[level-membership-for-hematology-oncology-and-palliative-medicine-category]
8 Pediatric Neuro-Oncology
Introduction
EPIDEMIOLOGY
The incidence of brain tumors among children (ages 0 to 14 years) is highest in the Scandinavian countries (31.4 per 106 per annum) followed by Western Europe, the United States of America, and New Zealand (24 to 27 per 106). The age-specific incidence for brain tumors is highest among children less than 5 years of age (36 per 106 per annum), then declines among those 5 to 9 years (31.9 per 106) and 10 to 14 years (24.6 per 106) to the 15-19 age group (20.2 per 106 annually).1–4
Age correlates with the relative site of origin among cancers of the brain, in a pattern that is quite different than that observed among adults. Brainstem gliomas (BSG) are more common in children between 5 and 9 years of age (5.9 per 106 per annum) than among preschoolers (4.7 per 106), adolescents of 10 to 14 years (2.8 per 106), or older teenagers (1.7 per 106 per annum). Cerebellar neoplasms are similarly distributed, occurring most frequently between 5 and 9 years of age (9.7 per 106). By comparison, the relative distribution of tumors arising in the cerebrum gradually increases following the first 5 years of life (5.8 per 106 per annum) through adolescence (15 to 19 years of age) (7 per 106).4 In contrast, during adulthood, the vast majority of primary CNS neoplasms are supratentorial in origin.
Reliable epidemiologic data confirm an increase in the incidence of children with gliomas between 1973 and 1992. This increase has been restricted to the AST (World Health Organization Grades I and II), with an average yearly rise of 3%. The trend has been more notable among females, in whom there has been an average annual increase of 3.9%. During this period, there was no change in the relative incidence of the other “classic” childhood CNS tumors, such as MBL and EPD.5
Among children and adolescents, tumors of astrocytic lineage constitute about 52% of reported cases. Other types of gliomas, such as gangliogliomas or mixed AST-OLG, contribute an additional 15.5%.4 There are two clearly defined peaks in relative incidence, observed at five years (20.7 per 106 per annum) and thirteen (19.7 per 106) years of age.3 After puberty, the incidence of AST and malignant astrocytomas (MA) falls to 12.3 per 106 annually.1
The PNETs comprise neoplasms considered “embryonal” in their pathogenetic derivation, and include the MBL, medulloepithelioma, pineoblastoma and the “cerebral neuroblastoma.” Overall, this family of cancers comprises only 3.6% of all brain tumors.6 As a group, the PNET constitute about 22% of primary CNS neoplasms occurring during childhood and adolescence.4 Their relative incidence declines throughout childhood, from the peak seen among children less than 5 years of age (9.6 per 106 per annum), through those of 5 to 9 years of age (7.4 per 106), 10 to 14 years (4 per 106), and older adolescents (2.5 per 106 annually).4 According to the National Cancer Institute’s SEER data, the incidence of MBL-PNET rose 23% between 1973-1976 and 1993-1998, that is from 4 to 4.9 per 106 person-years (PY).10
Ependymomas constitute about 9% of tumors arising within the CNS during childhood and adolescence, but represent only 3% of brain tumors overall.4,9 The relative incidence varies among age cohorts, being highest among infants, toddlers, and preschoolers (5.6 per 106 per annum), then falling to 1.1-1.6 per 106 annually among children older than 5 years of age.1 The peak age at diagnosis is during the second year of life (8.6 per 106).4
Germ cell tumors present in three principal age periods. Congenital GCTs are typically benign teratomas, and during infancy (1 month to 3 years), endodermal sinus tumors and/or malignant teratomas predominate. Adolescence and young adulthood are usually associated with endodermal sinus tumors and embryonal carcinomas, often with teratomatous elements. In contrast to the prevalence of these nongerminomatous GCTs (NG-GCT) among the young, the majority of adult GCTs are pure germinomas.8 In Japan, Taiwan, and South Korea, GCTs comprise 2.1% to 9.4% of primary intracranial neoplasms.9 This is consistently higher than the 0.4% to 3.4% reported in western series.10 In North America, origin within the brain accounts for 14% of all GCTs occurring among persons less than 20 years of age. Their relative incidence has steadily risen from 0.5 per 106 per annum in the 1970s to 1.5 per 106 in the late 1990s, which, within the industrialized countries, has suggested an environmental influence for their pathogenesis.
The observed survival rates for malignant CNS tumors diagnosed between 1973 and 1999 were grim: 50.0% at 1 year, 35.0% at 2 years, 25.6% at 5 years, and only 19.5% at 10 years from diagnosis. This translates to a mortality rate of 5.3/100,000 PY for males and 3.6/100,000 PY among females.2
PROPOSED ETIOLOGIC FACTORS
There is an approximately three- to nine-fold increased risk of developing a brain tumor should a family member be so affected; however, the nature of this association is not understood.11 Allegations have been raised regarding electromagnetic waves, nitrosourea derivatives in processed meats, pesticides, and unique occupational hazards. However, no chemical agent not already known to be carcinogenic for other human tissues has been found to be uniquely associated with cancer of the brain. Neuroectodermal tissue is vulnerable to the carcinogenic effects of external-beam radiation therapy (EBRT). A strong dose-response relationship exists, as exposures higher than 2.5 Gray (Gy) are associated with a 20-fold increased carcinogenic potential.12 The latency period for postradiation neuro-oncogenesis stretches from 12 years to 15 years (range 4 to 30 years).13
CLINICAL PRESENTATION
Low-Grade and Malignant Glioma
The child afflicted with a cerebral neoplasm complains of symptoms referable to its location. Cognitive impairment (15% to 20%) and seizures, either at presentation (about one third) or during the course of the illness (50% to 70%), are common problems that bring the patient to medical attention.14 Superficial cerebral tumors may produce partial, complex partial, or secondarily generalized seizures. Symptoms of dysphasia, mental confusion, or motor deficit are predictive of significantly impaired physical, cognitive, emotional, and social function.15 Motor or sensory compromise will reflect proximity to eloquent cortex. Deeply seated neoplasms, or those situated so as to obstruct the drainage of cerebrospinal fluid (CSF), produce headache, altered mental status, and, possibly, visual obscuration with or without localizing corticospinal tract findings. The pattern of headache, such as those which awaken the patient from sleep (10% to 32%), early morning occurrence followed by vomiting, as well as the influence of position, posture, or exercise (20% to 32%), contribute to the recognition of increased intracranial pressure. Recent onset, evolution of the headache’s intensity, papilledema and/or focal deficits on exam also expedite a diagnostic investigation.16 Brainstem gliomas are recognized by the combination of multiple cranioneuropathies, corticospinal tract findings, ataxia, and/or obstructive hydrocephalus.
Medulloblastoma
Park et al.17 reviewed 144 patients treated at a major Canadian referral center between 1950 through 1980. The prediagnosis symptomatic interval was less than 6 weeks in 51%, and less than 12 weeks in 76%. The age at the time of diagnosis ranged from 7 weeks to 15 years. Eighty-one percent of patients were between 1 and 10 years of age. The clinical presentation was that of a posterior fossa mass, i.e., intracranial hypertension from hydrocephalus (Figure 8-1). Seventy-six percent had papilledema and 65% complained of headache. Recurrent vomiting occurred in 47%. Axial ataxia (62%), nystagmus (44%), and appendicular dysmetria (35%) were common neurologic signs. Hemorrhage into the primary tumor occurred in four, who presented with abrupt neurological deterioration.17
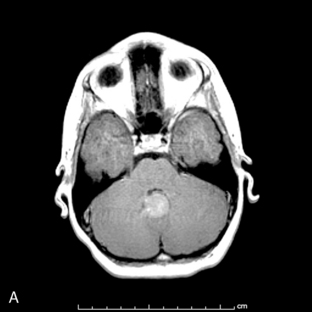
Staging evaluation is an important aspect in establishing the clinical diagnosis. Harisiadis and Chang18 stratified patients based on tumor size (“T”) and extent of metastatic spread (“M”). Spinal subarachnoid dissemination has been identified in 36% to 43% of prospectively studied patients.19 Dissemination to the bone marrow has been reported to be as high as 18% at the time of diagnosis.19 The frequency of this complication at recurrence rises to approximately 35%.20
The “atypical teratoid/rhabdoid tumor” has previously been confused with the MBL. This early childhood neoplasm of the CNS arises either supra- or infra-tentorially and is composed of rhabdoid cells and undifferentiated small cells, with epithelial, mesenchymal, and neural elements. Overall survival (OS) periods of only 11 to 13 months have been reported following surgery and EBRT.21,22
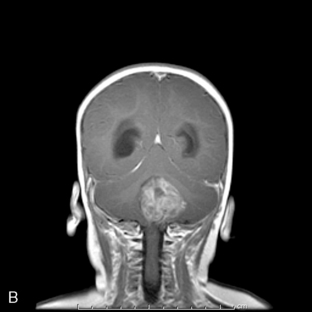
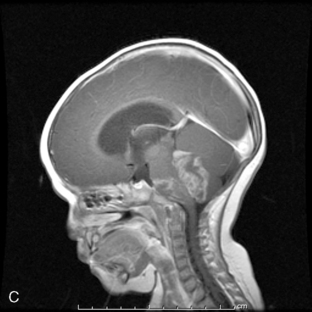
Ependymoma
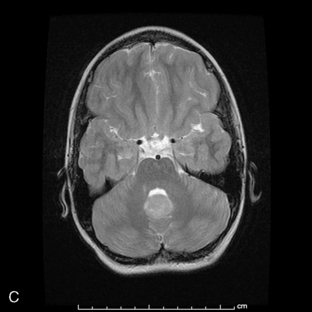
Supratentorial origin is more likely to be associated with corticospinal tract deficits and increased intracranial pressure at the time of presentation. Intratentorial location is frequently identified because of cranioneuropathies, ataxia, hydrocephalus, and a briefer prodrome because of the higher grade of malignancy encountered (Figure 8-2). Spinal EPDs present with back and/or radicular pain as well as lower motor neuron signs. The overall incidence of subarachnoid dissemination with intracranial EPD is reported to be approximately 10% to 22%, but this is influenced by site of origin and tumor grade. Anaplastic infratentorial tumors have a rate approaching 30%, while the incidence of spinal dissemination among the supratentorial EPD is negligible.23
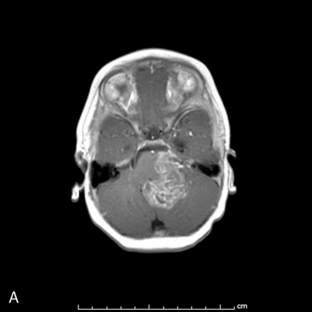
Oligodendroglioma
A series of 37 patients demonstrated that the common presenting symptoms were seizures (43%), headache (38%), motor deficits (38%), and to a lesser degree, behavioral changes (16%). The most common site of origin was the frontal lobe (43%). Low grade histopathology (WHO grades I and II) was documented in 60%, with the remainder being grade III or IV disease.24
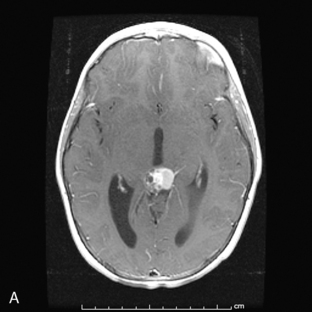
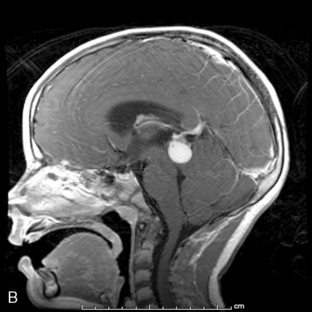
Germ Cell Tumor
Ninety-five percent of primary intracranial GCTs arise along an axis from the suprasellar cistern (37%) to the pineal gland (48%) (Figure 8-3). In one study, germinomas were found to preferentially involve the suprasellar region (57%), while 68% of NG-GCT’s originated in the pineal recess (P < .0001). Germ-cell tumors were found in the suprasellar region in 75% of female patients; pineal involvement was more frequent (67%) among males (P = .0001). The age distribution peaked during early puberty. Nongerminomatous GCTs (24%) were more frequently diagnosed between birth and 9 years than were germinomas (11%) (P < .0001). Among patients with germinomas, 35% were reported to be symptomatic for 6 months or longer, with half of these in excess of 24 months. The prodrome was much shorter among patients with NG-GCT (P = .0007).8
Patients with germinomas, which were commonly suprasellar, presented with chiasmal visual field defects (33%), diabetes insipidus (41%), and hypothalamic-pituitary dysfunction (33%). These neuroendocrine deficits included delay or regression of sexual development (16%), hypopituitarism (16%), and growth failure (9%). Presenting symptoms and signs among the NG-GCTs, which usually localized the lesion to the pineal recess, included hydrocephalus (47%), Parinaud’s sign (34%), obtundation (26%), pyramidal tract findings (21%), and ataxia (19%). Spinal cord metastases were more prevalent among patients with germinomas (11%) and endodermal sinus tumor (23%). Abdominal and pelvic metastases developed in approximately 10% of the 106 patients who were known to have received ventriculoperitoneal shunting.8
Brain Tumors Arising During Infancy
Primary CNS neoplasms during the first 2 years of life are considered quite uncommon in individual practice. However, they actually constitute 13% of all childhood brain tumors. The ratio between malignant (47%) and low-grade tumors is nearly equal in this age-group. Symptoms and signs are related to the site of origin, which is infratentorial in most (40% to 57%).25 The most common symptom has been vomiting (43%), with early morning occurrence in only a third of infants. Headache may be suspected in 33% because of head banging and irritability. Gait ataxia (38%) and macrocephaly (19%) were other findings. The nonspecific nature of these symptoms contributes to the frequent delay in diagnosis. Tonsillar herniation has been documented in 43% at the time of diagnosis, presenting with head tilt (one third) or opisthotonic arching (one third), the latter has been misinterpreted as tonic seizure activity. As many as 10% of these children have presented as failure to thrive secondary to hypothalamic involvement, termed “the diencephalic syndrome.”25
PROGNOSTIC VARIABLES
Malignant Glioma
It has proven difficult to identify biologic determinants of survival that can be conveniently assayed in a timely manner at the time of diagnosis in order to better guide the intensity of therapy. However, some attempts have been encouraging. For example, identification of the labeling index has shown an encouraging prognostic correlation with the histopathologic grade of MG, which in the future may allow stratification of pediatric subjects in clinical trials by relative risk.26 The literature also suggests that expression of the multiple drug resistance (PgP/MDR) proteins and intratumoral hypoxia may also influence outcome.
