially mask the underlying disorder in patients with PJS.
The ruby laser (Q-switched and short pulsed) has been used in the treatment of labial macules without sequelae or recurrences. It may be suitable for children with PJS, since anesthesia is usually not required and no wound care is necessary. The CO2, alexandrite, and argon lasers, as well as intense pulsed light, have also been effective in the treatment of labial macules. Cryosurgery, surgical excision, electrodesiccation, and dermabrasion may lead to scarring and dyspigmentation, often without complete removal of the pigmented macules. Similarly, trichloroacetic acid may not produce total resolution.
Histology (if diagnosis is in question)
Gastrointestinal evaluation
Genetic testing (in some cases)
Psychosocial evaluation
Korsse SE, Dewint P, Kuipers EJ, van Leerdam ME. Best Pract Res Clin Gastroenterol 2012; 26: 263–78.
This review provides a systematic analysis of endoscopic techniques used to examine the small bowel in patients with PJS.
Small bowel surveillance is recommended in patients with PJS every 2 to 3 years from the age of 8–10 years. Visualization of the small bowel is technically challenging. A gold standard method has not been established.
Beggs AD, Latchford AR, Vasen HF, Moslein G, Alonso A, Aretz S, et al. Gut 2010; 59: 975–86.
This review covers current genotype–phenotype studies and an outline of consensus recommendations for screening and follow-up from a group of European experts who previously produced guidelines for the management of Lynch syndrome and familial adenomatous polyposis.
Surveillance in PJS aims to (a) reduce polyp burden and the incidence of intussusceptions in young patients, and (b) diminish the burden of cancer. Capsule endoscopy allows for thorough, mildly invasive surveillance of the small bowel for polyposis. Evaluation of the small bowel with upper gastrointestinal endoscopy, colonoscopy, and double-balloon enteroscopy has been useful in preventing the need for subsequent surgical polypectomy. Since double-balloon enteroscopy is often traumatic, it is reserved as a treatment modality.
van Lier MG, Mathus-Vliegen EM, van Leerdam ME, Kuipers EJ, Looman CW, Wagner A, et al. Clin Genet 2010; 78: 219–26.
A cross-sectional study in order to assess the quality of life and psychological distress in PJS patients compared to the general population.
Compared with the general population, PJS patients reported a lower general health perception, more limitations due to emotional problems, and a lower mental well-being.
Chen HM, Fang JY. Int J Colorectal Dis 2009; 24: 865–74.
This work is a useful discussion of hamartoma syndromes, including clinical features and molecular diagnostic modalities which help differentiate between them and identify at-risk patients.
Heymann WR. J Am Acad Dermatol 2007; 57: 513–14.
This useful synopsis of the syndrome correlates molecular advances with the cutaneous, malignant, and endocrinologic features of the disorder.
Lim W, Hearle N, Shah B, Murday V, Hodgson SV, Lucassen A, et al. Br J Cancer 2003; 89: 308–13.
This genotype–phenotype analysis demonstrates significant genetic heterogeneity in PJS. The authors demonstrated an elevated relative risk of cancer development in patients with PJS. They also demonstrated that PJS may occur either as a result of several types of mutations within the above gene or even in the absence of a mutation.
Leggett BA, Young JP, Barker M. Exp Rev Anticancer Ther 2003; 3: 518–24.
Diagnostic and predictive testing of causative mutations in the STK11/LKB1 gene is available in routine practice and allows for early recognition of young at-risk patients, thus reducing disease burden and morbidity.
Mehenni H, Blouin JL, Radhakrishna U, Bhardwaj SS, Bhardwaj K, Dixit VB, et al. Am J Hum Genet 1997; 61: 1327–34.
The authors described the location of the most prevalent mutation in PJS, the LKB1/STK11 gene at location 19p13.3, and identified an additional locus.
Most patients with PJS have associated LKB1/ STK11 mutations at location 19p13.3; however, a second disease locus at 19q13.4 has also been demonstrated. Some patients have no mutation at either site, which serves as a limitation to genetic testing for diagnosis.
In: Löser C, Plewig G, Burgdorf W, eds. Pantheon of Dermatology. Heidelberg: Springer Verlag, 2013; 553–6.
A biography of Harold Jeghers is presented with a review of this syndrome which carries his name.
Xi Z, Hui Q, Zhong L. Dermatol Surg 2009; 35: 1084–8.
Fourteen cases of oral labial lentigines in patients with PJS were managed using a single treatment with Q-switched alexandrite laser with a 3 mm hand piece and a fluence of 4.0–9.0 J/cm2. All exhibited successful elimination without scarring or recurrence at median 2-year follow-up.
