Chapter 38 Peroxisomal Disorders
Disorders of the peroxisome – organelles found in all eukaryotic cells – are characterized by alterations in their unique metabolic functions in the cell and tissues. The pervasive presence of the peroxisome leads to far-reaching consequences of these genetic disorders. Peroxisomal disorders are divided into two major categories. In the first, the organelle fails to develop normally, leading to disruption of multiple peroxisomal enzymes. The second category consists of those disorders in which the peroxisome structure is normal but functioning of a single peroxisomal enzyme is defective. Box 38-1 lists the known peroxisomal disorders; their combined incidence is estimated at 1 in 25,000 or higher [Heymans, 1984]. Because peroxisomal disorders are genetically determined, with a majority readily identifiable by biochemical means, including prenatal testing, and nearly all affecting the nervous system, knowledge of these diseases is important. As the pathophysiology of these disorders is better understood, novel therapeutic strategies may be developed, which gives new hope for advances not only in the diagnosis but also in the management and outcome of peroxisomal disorders.
Box 38-1 Peroxisomal Disorders













CADDS, contiguous ABCD1 DXS1357E deletion syndrome; CoA, coenzyme A; DHAP, dihydroxyacetone phosphate.
Historical Overview
The organelle was identified in 1954 by Rhodin and named the “microbody” [Rhodin, 1954]. In 1960, it was found that this structure contained urate, d-amino acid oxidase, and catalase; it was named the peroxisome by de Duve and Baudhuin [1966]. Recognition of its function in active cellular processes came with the demonstration of its role in the β-oxidation of fatty acids (Figure 38-1) [Lazarow, 1978] and plasmalogen synthesis [Hajra and Bishop, 1982]. That change in peroxisome function results in disease was not evident until 1973, when Goldfischer and colleagues determined its absence in Zellweger’s syndrome [Goldfischer et al., 1973]. The link between peroxisomes and human disease was not made until that time, even though peroxisomal disorders had been described much earlier, including X-linked adrenoleukodystrophy in 1923 [Siemerling and Creutzfeldt, 1923], acatalasemia in 1948 [Takahara and Migamoto, 1948], and Zellweger’s syndrome in 1964 [Bowen et al., 1964]. From the initial three enzymes identified, more than 40 enzymes have been localized to the peroxisomes [Tolbert, 1981].

Structure and Function of Peroxisomes
The peroxisome is bound by a single membrane and contains a fine granular matrix. Histologically, these organelles are identified by the presence of catalase, are present in all human tissues except mature erythrocytes, and demonstrate variation in size and number. In liver and kidney, where they are abundant, the average diameter is 500 nm, whereas in the nervous system, fibroblasts, and amniocytes, they measure 100–250 nm [Hruban and Rechcigl, 1969]. They do not contain DNA and appear to be devoid of glycoproteins. The membrane is 6.5–7 nm thick and has a trilaminar appearance and a unique protein composition. Peroxisome membrane proteins with molecular masses of 22, 26, 27, 41, 57, 68, and 70 kDa have been identified [Hashimoto et al., 1986; Imanaka et al., 1991]. The peroxisomal membrane also contains four ATP binding cassette (ABC) proteins. Although the precise role of these proteins in the peroxisomal membrane is still under investigation, it is known that this family of proteins has important intracellular roles in transport and other functions. As discussed later on, the ABC protein, ABCD1, is defective in X-linked adrenoleukodystrophy, the most prevalent peroxisomal disorder [Wanders et al., 2007].
The process of peroxisomal biogenesis is highly conserved in all eukaryotic organisms, which has permitted the study of yeast to identify the cellular mechanism for the assembly of the organelle and targeting of proteins to the developing vesicle [Weller et al., 2003]. The early concept of peroxisome biogenesis by budding of the endoplasmic reticulum has undergone significant revision. Peroxisomal proteins are encoded by nuclear genes, synthesized on free polyribosomes, and discharged into the cytosol in the mature form. Work in yeast has identified more than 20 genes labeled PEX whose products, peroxins, are required for the incorporation of peroxisome membrane proteins and matrix protein importation. Peroxins are required for the proper importation and have roles in receptor docking, stability, and translocation across the membrane [Weller et al., 2003; Ma and Subramani, 2009].
Targeting information directing matrix proteins into the peroxisomes is inherent in the mature polypeptide. A majority of proteins destined for the peroxisome use peroxisome targeting sequence 1 (PTS1), which consists of a terminal tripeptide of serine-lysine-leucine (-SKL) that is recognized by the soluble receptor Pex5p [Gould et al., 1987, 1988, 1990; Keller et al., 1991; Miyazawa et al., 1989]. Not all matrix proteins contain the carboxyl-terminal PTS1 signal. Peroxisomal 3-ketoacyl-coenzyme A (CoA) thiolase and phytanoyl-CoA hydroxylase have a different peroxisomal targeting sequence (PTS2). PTS2 consists of a nine-residue signal located at the amino terminus. It directs the import of a smaller number of proteins using the soluble receptor Pex7p [Swinkels et al., 1991].
Both Pex5p and Pex7p receptors bind their targeted proteins in the cytoplasm outside the peroxisome. The present model has the receptor-peptide complex dock at the peroxisome surface by means of membrane-associated complexes containing other peroxisome assembly proteins including Pex3p, Pex13p, Pex14p, and Pex17p. Other peroxins appear to function later in the process of translocation. Several are zinc-binding proteins acting downstream of the docking complex and are postulated to constitute the translocation complex involved in matrix protein import. Recent evidence has demonstrated that Pex5p is translocated into the peroxisome and is capable of returning to the cytoplasm – an extended shuttle model [Dammai and Subramani, 2001].
This import method has no parallel with any other organelle. Walton and colleagues demonstrated that peroxisome import allows the uptake of folded, oligomerized proteins and even the import of non-PTS-containing substances, so long as it was with PTS-containing cargo [Walton et al., 1995]. This mechanism is in contrast with that in the mitochondria and lysosome, which have a translocon mechanism with structural modification occurring within the targeted organelle [Lanyon-Hogg et al., 2010].
In contrast with the proteins targeted for the matrix, the proteins used in the membrane utilize another mechanism. Jones and associates examined PMP34 and found that it contained at least two targeting regions. Examining another peroxisomal membrane protein, Pex13, these investigators again found that it had multiple nonoverlapping targeting signals [Jones et al., 2001]. These and other peroxisomal membrane proteins did not share targeting regions. Regions were relatively long and contained at least one membrane-spanning domain.
Peroxisomal number and division appear to be under metabolic control. Hypolipidemic agents, such as clofibrate and industrial phthalate plasticizers, result in the proliferation of liver peroxisomes [Hess et al., 1965; Lazarow et al., 1985]. These agents induce a 20- to 30-fold increase in the activity of the fatty acid β-oxidation system by the rapid coordinated increase of peroxisomal acyl coenzyme A (acyl-CoA) oxidase and bifunctional enzyme [Reddy et al., 1986]. This effect is mediated by activation of peroxisome proliferator-activated receptorα (PPARα), a binding protein related to the steroid hormone receptor superfamily [Issemann and Green, 1990; Lalwani et al., 1987]. Activated PPARα heterodimerizes with a second member of the nuclear receptor superfamily, retinoid X receptor, to form an active transcription factor [Weller et al., 2003]. Peroxisomes also are induced by high-fat diets [Neat et al., 1980], by adrenocorticotropic hormone [Black and Russo, 1980], by thyroid hormone [Fringes and Reith, 1982], and by the agent 4-phenylbutyrate [Wei et al., 2000]. Peroxisomal function is therefore influenced by metabolic state, nutrition, and pharmaceutical agents.
Reflecting this genetic and metabolic control, peroxisomes vary in tissues throughout the body and during development. In the nervous system of the rat, the peroxisomes are smaller, approximately 140 nm, and fewer than in the liver. They predominate in the first 2 weeks of life in the glia and neurons of the cerebrum, cerebellum, locus coeruleus, and spinal cord, but are fewer in the neurons of adult animals [Nagase et al., 2004]. They are rare in oligodendrocytes in neonatal or adult animals, but become prominent during myelin formation, and may lie adjacent to the outer lamellae that form the myelin sheath [Arnold and Holtzman, 1978]. The biochemical activities follow a similar pattern wherein the activities of catalase and peroxisomal acyl-CoA oxidase and oxidation of lignoceric acid in brain reach a peak at postnatal days 10–16 and then decline. This decline suggests that peroxisomes play a vital role during brain development and myelinogenesis, which may account for the severe brain abnormalities noted in the neonatal and infantile forms of these disorders, including neuronal migration defects.
Metabolic Function of Peroxisomes
Peroxisomes were named for the presence of hydrogen peroxide and catalase, which decomposes the hydrogen peroxide. The seminal observation by Lazarow and de Duve in 1976 that rat liver peroxisomes oxidized palmityl-CoA, followed in 1978 by the demonstration of enzymes of β-oxidation and production of acetyl-CoA by Lazarow and Fujiki [1985], led to the recognition of the importance of this organelle in biologic processes. It is now known that more than 40 enzymatic functions are found in the peroxisome. Some peroxisomal activities, such as oxidation of fatty acids and cholesterol synthesis, can occur in other cellular compartments as well. Certain reactions, however, occur exclusively in the peroxisome. These reactions include oxidation of very long chain fatty acids and pipecolic acid and certain steps in the synthesis of plasmalogens and bile acids. These reactions are abnormal in many peroxisomal disorders. The composition of enzymes within the peroxisome varies among species and within tissues in a species, as well as with maturation, metabolic state, and environmental factors. The following discussion focuses on those metabolic pathways that are unique and are used as markers in diagnosis.
Peroxisomal Fatty Acid Oxidation
Peroxisomal β-oxidation enzymes are distinct from their mitochondrial counterparts. The biochemical steps are similar, involving several distinct enzymes including an oxidase, multifunctional enzyme (displaying enoyl-CoA hydratase and 3-hydroxy acyl-CoA dehydrogenase activity), and 3-oxo acyl-CoA thiolase. It recently has become clear that several distinct oxidases, multifunctional proteins, and enzymes catalyzing the thiolytic cleavage exist. The two pathways, l– and d-specific, are depicted in Figure 38-1.
Long-chain acyl-coenzyme a ligase and lignoceroyl-coenzyme a ligase
Initially it was thought that a single acyl-CoA ligase that activated long-chain (C10–C18) fatty acids was involved in oxidation of fatty acids and that it was common to mitochondria, microsomes, and peroxisomes [Miyazawa et al., 1985]. This enzyme is localized to the cytoplasmic side of the peroxisomal membrane, unlike other peroxisomal enzymes, which are in the matrix [Mannaerts et al., 1982]. Subsequently, a series of investigations led to the recognition of a closely related but separate enzyme, which is a ligase for very long chain fatty acids and is referred to as lignoceryl-CoA ligase or synthetase [Singh et al., 1988]. This enzyme is active toward lignoceric acid (C24:0) and hexacosanoic acid (C26:0). Unlike long-chain ligase, very long chain fatty acid ligase is absent in mitochondria. Singh et al. [1984] demonstrated that lignoceric acid is oxidized exclusively in the peroxisome. It is unclear if the very long chain fatty acid ligase plays any role in the pathogenesis of peroxisomal disorders.
Acyl-coenzyme a oxidases
The straight-chain acyl-CoA oxidase is synthesized in the mature form containing the carboxyl-terminal PTS1 [Miyazawa et al., 1987]. The oxidase is most active toward saturated and unsaturated fatty acids with 12- to 18-carbon chain lengths. The oxidase for bile acid intermediates and branched-chain fatty acids is a separate enzyme in humans [Casteels et al., 1990; Ferdinandusse et al., 2003; Scheperse et al., 1990].
Bifunctional or multifunctional enzymes
In both peroxisomal β-oxidation systems, the hydration step (enoyl-CoA hydratase) and the dehydrogenation step (3-hydroxyacyl-CoA dehydrogenase) are catalyzed by multifunctional enzymes (MFEs). The originally identified “bifunctional enzyme,” known as either MFE1 or l-bifunctional protein (l-BP), is l-specific. Human deficiency of the d-specific enzyme, referred to as either MFE2 or d-bifunctional protein (d-BP), causes a failure in the oxidation of very long chain fatty acids and branched-chain fatty acids. MFE2 was found to be identical to 17-hydroxysteroid dehydrogenase type 4 and has a domain at its carboxyl terminus that resembles sterol carrier protein 2 (SCP2), an important intracellular sterol and lipid-binding and transport protein [van Grunsven et al., 1999].
Thiolases
3-Ketoacyl-CoA thiolase is the enzyme for the l-specific pathway and was characterized and cloned first [Bout et al., 1988; Hashimoto, 1982]. Unlike other peroxisomal enzymes but similar to mitochondrial enzymes, it is synthesized as a precursor and converted to the mature form by cleavage of a leader sequence. The peroxisomal targeting sequence for thiolase is located at the amino-terminal end [Swinkels et al., 1991] and was the first protein in which the PTS2 signal was characterized. The thiolytic cleavage in the d-specific pathways is catalyzed by sterol carrier protein X (SCPX). This 58-kDa protein has an amino-terminal thiolase domain that is a phylogenetic equivalent of the mitochondrial and peroxisomal thiolases [Igual et al., 1992].
Oxidation of Unsaturated Fatty Acids
Degradation of monounsaturated and polyunsaturated fatty acids has been demonstrated to occur in peroxisomes, in addition to chain shortening of long-chain dicarboxylic acids [Ferdinandusse et al., 2001; Hiltunen et al., 1986; Kolvraa and Gregersen, 1983; Schulz and Kunau, 1987]. An example of a polyunsaturated fatty acid that requires peroxisomal oxidation is docosahexaenoic acid, which has an important role in retina and brain development, and is known to be deficient in certain peroxisomal disorders [Moser et al., 1999; Watkins et al., 2001].
Branched-Chain Fatty Acid Oxidation and Phytanic Acid α-Oxidation
Branched-chain fatty acids are channeled into the d-specific pathway. The relevant enzymes include branched-chain acyl-CoA oxidase, MFE2, and sterol carrier protein, which functions as thiolase (see Figure 38-1). Phytanic acid has a β-methyl group that blocks β-oxidation and therefore must be α-oxidized first. After activation to its CoA derivative, phytanic acid is α-hydroxylated by phytanoyl CoA α-hydroxylase, the enzyme defective in classic Refsum’s disease [Jansen et al., 1997; Mihalik et al., 1997]. The α-hydroxy phytanoyl CoA is then degraded by a lyase to pristanic acid and formate [Croes et al., 1997]. The pristanic acid then can enter β-oxidation through the branched-chain pathway.
Bile Acid Synthesis
Cholesterol is converted in the liver to cholic acid and chenodeoxycholic acid by a series of enzymatic steps localized to several subcellular compartments. Pedersen and Gustafsson [1980] first demonstrated that the peroxisomal fraction of rat liver catalyzes the conversion of an intermediate product of this pathway, tri- or tetrahydroxy-5-β-cholestanoic acid (trihydroxycholestanoic acid), to cholic acid. Subsequently, Kase et al. [1986] reported that the conversion of trihydroxycholestanoic acid to cholic acid, and of dihydrocholestanoic acid to chenodeoxycholic acid also took place in peroxisomes and was deficient in patients with Zellweger’s syndrome. Shortening of the cholesterol side chain by β-oxidation resulting in the formation of bile acids is analogous to fatty acid oxidation and uses the enzymes of the d-specific peroxisomal β-oxidation pathway. The oxidase for trihydroxycholestanoic acid–CoA is separate from that of fatty acids and in humans also is believed to function as a branched-chain fatty acid oxidase (see Figure 38-1). A cytochrome P-450 catalyzing the hydroxylation of the C26/27 carbon of cholesterol in bile acid synthesis and formation of taurine conjugates of bile acid also has been localized to peroxisomes [Gutierrez et al., 1988; Kase and Bjorkheim, 1989]. In peroxisomal disorders, an increase in plasma and urine bile acid intermediates can be used as a diagnostic criterion.
Plasmalogen Synthesis
Plasmalogens are ether phospholipids and constitute 5–20 percent of phospholipids in mammalian cell membranes [Snyder, 1972]. They are abundant in myelin, in which they constitute one-third of the myelin phospholipids [Norton and Autilio, 1966]. Although the role of ether phospholipids is not fully elucidated, they appear to have important roles as antioxidants in membrane dynamics, storage of polyunsaturated fatty acids, and signal transduction [Brites et al., 2004].
Plasmalogen synthesis is initiated by the acylation of dihydroxyacetone phosphate by dihydroxyacetone phosphate acyltransferase. Experimental studies in cultured skin fibroblasts demonstrate that ATP is required to overcome the latency of dihydroxyacetone phosphate acyltransferase, a finding that is compatible with an ATP-linked translocation of the substrate across the peroxisomal membrane [Wolvetang et al., 1990]. The second enzyme, alkyl-dihydroxyacetone phosphate synthase, replaces the acyl group with a long-chain alcohol [Hajra and Bishop, 1982]. Both enzymes involved in these initial steps of introducing the ether bond into ether phospholipids are located on the inner surface of peroxisome membranes. The third step is the reduction of the alkyl-dihydroxyacetone phosphate to 1-alkyl glycerol-3-phosphate by the enzyme acyl/alkyl-dihydroxyacetone phosphate reductase, which is present in both peroxisomes and microsomes [Datta et al., 1990]. All subsequent steps of synthesis occur in the microsomes. A method developed by Roscher et al. [1985] determines the ratio of the activities in peroxisomal and microsomal components, and has proved to be of value in the study of peroxisomal disorders, many of which feature a marked reduction in plasmalogen synthesis.