Histopathologic Grade and Age among Malignant Astrocytomas
The Children’s Cancer Group (CCG) #943 study showed the median survival among children with anaplastic astrocytomas (AAST) was greater than 6 years; among GBM patients, overall survival averaged less than 15 months (P = .012).27 Age and histopathologic grade jointly influenced outcome. The CCG #921 study found that among children less than 24 months old, the 3-year progression-free survival (3Y-PFS) was 36%. The 3Y-PFS for patients with AAST was 44%, which was dramatically better than that for GBM (0%) (Table 8-1).28
Multidrug Resistance Phenotype
The PgP and MRP proteins are thought to function as ATP-dependent transmembrane transporters, which nonspecifically remove large molecular weight solutes from the brain and/or prevent hematogenously borne chemicals from entering the CNS.29 PgP/MDR-1-associated resistance is hypothesized to function at the interface between the blood-brain and blood-tumor barriers as well as within the neoplastic cells themselves. Despite their association with progressive anaplasia and an adverse prognosis, there is actually little evidence that the PgP/MDR1 is a major mechanism of acquired resistance among MG.29–31 Resistance to therapy appears to be a more complex phenomenon which disrupts the mechanisms of repair of genotoxic injury and/or induction of apoptosis.
The O6-methylguanine-DNA methyltransferase (MGMT) enzyme’s function is to counteract the cytotoxic effects of alkylating agents; its expression correlates inversely with survival among adult MG patients. Archival histopathologic specimens from 109 patients treated on the CCG Study #945 were analyzed, and 11% had overexpression of MGMT. The 5Y-PFS was 8.3% among these patients, in contrast to those whose tumors did not show overexpression of MGMT (5Y-PFS of 42%, P = .017). This adverse correlation was independent of age, histopathologic grade, tumor location, or extent of residual disease.32
Hypoxia and the Abrogation of Apoptosis
Intratumoral hypoxia is known to be the most significant predictor of radiotherapeutic resistance.33 Radiation therapy has only one third the effectiveness against hypoxic tumor cells that it does against well-oxygenated cells. It was previously thought that radiation-induced DNA damage required the presence of oxygen radicals. Instead, normal cells, when deprived of oxygen, will arrest in late G1 and early S phase, in concert with hypophosphorylation of the retinoblastoma gene protein, pRB, and die through induction of apoptosis.33 Arrest of the DNA-damaged cell at the G1/S interface is associated with stabilization of intact wild-type TP53 (TP53wt) expression, inhibition of replicative DNA synthesis, and possibly the initiation of repair. TP53wt appears to achieve G1/S arrest through transcriptional regulation of at least one critical downstream target, the Cdk inhibitor p21WAF1/CIP1. This protein inactivates cyclin D/Cdk4, D/Cdk6, and E/Cdk2 complex activity and directly inhibits PCNA, a regulatory subunit of DNA polymerase (the principal replicative DNA polymerase).34
Ionizing radiation also produces G1 arrest in cells with TP53wt and increases TP53 protein expression.35 In contrast, the hypoxic regions within the tumor actually create a reservoir for malignantly transformed cells, which do not arrest at the G1/S interface, are more likely to have mutations in the TP53 gene (TP53 m), and become resistant to TP53-induced apoptosis (Graeber et al., 1996).36 Stable cytoplasmic TP53wt expression has been shown to a favorable predictor for survival among AAST patients (P = .015). By comparison, such expression was not found among the GBM (P = .8), which by definition contain areas of necrosis.37 Intratumoral hypoxia thus constitutes a key mechanism of clonal selection.
Furthermore, hypoxia has also been correlated with insensitivity to carmustine (BCNU) and cisplatin, independently of the putative drug resistance genes MDR1, MRP, O6MGMT, or ERCC.38 Here too, the acquisition of resistance appears be mediated through TP53 m. For example, 86% of TP53wt positive MG cell lines have been shown to be responsive to currently available chemotherapy drugs. In contrast, there were no effective cytotoxic agents against 75% of the glioma cultures expressing TP53 m (P = .0006). Supporting this specificity, the resistance conferred by TP53 m is restricted to alkylators and platinators, which damage DNA, rather than against the microtubular toxins.39
The Molecular Basis for Multiple Models of Glioma Progression: The Tumor Suppressor Genes
There is a large literature on the nonrandom cytogenetic aberrations observed among MGs, which include gains or losses of chromosomes, as well as rearrangements. Some of these changes, such as 10q, 9p, 11p15.5-pter and 19q may be found only among AASTs and/or GBMs.40,41 Loss of heterozygosity (LOH) analysis has correlated 17p deletions with allelic loss of the tumor suppressor gene (TSG), TP53.42 Hence, it has been hypothesized that such nonrandom patterns of genetic loss can be exploited to predict the location of other as yet unknown TSGs. This in turn has generated the concept of modeling mechanisms for tumor progression.
TP53
One pathway in the evolution of the MG involves TP53 m, which occurs in 14% to 46% of adult patients and is preferentially associated with development of the glioma (often an AAST) between 18 and 45 years of age.42,43 Clonal expansion of TP53 m cells have been observed from a small subpopulation within a primary AST to become the dominant cell type within a recurrent, or “secondary” MG.44
Alternate means of TP53 wt inactivation exist as TP53 wt may be complexed by the MDM2 oncogene product. In 8% to 10% of GBM, the MDM2 gene is amplified and overexpressed, supporting the importance of TP53 wt in glial cell cycle dysregulation. A more recent study has identified that about 35% of gliomas demonstrate either TP53 m or methylation of p14(ARF), suggesting that TP53 wt is controlled by down-regulation of p14(ARF).45 Inactivation of the astrocyte’s apoptotic response may therefore be effected either by deletion of 17p, mutation of TP53, MDM2 overpression, or hypermethylation of p14(ARF), thus unmasking multiple points of vulnerability along a common pathway that permit further progression towards the endpoint of malignant transformation.45
PTEN
Another fraction of about 30% (23% to 62%) of adult GBM patients has been characterized by the association of LOH of chromosome 10 with epidermal growth factor receptor (EGFR) amplification. In contrast to the previous group, these patients (typically older individuals) appear to present de novo with a “primary” GBM. The LOH at 10q23 has been demonstrated in approximately 70% of GBMs, and therefore predicted to be the site of another glial TSG.42,43 The “Mutated in Multiple Advanced Cancers” and “Phosphatase and Tensin” gene (MMAC/PTEN) has been localized to chromosome 10q25 and shown to function as a phosphatidylinositol 3,4,5-triphosphate phosphatase, which modulates cell growth as well as apoptosis.46,47
Patients with GBM have demonstrated a significantly higher incidence of LOH at the MMAC/PTEN locus (72%) than did patients with AAST (29%)(P < .0001). The clinical relevance is that patients with LOH at the MMAC/PTEN locus have a more adverse prognosis (P < .0001), even when controlled for age at surgery and histologic grade.48 Conversely, Kaplan-Meier analysis has demonstrated significantly better survival for patients whose MG exhibited high MMAC/PTEN expression.49 These results have been interpreted to implicate alteration or loss of the MMAC/PTEN locus as a late marker of disease progression with ominous significance.
RB
About 15% to 44% of MG patients are reported to have loss of chromosome 13 as well as LOH or partial deletions of the 13q14 allele, which suggests the possibility that the RB gene is also a glioma TSG. Loss of heterozygosity at the RB 1.20 region has not been detected among low-grade gliomas, but occurred in 20% of AAST and 32% of GBM. The presence of LOH at the RB 1.20 region has also been associated with an adverse prognosis, implying that loss/inactivation of pRB contributes to disease progression.50
Investigation suggests that aberrant function of cyclin D, Cdk4, p16INK4A/MTS1, and/or pRB is critical for both the process of transformation and that of progression towards increasing anaplasia among glial neoplasms. Cyclin D1 was originally cloned from a human GBM cDNA library, where its transcript and 34 kD product were in abundant expression. Deletion of p16INK4A/MTS1 and/or amplification of its target Cdk4 occur in 50% of AAST and 85% of GBM.51,52 The coordinate deletion of both p15INK4B/MTS2 and p16INK4A/MTS1 among GBMs suggests that selection favors homozygous deletions of chromosome 9p21 as more efficient for the simultaneous inactivation of both Cdk inhibitors than independent intragenic mutations would be.51 This remarkable specificity suggests that knocking out p16INK4A/MTS1’s inhibition of cyclin D1/Cdk4-induced hyperphosphorylation of pRB marks a critical event in the progression to the most advanced grade of glioma, the GBM.52
Evidence is accumulating that cyclin D1 overexpression, absence of pRB expression, failure of pRB dephosphorylation, and/or pRB cleavage may also contribute to chemotherapeutic drug resistance as well, although the mechanism or mechanisms are not understood. Altered CDKN2/p16 function has been correlated with increased sensitivity to antimetabolite agents as opposed to the alkylators, topoisomerase inhibitors, and microtubular toxins.53
To conclude, the genetic mechanism of glial transformation in children is not understood. In one series of 20 pediatric GBMs, 25% were associated with TP53m, the majority of which presented with a brief prodromal period and were considered “primary.” Loss of p16CDKN2/INK4A/MTS1 was detected in 61% of these GBMs. Overexpression of EGFR was infrequent (11%) and was coincidental with TP53 m in one case. Of the four GBMs, which progressed from lower-grade tumors, only one contained TP53 m.54 In another series of 29 childhood MGs, 95% of cases were found to harbor an alteration in at least one member of the TP53/MDM2/p14ARF tumor suppressor pathway. Overexpression of MDM2, which downregulates TP53 transcriptional activity, was present in 65%, and loss of p14AFT, which inactivates the pathway through MDM2, was found in 10%.55
Among pediatric MGs, the presence of MMAC/PTEN mutations has also been confirmed as a marker of an adverse outcome that is independent of patient age or histopathologic grade (III vs. IV).56 In tumor specimens of 62 evaluable patients treated on the Children’s Cancer Group (CCG) #945 protocol for children with MG (vide infra), alteration of PTEN sequence was detected in just one, in conjunction with loss of chromosome 10. Deletions of PTEN without mutations were found in seven additional specimens. The PTEN alterations were more common among GBM than other histopathologic grades (P = .0056). Although 37% (14/38) of evaluable tumors had increased EGFR expression, only one exhibited amplification of the EGFR gene. This may be interpreted to indicate that pediatric MGs differ in the mechanisms of tumorigenesis from those of adulthood.57
Brainstem Glioma
Favorable prognostic features include (a) protracted symptoms, (b) origin within the optic tectum, the mesencephalon, or at the cervicomedullary junction, (c) lesions which are cystic, focal, and/or dorsally exophytic, (d) onset in adulthood, and (e) neurofibromatosis type I (NF1). By comparison, the adverse markers for disease progression are diffuse pontine infiltration, a high mitotic index, a brief symptomatic prodrome, and multiple cranioneuropathies at presentation.58
Low-Grade Astrocytoma
Low-grade glial neoplasms (LGGN), which include AST, mixed astrocytoma-oligodendroglioma (AST-OLG), and pure OLG, constitute a heterogenous group of tumors without a predictable natural history. The 5Y-OS and 10Y-OS rates for LGGN following resection and EBRT are reported to range between 40% to 70% and 11% to 50%, respectively.59 In a review of 461 AST patients, the 5Y-OS rate was 36%; that is a third of the predicted actuarial survival for a comparable population of unaffected individuals.59 Only 16% of LGGN patients of any age were alive at 15 years following diagnosis.60 Otherwise stated, the long-term survival rate among patients with LGGN is less than that of their peers suffering from acute lymphocytic leukemia.
The literature suggests that age at presentation, seizures as the sole presenting symptom, site of origin, favorable Karnofsky status, absence of contrast enhancement on imaging, histopathologic grade (i.e., pilocytic vs. diffuse) (Figure 8-4), initial tumor dimensions, extent of resection (i.e., unresectable tumor), presence of hydrocephalus, or coexistence of NF1 are the important variables that influence quality of life as well as survival.61 The prognostic value of the labeling index has been previously reviewed.26
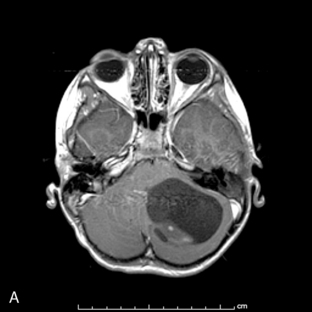
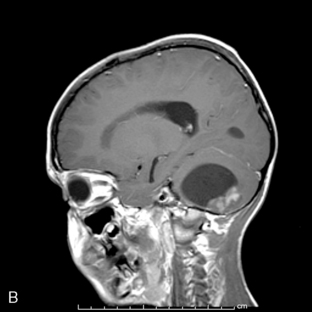
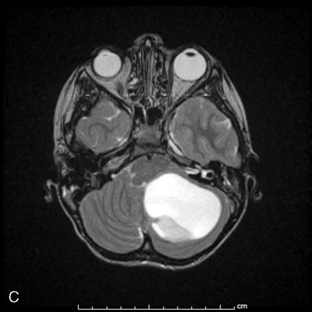
Site of Origin
It is accepted that the biology of LGGN arising within the hypothalamus, optic tectum, and cervico-medullary junction clearly differs from those arising within the pons and cerebrum. Consensus exists that gross total resection of an AST or mixed LGGN during childhood is adequate therapy and does not require immediate postoperative irradiation (Nishio et al., 1989).62 Univariate analysis has demonstrated that optic chiasmal origin and only partial resection are associated with a worsened prognosis.63 This has influenced the controversy regarding EBRT (vide infra).
