[level-membership-for-neurology-category]
Chapter 20 Perinatal Metabolic Encephalopathies
Introduction
Box 20-1 Predominant Presenting Clinical Features in Neonatal Genetic Metabolic Disorders


























A brief synopsis of each condition is provided, with emphasis on distinctive clinical features, diagnostic approach, and management. Comprehensive discussion of the molecular biology, pathophysiology, and genetics of these disorders may be found in several excellent reviews [Scriver et al., 2001; Hoffmann et al., 2010].
General Approach
Metabolic disease should be considered in the differential diagnosis of any newborn demonstrating symptoms or signs of encephalopathy, which are summarized in Table 20-1. While none of these abnormalities is specific to metabolic disease, the temporal pattern and specific constellation of symptoms taken together often suggest a metabolic disease. Encephalopathies caused by acute correctable metabolic perturbations, such as in hypoglycemic encephalopathy, typically follow a monophasic course that resolves in proportion to the timeliness of correction of the metabolic perturbation. The tempo of genetically determined metabolic diseases is more variable than acquired acute injury such as hypoxia or trauma. It often has a delayed or subacute presentation with unexplained disturbances in state of arousal or neonatal behaviors, such as feeding or visual attentiveness, or with the emergence of myoclonic seizures or movement disorder. Chronic encephalopathies that initially manifest in the newborn period are increasingly being linked to determine metabolic disease genetically. These disorders may have few or no features that distinguish them as metabolic, presenting with nonspecific signs such as hypotonia, depressed neonatal behaviors, or poor feeding. Nervous system manifestations of metabolic disease are generalized in nature, with depression of consciousness or irritability. Motor abnormalities and brainstem dysfunction parallel the severity of disturbed consciousness in a nonlocalizing manner. Suspicion of an inborn error of metabolism should be elevated by any of the following patterns:
Table 20-1 Neurologic Signs and Symptoms of Neonatal Metabolic Disease
| Neurologic Examination Category | Finding in Affected Infant with Metabolic Disease |
|---|---|
| Alterations in consciousness Mild to moderate Severe |
Excessively irritable, or somnolent but awakens with light stimulation Unarousable, but withdraws purposefully Comatose, responds to pain only by posturing, or not at all |
| Brainstem dysfunction | Pupillary abnormalities Oculomotor deficits Depressed or disordered suck and swallow function Disordered control of breathing (hyperpnea, apnea) High-pitched (“cerebral”) cry |
| Cortical special senses | Absent arousal to sound, poor or absent visual fixation |
| Tone and movement abnormalities | Generalized hypotonia or hypertonia Nonepileptic myoclonus Dystonia, rigidity, opisthotonus, oculogyric crises |
| Reflex abnormalities | Exaggerated deep tendon reflexes Disturbed neonatal reflex behaviors (e.g., Moro, grasp) |
| Seizures | Myoclonic, clonic, tonic, subtle, apnea |
| Neuromuscular abnormalities | Diffusely absent tendon reflexes, bilateral symmetric facial or limb weakness, respiratory insufficiency |
| Autonomic dysfunction | Temperature instability, gastrointestinal dysmotility |
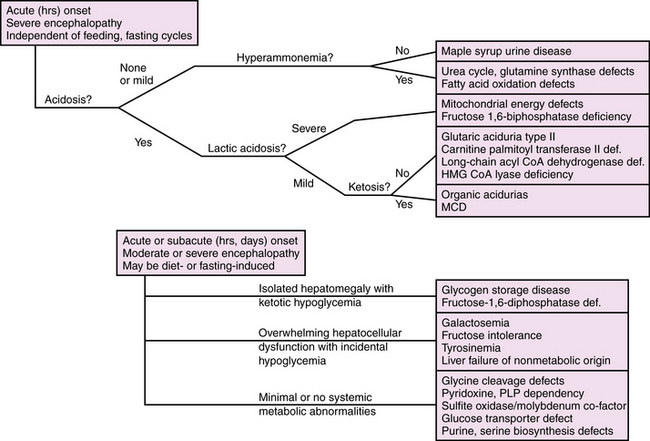
Diagnosis is further delineated by appropriate staged laboratory studies, as shown in Table 20-2. The aim of the initial evaluation is to narrow down the differential diagnosis and simultaneously treat potentially life-threatening or handicapping conditions as quickly as possible. The results of “first-stage” studies will reveal abnormalities requiring immediate correction, such as hypoglycemia, hyponatremia, hypocalcemia, or hypoxia. They will further define other organ involvement, such as renal, cardiac, or hepatocellular dysfunction. A constellation of abnormalities on the initial screening testing may suggest a particular inborn error of metabolism (Figure 20-1). If the results of first-stage assessment, combined with clinical history, examination, and neuroimaging, do not provide a specific diagnosis, they should provide the rationale for choosing appropriate “second-stage” studies. These are aimed at identifying the category of metabolic defect, and in some cases provide a probable specific diagnosis that can be verified in “third-stage” studies, in which DNA or tissue sampling provides material for enzyme quantification or genetic analysis. A careful family history should be obtained to ascertain consanguinity, known inherited metabolic disease, excess fetal loss, or infant demise in a sibling under similar circumstances.
Table 20-2 Laboratory Evaluation of the Infant with Suspected Metabolic Encephalopathy
| First Stage Survey and Screening |
Second Stage Identify Category of Metabolic Defect |
Third Stage Verify Enzyme or Gene Defect |
|---|---|---|
| Blood Glucose Electrolytes Mg++, Ca++, PO4 BUN, creatinine Liver enzymes CBC, platelets, PT, PTT Arterial blood gas Ammonia Lactate, pyruvate |
Blood Quantitative plasma amino acids Very long chain fatty acids Acyl carnitine profile Isoelectric transferrin point Copper, ceruloplasmin levels Cholesterol Uric acid |
Tissue biopsy (muscle, liver) for enzyme assay Skin fibroblast culture for enzyme assay DNA studies and specific gene mutation probes Family studies |
| Urine Ketone bodies, reducing substances Protein, blood Cells |
Urine Quantitative amino acids Organic acids Galactose Sulfites Xanthine, hypoxanthine Uric acid Ribosides |
|
| CSF Glucose, protein Cell count Microbiology |
CSF Quantitative amino acids Neurotransmitters – Biopterin metabolites Lactate, pyruvate |
BUN, blood urea nitrogen; CBC, complete blood count; CSF, cerebrospinal fluid; PT, prothrombin time; PTT, partial thromboplastin time.
Correctable Disturbances of Glucose and Salt Balance
Hypoglycemia
Hypoglycemia is common among critically ill newborns and is a frequent manifestation of inborn errors of metabolism. The definition of hypoglycemia is controversial. Blood glucose values reflect a dynamic balance between substrate availability and utilization, and in healthy newborns vary across a broad range, typically >2.2 μM (36 mg/dL), but reported as low as 1.3 μM (23 mg/dL) in healthy breastfed infants in the first few days of life. The risk of neurologic injury from hypoglycemia depends on multiple factors, including gestational age, availability of alternate substrates, metabolic rate, maturity and efficiency of glucose transport systems, status of oxidative metabolism, and differences in regional brain metabolic activity. As such, clinical factors such as coexistent hypoxic-ischemic injury, seizures, and the presence of hyperinsulinism strongly influence the occurrence and nature of hypoglycemia-related neuronal injury. These observations have led a number of investigators to propose an “operational” definition, whereby threshold blood glucose value, for example, 2.5 mmol/L (46 mg/dL), prompts treatment and further evaluation. This threshold may be modified up or down in individual cases, depending on clinical symptoms and factors likely to affect brain substrate demand, such as seizures or hypoxia, and alternative substrate availability, such as lactate or ketones or the effects of hyperinsulinism. Presently, there is insufficient evidence to define specific threshold values as predictive of neurologic injury in all clinical settings, and further research is needed on this point [Hay et al., 2009].
Disorders leading to neonatal hypoglycemia may be grouped into four broad categories: maladaptation to extrauterine life, hyperinsulinism, increased glucose consumption due to prior or intercurrent illness, and inborn errors of metabolism (Table 20-3). Maladaptation to extrauterine life affects premature infants and small-for-gestational-age infants due to low stores of fat, protein, and glycogen; inefficient ketogenesis and gluconeogenesis; or diminished tolerance of feedings. Hyperinsulinism is associated with maternal diabetes and gene defects affecting regulation of insulin production [Kapoor et al., 2009], and should be suspected when severe hypoglycemia (<2.6 mmol/L) without ketonuria in a well-nourished infant persists beyond the first 2 postnatal hours despite initiation of enteral feeds. Hypoglycemia from increased substrate utilization occurs during hypoxia or ischemia due to a shift from aerobic to anaerobic metabolism, sometimes compounded by decreased hepatic gluconeogenesis and ketogenesis. Brain glucose consumption is dramatically increased by seizures from any cause, and may exceed the capacity of cerebral energy metabolism in brain regions compromised by a recent ischemic insult. Inborn errors of metabolism may cause hypoglycemia due to enzyme deficiencies in glycogenolysis or gluconeogenesis, or as a result of hepatocellular dysfunction. The neurologic manifestations in these cases represent combined effects of the hypoglycemia and the metabolic disorder. Table 20-4 lists inborn errors of metabolism commonly associated with neonatal hypoglycemia. Neonatal hypoglycemia may be associated with congenital endocrinopathies affecting the hypothalamic-pituitary-adrenal axis as a result of major brain malformations of the hypothalamic-pituitary region, congenital adrenal hypoplasia, or hypothyroidism.
| Category | Specific Disorders |
|---|---|
| Disorders of adaptation to extrauterine life | Prematurity Intrauterine growth retardation |
| Hyperinsulinism | Infants of diabetic mothers Maternal isoimmune disease Genetic defects of insulin secretion ABCC8, ATP binding cassette C KCNJ11, potassium channel J11 GLUD1, glutamate dehydrogenase GCK, glucokinase HADH, OH–acyl-CoA dehydrogenase SLC16A1, solute carrier 16-1 HNF4A, hepatocyte nuclear factor 4 alpha Beckwith–Wiedemann syndrome Maternal isoimmune disease Congenital disorders of glycosylation |
| Elevated glucose consumption | Hypoxia Global ischemia Seizures |
| Acquired or transient hepatic dysfunction | Post hypoxic-ischemic hepatocellular injury Infectious hepatitis Any cause of liver failure |
| Primary endocrine disorders | Hypopituitarism due to brain malformation Congenital adrenal insufficiency Adrenal hemorrhage Hypothyroidism |
| Inborn errors of metabolism | See Table 20-4 |
Table 20-4 Inborn Errors of Metabolism in which Neonatal Hypoglycemia is a Common Symptom
| Category | Specific Disorders |
|---|---|
| Disorders of carbohydrate metabolism | Hereditary fructose intolerance Fructose 1,6-biphosphatase deficiency Glycogen storage diseases, esp. I and III |
| Fatty acid oxidation defects | Medium-chain and long-chain acyl-CoA dehydrogenase deficiencies Carnitine palmitoyl transferase II deficiency Mitochondrial trifunctional protein deficiency Malonyl-CoA decarboxylase deficiency (MCD) |
| Defects in ketone body synthesis | HMG CoA lyase deficiency |
| Gluconeogenesis defects | Holocarboxylase synthetase deficiency (multiple carboxylase deficiency) Pyruvate carboxylase deficiency |
| Disorders of branched chain amino acid catabolism and related organic acidurias | Maple syrup urine disease Propionic acidemia Methylmalonic acidemia Isovaleric acidemia Ethylmalonic aciduria 3-methyl-glutaconic aciduria 2-methyl-3-OHbutyryl-CoA dehydrogenase deficiency (MHBD) |
The clinical signs and symptoms of hypoglycemia in the neonate are variable and nonspecific, exemplified by infants with hyperinsulinism. Signs and symptoms in the early stages may be dominated by signs of elevated circulating catecholamines: tachycardia, pallor, diaphoresis, irritability, jitteriness or tremor, and exaggerated tendon reflexes and neonatal reflexes (e.g., Moro reflex). Progressive depression of consciousness occurs as cerebral glucose stores are exhausted, associated with hypotonia, depressed reflexes, hypothermia, and often seizures. At its worst, uncorrected hypoglycemia at levels <1.0 μM in the absence of circulating ketone bodies will progress to coma, characterized by isoelectric electroencephalogram (EEG), cerebral edema, flaccid unresponsiveness, apnea, loss of brainstem reflexes, and circulatory collapse. Mild and moderate degrees of symptoms are rapidly reversed with glucose administration, whereas severe encephalopathy reflecting irreversible neuronal injury is not. Hypoglycemic coma is associated with irreversible neuronal necrosis and lasting neurologic deficits, with a predilection for parietal-occipital cortex and periventricular white matter [Burns et al., 2008]. Long-term sequelae may include microcephaly, cortical visual impairment, combined motor and cognitive handicap, and, in some cases, epilepsy [Tam et al., 2008].
