Peripheral nerve sheath neoplasms
 GENETICS OF NEUROFIBROMATOSIS TYPE 1 (NF1)
GENETICS OF NEUROFIBROMATOSIS TYPE 1 (NF1)



SCHWANNOMAS
Schwannomas are slowly growing neoplasms composed of Schwann cells. Solitary schwannomas with effects on the CNS occur on cranial (Table 42.1, Fig. 42.1) and spinal nerve roots; rarely they are found in the substance of the brain or spinal cord. Melanotic schwannomas have a predilection for spinal nerve roots. The plexiform schwannoma is rare and occurs in the skin. Unlike the plexiform neurofibroma, it is not associated with NF.
Table 42.1
The commonest neoplasm of the cerebellopontine angle is a schwannoma of the vestibulocochlear nerve
| Cerebellopontine angle neoplasm | % |
| Schwannoma | 85 |
| Meningioma | 10 |
| Others (cholesteatoma, glioma, medulloblastoma, metastatic carcinoma, paraganglioma, hemangioma, AT/RT) | 5 |
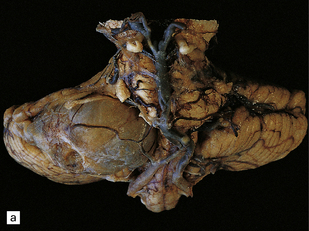
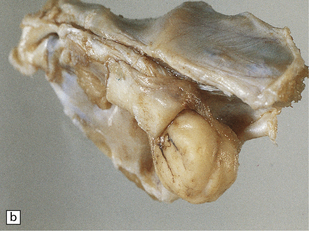
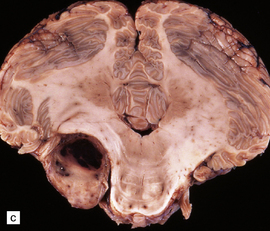
42.1 Vestibulocochlear nerve schwannomas.
(a) A partly cystic schwannoma in the cerebellopontine angle indents the brain stem. Flattened fascicles of vestibulocochlear nerve run over the surface of the neoplasm. (b) A schwannoma sits at the mouth of the internal auditory meatus. (c) A hemorrhage occupies part of this schwannoma, which has caused compression and distortion of the brain stem.
 NF1
NF1






Clinical criteria for diagnosing NF1
Two or more of the following clinical features must be present to fulfil current diagnostic criteria for NF1:
 At least two solitary neurofibromas or one plexiform neurofibroma.
At least two solitary neurofibromas or one plexiform neurofibroma.

 At least two Lisch nodules (iris hamartomas).
At least two Lisch nodules (iris hamartomas).
 Optic glioma (pilocytic astrocytoma).
Optic glioma (pilocytic astrocytoma).
 Osseous lesions (sphenoid dysplasia, pseudoarthrosis, or spinal deformity).
Osseous lesions (sphenoid dysplasia, pseudoarthrosis, or spinal deformity).