Neurofibromatosis
The 5Y-OS and 10Y-OS for patients with optic nerve gliomas and NF1 has been reported to be 93% and 81%, respectively. This compares favorably to survival rates of 83% and 76% at 5 and 10 years, respectively, for those children without evidence of NF1. The mean time to disease progression (TDP) was significantly different between the two groups (P < .01) in favor of those with NF1, although this did not impact OS.64 However, this relative advantage does not appear to apply to patients with von Recklinghausen disease who suffer from gliomas of the cerebrum.65
Histopathologic Grade and Malignant Progression among Low-Grade Astrocytomas
The process of malignant transformation was studied among 11 of 65 children with LGGN. The median latency was 5.1 years, and the 15-year cumulative incidence estimate was 6.7 %. Overexpression of TP53 was more common after progression to higher histopathologic grade. Deletions of RB1 and/or CDKN2A were observed in 71% of the LGGN and 90% of their malignant successors. There were PTEN pathway abnormalities found in 76% of patients.66
Medulloblastoma and Primitive Neuroectodermal Tumor
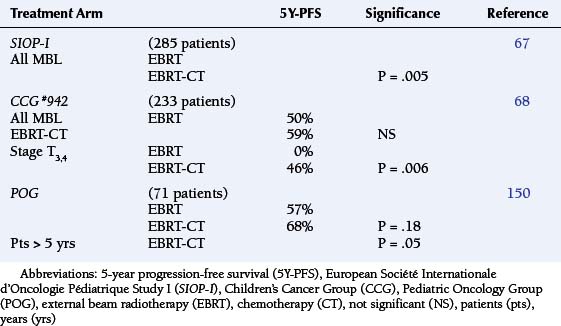
Staging evaluation stratifies patients by tumor size (“T”) and extent of metastatic spread (“M”).18 Table 8-2 summarizes its prognostic significance as derived from the European Societie Internationale Oncologie Pediatrie (SIOP) and the North American CCG study #942.67,68 Stepwise analysis of survival among randomized patients demonstrated earlier age of onset to be the dominant adverse variable (P = .034). There were significant interactions between treatment and M stage, which support postoperative irradiation and chemotherapy among patients with advanced disease, i.e., high T and M stages (P = .004) (Table 8-2).68
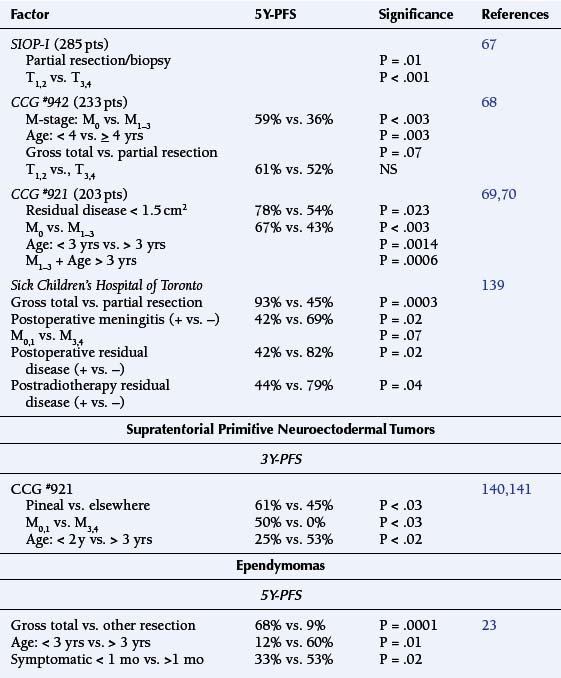
The subsequent CCG #921 study of children with MBL (203 patients) demonstrated the significance of residual disease prior to EBRT, whether determined by extent of surgical resection (P = .023) or by evidence of metastatic tumor (M1–3) (P < .003) (Table 8-2).69,70
Pathologic Studies
A series of 330 cases studied by the Pediatric Oncology Group (POG) found significant anaplasia among 24%, which was strongly associated with an unfortunate outcome. Diffuse or extensive anaplasia was worse than focal involvement. In contrast, extensive nodularity conferred a more favorable course.71 Reevaluation of 347 MBL biopsies treated under the SIOP II trial confirmed that severe anaplasia conferred an adverse prognostic effect on 5-year progression-free survival (49.5%) relative to mild-moderate change (65.4%) (P = .001). This effect was magnified when the presence or absence of extensive apoptosis (P = .00216) was factored in.72
Drug Resistance Genes
There is evidence that the expression of the MDR1, MRP1, LRP and BCRP genes does not correlate with overall outcome among patients with MBL/PNET.73
Biologic Determinants of Survival
Established chromosome abnormalities include amplication of isochromosome 17q, novel amplicons, and losses as well as gains of chromosomes; these are known to occur in greater than 20% of MBL. Copy number abnormalities have been identified in specific regions of chromosomes 1, 8p, 10q, 11p, and 16q, which occur frequently among MBLs and may identify distinct subsets.74 Furthermore, differences in DNA copy number at chromosome regions 1p12-22.1, 9p21, 19p, and chromosome 17 have been used to segregate between MBL and supratentorial PNET. The 9p21 deletions correlated with loss of CDKN2A protein expression more frequently among the PNET (P < .001). Gains of 19p were also more evident among the PNET (P = .02), whereas gains of 17q were more common among the MBL (P = .02).75
(i) Tumor suppressor genes
Haploinsufficiency of the 17p13.3 region is the most commonly found disrupted genetic site (35-50%) among MBLs, which may indicate the site of yet another TSG(s). This chromosomal site contains the “Hypermethylated in Cancer” gene (HIC1), a transcriptional repressor, which is a frequent target of epigenetic gene silencing in MBL. HIC1 is a direct transcriptional repressor of “Atonal Homolog 1” gene (ATOH1), a proneural transcription factor essential for cerebellar development. ATOH1 is a putative target of “(Sonic) Hedgehog” (HH) signaling and its expression has been shown to be required for human MBL cell growth in vitro. The PTCH gene, an inhibitor of SHH signaling, is among the characterized TSG in MBL; however, fewer than 20% of MBLs have mutations in this gene. Recent work suggests that the HIC1 and PTCH1 TSG cooperate to silence ATOH1 expression during a critical phase of granule cell precursor differentiation in the cerebellum to contribute to the malignant progression to MBL.76
Specimens of MBL collected from 65 children treated in the SIOP/United Kingdom Children’s Cancer Group PNET3 trial were segregated into the histopathologic groups such as the large cell/anaplastic phenotype and nodular/desmoplastic variant, among others. Loss of 17p13.3 was found among 38% of samples of all variants, whereas MYCC/MYCN amplification (6%/8% respectively of MBLs) was significantly associated with the large cell/anaplastic phenotype. Loss of 9q22 coincided with the nodular/desmoplastic type. Together with metastatic disease at diagnosis, the large cell/anaplastic phenotype, 17p13.3 loss, or high frequency MYC amplification defined a high-risk group of children whose outcome was significantly worse than those without such tumor characteristics (P = .0002).77 However, the relationships between chromosome 17 lesions with anaplastic/large cell MBL and the abnormalities in the Sonic Hedgehog/PATCH (SHH/PTCH) pathway with the desmoplastic variant remain controversial.78
The SHH antagonist, cyclopamine, blocked expression of the SHH pathway targets PTCH1 and GLI1, lowered Bcl2 levels, and increased apoptosis in MBL cells in vitro. Blockade of the SHH pathway sensitized MBL cells to lovastatin, a proapoptotic agent used for lowering cholesterol levels. The combination of cyclopamine and lovastatin target pathways appear crucial for MBL cell survival.79 Agents targeting the SHH pathway are in clinical trials for the therapy of medulloblastomas.
Activation of the canonical WNT/Wingless (WNT/WG) signaling pathway occurs in up to 25% of cases of primary MBL and is associated with a favorable prognosis. Activation of WNT/WG was determined by evidence of CTNNB1 mutations and/or beta-catenin nuclear stabilization. Loss of chromosome 6 has been correlated with WNT/WG active tumors (P < .001), but few other cytogenetic aberrations including chromosome 17. In contrast, WNT/WG-negative MBLs were found to have losses of chromosomes 17p, 8, 10, and 16, with gains of chromosomes 7 and 17q. This supports the hypothesis of independent pathways of tumorigenesis among MBL, which are of potential clinical relevance.80
TP73 is a member of the TP53 TSG family that is overexpressed in a variety of tumors and mediates apoptotic responses to genotoxic stress. Biopsy samples of MBL and MBL cell lines have been reported to contain elevated levels of TP73 RNA and increased expression of the TAp73 and DeltaNp73 protein products. Overexpression of these induced apoptosis among cultured MBL cells in vitro and sensitized them, resulting in cell death upon exposure to chemotherapeutic agents. TAp73 RNA overexpression within biopsy samples was determined to correlate with a favorable PFS by Kaplan-Meier analysis.81
The 10q23.3 chromosomal region is subject to frequent allelic losses in MBL, which is the locus of the PTEN gene. Activation of the phosphoinosityl 3-kinase/ AKT (P13 k/AKT) signaling pathway appears to be associated with alterations of the PTEN gene. Proliferation of MBL cell lines has been shown to be dependent upon P13 k/AKT signaling and inhibited by a P13K antagonist as well as by AKT overexpression. Reduction of PTEN mRNA and protein expression has been found to be correlated with PTEN promoter hypermethylation in 50% of 22 MBL tissue samples. This suggests that PTEN loss or dysregulation by the P13 k/AKT signaling pathway may be an important mechanism of tumorigenesis for a subset of MBL.82 In contrast, PTEN deletion has not been found among supratentorial PNET biopsies or cell lines.83
(ii) The Putative Oncogenes.
The Duke group studied 31 MBL specimens for MYCC, MYCN and TRKC expression and correlated this with clinical outcome and histopathologic grading. The presence of MYCC mRNA was associated with shorter survival (P = .04) as well as with anaplasia. Regulation of MYCC is influenced by WNT signaling and MXI1 mutations. Nuclear translocation of beta-catenin, a marker of WNT pathway activation, was more common among the MBLs with high MYCC. No MXI1 mutations were detected in the 22 cases examined.84
Archival formalin-fixed, paraffin-embedded MBL samples from 78 patients treated on the prospective European multicenter HIT’91 protocol were studied for DNA amplification of C-MYC and N-MYC and mRNA expression of C-MYC and TRKC. TRKC and C-MYC mRNA expression were identified as independent prognostic factors on multivariate analysis. A favorable-risk group (eight patients), with a 7Y-PFS of 100%, possessed elevated TRKC and reduced C-MYC expression. The poor-risk group (15 patients) had metastatic disease, with high C-MYC and low TRKC mRNA expression. Their 7Y-PFS was only 33%. An intermediate-risk group of the remaining subjects showed a 7Y-PFS of 65%.85 Not all investigators have made similar correlations.86
Ependymoma
Pathologic Grading
The prognostic utility of pathologic grading of ependymomas has engendered a long-standing controversy, as there has been no clear distinction in 5Y-OS rates between “benign” and “malignant” EPD. However, one recent blinded review demonstrated that the PFS at 2 years following postoperative irradiation was 84% for differentiated EPD, and 32% for specimens considered anaplastic.87
Site of Origin
There is a correlation between site of origin and histopathologic grade of malignancy. Approximately two thirds of supratentorial EPDs are high-grade at diagnosis, whereas those arising in the IVth ventricle tend to have a better prognosis.88 A series of 49 patients found that 78% originated infratentorially and exhibited low grade histology.89 Those arising in the spine and cauda equina are usually low-grade and/or myopapillary.
Extent of Surgical Resection
The CCG #921 study accessioned 20 EPD and 12 anaplastic EPD patients, of whom only three had metastatic disease at diagnosis. The prognostic variables of significance included the extent of resection (gross total vs. other, P = .0001) and postoperative residual disease of < 1.5 cm2 (P < .0001), in contrast to age, staging, or treatment, emphasizing the importance of local disease control.90
The French Society of Pediatric Oncology has studied the efficacy of postoperative chemotherapy, with the intent of avoiding EBRT, among 73 children less than 5 years of age with intracranial EPD. The favorable prognostic variables included supratentorial origin (P = .0004) and complete resection (P = .0009). Patients with gross total resection demonstrated a 4Y-OS of 74% in contrast to those with residual disease (35%).91
Univariate and Multivariate Analysis
Three prognostic factors have been identified to have a significant correlation with the 5Y-PFS. These included the extent of surgical resection (P < .0001), age at diagnosis (P = .003), and the prediagnosis symptomatic interval (P = .02).23 Another multicenter retrospective study of 83 children with EPD confirmed that age of less than 3 years, identifiable postoperative residual disease, and Grade III histology were significant adverse factors for PFS in both univariate and multivariate analysis.92
Biologic Determinants of Survival
Overexpression of specific genes (YAP and LOC374491) and downregulation of others such as SULT4A1, NF-[kappa]B2, and PLEK have been implicated in determining the age of onset, relapse potential, and tumor location of pediatric EPD.93,94 The catalytic subunit of telomerase, the human telomere reverse transcriptase (hTERT), which aids in uncontrolled cell proliferation, has been shown to correlate with prognosis among 65 children with EPD. Study of 87 tumors from these patients found the presence of hTERT to be adversely correlated with 5Y-OS (41% vs. 84% in its absence).95
Oligodendroglioma
The accepted favorable prognostic variables for OLG include youth (less than 21 years), low histopathologic grade, and extent of resection.96 Multivariate analysis in a series of 51 patients aged 5 to 75 years found younger age and presentation with seizures alone to be statistically significant for a favorable outcome. Approximately one third of patients of all ages appear to be cured by aggressive treatment including surgical resection, EBRT, and chemotherapy.96
Biologic Determinants of Survival
In several studies of LGGN, the classic histology of the OLG has been strongly associated with 1p deletion (P = .002), loss of 19q (P < .0001), or loss of both (P < .0001).97 Deletion of 9p was found in 36% (8/22), always in association with tumor necrosis and/or microvascular proliferation. In addition, epigenetic alterations of CDKN2A were observed in 71% of these 1p/19q/9p deleted OLG, suggesting that it may have a role in microvascular proliferation.98 A collaborative study of 162 diffuse gliomas (52 OLG, 79 AST, and 31 AST-OLG) has demonstrated that the combined loss of 1p/19q is a statistically significant predictor (P < .0001) of prolonged survival among patients with pure OLG, even after adjusting for patient age and tumor grade (P < .01).97
Allelic deletions on the short arm of chromosome 1 have been correlated with chemosensitivity and a better prognosis for patients with high-grade OLG. The p18INK4C gene is considered to be a good candidate for the putative TSG located at the chromosome 1p32 locus. The incidence of LOH at 1p is 50% among primary OLG; mutations in the gene have been found among recurrent tumors, implicating it as a possible progression factor as well.99 The 1p loss has been shown to be inversely related to deletions of the CDKN2A gene on 9p, which encodes a key cell cycle regulatory molecule p16INK41.100 The 19q13.3 locus is deleted in 50% to 80% of OLG. This region codes for the p190RhoGAP gene, which appears to suppress gliomagenesis by inducing a differentiated glial phenotype.101
Germ Cell Tumors
Histopathologic diagnosis
Matsutani et al.102 performed a clinical analysis of 153 cases treated between 1963 and 1994 at the University of Tokyo Hospital, which revealed pathologic diagnosis to be the dominant prognostic determinant. The 10-year overall survival rate (10Y-OS) for germinomas was 93%. The 10Y-OS rates for mature and “malignant” teratoma were 93% and 71%, respectively. The diagnoses of embryonal carcinoma, endodermal sinus tumor, and choriocarcinoma had a 3Y-OS of 27%.102
Biomarker Expression
The First International Germ Cell Tumor Study observed, in a series of 71 patients, that elevated β-HCG was associated with increased risk of disease progression (P = .06), but did not affect OS.103 In a Japanese study, 33 GCTs (16 germinomas, 11 β-HCG positive germinomas, 3 mixed teratoma-germinoma and 3 NG-GCT) were treated with preradiation chemotherapy. Patients with “pure germinomas” had a 86% 5Y-PFS, while germinoma patients with measurable β-HCG expression exhibited only a 44% 5Y-PFS.104
THERAPEUTIC EFFECTIVENESS AND CONSEQUENT PROGNOSIS
The essential clinical problem of neuro-oncology has been succinctly stated. The patient typically presents with a tumor volume of 30 to 60 gm at the time of diagnosis. This represents 3 to 6 × 1010 cells within the mass, of which the neurosurgeon may be able to resect 20% to 90%. Therefore, a “best-case” postoperative scenario is a residual tumor burden of 3 to 6 × 109 cells. EBRT achieves an approximate two-log cell kill in the treatment of MG. Conventionally dosed chemotherapy may provide an additional one-log cytoreduction. Thus the “ideal” patient completes multimodality therapy with a repository of approximately 3 to 6 × 106 malignant cells, which are presumably then radio- and chemo-resistant.105 The treatment intensity concept may help to explain the patterns of failure when the strategic plan is critiqued for the relative degree of cytoreduction achieved at each sequential stage of multimodality intervention.