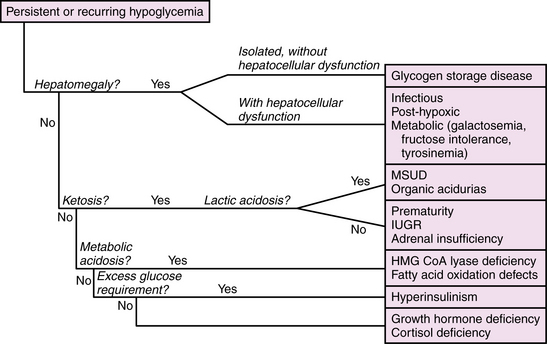
The management of neonatal hypoglycemia remains a controversial subject [Hay et al., 2009]. High-risk infants should be screened for hypoglycemia, and provided exogenous feeds, intravenous or enteral, at rates and concentrations sufficient to maintain adequate blood glucose concentrations. Bedside screening for low blood glucose should be performed in any infant who develops signs and symptoms of acute encephalopathy. Symptomatic infants should be treated while the first-stage evaluation proceeds, as outlined in Table 20-2. If ketones are absent and the exogenous glucose requirement is high (>10 mg/kg/min), then hyperinsulinism should be suspected and further evaluated by measuring insulin levels. A paradigm for assessment of hypoglycemia in neonates is shown in Figure 20-2. According to the “operational” approach to the definition of hypoglycemia, treatment of acute symptomatic hypoglycemia should proceed immediately for any infant with a blood glucose <2.5 mmol/L (46 mg/dL), with a minibolus of 200 mg/kg using 10 percent dextrose infused over 2 minutes, followed by continuous infusion of 10 percent dextrose starting at 8 mg/kg/min and increasing as necessary to maintain stable blood glucose levels >2.6 mmol/L. Monitoring the effect of glucose administration on clinical symptoms, particularly the level of consciousness, can help clarify the neurologic significance of the hypoglycemia, and the need for evaluation for other causes of encephalopathy.
Disturbances of Sodium Balance
Hyponatremia
Hyponatremia occurs when sodium losses exceed sodium or water intake, or reabsorption exceeds water excretion, or both. Hyponatremia is common in sick neonates due to immature or damaged renal mechanisms of salt and water balance or to perturbations in the hypothalamic-pituitary mechanisms regulating volume and osmolar status. Hyponatremia during the first 1–2 weeks of life in sick neonates is most commonly due to excess arginine vasopressin (AVP) secretion in association with other diseases such as lung disease, intracranial hemorrhage, pain, and surgical procedures, and rarely is due to congenital adrenal insufficiency. Causes of hypo- or hypernatremia are summarized in Box 20-2.
Box 20-2 Causes of Hyponatremia and Hypernatremia in Neonates
Causes of Hyponatremia


































The management of hyponatremic encephalopathy in neonates depends on the cause, the chronicity, and the severity of symptoms. Subacute or chronic (days to weeks) hyponatremia of mild degree (>120 mEq/L) should be treated by determining and correcting the underlying cause. Controversy surrounds the treatment of acute severe hyponatremic encephalopathy, which is usually associated with serum sodium concentrations <120 mEq/L. In such cases, seizures are usually resistant to standard anticonvulsants, and the encephalopathy frequently depresses respiratory drive and airway protective reflexes, all of which constitute a degree of urgency in favor of rapid correction. Rapid correction of hyponatremic seizures in infants and young children can be accomplished without neurologic complications by the use of 3 percent saline [Sarnaik et al., 1991]. The fact remains, however, that very little is known about neurologic complications from rapid correction of acute symptomatic hyponatremia in premature infants and critically ill term neonates who are already at high risk for neurologic injury from other causes. Until better data are available in this population, treatment should proceed with caution.
Hypernatremia
Neurologic manifestations of hypernatremia arise from hypertonic dehydration of neurons and glia, and may be compounded by secondary ischemic injury from hyperviscosity, cerebral venous thrombosis, or intracranial hemorrhage. Mild symptoms accompany mild elevations of serum sodium, generally <160 mEq/L, while severe encephalopathy usually occurs with elevations >170 mEq/L. The causes of hypernatremia are the inverse of those causing hyponatremia: excess sodium intake relative to sodium losses, or excess water losses relative to water intake or reabsorption, or both (see Box 20-2). Excess sodium intake occurs with administration of hypertonic intravenous solutions or from hypertonic oral feedings. Negative water balance is the most common cause of hypernatremia, caused by excess losses of hypotonic body fluids, defective renal water absorption due to nephrogenic diabetes insipidus, deficient pituitary secretion of AVP, or cerebral salt wasting. Hypothalamic-pituitary insufficiency is caused by acquired brain insults, such as meningitis or hypoxic-ischemic encephalopathy, and by midline brain malformations. Management of hypernatremia begins with assessment and correction of intravascular volume deficits with normotonic solutions or plasma expanders in sufficient amounts to restore perfusion to normal levels and to maintain urinary output. The guiding principle in treatment is to prevent cerebral edema by correcting the hypernatremia over 48–72 hours, decreasing the serum sodium concentration at a rate not exceeding 10–15 mEq/L per 24 hours.
Inborn Errors of Metabolism
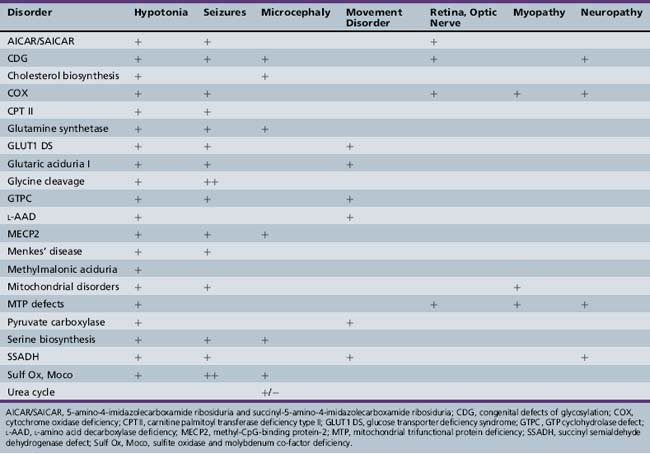
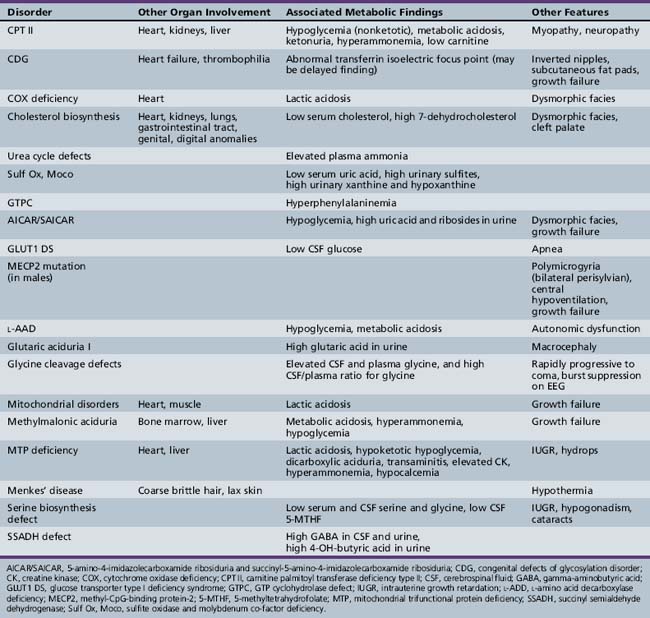
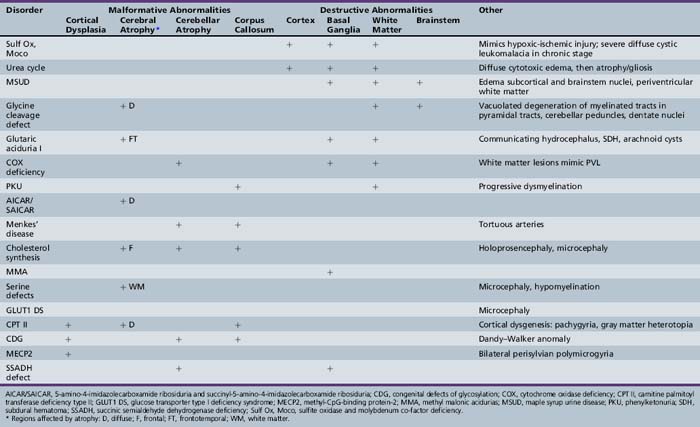
Patterns of clinical features may be helpful in recognizing characteristic patterns associated with specific disorders, as shown in Table 20-5 and Table 20-6. Specific diagnoses can be suspected based on the predominant clinical features and results of first-stage assessments, then verified by definitive second- or third-stage studies, as described in Table 20-2. Many of the subacute epileptic encephalopathies and chronic disorders are less common and less well characterized. The full range of phenotypic variation remains to be fully elucidated in more recently described disorders. The widespread availability of MRI has provided new insights concerning the nature and anatomic distribution of brain abnormalities in these disorders, as well as in more classic metabolic diseases. Some disorders are associated with distinctive MRI findings, such as the cavitary leukomalacia seen in sulfite oxidase deficiency (Table 20-7). A brief summary follows for genetically determined metabolic encephalopathies that are reported to present predominantly, or in variant form, in the neonatal period. The reader is referred to excellent reviews for comprehensive discussions of inherited metabolic diseases [Saudubray et al., 2002].