Prostaglandin Degradation
Peroxisomes are involved in the chain shortening of prostaglandins F2a [Diczfalusy and Alexson, 1988] and E2 [Schepers et al., 1986]. Tiffany et al. [1991] demonstrated that basal and interleukin-1-stimulated synthesis of prostaglandin E2 increased in a group of patients with X-linked adrenoleukodystrophy.
Amino Acid Metabolism
D-Amino acid oxidase
d-Amino acid oxidase is a flavoprotein with stereospecific activity toward d-amino acids and greatest activity toward d-proline. The kidney has the highest level of activity, although the enzyme is widely distributed in vertebral tissues. The role of d-amino acids as neurotransmitters has recently attracted attention [Errico et al., 2009].
Alanine-glyoxalate aminotransferase
Alanine-glyoxalate aminotransferase is a pyridoxal phosphate-dependent enzyme that catalyzes the transamination of glyoxalate to glycine, with alanine serving as the amino group donor. The subcellular localization of alanine-glyoxalate aminotransferase is species-dependent; it is present in peroxisomes in humans, rabbits, guinea pigs, and macaques, whereas it is mitochondrial in cats and dogs. It is present in both organelles in the rat, mouse, and hamster. The complementary DNA (cDNA) sequence of alanine-glyoxalate aminotransferase has been determined, and the subcellular localization appears to be determined by the targeting signal at the amino terminus [Danpure, 1995; Noguchi and Takada, 1979; Noguchi et al., 1978]. Of great interest is the observation that the mitochondrial targeting signal active in the rat enzyme is not expressed in humans. In some patients with hyperoxaluria type 1, this is altered, resulting in targeting of the alanine-glyoxalate aminotransferase to mitochondria instead of to the peroxisomes [Purdue et al., 1990]. In hyperoxaluria, the defective alanine-glyoxalate aminotransferase results in the conversion of glyoxalate to oxalic acid, which causes oxaluria and nephrocalcinosis.
Pipecolic acid oxidase
l-Pipecolic acid is a component of an alternate lysine degradation pathway and is oxidized by l-pipecolic acid oxidase to α-aminoadipic acid. This enzyme contains flavin, and its subcellular localization is species-dependent. The major site of oxidation is peroxisomal in humans and monkeys, and mitochondrial in rabbits [Mihalik and Rhead, 1989]. The enzyme has been identified in human liver and has been reported to be deficient in Zellweger’s syndrome [Mihalik et al., 1989; Wanders et al., 1988a]. The human enzyme most closely resembles the bacterial monomeric sarcosine oxidases [Dodt et al., 2000]. Isolated increases in pipecolic acid have been reported in normal adults and may be a benign trait [Vallat et al., 1996]. Pipecolic acid elevation has also been seen in pyridoxine-responsive seizures, but this is due not to defects in the oxidase, but rather to the antiquitin (ALDH7A1) gene, which affects other aspects of the pathway [Bennett et al., 2009].
Classification of Peroxisomal Disorders
Peroxisomal disorders may be divided into two categories:
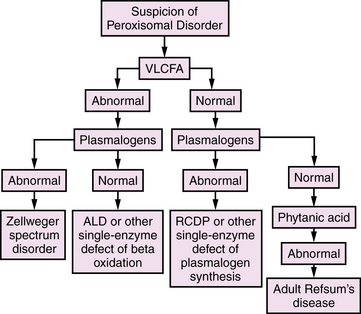
All of these disorders are, in actuality, single-gene and ultimately single-protein deficiencies, but the downstream consequences of the first group affect more than one peroxisomal pathway, resulting in multiple diagnostic abnormalities. With the emergence of DNA-based diagnosis, the utility of this division may require reassessment. An overview of diagnostic evaluation of peroxisomal disorders is provided in Figure 38-2.
Conditions Resulting from Defective Peroxisome Biogenesis
Conditions resulting from defective peroxisome biogenesis are listed in Box 38-1; Zellweger’s syndrome and rhizomelic chondrodysplasia punctata are the respective prototypes. Clinical and biochemical variation between these two types of assembly defects still makes it useful to discuss them separately, although it is important to recognize that these disorders were described on a clinical basis before details of the cell and molecular biology of peroxisomal disorders was known, and hence they were assigned names based on the clinical features, pathologic findings, or biochemical defects that identified them.
Molecular Etiology of Disorders of Peroxisome Assembly
Cell complementation studies were instrumental in the early understanding of the genetics of peroxisomal assembly disorders. Identification of complementation groups provided cell lines that are homogeneous with respect to their gene defect. In conjunction with the use of other molecular biology tools, including the study of yeast and other cell types and the emergence of computerized genomic databases, a new understanding of the molecular underpinnings of the peroxisome assembly disorders has been attained [Lazarow, 1995]. These disorders result from defects in the PEX genes. A complex interaction of peroxins is necessary for the biogenesis of peroxisomes, and a defect in any of these proteins impairs the process. The final common pathway is peroxisomal dysfunction, with the respective clinical syndromes.
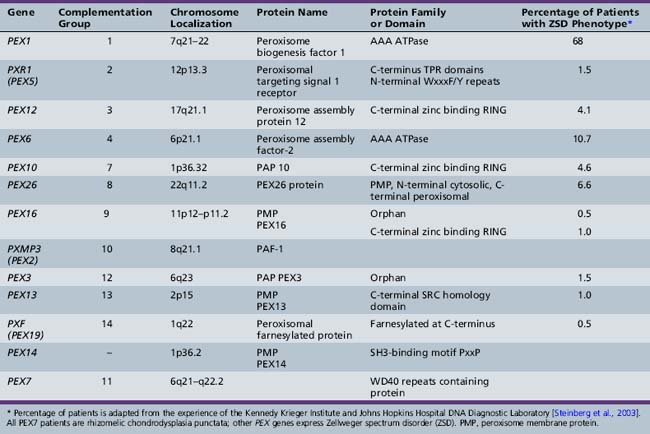
From the study of complementation groups, it was determined that peroxisomal disorders comprise up to 16 complementation groups [Moser et al., 1995]. A majority of the identified groups included persons with Zellweger’s syndrome and milder variations of that condition. One complementation group, however, contained all of the patients with rhizomelic chondrodysplasia punctata, and no overlap between this clinical group and the others was identified. It has subsequently been determined that Zellweger spectrum disorders are secondary to PTS1-mediated pathways including PEX5, and that rhizomelic chondrodysplasia punctata is secondary to mutations in PEX7, the receptor for PTS2 proteins.
The most common causes of Zellweger spectrum disorders are mutations in either PEX1 or PEX6, although to date, 13 PEX genes have been identified as potential causes of these disorders (Table 38-1). PEX1 and PEX6 encode AAA ATPases. Their exact role in matrix protein importation is not certain, but it is evident that they interact. Overexpression of PEX6 or PEX1 can correct mild deficiencies of the other [Germain-Lee et al., 1997; Reuber et al., 1997]. PEX1 defects account for more than 65 percent of cases of such defects [Steinberg et al., 2003; Weller et al., 2003]. A degree of phenotype correlation with the causative mutation has been noted. A single base-pair deletion results in a severe phenotype, and G843D allows a milder phenotype [Collins and Gould, 1999]. Because it is known that PEX1 and PEX6 interact, the mutation G843D affects this interaction [Weller et al., 2003].
Zellweger Spectrum Disorders
Zellweger’s syndrome (cerebrohepatorenal syndrome) was first described by Bowen and associates [Bowen et al., 1964; Opitz, 1985]. Subsequently, the disorders neonatal adrenoleukodystrophy and infantile Refsum’s disease were described as separate entities. Although classic Zellweger’s syndrome is the most severe form with a characteristic phenotype, clinical overlap exists between it and the other forms. All of the Zellweger spectrum disorders share morphologic and biochemical abnormalities, and genetic understanding of the underlying mutations has been obtained in many cases. In view of this overlap, it appears prudent at this time to retain a portion of this clinical nomenclature and refer to the group as Zellweger spectrum disorders.
Clinical and Pathologic Features
Zellweger’s syndrome
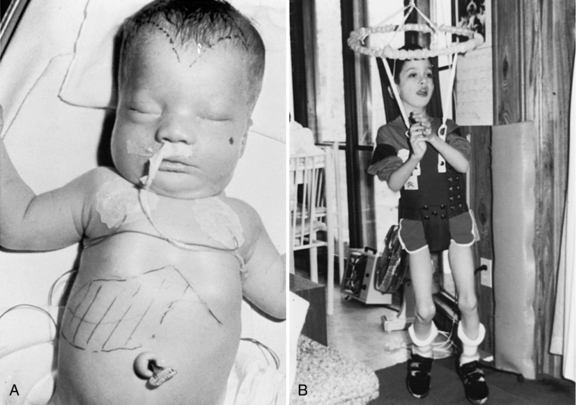
Zellweger’s syndrome is a multiple congenital anomaly syndrome characterized by craniofacial abnormalities, eye abnormalities, neuronal migration defects, hepatomegaly, chondrodysplasia punctata, and near-complete absence of peroxisomes. The craniofacial features include a high forehead, hypoplastic supraorbital ridges, epicanthal folds, midface hypoplasia, and a large fontanel (Figure 38-3). The head circumference usually is normal. Reported ocular abnormalities include cataracts, glaucoma, corneal clouding, Brushfield spots, optic nerve hypoplasia, and pigmentary retinal abnormalities. Severe weakness and hypotonia manifest in the newborn period, often accompanied by seizures and apnea [Heymans, 1984; Wilson et al., 1986]. Most affected infants have oromotor dysfunction and require tube feeding. Little psychomotor development ensues, and the average life span is limited, with most affected children surviving for 12–24 months. The facial appearance, Brushfield spots, and profound hypotonia may lead to a consideration of Down syndrome, although the chromosomal determination will eliminate that as a consideration.
Striking abnormalities of neuronal migration unique to Zellweger’s syndrome are evident in the cerebral hemispheres as areas of pachygyria or polymicrogyria localized to the opercular region. In the cerebellum, the Purkinje cells form scattered heterotopias throughout the cortex and in the granule cell layer. Laminar discontinuities involving the olivary nucleus are noted, which also are unique to Zellweger’s syndrome [Evrard et al., 1978; Volpe and Adams, 1972]. Studies of PEX gene knockout mice have demonstrated similar neuropathologic changes [Baes et al., 1997; Faust, 2003; Faust et al., 2001; Gressens et al., 2000; Janssen et al., 2000, 2003]. N-methyl-d-aspartate (NMDA)-mediated abnormalities may contribute to the clinical findings seen in the most severely affected persons [Gressens et al., 2000].
Multiple other abnormalities have been reported. The eyes demonstrate loss of retinal ganglion cells and gliosis of the optic nerve [Cohen et al., 1983]. Retinal pigmentary degenerative changes are associated with absent electroretinograms [Hittner et al., 1981]. Hepatomegaly (present in 78 percent of patients) with periportal fibrosis may result in significant cholestasis (in 59 percent) and jaundice, micronodular cirrhosis (in 37 percent), and hypoprothrombinemia [Heymans, 1984]. Excessive iron deposition has been noted, which diminishes with age [Gilchrist et al., 1976]. Renal cortical cysts of varied sizes are present in 97 percent of patients studied pathologically but may be missed by ultrasound analyses [Bernstein et al., 1974; Heymans, 1984]. The adrenal gland demonstrates changes similar to those in X-linked adrenoleukodystrophy, with cytoplasmic lamellar inclusions consisting of cholesterol esterified with very long chain fatty acids [Goldfischer et al., 1983]. Skeletal abnormalities include clubfoot, thumb rotation, and stippled chondral calcification of the patella and acetabulum in 50 percent of patients [Heymans, 1984].
Neonatal adrenoleukodystrophy and infantile refsum’s disease
The neonatal form of adrenoleukodystrophy was described by Ulrich et al. [1978] in a male infant with neonatal seizures and severe developmental delay. Postmortem examination at 18 months revealed adrenal atrophy and cytoplasmic inclusions typically seen in X-linked adrenoleukodystrophy; therefore, the disease was considered to be a connatal form of adrenoleukodystrophy. The multiple peroxisomal defects were first recognized in 1982 by Mobley and co-workers and substantiated later by others [Kelley et al., 1986; Partin and McAdams, 1983; Wanders et al., 1987]. Hyperpipecolicacidemia was described in 1968 by Gatfield and associates in a male infant with neurodegenerative disease and an enlarged liver, who died at 27 months of age. A marked excess of pipecolic acid was noted in the tissues and body fluids. In 1975, Danks and colleagues demonstrated excess pipecolic acid in Zellweger’s syndrome, and in 1988, Wanders and associates described a generalized peroxisomal dysfunction in that disorder, including the original case of Gatfield and associates [Wanders et al., 1988c]. The term infantile phytanic acid storage disease, a possible variant of Refsum’s disease, was used by Scotto et al. [1982] to describe the disorder in three unrelated males aged 3–6 years with hepatomegaly, dysmorphism, mental retardation, sensorineural hearing loss, and retinitis pigmentosa. The investigators noted that the liver biopsy specimen contained inclusions similar to plant chloroplasts that included phytol, which led to the measurement and recognition of increased phytanic acid levels, as in classic Refsum’s disease. The generalized peroxisomal dysfunction was documented by Poulos et al. [1984, 1986], and later by Poll-The et al. [1986a, b] and Wanders et al. [1986b].
Patients with milder forms may present in the neonatal period with mild to moderate dysmorphism, hypotonia, poor feeding, and hepatomegaly with micronodular cirrhosis. Motor and cognitive development usually is delayed. Even with hypotonia, patients may be able to walk, although gait often is ataxic. Retinal pigmentary degeneration may not become evident until the age of 4–6 months and often results in visual loss in the first years of life. Electroretinograms show profound abnormalities in nearly all affected persons and do not correlate well with vision in this population. Sensorineural hearing loss is associated with limited language development [Aubourg et al., 1986; Boltshauser et al., 1982; Budden et al., 1986; Poll-The et al., 1987; Scotto et al., 1982; Torvik et al., 1988; Wanders et al., 1990a; Weleber et al., 1984]. Adrenal dysfunction may develop with age [Govaerts et al., 1989; Poll-The et al., 1987]. Liver dysfunction often is present and detectable by persistent elevation of liver enzymes. A bleeding diathesis that responds to vitamin K also may develop, and in several children, esophageal varices were observed, consistent with portal hypertension. Several investigators have reported postmortem studies in patients aged 8 months to 12 years [Challa et al., 1983; Chow et al., 1992; Powers and Moser, 1998; Torvik et al., 1988]. The liver demonstrated micronodular cirrhosis, and the adrenal glands were hypoplastic. Lipid-laden macrophages were present in the liver, lymph nodes, and certain areas of the cerebral white matter. The only cerebral malformation was a variable hypoplasia of the cerebellar granular layer and a migrational defect of the Purkinje cells. Retinal abnormalities include a loss of ganglion cells, gliosis of the nerve fiber layer, and bileaflet inclusions in the pigment epithelium and macrophages [Cohen et al., 1983].
Optic atrophy seen in patients with neonatal adrenoleukodystrophy/infantile Refsum’s disease has been confused with Leber’s optic atrophy [Ek et al., 1986], and the association of retinal pigmentary degeneration and sensorineural hearing loss has led to misdiagnosis as Usher’s syndrome [Kelley et al., 1986; Noetzel et al., 1983].
Life span is variable, and patients have survived to adulthood [Kelley et al., 1986; Poll-The et al., 1987]. Several older patients have now been described. All identified patients have been visually impaired, with sensorineural hearing loss.
After analyzing 40 cases, Theil et al. [1992] subdivided the clinical characteristics into major and minor features. They suggest that the combined presence of three major features (present in more than 75 percent of patients) and one or more minor features (present in 50–75 percent) warrants biochemical investigation for peroxisomal disease. Other investigators have pointed out that, in view of the lack of specificity and ease of testing, strict screening criteria are not warranted, and the possibility of neonatal adrenoleukodystrophy and infantile Refsum’s disease should be considered in more children with previously undiagnosed developmental delay [Moser and Raymond, 1998].
All of the disorders in this group are inherited in an autosomal-recessive fashion, so it is important for genetic counseling that an accurate diagnosis be arrived at expeditiously. Carrier detection is possible only by DNA analysis if the molecular defect has been identified in the index case [Shimozawa et al., 1992]. Carrier detection is not possible by biochemical determination in blood or fibroblasts.
Laboratory Diagnosis
The diagnostic abnormalities present in patients with Zellweger spectrum disorders are listed in Table 38-2. A major diagnostic biochemical abnormality is the increased amount of very long chain fatty acids, which are fatty acids with carbon chains of more than 22. They also are elevated in single-enzyme defects of peroxisomal β-oxidation, so their presence is nonspecific, and other studies are required for this diagnosis. Very long chain fatty acid accumulation here is due to the reduction of peroxisomal β-oxidation, which is greater in Zellweger’s syndrome than in milder forms. A virtual absence of peroxisomal acyl-CoA oxidase and bifunctional enzyme has been demonstrated in the liver of patients with Zellweger’s syndrome [Suzuki et al., 1986; Tager et al., 1985]. It should be noted, however, that very long chain fatty acids are not elevated in rhizomelic chondrodysplasia punctata and other peroxisomal disorders (e.g., acatalasemia) that do not involve lipid oxidation, and normal levels do not exclude all peroxisomal disorders.