Malignant Gliomas
Surgical Intervention
Among newly diagnosed pediatric MG patients, the CCG #943 study demonstrated the impact of the following factors on progression free-survival. Deeply seated diencephalic MG demonstrated a median survival being less than 10 months. Patients receiving only a biopsy experienced a median survival of less than 8 months regardless of subsequent therapy. Partial resection and gross total resection were associated with better but similar long-term outcomes (Table 8-1).27 Among children with MG treated on the CCG phase III study #945, radical surgical resection doubled the 5Y-PFS rate (P = .006) (Table 8-1).106 Unfortunately, it has only been possible to achieve minimal postoperative residual tumor burden for 33% (19% to 45%) of newly diagnosed pediatric MG patients treated in contemporary series.106–109
Radiotherapy
The conventional radiotherapeutic prescription for MG usually consists of 2 Gray (Gy) fractions per day, 5 days/week to achieve total dosages of 40 to 60 Gy. (One Gray equals 100 rad.) The initial Brain Tumor Cooperative Group (BTCG) #6901 trial proved, for the first time, that surgery and conventionally fractionated EBRT (1.8 to 2 Gy) improved the median survival among adult MG patients (Table 8-3). Unfortunately, subsequent studies concluded that irradiation was therapeutic rather than curative. Responses were not improved by altering fractionation schedules or adding the radiosensitizing agents then available.110 These studies have not been replicated among children with MG.
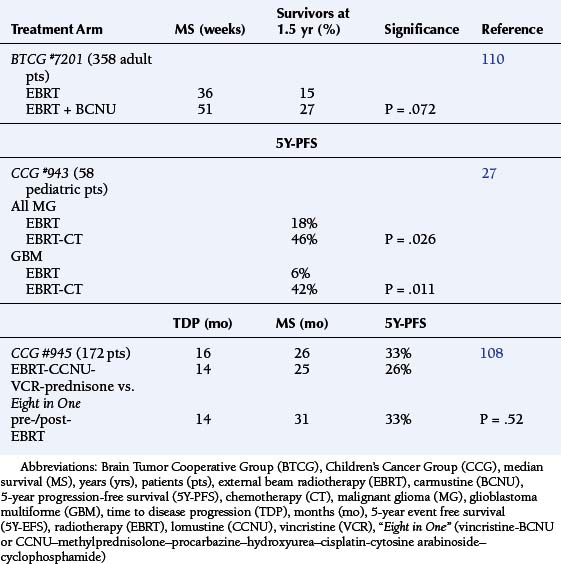
Chemotherapy Trials: Phase III and Phase II, Dose Intensification of Chemotherapy with Hematopoietic Support, and Chemo-radiotherapy
The Phase III Trials
The BTCG #7201 study showed that the addition of a nitrosourea, carmustine (BCNU), further improved median survival to 51 weeks (Table 8-3). One may critique the subsequent BTCG experience (studies #7501, 7702, 8001, 8301, 8420A) to say that nitrosourea-based adjuvant chemotherapy has provided a modest improvement in survival among adult patients, which was comparable to that of other single drugs or multidrug regimes. The multiagent schedules, however, had a correspondingly higher toxicity rate.110,111
The pediatric neuro-oncologic experience with the adjuvant chemotherapy of MG is less comprehensive and methodical, although more encouraging. The CCG #943 study of newly diagnosed AAST and GBM contrasted surgery with conventional EBRT against surgery, EBRT, and lomustine (CCNU) –vincristine−prednisone. The chemotherapy arm achieved a significantly better 5Y-PFS than the EBRT-only control group (P < .05). The impact of chemotherapy on the treatment of GBM was proven by comparison to the irradiated arm (P = .01).27 These data remain the best results yet achieved with conventional EBRT and nitrosoureas for MG patients of any age (Table 8-3).
Single Agent Phase II Studies
A variety of Phase II studies have been directed at the treatment of newly diagnosed, progressive or recurrent MGs to identify promising agents for further development.111
Rationale for Multiagent Adjuvant Chemotherapy Regimens
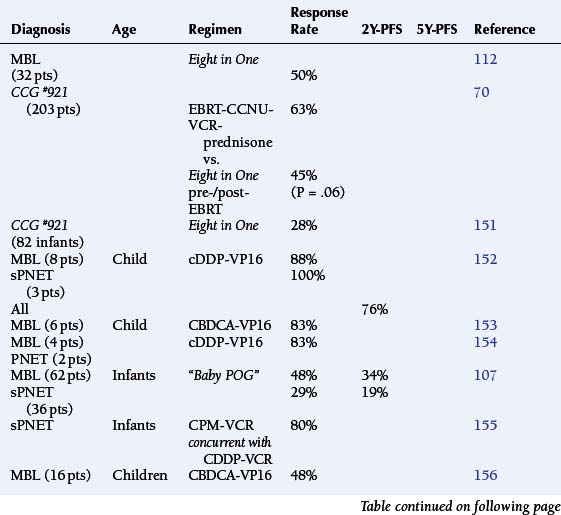
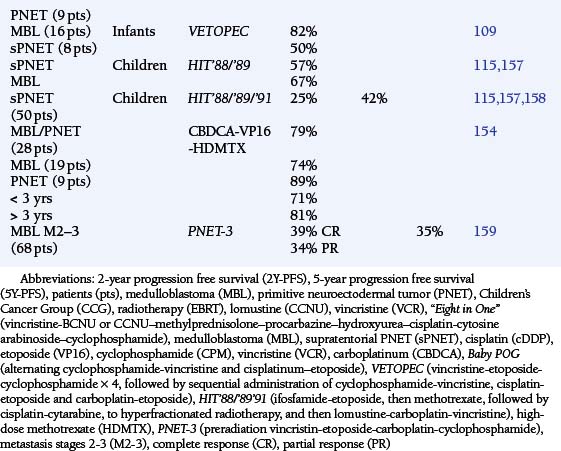
The theoretic rationale for the multidrug protocols is to combine agents with activity against tumor cells traversing different phases of the cell cycle, in order to elicit an additive or synergistic cytotoxic effect. The enthusiasm for the Eight in One [vincristine−BCNU or CCNU–methylprednisolone–procarbazine–hydroxyurea–cisplatin−cytosine arabinoside−cyclophosphamide] chemotherapy regimen was based upon the early response rate among those children treated, following resection and prior to EBRT.112 The CCG #945 trial for the treatment of newly diagnosed children with MA, contrasted EBRT followed by CCNU-vincristine-prednisone against the Eight in One chemotherapy regimen, which was administered for two courses before EBRT and with eight subsequent courses. Comparison of this complex protocol structure with a single nitrosourea found no statistical difference in outcome between the two arms, although the Eight in One regimen was clearly more toxic (Table 8-3).108,113 During this period, the Eight in One regimen continued to be studied among infants and younger children in order to defer EBRT. (Table 8-4 gives the response rates when used in a neoadjuvant setting.)28,107,109,113–116
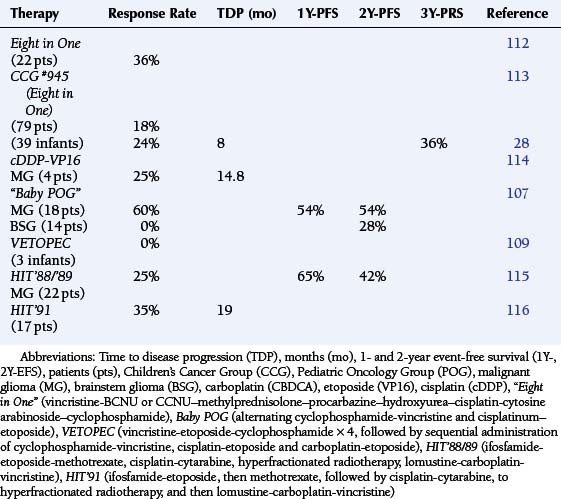
Chemotherapy with Putatively Synergistic Drug Combinations
Platinators and topoisomerase inhibitors are thought to have more than an additive interaction. Clinical trials against pediatric CNS tumors with a variety of drug combinations have not confirmed this, exhibiting response rates of roughly 25% (0 to 60%) and 2Y-PFS rates of 28% to 54% (Table 8-4). Further experience with multiagent chemotherapy regimens suggested greater dose-intensification prior to proceeding to irradiation. The German HIT’88/’89 protocol sequentially combined procarbazine, followed by ifosfamide and etoposide, then methotrexate, and finally cisplatin with cytarabine administered over a 7 week period. Two complete courses were administered prior to EBRT. However, the response rate among children and young adults with MA remained 25% (3/12) with a 5Y-PFS rate of 36% (Table 8-4).115
Myeloablative Chemotherapy with Bone Marrow Rescue
The introduction of myeloablative chemotherapy followed by autologous bone marrow transplant (ABMT) for MA has been justified by the availability of drugs exhibiting steep dose/response curves and limited nonhematologic toxicity, as well as by the rarity of metastatic spread to the bone marrow. The most recently published series showed that no significant improvement in long-term survival could be demonstrated for adult MA patients treated with myeloablative therapy and ABMT.117 The results among pediatric MA patients have not been significantly better, although an objective critique is complicated by the small numbers of patients, heterogeneous induction regimens (even within a small series), lack of stratification by known risk factors, and equating ABMT and peripheral blood stem cell (PBSC) harvesting techniques (Table 8-5).118–122
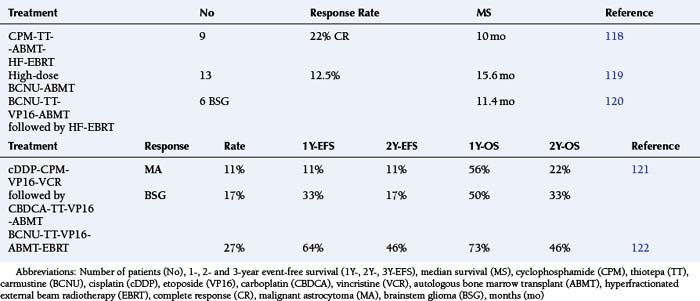
Dose-Intensive Chemotherapy with Peripheral Blood Stem Cell Rescue.
Multiple investigators have demonstrated that dose-dependent cytoreduction is achievable only if the inherent dose-limiting toxicity of myelosuppression can be overcome. The relatively low mitotic index of solid tumors limits the effectiveness of phase-specific agents administered intensively over a brief exposure period. Hence the ABMT regimens, which maximize the peak dose by administering ultra-high doses over a short time, have shown little in the way of sustained efficacy among MAs. Repetitive chemotherapy protocols that emphasize the concepts of peak-dose and time-intensity, have now become feasible with the development of PBSC methodology. A dose-escalation study of the chemotherapy combination of procarbazine–CCNU–vincristine with PBSC support allowed the 1.7- to 1.8-fold intensification among patients aged 2 to 35 years (Table 8-6).123,124
TABLE 8-6 Results with Dose Intensification with Peripheral Stem Cell Support Among Newly Diagnosed Pediatric Malignant Glioma Patients
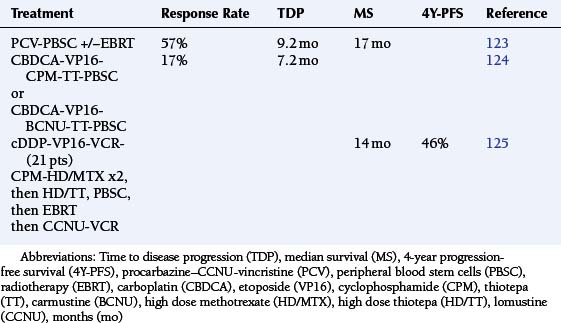
The Milan group reported 21 children with MG who were treated with induction chemotherapy consisting of cisplatin–etoposide–cyclophosphamide–vincristine–high dose methotrexate for two courses followed by high dose thiotepa with PBSC, then by focal EBRT and maintenance with CCNU-vincristine for a total duration of one year. The median time to disease progression and 4Y-PFS are shown in Table 8-6.125
Phase-specific Chemotherapy Agents as Radiosensitizers—“Chemoradiotherapy.”
The Goldie-Goldman model predicts that concomitant administration should minimize the emergence of resistance to chemotherapy and EBRT. Sequential radiochemotherapy was administered in the HIT’91 regimen to 17 children and young adults. Induction chemotherapy consisted of two courses of ifosfamide-etoposide-methotrexate and cisplatin-cytarabine, delivered over a 4-week period. Hyperfractionated EBRT was begun on day 12 of the first course and continued into the second. Maintenance chemotherapy consisted of CCNU, carboplatin, and vincristine administered every 6 weeks for eight courses. Among 11 patients with MA, there were one complete and two partial responses; overall results were not very different from the other strategies discussed above. There was no additive, acute toxicity due to the synchronous administration of radiochemotherapy among the MA patients, who had received only involved field radiotherapy (Table 8-4).116
Brainstem Glioma
Surgical Intervention
A de facto consensus has emerged within the CCG and POG that a biopsy is not warranted in cases of diffuse pontine glioma (DPG) with a “classical” presentation and a diagnostic MRI appearance, as the therapeutic approach employed by current multicenter protocols has not been altered by a specific pathologic diagnosis.126
Previous Combination Therapy Trials
Adjuvant chemotherapy with nitrosoureas has not improved the outcome among children with BSG receiving EBRT. However, pilot studies with neo-adjuvant multiagent chemotherapy did achieve responses in terms of tumor reduction.126,127 The POG-8833 study of preirradiation chemotherapy and HFEBRT for BSG treated 32 evaluable, newly diagnosed patients with four induction cycles of cisplatin and cyclophosphamide followed by HFEBRT (66 Gy). While clinical responses were reported in 65%, the 1-year PFS was 16%; the median survival of 9 months was not significantly different from the previous POG experience with HFEBRT alone.126
The CCG Phase II trial #9941 evaluated the efficacy and toxicity of two arms of induction chemotherapy administered prior to HFEBRT. Neither the difference in postinduction response rate nor the difference in postinduction/HFEBRT response rate was statistically significant. The PFS was 17% at one year and 6% at two years. Hyperfractionated radiotherapy was also not found to consolidate the response to induction chemotherapy among those who did achieve a partial or minor response.128
Low-grade Glial Neoplasms
Surgical Intervention
Operative resection is the accepted standard of care for actively symptomatic patients or those with progressive disease. Analysis of 70 children with LGGN treated in a single institution demonstrated that extent of surgical resection was the dominant predictor for PFS (P = .015) and OS (P = .013). Actuarial PFS rates were 88%, 79%, and 76% at 5, 10, and 20 years, respectively, following complete removal.129
About 80% of infratentorial and only 18% of supratentorial LGGN are accessible enough to allow such radical resection.59 A retrospective review of cerebellar AST demonstrated postoperative residual disease in as many as 35%.130 Realistically, a significant percentage of children with LGGN have postoperative residual disease and are at risk for disease progression or recurrence.
