The perinatal environment influences saccular and alveolar lung development. Mammalian lung development proceeds through five stages that culminate in the formation of a dichotomously branched organ with many uniformly sized alveoli. Perinatal influences (blue arrows) alter bronchoalveolar development, often resulting in shorter, muscularized airways that terminate in heterogeneously sized alveoli. Such changes perturb efficiency of gas exchange, ability to defend against inhaled toxins and microorganisms, and regeneration of injured regions of the adult lung.
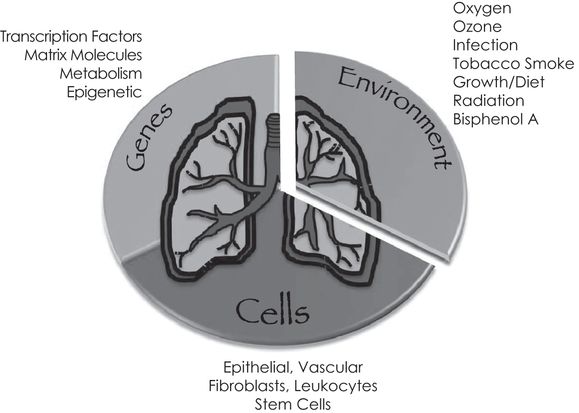
Gene–environment interactions on specific types of cells contribute to lung development. Representative types of genes, environmental influences, and specific cells that work coordinately to create the lung discussed in this chapter are depicted.
Perinatal Influences on Lung Development
Unlike organs such as the heart and brain, the lung is unique in that it is not required for fetal development or survival until birth, when it is called on to replace the placenta’s role in gas exchange. But unlike the placenta that is protected within the mother, the lung is exposed to the external environment. The newborn lung must therefore defend itself and other internal organs against inhaled toxins and pathogens. It does so using anatomical and physiological barriers, innate and adaptive immunity, antioxidant defenses, and a high repair/regenerative capacity (22–24). Anatomical and physiological barriers serve to physically impede entry and enhance removal of harmful agents from the lung. This includes airway constriction, mucociliary clearance, and cough. Innate and adaptive immunity act when the anatomical and physiological barriers fail. Innate defenses are an ancestral form of host defense that uses a limited set of cell surface receptors, including Toll receptors and complement, to detect invading pathogens. Adaptive immunity uses an abundant and diverse set of randomly generated receptors, such as immunoglobulins to detect pathogens and activate cell-mediated killing of infected or damaged cells. Antioxidant defenses mature during the last 15% of lung development and are thought to protect the lung from oxidative damage formed as it transitions out of a low hypoxic in utero environment to air at birth (24). The ability to properly regenerate injured cells and restore normal lung function is perhaps the final level of protection. Accordingly, the same environmental exposures during critical developmental windows may affect proliferation, differentiation, and survival of progenitor/stem cells, thereby altering the balance of cells used to build the lung (Figure 15-3). Successful transition to air at birth therefore requires that the newborn lung is capable of exchanging oxidant gases, defending against oxidative stress and inhaled microbes, and repairing cell injury or regenerating tissue when damaged.
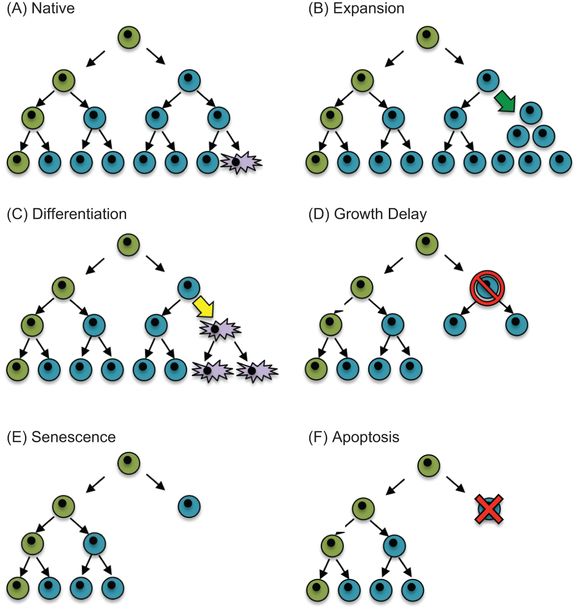
Hypothetical model of how perinatal stress might influence the balance of cells in a tissue. (A) A putative stem cell (green) divides asymmetrically to produce itself and a committed daughter cell (blue) that subsequently divides symmetrically or differentiates into a mature cell (purple). Perinatal stresses may alter the balance of cells in a tissue by (B) stimulating expansion of differentiated cells (green arrow), (C) stimulating differentiation of a committed cell (yellow arrow), (D) transiently inhibiting proliferation (red slashed circle), (E) activating senescence programs that terminate cellular expansion, or (F) inducing cell death (red X).