Acute Fulminant Metabolic Diseases
Maple Syrup Urine Disease
Maple syrup urine disease (MSUD) is an autosomal-recessive disorder due to deficient activity of branched chain keto-acid dehydrogenase (BCKAD); it causes accumulation in plasma of leucine, isoleucine, and valine, and their corresponding keto-acids (Figure 20-3). Of the five clinical phenotypes, the severe neonatal form is the most common [Morton et al., 2002]. After a normal interval of 4–7 days after birth, affected infants develop progressive lethargy and hypotonia, evolving to coma and decerebrate posturing, sometimes with seizures. Cerebral edema, particularly involving white matter, appears in the early stages, giving rise to a distinctive pattern of abnormalities on MRI, with pronounced T2 hyperintensity and restricted diffusion in the brainstem, cerebellar dentate nuclei, deep gray nuclei, and periventricular white matter [Jan et al., 2003]. Biochemical abnormalities include ketoacidosis, ketonuria, and hypoglycemia, often complicated by hyponatremia. Neuropathology consists of spongiform degeneration or delayed myelination of white matter with oligodendroglial cell loss, and cerebellar granular cell necrosis. The EEG during metabolic decompensation may show characteristic central sharp waves, or diffuse slowing to burst suppression as the symptoms progress. Diagnosis rests on the detection of marked elevations of plasma leucine (up to 1000–5000 μM during acute decompensation), isoleucine, and valine. Detection of l-alloisoleucine in plasma is pathognomonic for this disorder. An elevated ratio of leucine:alanine has been described as a sensitive and specific marker for MSUD, along with appreciation of the distinctive maple syrup odor in cerumen, allowing for earlier diagnosis of newborns in the presymptomatic stage. Urine organic acid profiles reveal elevated 3-hydroxyvaleric acid. BCKAD assay in cultured skin fibroblasts reveals activity at less than 2 percent of normal levels, and confirms the diagnosis. Treatment for acute severe decompensation requires metabolite removal via peritoneal dialysis or hemodialysis, along with supportive and nutritional measures to induce an anabolic state. Morton et al. reported that infants diagnosed in the first 72 hours of life, before becoming symptomatic, can be managed readily with enteral MSUD formula, thereby preventing acute metabolic decompensation and the associated neurologic injury [Morton et al., 2002]. Long-term management consists of dietary restriction of leucine, good nutritional support, and avoidance of catabolic stresses. Long-term outcome is related to the number and severity of metabolic decompensations.
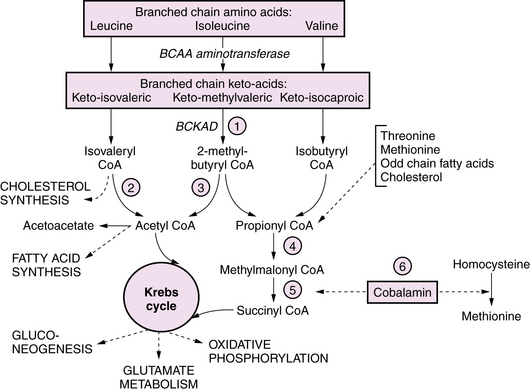
The pathophysiology of the neurologic manifestations is incompletely understood, and is likely multifactorial and related to the roles of branched chain amino acids and their metabolites in cellular energetics, glutamate and GABA metabolism, fatty acid and cholesterol synthetic pathways, and the regulation of neuronal cytoskeleton development [Pessoa-Pureur and Wajner, 2007]. It has been suggested that acute symptoms reflect toxic effects of the keto-isocaproic acid (KIC) metabolite on glutamate, glutamine, and GABA signaling pathways and energy metabolism. Long-term effects of KIC may be related to altered phosphorylation of cytoskeletal proteins, which is influenced by glutamatergic and GABAergic receptor activation in a developmentally regulated manner. A further potential mechanism for toxicity of branched chain keto-aciduria (BCKAs) is via inhibition of glutamate reuptake (see Figure 20-3). Deficiencies in transmitter pools of glutamate, aspartate, and GABA, along with deficient GABA receptors, have been demonstrated in an animal model [Dodd et al., 1992], while human neuropathologic analysis has revealed reduced myelin lipids in untreated patients.
Organic Acidopathies due to Defects in Branched Chain Amino Acid Metabolism
BCKAs are autosomal-recessive disorders due to defects in the catabolism of branched chain amino acids. MSUD is a prominent example of an organic aciduria, as described in the preceding section. Defects in the downstream degradation of branched chain ketoacids of leucine, valine, and isoleucine cause accumulation of one or more branched chain organic acid intermediates (see Figure 20-3). While isovaleric, propionic, and methylmalonic aciduria are the best known of these disorders, several others have been described (Table 20-8). Clinical symptoms first appear from the neonatal period to early infancy, characterized by acute onset of severe metabolic acidosis with ketoacidosis, hypoglycemia, and hyperammonemia, which may progress to seizures and coma. Associated features may include growth failure, vomiting, diarrhea, hepatic dysfunction, circulatory collapse, neutropenia, and thrombocytopenia. Pathogenesis of clinical symptoms has been attributed to a combination of direct toxic effects of accumulated metabolites and compensatory responses. Energy metabolism may be compromised as a result of deficient substrates for gluconeogenesis and limited availability of free CoA for mitochondrial fatty acid oxidation. Accumulation of acyl-CoA intermediates inhibits the urea cycle, resulting in hyperammonemia.
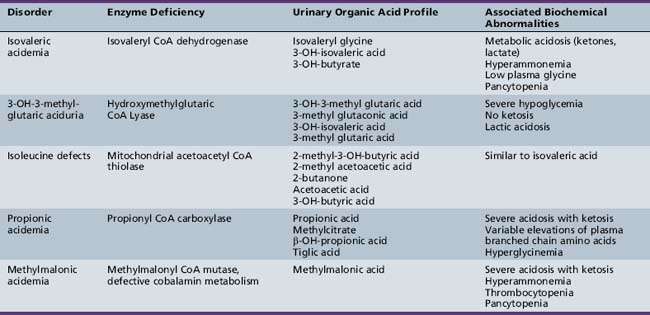
Diagnosis should be suspected when first-stage screening reveals prominent metabolic acidosis with variable combinations of ketosis, lactic acidosis, hypoglycemia, and hyperammonemia. Specific diagnosis rests on specific urinary organic acid profiles, and can be confirmed by enzyme assay in cultured skin fibroblasts (see Table 20-8). Management consists of glucose administration, with insulin if necessary, to reverse catabolic states, and protein restriction, along with respiratory and circulatory support. Peritoneal dialysis may be necessary in the most severe cases with acute metabolic coma. l-carnitine and N-carbamylglutamate (NCG) supplementation respectively have been used to mitigate excessive ketogenesis and the inhibitory effects of high propionic acid levels on urea cycle function [Tuchman et al., 2008]. Carnitine and glycine supplementation may improve clearance of toxic metabolites in isovaleric acidemia. Co-factor supplementation can be helpful in subgroups characterized by primary defects in the co-factor metabolism using pharmacologic doses of cobalamin in some cases of methylmalonic acidemia, or biotin in some cases of propionic acidemia. Long-term management includes protein-restricted, high-carbohydrate diets and avoidance of catabolic stresses. Several case reports and case series have described improved long-term outcome following liver transplantation for some organic acidopathies [Morioka et al., 2007]. Long-term outcome can be normal if decompensations are minimized in frequency and treated aggressively when they occur.
Primary Lactic Acidosis due to Defects in Oxidative Phosphorylation
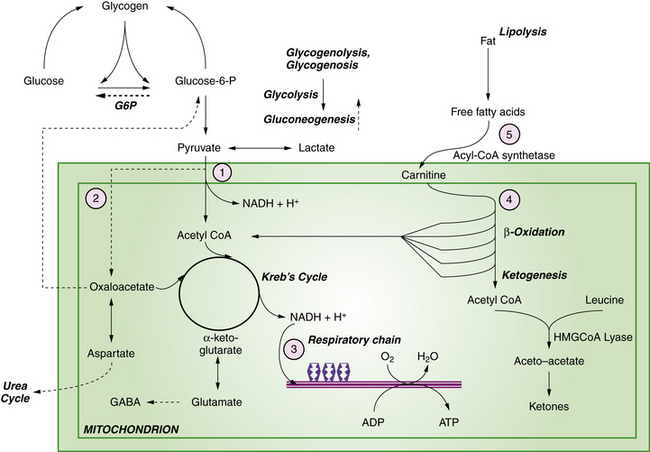
Primary lactic acidosis causes a severe metabolic encephalopathy in neonates with several types of defects in energy metabolism, including defects of pyruvate dehydrogenase complex, pyruvate carboxylase, and the respiratory chain. Figure 20-4 illustrates these pathways in relation to cellular energy and lactate metabolism. Secondary lactic acidosis is common in hypoxia or ischemia, and in other inborn errors of metabolism that secondarily affect energy metabolism, including organic acidopathies, urea cycle disorders, and fatty acid oxidation defects. Changes in the flux of citric acid cycle intermediates produce a multitude of downstream effects on energy substrate availability, precursors of neurotransmitters, and protein synthesis pathways. In the developing brain, these metabolic shifts have profound effects on immediate cellular functional integrity, as well as transcriptional and translational processes central to cell proliferation and differentiation. Advances in the understanding of the role of cellular energetics in the regulation of the cell cycle and differentiation have shed new light on the mechanisms of neurologic effects of these disorders (Figure 20-5). Energy substrate deficiency shifts the ratio of adenosine monophosphate (AMP):adenosine triphosphate (ATP), which is a key regulator of the intracellular signaling system involving mammalian target of rapamycin (mTOR). The state of activation of mTOR broadly modulates protein synthesis and cell proliferation [Fingar and Blenis, 2004]. Under conditions of energy substrate deprivation, changes in mTOR signaling shift protein metabolism from an anabolic to a catabolic state, initiating a cascade of events with major implications for subsequent growth and development.
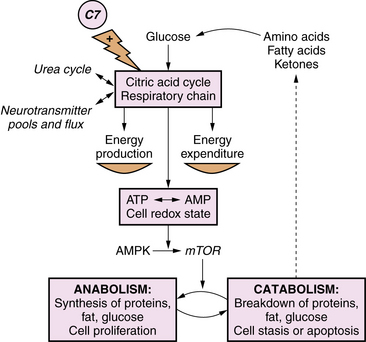
Improved understanding of the molecular and cellular pathophysiology of metabolic disease has led to the development of “anaplerotic” therapeutic strategies [Roe and Mochel, 2006]. These interventions aim to restore a more physiologic state of cellular energetics and protein synthesis, and go beyond replacement of deficient metabolites or removal of toxins by providing alternative substrates for the citric acid cycle and respiratory chain. This approach is exemplified by use of the triglyceride triheptanoin as an alternative source of citric acid cycle substrates in the treatment of pyruvate carboxylase deficiency, further discussed below. As such, these strategies are most relevant for defects of oxidative phosphorylation and the respiratory chain, but can theoretically play a role in any metabolic disorder that disrupts energy metabolism.