 A family history of a first-degree relative with NF1 fulfilling the above criteria.
A family history of a first-degree relative with NF1 fulfilling the above criteria.
MACROSCOPIC APPEARANCES
Schwannomas are composed of nodular rubbery tissue, which has a variegated cut surface. Yellow and gray areas may be interspersed with hemorrhagic foci or cysts. The neoplasm has a capsule and the nerve from which the schwannoma arises may be splayed over the surface of the neoplasm (Figs 42.2, 42.3).
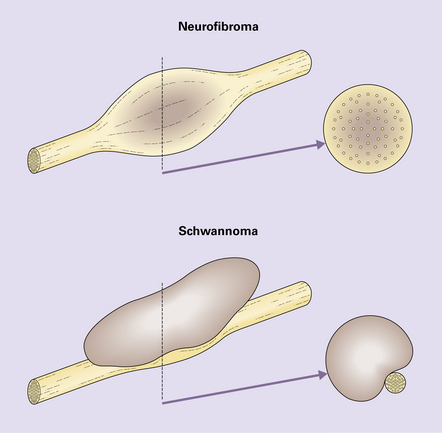
42.2 The relationship of neurofibroma and schwannoma to associated nerve.
The neurofibroma incorporates axons, but the schwannoma displaces normal elements of the nerve to one side (see Fig. 42.3).
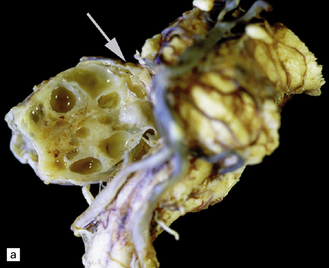
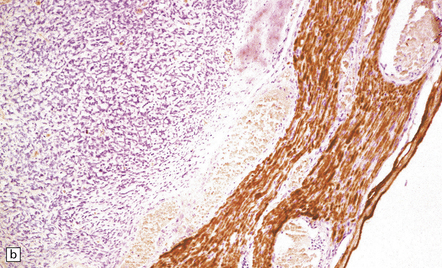
42.3 The relationship between schwannoma and peripheral nerve.
(a) A cystic schwannoma arising from the eighth nerve (arrow) distorts the brain stem. The tumor has been hemisected to reveal its multicystic architecture. (b) An antibody to neurofilament protein labels axons in a nerve alongside a schwannoma.
MICROSCOPIC APPEARANCES
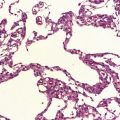
Two histologic patterns predominate (Figs 42.4–42.6):
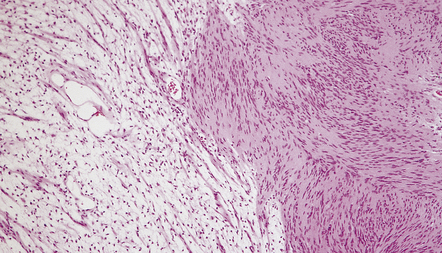
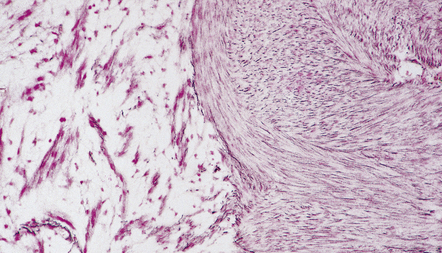
42.5 Schwannoma.
Abundant pericellular reticulin in an Antoni A area. Section adjacent to that in Figure 42.4.
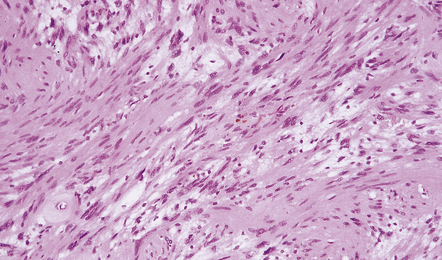
42.6 Schwannoma.
Fascicles of spindle-shaped cells are interrupted by small myxoid areas. Note the small blood vessel with a markedly thickened wall.
 Antoni A areas, in which spindle-shaped cells with rod-shaped nuclei and dense pericellular reticulin are arranged in compact intertwining fascicles.
Antoni A areas, in which spindle-shaped cells with rod-shaped nuclei and dense pericellular reticulin are arranged in compact intertwining fascicles.

 GENETIC ASPECTS OF NERVE SHEATH NEOPLASMS
GENETIC ASPECTS OF NERVE SHEATH NEOPLASMS






 GENETICS OF NEUROFIBROMATOSIS TYPE 2 (NF2)
GENETICS OF NEUROFIBROMATOSIS TYPE 2 (NF2)