Oxygen Environment at birth
The transition to breathing oxygen is perhaps one of the most profound environmental changes one will ever experience. At term, the human fetal lung is bathed in amniotic fluid with a pO2 of about 11 mm Hg or less than 1% oxygen (25). Focal hypoxia created by the lower oxygen levels in amniotic fluid relative to arterial levels may stimulate hypoxic signaling of hypoxia-inducible factor (HIF) and vascular endothelial growth factor (VEGF) necessary for promoting vascular growth and lung development (26). Indeed, expression of the transcription factor hypoxia inducible factor (HIF)-1α, HIF-1β, or HIF-2 are required for proper maturation of surfactant production and ability to survive at birth (27,28). Hydroxylation of prolines on HIF by HIF-dependent prolyl hydroxylases stimulates HIF degradation. Inhibition of HIF prolyl hydroxylases in preterm baboons increases HIF expression and improves lung growth and function (29, 30). Birth rapidly creates a hyperoxic environment in the airspace that may influence lung development by terminating HIF signaling. Maturational expression of antioxidant enzymes supports the idea that birth is an oxidizing event that requires protection against reactive oxygen species (ROS) produced during the transition to air (31). Although this implies the oxygen environment at birth is toxic, it may also serve to regulate normal postnatal lung growth. However, there are few studies suggesting that ambient levels of oxygen affect lung development. Perhaps the best example that birth may influence genes controlling postnatal lung development comes from a study showing how expression of the transcription factor Klf4 increases in mice two hours after birth (32). Mice lacking Klf4 display excessive proliferation and apoptosis in the respiratory epithelium and loss of α-smooth muscle actin in septal tips. This implies that the elevated expression of Klf4 at birth is required for the proper transition to air. Another example comes for a recent paper showing how oxygen-induced DNA damage signaling may influence proliferation of cardiomyocytes in the newborn mouse (33). Whether this is directly related to the oxygen environment in the lung or some form of oxidative stress created in the heart as the pAO2 changes remains to be determined.
Although it remains unclear whether the transition to air regulates proper lung development, excessive levels clearly disrupt lung development. Excess levels are often given to preterm infants whose lungs are not adequately prepared to breathe air. Such infants often also have immature antioxidant defense that make them hypersensitive to oxygen-induced injury (34–36). Prolonged oxygen exposure can contribute to bronchopulmonary dysplasia (BPD), a chronic form of lung disease characterized by restrictive airways, inflammation, mild fibrosis, and reduced angiogenesis, increased elastogenesis, and alveolar simplification (34,37). Treatment strategies are generally supportive and designed to enhance oxygen delivery, improve monitoring of oxygen saturations, minimize tissue injury, and stimulate lung growth (for review see (38)). This includes the use of exogenous surfactant, milder ventilation strategies, inhaled nitric oxide, and retinoic acid. Such treatments have reduced infant mortality as well as increase survival of preterm infants born as young as twenty-three to twenty-four weeks gestation. However, these advances have changed the pathology of BPD from a scarring disease seen radiographically to one that requires continuous ventilation and supplemental oxygen treatment. This new BPD appears to be a form of disrupted lung development and impaired lung function that can be recapitulated in newborn animals exposed to high oxygen. Hence, there has been an increasing interest in understanding how oxygen influences lung development with a focus on the potential use of antioxidants and anti-inflammatory agents as therapeutic agents (24,39). But, recombinant human CuZn superoxide dismutase delivered intratracheally to very low-birth-weight preterm infants did not influence the incidence of BPD (40,41). It did, however, improve respiratory health at one year of corrected age and the incidence of retinopathy in preterm infants born at extremely low gestational age. It also improved oxygenation and development of the pulmonary vasculature in a preterm sheep model of prematurity (42). Unfortunately, additional studies testing the efficacy of antioxidant strategies have not gone forward, perhaps because there is some concern that antioxidants may adversely influence signaling pathways required for development of other organs such as brain.