Pyruvate carboxylase (PC) converts pyruvate to oxaloacetate. It plays a critical role in hepatic gluconeogenesis and in maintaining a stable supply of citric acid cycle intermediates influencing energy metabolism, the function of the urea cycle, and substrate availability for neurotransmitter pools of glutamate and GABA. PC deficiency has several subtypes with a broad spectrum of clinical manifestations. PC deficiency type B (the French subtype) is the severe neonatal form, and presents with a fulminant neonatal encephalopathy with permanent severe lactic acidosis, hypoglycemia, citrullinemia, and hyperammonemia, and is usually fatal in early infancy. The neurologic symptoms include variably depressed mental status, hypotonia, high-amplitude tremors and dystonic movements, and a variety of paroxysmal eye movement abnormalities (gaze defects, pendular nystagmus) [Garcia-Cazorla et al., 2006]. Imaging shows cystic periventricular leukomalacia. Autopsies in these patients show widespread neuronal death and arrest of myelination, leading to cortical and white matter atrophy or cystic encephalomalacia. Treatment strategies with high carbohydrate diets have been used in these patients to avoid dependence on the defective gluconeogenic pathways. Early administration of triheptanoin as a form of anaplerotic therapy may rapidly reverse the metabolic crisis and ameliorate neurologic symptoms in symptomatic newborns.
Glutamine Synthetase Deficiency
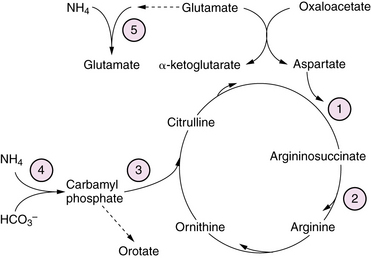
Glutamine synthetase (GS) forms glutamine from glutamate and ammonia (Figure 20-6). Glutamine is the most abundant amino acid in plasma, and functions as an amino-group donor in the synthesis of several amino acids and nucleotides. GS plays a critical role in the brain to detoxify ammonia and regulate the concentration and compartmentalization of neurotransmitter pools of glutamate and GABA (see Figure 20-6). GS deficiency has been described as a rare autosomal-recessive condition manifest as profound neonatal encephalopathy with coma, quadriparesis, severe bulbar dysfunction, lissencephaly, and death shortly after birth due to multi-organ failure [Haberle et al., 2006]. Diagnosis rests on the finding of moderate hyperammonemia, and very low or absent glutamine concentration in blood, cerebrospinal fluid (CSF), and urine, and is confirmed by severely deficient enzyme activity in cultured fibroblasts. There is no effective treatment.
Fatty Acid Oxidation Defects
Fatty acid oxidation defects comprise a broad spectrum of disorders, most of which present beyond the neonatal period as acute encephalopathies with nonketotic hypoglycemia provoked by fasting or catabolic stress. In these disorders, one of several enzymes is deficient in the pathway for degradation of lipids to fatty acids to acetyl CoA or ketone body production (see Figure 20-4). Several of these disorders have been reported with prominent and distinctive presentations in neonates. These include carnitine palmitoyl transferase II (CPT II) deficiency, mitochondrial trifunctional protein deficiency (MTP) and malonyl CoA decarboxylase deficiency (MCD). Acyl CoA dehydrogenase deficiencies (long-chain or LCAD, medium-chain or MCAD, short-chain or SCAD) more typically present outside the neonatal period. These disorders share a constellation of clinical features, which include the rapid onset of a fulminant metabolic encephalopathy with hypoglycemia, moderate hyperammonemia, low or moderate ketonuria (disproportionately low for the degree of hypoglycemia), and metabolic acidosis. Associated clinical features may include hepatomegaly, cardiomyopathy, and myopathy. Patients with MCD deficiency have been reported to have cortical dysgenesis, manifest as pachygyria, white matter atrophy, and gray matter heterotopias. The pathogenesis of neurologic symptoms has been attributed to the combined effects of direct toxicity from accumulated free fatty acids, as well as insufficient glucose and ketones to fuel the Krebs cycle, and inhibition of mitochondrial energy production by the disturbed ratio of acyl CoA:free CoA. The characteristic clinical features should suggest the diagnosis, which can be further supported by finding decreased total plasma carnitine levels and specific patterns of elevated plasma acylcarnitine intermediates as follows: C16 and C18 species in CPT II deficiency; C14 species in LCAD; C6–C8 species in MCAD; C4 species in SCAD. MCD deficiency is identified by the finding of elevated urinary malonic acid. Definitive diagnosis rests on assay of enzyme activity from cultured skin fibroblasts, and identification of specific mutations by genomic sequence analysis. Treatment involves supportive measures, glucose administration, and carnitine supplementation, and modification of feeding schedules to minimize fasting states. Creatine supplements have been suggested for patients with MTP deficiency. Recurrent decompensations can be minimized by early initiation of intravenous glucose during intercurrent illnesses and avoidance of fasting. Long-term outcome depends on the frequency and severity of decompensations.
MTP is distinguished among these disorders by the high proportion of cases (about 50 percent) presenting in the neonatal period [den Boer et al., 2003]. It has two types of presentations:
CPT II deficiency has several phenotypes, with onset in adulthood, infancy, or the neonatal period. The latter is the least common and most severe form [Sigauke et al., 2003], and is reported to be universally fatal. Affected infants often have prenatally detected cerebral lesions and malformations, including ventriculomegaly, agenesis of the corpus callosum, polymicrogyria, periventricular cystic leukomalacia, and subependymal hemorrhages. Neurologic symptoms appear at birth or within the first few days of life, including seizures, depressed consciousness, and hypotonia with myopathic features. Cardiomyopathy leading to circulatory failure is common. The metabolic findings are typical of fatty acid oxidation defects, as described above. Management is supportive and symptomatic, but ineffective in most cases. Definitive biochemical diagnosis is a prelude to genetic testing, which can provide a basis for family counseling and management of future pregnancies. A number of US states have incorporated CPT II in neonatal metabolic screening programs in hopes of promoting early diagnosis.
Urea Cycle Disorders
Urea cycle defects presenting in the neonate are characterized by the onset, between 1 and 4 days of age, of severe hyperammonemia with rapidly progressive lethargy and vomiting, progressing to coma with signs of cerebral edema [Summar et al., 2008]. Associated features may include respiratory alkalosis and hypothermia. First-stage laboratory assessments reveal no other major abnormalities. A presumptive diagnosis can be made from results of plasma amino acid, urine organic acid, and urinary orotate excretion profiles (see Figure 20-6). Elevated plasma glutamine levels are common to all the urea cycle disorders. Organic acidopathies and congenital lactic acid disorders that may cause hyperammonemia can be distinguished from urea cycle disorders by the presence in the former of acidosis, ketosis, or lactic acidosis, as well as by specific metabolites in urinary organic acid profiles, as described in previous sections. Transient hyperammonemia of the newborn is a poorly understood disorder, characterized by severe transient hyperammonemia associated with respiratory distress syndromes or herpes simplex infection. It is distinguished from the genetically determined enzyme defects by its earlier onset (first 24 hours of life) and its association with prematurity and pulmonary disease. The pathophysiology of hyperammonemic encephalopathy is complex, involving astrocyte swelling and dysfunction due to excessive glutamine accumulation [Takahashi et al., 1991] and disturbed regulation of nitric oxide synthesis [Nagasaka et al., 2009]. Treatment of acutely symptomatic infants includes administration of sodium benzoate, phenylacetate, and arginine. Infants unresponsive to these measures or those presenting with acute severe hyperammonemic coma are treated with hemodialysis to reduce plasma ammonia levels rapidly [Enns et al., 2007]. Use of pressors and volume to maintain adequate cerebral perfusion pressure is preferable to the use of hyperventilation or osmotic diuresis in managing intracranial hypertension. Careful protein restriction, providing adequate calories to prevent catabolic state, and administration of drugs to enhance excretion of excessive metabolites are tailored according to the specific enzyme defect. Orthotopic liver and liver cell transplantation have been reported with some success in stabilizing the disease process and improving outcome in some cases [Meyburg et al., 2009]. Long-term outcome is variable, with motor and cognitive deficits dependent on the severity and frequency of hyperammonemic decompensations.
Subacute Epileptic Encephalopathies
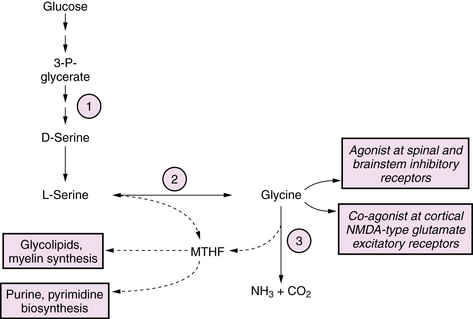
Glycine Cleavage Defects
Glycine cleavage defects are a group of autosomal-recessive disorders that lead to accumulation of glycine (Figure 20-7). The most common form is a neonatal-onset progressive encephalopathy with depressed consciousness, apnea, and seizures, usually myoclonic, associated with a pattern of burst suppression on EEG. A transient form of nonketotic hyperglycinemia has been reported, in which symptoms similar to the classic form appear in the neonatal period, but which may resolve at 1–3 months of age when the glycine levels spontaneously normalize. In such children, enzyme activity measured from liver biopsy is normal, and long-term outcome is normal in the majority of individuals [Lang et al., 2008]. Glycine toxicity appears to be maximal in the neonatal period, and may spontaneously diminish after 1 month of age. Lifelong severe cognitive and motor handicap and intractable epilepsy are the rule among survivors, although there is considerable variability. Factors that predict a poorer outcome include earlier age of symptom onset, presence of cerebral dysgenesis, and degree of glycine elevation. Neurologic manifestations occur as a result of excessive stimulation of CNS glycine receptors, which are inhibitory in spinal cord and brainstem and excitatory in brain via a co-agonist effect at the N-methyl-d-aspartate (NMDA) subtype of glutamate receptors. Neuroimaging findings are varied, and may include cerebral dysgenesis of prenatal origin related to disturbed neuronal proliferation and differentiation, evolving to a progressive vacuolating myelinopathy in the postnatal period [Mourmans et al., 2006]. Biochemical abnormalities are limited to the finding of elevated glycine levels in plasma (1–8-fold above normal) and CSF (15–30-fold above normal). In cases with equivocal plasma elevations of glycine, the diagnosis can be made by finding a CSF:plasma ratio >0.06. Definitive diagnosis can be made by demonstrating absent or very low activity of the glycine cleavage system enzyme in liver biopsy or autopsy. Glycine cleavage defects can be distinguished from the hyperglycinemia that accompanies organic acidopathies by the presence of ketosis in the latter, and by characteristic organic acid excretion profiles. There are no proven therapies. Seizures have generally been resistant to standard anticonvulsants. Treatment with dextromethorphan, an NMDA-receptor antagonist, or with ketamine and benzoate has been reported in a small number of cases with limited success [Korman et al., 2006].
Pyridoxine and Pyridoxal Phosphate Dependency Epileptic Encephalopathies
Pyridoxine is the precursor for pyridoxal-5-phosphate (PLP), an essential co-factor for multiple enzymes in brain metabolism. There are two known enzyme defects with distinct gene mutations that result in depletion or deficient production of PLP. These are pyridoxine-dependent epilepsy (PDE) and pyridoxal phosphate-dependency disorder, known as PNPO deficiency. The biochemical and genetic basis for PDE has been defined, and involves depletion of PLP as a result of binding with 1-piperideine 6-carboxylate, a byproduct of defective lysine metabolism (Figure 20-8). The defect in the lysine pathway enzyme, alpha-aminoadipic semialdehyde dehydrogenase (AASAD), arises from mutation of the gene antiquitin (ALDH7A1) [Mills et al., 2010]. Pyridoxal phosphate-dependency disorder involves mutations in the gene for pyridox(am)ine-5′-phosphate oxidase (PNPO), which converts pyridoxine to PLP [Bagci et al., 2008]. The pathophysiology of the encephalopathy is not fully defined, but likely involves multiple pyridoxine-dependent pathways in brain metabolism. Prominent among these is glutamic acid decarboxylase (GAD), which converts glutamate to GABA, the major inhibitory neurotransmitter in mammalian brain (Figure 20-9). A disruption of this pathway alters GABA-related neurotransmission, and may contribute to the clinical manifestations of intractable seizures and severe cortical dysfunction.