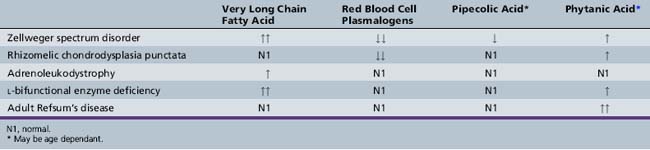
Because the first two steps of plasmalogen synthesis are peroxisomal functions, a reduction of plasmalogen levels is noted in patients with peroxisome biogenesis defects. Deficient levels of dihydroxyacetone phosphate acyltransferase have been used for diagnostic purposes in skin fibroblasts, amniocytes, chorionic villus samples, red blood cells, leukocytes, and platelets [Besley and Broadhead, 1987; Datta et al., 1984; Schutgens et al., 1985; Wanders et al., 1985, 1986a]. Webber et al. [1987] have demonstrated that the Km values and other properties of the residual enzyme are normal, suggesting that the enzyme is not defective but is abnormally labile in the cytosol because of failure of transport into the peroxisome. Plasmalogens are reduced to 5 percent of control values in Zellweger’s syndrome [Heymans et al., 1984; Wanders et al., 1986a].
An age-related increase in the levels of phytanic acid occurs in all of the peroxisome biogenesis disorders, including rhizomelic chondrodysplasia punctata, although not to the extent seen in classic adult Refsum’s disease. The accumulation of phytanic acid was demonstrated first in infantile Refsum’s disease [Scotto et al., 1982] and later in Zellweger’s syndrome [Poulos et al., 1984]. Poulos et al. [1988] demonstrated that pristanic acid is elevated in disorders of peroxisome biogenesis. This elevation was later confirmed to be due to an impaired capacity to oxidize pristanic acid [Singh et al., 1990]. These findings are unlike those in classic Refsum’s disease, in which the defect involves the conversion of phytanic acid to pristanic acid. Thus, oxidation of pristanic acid is a peroxisomal function that is disrupted in disorders of peroxisome biogenesis [Watkins et al., 1990].
Normally, bile acid intermediates, such as trihydroxycholestanoic acid and dihydroxycholestanoic acid, are absent or present in low concentrations. In Zellweger spectrum disorders, however, they constitute 30–50 percent of total plasma bile acids [Clayton et al., 1987; Eyssen et al., 1985; Gustafsson et al., 1983; Hanson et al., 1979; Mathis et al., 1980; Monnens et al., 1980; Poulos and Whiting, 1985]. Bile acid intermediates also may be abnormal in single-enzyme defects, such as peroxisomal bifunctional enzyme deficiency.
The impaired activity of l-pipecolic acid oxidase results in increased accumulation of pipecolic acid in plasma and increased excretion of pipecolic acid in urine of patients with Zellweger spectrum disorders [Kelley and Moser, 1984; Mihalik et al., 1989; Wanders et al., 1988a]. Pipecolic acid levels in blood and urine are age-dependent, but an excess can be demonstrated in all patients, irrespective of age [Dancis and Hutzler, 1986]. Medium- and long-chain dicarboxylic acids accumulate and are excreted in urine, suggestive of a partial block in the degradation of long-chain dicarboxylic acids [Bjorkheim et al., 1984; Rocchiccioli et al., 1986].
In Zellweger’s syndrome, the number of catalase-containing particles in liver or kidney biopsy specimens is reduced [Goldfischer et al., 1973; Small et al., 1988], although today biopsy rarely is needed for diagnosis. A reduction in the number of peroxisomes also occurs in neonatal adrenoleukodystrophy and infantile Refsum’s disease but, as expected, is less severe than in Zellweger’s syndrome. The catalase is cytosolic rather than particulate in catalase-containing particles. Although, in the past, peroxisomes were considered absent, Santos et al. [1988] demonstrated membranous structures called peroxisomal ghosts that contain peroxisomal membrane proteins. This finding has been substantiated by other investigators, who demonstrated the presence of major membrane proteins in skin fibroblasts from patients with Zellweger’s syndrome, some of which were quite abundant [Gaertner et al., 1991; Suzuki et al., 1989]. These ghosts lack the matrix enzyme catalase and some or all of the matrix proteins. Schram et al. [1986] have reported that peroxisomal β-oxidation enzymes, which normally are located in the peroxisome matrix, are formed at a normal rate but are rapidly degraded in the cytosol. The mitochondrial abnormalities observed are considered secondary [Trijbels et al., 1983].
Prenatal Diagnosis
Cultured amniocytes or chorionic villus cells can be used to diagnose all of the disorders of peroxisome biogenesis. A variety of biochemical strategies have been used and focus on the type and degree of biochemical abnormality. Two independent techniques, the measurement of very long chain fatty acid β-oxidation [Moser et al., 1984a; Solish et al., 1985] and plasmalogen synthesis [Roscher et al., 1985; Schutgens et al., 1985], have demonstrated accurate diagnosis when performed by laboratories experienced in these methodologies. Molecular genetic techniques may be used in families in which the mutation has been identified and pre-implantation genetic diagnosis has been performed [Al-Sayed et al., 2007].
Therapy
Attempts to normalize some of the biochemical abnormalities include oral ether lipid therapy [Holmes et al., 1987; Poulos et al., 1990; Wilson et al., 1986] and dietary restriction of very long chain fatty acids [Van Duyn et al., 1984] and phytanic acid [Greenberg et al., 1987; Steinberg, 1989]. These approaches may normalize plasma levels of very long chain fatty acids, phytanic acid, and red blood cell plasmalogens. The clinical effectiveness of these interventions, however, has yet to be demonstrated because of the phenotypic variability of these disorders. Ursodeoxycholic acid can reduce levels of bile acid intermediates and has the potential to prevent liver damage [Colombo et al., 1990]. Dietary therapy with docosahexaenoic acid replaces low levels in plasma and red blood cells of patients with Zellweger’s syndrome, neonatal adrenoleukodystrophy, and infantile Refsum’s disease [Martinez et al., 1993, 1994]; however, it is ineffective in improving clinical function [Mahmood et al., 2010]. Peroxisome proliferators, such as clofibrate, do not induce the formation of catalase-containing peroxisomes or improve clinical status [Lazarow et al., 1985].
Rhizomelic Chondrodysplasia Punctata
Rhizomelic chondrodysplasia punctata is characterized by severe shortening of limbs, mental retardation, and early death [Spranger et al., 1971]. A striking reduction in plasmalogen synthesis exceeding that seen in Zellweger’s syndrome led to the recognition of a peroxisomal defect in this disorder [Heymans et al., 1985, 1986]. Subsequent studies have revealed the following triad of biochemical defects:
The levels of very long chain fatty acids are normal, suggesting that the immature thiolase is catalytically active. Despite the presence of catalase in the particulate fraction, peroxisomal structure may not always be normal. Heymans et al. [1986] described two patients in whom peroxisomes were absent in some hepatocytes, but large or irregularly shaped in others.
The clinical features (Figure 38-4A) include disproportionate shortening of the proximal portions of the extremities, short stature, microcephaly, dysmorphic facial appearance, cataracts, ichthyosis, and severe mental retardation. Radiologic highlights include shortening of the proximal limbs, metaphyseal cupping, and disturbed ossification with epiphyseal and extra-epiphyseal calcification (see Figure 38-4B). Lateral views of the spine reveal coronal clefts of the vertebral bodies (see Figure 38-4C). Epiphyseal stippling involves mainly the knee, hip, elbow, and shoulder, and is uncommon in the vertebral column. The clinical features resemble those seen in warfarin embryopathy and should be differentiated from those of the milder autosomal-dominant form (Conradi–Hunermann type) and the X-linked forms of chondrodysplasia punctata, in which no biochemical abnormalities of peroxisomal function are detectable [Schutgens et al., 1988].
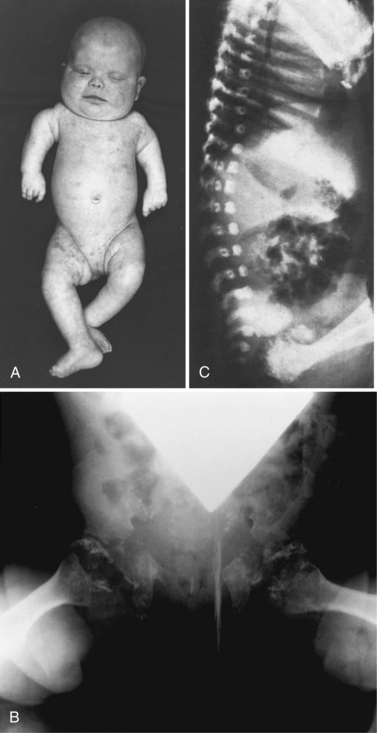
Fig. 38-4 Rhizomelic chondrodysplasia punctata
(A and B, Courtesy of Dr. B McGillivary, Vancouver, British Columbia. C, Courtesy of Dr. J Dorst, Johns Hopkins Hospital, Baltimore, Maryland.)
Rhizomelic chondrodysplasia punctata in a 9-month-old female with the classic triad of biochemical abnormalities, but with less severe disability, has been recognized. Her mentation and limb length were normal, despite radiographic evidence of chondrodysplasia punctata [Poll-The et al., 1991]. Other mildly affected patients have been identified, and many persons with an adult Refsum phenotype have alterations in PEX7 [Braverman et al., 2002; van den Brink et al., 2003].
The immature form of thiolase appears to be enzymatically active in patients with rhizomelic chondrodysplasia punctata because the levels of very long chain fatty acids are not increased and very long chain fatty acid oxidation is normal. Alternatively, small amounts of mature enzyme undetected by the immunoblot technique may be present. Balfe et al. [1990] reported that, in rhizomelic chondrodysplasia punctata and in some cases of Zellweger’s syndrome, the immature thiolase is present in a subcellular particle with a lower density than in normal peroxisomes or mitochondria, and speculated that this density difference may be related to peroxisome ghosts described by Santos et al. [1988].
The diagnosis is confirmed by the impaired plasmalogen synthesis and elevated phytanic acid with reduced phytanic acid oxidation. Prenatal diagnosis using cultured amniocytes or chorionic villus samples is available. Duff et al. [1990] have reported on the prenatal diagnosis of rhizomelic chondrodysplasia punctata by ultrasound analysis. As suggested by the presence of the biochemical triad with normal intelligence and normal limb length reported by Poll-The et al. [1991], the phenotype of rhizomelic chondrodysplasia punctata is more varied than was previously recognized and cautions against the previous assumptions of these diagnostic criteria for prenatal diagnosis.
Rhizomelic chondrodysplasia punctata is inherited in an autosomal-recessive manner. The disease results from a defect in PEX7, which encodes the receptor for PTS2. Several mutations have been noted in the PEX7 gene in patients with rhizomelic chondrodysplasia punctata; however, the L292ter found in more than half of affected genes probably indicates a Northern European founder effect [Braverman et al., 2000, 2002].
Treatment is very limited and consists of palliative orthopedic care and cataract removal. Although dietary restriction of phytanic acid has been proposed, no evidence indicating that it affects outcome is available. The recent development of a mouse model offers the potential to understand the pathogenesis and attempt therapies in this serious disorder [Braverman et al., 2010].
Defects of Single Peroxisomal Enzymes
Defects of Single Peroxisomal β-Oxidation Enzymes
Adrenoleukodystrophy
The first clinical cases of what is known as X-linked adrenoleukodystrophy were described in 1923 by Siemerling and Creutzfeldt. The X-linked mode of inheritance was suggested by Fanconi et al. [1963]. A key observation of characteristic lamellar lipid-soluble cytoplasmic inclusions in the adrenal cortical cells and brain macrophages of patients with childhood adrenoleukodystrophy was made by Powers et al. [1980]. Similar inclusions were observed in men with Addison’s disease and spastic paraparesis by Budka et al. [1976], and by Griffin et al. [1977], and the related condition was named adrenomyeloneuropathy. Similar histopathologic changes observed in a symptomatic child and an adult within a kindred strengthened the assertion that, despite the phenotypic variation, the same disease process was present. Igarashi et al. [1976] defined the biochemical defect by demonstrating that the inclusions in the adrenal cortex and cerebral white matter consisted of cholesterol esters with a striking excess of saturated unbranched fatty acids of 24- to 30-carbon chain length, of which C26:0 and C25:0 were the most abundant. This excess of very long chain fatty acids in cultured skin fibroblasts, plasma, or red blood cells is useful for diagnostic purposes [Kawamura et al., 1978; Moser et al., 1981; Tanaka et al., 1986, 1990; Tsuji et al., 1981].
Biochemical and molecular basis
The peroxisomal β-oxidation pathway consists of four enzymatic steps (as outlined in Figure 38-1). Before the substrates can enter this pathway, the acyl residues require activation by transfer of a coenzyme A. For very long chain fatty acids entering the l-specific β-oxidation pathway, lignoceryl-CoA ligase (also referred to as lignoceryl-CoA synthetase) is the activating enzyme. Deficiency of this enzyme initially was implicated as the basis of the very long chain fatty acid accumulation in X-linked adrenoleukodystrophy. Singh and co-workers [1984] demonstrated that the capacity to oxidize the C24:0 and C26:0 fatty acids was reduced in patients with X-linked adrenoleukodystrophy. Studies by Wanders et al. [1988c], indicated that the defect involves the capacity to form the CoA derivative of very long chain fatty acid, because the capacity to oxidize lignoceryl-CoA is intact. It is known that the genetic basis for X-linked adrenoleukodystrophy is mutations of the ABCD1 gene, which encodes a PMP, an ATP-binding cassette protein [Mosser et al., 1993], and the gene for this protein is located on the X chromosome in the Xq28 region. It remains unclear how mutations in this nonenzyme membrane protein affect the function of lignoceryl-CoA ligase [Wanders et al., 2010]. The following other peroxisomal proteins with homology to ABCD1 have been described:
All of these proteins are located within the peroxisomal membrane and contain an ATP-binding cassette motif. It has been hypothesized that they are involved in the transport of other proteins or possibly substrates for peroxisomal pathways across the peroxisome membrane. Experimental data indicate that these proteins form homodimers and heterodimers [Valle and Gartner, 1993]. Multiple mutations in ABCD1 have been identified in patients with X-linked adrenoleukodystrophy, and no phenotype correlation has been identified [Kemp et al., 2001; Kok et al., 1995]. Several mouse models of X-linked adrenoleukodystrophy are now available [Lu et al., 1997].
Clinical and pathologic features of X-linked adrenoleukodystrophy and adrenomyeloneuropathy
Table 38-3 summarizes the different phenotypes of patients with X-linked adrenoleukodystrophy. These patients all demonstrate accumulation of very long chain fatty acid, and molecular studies have confirmed ABCD1 gene mutations. Various phenotypes have been recognized as occurring within the same pedigree, so neither the genetic mutation nor the biochemical abnormality predicts the clinical presentation.
Table 38-3 Phenotypes among X-Linked Adrenoleukodystrophy Hemizygotes
| Type | Frequency (%) |
|---|---|
| Childhood cerebral | 33–39 |
| Adolescent cerebral | 4–7 |
| Adult cerebral | 2–3 |
| AMN | 26–32 |
| Addison’s only | 13–17 |
| Asymptomatic | 7–18 |
AMN, adrenomyeloneuropathy.
(Adapted from the North American data of the Kennedy Krieger Institute [Bezman and Moser, 1998].)
Childhood Cerebral Form of Adrenoleukodystrophy
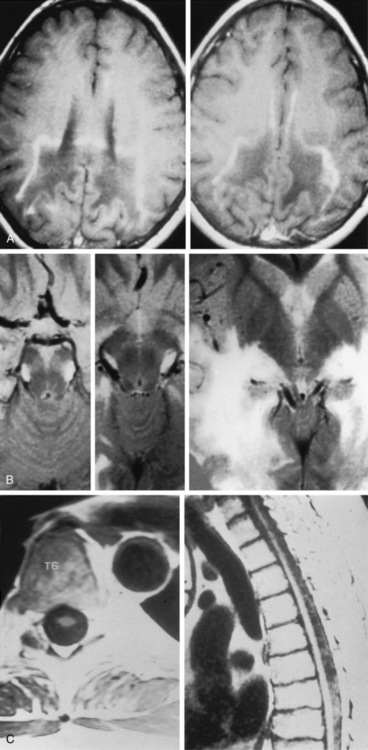
The childhood cerebral variant is the most common and fulminant form of X-linked adrenoleukodystrophy. Affected boys are normal until 4–8 years of age, when they manifest behavior problems and failure in school as a result of rapid regression of auditory discrimination, spatial orientation, speech, and writing. Seizures occur in 30 percent of patients and in rare instances may be the initial sign. The magnetic resonance imaging (MRI) scan reveals parieto-occipital white matter lesions (in 85 percent of patients) or frontal lesions (in 15 percent) at this stage, with contrast accumulation at the leading edge of the lesion [Duda and Huttenlocher, 1976; Eiben and DiChiro, 1977; Kumar et al., 1987] (Figure 38-5). Rapid clinical deterioration leads to spastic quadriparesis, swallowing difficulty, and visual loss, culminating in a vegetative state usually within 2 years of the initial signs and symptoms. Although males come to medical attention because of the neurologic deficits, impaired cortisol response can be identified by adrenocorticotropic hormone stimulation in 85 percent of this group [Moser et al., 1991b].
Adult Cerebral Form of Adrenoleukodystrophy
In the adult cerebral form of adrenoleukodystrophy, dementia, psychiatric disturbances, seizures, and spastic paraparesis develop after age 21 [Bresnan and Richardson, 1979; Esiri et al., 1984; James et al., 1984; Kitchin et al., 1987; Sereni et al., 1987; Turpin et al., 1985]. Patients may be misdiagnosed as having multiple sclerosis, brain tumor, or schizophrenia, and demonstrate rapid deterioration similar to that in the childhood cerebral form. Therefore, patients with these clinical presentations and adrenal insufficiency or leukodystrophy should be evaluated for a positive family history; in addition, measurement of plasma very long chain fatty acids is warranted.