This raises the related controversy of “early vs. late” surgery. This issue has been difficult to answer in the setting of multi-institutional group trials due to the selection bias toward operating on readily accessible lesions. What data exists would suggest that patients presenting with epilepsy as their sole symptom have a favorable prognosis, which is not adversely affected by waiting until disease progression.131
Radiotherapy
While EBRT has been the “accepted” treatment for unresectable and/or progressive disease, a quantitative determination of its contribution to disease control remains problematic.129,132 Postoperative irradiation for adults with incompletely resected LGGN has been associated with 5Y-OS of approximately 48% and 10Y-OS of 26%, which are twice the 26% and 12% rates, respectively, reported for patients receiving surgery alone.133 The addition of EBRT has been found to favorably influence disease control among patients older than 35 years (P = .008). However, this association between younger age at diagnosis with a lower grade of glioma and better prognosis has long been known and compromises interpretation of this data.
Recent analysis of the literature has concluded that for gliomas of the visual pathway, local tumor control with stable or improved visual function is achieved with EBRT in approximately 90% of cases. There is a consensus to employ radiotherapy in older children with progressive disease, regardless of location or histopathologic subtype.134 Given the absence of compelling data for the use of EBRT among children, there has been increasing interest in the application of chemotherapy for unresectable, residual, or progressive disease.
Chemotherapy
Among children, single chemotherapy agents have largely been considered ineffective. The Washington group piloted a 10-week induction regimen of carboplatinum-vincristine for children with LGGN. Patients with either SD or a cytoreductive response were additionally treated with twelve cycles of carboplatinum-vincristine. Among 37 newly diagnosed patients, the overall response rate was 62%. In a subsequent report of 70 children less than 5 years of age who were treated with this combination, 57% experienced objective responses, with a PFS rate of 85% at one year and 76% at two years.135
The San Francisco group has proposed the TPCV combination (thioguanine, procarbazine, lomustine, and vincristine) in order to defer EBRT among younger children with chiasmal and hypothalamic AST. Among 41 children with a median age of 5.2 years, the median TDP was delayed to 30 months with a 5Y-OS of 83%.136
Medulloblastomas and Primitive Neuroectodermal Tumors
Surgical Resection
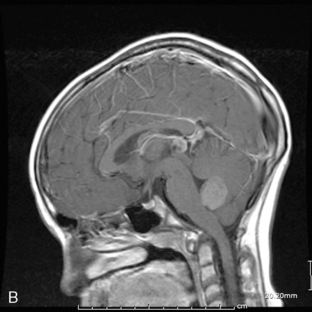
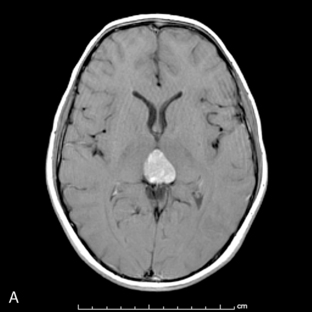
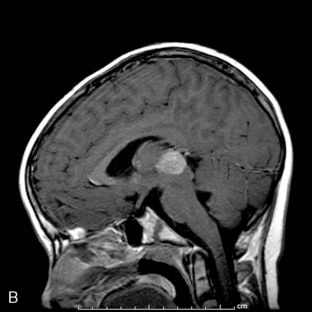
The studies reviewed in Table 8-2 have demonstrated the advantage of aggressive surgical debulking to achieve minimal residual disease among children with MBL. Several of these reach statistical significance.67–70,137 Jakacki et al.138 and Albright et al.139 investigated the results of the CCG #921 study for children with pineal (Figure 8-5) and other supratentorial PNETs. Among these other supratentorial PNETs, postoperative tumor burden of greater than 1.5 cm2 was associated with a 4Y-OS of only 13%. This was much worse than that of children with a residual of less than 1.5 cm2, 40% of whom were alive after 4 years.139
Radiotherapy
For children older than 3 to 4 years of age with minimal postoperative residual disease and no evidence of dissemination, treatment with surgical resection and conventional craniospinal EBRT (tumor dose: 54 Gy; whole brain and spinal dose 36 Gy) has achieved a 5Y-PFS rate of approximately 63% (±5) (Table 8-7).70
Lower Craniospinal EBRT Dosimetry for Standard-Risk Therapy
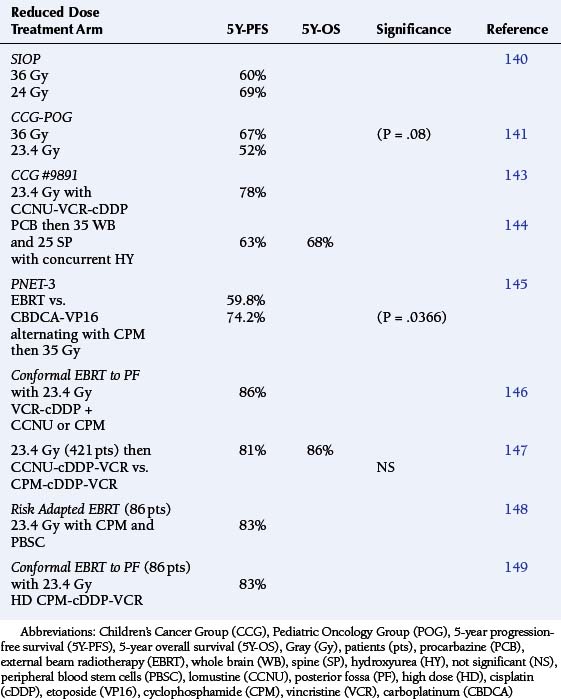
There has been increasing concern regarding the neurologic and endocrinologic toxicity of neuraxis irradiation for younger children with standard risk MBL, and attempts have been made to reduce the extratumoral dosage. The SIOP sponsored a study that compared 36 Gy vs. 24 Gy craniospinal EBRT and found the 5Y-PFS was actually better for the reduced dose arm, although some patients did receive preradiation chemotherapy (Table 8-7).140
A joint CCG-POG trial was performed between 1985 and 1991 to determine whether the craniospinal EBRT prescription could safely be reduced from 36 Gy to 23.4 Gy. The study was halted when preliminary information suggested that the reduced dose arm was associated with a higher early relapse and “distant failure” rate. However, subsequent follow-up studies have found that the 5Y-PFS for the standard radiation prescription arm was greater but not statistically significant when compared to the reduced dose arm (P = .08). These results remained durable at 8 years following treatment, as well.141
Encouraging results with adjuvant chemotherapy had been reported around this time by Packer et al.142 for the treatment of “high-risk” MBL patients. The addition of eight cycles of lomustine-vincristine-cisplatinum increased the 5Y-PFS to 85%. This exceeded the survival rates being reported among “standard-risk” patients treated with EBRT alone, at the time. Therefore, the CCG initiated trial #9892 for children without significant postoperative or metastatic disease, aged 3 to 10 years, using the same adjuvant chemotherapy but with a decrease in the neuraxis radiation prescription (23.4 Gy). The 5Y-PFS of this regimen was a remarkable 78% (Table 8-7).143
A series of trials then examined the same reduced EBRT dosage and randomized for adjuvant chemotherapy. These results are summarized in Table 8-7, but suffice it to say that the 5Y-PFS has remained at a plateau of 80% (±6). In one study, patients with average-risk medulloblastoma were treated with 23.4 Gy of CSRT, 55.8 Gy of posterior fossa RT, plus one of two adjuvant chemotherapy regimens: lomustine (CCNU), cisplatin, and vincristine; or cyclophosphamide, cisplatin, and vincristine. Five-year event-free survival and survival was 81% ± 2.1% and 86% ± 9%, respectively. These results were comparable to those obtained with higher doses of irradiation, suggesting that reduced-dose craniospinal radiation may be feasible in children with average-risk medulloblastomas. 147
High-Risk Medulloblastoma
This cancer would appear the prototypical target for chemotherapy, as it is characterized by a high mitotic rate, has known radiosensitivity, and has a relatively high rate of meningeal dissemination (36% to 43%) at time of diagnosis.19
Phase III Trials regarding the Role of Chemotherapy in Treatment of Medulloblastoma
Two parallel-designed studies conducted by the European (SIOP-1) and North American CCG (#942) have been discussed (see Prognostic Variables). The SIOP-I enrolled patients between 0 and 16 years during the years 1975 through 1979, and randomized between surgery and irradiation vs. surgery, EBRT, and CCNU-vincristine. Analysis at 54 months demonstrated a statistically significant advantage in outcome for the combination therapy arm (P = .005) (Table 8-8).67 The CCG #942 accessioned patients between 2 and 16 years, during the years 1975 through 1981. Comparison was made between surgery and EBRT vs. surgery, irradiation, and adjuvant chemotherapy with CCNU-vincristine-prednisone. Among the high-risk patients, the PFS was significantly better for the EBRT-chemotherapy treated patients (P = .006) (Table 8-8).68 The third randomized trial was a POG study comparing conventional neuraxis irradiation with/without MOPP chemotherapy (1979 to 1986), which demonstrated no advantage to adjunctive pharmacotherapy among children less than 5 years of age (Table 8-8).150
Multiagent Combination Chemotherapy Regimens
Preliminary studies of the Eight in One combination chemotherapy regimen had been encouraging in terms of response rates among MBL/PNET patients (Table 8-9). The CCG #921 protocol was designed to compare the relative efficacy of adjuvant CCNU-vincristine-prednisone for high-risk patients with this more aggressive chemotherapy regimen before and after EBRT. This study demonstrated no significant advantage for the Eight in One polypharmacy combination (P= .06) (Table 8-9).70
Synergistic Chemotherapy Studies
Platinators and topoisomerase antagonists have become increasingly popular by virtue of a putative synergistic interaction in treatment of a number of pediatric brain tumors (Table 8-9). The German HIT’91 trial demonstrated that high dose, multiagent chemotherapy could be as effective in achieving a complete response (42-57% of patients) as irradiation (57-63%) among newly diagnosed children with MBL (Table 8-9).157,158
Dose Intensive Chemotherapy with Autologous Bone Marrow Transplant or Peripheral Stem Cell Rescue
Several groups have examined the feasibility, toxicity, and efficacy of dose-intensification using this approach. A collaborative study treated 53 children with newly diagnosed MBL/PNET, 19 of whom were high-risk, and 34 with average-risk disease. All patients were treated with craniospinal EBRT. Postradiotherapy chemotherapy consisted of four courses of high-dose cyclophosphamide-cisplatin-vincristine administered with PBSC or ABMT hematopoietic support. The high-risk patients additionally received topotecan in a 6-week phase II window between EBRT and this chemotherapy regimen. Early outcome analysis revealed a 2Y-PFS of 93.6% among the average-risk patients and 73.7% among high-risk subjects.160
The New York University Group employed five courses of cisplatin-etoposide-cyclophosphamide-methotrexate with leucovorin rescue for MBL patients with disseminated disease. Following this induction, eligible patients were treated with a single myeloablative course of chemotherapy and PBSC. The 81% complete response rate and 49% 3Y-PFS were considered encouraging enough to warrant additional trials with methotrexate.161
Ependymoma
Surgical Management
Historically, meaningful surgical debulking has been possible in about 42% to 62% of patients, usually those with tumors originating supratentorially or in the roof of the IVth ventricle. Not surprisingly, the 5Y-PFS rates have been poor, on the order of 23% to 45%. In recent studies, aggressive surgical resection has improved the 5Y-PFS estimates to 51% to 75%.23,89,90,92
Radiotherapy
There has been no study to establish the optimal dose or appropriate treatment volume for EBRT among pediatric or adult patients with EPD. Comparison between patients treated with surgery versus surgery plus EBRT has shown long-term survival rates of 17% and 40%, respectively.162 A dose-response relationship was documented.
The requirement for craniospinal EBRT among children with anaplastic EPD has been controversial. Goldwein et al.163 have shown that the 2Y-OS for children receiving neuroaxis irradiation was 52%, while focal irradiation was resulted in a 40% survival rate over the same period. From the perspective of long-term survival, five out of 11 children treated with craniospinal EBRT were alive at 6 years, compared to none of those treated with local EBRT.163 Paulino et al.89 cited the 10Y-OS rates to be 64% for patients receiving craniospinal EBRT, 60% for whole brain irradiation, and 65% for local field radiotherapy (P = 0.88).
Chemotherapy
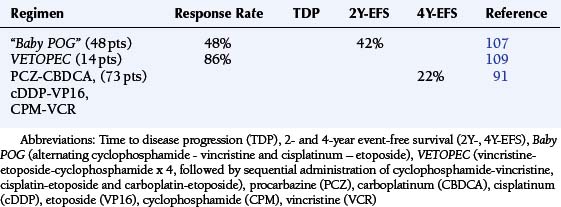
In retrospective analysis of 83 children with EPD, adjuvant chemotherapy did not significantly alter the PFS among patients older than 3 years of age, nor did craniospinal EBRT improve the PFS for M0 patients.92 Duffner et al.107 demonstrated responses and durable survival with two cycles of cyclophosphamide-vincristine among 25 infants less than 3 years of age. The VETOPEC regimen achieved an impressive initial response rate (Table 8-10).109 The French Society of Pediatric Oncology has studied the efficacy of postoperative chemotherapy, in hopes of avoiding EBRT among 73 children less than 5 years of age with intracranial EPD. The regimen consisted of seven cycles of three courses, alternating two drugs at each course (procarbazine–carboplatin, etoposide–cisplatin, cyclophosphamide−vincristine) over 1½ years. Unfortunately, there were no complete or partial responses observed. While the 4Y-OS was 59%, only 23% of those surviving evaded irradiation (Table 8-10).91 Disappointingly, dose intensification using PBSC with cyclophosphamide–etoposide−vincristine, with or without cisplatinum, has achieved response rates of only 16% among children with EPD less than 6 years of age.121
Oligodendroglioma
The combination of procarbazine-CCNU-vincristine (PCV) is regarded as the “gold standard” for chemotherapy due to the remarkable responses seen among anaplastic, progressive, and recurrent OLG. Neoadjuvant administration of PCV for anaplastic OLG has achieved meaningful cytoreduction for 52% to 70%; the remaining patients proceeded to EBRT because of chemoresistance or toxicity.164,165 Phase II examination of temozolamide has also resulted in impressive response rates (53%, with greater than 50% cytoreduction) among chemotherapy-naïve OLG patients at recurrence. The median time to further progression was 10.4 months for all patients, but 13.2 months for those responding to drug treatment.166
Germ Cell Tumors
Surgical Intervention
In Japan, it has been the practice to first irradiate a pineal tumor thought to be a germinoma with 20 Gy (“the radiation test”), and then, if the tumor regressed, to continue radiotherapy. If there was no reduction in tumor size, only then was surgical excision to be considered.167 Over time, aggressive surgical resection has proven itself to be an important determinant of survival, as evidenced in the largest single series of patients (number 153) reported by the University of Tokyo.102
The pendulum has swung back again, because of the increasing effectiveness of nonsurgical therapies. If the appropriate neuroradiographic findings and biomarker serologic studies are present, the Japanese and Korean Societies for Pediatric Neurosurgery advocate a minimally invasive surgical procedure, such as neuroendoscopic or stereotactic biopsy, to be followed by platinum-based chemotherapy and/or targeted EBRT.167 It is only among cases demonstrating a poor response to “trial therapy” that surgical intervention is then reconsidered to debulk the tumor and clarify the pathologic diagnosis.168
External Beam Radiotherapy
Among patients with known germinomas, combined surgical resection and radiotherapeutic intervention improved 10Y-OS rates from 69% to 93%.102 The typical radiotherapy prescription today consists of a primary tumor dose of 50 to 55 Gy, with 36 Gy to the neuraxis. Subsequent controversy regarding the treatment of germinomas has centered on the issues of dosimetry and the indication for craniospinal radiotherapy. As germinomas of the CNS proved to be as radiosensitive as those of the testis, it was shown that those without evident CSF dissemination could be controlled with involved field radiotherapy alone.102,169,170 In contrast, there is no debate regarding the necessity of treating NG-GCT with a full radiotherapy prescription to the tumor (50 to 55 Gy) and craniospinal axis (36 to 40 Gy) due to their significantly higher rate of metastasis and recurrence.8,168,169 Unfortunately, the NG-GCTs remain refractory to radiation prescriptions even greater than 50 Gy.