Q-switched lasers, in particular the alexandrite laser, are preferred in the treatment of benign melanocytic lesions, including oral labial lentigines in PJS patients; acute side effects included edema, erythema, and occasional bleeding.
Ashinoff R, Geronemus RG. J Am Acad Dermatol 1992; 27: 809–11.
The Q-switched ruby laser, with a wavelength of 694 nm and pulse duration of 40 ns, causes selective damage to pigmented cells. Three patients with labial lentigines treated with the Q-switched ruby laser noted dramatic clearing after one or two treatments with a fluence of 10 cm2.
Hanada K, Baba T, Sasaki C, Hashimoto I. J Dermatol 1996; 23: 263–6.
Six Japanese patients with melanosis had successful pulsed ruby laser therapy without recurrence or alteration of tissue texture.
Kato S, Takeyama J, Tanita Y, Ebina K. Eur J Pediatr 1998; 157: 622–4.
Two children with PJS and labial lentigines were treated successfully without sequelae or recurrence.
Raulin C, Schonermark MP, Greve B, Werner S. Ann Plast Surg 1998; 41: 555–65.
This review discusses indications for the use of the Q-switched ruby laser. These include tattoos, café-au-lait macules, and lentigines, especially those on the lips and eyelids.
Benedict LM, Cohen B. J Dermatol Surg Oncol 1991; 17: 954–5.
One patient with PJS had successful treatment of perioral lentigines with a CO2 laser.
Papadavid E, Walker NP. J Eur Acad Dermatol Venereol 2001; 15: 468–9.
Two patients with labial pigmented macules were treated with the Q-switched alexandrite laser. Recurrences were easily treated again.
Laugier–Hunziker syndrome, a disorder of mucocutaneous pigmentation similar to PJS, has no associated gastrointestinal abnormalities. Treatment of pigmented lesions in this syndrome may provide insight into the treatment of those seen in PJS.
Ohshiro T, Maruyama Y, Nakajima H, Mima M. Br J Plast Surg 1980; 33: 346–9.
Three PJS patients had successful treatment of pigmented oral macules with ruby and argon lasers.
Remington BK, Remington TK. Dermatol Surg 2002; 28: 1079–81.
A 10-year-old child had complete clearance of cosmetically concerning lentigines over the course of 12 treatment sessions to different facial regions. Most resolved with a single treatment.
This report demonstrated the first use of intense pulsed light in PJS and provided a viable alternative to laser treatment.
Yeh CJ. Clin Oral Maxillofac Surg 2000; 90: 12–13.
Simple cryotherapy for labial melanotic macules is discussed; a specific protocol is provided.
Cryosurgery needs no sophisticated equipment and is inexpensive. Recurrence is common, and inadequate destruction may occur.
Wei C, Amos CI, Zhang N, Zhu J, Wang X, Frazier ML. Cancer Lett 2009; 18: 149–54.
Rapamycin treatment of mice with LKB1 mutations led to a reduction in both polyp burden and polyp size, suggesting that rapamycin may be an option for chemoprevention in humans with PJS.
Wei C, Amos CI, Zhang N, Wang X, Rashid A, Walker CL, et al. Clin Cancer Res 2008; 14: 1167–71.
Mice with germ-line LKB1 mutations and polyposis were treated with rapamycin at the dose of 2 mg/kg/day for 2 months. Rapamycin decreased the tumor burden of large polyps (>8 mm).
Rapamycin effectively suppresses Peutz–Jeghers polyposis in a mouse model, suggesting that it and other inhibitors of the mammalian target of rapamycin (mTOR) may represent targeted therapy for this disorder.
Treatment of Skin Disease Comprehensive Therapeutic Strategies 4e
WhatsApp us
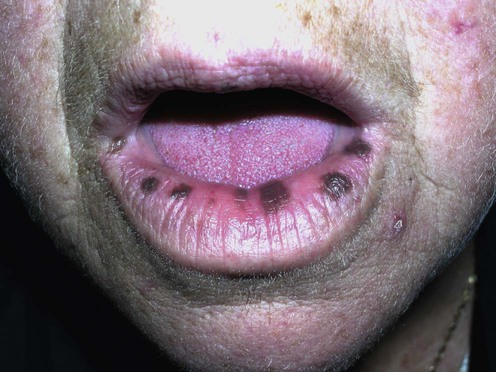





 Ruby and alexandrite lasers
Ruby and alexandrite lasers Other lasers
Other lasers Intense pulsed light
Intense pulsed light Cryotherapy
Cryotherapy