Adrenomyeloneuropathy
The neurologic manifestations of adrenomyeloneuropathy, an adult form of adrenoleukodystrophy, consist of an insidious onset and slow progression of spastic paraparesis, impaired vibratory sense in the lower extremity, and bladder or bowel dysfunction. Onset is typically in the third decade of life. The primary pathology involves the spinal cord with loss of myelinated axons in the corticospinal tracts, nucleus gracilis, and dorsal spinocerebellar tracts [Budka et al., 1983; Probst et al., 1980; Schaumburg et al., 1977]. Sural and peroneal nerves reveal a loss of large and small myelinated fibers.
The development of cerebral demyelination has been seen in approximately 15–20 percent of men with adrenomyeloneuropathy [van Geel et al., 2001]. This pathologic change needs to be differentiated from long tract findings on MRI [Loes et al., 2003]. Cerebral disease is similar in time course to the childhood form of the disease and leads to dementia, spasticity, blindness, and death. Approximately half of patients with adrenomyeloneuropathy appear to have some degree of cerebral involvement, with mild to moderate abnormalities noted on MRI in 46 percent [Kumar et al., 1995] (see Figure 38-5). These abnormalities consist most frequently of parietal-occipital white matter and optic radiation involvement.
Adrenoleukodystrophy with Addison’s Disease Only
The diagnosis of adrenoleukodystrophy with Addison’s disease only includes the patients who have isolated Addison’s disease in the absence of neurologic signs and symptoms, and is more common than was previously recognized. A study conducted in an endocrine clinic found that 5 of 8 male Addisonian patients had the biochemical defect of adrenoleukodystrophy [Sadeghi-Nejad and Senior, 1990]. Recognition of these patients is vital for genetic counseling and to monitor for the development of adrenomyeloneuropathy or cerebral symptoms later on. Onset of adrenal dysfunction does not correlate with neurologic involvement.
Symptomatic Heterozygotes
Between 10 and 20 percent of the women who are carriers for the adrenoleukodystrophy gene manifest myelopathy resembling that in adrenomyeloneuropathy between the third and fifth decades of life [Dooley and Wright, 1985; Heffungs et al., 1980; Morariu et al., 1982; Moser et al., 1980; Naidu et al., 1986; Noetzel et al., 1987; O’Neill et al., 1982, 1984; Penman, 1960; Pilz and Schiener, 1973]. Long tract signs and diminished vibration sense in the legs are present in two-thirds of the patients, although only 25 percent complain of symptoms. Approximately 14 percent have severe spinal involvement requiring assistance with ambulation, and 5 percent have dementia [Naidu and Moser, 1990]. In contrast with males, adrenal insufficiency rarely occurs in these women [El-Deiry et al., 1997].
Elevation of very long chain fatty acids in plasma or skin fibroblasts allowed identification of 85 percent of obligate heterozygotes in one series [Moser et al., 1983]. The remaining cases can be identified by molecular analysis for the gene defect. Mutation analysis helps identify heterozygotes once the mutation in the index case is known [Boehm et al., 1999]. Migeon et al. [1981] studied fibroblast clones from heterozygotes and identified two types of cells; one had a normal fatty acid pattern, and the pattern in the other cell type was similar to that in affected males, reflecting X-inactivation status. To determine if differential X-inactivation accounts for the clinical variability in heterozygotes, an indirect immunofluorescence analysis of ALDP, the protein defective in X-linked adrenoleukodystrophy, has been applied to skin fibroblasts. Previous studies have reported that 70 percent of males with X-linked adrenoleukodystrophy lack immunoreactivity for ALD protein (ALDP). As a result of differential X-inactivation, heterozygotes from these informative families have a mixed population of normal and ALDP-deficient cells. Despite considerable variation, a positive correlation is noted between immunonegative cells and clinical severity. No correlation is seen between immunoreactivity and either very long chain fatty acid levels or age alone. When age is considered, however, the correlation between severity and ALDP immunoreactivity is strengthened. Thus, differences in patterns of X-inactivation appear, in part, to contribute to the variable severity of the disease seen in the X-linked adrenoleukodystrophy heterozygotes [Naidu et al., 1997].
Pathogenesis of adrenoleukodystrophy
The main biochemical defect is the striking excess of very long chain fatty acids, which accumulate in the cholesterol ester fraction, primarily in the adrenal gland and cerebral white matter. Theda et al. [1987] noted that, in areas of brain where myelin was intact, there was a 20-fold excess of C26:0 in the phosphatidylcholine fraction, whereas the cholesterol ester fraction had a normal fatty acid composition. The enrichment with C26:0 in the cholesterol ester fraction reported by earlier investigators [Brown et al., 1983; Igarashi et al., 1976; Menkes and Corbo, 1977; Molzer et al., 1981; Ramsey et al., 1979; Reinecke et al., 1985; Taketomi et al., 1987] was present only in zones undergoing active demyelination and, to a lesser extent, where myelin had been replaced by glial tissue. They concluded that increased very long chain fatty acids in phosphatidylcholine could be the primary event and that the excess in the cholesterol ester fraction is due to myelin breakdown.
The accumulation of very long chain fatty acids in X-linked adrenoleukodystrophy is not as severe as in disorders of peroxisome biogenesis or single-peroxisomal β-oxidation enzyme defects. Red cell membrane microviscosity is increased in X-linked adrenoleukodystrophy [Knazek et al., 1983], which may impair functional capacity. This finding also is suggested by studies in human adrenal gland cultures, where the addition of C26:0 fatty acid to the culture medium resulted in an impaired cortisol response to adrenocorticotropic hormone [Whitcomb et al., 1988]. Cholesterol esterified with very long chain fatty acids is a poor substrate for cholesterol ester hydrolase, which may limit steroid production [Ogino and Suzuki, 1981].
The pathogenesis of the nervous system lesion has been more difficult to discern. It has been presumed that elevation of very long chain fatty acids results in the axonopathy seen in patients with adrenomyeloneuropathy and in women who are carriers. It is not clear, however, why rapid, inflammatory demyelination affecting the cerebral white matter tracts develops in certain persons. The levels of very long chain fatty acids in plasma or skin fibroblasts or the capacity to metabolize very long chain fatty acids in cultured fibroblasts [Boles et al., 1991] do not differ in males with the childhood form from those in men with adrenomyeloneuropathy or other forms. No correlation exists between severity of the neurologic disease and levels of very long chain fatty acids. Antoku et al. [1991], however, reported that patients with childhood cerebral adrenoleukodystrophy had higher levels of very long chain fatty acids in mononuclear cells than did patients with adrenomyeloneuropathy, even though levels in plasma and erythrocytes demonstrated no difference. The inability to distinguish the various forms prospectively hampers genetic counseling and choice of therapeutic modalities. These observations have led to the conclusion that very long chain fatty acid excess alone is not sufficient to explain cerebral demyelination and that other factors must play a role in the phenotypic variability.
The presence of the perivascular lymphocytic infiltration in the white matter of the cerebral forms provides evidence of immunologic involvement as one additional factor [Powers and Moser, 1998]. This is not seen in the adrenal gland or other leukodystrophies. Typing of the lymphocytic cells in brain from four patients with the cerebral form of adrenoleukodystrophy examined postmortem revealed 59 percent T cells, 34 percent T4 cells, 16 percent T8 cells, 24 percent B cells, and 11 percent monocytes [Griffin et al., 1985]. This pattern is similar to that found during a cellular immune response in the central nervous system. Bernheimer et al. [1983] reported increased levels of IgG and IgA in brain from patients with adrenoleukodystrophy, similar to that in patients with multiple sclerosis. Boutin et al. [1989] suggest that interleukin-2 plays an important role. McGuinness et al. [1995] demonstrated an elevation in tumor necrosis factor-α (TNFα) in the leading edge of areas of active demyelination. More recently, there is evidence that very long chain fatty acids are toxic to the microglia and their involvement in the disease cascade is being determined [Eichler et al., 2008]. The collective evidence implies that an immunologic mechanism contributes to the rapid progression of white matter lesions, possibly in response to the altered lipid composition in brain of patients with adrenoleukodystrophy. Segregation analyses suggest the existence of a modifier gene that potentially affects this immune response [Smith et al., 1991]. Further studies in this area may identify factors that contribute to the pathogenesis of the nervous system lesions.
Therapies for adrenoleukodystrophy
The two forms of therapy under intensive investigation are dietary restriction and bone marrow transplantation. Based on the observations of Kishimoto et al. [1980] that the very long chain fatty acids accumulating in the brain of patients with adrenoleukodystrophy were partly of dietary origin, a diet that restricted very long chain fatty acid intake was developed [Brown et al., 1982; Van Duyn et al., 1984]. This diet had no effect on the levels of plasma very long chain fatty acids or on the clinical course. Rizzo et al. [1986] reported that cultured skin fibroblasts synthesized saturated very long chain fatty acids, including C26:0, and that the rate of this synthesis was reduced by the addition of monounsaturated fatty acids, such as oleic acid. As saturated and monounsaturated fatty acids are elongated to very long chain fatty acids by the same microsomal elongation system, presumably the addition of oleic acid reduces synthesis of saturated very long chain fatty acids by competing for the same enzyme system [Bourre et al., 1976].
These findings led to clinical trials with oral glyceryl trioleate in combination with dietary restriction of very long chain fatty acids. This dual therapy achieved a 50 percent reduction of the levels of saturated very long chain fatty acids in plasma, as well as a statistically significant improvement in peripheral nerve function in patients with adrenomyeloneuropathy [Moser et al., 1991a; Rizzo et al., 1987]. Subsequently, erucic acid (C22:1) was demonstrated to be more effective in reducing synthesis of saturated very long chain fatty acids; therefore, glyceryl trioleate was combined with glyceryl trierucate. A dramatic normalization of the level of C26:0 in plasma occurred within a month [Moser et al., 1995; Rizzo et al., 1989], and the microviscosity of the red cell membranes returned to normal [Knazek et al., 1983].
Unfortunately, this regimen does not alter the course of the childhood cerebral form of adrenoleukodystrophy [Uziel et al., 1990]. In open, uncontrolled studies, the effects on the clinical course of the adult form have been uncertain but generally unimpressive [Aubourg et al., 1993; Kaplan et al., 1995]. Further studies are planned in presymptomatic males: controlled studies, in men with adrenomyeloneuropathy. Symptomatic heterozygotes have been managed with glyceryl trioleate/glyceryl trierucate and dietary restriction therapy without significant improvement, as in their male counterparts. Prophylactic treatment is not recommended for asymptomatic heterozygotes.
The results of bone marrow transplantation in mildly affected patients are encouraging [Aubourg et al., 1990; Lockman et al., 1993; Loes et al., 1994; Peters et al., 2004]. In patients with more advanced disease, transplantation has not resulted in halting the course and may be associated with worsening of the neurologic status immediately after the procedure; therefore, this procedure is not advised [Moser et al., 1984b; Weinberg et al., 1988]. In patients with advanced disease, neither dietary therapy nor bone marrow transplantation is effective. Immunosuppression with cyclophosphamide did not arrest the rapid progression of the illness [Naidu et al., 1988a; Stumpf et al., 1981]. Recently, gene therapy with a lentivirus vector has been successfully used in three boys with early cerebral disease. This advancement in the field potentially offers additional avenues of treatment for many individuals who could not previously undergo transplantation [Cartier et al., 2009].
Newborn screening for XALD
A biochemical assay using tandem mass spectroscopy and the standard newborn blood spot has been developed recently and has been found to be highly sensitive and specific. If this is subsequently incorporated into newborn screening programs, it will most likely change the proportion of males coming to attention in the presymptomatic phase, and will also improve their care and management [Hubbard et al., 2009].
Contiguous Deletion of the XALD Gene
Corzo and co-workers identified three males in the newborn period who demonstrated abnormalities suggestive of a defect of l-bifunctional enzyme [Corzo et al., 2002]. Very long chain fatty acids were elevated; other findings included hypotonia and liver disease. All three patients died in the first year of life. Further investigation demonstrated large deletions of ABCD1, including the promoter region, with extension of these 5′ deletions into contiguous genes, including the DNA marker DXS1356E. The investigators proposed the name “contiguous ABCD1 DXS1357E deletion syndrome” (CADDS) for this phenotype. The specific role of the additional genetic areas is uncertain at present. Two of the mothers were determined to be carriers for these deletions, indicating the importance of correct diagnosis of single-enzyme defects of peroxisomal β-oxidation.
Acyl-CoA Oxidase Deficiency
Two siblings and an unrelated female patient have been reported to have acyl-CoA oxidase deficiency [Kyllerman et al., 1990; Poll-The et al., 1988a]. A large deletion of the gene encoding this enzyme was reported in the siblings [Fournier et al., 1994]. Dysmorphism, hypotonia, feeding difficulties, and intractable neonatal seizures were the presenting features. Little motor or cognitive development occurred in the first 2 years of life. Ocular involvement with extinguished electroretinography and white matter involvement with abnormal contrast enhancement were present. Adrenal insufficiency was not evident, but elevated serum adrenocorticotropic hormone and low cortisol levels were present in one patient. No organomegaly was present. Although the clinical characteristics resembled those in the milder forms of peroxisome biogenesis disorders, the following two features distinguished it from the Zellweger spectrum disorders:
Immunoblot studies revealed absence of acyl-CoA oxidase in the liver, and other peroxisomal β-oxidation enzymes were normal [Poll-The et al., 1988a]. Cultured skin fibroblasts had a reduced capacity to oxidize lignoceric acid and peroxisomal palmitoyl-CoA oxidase. The first prenatal diagnosis of isolated acyl-CoA oxidase deficiency was made by Wanders et al. [1990b]. In amniotic fluid, measurements of very long chain fatty acids were significantly elevated, but levels of bile acid intermediates were normal. In amniocytes, very long chain fatty acids were markedly increased, but plasmalogen biosynthesis was normal. Immunoblot analysis of amniocytes and the aborted fetal liver tissues demonstrated that all components of acyl-CoA oxidase were absent, whereas the bifunctional protein and mature form of thiolase were normal.
Bifunctional Enzyme Deficiency
Watkins et al. [1988] described a patient without dysmorphism or organomegaly who had significant hypotonia and neonatal seizures. Mental retardation was severe, and death occurred at 5½ months. Neuropathologic abnormalities included polymicrogyria and focal cortical heterotopias, similar to those in peroxisome biogenesis disorders [Kaufmann et al., 1996; Naidu et al., 1988b]. The liver had normal-appearing peroxisomes. Biochemical studies revealed elevation of very long chain fatty acids and bile acid intermediates; all other peroxisomal functions were normal. Postmortem immunoblot studies of liver demonstrated the absence of the bifunctional protein; other peroxisomal β-oxidation enzymes were present. The mRNA for the bifunctional protein was normal, indicating defective translation of the mRNA or abnormal post-translational processing, such as defective peroxisomal import mechanisms.
Although bifunctional enzyme deficiency bears some clinical resemblance to the acyl-CoA deficiency with elevation of very long chain fatty acids, a significant difference is the additional accumulation of bile acid intermediates (trihydroxycholestanoic acid) as well [Watkins et al., 1995]. The reason for trihydroxycholestanoic acid accumulation is evident in Figure 38-1, which demonstrates that the initial oxidation step for bile acid intermediates and very long chain fatty acids occurs by separate enzymes, whereas the subsequent step at the level of the bifunctional enzyme is common to both systems. Another patient with similar clinical features was found to have a functionally inactive bifunctional protein, although immunologic studies revealed normal amounts of the enzyme [Wanders et al., 1990c]. This patient is presumed to have a different mutation from the one described previously.
3-Oxoacyl-CoA Thiolase Deficiency
The entity 3-oxoacyl-CoA thiolase deficiency was first described by Goldfischer and co-workers in 1986 and called pseudo-Zellweger syndrome because of the presence of normal peroxisomes in the liver despite a clinical resemblance to Zellweger’s syndrome. Biochemically, the levels of very long chain fatty acids and bile acid intermediates were increased, but phytanic acid level was normal. A re-examination of material from one of these cases demonstrated that the infant, in fact, had a defect in bifunctional enzyme. The clinical existence of a thiolase deficiency is therefore not proved [Ferdinandusse et al., 2002].
Trihydroxycholestanoic Acid-CoA Oxidase Deficiency
Christensen et al. [1990] described an unusual female patient who demonstrated normal development until 18 months of age, when she deteriorated, with clinical manifestations including ataxia, dysarthria, and dry skin. By 5 years of age, she was severely incapacitated, with hearing loss and mental retardation. At this stage she had normal findings on computed tomography (CT) scan, absence of retinal degeneration, and normal liver function and nerve conduction velocities. Because the phytanic acid level was elevated, she was placed on a diet low in phytanate, after which she demonstrated significant improvement in gait, hearing, and speech. The most prominent biochemical abnormality, however, was an elevation in bile acid intermediates with normal very long chain fatty acid concentration, presumed to be secondary to a defect in trihydroxycholestanoic acid-CoA oxidase. Because phytanic acid oxidase levels were normal in cultured fibroblasts, the elevation in phytanic acid is believed to result from inhibition of phytanate oxidation by the accumulating bile acid intermediates.
Peroxisomal α-Methylacyl-CoA Racemase Defect
Several patients with adult-onset neurologic findings, including spasticity, sensory motor neuropathy, seizures, and retinitis pigmentosa, have been described. Very long chain fatty acid levels were normal, but elevations in C27 bile acid intermediates and pristanic acid were found. Phytanic acid was mildly elevated. Because pristanic acid β-oxidation was reduced, the activity of α-methylacyl-CoA racemase (AMACR) was examined and found to be deficient. AMACR is involved in the interconversion of (R)- and (S)-stereoisomers of α-methyl branched-chain fatty acyl-CoA esters, including pristanoyl-CoA, and the interconversion of the CoA esters of dihydroxycholestanoic acid and trihydroxycholestanoic acid. Its physiologic function is to produce the (S)-stereoisomers because only these serve as substrate for branched-chain acyl-CoA oxidase [Ferdinandusse et al., 2000].