Chemotherapy
A number of reports investigating the role of chemotherapy in the management of GCT have been encouraging (Table 8-11).103,171–174
TABLE 8-11 Complete Response Rates to Induction Chemotherapy among Newly Diagnosed Germinomas and Non-Germinomatous Germ Cell Tumor Patients
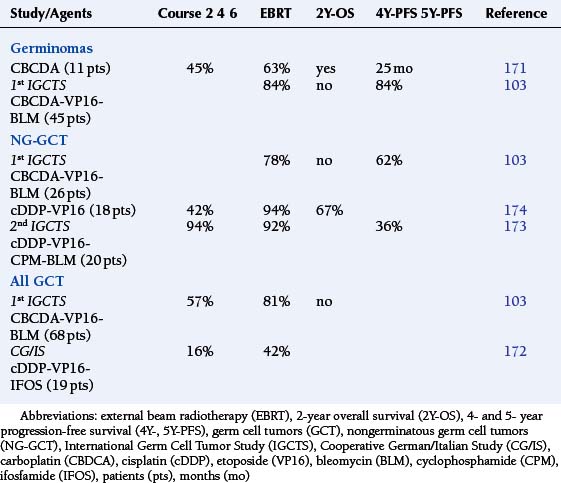
The practice of the University of Tokyo has been to rank GCT patients by relative risk: (i) mixed germinoma and teratoma, (ii) mixed GCT with predominance of germinoma or teratoma with some “pure malignant tumor” (embryonal carcinomas, endodermal sinus tumors, and choriocarcinomas), and (iii) mixed tumors with predominance of “pure malignant tumor.” Surgery and radiotherapy produce a 10Y-OS rate of 91.7% among germinoma patients. In the intermediate prognostic group, combination chemotherapy (cisplatin-vinblastine-bleomycin, cisplatin-etoposide, or carboplatin-etoposide) and EBRT has been shown to significantly reduce the risk of disease recurrence when compared to irradiation alone (P = .049). The high-risk patients did relatively better with chemotherapy than with only EBRT, although the difference was not statistically significant.102
The First International Germ Cell Tumor Study proposed a chemotherapy-only regimen of carboplatin, etoposide, and bleomycin. This study accessioned 45 patients with germinoma and 26 with NG-GCT, of whom 68 were considered evaluable. The protocol for germinoma patients, who achieved a complete response after four induction courses, prescribed two additional cycles. Those subjects with a lesser response were treated with a chemotherapy regimen fortified by cyclophosphamide then followed by EBRT. A complete response was achieved in 57% of patients after four induction courses of chemotherapy, and an additional 24% were left with no evident disease after intensified chemotherapy or “second-look surgery.” Thus, a significant majority were rendered disease-free without irradiation (Table 8-11). Thirty-nine percent of patients remained in complete remission over a median follow-up of 31 months.103
The Cooperative German/Italian Study accepted the classic neuroradiographic appearance with elevated biomarkers (β-HCG and/or α-FP) as diagnostic entry criteria. Nineteen patients (16 males) were placed into the study. The therapeutic design consisted of two induction courses of PEI (cisplatinum–etoposide-ifosfamide); patients responding to chemotherapy were to receive an additional two courses. Nonresponders and those with progressive disease were to be advanced to surgical resection, if feasible, prior to craniospinal EBRT (30 Gy with a tumor boost of 24 Gy). Thirteen of 16 with elevated α-FP and/or β-HCG had normalization of biomarker levels following the second course of PEI induction chemotherapy, which paralleled objective neuroradiographic evidence of a cytoreductive effect in ten. Increasing response rates were seen with further chemotherapy (Table 8-11). Seventeen of the patients survived, of whom 81% remain in remission over a median follow-up interval of 11 months (range 7 to 39 months).172
The Second International CNS Germ Cell Study Group employed two courses of cisplatin–etoposide–cyclophosphamide–bleomycin to assess chemosensitivity in a group of 20 patients with NG-GCT. The study design planned that participants achieving a complete response would receive two additional courses of carboplatin–etoposide–bleomycin and another cycle of the original treatment regimen. Those not reaching or sustaining this underwent “second-look surgery” and/or EBRT. A greater than 50% tumor cytoreduction was achieved in a remarkable majority of patients (Table 8-11). The median PFS for subjects with a complete response was 62 months compared with patients with lesser responses, who demonstrated a mean PFS of 23 months. Regrettably, 69% of evaluable patients suffered from progressive disease during or following chemotherapy.173
Infant Brain Tumors
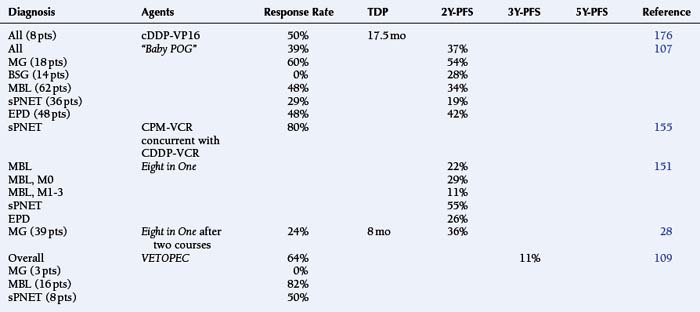
Long-term survival among infants with malignant tumors has been approximately 24%, in comparison those with “benign” tumors fare better (73%).175 While effective at disease control, the neurotoxicity of irradiation is unacceptable. In one study of 78 infants, 80% of those treated with surgery and chemotherapy had a satisfactory functional outcome in contrast to those who required irradiation prior to 24 months of age (42%). Only 21 of 39 survivors had little or no neurologic/cognitive deficit.175 An early study among babies with MG, supratentorial PNET, and anaplastic EPD showed an encouraging response rate to cisplatinum-etoposide with a median survival of 34 months (Table 8-12).176
TABLE 8-12 Results with Neoadjuvant, Synergistic Chemotherapy Agents among Infants with Malignant Brain Tumors

Studies of infants with Malignant Astrocytomas
The “Baby POG” protocol design consisted of alternating cycles of cyclophosphamide-vincristine and cisplatinum-etoposide.107 This combination yielded “very encouraging” results among infants with MA and DPG, which exceeded the responses seen at the time among older children treated with postoperative EBRT alone (PFS 20%), irradiation with CCNU–vincristine−prednisone, or the Eight in One regimen before and after EBRT (Table 8-4).27,107 Experience with Eight in One combination, the VETOPEC regimen (vincristine-etoposide-cyclophosphamide for four courses, followed by sequential administration of cyclophosphamide-vincristine, cisplatin-etoposide, and carboplatin-etoposide), and the more recently reported BBSFOP Protocol (seven cycles of three drug pairs: carboplatin-procarbazine, cisplatin-etoposide, cyclophosphamide-vincristine) is shown in Table 8-12.28,109,177 None of these has emerged as clearly superior.
Infants with Standard and High-Risk Medulloblastoma, Primitive Neuroectodermal Tumor and Ependymoma
The “Baby POG” study of alternating cyclophosphamide-vincristine and cisplatinum-etoposide yielded both responses and durable remissions for young children with MBL (Table 8-12).107 The CCG study #921 administered the Eight in One regimen; many of the infants, with different tumor types, were able to avoid irradiation (Table 8-12).151 The concurrently-administered combination of cisplatin-cyclophosphamide-etoposide-vincristine has been considered very active in infants with newly diagnosed CNS tumors, particularly supratentorial PNETs, with a response rate of approximately 80% in this high risk subgroup (Table 8-12).155 More recent studies have acknowledged 3Y-PFS rates of less than 50% with the Eight in One regimen and the VETOPEC regimen among babies with MBL and PNET.109,159 The addition of intrathecal and intravenous methotrexate to the platinator-cyclophosphamide-etoposide-vincristine structure appeared to make a difference among the younger infants with identifiable postoperative disease, however (Table 8-12).178
The POG study #9233 was the first to compare conventionally dosed versus dose-intensive induction chemotherapy among infants with newly diagnosed MBL (101 patients) and EPD (84 eligible patients). Induction consisted of cyclophosphamide-vincristine and cisplatin-etoposide, with the investigational arm receiving a relative dose intensification of 1.8. The MBL patients showed no difference in PFS (P = .67) between the two arms. The subjects with EPD found that dose intensification was advantageous in terms of PFS (P = .003).179
Dose-Intensive Chemotherapy with Autologous Stem Cell Rescue
A series of 15 children less than 38 months of age with newly diagnosed malignant CNS tumors was reported using high-dose chemotherapy with PBSC followed by selective EBRT. Three courses of cisplatin-cyclophosphamide-etoposide-vincristine were succeeded by three cycles of carboplatin-thiotepa with reinfusion of PBSC. Ten patients are disease-free at a median of 18 months; five received EBRT (Table 8-12). There were no toxicity-related deaths.180
The “Head Start” I and II protocols administered postoperative induction chemotherapy with vincristine-cisplatin-cyclophosphamide-etoposide for five cycles. Myeloablative chemotherapy with carboplatin-thiotepa-etoposide was supported with PBSC. Irradiation was used only at relapse. A series of 21 children with nonmetastatic MBL were studied. The 5Y-PFS rates are shown in Table 8-12; gross total resection and desmoplastic morphology were associated with especially favorable results. Most survivors (71%) and 52% of the whole cohort avoided EBRT. Mean intellectual function among these patients remained within the average range for the majority tested.181
LONG-TERM COMPLICATIONS OF DISEASE AND THERAPY
Cognitive, Behavioral, and Functional Sequelae
Hirsch et al.182 noted that only 12% of MBL survivors retained IQs above 90; in addition, 93% displayed behavioral disorders. Especially severe deficits appear to be associated with earlier age at irradiation and adjuvant chemotherapy.183 The frequency and causation of intellectual deficits of such magnitude consequent to therapeutic irradiation have been disputed.184 Comparable deficits are not seen among children with posterior fossa AST, for whom surgical resection constituted adequate therapy.185
Prospective evaluation of PNET/MBL patients who received whole brain EBRT has demonstrated progressive decrease of 14 points in full-scale intelligence quotient (FSIQ) during the two years after treatment (P = .001). Children less than 7 years at time of diagnosis have declined to a median FSIQ of 82 (range 50-98), compared to 103 (range 92-133) for the older children. Children less than 5 years of age suffered a mean reduction of 25 points in FSIQ (P < .02).183
One series of 34 children with MBL (30) and EPD (4) treated with EBRT examined the long-term cognitive effects of therapy. Twelve children received reduced-dose and 21 patients conventionally dosed cranial irradiation; all received an additional boost to the posterior fossa. Standardized neuropsychological testing revealed a 2-4 point annual decline in intelligence scores. Intellectual function declined more rapidly during the first few years after therapy and more gradually thereafter. Significant deterioration in visual-motor integration, visual memory, verbal fluency, and executive functioning were the dominant findings.186 Another investigation examined the late effects of reduced dose neuraxis EBRT (23.4 Gy craniospinal with 32.4 Gy boost to the posterior fossa) with adjuvant chemotherapy among 43 children with standard risk MBL. The rate of decline in FSIQ was −4.3 points per year, −4.2 points for verbal and −4.0 for nonverbal function annually (P < .001 for all three). Patients less than seven years of age (P = .016), females (P = .008), and children with higher baseline scores were more adversely affected.187
Retrospective investigation of 1,607 patients who survived a brain tumor during childhood demonstrated that 17% had a neurosensory impairment. Relative to their siblings, there was a significant increased risk for hearing impairment (P < .0001), legal blindness in one or both eyes (P < .0001), cataracts (P< .0001), and diplopia (P < .0001). Incoordination (49%), motor control difficulties (26%), and seizure disorders (25%) were other common neurologic problems. Irradiation of the frontal lobe to 50 or more Gy increased the risk for motor deficits (P < .05). Cerebral irradiation to more than 30 Gy doubled the risk of a subsequent seizure disorder.188
Radiation Toxicity and Necrosis
Of the long-term effects, radionecrosis is progressive, irreversible, dose-dependent, and potentially fatal. It occurs in about 3% to 14% of EBRT-treated patients; the incidence is increasing as duration of survival and imaging techniques improve. The latency interval is between 6 and 36 months in 78% of cases (range: 2 months to 19 years). Clinical features include focal seizures, encephalopathy, dementia, signs of increased intracranial pressure, and new focal neurologic deficits.189 Positron emission tomography is the best noninvasive test currently available for distinguishing radionecrosis from recurrent neoplasm.190
Cerebrovascular Disease
Another late sequela of EBRT that is recognized among children is the occurrence of cerebrovascular accidents. The latency for ischemic complications may vary between 2 and 24 years. Radiation-induced thrombosis of the carotid and intracranial arteries may present as transient ischemic attacks as well as acute infarction.189,191 Long-term survivors of pediatric brain tumors are forty times more likely to experience a stroke than siblings.192
Neuroendocrine Sequelae
Children treated with craniospinal EBRT are at risk for growth failure secondary to growth hormone deficiency, as well as primary, secondary, and/or tertiary hypothyroidism.193 Growth hormone deficiency is the most common form of pituitary insufficiency to follow irradiation.194 Chemical evidence of hypothyroidism occurs in 25% to 50% of patients following EBRT to the head and neck, although not all will be symptomatic. Primary hypothyroidism was found in 24% (71% of which had received craniospinal treatment), compensated hypothyroidism in 73%, and central hypothyroidism in 6%. Chemotherapy was not found to affect thyroid function.195 This may be a progressive process which requires replacement years following EBRT.