Inflammation created by oxygen damage to the lung or by maternal and newborn infections may also contribute to BPD. But, a study in fetal sheep infected with Ureaplasma parvum, a common microorganism found in women with chorioamnionitis, showed that the inflammatory response to infection alone did not disrupt lung airspace or vascular development (43). On the other hand, maternal exposure of mice to lipopolysaccharide (LPS) and neonatal hyperoxia disrupts cardiac structure and function to the extent that it causes cardiac failure in adults (44). These studies suggest inflammation alone may not be sufficient to promote BPD. Inflammation may enhance oxygen-dependent changes in lung development and hence the pathogenesis of BPD. So, although there has been great interest in the role of oxidants and inflammation to mediate high oxygen-dependent changes in lung development, current antioxidant and anti-inflammatory strategies have yet to prove effective. Perhaps a combination therapy is needed.
Despite reduced mortality of preterm infants, survivors develop persistent pulmonary disease (PPD) later in life, and antioxidant therapy can improve some of these adverse outcomes. Children born preterm often display reduced lung function, are often rehospitalized following respiratory viral infections, and have increased risk for developing asthma (45–49). There is also a growing concern that premature infants will have neurodevelopmental delay and may be at risk for high blood pressure and heart disease as adults (50,51). These observations suggest that many infants born prematurely will require costly lifelong health care. Indeed, according to the National Heart Lung and Blood Institute (NHLBI) web site (http://www.nhlbi.nih.gov/new/press/06-07-26.htm), the annual costs of treating children born prematurely in 2005 were $26.2 billion dollars, of which 10% was just for treating infants with BPD. The high cost of treating prematurity is second to the treatment of asthma and far exceeds costs of treating cystic fibrosis. Hence, there is an urgent need to better understand how prematurity, and more specifically, how early-life exposure to oxygen, behaves as an environmental risk factor for promoting disease later in life (52).
A number of clinical trials have attempted to define an acceptable level of oxygen use in preterm infants that minimizes harm. Early clinical trials showed an increase in the incidence of retinopathy of prematurity (ROP) with the unrestricted use of oxygen (53). However, the Supplemental Therapeutic Oxygen for Pre-threshold Retinopathy of Prematurity (STOP-ROP) trial found high oxygen saturation levels did not increase the severity of ROP in those infants with prethreshold ROP (54). Subsequently, the Benefits of Oxygen Saturation Targeting (BOOST) trial showed how a higher oxygen-saturation range prolonged oxygen dependence (55). These and other studies identified the need to know whether the incidence of ROP was less in infants treated with a lower oxygen saturation target. The Surfactant, Positive Pressure, and Pulse Oximetry Randomized Trial (SUPPORT) compared the incidence of ROP in 1300 infants born between twenty-four and twenty-eight weeks gestation who were treated with the use of a low (85–89%) or high (91–95%) oxygen saturation target (56). While infants treated with a low oxygen saturation target had reduced ROP, mortality was increased when compared to infants treated with a high oxygen saturation target. Similar findings were recently reported in the BOOST II trial, which enrolled 2,400 infants at fifty-four hospitals in the United Kingdom, Australia, and New Zealand (57). While none of these clinical trials established an acceptable level of oxygen use in preterm infants, they collectively suggest preterm infants should be treated with oxygen saturations in the 90–95% range despite the increased risk for developing ROP. An alternative approach has been to define oxygen exposure as a cumulative dose over the first three days of life (58). Infants whose cumulative oxygen exposure was in the highest quartile were two to three times more likely to experience symptomatic airway dysfunction than infants in the lowest quartile. This implies the environmental load of inhaled oxygen is sufficient to predict risk of oxygen-related respiratory morbidity.
Because we recently wrote a review on animal models of oxygen-induced BPD (16), we will briefly summarize some of the main points on how high oxygen exposure at birth influences lung development in animal models. High oxygen exposure in newborn rodents promotes alveolar simplification and abnormal vascular development (59,60). Failure to produce vessels has been attributed to the oxygen-dependent loss of VEGF (61–63). Inhibiting VEGF signaling disrupts postnatal alveolar development (64–66), while overexpression of VEGF can partially protect the developing rat lung exposed to neonatal hyperoxia (67,68). In addition, neonatal hyperoxia suppresses the number of circulating endothelial cell precursors detected in blood and lung (69) and when restored can attenuate murine models of BPD (70,71). These changes may contribute to pulmonary hypertension and cardiovascular disease observed in human adolescents born preterm (72) and in adult rodents exposed to hyperoxia as neonates (73,74).