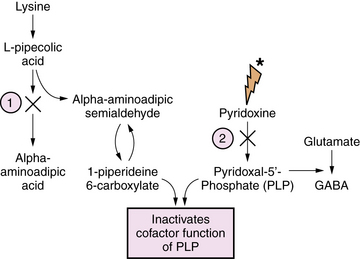
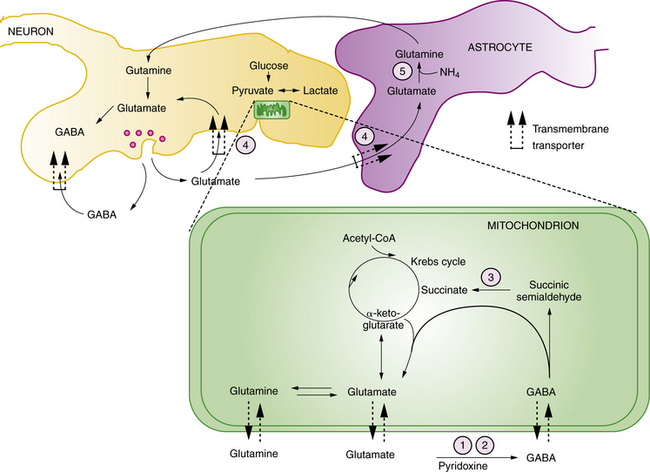
Affected infants with either disorder have a similar clinical presentation, dominated by neonatal-onset seizures, or in some cases in utero-onset seizures, and chronic encephalopathy, with no other metabolic abnormalities. Seizures are usually unresponsive to standard anticonvulsants. EEG is typically severely abnormal, with any of several types of epileptiform patterns including hypsarrhythmia, burst suppression, or generalized spike-wave activity. The diagnosis of either disorder starts with clinical suspicion in the setting of refractory neonatal-onset seizures. Diagnosis of PDE can be confirmed by finding elevated levels of α-aminoadipic semialdehyde and pipecolic acid in urine, blood, and CSF. It can be further defined by mutation analysis of the antiquitin gene in affected children and their parents [Mills et al., 2006, 2010]. Diagnosis of pyridoxal phosphate-dependency disorder should be considered in infants with the clinical features of pyridoxine dependency, but who are unresponsive to pyridoxine, lack the confirmatory biochemical markers (elevated urinary AASA), and who respond to PLP.
Folinic acid-responsive neonatal epileptic encephalopathy resembles PDE clinically and electrographically. Gallagher et al. demonstrated that folinic acid-responsive epileptic encephalopathy is identical biochemically and genetically to PDE [Gallagher et al., 2009]. The biochemical basis for the favorable response to folinic acid is unknown.
Sulfite Oxidase and Molybdenum Co-factor Deficiency
Molybdenum co-factor deficiency and isolated sulfite oxidase deficiency are closely related autosomal-recessive diseases involving defects in xanthine metabolism and sulfite degradation pathways [Schwarz et al., 2009]. Molybdenum co-factor (Moco) is essential for the function of xanthine dehydrogenase and sulfite oxidase. Synthesis of Moco involves conversion of guanosine triphosphate (GTP) to a pterin-containing intermediate (cPMP) (Figure 20-10). The mechanism of the brain injury is multifactorial, involving direct toxic effects of xanthines and sulfite metabolites on cellular architecture, damage to mitochondria, thiamine degradation, and impaired mucopolysaccharide synthesis. Moco deficiency leads to accumulation of S-sulfocysteine, which activates the glutamate receptor, and may contribute to the prominent epileptic features of affected patients.
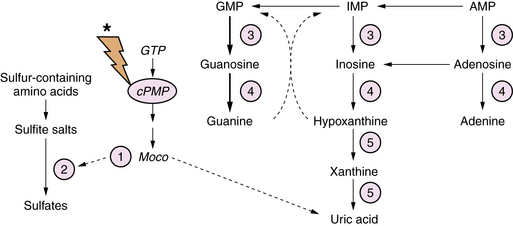
Diagnostic biochemical characteristics include increased urinary excretion of sulfites, thiosulfate, S-sulfocysteine, and taurine. Patients with Moco deficiency have low serum and urinary uric acid levels, and increased urinary xanthine and hypoxanthine levels. These findings may vary according to protein intake and nutritional status. Patients with isolated sulfite oxidase deficiency have normal uric acid metabolite levels. The diagnosis may be confirmed by enzyme assay of biopsied liver or cultured skin fibroblasts. Treatment is supportive with an emphasis on optimizing anticonvulsant therapy. Dietary intervention with cysteine and thiamine supplementation and methionine restriction has been reported to be beneficial in isolated cases [Boles et al., 1993]. Administration of cPMP in animal models of Moco deficiency has been effective in normalizing the phenotype, but has not been evaluated in humans [Schwarz et al., 2004].
Menkes’ disease
Menkes’ disease is an X-linked recessive disorder of the ATP7A gene, which results in defective function of the intestinal copper transport protein [Tumer and Moller, 2010]. This causes low tissue levels of copper and secondarily deficient function of numerous enzymes for which copper is a co-factor, including dopamine-β-hydroxylase, cytochrome c oxidase, and lysyl oxidase. The clinical phenotype invariably consists of severe neonatal-onset chronic encephalopathy with refractory epilepsy. Infants typically have central hypotonia and developmental arrest with microcephaly. Distinguishing findings on examination, outside of neurologic abnormalities, include redundant and hyperelastic skin, coarse brittle hair, thin or absent eyebrows and eyelashes, and poor temperature stability. When examined under a microscope, hair fibers display the classic appearance of pili torti. Non-neurologic manifestations include vasculopathy with excess tortuosity and fragility, leading to thromboembolic stroke or subdural hemorrhage. This disease should be suspected in the setting of early infantile epileptic encephalopathy with the characteristic physical stigmata, and is supported by the biochemical findings of low ceruloplasmin level and low serum copper. Confirmation of the diagnosis based on serum ceruloplasmin and copper markers is unreliable in the newborn period. Distinctive abnormalities in serum profiles of catecholamines, on the other hand, provide a sensitive and specific diagnostic approach in the presymptomatic neonates [Kaler et al., 2008]. Correction of serum copper levels with dietary supplements may be helpful if started in presymptomatic infants. Otherwise, treatment is supportive with an emphasis on anticonvulsant management.
Glucose Transporter Defects
DeVivo et al. characterized a disorder arising from mutations in the neuronal glucose transporter, known as GLUT1 deficiency syndrome (GLUT1 DS) [De Vivo et al., 2002]. The disease manifests as early infantile-onset epileptic encephalopathy associated with a low CSF glucose concentration (mean 30 mg/dL). Seizures have their onset from age 4 weeks to 18 months, with a mean of 5 months, include all clinical seizure types (focal, generalized, myoclonic), and are resistant to anticonvulsant drugs. Affected infants have neurodevelopmental impairment of variable severity, and acquired microcephaly. The cardinal biochemical feature is a decreased ratio of CSF glucose relative to plasma glucose, typically <33 percent, which contrasts with the relatively high ratio of CSF/blood glucose seen in term and premature newborns of 70–80 percent, accompanied by low levels of the GLUT1 glucose transporter assayed in red blood cells. Conventional anatomical neuroimaging with CT or MRI is typically normal, while metabolic imaging with 18F-fluorodeoxyglucose positron emission tomography (FDG-PET) reveals a distinctive pattern of hypometabolism in the thalami and mesial temporal regions [Pascual et al., 2002]. The ketogenic diet is the mainstay of treatment, resulting in good control of seizures in most patients but limited benefit for the neurodevelopmental disability. Long-term outcome is variable, with most children affected by some degree of cognitive impairment.
Serine Biosynthesis Defects
Defects in serine biosynthesis have been described that present as neonatal-onset chronic encephalopathies with prominent refractory epilepsy [de Koning and Klomp, 2004]. Affected infants are neurologically abnormal at birth with intrauterine growth retardation (IUGR), congenital microcephaly, cataracts, seizures, and neurodevelopmental impairment. A distinctive pattern of leukoencephalopathy is seen on MRI, characterized by hypomyelination, vacuolar changes, and gliosis, which may improve after treatment with serine and glycine supplementation. The disease involves defective serine biosynthesis due to 3-phosphoglycerate dehydrogenase deficiency (see Figure 20-7). Clinical manifestations have been attributed to multiple mechanisms, including loss of neurotrophic effects of serine, impaired synthesis of glycine, which acts as a glutamate receptor co-agonist, and impaired synthesis of 5-methyltetrahydrofolate (5-MTHF), which is a co-factor for numerous brain enzyme pathways. Diagnosis can be made by finding low CSF concentrations of serine, glycine, and 5-MTHF. In contrast to many inborn errors of metabolism, this disorder is potentially treatable by high-dose dietary supplementation of serine (200–600 mg/kg/day) and glycine (200 mg/kg/day).
Purine Biosynthesis Defects
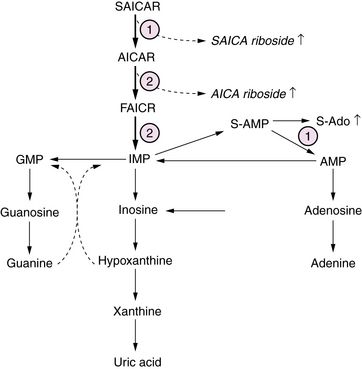
Defects of purine biosynthesis have recently been described, with clinical presentation in the neonatal period involving adenylosuccinate lyase, or riboside transformylase enzyme deficiencies [Jurecka, 2009] (Figure 20-11). These infants are typically born uneventfully at term, and manifest a severe neonatal encephalopathy with hypotonia, seizures, and mildly dysmorphic facies. Neuroimaging initially may be normal, and over time discloses diffuse atrophy. The long-term course is characterized by persistent severe static encephalopathy with profound mental retardation, blindness due to optic atrophy, refractory epilepsy, and growth failure. The cardinal biochemical features include massive urinary excretion of the riboside metabolites 5-amino-4-imidazolecarboxamide ribosiduria (AICA) and succinyl-5-amino-4-imidazolecarboxamide ribosiduria (SAICA) in urine and CSF. These metabolites give a positive result on the Bratton–Marshall test, which screens for high concentrations of succinyl purines. Diagnosis is confirmed by high-resolution thin-layer chromatography analysis of urine for succinyl purines. In addition, affected infants may have disturbed glucose and lipid metabolism as a result of impaired hepatic gluconeogenesis, fatty acid, and cholesterol synthesis. Management is symptomatic and supportive, as there is no definitive or curative treatment.
l-Amino Acid Decarboxylase Deficiency
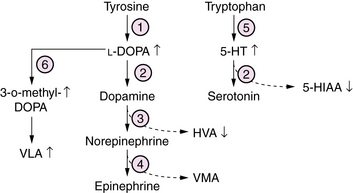
l-ADD is a defect of biogenic amine neurotransmitter metabolism, causing a combined deficiency of brain dopamine, serotonin, norepinephrine, and epinephrine [Swoboda et al., 2003] (Figure 20-12). Patients commonly present in the first weeks of life with a progressive disorder affecting multiple levels of the nervous system. Symptoms and signs include lethargy, hypotonia, suck/swallow dysfunction, and seizures, sometimes associated with hypoglycemia and acidosis. Autonomic dysfunction leads to ptosis, hypotension, gastric and intestinal dysmotility, and poor temperature regulation. With advancing age, a distinctive movement disorder appears, consisting of dystonia, athetosis, oculogyric crises, and nonepileptic myoclonus. Diagnosis rests on evaluation of CSF neurotransmitter profiles, which show increased levels of biogenic amine precursors L-DOPA and 5-hydroxytryptamine, and decreased levels of the neurotransmitter metabolites homovanillic acid (HVA) and 5-hydroxy-indole-acetic acid (5-HIAA). Similar profiles of neurotransmitter amino acids are measured in plasma and urine. A clue to this disorder would be the finding on a urine organic acid profile of elevated vanillactic acid (VLA), a breakdown product from an alternate degradative pathway for L-DOPA. Management is symptomatic and supportive. Attempts to treat by replacing deficient neuroactive amines have been hindered by the lack of an effective targeted delivery system. Outcome is poor in most patients, who develop mixed severe motor and cognitive disability and chronic movement disorders that are refractory to symptomatic treatment.