Single-Enzyme Defects of Plasmalogen Synthesis
Patients with defects in acyl-dihydroxyacetone phosphate transferase [Wanders et al., 1992] or alkyl-dihydroxyacetone phosphate synthase [Wanders et al., 1994] have a marked deficiency of plasmalogens. They differ from patients with defects of PEX7 because they metabolize phytanic acid and process thiolase normally. In most instances, however, the clinical picture resembles that in rhizomelic chondrodysplasia punctata. This similarity has led to the consideration that all of the clinical abnormalities in rhizomelic chondrodysplasia punctata are attributable to a deficiency of plasmalogens. Isolated dihydroxyacetone phosphate-AT deficiency also may manifest with a milder phenotype of rhizomelic chondrodysplasia punctata [Clayton et al., 1994].
Acatalasemia
Acatalasemia demonstrates clinical heterogeneity, with presentations ranging from absence of symptoms (in the Swiss variant) to oral ulcers, presumed to be secondary to peroxide-generating bacteria, in a severe Japanese variant. The Japanese patients have greatly reduced catalase activity; however, the electrophoretic mobility of the residual enzyme is normal. By contrast, the symptom-free patients with the Swiss variant have a large amount of catalase, but it demonstrates greater than normal heat lability. Studies by Crawford et al. [1988] report that the Japanese variant involves a regulatory mutation in which the gene is not transcribed, whereas in the Swiss variant, a structural mutation renders the enzyme unstable. The human catalase gene has been localized to 11p13 and cloned [Junien et al., 1980; Quan et al., 1986].
Hyperoxaluria Type I
Primary hyperoxaluria type I does not involve the nervous system, but does highlight an important pathogenic mechanism in peroxisomal disorders. It is inherited as an autosomal- recessive trait. Clinical onset is in childhood or adolescence. Patients present with urolithiasis and renal failure. Increased urinary excretion of oxalate (more than 50 mg/1.73 m2 of body surface per day), glycolate (more than 70 mg/1.73 2 per day), and occasionally glyoxylate is diagnostic. In 1986, Danpure and Jennings demonstrated that the enzyme alanine-glyoxalate aminotransferase, which converts glyoxylate to glycine, is deficient in patients with this disorder [Danpure and Jennings, 1986]. Latta and Brodhel [1990] noted that this disorder accounted for 2–2.7 percent of children with end-stage renal disease. In human liver, this enzyme is peroxisomal [Danpure and Jennings, 1986], and the activity varies in affected patients, correlating with the severity of the disease [Danpure and Jennings, 1988]. A milder pyridoxine-responsive form of the disease has a less severe reduction of the enzyme [Wanders et al., 1988b]. Liver biopsy establishes the enzyme defect, but peroxisomes are present and intact in the liver [Iancu and Danpure, 1969]. The functions of other peroxisomal enzymes are normal. Danpure et al. [1989a] and Cooper et al. [1988] noted that in one-third of the patients with diminished but not absent alanine-glyoxalate aminotransferase, the enzyme is misdirected to the mitochondria, instead of its normal localization in humans to the peroxisome. The alanine-glyoxalate aminotransferase cDNA has been sequenced from eight patients with hyperoxaluria type I in whom the alanine-glyoxalate aminotransferase was localized to the mitochondria, and three new mutations were identified. One of these encoded a glycine to arginine substitution at residue 170 and was unique to these patients, but the two other mutations also were present in 5–10 percent of the control population [Purdue et al., 1990]. Nishiyama et al. [1991] have reported a point mutation encoding a serine to proline mutation at residue 205 in a patient in whom alanine-glyoxalate aminotransferase activity was nearly absent and who thus differed from the other group.
Diagnosis is established by the demonstration of deficient alanine-glyoxalate aminotransferase activity in a liver biopsy specimen [Danpure and Jennings, 1986; Danpure et al., 1987]. Prenatal diagnosis has been achieved by second-trimester fetal liver biopsy [Danpure et al., 1989b] and, in the future, could be achieved by DNA analysis of amniocytes or chorionic villus sampling. The estimated incidence of this disorder is 1 in 5 million to 15 million children; family studies demonstrate an increased incidence of parental consanguinity [Latta and Brodhel, 1990].
An exciting therapeutic advance has been a combination of liver and kidney transplantation with extremely favorable results [McDonald et al., 1989; Morgan and Watts, 1989; Ruder et al., 1990; Watts et al., 1987]. Some of the mildly affected patients respond to pyridoxine [Wanders et al., 1988b; Watts et al., 1979]. MacCollin et al. [1991] have described a patient with normal intelligence, ataxia, peripheral neuropathy, and calcium oxalate stones. Peroxisomes were absent. A marked increase in very long chain fatty acid and pipecolic acid was documented, with catalase present in the cytosolic fraction. This patient’s case is the only recorded case of hyperoxaluria in association with abnormal peroxisome biogenesis and multiple peroxisomal enzyme involvement.
Peroxisomal Glutaryl-CoA Oxidase Deficiency
The first patient with peroxisomal glutaryl-CoA oxidase deficiency was identified at 11 months, when the child was presented for failure to thrive and vomiting (in particular, postprandial vomiting), by Bennett et al. [1991]. Urinary glutaric acid level was 500 mmol per mole of creatinine, whereas the normal level is less than 2 mmol. The glutaricaciduria was responsive to riboflavin 200 mg given twice daily. At the age of 5 years 8 months, the patient was judged to be developmentally normal, although this outcome may have been attributable to the vitamin therapy. Detailed metabolic investigations ruled out glutaricaciduria types I and II. The activity of peroxisomal glutaryl-CoA oxidase was reduced to less than 10 percent of control values. Of interest, loading studies demonstrated that the glutaric acid was derived from lysine, presumably through the same pathway in which pipecolic acid is an intermediate. This case represents a peroxisomal cause of glutaricaciduria distinct from the well-known mitochondrial disorders resulting in glutaricaciduria types I and II.
Classic Adult Refsum’s Disease
Classic adult Refsum’s disease is a rare disorder of autosomal-recessive inheritance; originally described by Refsum [1946], it is characterized by retinitis pigmentosa, peripheral polyneuropathy, cerebellar ataxia, and increased cerebrospinal fluid protein. Biochemical identification is by the increased plasma levels of phytanic acid with normal pristanic acid. In this condition, the capacity for hydroxylation of phytanoyl-CoA has been lost. Low phytanic acid oxidation also is seen in human cells lacking PEX7, the receptor for PTS2, suggesting that the enzyme defective in Refsum’s disease is targeted to peroxisomes by a PTS2.
The enzyme deficiency of phytanoyl-CoA hydroxylase A and the gene defect in this disease have been identified conclusively [Jansen et al., 1997; Mihalik et al., 1997] and are the primary cause of adult Refsum’s disease. The gene PAHX is localized to chromosome 10, and patients with Refsum’s disease are homozygous for inactivating mutations in PAHX. It must be emphasized, however, that a substantial number of adult patients with the same phenotypic features have now been identified as having mutations in PEX7 [Jansen et al., 1997], and mutational analysis is required for definitive diagnosis. Therapy consists of restriction of dietary phytanic acid and symptomatic treatment.
Mulibrey Nanism
Mulibrey nanism is a rare disorder of autosomal-recessive inheritance that manifests with prenatal and postnatal growth failure, hepatomegaly, pericardial constriction, and characteristic facial features. Mulibrey stands for “muscle-liver-brain-eye,” and features of this disorder include yellow dots on the fundi, muscular weakness, enlarged cerebral ventricles, increased incidence of Wilms’ tumor, and cutaneous nevus flammeus [Kallijarvi et al., 2002; Lapunzina et al., 1995]. Patients have normal intelligence. It is more common in the Finnish population. It is due to defects in TRIM37 located on 17q22–q23. This gene encodes RING-B-box-coiled-coil protein, which is localized to the peroxisome. Patients with TRIM37 mutations do have normal peroxisome morphology, and other PTS1-containing matrix proteins are not affected. No abnormality is seen in very long chain fatty acid concentration or other biochemical measures. TRIM37 apparently lacks a PTS1 signal, but uptake of this protein was lacking in cell lines defective for PEX1 and PEX5. The exact role in peroxisomal function is not known at present [Avela et al., 2000; Kallijarvi et al., 2002]. The demonstration of this disorder as involving the peroxisome highlights an important point in going forward in the identification of peroxisomal disorders. Current screening strategies have focused on known biochemical features, and expansion of our understanding will require newer diagnostic modalities.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Al-Sayed M., Al-Hassan S., Rashed M., et al. Preimplantation genetic diagnosis for Zellweger Syndrome. Fertil Steril. 2007;87:1468.
Antoku Y., Koike F., Othsuka, et al. Adrenoleukodystrophy: A correlation between saturated very long chain fatty acids in mononuclear cells and phenotype. Ann Neurol. 1991;30:101.
Arnold G., Holtzman E. Microperoxisomes in the central nervous system of the postnatal rat. Brain Res. 1978;155:1.
Aubourg P., Adamsbaum C., Lavallardrousseau M.C., et al. A 2-year trial of oleic and erucic acids (Lorenzo oil) as treatment for adrenomyeloneuropathy. N Engl J Med. 1993;329:745.
Aubourg P., Blanche S., Jambaque I., et al. Reversal of early neurologic and neuroradiologic manifestations of X-linked adrenoleukodystrophy by bone marrow transplantation. N Engl J Med. 1990;322:1860.
Aubourg P., Scotto J., Rocchiccioli F., et al. Neonatal adrenoleukodystrophy. J Neurol Neurosurg Psychiatry. 1986;49:77.
Avela K., Lipsanen-Nyman M., Idanheimo N., et al. Gene encoding a new RING-B-box-Coiled-coil protein is mutated in mulibrey nanism. Nat Genet. 2000;25:298.
Baes M., Gressens P., Baumgart E., et al. A mouse model for Zellweger syndrome. Nat Genet. 1997;17:49.
Bennett Cl, Chen Y., Hahn S., et al. Prevalence of ALDH7A1 mutations in 18 North American pyridoxine-dependent seizure (PDS) patients. Epilepsia. 2009;50:1167-1175.
Bennett M.J., Pollit R.J., Goodman S.I., et al. Atypical riboflavin-responsive glutaric aciduria, and deficient peroxisomal glutaryl-CoA oxidase activity: A new peroxisomal disorder. J Inherit Metab Dis. 1991;14:165.
Bernheimer H., Budka H., Muller P. Brain tissue immunoglobulins in adrenoleukodystrophy: A comparison with multiple sclerosis and systemic lupus erythematosus. Acta Neuropathol (Berl). 1983;59:95.
Bernstein J., Brough A.J., McAdams A.J. The renal lesion in syndromes of multiple congenital malformations: Cerebrohepatorenal syndrome; Jeune asphyxiating thoracic dystrophy; tuberous sclerosis; Meckel syndrome. Birth Defects Orig Artic Ser. 1974;10(4):35-43.
Besley G.T.N., Broadhead D.M. Dihydroxyacetone phosphate acyltransferase deficiency in peroxisomal disorders. J Inherit Metab Dis. 1987;10:236.
Bezman L., Moser H.W. Invited editorial comment – incidence of X-linked adrenoleukodystrophy and the relative frequency of its phenotypes. Am J Med Genet. 1998;76:415.
Bjorkheim I., Blomstrand S., Haga P., et al. Urinary excretion of dicarboxylic acids from patients with the Zellweger syndrome. Biochim Biophys Acta. 1984;795:15.
Black V.H., Russo J.J. Stereological analysis of the guinea pig adrenal: Effect of dexamethasone and ACTH treatment with emphasis on the inner cortex. Am J Anat. 1980;159:85.
Boehm C.D., Cutting G.R., Lachtermacher M.B., et al. Accurate DNA-based diagnostic and carrier testing for X-linked adrenoleukodystrophy. Mol Genet Metab. 1999;66:128.
Boles D.J., Craft D.A., Padgett D.A., et al. Clinical variation in X-linked adrenoleukodystrophy – fatty acid and lipid metabolism in cultured fibroblasts. Biochem Med Metab Biol. 1991;45:74.
Boltshauser E., Spycher M.A., Steinman B., et al. Infantile phytanic acid storage disease: A variant of Refsum’s disease? Eur J Pediatr. 1982;39:317.
Bourre J.M., Daudu O., Baumann N. Nervonic acid biosynthesis by erucyl-CoA elongation in normal and quaking mouse brain microsomes. Elongation of other unsaturated fatty acyl-CoAs (mono and polyunsaturated). Biochim Biophys Acta. 1976;424:1.
Bout A., Teunissen Y., Hashimoto T., et al. Nucleotide sequence of human peroxisomal 3-oxoacyl CoA thiolase. Nucleic Acids Res. 1988;16:10369.
Boutin B., Matsuguchi L., Lebon P., et al. Immunohistochemical analysis of brain macrophages in adrenoleukodystrophy. Neuropediatr. 1989;20:202.
Bowen P., Lee C.S., Zellweger H., et al. A familial syndrome of multiple congenital defects. Bull Johns Hopkins Hosp. 1964;114:402.
Braverman N., Chen L., Lin P., et al. Mutation analysis of PEX7 in 60 probands with rhizomelic chondrodysplasia punctata and functional correlations of genotype with phenotype. Hum Mutat. 2002;20:284.
Braverman N., Steel G., Lin P., et al. PEX7 gene structure, alternative transcripts, and evidence for a founder haplotype for the frequent RCDP allele, L292ter. Genomics. 2000;63:181.
Braverman N., Zhang R., Chen L., et al. A Pex7 hypomorphic mouse model for plasmalogen deficiency affecting the lens and skeleton. Mol Genet Metab. 2010;99:408-416.
Bresnan M.J., Richardson E.P.Jr. Case records of the Massachusetts General Hospital Case 18-1979. N Engl J Med. 1979;300:103.
Brites P., Waterham H.R., Wanders R.J. Functions and biosynthesis of plasmalogens in health and disease. Biochim Biophys Acta. 2004;1636:219.
Brown F.III, Chen W.W., Kirschner D.A., et al. Myelin membrane from adrenoleukodystrophy brain white matter – biochemical properties. J Neurochem. 1983;41:341.
Brown F.R.III, Van Duyn M.A., Moser A.B., et al. Adrenoleukodystrophy: Effects of dietary restriction of very long chain fatty acids and of administration of carnitine and clofibrate on clinical status and plasma fatty acids. Johns Hopkins Med J. 1982;151:164.
Budden S.S., Kennaway N.G., Buist N.R.M., et al. Dysmorphic syndrome with phytanic acid oxidase deficiency, abnormal very long chain fatty acids, and pipecolic acidemia: Studies in four children. J Pediatr. 1986;108:33.
Budka H., Molzer B., Bernheimer H., et al. Clinical, morphological and neurochemical findings in adrenoleukodystrophy and its variants. Neuropathology (Tokyo). 1983;1:209.
Budka H., Sluga E., Heiss W.D. Spastic paraplegia associated with Addison’s disease: Adult variant of adrenoleukodystrophy. J Neurol. 1976;213:237.
Cartier N., Hacein-Bey-Abina S., Bartholomae C.C., et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818.
Casteels M., Schepers L., Van Veldhoven P., et al. Separate peroxisomal oxidases for fatty acyl-CoAs and trihydroxycoprostanoyl-CoA in human liver. J Lipid Res. 1990;31:1865.
Challa V.R., Geisinger K.R., Burton B.K. Pathologic alterations in the brain and liver in hyperpipecolic acidemia. J Neuropathol Exp Neurol. 1983;42:627.
Chow C.W., Poulos A., Fellenberg A.J., et al. Autopsy findings in two siblings with infantile Refsum disease. Acta Neuropathol. 1992;83:190.
Christensen E., van Eldere J., Brandt N.J., et al. A new peroxisomal disorder: Di- and trihydroxycholestanoic acidemia due to presumed TCHA-CoA oxidase deficiency. J Inherit Metab Dis. 1990;13:363.
Clayton P.T., Eckhardt S., Wilson J. Isolated dihydroxyacetonephosphate acyltransferase deficiency presenting with developmental delay. J Inherit Metab Dis. 1994;17:315.
Clayton P.T., Lake B.D., Shortland D.B., et al. Plasma bile acids in patients with peroxisomal dysfunction syndromes: Analysis by capillary gas chromatography–mass spectrometry. Eur J Pediatr. 1987;146:166.
Cohen S.M.Z., Martyn L., Brown F.R.III, et al. Ocular histopathologic and biochemical studies of the cerebro-hepato-renal (Zellweger) syndrome and its relationship to neonatal adrenoleukodystrophy. Am J Ophthalmol. 1983;96:448.
Collins C.S., Gould S.J. Identification of a common PEX1 mutation in Zellweger syndrome. Hum Mutat. 1999;14:45.
Colombo C., Setchell K.D.R., Padda M., et al. Effects of ursodeoxycholic acid therapy for liver disease associated with cystic fibrosis. J Pediatr. 1990;117:482.
Cooper P.J., Danpure C.J., Wise P.J., et al. Immunocytochemical localization of human hepatic alanine glyoxylate aminotransferase in control subject and patients with primary hyperoxaluria type 1. J Histochem Cytochem. 1988;36:1285.
Corzo D., Gibson W., Johnson K., et al. Contiguous deletion of the X-linked adrenoleukodystrophy gene (ABCD1) and DXS1357E: A novel neonatal phenotype similar to peroxisomal biogenesis disorders. Am J Hum Genet. 2002;70:1520.