The effect of hyperoxia on the respiratory epithelium is equally complex. Alveolar epithelial type II cells express VEGF. Hyperoxia may directly inhibit expression of VEGF and indirectly by affecting the expansion of type II cells (59,75,76). Exposure of newborn rodents to hyperoxia for several days inhibits proliferation of type II cells (75). Growth arrest may be mediated by cell cycle checkpoints activated in response to oxidative DNA damage (77). The observation that overexpression of extracellular superoxide dismutase in type II cells attenuates oxygen-dependent changes in newborn lung development and DNA strand breaks supports this idea (78,79). Additional DNA damage may occur when leukocytes are recruited to the injured lung (80). Cell cycle checkpoints are thought to prevent replication of damaged DNA and also allow time for repair to take place. As seen in adult mice lacking the cell cycle inhibitor p21 (cdkn1), cell cycle arrest may limit the extent of oxidative stress and damage created by early-life oxygen exposure (81). On the other hand, we recently showed how short-term hyperoxia stimulates expansion of type II cells in newborn mice, which are subsequently growth arrested with continued exposure (82). Hyperplasia of type II cells has also been observed in a preterm baboon model of BPD and in adult rats exposed to sublethal doses of oxygen (4, 27). Although the relevance to lung disease remains to be determined, excessively expanded type II cells produced during neonatal hyperoxia are slowly pruned or depleted over several weeks when mice are returned to room air (82). Because type II cells produce surfactant necessary for reducing alveolar surface tension, express innate immune and other inflammatory genes, and function as transient amplifying precursors for type I cells, their depletion in adult mice exposed to neonatal hyperoxia may contribute to the altered lung function and host response to respiratory viral infection observed later in life (83,84).
Although early-life high oxygen exposure clearly perturbs normal lung development, sadly, a codified model of oxygen exposure in animal models has yet to be defined or accepted by the research community. This makes it difficult to compare outcomes between investigators who have used different doses and durations of hyperoxia on different developmental windows (16). Sex and strain of the animal can also influence how the lung responds to hyperoxia (85,86). We have taken two approaches to address this issue. First, preterm human infants are often born and exposed to excess oxygen during the saccular stage of lung. They typically go home breathing room air. Because the saccular stage of lung development in mice is between e17.5 and pnd 4, we have exposed mice to hyperoxia between birth and pnd4. The oxygen-exposed mice are then returned to room air so that we can study the long-term effects of oxygen exposure during the saccular phase of lung development. We found that exposure to 100% oxygen alters the balance of alveolar epithelial type I and II cells, promotes alveolar simplification, and alters the host response to influenza A virus infection (87). Exposure to 60% oxygen for four days altered lung development but not the host response to infection. Second, by exposing newborn mice to different levels of oxygen that produced the same cumulative dose, we were able to confirm the idea that quantifying neonatal cumulative excess oxygen can predict risk for respiratory morbidity later in life (88). Quantifying oxygen exposure as cumulative dose over different developmental windows may therefore help interpret findings from other studies that used different doses and durations of exposure. For example, adult mice or rats exposed to 100% oxygen as neonates develop cardiovascular disease (73,74). Perhaps the cumulative oxygen model can predict risk for high blood pressure such as seen in adolescent humans who were born preterm (72).