Chronic Encephalopathies without Multi-Organ Involvement
Hyperphenylalaninemia
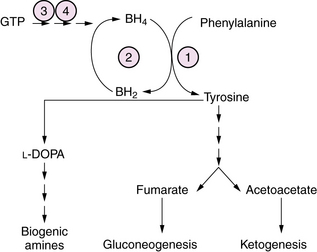
Hyperphenylalaninemia causes a chronic encephalopathy with neonatal onset due to one of several defects in the metabolism of phenylalanine. These include phenylalanine hydroxylase (PAH) deficiency, tetrahydrobiopterin (BH4) synthesis deficiency, and GTP cyclohydrolase deficiency (Figure 20-13). In classical phenylketonuria (PKU) caused by PAH deficiency, plasma phenylalanine levels exceed 1000 μM, and PAH activity in liver biopsy is severely deficient (<1 percent of normal). In a milder form of the disease, designated non-PKU hyperphenylalaninemia, plasma phenylalanine levels are <1000μM and PAH activity is less severely deficient (5–30 percent of normal). Clinical symptoms in untreated classical PKU evolve with age to include irritability, hyperkinesis, acquired microcephaly, and severe cognitive deficiency. Infants with GTP cyclohydrolase deficiency have a neonatal-onset chronic encephalopathy with severe hypotonia, bulbar dysfunction, and seizures. Symptoms may evolve over time to a pattern of rigidity, hyperkinesia, and oculomotor abnormalities. Treatment with L-DOPA has been beneficial in some patients [Nardocci et al., 2003]. Diagnosis is made by newborn metabolic screening in most countries. After identification of newborns with hyperphenylalaninemia, further quantitation of plasma phenylalanine and tyrosine, and of urinary and CSF biopterin metabolites at baseline and after phenylalanine loading is necessary to distinguish classical PKU from BH4 and GTP cyclohydrolase disorders. Treatment consists of the use of a semisynthetic diet low in phenylalanine that is carefully titrated according to plasma amino acid levels. Patients with BH4 disorders require folate and BH4 replacement. A subset of patients with milder forms of PKU, and some residual PAH enzyme activity, may respond to BH4 supplementation. In these patients, BH4 responsiveness is associated with missense mutations of PAH [Sarkissian et al., 2009]. A trial of L-DOPA and 5-hydroxytryptophan replacement to restore brain monoamine levels may prove helpful in some patients for symptoms of the dystonia and rigidity. Gene therapy and enzyme replacement therapy, while conceivable, are not yet clinically available, and are the focus of intensive experimental study.
Succinic Semialdehyde Dehydrogenase Deficiency
Succinic semialdehyde dehydrogenase deficiency is a defect of GABA degradation that results in elevated brain GABA levels (see Figure 20-9). The clinical presentation is variable. Some patients present in the neonatal period with a chronic encephalopathy and later symptoms of diffuse hypotonia, neurodevelopmental impairment, and epilepsy. An important clue to the diagnosis in this otherwise nonspecific clinical pattern is the finding on brain MRI of T2 hyperintensity in the globus pallidus. Diagnosis rests on the finding of elevated urinary 4-hydroxybutyric acid, and is associated with high levels of GABA in CSF and urine. Treatment with vigabatrin, a selective inhibitor of GABA transaminase, has been suggested, though results are mixed [Pearl et al., 2009].
Chronic Encephalopathies with Multi-Organ Involvement
Congenital Disorders of Glycosylation
Congenital disorders of glycosylation (CDG) comprise a group of genetically determined metabolic diseases characterized by abnormalities in protein glycosylation, which is a cotranslational modification step in the synthesis of most secretory and membrane-bound proteins (see Chapter 35). The nervous system is prominently affected, in association with abnormalities in multiple other organ systems, including liver, kidney, heart, and bone [Haeuptle and Hennet, 2009]. Two main subgroups have been described: type I, involving defects in assembly of the oligosaccharide precursors; and type II’ involving processing of the oligosaccharide. CDG Ia is the most common and best described of these disorders, and has two types of presentations: a neurologic and a multisystem pattern. The neurologic presentation is one of chronic severe neurologic disability punctuated by episodic acute deterioration resembling stroke. There is usually prominent cerebellar dysfunction, with severe cerebellar atrophy detected on brain MRI. Usually normal at birth, some patients present with hypotonia and oculomotor abnormalities. Over time, patients display persisting ataxic hypotonic motor impairment and severe cognitive deficiency, retinopathy, epilepsy, acquired microcephaly, thromboembolic strokes, and weakness due to polyneuropathy. Associated non-neurologic findings include growth failure, protein-losing enteropathy, obstructive cardiomyopathy, nephrotic syndrome, deficient antithrombin III or protein C or S. Inverted nipples and subcutaneous fat pads are distinctive physical stigmata that may serve as clues to the disorder. Diagnosis is made by measuring transferrin isoelectric focusing using mass spectrometry, which may not become abnormal until the second or third week of life. Associated biochemical findings include elevated liver transaminases, low serum proteins, anemia, leukopenia, and low serum cholesterol. Treatment with high-dose mannose supplementation may ameliorate systemic symptoms, but the benefit for neurologic and neuromuscular manifestations is uncertain [Sparks and Krasnewich, 2009].
Peroxisomal Disorders
Peroxisomal disorders comprise a group of progressive neurologic diseases with variable age of onset and severity (see Chapter 38). Neonatal forms with prominent neurologic involvement include Zellweger’s syndrome and neonatal adrenoleukodystrophy, both of which are inherited as autosomal-recessive disorders [Steinberg et al., 2006]. Infants with classic Zellweger’s syndrome have multi-organ involvement with typical facial dysmorphic features, ocular anomalies (cataracts, glaucoma, pigmentary retinopathy), hepatic fibrosis, cystic kidney disease, subclinical adrenocortical insufficiency, and cardiac anomalies. Neurologic features include seizures, cranial nerve dysfunction, optic atrophy, and diffuse myopathic weakness. Survivors are multiply and profoundly handicapped. Neuroimaging and pathologic studies have revealed cortical and cerebellar migrational abnormalities and a central white matter demyelinating process. The pathogenesis relates to near-complete failure of peroxisomal function as a result of defective transport of proteins into the peroxisomal matrix. This results in defective oxidation and abnormal accumulation of very long chain fatty acids (VLCFAs), phytanic acid, pipecolic acids, and deficient synthesis of plasmalogens. The typical clinical features should suggest the diagnosis, which is supported by finding markedly elevated plasma VLCFA and decreased plasmalogen levels. Neonatal adrenoleukodystrophy resembles Zellweger’s syndrome in many respects, but is less severe. There are no proven definitive treatments. Trials are under way to evaluate strategies aimed at modifying lipid and fatty acid composition of the diet.
Cholesterol Biosynthesis Defects (Smith–Lemli–Opitz Syndrome)
The molecular defect in Smith–Lemli–Opitz syndrome was recently characterized as a disorder of cholesterol biosynthesis. This condition presents in the neonatal period with a constellation of multiple congenital anomalies and a chronic static encephalopathy [Herman, 2003]. Infants present with microcephaly, abnormal facies with ptosis and micrognathia, genital anomalies, and growth retardation. Less frequent abnormalities include cataracts, congenital heart defects, digital anomalies, and cleft palate. Neurologic features include hypotonia, sensorineural deafness, feeding and swallowing dysfunction, and severe cognitive deficiency. Cerebral malformations are common, including holoprosencephaly, agenesis of the corpus callosum, frontal hypoplasia, and cerebellar hypoplasia. Serum cholesterol is low in 90 percent of cases, but definitive diagnosis rests on finding elevated serum levels of 7- and 8-dehydrocholesterol measured by GC mass spectrometry. Treatment is supportive and symptomatic. Enteral supplements of cholesterol may mitigate some of the symptoms.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Bagci S., Zschocke J., Hoffmann G.F., et al. Pyridoxal phosphate-dependent neonatal epileptic encephalopathy. Arch Dis Child Fetal Neonatal Ed. 2008;93(2):F151-F152.
Boles R.G., Ment L.R., Meyn M.S., et al. Short-term response to dietary therapy in molybdenum cofactor deficiency. Ann Neurol. 1993;34(5):742-744.
Burns C.M., Rutherford M.A., Boardman J.P., et al. Patterns of cerebral injury and neurodevelopmental outcomes after symptomatic neonatal hypoglycemia. Pediatrics. 2008;122(1):65-74.
de Koning T.J., Klomp L.W. Serine-deficiency syndromes. Curr Opin Neurol. 2004;17(2):197-204.
De Vivo D.C., Wang D., Pascual J.M., et al. Glucose transporter protein syndromes. Int Rev Neurobiol. 2002;51:259-288.
den Boer M.E., Dionisi-Vici C., Chakrapani A., et al. Mitochondrial trifunctional protein deficiency: a severe fatty acid oxidation disorder with cardiac and neurologic involvement. J Pediatr. 2003;142(6):684-689.
Dodd P.R., Williams S.H., Gundlach A.L., et al. Glutamate and GABA neurotransmitter systems in the acute phase of maple syrup urine disease and citrullinemia encephalopathies in newborn calves. J Neurochem. 1992;59:582-590.
Enns G.M., Berry S.A., Berry G.T., et al. Survival after treatment with phenylacetate and benzoate for urea-cycle disorders. N Engl J Med. 2007;356(22):2282-2292.
Fingar D.C., Blenis J. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene. 2004;23(18):3151-3171.
Gallagher R.C., Van Hove J.L., Scharer G., et al. Folinic acid-responsive seizures are identical to pyridoxine-dependent epilepsy. Ann Neurol. 2009;65(5):550-556.
Garcia-Cazorla A., Rabier D., Touati G., et al. Pyruvate carboxylase deficiency: metabolic characteristics and new neurological aspects. Ann Neurol. 2006;59(1):121-127.