Conclusions
Irradiation and chemotherapy are now the “standard of care” for “high-risk” pediatric brain tumor patients with an appropriate functional status. The early excitement (and hubris) of Phase III trials has faded, with more interest being subsequently paid to combination and neoadjuvant chemotherapy. The concept of dose-intensive escalation with alkylators and epiphyllotoxins for disease control continues to hold theoretic promise but must be regarded as unproven, given the paucity of trials utilizing protracted treatment schedules with PBSC support. Short-term myeloablative chemotherapy with ABMT rescue appears to have little or nothing to offer in the treatment of neoplastic diseases with a relatively low mitotic index. Sequential and/or alternating “chemoradiotherapy” appears attractive as a means of locoregional control but there has been relatively little experience with this approach. However, there are a number of effective chemotherapeutic agents, which may also be valid radiosensitizers in the appropriate setting. Antagonizing angiogenesis seems intuitively appealing; however, intratumoral hypoxia constitutes a very important mechanism for promoting resistance to treatment against both chemotherapy and EBRT. The intrinsic and selected-for mechanisms of therapeutic resistance are poorly understood and appear intimately related to the pathogenetic events of malignant transformation itself. These are obvious fields for investigation in order to develop a future generation of genetically based therapies.196
1. M.A. Smith, J.G. Gurney, L.A.G. Gloeckler, et al. Cancer among adolescents 15–19 years old. In: Reis, M.A. Smith, J.G. Berney, et al. Cancer Incidence and Survival Among Children and Adolescents: United States SEER Program 1975–1995. Bethesda MD: National Cancer Institute SEER Program; 1999:157-164. NIH Pub. No. 99–4649
2. CBTRUS. Statistical Report: Primary Brain Tumors in the United States, 1995–1999. Published by the Central Brain Tumor Registry of the United States, 2002.
3. L.A.G. Ries, C.L. Bercy, G.R. Bunin. L. AG. Reis, M.A. Smith, J.G. Berney, et al, editors. Cancer Incidence and Survival Among Children and Adolescents: United States SEER Program 1975–1995. Bethesda MD: National Cancer Institute SEER Program. 1999:1-15. NIH Pub. No. 99–4649
4. J.C. Gurney, M.A. Smith, G.R. Bunin. CNS and miscellaneous intracranial and intraspinal neoplasms. In: L.AG. Reis, M.A. Smith, J.G. Berney, et al, editors. Cancer Incidence and Survival Among Children and Adolescents: United States SEER Program 1975–1995. Bethesda MD: National Cancer Institute SEER Program; 1999:51-63. NIH Pub. No. 99–4649
5. U. Hjalmars, M. Kulldorff, Y. Wahlqvist, et al. Increased incidence rates but no space-time clustering of childhood astrocytoma in Sweden, 1973–1992: a population-based study of pediatric brain tumors. Cancer. 1999;85:2077-2090.
6. A.P. Polednak, J.T. Flannery. Brain, other central nervous system, and eye cancer. Cancer. 1995;75:330-337.
8. M.T. Jennings, R. Gelman, F. Hochberg. Intracranial germ cell tumors: natural history and pathogenesis. J Neurosurg. 1985;63:155-167.
9. C. Araki, S. Matsumoto. Statistical reevalution of pinealoma and related tumors in Japan. J Neurosurg. 1969;30:146-149.
10. K. Jellinger. Primary intracranial germ cell tumours. Acta Neuropathol. 1973;25:291-306.
11. S.A. Grossman, M. Osman, R. Hruban, et al. Central nervous system cancers in first-degree relatives and spouses. Cancer Investig. 1999;17:299-308.
12. E. Ron, B. Modan, J.D. Boice, et al. Tumors of the brain and nervous system after radiotherapy in childhood. N Engl J Med. 1988;319:1033-1039.
13. S. Shapiro, J. MealyJr, C. Sartorius. Radiation-induced intracranial malignant gliomas. J Neurosurg. 1989;71:77-82.
14. R.O. McKeran, D.G.T. Thomas. The clinical study of gliomas. D.GT. Thomas, D.L. Graham, editors. Brain Tumors: Scientific Basis, Clinical Investigation and Current Therapy. 1980:194-230. Baltimore;
15. D. Osoba, N.K. Aaronson, M. Muller, et al. Effect of neurological dysfunction on health-related quality of life in patients with high-grade glioma. J Neuro-Oncol. 1997;34:263-278.
16. P. Forsythe, J.B. Posner. Headaches in patients with brain tumors: a study of 111 patients. Ann Neurol. 1992;32:289. (Abstr.)
17. T.S. Park, H.J. Hoffman, E.B. Hendrick, et al. Medulloblastoma: clinical presentation and management. J Neurosurg. 1983;58:543-552.
18. L. Harisiadis, C.H. Chang. Medulloblastoma in children: a correlation between staging and results of treatment. Int J Radiat Oncol Biol Phys. 1997;2:833-841.
19. J.C. Allen, F. Epstein. Medulloblastoma and other primary malignant neuroectodermal tumors of the CNS. The effect of patients’ age and extent of disease on prognosis. J Neurosurg. 1982;57:446-451.
20. E.M. Chatty, K.M. Earle. Medulloblastoma. A report of 201 cases with emphasis on the relationship of histologic variants to survival. Cancer. 1971;28:977-983.
21. P.C. Burger, I-T. Yu, T. Tihan, et al. Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a Pediatric Oncology Group study. Amer J Surg Pathol. 1998;22:1083-1092.
22. R.J. Packer, J.A. Biegel, S. Blaney, et al. Atypical teratoid/rhabdoid tumor of the central nervous system: report on workshop. J Ped Hematol/Oncol. 2002;24:337-342.
23. I.F. Pollack, P.C. Gerszten, A.J. Martinez, et al. Intracranial ependymomas of childhood: long-term outcome and prognostic factors. Neurosurgery. 1995;37:655-666.
24. A. Allam, A. Radwi, A. El Weshi, et al. Oligodendroglioma: an analysis of prognostic factors and treatment results. Amer J Clin Oncol. 2000;23:170-175.
25. G.S. Gordon, S.J. Wallace, J.W. Neal. Intracranial tumours during the first two years of life: presenting features. Arch Dis Child. 1995;73:345-347.
26. I.F. Pollack, R.L. Hamilton, J. Burnham, et al. Impact of proliferation index on outcome in childhood malignant gliomas: results in a multiinstitutional cohort. Neurosurgery. 2002;50:1238-1244.
27. R. Sposto, I.J. Ertel, R.DT. Jenkin, et al. The effectiveness of chemotherapy for treatment of high grade astrocytoma in children: results of a randomized trial. J Neuro-Oncol. 1989;7:165-177.
28. J.R. Geyer, J.L. Finlay, J.M. Boyett, et al. Survival in infants with malignant astrocytomas. A report of the Children’s Cancer Group. Cancer. 1995;75:1045-1050.
29. S.M. Ashmore, D.GT. Thomas, J.L. Darling. Does p-glycoprotein play a role in clinical resistance of malignant astrocytoma? Anticancer Drugs. 1999;10:861-872.
30. L.J. Goldstein. Clinical reversal of drug resistance. Curr Probl Cancer. 1995;19:65-124.
31. E.T. Valera, A.K. Lucio-Eterovic, L. Neder, et al. Quantitative PCR analysis of the expression profile of genes related to multiple drug resistance in tumors of the central nervous system. J Neuro-Oncol. 2007;85:1-10.
32. I.F. Pollack, R.L. Hamilton, R.W. Sobol, et al. O6-methylguanine-DNA methyltransferase expression strongly correlates with outcome in childhood malignant gliomas: results from the CCG-945 cohort. J Clin Oncol. 2006;24:3431-3437.
33. S.L. Green, A.J. Giaccia. Tumor hypoxia and the cell cycle: implications for malignant progression and response to therapy. The Cancer Journal. 1998;4:214-223.
34. P. Hainaut. The tumor suppressor protein p53: a receptor to genotoxic stress that controls cell growth and survival. Curr Opin Oncol. 1995;7:76-82.
35. M.B. Kastan, N. Onyekwere, D. Sidransky, et al. Participation of p53 protein in cellular response to DNA damage. Cancer Res. 1991;51:6304-6311.
36. T.C. Graeber, C. Osmanian, T. Jacks, et al. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumors. Nature. 1996;379:88-91.
37. A.P. Kyritsis, M.L. Bondy, K.R. Hess, et al. Prognostic significance of p53 immunoreactivity in patients with glioma. Clin Cancer Res. 1995;1:1617-1622.
38. B.C. Liang. Effects of hypoxia on drug resistance phenotype and genotype in human glioma cell lines. J Neuro-Oncol. 1996;29:149-155.
39. Y. Iwadate, M. Tagawa, S. Fujimoto, et al. Mutation of the p53 gene in human astrocytic tumours correlates with increased resistance to DNA-damaging agents but not to antimicrotubule anti-cancer agents. Brit J Cancer. 1998;77:547-551.
40. S.H. Bigner, J. Mark, D.E. Bullard, et al. Chromosomal evolution in malignant human gliomas starts with specific and usually numerical deviations. Cancer Genet Cytogenet. 1986;22:121-135.
41. D.T. Ransom, S.R. Ritland, C.A. Moertel, et al. Correlation of cytogenetic analysis and loss of heterozygosity studies in human diffuse astrocytomas and mixed oligo-astrocytomas. Genes Chromosomes Cancer. 1992;5:357-374.
42. F.F. Lang, D.C. Miller, M. Koslow, et al. Pathways leading to glioblastoma multiforme: a molecular analysis of genetic alterations in 65 astrocytic tumors. J Neurosurg. 1994;81:427-436.
43. A. von Deimling, D.N. Louis, K. von Ammon, et al. Molecular genetic evidence for two distinct subsets of glioblastoma multiforme. Clin Neuropathol. 1992;11:265-266.
44. D. Sidransky, T. Mikkelsen, D. Schwechheimer, et al. Clonal expansion of p53 mutant cells is associated with brain tumour progression. Nature. 1992;355:846-847.
45. D. Yin, D. Xie, W.K. Hofmann, et al. Methylation, expression, and mutation analysis of the cell cycle control genes in human brain tumors. Oncogene. 2002;21:8372-8378.
46. J. Li, C. Yen, D. Liaw, et al. PTEN, a putative protein tyrosine phosphate gene mutated in human brain, breast and prostate cancer. Science. 1997;275:1943-1947.
47. P.A. Steck, M.A. Pershouse, S.A. Jasser, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 thi is mutated in multiple advanced cancers. Nature Genet. 1997;15:356-362.
48. H. Lin, M.L. Bondy, L.A. Langford, et al. Allelic deletion analyses of MMAC/PTEN and DMBT1 loci in gliomas: relationship to prognostic significance. Clin Cancer Res. 1998;4:2447-2457.
49. T. Sano, H. Lin, X. Chen, et al. Differential expression of MMAC/PTEN in glioblastoma multiforme: relationship to localization and prognosis. Cancer Res. 1999;59:1820-1824.
50. J.W. Henson, B.L. Schnitker, K.M. Correa, et al. The retinoblastoma gene is involved in malignant progression of astrocytomas. Ann Neurol. 1994;36:714-721.
51. A. Kamb, N.A. Gruis, J. Weaver-Feldhaus, et al. A cell cycle regulator potentially involved in the genesis of many tumor types. Science. 1994;264:436-440.
52. E.E. Schmidt, K. Ichimura, G. Reifenberger, et al. CDKN2 (p16/MTS1) gene deletion or CDK4 amplification occurs in the majority of glioblastomas. Cancer Res. 1994;54:6321-6324.
53. Y. Iwadate, S. Mochizuki, S. Fujimoto, et al. Alteration of CDKN2/p16 in human astrocytic tumors is related with increased susceptibility to antimetabolite anticancer agents. Int J Oncol. 2000;17:501-505.
54. U. Sure, D. Ruedi, O. Tachibana, et al. Determination of p53 mutations, EGFR overexpression, and loss of p16 expression in pediatric glioblastomas. J Neuropathol & Exper Neurol. 1997;56:782-789.
55. T. Sung, D.C. Miller, R.L. Hayes, et al. Preferential inactivation of the p53 tumor suppressor pathway and lack of EGFR amplication distinquish de novo high grade pediatric astrocytomas from de novo adult astrocytomas. Brain Pathol. 2000;10:249-259.
56. C. Raffel, L. Frederick, J.R. O’Fallor, et al. Analysis of oncogene and tumor suppressor gene alterations in pediatric malignant astroytomas reveals reduced survival for patients with PTEN mutations. Clin Cancer Res. 1999;5:4085-4090.
57. I.F. Pollack, R.L. Hamilton, C.D. James, et al. Rarity of PTEN deletions and EGFR amplification in malignant gliomas of childhood: results from the Children’s Cancer Group 945 cohort. J Neurosurg. 2006;105(Suppl. 5):418-424.
58. A.L. Albright, A.N. Guthkelch, R.J. Packer, et al. Prognostic factors in pediatric brainstem gliomas. J Neurosurg. 1986;65:751-775.
59. E.R. Laws, W.R. Taylor, E.J. Bergstralh, et al. The neurosurgical management of low-grade astrocytoma. Clin Neurosurg. 1985;33:575.
60. G.E. Sheline. The role of radiation therapy in the treatment of low-grade gliomas. Clin Neurosurg. 1986;33:563.
61. P. Janny, H. Cure, M. Mohr, et al. Low grade supratentorial astrocytomas: management and prognostic factors. Cancer. 1994;73:1937-1945.
62. S. Nishio, I. Takeshia, K. Fujii, et al. Supratentorial astrocytic tumours of childhood: a clinicopathologic study of 41 cases. Acta Neurochir (Wien). 1989;101:3-8.
63. C. Fernandez, D. Figarella-Branger, N. Girard, et al. Pilocytic astrocytomas in children: prognostic factors – a retrospective study of 80 cases. Neurosurgery. 2003;53:544-555.
64. A.V. Deliganis, J.R. Geyer, M.S. Berger. Prognostic significance of type 1 neurofibromatosis (von Recklinghausen disease) in childhood optic glioma. Neurosurgery. 1996;38:1114-1118.
65. R. Micheli, L. Giordano, M.R. Balestrini. Cerebral tumors in children with neurofibromatosis type 1. Minerva Pediatr. 1996;48:89-97.
66. A. Broniscer, S.J. Baker, A.N. West, et al. Clinical and molecular characteristic of malignant transformation of low-grade glioma in children. J Clin Oncol. 2007;25:682-689.
67. D.M. Tait, H. Thorton-Jones, H.J.G. Bloom, et al. Adjuvant chemotherapy for medulloblastoma: the first multi-centre control trial of the International Society of Paediatric Oncology (SIOP). Eur J Cancer. 1990;26:464-469.
68. A.E. Evans, R.D.T. Jenkin, R. Sposto, et al. The treatment of medulloblastoma: results of a prospective randomized trial of radiation therapy with and without CCNU, vincristine and prednisone. J Neurosurg. 1990;72:572-582.
69. A.L. Albright, J.H. Wisoff, P.M. Zeltzer, et al. Effects of medulloblastoma resections on outcome in children: a report from the Children’s Cancer Group. Neurosurgery. 1996;38:265-271.
70. P.M. Zeltzer, J.M. Boyett, J.L. Finlay, et al. Metastasis stage, adjuvant treatment, and residual tumor are prognostic factors for medulloblastoma in children: conclusions from the Children’s Cancer Group 921 randomized phase III study. J Clin Oncol. 1999;17:832-845.
71. C.G. Eberhart, J.L. Kepner, P.T. Goldthwaite, et al. Histologic grading of medulloblastomas. A Pediatric Oncology Group study. Cancer. 1992;94:552-560.