While preterm infants are often exposed to high oxygen at birth, people born at high altitude are born into low partial pressures of oxygen or hypoxia. It has been known for several decades that high altitude native Tibetans have a depressed response to hypoxia when at sea level (89). Whereas hypoxia stimulates erythropoietin and hemoglobin concentrations, natives born at high altitude develop larger lungs with increased functional capacity that is better suited for breathing lower partial pressures of oxygen (90). Hypoxia-induced lung growth is not seen after the age of ten, suggesting that the low oxygen-environment at birth affects postnatal lung development. Lower levels of hemoglobin have also been reported in high-altitude Tibetans and Bolivian Aymara people (91). Although lower levels of hemoglobin may seem counterproductive under high altitude conditions, it may be protective because elevated hemoglobin concentrations typically observed in hikers at high altitude are associated with mountain sickness and cardiovascular disease. In 2010, several studies were published showing how lower hemoglobin levels correlated positively with haplotypes in the HIF prolyl hydroxylase EGLN1, HIF-2α, and PPARA (92–95). Interestingly, the pattern of variation in EGLN1 differed between Tibetans and Andean natives (95). The Tibetan PHD2 haplotype (D4E/C127S) diminishes interaction of PHD2 with the heat shock protein chaperone p23, thereby reducing hydroxylation activity and downregulation of HIF pathway (96). Higher blood flow and circulating nitric oxide products may also serve to protect against high-altitude hypoxia (97). Exposure of newborn mice to low oxygen has revealed other pathways that may help clarify how a low oxygen environment influences development of the lung and other organs. For example, exposure of newborn mice to 12% oxygen causes lung simplification and intriguingly alters many of the same signaling pathways (such as TGF-β, PPAR-γ) typically activated by high oxygen exposure (98,99). As defined by elevated tumor necrosis factor-α and interleukin-6, neonatal hypoxia promotes an inflammatory state in rat heart and skeletal muscle (100). Thus, the low oxygen environment, such as seen in people living at high altitude, permanently influences postnatal lung development.
The successful adaptation of the lung to hypoxia at birth may come at an increased cost of altered long-term health of the high altitude native (for review see (90)). When compared to low landers, high-altitude natives have increased risk for cardiovascular disease particularly related to cardiac hypertrophy. A zip code study of children born at high altitude in Colorado suggests that birth at high altitude increases rehospitalization following infection with respiratory syncytial virus (RSV) (101). There is also some evidence that living at high altitude reduces brain activity (102). High-altitude natives may have lower rates of obesity (103), but are often born small for gestational age and have transient growth delay with compensatory catchup growth (90, 104). Rats living at high altitude also develop hypertension and have a shortened life span that can be reversed when birthed into sea-level amounts of oxygen (105). Not only do these data imply birth into low oxygen can be an early antecedent of disease later in life, but they also surprisingly sound like diseases attributed to prematurity and exposure to too much oxygen at birth. In other words, being born too early and exposed to too much oxygen or being born at high altitude and thus less oxygen alters lung development, host response to respiratory viral infections, cognition, and overall growth. This implies that complete development of the lung and perhaps other organs is heavily influenced by the amount of oxygen present in the environment at birth.
So, why would the lung and other organs respond to different levels of oxygen at birth? The geological record suggests that the oxygen environment ranged from as low as 15% to as high as 35% over the past 300 million years (106). These fluctuating changes in oxygen levels may have substantially influenced development and evolution of the mammalian lung and perhaps other organs (106,107). In fact, the transition from aquatic to terrestrial habitation by vertebrates likely occurred during a time when atmospheric levels of oxygen were rising (108). The cutaneous respiration and inadequate removal of carbon dioxide by aquatic species was incompatible with life on land, leading to the evolution of a more sophisticated vascular and respiratory tree (108). As organismal size and complexity increased over time, these two systems became critical for the efficient uptake and transport of oxygen to tissues and organs, thus increasing chances for survival (109). In Drosophila melanogaster, high oxygen increases body weight and wing size, whereas low oxygen promotes smaller body weight and wing size (106). Changes in oxygen concentrations negatively correlate with tracheal diameter and cell size in these insects (106, 110). Hence, the oxygen environment at birth is a modifier of evolutionary plasticity for the developing lung and other organs.
Additional Old and New World Perinatal Influences to Lung Development
The developing lung is constantly exposed to other environmental influences, both as a fetus and as a newborn. If the modern lung evolved in response to a changing oxygen environment, perhaps it evolved in response to other ancient environmental influences as well. Ozone, radiation, diet, and pathogens like viruses and bacteria are additional examples of old world perinatal influences. Because the mammalian lung evolved in an environment containing these agents, it presumably is programmed to respond to them. Consistent with this argument, expression profiling studies have defined a pattern of gene expression that defines a “time-to-birth” program wherein developmental genes are expressed first and genes involved in oxygen transport, protection against reactive oxygen species, and host defense are expressed near birth (111). This type of genetic programming ensures that the lung is ready to breathe air and defend against environmental toxins and pathogens at birth. Cigarette smoke and bisphenol A are examples of new world pollutants because they are products whose abundance in the environment is caused by human activity. The response to these agents may be more primitive or less programmed because the lung did not evolve when they were present in the environment. The following briefly describes the pulmonary response to an old world pollutant, ozone, and the new world pollutants, tobacco smoke and bisphenol A. Regardless of the exposure material, the net outcome is an effort to develop a lung that is best suited to function in an environment containing that material.