Haberle J., Gorg B., Toutain A., et al. Inborn error of amino acid synthesis: human glutamine synthetase deficiency. J Inherit Metab Dis. 2006;29(2–3):352-358.
Haeuptle M.A., Hennet T. Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutat. 2009;30(12):1628-1641.
Hay W.W.Jr, Raju T.N., Higgins R.D., et al. Knowledge gaps and research needs for understanding and treating neonatal hypoglycemia: workshop report from Eunice Kennedy Shriver National Institute of Child Health and Human Development. J Pediatr. 2009;155(5):612-617.
Herman G.E. Disorders of cholesterol biosynthesis: prototypic metabolic malformation syndromes. Hum Mol Genet. 2003;12(Spec No 1):R75-R88.
Hoffmann G.F., Zschocke J., Nyhan W.L. Inherited Metabolic Diseases: A Clinical Approach. Heidelberg: Springer; 2010.
Jan W., Zimmerman R.A., Wang Z.J., et al. MR diffusion imaging and MR spectroscopy of maple syrup urine disease during acute metabolic decompensation. Neuroradiology. 2003;45(6):393-399.
Jurecka A. Inborn errors of purine and pyrimidine metabolism. J Inherit Metab Dis. 2009;32(2):247-263.
Kaler S.G., Holmes C.S., Goldstein D.S., et al. Neonatal diagnosis and treatment of Menkes disease. N Engl J Med. 2008;358(6):605-614.
Kapoor R.R., James C., Hussain K. Advances in the diagnosis and management of hyperinsulinemic hypoglycemia. Nat Clin Pract Endocrinol Metab. 2009;5(2):101-112.
Korman S.H., Wexler I.D., Gutman A., et al. Treatment from birth of nonketotic hyperglycinemia due to a novel GLDC mutation. Ann Neurol. 2006;59(2):411-415.
Lang T.F., Parr J.R., Matthews E.E., et al. Practical difficulties in the diagnosis of transient non-ketotic hyperglycinaemia. Dev Med Child Neurol. 2008;50(2):157-159.
Meyburg J., Schmidt J., Hoffmann G.F. Liver cell transplantation in children. Clin Transplant. 2009;23(Suppl 21):75-82.
Mills P.B., Struys E., Jakobs C., et al. Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat Med. 2006;12(3):307-309.
Mills P.B., Footitt E.J., Mills K.A., et al. Genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy (ALDH7A1 deficiency). Brain. 2010;133(7):2148-2159.
Morioka D., Kasahara M., Horikawa R., et al. Efficacy of living donor liver transplantation for patients with methylmalonic acidemia. Am J Transplant. 2007;7(12):2782-2787.
Morton D.H., Strauss K.A., Robinson D.L., et al. Diagnosis and treatment of maple syrup disease: a study of 36 patients. Pediatrics. 2002;109(6):999-1008.
Mourmans J., Majoie C.B., Barth P.G., et al. Sequential MR imaging changes in nonketotic hyperglycinemia. Am J Neuroradiol. 2006;27(1):208-211.
Nagasaka H., Tsukahara H., Yorifuji T., et al. Evaluation of endogenous nitric oxide synthesis in congenital urea cycle enzyme defects. Metabolism. 2009;58(3):278-282.
Nardocci N., Zorzi G., Blau N., et al. Neonatal dopa-responsive extrapyramidal syndrome in twins with recessive GTPCH deficiency. Neurology. 2003;60(2):335-337.
Pascual J.M., Van Heertum R.L., Wang D., et al. Imaging the metabolic footprint of Glut1 deficiency on the brain. Ann Neurol. 2002;52(4):458-464.
Pearl P.L., Gibson K.M., Cortez M.A., et al. Succinic semialdehyde dehydrogenase deficiency: lessons from mice and men. J Inherit Metab Dis. 2009;32(3):343-352.
Pessoa-Pureur R., Wajner M. Cytoskeleton as a potential target in the neuropathology of maple syrup urine disease: insight from animal studies. J Inherit Metab Dis. 2007;30(5):664-672.
Roe C.R., Mochel F. Anaplerotic diet therapy in inherited metabolic disease: therapeutic potential. J Inherit Metab Dis. 2006;29(2–3):332-340.
Sarkissian C.N., Gamez A., Scriver C.R. What we know that could influence future treatment of phenylketonuria. J Inherit Metab Dis. 2009;32(1):3-9.
Sarnaik A.P., Meert K., Hackbarth R., et al. Management of hyponatremic seizures in children with hypertonic saline: a safe and effective strategy. Crit Care Med. 1991;19:758-762.
Saudubray J.M., Nassogne M.C., de Lonlay P., et al. Clinical approach to inherited metabolic disorders in neonates: an overview. Semin Neonatol. 2002;7(1):3-15.
Schwarz G., Mendel R.R., Ribbe M.W. Molybdenum cofactors, enzymes and pathways. Nature. 2009;460(7257):839-847.
Schwarz G., Santamaria-Araujo J.A., Wolf S., et al. Rescue of lethal molybdenum cofactor deficiency by a biosynthetic precursor from Escherichia coli. Hum Mol Genet. 2004;13(12):1249-1255.
Scriver C.R., Beaudet A.L., Sly W.S., et al. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001.
Sigauke E., Rakheja D., Kitson K., et al. Carnitine palmitoyltransferase II deficiency: a clinical, biochemical, and molecular review. Lab Invest. 2003;83(11):1543-1554.
Sparks S.E., Krasnewich D.M. Congenital Disorders of Glycosylation Overview. In: Pagon R.A., Bird T.C., Dolan C.R., Stephens K., editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle, 2009. 1993–2005 Aug 15 [updated 2009 Sep 01]
Steinberg S.J., Dodt G., Raymond G.V., et al. Peroxisome biogenesis disorders. Biochim Biophys Acta. 2006;1763(12):1733-1748.
Summar M.L., Dobbelaere D., Brusilow S., et al. Diagnosis, symptoms, frequency and mortality of 260 patients with urea cycle disorders from a 21-year, multicentre study of acute hyperammonaemic episodes. Acta Paediatr. 2008;97(10):1420-1425.
Swoboda K.J., Saul J.P., McKenna C.E., et al. Aromatic L-amino acid decarboxylase deficiency: overview of clinical features and outcomes. Ann Neurol. 2003;54(Suppl 6):S49-S55.
Takahashi H., Koehler R.C., Brusilow S.W., et al. Inhibition of brain glutamine accumulation prevents cerebral edema in hyperammonemic rats. Am J Physiol. 1991;26:H825.
Tam E.W.Y., Widjaja E., Blaser S.I., et al. Occipital lobe injury and cortical visual outcome after neonatal hypoglycemia. Pediatrics. 2008;122(3):507-512.
Tuchman M., Caldovic L., Daikhin Y., et al. N-carbamylglutamate markedly enhances ureagenesis in N-acetylglutamate deficiency and propionic acidemia as measured by isotopic incorporation and blood biomarkers. Pediatr Res. 2008;64(2):213-217.
Tumer Z., Moller L.B. Menkes disease. Eur J Hum Genet. 2010;18(5):511-518.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 20 Perinatal Metabolic Encephalopathies
Introduction
Box 20-1 Predominant Presenting Clinical Features in Neonatal Genetic Metabolic Disorders
A brief synopsis of each condition is provided, with emphasis on distinctive clinical features, diagnostic approach, and management. Comprehensive discussion of the molecular biology, pathophysiology, and genetics of these disorders may be found in several excellent reviews [Scriver et al., 2001; Hoffmann et al., 2010].
General Approach
Metabolic disease should be considered in the differential diagnosis of any newborn demonstrating symptoms or signs of encephalopathy, which are summarized in Table 20-1. While none of these abnormalities is specific to metabolic disease, the temporal pattern and specific constellation of symptoms taken together often suggest a metabolic disease. Encephalopathies caused by acute correctable metabolic perturbations, such as in hypoglycemic encephalopathy, typically follow a monophasic course that resolves in proportion to the timeliness of correction of the metabolic perturbation. The tempo of genetically determined metabolic diseases is more variable than acquired acute injury such as hypoxia or trauma. It often has a delayed or subacute presentation with unexplained disturbances in state of arousal or neonatal behaviors, such as feeding or visual attentiveness, or with the emergence of myoclonic seizures or movement disorder. Chronic encephalopathies that initially manifest in the newborn period are increasingly being linked to determine metabolic disease genetically. These disorders may have few or no features that distinguish them as metabolic, presenting with nonspecific signs such as hypotonia, depressed neonatal behaviors, or poor feeding. Nervous system manifestations of metabolic disease are generalized in nature, with depression of consciousness or irritability. Motor abnormalities and brainstem dysfunction parallel the severity of disturbed consciousness in a nonlocalizing manner. Suspicion of an inborn error of metabolism should be elevated by any of the following patterns:
Table 20-1 Neurologic Signs and Symptoms of Neonatal Metabolic Disease
| Neurologic Examination Category | Finding in Affected Infant with Metabolic Disease |
|---|---|
| Alterations in consciousness Mild to moderate Severe |
Excessively irritable, or somnolent but awakens with light stimulation Unarousable, but withdraws purposefully Comatose, responds to pain only by posturing, or not at all |
| Brainstem dysfunction | Pupillary abnormalities Oculomotor deficits Depressed or disordered suck and swallow function Disordered control of breathing (hyperpnea, apnea) High-pitched (“cerebral”) cry |
| Cortical special senses | Absent arousal to sound, poor or absent visual fixation |
| Tone and movement abnormalities | Generalized hypotonia or hypertonia Nonepileptic myoclonus Dystonia, rigidity, opisthotonus, oculogyric crises |
| Reflex abnormalities | Exaggerated deep tendon reflexes Disturbed neonatal reflex behaviors (e.g., Moro, grasp) |
| Seizures | Myoclonic, clonic, tonic, subtle, apnea |
| Neuromuscular abnormalities | Diffusely absent tendon reflexes, bilateral symmetric facial or limb weakness, respiratory insufficiency |
| Autonomic dysfunction | Temperature instability, gastrointestinal dysmotility |
Diagnosis is further delineated by appropriate staged laboratory studies, as shown in Table 20-2. The aim of the initial evaluation is to narrow down the differential diagnosis and simultaneously treat potentially life-threatening or handicapping conditions as quickly as possible. The results of “first-stage” studies will reveal abnormalities requiring immediate correction, such as hypoglycemia, hyponatremia, hypocalcemia, or hypoxia. They will further define other organ involvement, such as renal, cardiac, or hepatocellular dysfunction. A constellation of abnormalities on the initial screening testing may suggest a particular inborn error of metabolism (Figure 20-1). If the results of first-stage assessment, combined with clinical history, examination, and neuroimaging, do not provide a specific diagnosis, they should provide the rationale for choosing appropriate “second-stage” studies. These are aimed at identifying the category of metabolic defect, and in some cases provide a probable specific diagnosis that can be verified in “third-stage” studies, in which DNA or tissue sampling provides material for enzyme quantification or genetic analysis. A careful family history should be obtained to ascertain consanguinity, known inherited metabolic disease, excess fetal loss, or infant demise in a sibling under similar circumstances.