 NF2
NF2








 CNS SCHWANNOMAS
CNS SCHWANNOMAS









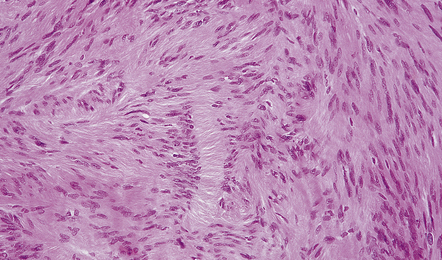
There may be conspicuous palisading of nuclei. Verocay bodies are a characteristic feature (Fig. 42.7). Thickened blood vessels with hyaline walls and clumps of hemosiderin-laden or foamy macrophages are common findings (Fig. 42.8). Mitoses, if present, are sparse.
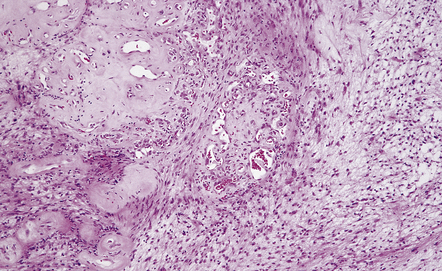
A small proportion of schwannomas contains melanin (Fig. 42.9), and melanotic schwannomas may contain epithelioid cells.
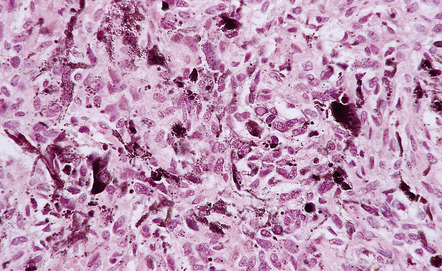
The presence of cells with misshapen hyperchromatic nuclei in many otherwise cytologically bland schwannomas is thought to be a manifestation of degenerative (or ‘ancient’) change (Fig. 42.10).
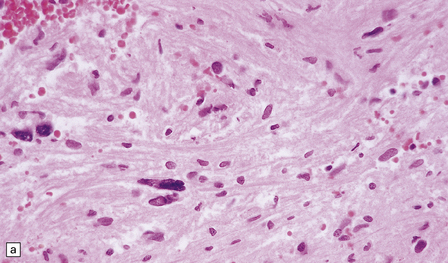
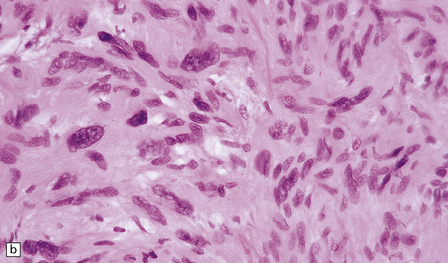
42.10 Nuclear hyperchromasia and pleomorphism in a schwannoma.
(a,b) These are common findings in schwannomas and, if there are no mitoses, are thought to be degenerative.
Cellular schwannomas rarely affect the CNS (Fig. 42.11). They show a marked preponderance of Antoni A areas that contain cells with a high nuclear:cytoplasmic ratio. Mitotic figures are evident. Xanthomatous foci may be seen. The cellular schwannoma can be distinguished from MPNSTs by its circumscribed shape and fibrous capsule, hyalinized blood vessels, and small but distinct myxoid areas.
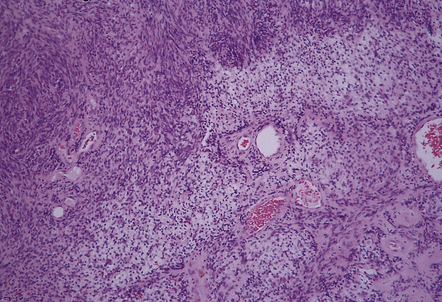
42.11 Cellular schwannoma.
A high nuclear:cytoplasmic ratio and scattered mitoses are present in some areas, but this neoplasm retains the architectural pattern of a schwannoma with Antoni A and B areas, and contains thick-walled blood vessels.
 CEREBELLOPONTINE ANGLE (CPA) NEOPLASMS
CEREBELLOPONTINE ANGLE (CPA) NEOPLASMS
Schwannomas make up the vast majority of CPA neoplasms (see Table 42.1), but two other CPA neoplasms (fibroblastic meningioma and astrocytoma of the brain stem or cerebellum) may be confused with schwannomas. Histologic features that distinguish these neoplasms include:



NEUROFIBROMAS


They rarely affect the CNS, and then usually in the context of NF (Fig. 42.12). Involvement of a plexus or large nerve trunks by plexiform neurofibromas is pathognomonic of NF1. In contrast, the rare plexiform schwannoma that occurs mainly in the skin is not associated with NF1. Neurofibromas grow slowly and arise within the endoneurium. They consist largely of Schwann cells, but fibroblasts and pericytes are also significant components.
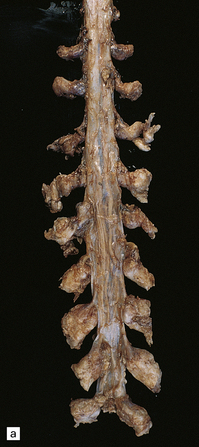
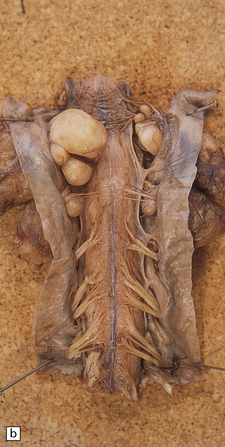
42.12 Neurofibromatosis type 1.
Multiple thoracolumbar (a) and cervical root (b) nerve sheath tumors in a patient with NF1.
MACROSCOPIC APPEARANCES
Solitary intraneural neurofibromas are usually oval, gray or tan-colored neoplasms with a smooth shiny surface covered by a delicate pseudocapsule. Neurofibromas are not cystic and do not contain the xanthomatous areas seen in schwannomas. Solitary neurofibromas incorporate and expand the nerve from which they arise, whereas schwannomas grow away from the edge of the nerve (see Fig. 42.2).
Plexiform neurofibromas expand several nerves in a plexus or fascicles in a nerve trunk (Fig. 42.13). Irregular swellings give the nerve trunks the appearance of a ginger root.
MICROSCOPIC APPEARANCES
Neurofibromas are circumscribed, but not encapsulated. Their wavy spindle-shaped cells lie in a mucoid matrix or between bundles of collagen (Fig. 42.14). Many cells have nuclei with a serpentine form.
42.14 Neurofibroma.
(a,b) This nerve sheath tumor is composed of haphazardly arranged cells with wavy nuclei and variable amounts of cytoplasm. Scattered lymphocytes are also present in this case.
Immunohistochemical and ultrastructural studies suggest that neurofibromas are composed of several types of cell: fibroblasts and pericytes are mixed with Schwann cells, and mast cells may be scattered through the neoplasm. Neoplastic cells infiltrate nerve fascicles rather than displacing them (Fig. 42.15).
42.15 Neurofibroma.
Axons in the infiltrated nerve are immunolabeled with an antibody to neurofilaments. The axons are widely separated by the neoplastic cells.
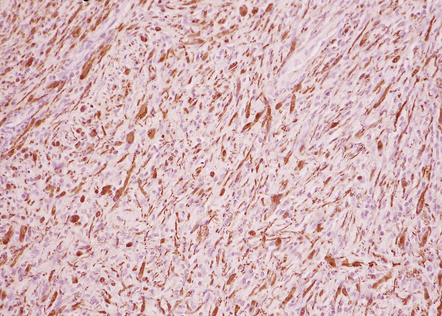
Neurofibromas are immunoreactive for vimentin, but S-100 immunoreactivity is patchy, reflecting the scattered distribution of Schwann cells (Fig. 42.16).
MALIGNANT NERVE SHEATH TUMORS
 DIFFERENTIAL DIAGNOSIS OF MPNST
DIFFERENTIAL DIAGNOSIS OF MPNST