The photolysis of oxygen into ozone in the upper atmosphere by UV radiation allowed for the massive expansion of life. Normally there is little ozone in the lower atmosphere because UV radiation is weak. However, ozone has emerged as an urban pollutant created by chemical reactions between nitrogen oxides and volatile organic compounds typically produced from industries producing electricity, motor vehicle exhaust, gasoline vapors, and chemical solvents. Epidemiologic evidence suggests that ozone is an airway irritant that provokes asthma in both adults and children (18). Children may be at greatest risk because their lungs are still developing and likely to have greater exposure to ozone and other air pollutants as they play outdoors in polluted air. Indeed, early-life episodic ozone exposures of 0.5 parts per millions (ppm) in nonhuman primates and rats shorten growth of distal airways (112,113). A single acute exposure of 1 ppm for three hours is sufficient to inhibit alveolar cell proliferation in three-day-old mice (114). But, it is important to remember that ozone is not the only component of air pollution and that pollutant effects can originate from maternal exposures. For example, maternal diesel exhaust exposure enhances ozone-dependent airway hyperreactivity in newborn mice (115). Nonetheless, ozone is an oxidant gas that may influence distal airway and alveolar structure through similar mechanisms as hyperoxia, namely oxidant injury and inflammation (17). By shortening the airway and increasing alveolar size, the lung may be attempting to limit the amount of surface exposed to a highly reactive oxidant gas and yet still be able to efficiently exchange oxygen and carbon dioxide.
Tobacco smoke exposure is a significant cause of lung disease in the elderly population and is the primary cause of COPD and lung cancer. It has also been linked to airway hyperreactivity and asthma in children (18). Studies in animal models reveal maternal and neonatal exposures are sufficient to permanently affect host defense and lung development. Prenatal nicotine exposure reduces lung function in newborn rhesus monkeys presumably via the binding of nicotine to the nicotinic acetylcholine receptor (116). Neonatal mice exposed to cigarette smoke have reduced innate immunity and slight deficits in alveolar development as adults (117). Interestingly, high oxygen exposure in newborn mice enhances COPD in adult mice exposed to tobacco smoke products (118). Given the large number of pollutants present in tobacco smoke, it will be challenging to figure out which ones are responsible for altering lung development and how this happens. However, changes in innate immunity and increased airway hyperreactivity may represent an attempt to build a lung that is best suited to exclude particulate matter whose origins may be less molecularly defined than oxidant gases (i.e., antioxidants) or pathogens (i.e., toll-receptors).
Bisphenol A (BPA) is an organic chemical used to make polycarbonate plastics and epoxy resins. Because it is used to produce materials for packaging and storing food, its effects on children’s health are a concern. Fetal exposures in rhesus macaques stimulate expression of Scgb1a1 and MUC5AC/5B in the proximal conducting airways (119). Increased expression of these proteins suggests BPA may be inducing airway mucin defenses. Fetal exposure of pregnant mice to BPA reduced the extent of influenza A virus-associated pulmonary inflammation and antiviral gene expression in the offspring (120). This implies maternal BPA slightly modulates innate immunity. Similarly, maternal administration of BPA enhanced the response to ovalbumin challenge in offspring as defined by increased serum IgE, airway eosinophilia, and airway hyperresponsiveness (121). But those findings were not confirmed in a related study, which actually observed a subtle suppression of response to ovalbumin (122). This may reflect differences in experimental design and the developmental window when BPA was administered. Nonetheless, the observation that the developing lung is permanently affected by maternal exposure to an organic compound that is unlikely to reach the fetus is provocative. It suggests that the fetal lung is sampling what the mother is being exposed to and using that information to modify lung development. Once again, the response seems to be to build a lung designed to exclude materials that would negatively impact its ability to efficiently exchange oxidant gases. But, this example also raises the question of why the developing lung and not every organ is so heavily influenced by perinatal exposures.