Crawford D.R., Mirault M.-E., Moret R., et al. Molecular defect in human acatalasia fibroblasts. Biochem Biophys Res Commun. 1988;153:59.
Croes K., Casteels M., Asselberghs S., et al. Formation of a 2-methyl-branched fatty aldehyde during peroxisomal alpha-oxidation. FEBS Lett. 1997;412:643.
Dammai V., Subramani S. The human peroxisomal targeting signal receptor, Pex5p, is translocated into the peroxisomal matrix and recycled to the cytosol. Cell. 2001;105:187.
Dancis J., Hutzler J. The significance of hyperpipecolatemia in Zellweger syndrome. Am J Hum Genet. 1986;38:707.
Danpure C.J. How can the products of a single gene be localized to more than one intracellular compartment? Trends Cell Biol. 1995;5:230.
Danpure C.J., Jennings P.R. Enzymatic heterogeneity in primary hyperoxaluria type 1 (hepatic peroxisomal alanine: glyoxylate aminotransferase deficiency). J Inherit Metab Dis. 1988;11:205.
Danpure C.J., Jennings P.R. Peroxisomal alanine: glyoxylate aminotransferase deficiency in primary hyperoxaluria type 1. FEBS Lett. 1986;201:20.
Danpure C.J., Jennings P.R., Penketh R.J., et al. Fetal liver alanine: glyoxylate aminotransferase and the prenatal diagnosis of primary hyperoxaluria type 1. Prenat Diagn. 1989;9:271.
Danpure C.J., Jennings P.R., Watts R.W.E. Enzymological diagnosis of primary hyperoxaluria type 1 by measurement of hepatic alanine glyoxylate aminotransferase activity. Lancet. 1987;1:289.
Datta N.S., Wilson G.N., Hajra A.K. Deficiency of enzymes catalyzing the biosynthesis of glycerol-ether lipids in Zellweger syndrome: A new category of metabolic disease involving the absence of peroxisomes. N Engl J Med. 1984;31:1080.
Datta S.C., Ghosh M.K., Hajra A.K. Purification and properties of acyl/alkyl dihydroxyacetone-phosphate reductase from guinea pig liver peroxisomes. J Biol Chem. 1990;265:8268.
de Duve C., Baudhuin P. Peroxisomes (microbodies and related particles). Physiol Rev. 1966;46:323.
Diczfalusy U., Alexson S.E.H. Peroxisomal chain-shortening of prostaglandin F(2). J Lipid Res. 1988;29:1629.
Dodt G., Kim D.G., Reimann S.A., et al. L-Pipecolic acid oxidase, a human enzyme essential for the degradation of L-pipecolic acid, is most similar to the monomeric sarcosine oxidases. Biochem J. 2000;345:487.
Dooley J.M., Wright B.A. Adrenoleukodystrophy mimicking multiple sclerosis. Can J Neurol Sci. 1985;12:73.
Duda E.E., Huttenlocher P.R. Computed tomography in adrenoleukodystrophy: Correlation of radiological and histological findings. Radiology. 1976;120:349.
Duff P., Harlass F.E., Milligan D.A. Prenatal diagnosis of chondrodysplasia punctata by sonography. Obstet Gynecol. 1990;76:497.
Eiben R.M., DiChiro G. Computer assisted tomography in adrenoleukodystrophy. J Comput Assist Tomogr. 1977;1:308.
Eichler F.S., Ren J.Q., Cossoy M., et al. Is microglial apoptosis an early pathogenic change in cerebral X-linked adrenoleukodystrophy? Ann Neurol. 2008;63:729.
Ek J., Kase B.F., Reith A., et al. Peroxisomal dysfunction in a boy with neurologic symptoms and amaurosis (Leber disease): Clinical and biochemical findings similar to those observed in Zellweger syndrome. J Pediatr. 1986;18:19.
El-Deiry S.S., Naidu S., Blevins L.S., et al. Assessment of adrenal function in women heterozygotes for ALD. J Clin Endocrinol Metab. 1997;82:856.
Errico F., Napolitano F., Nistico R., et al. D-aspartate: an atypical amino acid with neuromodulatory activity in mammals. Rev Neurosci. 2009;20:429-440.
Esiri M.M., Hyman N.M., Horton W.L., et al. Adrenoleukodystrophy: Clinical, pathological and biochemical findings in two brothers with the onset of cerebral disease in adult life. Neuropathol Appl Neurobiol. 1984;10:429.
Evrard P., Caviness V.S., Prats-Vinas J., et al. The mechanism of arrest of neuronal migration in the Zellweger malformation: An hypothesis based upon cytoarchitectonic analysis. Acta Neuropathol (Berl). 1978;41:109.
Eyssen H., Eggermont E., Van Eldere J., et al. Bile acid abnormalities and the diagnosis of cerebro-hepato-renal syndrome (Zellweger syndrome). Acta Pediatr Scand. 1985;74:538.
Fanconi A., Von Prader A., Isler W., et al. Morbus Addison mit himsklerose im kindesalter – ein hereditares syndrom mit X-chromosomaler vererbung? Helv Paediatr Acta. 1963;18:480.
Faust P.L. Abnormal cerebellar histogenesis in PEX2 Zellweger mice reflects multiple neuronal defects induced by peroxisome deficiency. J Comp Neurol. 2003;461:394.
Faust P.L., Su H.M., Moser A., et al. The peroxisome deficient PEX2 Zellweger mouse: Pathologic and biochemical correlates of lipid dysfunction. J Mol Neurosci. 2001;16:289.
Ferdinandusse S., Denis S., Clayton P.T., et al. Mutations in the gene encoding peroxisomal alpha-methylacyl-CoA racemase cause adult-onset sensory motor neuropathy. Nat Genet. 2000;24:188.
Ferdinandusse S., Denis S., Dacremont G., et al. Studies on the metabolic fate of n-3 polyunsaturated fatty acids. J Lipid Res. 2003;44:1992.
Ferdinandusse S., Denis S., Mooijer P.A.W., et al. Identification of the peroxisomal beta-oxidation enzymes involved in the biosynthesis of docosahexaenoic acid. J Lipid Res. 2001;42:1987.
Ferdinandusse S., van Grunsven E.G., Oostheim W., et al. Reinvestigation of peroxisomal 3-ketoacyl-CoA thiolase deficiency: Identification of the true defect at the level of d-bifunctional protein. Am J Hum Genet. 2002;70:1589.
Fournier B., Saudubray J.M., Benichou B., et al. Large deletion of the peroxisomal acyl-CoA oxidase gene in pseudoneonatal adrenoleukodystrophy. J Clin Invest. 1994;94:526.
Fringes B., Reith A. A time course of peroxisome biogenesis during adaptation to mild hyperthyroidism in rat liver. Lab Invest. 1982;47:19.
Gaertner J., Chen W.W., Kelley R.I., et al. The 22-kD peroxisomal integral membrane protein in Zellweger syndrome – presence, abundance, and association with a peroxisomal thiolase precursor protein. Pediatr Res. 1991;29:141.
Gatfield P.D., Taller E., Hinton G.G., et al. Hyperpipecolatemia: A new metabolic disorder associated with neuropathy and hepatomegaly. Can Med Assoc J. 1968;99:1215.
Germain-Lee E., Morrell J.C., Geisbrecht B., et al. Defects in PEX1, a peroxisome-associated ATPase that interacts with PEX6, are the most common cause of Zellweger syndrome, neonatal adrenoleukodystrophy and infantile Refsum disease. Am J Hum Genet. 1997;61:A54.
Gilchrist K.W., Gilbert E.F., Goldfarb S., et al. Studies of malformation syndromes of Man X1B: The cerebro-hepato-renal syndrome of Zellweger: Comparative pathology. Eur J Pediatr. 1976;121:99.
Goldfischer S., Collins J., Rapin I., et al. Pseudo-Zellweger syndrome: Deficiencies in several peroxisomal oxidative activities. J Pediatr. 1986;108:25.
Goldfischer S., Moore C.L., Johnson A.B., et al. Peroxisomal and mitochondrial defects in the cerebro-hepato-renal syndrome. Science. 1973;182:62.
Goldfischer S., Powers J.M., Johnson A.B., et al. Striated adrenocortical cells in cerebro-hepato-renal (Zellweger) syndrome. Virchows Archiv A. 1983;401:355.
Gould S.J., Keller G.A., Subramani S. Identification of a peroxisomal targeting signal at the carboxy terminus of firefly luciferase. J Cell Biol. 1987;105:2923.
Gould S.J., Keller G.A., Subramani S. Identification of peroxisomal targeting signals located at the carboxy terminus of four peroxisomal proteins. J Cell Biol. 1988;107:897.
Gould S.J., Krisans S., Keller G.A., et al. Antibodies directed against the peroxisomal targeting signal of firefly luciferase recognize multiple mammalian peroxisomal proteins. J Cell Biol. 1990;110:27.
Govaerts L., Sippell W.G., Monnens L. Further analysis of the disturbed adrenocortical function in the cerebro-hepato-renal syndrome of Zellweger. J Inherit Metab Dis. 1989;12:423.
Greenberg C.R., Hajra A.K., Moser A.B. Triple therapy of a patient with a generalized peroxisomal disorder. Am J Hum Genet. 1987;41(Suppl):A64.
Gressens P., Baes M., Leroux P., et al. Neuronal migration disorder in Zellweger mice is secondary to glutamate receptor dysfunction. Ann Neurol. 2000;48:336.
Griffin D.E., Moser H.W., Mendoza Q., et al. Identification of the inflammatory cells in the nervous system of patients with adrenoleukodystrophy. Ann Neurol. 1985;18:660.
Griffin J.W., Goren E., Schaumburg H., et al. Adrenomyeloneuropathy: A probable variant of adrenoleukodystrophy. Neurology. 1977;27:1107.
Gustafsson J., Gustavson K.H., Karlaganis G., et al. Zellweger’s cerebro-hepato-renal syndrome – variations in expressivity and in defects of bile acid synthesis. Clin Genet. 1983;24:313.
Gutierrez C., Okita R., Krisans S. Demonstration of cytochrome reductase in rat liver peroxisomes: Biochemical and immunochemical analysis. J Lipid Res. 1988;29:613.
Hajra A.K., Bishop J.E. Glycolipid biosynthesis in peroxisomes via the acyl dihydroxyacetone phosphate pathway. Ann N Y Acad Sci. 1982;386:170.
Hanson R.F., Szczepanik-van Leeuwen P., Williams G.C., et al. Defects of bile acid synthesis in Zellweger’s syndrome. Science. 1979;203:1107.
Hashimoto T. Individual peroxisomal beta-oxidation enzymes. Ann N Y Acad Sci. 1982;386:5.
Hashimoto T., Kuwabara T., Usuda N., et al. Purification of membrane polypeptides of rat liver peroxisomes. J Biochem. 1986;100:301.
Heffungs W., Hameisier H., Ropers H.H. Addison disease and cerebral sclerosis in an apparently heterozygous girl: Evidence of inactivation of the adrenoleukodystrophy locus. Clin Genet. 1980;18:184.
Hess R., Staubli W., Riess W. Nature of the hepatomegalic effect produced by ethylchlorophenoxyisobutyrate in the rat. Nature. 1965;208:856.
Heymans H.S.A.. Cerebro-hepato-renal (Zellweger) syndrome. Clinical and biochemical consequences of peroxisomal dysfunction. Ph.D. dissertation. Amsterdam: University of Amsterdam; 1984.
Heymans H.S.A., Oorthuys J.W.E., Nelck G., et al. Rhizomelic chondrodysplasia punctata: Another peroxisomal disorder. N Engl J Med. 1985;313:187.
Heymans H.S.A., Oorthuys J.W.E., Nelck G., et al. Peroxisomal abnormalities in rhizomelic chondrodysplasia punctata. J Inherit Metab Dis. 1986;9:328.
Heymans H.S.A., van den Bosch H., Schutgens R.B.H., et al. Deficiency of plasmalogens in the cerebro-hepato-renal (Zellweger) syndrome. Eur J Pediatr. 1984;142:10.
Hiltunen J.K., Karki T., Hassinen I.E., et al. β-Oxidation of polyunsaturated fatty acids by rat liver peroxisomes. J Biol Chem. 1986;261:16484.
Hittner H.M., Dretzer F.L., Mehta R.S. Zellweger syndrome: Lenticular opacities indicating carrier status and lens abnormalities characteristic of homozygotes. Arch Ophthalmol. 1981;99:1977.
Hoefler G., Hoefler S., Watkins P., et al. Biochemical abnormalities in rhizomelic chondrodysplasia punctata. J Pediatr. 1988;112:726.
Holmes R.D., Wilson G.N., Hajra A. Oral ether lipid therapy in patients with peroxisomal disorders. J Inherit Metab Dis. 1987;10:239.
Hruban Z., Rechcigl M.Jr. Microbodies and related particles. New York: Academic Press; 1969.
Hubbard W.C., Moser A.B., Liu A.C., et al. Newborn screening for X-linked adrenoleukodystrophy (X-ALD): validation of a combined liquid chromatography-tandem mass spectrometric (LC-MS/MS) method. Mol Genet Metab. 2009;97(3):212-220.
Iancu T.C., Danpure C.J. Primary hyperoxaluria type 1: Ultrastructural observations in liver biopsies. J Inherit Metab Dis. 1969;10:330.
Igarashi M., Belchis D., Suzuki K. Brain gangliosides in adrenoleukodystrophy. J Neurochem. 1976;27:327.
Igual J.C., Gonzalezbosch C., Dopazo J., et al. Phylogenetic analysis of the thiolase family – implications for the evolutionary origin of peroxisomes. J Mol Evol. 1992;35:147.
Imanaka T., Lazarow P., Takano T. A novel 57 kDa peroxisomal membrane polypeptide detected by monoclonal antibody (PXCM1a/207B). Biochim Biophys Acta. 1991;1062:264.
Issemann I., Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferates. Nature. 1990;347:645.
James A.C.D., Kaplan P., Lees A., et al. Schizophreniform psychosis and adrenomyeloneuropathy. J R Soc Med. 1984;77:882.
Jansen G.A., Ofman R., Ferdinandusse S., et al. Refsum disease is caused by mutations in the phytanoyl-CoA hydroxylase gene. Nat Genet. 1997;17:190.
Janssen A., Baes M., Gressens P., et al. Docosahexaenoic acid deficit is not a major pathogenic factor in peroxisome-deficient mice. Lab Invest. 2000;80:31.
Janssen A., Gressens P., Grabenbauer M., et al. Neuronal migration depends on intact peroxisomal function in brain and in extraneuronal tissues. J Neurosci. 2003;23:9732.
Jones J.M., Morrell J.C., Gould S.J. Multiple distinct targeting signals in integral peroxisomal membrane proteins. J Cell Biol. 2001;153:1141.
Junien C., Turleau C., de Grouchy J., et al. Regional assignment of catalase (CAT) gene to band 11p13: Association with the aniridia–Wilms’ tumor–gonadoblastoma (WAGR) complex. Ann Genet. 1980;23:165.
Kallijarvi J., Avela K., Lipsanen-Nyman M., et al. The TRIM37 gene encodes a peroxisomal RING-B-box-coiled-coil protein: Classification of mulibrey nanism as a new peroxisomal disorder. Am J Hum Genet. 2002;70:1215.
Kamijo K., Taketani S., Yokota S., et al. The 70-kDa peroxisomal membrane protein is a member of the Mdr (P-glycoprotein)-related ATP-binding protein superfamily. J Biol Chem. 1990;265:4534.
Kaplan P.W., Tusa R., Shankroff J., et al. Somatosensory-evoked potentials in adrenomyeloneuropathy patients on Lorenzo oil. Ann Neurol. 1995;38:351.
Kase B.F., Bjorkheim I. Peroxisomal bile acid-CoA: Amino acid N-acyltransferase in rat liver. J Biol Chem. 1989;264:9220.
Kase B.F., Prydz K., Bjorkheim I., et al. In vitro formation of bile acids from di- and trihydroxy-58-cholestanoic acid in human liver peroxisomes. Biochim Biophys Acta. 1986;877:37.
Kaufmann W.E., Theda C., Naidu S., et al. Neuronal migration abnormality in peroxisomal bifunctional enzyme defect. Ann Neurol. 1996;39:268.
Kawamura N., Moser A.B., Moser H.W., et al. High concentration of hexacosanoate in cultured skin fibroblast lipids from adrenoleukodystrophy patients. Biochem Biophys Res Commun. 1978;82:114.
Keller G.A., Krisans S., Gould S.J., et al. Evolutionary conservation of a microbody targeting signal that targets proteins to peroxisomes, glyoxysomes, and glycosomes. J Cell Biol. 1991;114:893.
Kelley R.I., Datta N.S., Dobyns W.B., et al. Neonatal adrenoleukodystrophy: New cases, biochemical studies, and differentiation from Zellweger and related peroxisomal polydystrophy syndromes. Am J Med Genet. 1986;23:869.
Kelley R.I., Moser H.W. Hyperpipecolic acidemia in neonatal adrenoleukodystrophy. Am J Med Genet. 1984;19:791.
Kemp S., Pujol A., Waterham H.R., et al. ABCD1 mutations and the X-linked adrenoleukodystrophy mutation database: Role in diagnosis and clinical correlations. Hum Mutat. 2001;18:499.
Kishimoto Y., Moser H.W., Kawamura N., et al. Evidence that abnormal very long chain fatty acids of brain cholesterol esters are of exogenous origin. Biochem Biophys Res Commun. 1980;96:69.
Kitchin W., Cohen-Cole S.A., Mickel S.F. Adrenoleukodystrophy: Frequency of presentation as a psychiatric disorder. Biol Psychiatry. 1987;22:1375.