MICROSCOPIC APPEARANCES
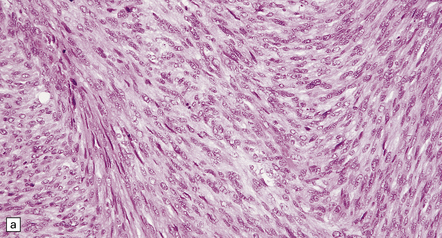
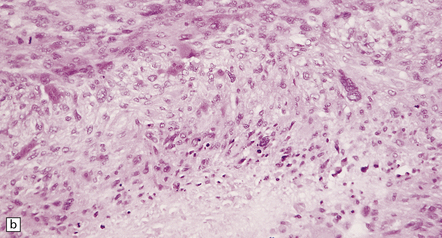
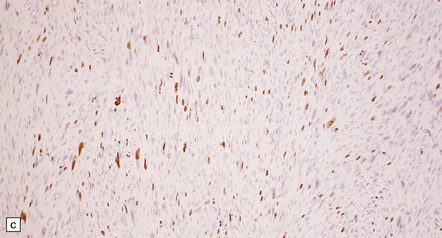
Fascicular and storiform patterns are typical. The nuclear:cytoplasmic ratio is high and mitotic figures are easily found (Fig. 42.17). Nuclear palisading is infrequent. Foci of necrosis are often evident. The waviness of the cell nuclei may be maintained, at least in some parts of the tumor.
42.17 MPNST.
(a) A higher nuclear:cytoplasmic ratio and mitotic count than in a schwannoma are found in this neoplasm. (b) Cytologic pleomorphism and micronecrosis are additional features. (c) The growth fraction, as indicated by Ki-67 immunoreactivity, is high.
About 10% of MPNSTs show heterologous differentiation. Exhibiting varying degrees of differentiation/cytologic malignancy, mesenchymal tissues – skeletal muscle (triton tumor), bone, and cartilage – may be present (Fig. 42.18).
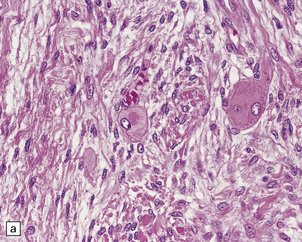
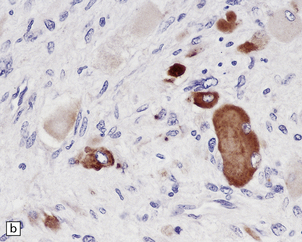
42.18 Triton tumor.
This variant of MPNST is characterized by a rhabdomyosarcomatous element. This manifests as large cells with deeply eosinophilic cytoplasm (a), in which cross-striations may occasionally be seen. These cells are immunoreactive for myoglobin (b).
Epithelial differentiation may result in the formation of clusters of glands (Fig. 42.19). An epithelioid variant accounting for 5% of cases and consisting of strings or nests of round cells with prominent nucleoli can be mistaken for a carcinoma or malignant melanoma (Fig. 42.20). Very occasionally, MPNSTs contain melanin, and rarely an MPNST may arise from a melanotic schwannoma.
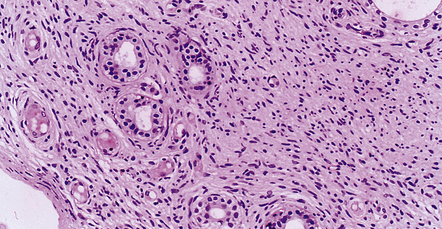
42.19 MPNST.
Epithelial differentiation, usually in the form of clusters of glands, is a rare finding in MPNST and must be distinguished from epithelial structures entrapped by the invading tumor.
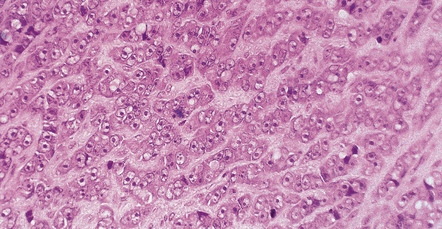
Approximately 50% of MPNSTs have the ultrastructural features of Schwann cell differentiation.
REFERENCES
Berg, J.C., Scheithauer, B.W., Spinner, R.J., et al. Plexiform schwannoma: a clinicopathologic overview with emphasis on the head and neck region. Hum Pathol.. 2008;39:633–640.
Brooks, J.S., Freeman, M., Enterline, H.T. Malignant Triton tumors. Natural history and immunohistochemistry of nine new cases with literature review. Cancer. 1985;55:2543–2549.
Brossier, N.M., Carroll, S.L. Genetically engineered mouse models shed new light on the pathogenesis of neurofibromatosis type I-related neoplasms of the peripheral nervous system. Brain Res Bull. 2011. [Epub:21855613].
Carroll, S.L., Ratner, N. How does the Schwann cell lineage form tumors in NF1? Glia. 2008;56:1590–1605.
Casadei, G.P., Scheithauer, B.W., Hirose, T., et al. Cellular schwannoma. A clinicopathologic, flow cytometric, DNA, and proliferation marker study of 70 patients. Cancer. 1995;75:1109–1119.
Colman, S.D., Williams, C.A., Wallace, M.R. Benign neurofibromas in type 1 neurofibromatosis (NF1) show somatic deletions of the NF1 gene. Nat Genet.. 1995;11:90–92.
Ducatman, B.S., Scheithauer, B.W. Malignant peripheral nerve sheath tumors with divergent differentiation. Cancer. 1984;54:1049–1057.
Ducatman, B.S., Scheithauer, B.W., Piepgras, D.G., et al. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986;57:2006–2021.
Evans, D.G., Huson, S.M., Donnai, D., et al. A clinical study of type 2 neurofibromatosis. Q J Med.. 1992;84:603–618.
Feany, M.B., Anthony, D.C., Fletcher, C.D. Nerve sheath tumours with hybrid features of neurofibroma and schwannoma: a conceptual challenge. Histopathology. 1998;32:405–410.