Environmental Influences on Developmental Plasticity as an Antecedent of Evolutionary Novelty
By now it should be apparent that prenatal and postnatal environmental influences on genes and cells contribute greatly to proper lung health and disease later in life. Environmental influences are often thought of as being bad because they change lung function often for the worse. Asthma in children of mothers who smoked tobacco during pregnancy is a classic example of how perinatal exposures cause respiratory morbidity later in life (18). Similarly, neonatal ozone exposure inhibits distal branching and promotes alveolar simplification (112,113). Likewise, exposure to hyperoxia causes airway hyperreactivity and alveolar simplification. On the other hand, increased lung development seen in children born at high altitude or under low levels of oxygen is another example of how the environment can influence lung development and health (90). Birth into low oxygen environment stimulates lung growth in order to increase alveolar surface area and hence capture more oxygen under conditions of limiting levels of oxygen. In this example, the developing lung is “functionally adapting” to a low oxygen environment (90). This raises the question of whether the developing lung is trying to functionally adapt to other environmental agents like tobacco smoke and bisphenol A. Hypothetically, some forms of asthma that are linked to an early-life environmental influence may be considered an adaptive response to an airway irritant. For example, childhood asthma caused by mothers who smoked tobacco products may represent a developmental response by an organ expecting to function in a dirty air environment. Increased mucous production and airway smooth muscle reflects the lungs’ first line of defense, namely to exclude toxins via mucociliary clearance, cough, and airway closure. Although this biology is considered productive against respiratory irritants, it is interpreted as morbid when observed in a clean air environment. Exposure to high oxygen at birth inhibits angiogenesis, which could be beneficial when the lung is saturated by oxygen and extensive vasculature is not needed. However, this becomes maladaptive when the lung returns to room air and requires an extensive microvascular network to efficiently capture oxygen at lower partial pressures. Long-term consequences of impaired vascular development include pulmonary hypertension and cardiac hypertrophy with risk for cardiac failure.
Environmental influences on the developing lung may therefore represent a process by which the lung is functionally adapting to that environment. Some changes may provide a selective advantage to an adverse environment and may be evolutionarily beneficial if inherited. Tibetans and Andean natives born at high altitude build larger lungs. Interestingly, Tibetans build larger lungs even if they are born at low altitude. Because Tibetans lived at high altitude for 60,000 years longer than Andeans, they may have evolved heritable changes that drive lung development independent of the oxygen environment. The idea that environmental pressures influence organ development in an evolutionary manner is well rooted in simpler models. The spadefoot toad is one example where the environment can profoundly influence organ development (123). Larvae typically live in small ponds with limited food resources. They develop small jaw muscles, smooth keratinized mouth parts, and an elongated gut. However, when ponds are larger and support growth of shrimp, larvae become carnivores and develop large jaw muscles, notched and keratinized parts of the mouth, and a shorter gut better suited for eating shrimp. The peppered moth in England is an example of how the environment influences survival of two highly related subspecies (124). The light-colored species was protected from predators because it could hide in light-colored trees and lichen. Air pollution produced during the Industrial Revolution killed lichen, and the trees became black from the soot. The light-colored subspecies became susceptible to predators, while the dark-colored species was better able to hide on trees and therefore flourished. As air quality improved over the past decade, the light-colored species has become more common. Although this serves as an example of how the environment affects two different subspecies, it represents how the environment influences the fitness of a species.
Environmental influences on developmental plasticity should therefore be considered an antecedent of evolutionary novelty. In other words, the developing lung is responding to environmental influences so that it is best suited to function in that environment. Rather than considering perinatal influences as disruptive to lung development, perhaps we should consider them as an integration of ecological and evolutionary developmental biology (eco-evo-devo) designed to generate novelty. Those environmental influences that are beneficial will become heritable over time (125). Others may be maladaptive, impede normal lung development, and increase risk for respiratory morbidity later in life. Such changes would not be considered evolutionarily beneficial to the species and would therefore not be inherited. Assuming this concept has some validity, it becomes less surprising that the research literature is replete with papers documenting how the environment alters expression of virtually every molecular pathway known to be involved in saccular and alveolar development. In other words, use all the available tools necessary to functionally adapt to the environment.