Table 20-2 Laboratory Evaluation of the Infant with Suspected Metabolic Encephalopathy
| First Stage Survey and Screening |
Second Stage Identify Category of Metabolic Defect |
Third Stage Verify Enzyme or Gene Defect |
|---|---|---|
| Blood Glucose Electrolytes Mg++, Ca++, PO4 BUN, creatinine Liver enzymes CBC, platelets, PT, PTT Arterial blood gas Ammonia Lactate, pyruvate |
Blood Quantitative plasma amino acids Very long chain fatty acids Acyl carnitine profile Isoelectric transferrin point Copper, ceruloplasmin levels Cholesterol Uric acid |
Tissue biopsy (muscle, liver) for enzyme assay Skin fibroblast culture for enzyme assay DNA studies and specific gene mutation probes Family studies |
| Urine Ketone bodies, reducing substances Protein, blood Cells |
Urine Quantitative amino acids Organic acids Galactose Sulfites Xanthine, hypoxanthine Uric acid Ribosides |
|
| CSF Glucose, protein Cell count Microbiology |
CSF Quantitative amino acids Neurotransmitters – Biopterin metabolites Lactate, pyruvate |
BUN, blood urea nitrogen; CBC, complete blood count; CSF, cerebrospinal fluid; PT, prothrombin time; PTT, partial thromboplastin time.
Correctable Disturbances of Glucose and Salt Balance
Hypoglycemia
Hypoglycemia is common among critically ill newborns and is a frequent manifestation of inborn errors of metabolism. The definition of hypoglycemia is controversial. Blood glucose values reflect a dynamic balance between substrate availability and utilization, and in healthy newborns vary across a broad range, typically >2.2 μM (36 mg/dL), but reported as low as 1.3 μM (23 mg/dL) in healthy breastfed infants in the first few days of life. The risk of neurologic injury from hypoglycemia depends on multiple factors, including gestational age, availability of alternate substrates, metabolic rate, maturity and efficiency of glucose transport systems, status of oxidative metabolism, and differences in regional brain metabolic activity. As such, clinical factors such as coexistent hypoxic-ischemic injury, seizures, and the presence of hyperinsulinism strongly influence the occurrence and nature of hypoglycemia-related neuronal injury. These observations have led a number of investigators to propose an “operational” definition, whereby threshold blood glucose value, for example, 2.5 mmol/L (46 mg/dL), prompts treatment and further evaluation. This threshold may be modified up or down in individual cases, depending on clinical symptoms and factors likely to affect brain substrate demand, such as seizures or hypoxia, and alternative substrate availability, such as lactate or ketones or the effects of hyperinsulinism. Presently, there is insufficient evidence to define specific threshold values as predictive of neurologic injury in all clinical settings, and further research is needed on this point [Hay et al., 2009].
Disorders leading to neonatal hypoglycemia may be grouped into four broad categories: maladaptation to extrauterine life, hyperinsulinism, increased glucose consumption due to prior or intercurrent illness, and inborn errors of metabolism (Table 20-3). Maladaptation to extrauterine life affects premature infants and small-for-gestational-age infants due to low stores of fat, protein, and glycogen; inefficient ketogenesis and gluconeogenesis; or diminished tolerance of feedings. Hyperinsulinism is associated with maternal diabetes and gene defects affecting regulation of insulin production [Kapoor et al., 2009], and should be suspected when severe hypoglycemia (<2.6 mmol/L) without ketonuria in a well-nourished infant persists beyond the first 2 postnatal hours despite initiation of enteral feeds. Hypoglycemia from increased substrate utilization occurs during hypoxia or ischemia due to a shift from aerobic to anaerobic metabolism, sometimes compounded by decreased hepatic gluconeogenesis and ketogenesis. Brain glucose consumption is dramatically increased by seizures from any cause, and may exceed the capacity of cerebral energy metabolism in brain regions compromised by a recent ischemic insult. Inborn errors of metabolism may cause hypoglycemia due to enzyme deficiencies in glycogenolysis or gluconeogenesis, or as a result of hepatocellular dysfunction. The neurologic manifestations in these cases represent combined effects of the hypoglycemia and the metabolic disorder. Table 20-4 lists inborn errors of metabolism commonly associated with neonatal hypoglycemia. Neonatal hypoglycemia may be associated with congenital endocrinopathies affecting the hypothalamic-pituitary-adrenal axis as a result of major brain malformations of the hypothalamic-pituitary region, congenital adrenal hypoplasia, or hypothyroidism.
| Category | Specific Disorders |
|---|---|
| Disorders of adaptation to extrauterine life | Prematurity Intrauterine growth retardation |
| Hyperinsulinism | Infants of diabetic mothers Maternal isoimmune disease Genetic defects of insulin secretion ABCC8, ATP binding cassette C KCNJ11, potassium channel J11 GLUD1, glutamate dehydrogenase GCK, glucokinase HADH, OH–acyl-CoA dehydrogenase SLC16A1, solute carrier 16-1 HNF4A, hepatocyte nuclear factor 4 alpha Beckwith–Wiedemann syndrome Maternal isoimmune disease Congenital disorders of glycosylation |
| Elevated glucose consumption | Hypoxia Global ischemia Seizures |
| Acquired or transient hepatic dysfunction | Post hypoxic-ischemic hepatocellular injury Infectious hepatitis Any cause of liver failure |
| Primary endocrine disorders | Hypopituitarism due to brain malformation Congenital adrenal insufficiency Adrenal hemorrhage Hypothyroidism |
| Inborn errors of metabolism | See Table 20-4 |
Table 20-4 Inborn Errors of Metabolism in which Neonatal Hypoglycemia is a Common Symptom
| Category | Specific Disorders |
|---|---|
| Disorders of carbohydrate metabolism | Hereditary fructose intolerance Fructose 1,6-biphosphatase deficiency Glycogen storage diseases, esp. I and III |
| Fatty acid oxidation defects | Medium-chain and long-chain acyl-CoA dehydrogenase deficiencies Carnitine palmitoyl transferase II deficiency Mitochondrial trifunctional protein deficiency Malonyl-CoA decarboxylase deficiency (MCD) |
| Defects in ketone body synthesis | HMG CoA lyase deficiency |
| Gluconeogenesis defects | Holocarboxylase synthetase deficiency (multiple carboxylase deficiency) Pyruvate carboxylase deficiency |
| Disorders of branched chain amino acid catabolism and related organic acidurias | Maple syrup urine disease Propionic acidemia Methylmalonic acidemia Isovaleric acidemia Ethylmalonic aciduria 3-methyl-glutaconic aciduria 2-methyl-3-OHbutyryl-CoA dehydrogenase deficiency (MHBD) |
The clinical signs and symptoms of hypoglycemia in the neonate are variable and nonspecific, exemplified by infants with hyperinsulinism. Signs and symptoms in the early stages may be dominated by signs of elevated circulating catecholamines: tachycardia, pallor, diaphoresis, irritability, jitteriness or tremor, and exaggerated tendon reflexes and neonatal reflexes (e.g., Moro reflex). Progressive depression of consciousness occurs as cerebral glucose stores are exhausted, associated with hypotonia, depressed reflexes, hypothermia, and often seizures. At its worst, uncorrected hypoglycemia at levels <1.0 μM in the absence of circulating ketone bodies will progress to coma, characterized by isoelectric electroencephalogram (EEG), cerebral edema, flaccid unresponsiveness, apnea, loss of brainstem reflexes, and circulatory collapse. Mild and moderate degrees of symptoms are rapidly reversed with glucose administration, whereas severe encephalopathy reflecting irreversible neuronal injury is not. Hypoglycemic coma is associated with irreversible neuronal necrosis and lasting neurologic deficits, with a predilection for parietal-occipital cortex and periventricular white matter [Burns et al., 2008]. Long-term sequelae may include microcephaly, cortical visual impairment, combined motor and cognitive handicap, and, in some cases, epilepsy [Tam et al., 2008].
The management of neonatal hypoglycemia remains a controversial subject [Hay et al., 2009]. High-risk infants should be screened for hypoglycemia, and provided exogenous feeds, intravenous or enteral, at rates and concentrations sufficient to maintain adequate blood glucose concentrations. Bedside screening for low blood glucose should be performed in any infant who develops signs and symptoms of acute encephalopathy. Symptomatic infants should be treated while the first-stage evaluation proceeds, as outlined in Table 20-2. If ketones are absent and the exogenous glucose requirement is high (>10 mg/kg/min), then hyperinsulinism should be suspected and further evaluated by measuring insulin levels. A paradigm for assessment of hypoglycemia in neonates is shown in Figure 20-2. According to the “operational” approach to the definition of hypoglycemia, treatment of acute symptomatic hypoglycemia should proceed immediately for any infant with a blood glucose <2.5 mmol/L (46 mg/dL), with a minibolus of 200 mg/kg using 10 percent dextrose infused over 2 minutes, followed by continuous infusion of 10 percent dextrose starting at 8 mg/kg/min and increasing as necessary to maintain stable blood glucose levels >2.6 mmol/L. Monitoring the effect of glucose administration on clinical symptoms, particularly the level of consciousness, can help clarify the neurologic significance of the hypoglycemia, and the need for evaluation for other causes of encephalopathy.
Disturbances of Sodium Balance
Hyponatremia
Hyponatremia occurs when sodium losses exceed sodium or water intake, or reabsorption exceeds water excretion, or both. Hyponatremia is common in sick neonates due to immature or damaged renal mechanisms of salt and water balance or to perturbations in the hypothalamic-pituitary mechanisms regulating volume and osmolar status. Hyponatremia during the first 1–2 weeks of life in sick neonates is most commonly due to excess arginine vasopressin (AVP) secretion in association with other diseases such as lung disease, intracranial hemorrhage, pain, and surgical procedures, and rarely is due to congenital adrenal insufficiency. Causes of hypo- or hypernatremia are summarized in Box 20-2.
Box 20-2 Causes of Hyponatremia and Hypernatremia in Neonates
Causes of Hyponatremia
The management of hyponatremic encephalopathy in neonates depends on the cause, the chronicity, and the severity of symptoms. Subacute or chronic (days to weeks) hyponatremia of mild degree (>120 mEq/L) should be treated by determining and correcting the underlying cause. Controversy surrounds the treatment of acute severe hyponatremic encephalopathy, which is usually associated with serum sodium concentrations <120 mEq/L. In such cases, seizures are usually resistant to standard anticonvulsants, and the encephalopathy frequently depresses respiratory drive and airway protective reflexes, all of which constitute a degree of urgency in favor of rapid correction. Rapid correction of hyponatremic seizures in infants and young children can be accomplished without neurologic complications by the use of 3 percent saline [Sarnaik et al., 1991]. The fact remains, however, that very little is known about neurologic complications from rapid correction of acute symptomatic hyponatremia in premature infants and critically ill term neonates who are already at high risk for neurologic injury from other causes. Until better data are available in this population, treatment should proceed with caution.
Hypernatremia
Neurologic manifestations of hypernatremia arise from hypertonic dehydration of neurons and glia, and may be compounded by secondary ischemic injury from hyperviscosity, cerebral venous thrombosis, or intracranial hemorrhage. Mild symptoms accompany mild elevations of serum sodium, generally <160 mEq/L, while severe encephalopathy usually occurs with elevations >170 mEq/L. The causes of hypernatremia are the inverse of those causing hyponatremia: excess sodium intake relative to sodium losses, or excess water losses relative to water intake or reabsorption, or both (see Box 20-2
[/not-level-membership-for-neurology-category]