Fletcher, C.D., Davies, S.E., McKee, P.H. Cellular schwannoma: a distinct pseudosarcomatous entity. Histopathology. 1987;11:21–35.
Hornick, J.L., Fletcher, C.D. Soft tissue perineurioma: clinicopathologic analysis of 81 cases including those with atypical histologic features. Am J Surg Pathol.. 2005;29:845–858.
Kourea, H.P., Cordon-Cardo, C., Dudas, M., et al. Expression of p27(kip) and other cell cycle regulators in malignant peripheral nerve sheath tumors and neurofibromas: the emerging role of p27(kip) in malignant transformation of neurofibromas. Am J Pathol.. 1999;155:1885–1891.
Kudo, M., Matsumoto, M., Terao, H. Malignant nerve sheath tumor of acoustic nerve. Arch Pathol Lab Med.. 1983;107:293–297.
Kurtkaya-Yapicier, O., Scheithauer, B., Woodruff, J.M. The pathobiologic spectrum of Schwannomas. Histol Histopathol.. 2003;18:925–934.
Laskin, W.B., Weiss, S.W., Bratthauer, G.L. Epithelioid variant of malignant peripheral nerve sheath tumor (malignant epithelioid schwannoma). Am J Surg Pathol.. 1991;15:1136–1145.
Louis, D.N., Ramesh, V., Gusella, J.F. Neuropathology and molecular genetics of neurofibromatosis 2 and related tumors. Brain Pathol.. 1995;5:163–172.
McMenamin, M.E., Fletcher, C.D. Expanding the spectrum of malignant change in schwannomas: epithelioid malignant change, epithelioid malignant peripheral nerve sheath tumor, and epithelioid angiosarcoma: a study of 17 cases. Am J Surg Pathol.. 2001;25:13–25.
Nascimento, A.F., Fletcher, C.D. The controversial nosology of benign nerve sheath tumors: neurofilament protein staining demonstrates intratumoral axons in many sporadic schwannomas. Am J Surg Pathol.. 2007;31:1363–1370.
Nielsen, G.P., Stemmer-Rachamimov, A.O., Ino, Y., et al. Malignant transformation of neurofibromas in neurofibromatosis 1 is associated with CDKN2A/p16 inactivation. Am J Pathol.. 1999;155:1879–1884.
Scheithauer, B.W., Erdogan, S., Rodriguez, F.J., et al. Malignant peripheral nerve sheath tumors of cranial nerves and intracranial contents: a clinicopathologic study of 17 cases. Am J Surg Pathol.. 2009;33:325–338.
Stratakis, C.A. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit (PRKAR1A) in patients with the complex of spotty skin pigmentation, myxomas, endocrine overactivity, and schwannomas (Carney complex). Ann N Y Acad Sci.. 2002;968:3–21.
Stratakis, C.A., Kirschner, L.S., Carney, J.A. Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab.. 2001;86:4041–4046.
Swanson, P.E., Scheithauer, B.W., Wick, M.R. Peripheral nerve sheath neoplasms. Clinicopathologic and immunochemical observations. Pathol Annu.. 1995;30:1–82.
von Deimling A, Krone W, Menon AG. Neurofibromatosis type 1995; 1: pathology, clinical features and molecular genetics. Brain Pathol. 5:153–162.
Wanebo, J.E., Malik, J.M., VandenBerg, S.R., et al. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 28 cases. Cancer. 1993;71:1247–1253.
Woodruff, J.M., Erlandson, R.A., Scheithauer, B.W. Nerve sheath tumors. Am J Surg Pathol.. 1995;19:608–611.
Woodruff, J.M., Scheithauer, B.W., Kurtkaya-Yapicier, O., et al. Congenital and childhood plexiform (multinodular) cellular schwannoma: a troublesome mimic of malignant peripheral nerve sheath tumor. Am J Surg Pathol.. 2003;27:1321–1329.
Zhou, H., Coffin, C.M., Perkins, S.L., et al. Malignant peripheral nerve sheath tumor: a comparison of grade, immunophenotype, and cell cycle/growth activation marker expression in sporadic and neurofibromatosis 1-related lesions. Am J Surg Pathol.. 2003;27:1337–1345.