Knazek R.A., Rizzo W.B., Schulman J.D., et al. Membrane microviscosity is increased in the erythrocytes of patients with adrenoleukodystrophy and adrenomyeloneuropathy. J Clin Invest. 1983;72:245.
Kok F., Neumann S., Sarde C.O., et al. Mutational analysis of patients with X-linked ALD. Hum Mutat. 1995;6:104.
Kolvraa S., Gregersen N. In vitro studies on the oxidation of medium chain dicarboxylic acids in rat liver. Biochim Biophys Acta. 1983;876:515.
Kumar A.J., Kohler W., Kruse B., et al. MR findings in adult-onset ALD. AJNR. 1995;16:1227.
Kumar A.J., Rosenbaum A.E., Naidu S., et al. Adrenoleukodystrophy: Correlating MR imaging with CT. Radiology. 1987;165:496.
Kyllerman M., Blomstrand S., Mansson J.E., et al. Central nervous system malformations and white matter changes in pseudo-neonatal adrenoleukodystrophy. Neuropediatrics. 1990;21:199.
Lalwani N.D., Alvarez K., Reddy M.K., et al. Peroxisome proliferator binding protein: Identification and partial characterization of nafenopin, clofibric acid and ciprofibrate-binding protein from rat liver. Proc Natl Acad Sci USA. 1987;84:5342.
Lanyon-Hogg T., Warriner S.L., Baker A. Getting a camel through the eye of a needle: the import of folded proteins by peroxisomes. Biol Cell. 2010;102:245-263.
Lapunzina P., Rodriguez J.I., Dematteo E., et al. Mulibrey nanism – 3 additional patients and a review of 39 patients. Am J Med Genet. 1995;55:349.
Latta K., Brodhel J. Primary hyperoxaluria Type 1. Eur J Pediatr. 1990;149:518.
Lazarow P.B, DeDuve C. A fatty acyl-CoA oxidising system in rat liver peroxisomes; enhancement by clofibrate, a hypolipidemic drug. Proc Natl Acad Sci USA June. 1976;73(6):2043-2046.
Lazarow P.B. Peroxisome structure, function, and biogenesis – human patients and yeast mutants show strikingly similar defects in peroxisome biogenesis. J Neuropathol Exp Neurol. 1995;54:720.
Lazarow P.B. Rat liver peroxisomes catalyze the β-oxidation of fatty acids. J Biol Chem. 1978;253:1522.
Lazarow P.B., Black V., Shio H., et al. Zellweger syndrome: Biochemical and morphological studies on two patients treated with clofibrate. Pediatr Res. 1985;19:1356.
Lazarow P.B., Fujiki Y. Biogenesis of peroxisomes. Ann Rev Cell Biol. 1985;1:489.
Lockman L.A., Shapiro E.G., Krivit W. Studies of 8 ALD patients at least 1 year after BMT. Ann Neurol. 1993;34:447.
Loes D.J., Fatemi A., Melhem E.R., et al. Analysis of MRI patterns aids prediction of progression in X-linked adrenoleukodystrophy. Neurology. 2003;61:369.
Loes D.J., Stillman A.E., Hite S., et al. Childhood cerebral form of adrenoleukodystrophy: Short term effect of bone marrow transplantation on brain MR observations. Am J Neuroradiol. 1994;15:1767.
Lu J.F., Lawler A.M., et al. A mouse model for X-linked adrenoleukodystrophy. Proc Natl Acad Sci USA Aug 19. 1997;94(7):9366-9367.
Ma C., Subramani S. Peroxisome matrix and membrane protein biogenesis. IUBMB Life. 2009;61(7):713-722.
MacCollin M., DeVivo D.C., Moser A.B., et al. Ataxia and peripheral neuropathy: A benign variant of peroxisome dysgenesis. Ann Neurol. 1991;28:833.
Mahmood Paker A., Sunness J., Brereton N.H., et al. Docosahexaenoic acid therapy in peroxisomal diseases: a double blind randomized trial. Neurology. 2010. (in press)
Mannaerts G.P., van Veldhoven P., van Broekhoven A., et al. Evidence that peroxisomal acyl-CoA synthetase is located at the cytoplasmic side of the peroxisomal membrane. Biochem J. 1982;204:17.
Martinez M., Mougan I., Roig M., et al. Blood polyunsaturated fatty acids in patients with peroxisomal disorders. A multicenter study. Lipids. 1994;29:273.
Martinez M., Pineda M., Vidal R., et al. Docosahexaenoic acid: A new therapeutic approach to peroxisomal patients. Experience with two cases. Neurology. 1993;43:1389.
Mathis R.K., Watkins J.B., Szczepanik-Van Leeuween P., et al. Liver in the cerebro-hepato-renal syndrome: Defective bile acid synthesis and abnormal mitochondria. Gastroenterology. 1980;79:1311.
McDonald J.C., Landrenaeau M.D., Rohr M.S., et al. Reversal by liver transplantation of the complications of primary hyperoxaluria as well as the metabolic defect. N Engl J Med. 1989;321:1100.
McGuinness M.C., Griffin D.E., Raymond G.V., et al. Tumor-necrosis-factor-alpha and X-linked adrenoleukodystrophy. J Neuroimmunol. 1995;61:161.
Menkes J.H., Corbo L.M. Adrenoleukodystrophy: Accumulation of cholesterol esters with very long chain fatty acids. Neurology. 1977;27:928.
Migeon B.R., Moser H.W., Moser A.B., et al. Adrenoleukodystrophy: Evidence for X-linkage, inactivation and selection favoring the mutant allele in heterozygous cells. Proc Natl Acad Sci USA. 1981;78:5066.
Mihalik S.J., Morrell J.C., Kim D., et al. Identification of PAHX, a Refsum disease gene. Nat Genet. 1997;17:185.
Mihalik S.J., Moser H.W., Watkins P.A., et al. Peroxisomal L-pipecolic acid oxidation is deficient in liver from Zellweger syndrome patients. Pediatr Res. 1989;25:548.
Mihalik S., Rhead W.J. L-Pipecolic acid oxidation in the rabbit and cynomolgus monkey. J Biol Chem. 1989;264:2509.
Miyazawa S., Hashimoto T., Yokota S. Identity of long-chain acyl-coenzyme A synthetase of microsomes, mitochondria, and peroxisomes in rat liver. J Biochem. 1985;98:723.
Miyazawa S., Osumi T., Hashimoto T., et al. Peroxisome targeting signal of rat liver acyl-coenzyme A oxidase resides at the carboxy terminus. Mol Cell Biol. 1989;9:83.
Miyazawa W., Hayashi H., Hijikata M., et al. Complete nucleotide sequence of cDNA and predicted amino acid sequence of rat acyl-CoA oxidase. J Biol Chem. 1987;262:8131.
Molzer B., Bernheimer H., Budka H., et al. Accumulation of very long chain fatty acids is common to 3 variants of adrenoleukodystrophy. J Neurol Sci. 1981;51:301.
Monnens L., Bakkeren J., Parmentier G., et al. Disturbances in bile acid metabolism of infants with the Zellweger (cerebro-hepato-renal) syndrome. Eur J Pediatr. 1980;133:31.
Morariu M.A., Chasan J.L., Norum R.A., et al. Adrenoleukodystrophy variant in a heterozygous female. Neurology. 1982;32:A81.
Morgan S.H., Watts R.W.E. Perspectives in the assessment and management of patients with primary hyperoxaluria type 1. Adv Nephrol. 1989;18:95.
Moser A.B., Jones D.S., Raymond G.V., et al. Plasma and red blood cell fatty acids in peroxisomal disorders. Neurochem Res. 1999;24:187.
Moser A.B., Rasmussen M., Naidu S., et al. Phenotype of patients with peroxisomal disorders subdivided into sixteen complementation groups. J Pediatr. 1995;127:13.
Moser A.B., Singh I., Brown F.R.III, et al. The cerebro-hepato-renal (Zellweger) syndrome: Increased levels and impaired degradation of very long chain fatty acids, and prenatal diagnosis. N Engl J Med. 1984;310:1141.
Moser H.W., Tutschka P.J., Brown F.R.III, et al. Bone marrow transplant in adrenoleukodystrophy. Neurology. 1984;34:1410.
Moser H.W., Aubourg P., Cornblath D., et al. Therapy for X-linked adrenoleukodystrophy. In: Desnick R.J., editor. Treatment of genetic diseases. New York: Churchill Livingstone, 1991.
Moser H.W., Bergin A., Naidu S., et al. Adrenoleukodystrophy: New aspects of adrenal cortical disease. In: Nelson D., editor. Endocrinology and metabolism clinics of North America. Philadelphia: WB Saunders, 1991.
Moser H.W., Moser A.B., Frayer K.K., et al. Adrenoleukodystrophy: Increased plasma content of saturated very long chain fatty acids. Neurology. 1981;31:1241.
Moser H.W., Moser A.B., Kawamura N., et al. Adrenoleukodystrophy: Studies of the phenotype, genetics and biochemistry. Johns Hopkins Med J. 1980;147:217.
Moser H.W., Moser A.B., Trojak J.E., et al. The identification of female carriers for adrenoleukodystrophy. J Pediatr. 1983;103:54.
Moser H.W., Powers J.M., Smith K.D. Adrenoleukodystrophy: Molecular genetics, pathology, and Lorenzo’s oil. Brain Pathol. 1995;5:259-266.
Moser H.W., Raymond G.V. Genetic peroxisomal disorders: Why, when, and how to test. Ann Neurol. 1998;44:713.
Mosser J., Douar A.M., Sarde C.O., et al. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with Abc transporters. Nature. 1993;361:726.
Nagase T., Shimozawa N., Takemoto Y., et al. Peroxisomal localization in the developing mouse cerebellum: Implications for neuronal abnormalities related to deficiencies in peroxisomes. Biochim Biophys Acta. 2004;1671:26.
Naidu S., Bresnan M.J., Griffin D., et al. Childhood adrenoleukodystrophy: Failure of intensive immunosuppression to arrest neurologic progression in childhood adrenoleukodystrophy. Arch Neurol. 1988;45:846.
Naidu S., Hoefler G., Hoefler S., et al. Neonatal seizures and retardation in a female with biochemical changes resembling X-linked ALD: A probable new peroxisomal disease entity. Neurology. 1988;38:1100.
Naidu S., Cornblath D., Moser A., et al. Neurological abnormalities in ALD heterozygotes. Muscle Nerve. 1986;9:129.
Naidu S., Moser H.W. Peroxisomal disorders. Neurol Clin. 1990;8:507.
Naidu S., Washington C., Thirumalai S., et al. X-chromosome inactivation in symptomatic heterozygotes of X-linked adrenoleukodystrophy. Ann Neurol. 1997;42:498.
Neat C.E., Thomassen M.S., Osmundsen H. Induction of peroxisomal beta-oxidation in rat liver by high-fat diets. Biochem J. 1980;186:369.
Nishiyama K., Funai T., Katakfuchi R., et al. Primary hyperoxaluria type 1 due to a point mutation of T to C in the coding region of the serine:pyruvate aminotransferase gene. Biochem Biophys Res Commun. 1991;176:1093.
Noetzel M.J., Clark H.B., Moser H.W. Neonatal adrenoleukodystrophy with prolonged survival. Ann Neurol. 1983;14:380.
Noetzel M.J., Landau W.M., Moser H.W. Adrenoleukodystrophy carrier state presenting as a chronic nonprogressive spinal cord disorder. Arch Neurol. 1987;44:566.
Noguchi T., Okuno E., Takada Y., et al. Characteristics of hepatic alanine-glyoxylate aminotransferase in different mammalian species. Biochem J. 1978;169:113.
Noguchi T., Takada Y. Peroxisomal localization of alanine-glyoxylate aminotransferase in human liver. Arch Biochem Biophys. 1979;196:645.
Norton W., Autilio L.A. The lipid composition of purified bovine brain myelin. J Neurochem. 1966;13:213.
O’Neill B.P., Moser H.W., Saxena K.M., et al. Adrenoleukodystrophy (ALD): Neurological disease in carriers and correlation with very long chain fatty acid (VLCFA) concentrations in plasma and cultured skin fibroblasts. Neurology. 1982;32:A216.
O’Neill B.P., Moser H.W., Saxena K.M., et al. Adrenoleukodystrophy: Clinical and biochemical manifestations in carriers. Neurology. 1984;34:789.
Ogino T., Suzuki K. Specificities of human and rat brain enzymes of cholesterol ester metabolism toward very long chain fatty acids: Implications for biochemical pathogenesis of adrenoleukodystrophy. J Neurochem. 1981;36:776.
Opitz J.M. The Zellweger syndrome: Book review and bibliography. Am J Med Genet. 1985;22:419.
Partin J., McAdams A.J. Absence of hepatic peroxisomes in neonatal onset adrenoleukodystrophy. Pediatr Res. 1983;17:294a.
Pedersen J.I., Gustafsson J. Conversion of 3 alpha, 7 alpha, 12 alpha-trihydroxy-5 beta-cholestanoic acid into cholic acid by rat liver peroxisomes. FEBS Lett. 1980;121:345.
Penman R.W.B. Addison’s disease in association with spastic paraplegia. BMJ. 1960;1:402.
Peters C., Charnas L.R., Tan Y., et al. Cerebral X-linked adrenoleukodystrophy: The international hematopoietic cell transplantation experience from 1982 to 1999. Blood. 2004;104:881.
Pilz P., Schiener P. Kombination von morbus Addison und morbus Schilder bei einer 43 Jahrigen Frau. Acta Neuropathol. 1973;26:357.
Poll-The B.T., Maroteaux P., Narcy C., et al. A new type of chondrodysplasia punctata associated with peroxisomal dysfunction. J Inherit Metab Dis. 1991;14:361.
Poll-The B.T., Ogier H., Saudubray J.M., et al. Impaired plasmalogen metabolism in infantile Refsum’s disease. Eur J Pediatr. 1986;144:513.
Poll-The B.T., Saudubray J.M., Ogier H., et al. Infantile Refsum’s disease: Biochemical findings suggesting multiple peroxisomal dysfunction. J Inherit Metab Dis. 1986;9:169.
Poll-The B.T., Roels F., Ogier H., et al. A new peroxisomal disorder with enlarged peroxisomes and a specific deficiency of acyl-CoA oxidase (pseudo-neonatal adrenoleukodystrophy). Am J Hum Genet. 1988;42:422.
Poll-The B.T., Saudubray J.M., Ogier H., et al. Infantile Refsum’s disease: An inherited peroxisomal disorder. Comparison with Zellweger syndrome and neonatal adrenoleukodystrophy. Eur J Pediatr. 1987;146:477.
Poulos A., Robertson E.F., Johnson D.W., et al. Use of stable-isotope-labeled tracer to monitor ether lipid dietary supplementation in peroxisomal disease patient. In: Stable isotopes in paediatric nutritional and metabolic research. Hampshire, UK: Intercept, Ltd; 1990.
Poulos A., Sharp P., Whiting M. Infantile Refsum’s disease (phytanic acid storage disease): A variant of Zellweger’s syndrome? Clin Genet. 1984;26:579.
Poulos A., Singh H., Paton B., et al. Accumulation and defective β-oxidation of very long chain fatty acids in Zellweger’s syndrome, adrenoleukodystrophy and Refsum’s disease variants. Clin Genet. 1986;29:397.
Poulos A., Whiting M.J. Identification of 3 alpha, 7 alpha, 12 alpha-trihydroxy-5 beta-cholestan-26-oic acid, an intermediate in cholic acid synthesis, in the plasma of patients with infantile Refsum’s disease. J Inherit Metab Dis. 1985;8:13.
Powers J.M., Moser H.W. Peroxisomal disorders: Genotype, phenotype, major neuropathologic lesions, and pathogenesis. Brain Pathol. 1998;87:101.
Powers J.M., Schaumburg H.H., Johnson A.B., et al. A correlative study of the adrenal cortex in adrenoleukodystrophy: Evidence for a fatal intoxication with very long chain fatty acids. Invest Cell Pathol. 1980;3:353.
Probst A., Ulrich J., Heitz P.U., et al. Adrenomyeloneuropathy: A protracted, pseudosystematic variant of adrenoleukodystrophy. Acta Neuropathol. 1980;49:105.
Purdue P.E., Takada T., Danpure C.J. Identification of mutations associated with peroxisome-to-mitochondrion mistargeting of alanine/glyoxylate aminotransferase in primary hyperoxaluria Type 1. J Cell Biol. 1990;111:2341.
Quan F., Korneluk R.G., Tropak M.B., et al. Isolation and characterization of the human catalase gene. Nucleic Acids Res. 1986;14:531.
Ramsey R.B., Banik N.L., Davison A.N. Adrenoleukodystrophy brain cholesteryl esters and other neutral lipids. J Neurol Sci. 1979;40:189.
Raymond G.V. Peroxisomal disorders. Curr Opin Neurol. 2001;14:783.
Reddy J.K., Goel S.K., Nemali M.R., et al. Transcriptional regulation of peroxisomal fatty acyl-CoA oxidase and enoyl-CoA hydratase/3 hydroxyacyl-CoA dehydrogenase in rat liver by peroxisome proliferators. Proc Natl Acad Sci USA. 1986;83:1747.
Refsum S. Heredopathia atactica polyneuritiformis. A familial syndrome not hitherto described. Acta Psychiatr Scand. 1946;38(Suppl):1.
Reinecke C.J., Knoll D.P., Pretorius P.J., et al. The correlation between biochemical and histopathological findings in adrenoleukodystrophy. J Neurol Sci. 1985;70:21.