Research Opportunities
The widespread acceptance that gene–environment interactions during critical developmental windows profoundly alter lung development and respiratory health over a lifetime provides the foundation to define new questions and research opportunities. Although the following examples reflect those by the author, they hopefully will stimulate additional ideas that will help clarify how early-life environmental exposures affect respiratory health later in life.
1. In the early sixteenth century, Paracelsus proclaimed, “All things are poison and nothing is without poison; only the dose permits something not to be poisonous.” When studying how a single agent or mixture of environmental factors influences lung development, the dose and duration of exposure needs to be considered. A super physiologic or prolonged dose may generate experimental data, but whether those outcomes are grounded in reality of how lower real-world doses affect the lung needs to be considered when designing experiments.
2. Pediatricians often say, “Children are not little adults” because children respond to drugs and disease differently than adults. But even children are not fetuses. Future studies therefore need to define prenatal and postnatal developmental windows within which specific environmental factors influence lung development. Identifying developmental windows of sensitivity or vulnerability will help clarify mechanisms of action and potentially guide development of age-specific interventions.
3. It is often written in review articles that the normal adult mammalian lung contains approximately forty different cell types, yet the origin of this statement seems to have disappeared in the historical literature. However, it should not be surprising to find that this is a gross underestimation when one considers how expression of cell surface receptors has markedly increased the diversity of leukocytes present in the lung (126). The emerging use of microfluidic single-cell RNA sequencing is also uncovering an equally rich diversity among the nonhematopoietic population of cells (127,128). Pulse-chase labeling with 3H-thymidine or cell-restricted fluorescent reporter genes and cell-specific ablation with toxins has identified region-specific niches containing stem cells required for proper lung development and repair (129). Having more than one stem cell niche may seem excessive or redundant until one appreciates that deposition of pollutants (particles and gases) in the airspace varies according to size, shape, and chemical reactivity (130). Unique specific stem cell niches may therefore have evolved to facilitate repair of specific areas of the lung damaged by region-specific pollutants. The molecular and cellular tools being used to identify and characterize the “omics” of stem cells need to be used to understand how the environment influences cell-specific pathways and processes controlling lung development.
4. It may equally prudent to identify hallmarks of perinatal influences to lung development much like the cancer field has identified hallmarks of cancer progression. This includes changes in proliferative signaling, evasion of growth suppressors, resistance to cell death, replicative immortality, increased angiogenesis, enhanced cell invasion and metastasis, reprogramming of energy metabolism, and evading immune destruction (131). Underlying these changes is genomic instability and inflammation. Interestingly, the same environmental stressors and chemicals that promote cancer also affect postnatal lung development (132,133). Except for genomic instability, some of the hallmarks defined in cancer progression may therefore also be involved in reprogramming cell fate and hence organ development.
5. Environmental factors alter development by epigenetically altering cells toward different fates. Because epigenetics reflects heritable genetic changes that are not caused by changes in the DNA sequence, more research is needed to know whether perinatal influences on lung development can be reversed and whether they are heritable.
6. As an open conduit to the environment, the lung is uniquely poised to act as a sensor of the environment and transfer that information to other organs. Because perinatal influences often affect development of other organs, future studies should also consider the lung as a portal for how other organs respond to a changing environment.
Disclosures
The authors report no proprietary or commercial interest in any product mentioned or concept discussed in this article.
Acknowledgments
A single review cannot capture all the information in the literature related to that topic. I apologize to those whose work was not included. I am grateful to my colleagues and the past and present members of my laboratory who have helped me understand how early-life oxygen exposure alters lung development and function. This work was supported in part by National Institutes of Health Grants HL-067392, HL-091968, and HL-097141 (M. A. O’Reilly).
References
Related posts:
 Lung Growth Through the “Life Course” and Predictors and Determinants of Chronic Respiratory Disorders
Lung Growth Through the “Life Course” and Predictors and Determinants of Chronic Respiratory Disorders
 Alveolarization into Adulthood
Alveolarization into Adulthood
 Chronic Neonatal Lung Injury and Care Strategies to Decrease Injury
Chronic Neonatal Lung Injury and Care Strategies to Decrease Injury
 Perinatal Modifiers of Lung Structure and Function
Perinatal Modifiers of Lung Structure and Function
 Surfactant During Lung Development
Surfactant During Lung Development
 Congenital Malformations of the Lung
Congenital Malformations of the Lung