Reuber B.E., Germain-Lee E., Morrell J.C., et al. Defects in PEX1, a peroxisome-associated ATPase that interacts with PEX5 and PEX6, disrupt peroxisomal matrix protein import and are the most common cause of the peroxisome biogenesis disorders. Mol Biol Cell. 1997;8:668.
Rhodin J.. Correlation of ultrastructural organization and function in normal and experimentally changed proximal convoluted tubule cells of the mouse kidney. Ph.D. dissertation. Stockholm: Actiebolaget Godvil; 1954.
Rizzo W.B., Leshner R.T., Odone A., et al. Dietary erucic acid therapy for X-linked adrenoleukodystrophy. Neurology. 1989;39:1415.
Rizzo W.B., Phillips M.W., Dammann A.L., et al. Adrenoleukodystrophy: Dietary oleic acid lowers hexacosanoate levels. Ann Neurol. 1987;21:232.
Rizzo W.B., Watkins P.A., Phillips M.W., et al. Adrenoleukodystrophy: Oleic acid lowers fibroblast saturated C22-C26 fatty acids. Neurology. 1986;36:357.
Rocchiccioli F., Aubourg P., Bougneres P.F. Medium- and long-chain dicarboxylic aciduria in patients with Zellweger syndrome and neonatal adrenoleukodystrophy. Pediatr Res. 1986;20:62.
Roscher A., Molzer B., Bernheimer H., et al. The cerebrohepatorenal (Zellweger) syndrome: An improved method for the biochemical diagnosis and its potential value of prenatal detection. Pediatr Res. 1985;19:930.
Ruder H., Otto G., Schutgens R.B.H., et al. Excessive urinary oxalate excretion after combined renal and hepatic transplantation for correction of hyperoxaluria type 1. Eur J Pediatr. 1990;150:56.
Sadeghi-Nejad A., Senior B. Adrenomyeloneuropathy presenting as Addison’s disease in childhood. N Engl J Med. 1990;322:13.
Santos M.J., Imanaka T., Shio H., et al. Peroxisomal membrane ghosts in Zellweger syndrome – aberrant organelle assembly. Science. 1988;239:1536.
Schaumburg H., Powers J.M., Raine C.S., et al. Adrenomyeloneuropathy: A probable variant of adrenoleukodystrophy. II. General pathologic, neuropathologic, and biochemical aspects. Neurology. 1977;27:1114.
Schepers L., Casteels M., Vamecq J., et al. β-Oxidation of the carboxyl side chain of prostaglandin E(2) in rat liver peroxisomes and mitochondria. J Biol Chem. 1986;263:2724.
Schepers L., Van Veldhoven P.P., Casteels M., et al. Presence of three Acyl-CoA oxidases in rat liver peroxisomes. An inducible fatty acyl-CoA oxidase, a noninducible fatty acyl-CoA oxidase, and a noninducible trihydroxycoprostanoyl-CoA oxidase. J Biol Chem. 1990;265:5242.
Schram A., Strijland A., Hashimoto T., et al. Biosynthesis and maturation of peroxisomal β-oxidation enzymes in fibroblasts in relation to the Zellweger syndrome and infantile Refsum disease. Proc Natl Acad Sci USA. 1986;83:6156.
Schulz H., Kunau W.-H. Beta-oxidation of unsaturated fatty acids: A revised pathway. Trends Biochem Sci. 1987;12:403.
Schutgens R.B.H., Heymans H.S.A., Wanders R.J.A. Prenatal diagnosis of the cerebro-hepato-renal (Zellweger) syndrome by detection of an impaired plasmalogen biosynthesis. J Inherit Metab Dis. 1985;8:153.
Schutgens R.B.H., Heymans H.S.A., Wanders R.J.A., et al. Multiple peroxisomal enzyme deficiencies in rhizomelic chondrodysplasia punctata: Comparison with Zellweger syndrome, Conradi-Hunermann syndrome and the X-linked dominant type of chondrodysplasia punctata. Adv Clin Enzymol. 1988;6:57.
Scotto J.M., Hadchouel M., Odievre M. Infantile phytanic acid storage disease: A possible variant of Refsum’s disease: Three cases, including ultrastructural studies of the liver. J Inherit Metab Dis. 1982;5:83.
Sereni C., Ruel M., Iba-Zizen T., et al. Adult adrenoleukodystrophy: A sporadic case? J Neurol Sci. 1987;80:121.
Shimozawa N., Tsukamoto T., Suzuki Y., et al. A human gene responsible for Zellweger syndrome that affects peroxisome assembly. Science. 1992;255:1132.
Siemerling E., Creutzfeldt H.G. Bronzekrankheit und sklerosierende Encephalomyelitis. Arch Psychiatr Nervenkr. 1923;68:217.
Singh H., Usher S., Johnson D., et al. A comparative study of straight chain and branched chain fatty acid oxidation in skin fibroblasts from patients with peroxisomal disorders. J Lipid Res. 1990;31:217.
Singh I., Johnson G.H., Brown F.R.III. Peroxisomal disorders: Biochemical and clinical diagnostic considerations. Am J Dis Child. 1988;142:1297.
Singh I., Moser A.B., Goldfischer S., et al. Lignoceric acid is oxidized in the peroxisomes: Implications for the Zellweger cerebro-hepato-renal syndrome and adrenoleukodystrophy. Proc Natl Acad Sci USA. 1984;81:4203.
Small G.M., Santos M.J., Imanaka T., et al. Peroxisomal integral membrane proteins in livers of patients with Zellweger syndrome, infantile Refsum’s disease and X-linked adrenoleukodystrophy. J Inherit Metab Dis. 1988;11:358.
Smith K.D., Sack G., Beaty T., et al. A genetic basis for multiple phenotypes of X-linked adrenoleukodystrophy. Am J Hum Genet. 1991;49:864A.
Snyder F. Ether lipids. New York: Academic Press; 1972.
Solish G.I., Moser H.W., Ringer L.D., et al. The prenatal diagnosis of the cerebro-hepato-renal syndrome of Zellweger. Prenat Diagn. 1985;5:27.
Spranger J.W., Opitz J.M., Bidder U. Heterogeneity of chondrodysplasia punctata. Humangenetik. 1971;11:190.
Steinberg D. Refsum disease. In: Scriver C.R., Beaudet A.L., Sly W., et al, editors. The metabolic basis of inherited disease. New York: McGraw-Hill, 1989.
Steinberg S.J., Raymond G.V., Braverman N.E., et al. Peroxisome biogenesis disorders, Zellweger syndrome spectrum. Gene reviews at gene tests: Medical genetics information resource [Database online]. December 2003. Copyright University of Washington, Seattle, 1997-2004. Available at: http://www.genetests.org
Stumpf D.A., Hayward A., Haas R., et al. Adrenoleukodystrophy. Failure of immunosuppression to prevent neurological progression. Arch Neurol. 1981;38:48.
Suzuki Y., Orii T., Hashimoto T. Biosynthesis of peroxisomal β-oxidation enzymes in infants with Zellweger syndrome. J Inherit Metab Dis. 1986;9:292.
Suzuki Y., Shimozawa N., Orii T., et al. Major peroxisomal membrane polypeptides are synthesized in cultured skin fibroblasts from patients with Zellweger syndrome. Pediatr Res. 1989;26:150.
Swinkels B.W., Gould S.J., Bodnar A.G., et al. A novel, cleavable peroxisomal targeting signal at the amino-terminus of the rat 3-ketoacyl-CoA thiolase. EMBO J. 1991;10:3255.
Tager J.M., Harmsen T., van der Beek W.A., et al. Peroxisomal beta-oxidation enzyme proteins in the Zellweger syndrome. Biochem Biophys Res Commun. 1985;126:1269.
Takahara S., Migamoto H. Three cases of progressive oral gangrene due to lack of catalase. J Otorhinolaryngol Soc. 1948;51:165.
Taketomi T., Hara A., Kitazawa N., et al. An adult case of adrenoleukodystrophy with features of olivo-ponto-cerebellar atrophy: 2. Lipid abnormalities. Jpn J Exp Med. 1987;57:59.
Tanaka K., Nishizawa K., Yamamoto H., et al. Analysis of very long chain fatty acids and plasmalogen in the erythrocyte membrane—a simple method for the detection of peroxisomal disorders and discrimination between adrenoleukodystrophy and Zellweger syndrome. Neuropediatrics. 1990;21:119.
Tanaka K., Shimada M., Naruto T., et al. Very long-chain fatty acids in erythrocyte membrane sphingomyelin: Detection of ALD hemizygotes and heterozygotes. Neurology. 1986;36:791.
Theda C., Moser A., Moser H.W., et al. Temporal evolution of brain biochemical changes in adrenoleukodystrophy. J Neurochem. 1987;48:S35.
Theil A.C., Schutgens R.B.H., Wanders R.J.A., et al. Clinical recognition of patients affected by a peroxisomal disorder: A retrospective study in 40 patients. Eur J Pediatr. 1992;151:117.
Tiffany C.W., Hoefler S., Moser H.W., et al. Arachidonic acid metabolism in fibroblasts from patients with peroxisomal diseases. Biochim Biophys Acta. 1991;1096:41.
Tolbert N.E. Metabolic pathways in peroxisomes and glyoxysomes. Annu Rev Biochem. 1981;50:133.
Torvik A., Torp S., Kase B.F., et al. Infantile Refsum’s disease: A generalized peroxisomal disorder. Case report with postmortem examination. J Neurol Sci. 1988;85:39.
Trijbels J.M.F., Berden J.A., Monnens L.A.H., et al. Biochemical studies in the liver and muscle of patients with Zellweger syndrome. Pediatr Res. 1983;17:514.
Tsuji S., Suzuki M., Ariga T., et al. Abnormality of long-chain fatty acids in erythrocyte membrane sphingomyelin from patients with adrenoleukodystrophy. J Neurochem. 1981;36:1046.
Turpin J.C., Paturneau-Jouas M., Sereni C., et al. Revelation a l’age adulte d’un cas d’adrenoleucodystrophie familiale. Rev Neurol (Paris). 1985;141:289.
Ulrich J., Hershkowitz N., Heits P., et al. Adrenoleukodystrophy: Preliminary report of a connatal case, light- and electron microscopical, immunohistochemical and biochemical findings. Acta Neuropathol (Berl). 1978;43:77.
Uziel G., Bertini E., Rimoldi M., et al. Italian multicentric dietary therapeutical trial in adrenoleukodystrophy. Adrenoleukodystrophy and other peroxisomal disorders: Clinical, biochemical, genetic and therapeutic aspects. Excerpta Medica International Congress Series. New York: Elsevier Science; 1990.
Vallat C., Denis S., Bellet H., et al. Major hyperpipecolataemia in a normal adult. J Inherit Metab Dis. 1996;19:624.
Valle D., Gartner J. Penetrating the peroxisome. Nature. 1993;361:682.
van den Brink D.M., Brites P., Haasjes J., et al. Identification of PEX7 as the second gene involved in Refsum disease. Adv Exp Med Biol. 2003;544:69.
Van Duyn M.A., Moser A.B., Brown F.R.III, et al. The design of a diet restricted in saturated very long chain fatty acids: Therapeutic application in adrenoleukodystrophy. Am J Clin Nutr. 1984;40:277.
van Geel B.M., Bezman L., Loes D.J., et al. Evolution of phenotypes in adult male patients with X-linked adrenoleukodystrophy. Ann Neurol. 2001;49:186.
van Grunsven E.G., van Berkel E., Mooijer P.A., et al. Peroxisomal bifunctional protein deficiency revisited: Resolution of its true enzymatic and molecular basis. Am J Hum Genet. 1999;64:99.
Volpe J.J., Adams R.D. Cerebro-hepato-renal syndrome of Zellweger: An inherited disorder of neuronal migration. Acta Neuropathol. 1972;20:175.
Walton P.A., Hill P.E., Subramani S. Import of stably folded proteins into peroxisomes. Mol Biol Cell. 1995;6:675.
Wanders R.J.A., Boltshauser E., Steinmann B., et al. Infantile phytanic acid storage disease, a disorder of peroxisome biogenesis: A case report. J Neurol Sci. 1990;98:1.
Wanders R.J.A., van Roermund C.W.T., Schelen A., et al. A bifunctional protein with deficient enzyme activity: Identification of a new peroxisomal disorder using novel methods to measure the peroxisomal beta-oxidation enzyme activities. J Inherit Metab Dis. 1990;13:375.
Wanders R.J.A., Decker C., Horvath V.A.P., et al. Human alkyldihydroxyacetone phosphate synthase deficiency: A new peroxisomal disorder. J Inherit Metab Dis. 1994;17:315.
Wanders R.J.A., Purvis Y.R., Heymans H.S.A., et al. Age-related differences in plasmalogen content of erythrocytes from patients with the cerebro-hepato-renal (Zellweger) syndrome: Implications for postnatal detection of the disease. J Inherit Metab Dis. 1986;9:335.
Wanders R.J.A., Schutgens R.B.H., Schrakamp G., et al. Infantile Refsum disease: Deficiency of catalase-containing particles (peroxisomes), alkyldihydroxyacetone phosphate synthase and peroxisomal β-oxidation enzyme proteins. Eur J Pediatr. 1986;145:172.
Wanders R.J.A., Romeyn G.J., van Roermund C.W.T., et al. Identification of l-pipecolate oxidase in human liver and its deficiency in the Zellweger syndrome. Biochem Biophys Res Commun. 1988;154:33.
Wanders R.J.A., van Roermund C.W.T., Jurrians S., et al. Diversity in residual alanine glyoxylate aminotransferase activity in hyperoxaluria type 1: Correlation with pyridoxine responsiveness. J Inherit Metab Dis. 1988;11:208.
Wanders R.J.A., van Roermund C.W.T., van Wijland M.J.A., et al. Peroxisomes and peroxisomal functions in hyperpipecolic acidemia. J Inherit Metab Dis. 1988;11:161.
Wanders R.J.A., Schumacher H., Heikoop J., et al. Human dihydroxyacetone phosphate acyltransferase deficiency: A new peroxisomal disorder. J Inherit Metab Dis. 1992;15:389.
Wanders R.J.A., Schutgens R.B.H., Schrakamp G., et al. Neonatal adrenoleukodystrophy: Impaired plasmalogen biosynthesis and peroxisomal β-oxidation due to a deficiency of catalase-containing particles (peroxisomes) in cultured skin fibroblasts. J Neurol Sci. 1987;77:331.
Wanders R.J.A., van Weringh G., Schrakamp G., et al. Deficiency of acyl-CoA:dihydroxyacetone phosphate acyltransferase in thrombocytes of Zellweger patients: A simple postnatal diagnostic test. Clin Chim Acta. 1985;151:217.
Wanders R.J., Ferdinandusse S., Brites P., et al. Peroxisomes, lipid metabolism and lipotoxicity. Biochim Biophys Acta. 2010;1801(3):272-280.
Wanders R.J., Visser W.F., van Roermund C.W., et al. Transporter family. Pflugers Arch. 2007;453(5):719-734.
Watkins P.A., Chen W.W., Harris C.J., et al. Peroxisomal bifunctional enzyme deficiency. J Clin Invest. 1988;83:771.
Watkins P.A., Hamilton J.A., Leaf A., et al. Brain uptake and utilization of fatty acids – applications to peroxisomal biogenesis diseases. J Mol Neurosci. 2001;16:87.
Watkins P.A., McGuinness M.C., Raymond G.V., et al. Distinction between peroxisomal bifunctional enzyme and acyl-CoA oxidase deficiencies. Ann Neurol. 1995;38:472.
Watkins P.A., Mihalik S.J., Skjeldal O.H. Mitochondrial oxidation of phytanic acid in human and monkey liver – implication that Refsum’s disease is not a peroxisomal disorder. Biochem Biophys Res Commun. 1990;67:580.
Watts R.W., Calne R.Y., Rolles K., et al. Successful treatment of primary hyperoxaluria type 1 by combined hepatic and renal transplantation. Lancet. 1987;2:474.
Watts R.W., Chalmers R.A., Gibbs D.A., et al. Studies on some biochemical treatments of primary hyperoxaluria. Q J Med. 1979;48:259.
Webber K.O., Datta N.S., Hajra A.K. Properties of the enzymes catalyzing the biosynthesis of lysophosphatidate and its ether analog in cultured fibroblasts from Zellweger syndrome patients and normal controls. Arch Biochem Biophys. 1987;254:611.
Wei H., Kemp S., McGuinness M.C., et al. Pharmacological induction of peroxisomes in peroxisome biogenesis disorders. Ann Neurol. 2000;47:286.
Weinberg K., Moser A., Watkins P., et al. Bone marrow transplantation (BMT) for adrenoleukodystrophy (ALD). Pediatr Res. 1988;23:334A.
Weleber R.G., Tongue A.C., Kennaway N.G., et al. Ophthalmic manifestations of infantile phytanic acid storage disease. Arch Ophthalmol. 1984;102:1317.
Weller S., Gould S.J., Valle D. Peroxisome biogenesis disorders. Annu Rev Genomics Hum Genet. 2003;4:165.
Whitcomb R.W., Linehan W.R., Knazek R.A. Effects of long-chain, saturated fatty acids on membrane microviscosity and adrenocorticotropin responsiveness of human adrenocortical cells in vitro. J Clin Invest. 1988;81:185.
Wilson G.N., Holmes R.G., Custer J., et al. Zellweger syndrome: Diagnostic assays, syndrome delineation, and potential therapy. Am J Med Genet. 1986;24:69.
Wolvetang E.J., Tager J.M., Wanders R.J. Latency of the peroxisomal enzyme acyl-CoA:dihydroxyacetonephosphate acyltransferase in digitonin permeabilized fibroblasts: The effect of ATP and ATPase inhibitors. Biochem Biophys Res Commun. 1990;170:1135.