72. F. Giangaspero, S. Wellek, J. Masuoka, et al. Stratification of medulloblastoma on the basis of histopathological grading. Acta Neuropath. 2006;112:5-12.
73. E.T. Valera, A.K. Lucio-Eterovic, L. Neder, et al. Quantitative PCR analysis of the expression profile of genes related to multiple drug resistance in tumors of the central nervous system. J Neuro-Oncol. 2007;85:1-10.
74. K.C. Lo, M.R. Rossi, C.G. Eberhart, et al. Genome wide copy number abnormalities in pediatric medulloblastoma as assessed by array comparative genome hybridization. Brain Pathol. 2007;17:282-296.
75. S. Pfister, M. Remke, G. Toedt, et al. Supratentorial primitive neuroectodermal tumors of the central nervous system frequently harbor deletions of the CDKN2A locus and other genomic aberrations distinct from medulloblastomas. Genes Chromosome Cancer. 2007;46:839-851.
76. K.J. Briggs, I.M. Corcoran-Schwartz, W. Zhang, et al. Cooperation between the Hic1 and Ptch1 tumor suppressors in medulloblastoma. Genes Dev. 2008;22:770-785.
77. J.M. Lamont, C.S. McManamy, A.D. Pearson, et al. Combined histopathological and molecular cytogenetic stratification of medulloblastoma patients. Clin Cancer Res. 2004;10:5482-5493.
78. E. Ellison. Classifying the medulloblastoma: insights from morphology and molecular genetics. Neuropathol Appl Neurobiol. 2002;278:257-282.
79. E.E. Bar, D. Stearns. New developments in medulloblastoma treatment: the potential of a cycopamine-lovastatin combination. Expert Opinion Investig Drugs. 2008;17:185-195.
80. S.C. Clifford, M.E. Lusher, J.C. Lindsey, et al. Wnt/Wingless pathway activation and chromosome 6 loss characterize a distinct molecular subgroup of medulloblastomas associated with a favorable prognosis. Cell Cycle. 2006;5:2666-2670.
81. R.C. Castellino, M. De Bortoli, L.L. Lin, et al. Overexpressed TP73 induces apoptosis in medulloblastoma. BMC Cancer. 2007;7:127.
82. W. Hartmann, B. Digon-Sontgerath, A. Koch, et al. Phosphatidyl 3’±-kinase/AKT signaling is activated in medulloblastoma proliferation and associated with reduced expression of PTEN. Clinical Cancer Res. 2006;12:3019-3027.
83. M.M. Inda, J. Mercapide, J. Munoz, et al. PTEN and DMBT1 homozygous deletion and expression in medulloblastoma and primitive neuroectodermal tumors. Oncol Reports. 2004;12:1341-1347.
84. C.G. Eberhart, J. Kratz, Y. Wang, et al. Histopathological and molecular prognostic markers in medulloblastoma: c-myc, N-myc, trkc, and anaplasia. J Neuropath Exp Neurol. 2004;63:441-449.
85. M.A. Grotzer, K. von Hoff, A.O. von Buieren, et al. Which clinical and biological tumor markers proved predictive in the prospective multicenter trial HIT’91 – implications for investigating childhood medulloblastoma. Klin Padiatr. 2007;219:312-317.
86. A. Gajjar, R. Hernan, M. Kocak, et al. Clinical, histopathologic, and molecular markers of prognosis: toward a new disease risk stratification system for medulloblastoma. J Clin Oncol. 2004;22:984-993.
87. T.E. Merchant, J.J. Jenkins, P.C. Burger, et al. The influence of histology on the time to progression after irradiation for localized ependymoma in children. Int J Radiat Oncol Biol Phys. 1999;45:235. (Abstr. 169)
88. G.B. Nazar, H.J. Hoffman, L.E. Becker, et al. Infratentorial ependymomas in childhood: prognostic factors and treatment. J Neurosurg. 1990;72:408-417.
89. A.C. Paulino, B-C Wen, J.M. Buatti, et al. Intracranial ependymomas: an analysis of prognostic factors and patterns of failure. Amer J Clin Oncol. 2002;25:117-122.
90. P.L. Robertson, P.M. Zeltzer, J.M. Boyett, et al. Survival and prognostic factors following radiation therapy and chemotherapy for ependymomas in children: a report of the Children’s Cancer Group. J Neurosurg. 1998;88:695-703.
91. J. Grill, M-C Le Deley, D. Gambarelli, et al. Postoperative chemotherapy without irradiation for ependymoma in children under 5 years of age: a multicenter trial of the French Society of Pediatric Oncology. J Clin Oncol. 2001;19:1288-1296.
92. B. Horn, R. Heideman, R. Geyer, et al. A multi-institutional retrospective study of intracranial ependymoma in children: identification of risk factors. J Pediatr Hemat Oncol. 1999;21:203-211.
93. K. Sowar, J. Straessle, A.M. Donson, et al. Predicting which children are at risk for ependymoma relapse. J Neurooncol. 2006;78:41-46.
94. P. Modena, E. Lualdi, F. Facchinetti. Identification of tumor-specific molecular signatures in intracranial ependymoma and association with clinical characteristics. J Clin Oncol. 2006;24:5223-5233.
95. U. Tabori, J. Ma, M. Carter, et al. Human telomere reverse transcriptase expression predicts progression and survival in pediatric intracranial ependymoma. J Clin Oncol. 2006;24:1522-1528.
96. S.J. Feigenberg, R.J. Amdur, C. Morris, et al. Oligodendroglioma: does deferring treatment compromise outcome? Amer J Clin Oncol. 2003;26:e60-e66.
97. J.S. Smith, A. Perry, T.J. Borell, et al. Alterations of chromosome arms 1p and 19q as predictors of survival in oligodendrogliomas, astrocytomas and mixed oligoastrocytomas. J Clin Oncol. 2000;18:636-645.
98. C. Godfraind, E. Rousseau, M-M Ruchoux, et al. Tumour necrosis and microvascular proliferation are associated with 9p deletion and CDKNA2 alterations in 1p/19q-delected oligodendrogliomas. Neuropathol Appl Neurobiol. 2003;29:462-471.
99. J. He, K. Hoang-Xuan, Y. Marie, et al. P18 tumor suppressor gene and progression of oligodendrogliomas to anaplasia. Neurology. 2000;55:867-869.
100. U. Pohl, J.G. Cairncross, D.N. Louis. Homozygous deletions of the CDKN2C/pinh4c gene on the short arm of chromosome 1 in anaplastic oligodendrogliomas. Brain Pathology. 1999;9:639-643.
101. R.M. Wolf, N. Draghi, X. Liang, et al. P190rhogap can act to inhibit PDGF-induced gliomas in mice: a putative tumor suppressor encoded on human chromosome 19q13.3. Genes Dev. 2003;17:476-487.
102. M. Matsutani, K. Sano, K. Takakura, et al. Primary intracranial germ cell tumors: a clinical analysis of 153 histologically verified cases. J Neurosurg. 1997;86:446-455.
103. C. Balmaceda, G. Heller, M. Rosenblum, et al. Chemotherapy without irradiation – a novel approach for newly diagnosed CNS germ cell tumors: results of an international cooperative trial. J Clin Oncol. 1996;14:2908-2915.
104. H. Aoyama, H. Shirato, J. Ikeda, et al. Induction chemotherapy followed by low-dose involved field radiotherapy for intracranial germ cell tumors. J Clin Oncol. 2002;20:857-865.
105. W.R. Shapiro. Treatment of neuroectodermal brain tumors. Ann Neurol. 1982;12:231-237.
106. J.H. Wisoff, J.M. Boyett, M.S. Berger, et al. Current neurosurgical management and the impact of the extent of resection in the treatment of malignant gliomas of childhood: a report of the Children’s Cancer Group trial No. CCG-945. J Neurosurg. 1998;90:1147-1148.
107. P.K. Duffner, M.E. Horowitz, J.P. Krischer, et al. Postoperative chemotherapy and delayed radiotherapy in children less than three years of age with malignant brain tumors. N Engl J Med. 1993;328:1725-1731.
108. J.L. Finlay, J.M. Boyett, A.J. Yates, et al. Randomized phase III trial in childhood highgrade astrocytoma comparing vincristine, lomustine, and prednisone with the eight drugs in 1 day regimen. J Clin Oncol. 1995;13:112-123.
109. L. White, S. Kellie, E. Gray, et al. Postoperative chemotherapy in children less than 4 years of age with malignant brain tumors: promising initial response to a VETOPEC-based regimen. J Ped Hem Onc. 1998;20:125-130.
110. W.R. Shapiro. Therapy of adult malignant brain tumors: what have the clinical trials taught us? Seminars Oncol. 1986;13:38-45.
111. M.T. Jennings, S. Iyengar. Pharmacotherapy of malignant astrocytomas of children and adults. CNS Drugs. 2001;15:719-743.
112. T.W. Pendergrass, J.M. Milstein, J.R. Geyer, et al. Eight drugs in one day chemotherapy for brain tumors: experience with 107 children and rationale for preradiation chemotherapy. J Clin Oncol. 1987;5:1221-1231.
113. J.L. Finlay, J.R. Geyer, P.A. Turski, et al. Pre-irradiation chemotherapy in children with high-grade astrocytoma: tumor response to two cycles of the “8-drugs-in-1-day” regimen. A Children’s Cancer Group study, CCG-945. J Neuro-Oncol. 1994;21:255-265.
114. S.J. Kellie, E.H. Kovnar, L.E. Kun, et al. Neuraxis dissemination in pediatric brain tumors: response to preirradiation chemotherapy. Cancer. 1992;69:1061-1066.
115. J. Kühl, H.L. Müller, F. Berthold, et al. Preradiation chemotherapy with children and young adults with malignant brain tumors: results of the German pilot trial HIT‘88/’89. Klin Paediatr. 1998;210:227-233.
116. C. Urban, M. Benesch, B. Pakisch, et al. Synchronous radiochemotherapy in unfavorable brain tumors of children and young adults. J Neuro-Oncol. 1998;39:71-80.
117. L.E. Abrey, M.K. Rosenblum, E. Papadopoulos, et al. High dose chemotherapy with autologous stem cell rescue in adults with malignant primary brain tumors. J Neuro-Oncol. 1999;44:147-153.
118. A. Kedar, B.L. Maria, J. Graham-Pole, et al. High-dose chemotherapy with marrow reinfusion and hyperfractionated irradiation for children with high-risk brain tumors. Med Pediatr Oncol. 1994;23:428-436.
119. E. Bouffet, F. Khelfaoui, I. Philip, et al. High-dose carmustine for high-grade gliomas in childhood. Cancer Chemother Pharmacol. 1997;39:376-379.
120. I.J. Dunkel, J.H. GarvinJr, S. Goldman, et al. High dose chemotherapy with autologous bone marrow rescue for children with diffuse pontine gliomas. J Neuro-Oncol. 1998;37:67-73.
121. W.P. Mason, A. Grovas, S. Halpern, et al. Intensive chemotherapy and bone marrow rescue for young children with newly diagnosed malignant brain tumors. J Clin Oncol. 1998;16:210-221.
122. A.C. Grovas, J.M. Boyett, K. Lindsley, et al. Regimen-related toxicity of myeloablative chemotherapy with BCNU, thiotepa and etoposide followed by autologous stem cell rescue for children with newly diagnosed glioblastoma multiforme: report from the Childrens Cancer Group. Med Pediatr Oncol. 1999;33:83-87.
123. R.I. Jakacki, C. Jamison, V.P. Mathews, et al. Dose-intensification of procarbazine, CCNU (lomustine), vincristine (PCV) with peripheral blood stem cell support in young patients with gliomas. Med Pediatr Oncol. 1998;31:483-490.
124. R.I. Jakacki, C. Jamison, S.A. Heifetz, et al. Feasibility of sequential high-dose chemotherapy and peripheral blood stem cell support for pediatric central nervous system malignancies. Med Pediatr Oncol. 1997;29:553-559.
125. M. Massimino, L. Gandola, R. Luksch, et al. Sequential chemotherapy, high dose thiotepa, circulating progenitor cell rescue and radiotherapy for childhood high grade glioma. Neuro-Oncol. 2005;7:41-48.
126. C.S. Kretschmar, N.J. Tarbell, P.D. Barnes, et al. Pre-irradiation chemotherapy and hyperfractionated radiation therapy 66 Gy for children with brain stem tumors. Cancer. 1993;72:1404-1413.
127. B. Pakisch, C. Urban, I. Slavc, et al. Hyperfractionated radiotherapy and polychemotherapy in brain stem tumors in children. Child’s Nerv Syst. 1992;8:215-218.
128. M.T. Jennings, R. Sposto, L.G. Vezina, et al. Preradiation Chemotherapy in Primary High Risk Brain Stem Tumors: CCG-9941, a Phase II Study of the Children’s Cancer Group. J Clin Oncol. 2002;20:3431-3437.
129. I.F. Pollack, D. Claassen, Q. al-Shboul, et al. Low-grade gliomas of the cerebral hemispheres in children: an analysis of 71 cases. J Neurosurg. 1995;82:536-547.
130. D.W. Smoots, J.R. Geyer, D.M. Lieberman, et al. Predicting disease progression in childhood cerebellar astrocytoma. Childs Nerv Syst. 1998;14:636-648.
131. S. Rudoler, B.W. Corn, M. Werner-Wasik, et al. Patterns of tumor progression after radiotherapy for low-grade gliomas: analysis from the computed tomography/magnetic resonance imaging era. Amer J Clin Oncol. 1998;21:23-27.
132. C.GH. West, R. Gattamaneni, V. Blair. Radiotherapy in the treatment of low-grade astrocytomas. I. A survival analysis. Child’s Nerv Syst. 1995;11:438-442.
133. M.S. Berger, S.A. Leibel, J.M. Bruner, et al. Primary central nervous system tumors of the supratentorial compartment. In: V.A. Levin, editor. Cancer in the Nervous System. New York: Churchill Livingstone, 1996.
134. R.D. Kortmann, B. Timmermann, R.E. Taylor, et al. Current and future strategies in radiotherapy of childhood low-grade glioma of the brain. Part I: Treatment modalities of radiation therapy. Strahlentherapie und Onkologie. 2003;179:509-520.
135. R.J. Packer, J.C. Allen, P.C. Phillips, et al. Efficacy of chemotherapy for children with newly diagnosed progressive low-grade gliomas. San Francisco: Child Neurology Society Meeting, 1994. (Abstr. 435)
136. M. Prados, V. Levin, M.SB. Edwards, et al. Combination chemotherapy as primary therapy for pediatric low grade gliomas. BTRC Protocol 8422, Sixth Internatl Symposium on Pediatric Neuro-Oncology, Houston. 1994. (Abstr.)
137. R.DT. Jenkin, K. Goddard, D. Armstrong, et al. Posterior fossa medulloblastoma in childhood: treatment results and a proposal for a new staging system. Int J Radiat Oncol Biol Phys. 1990;19:165-274.
138. R.I. Jakacki, P.M. Zeltzer, J.M. Boyett, et al. Survival and prognostic factors following radiation and/or chemotherapy for primitive neuroectodermal tumors of the pineal region in infants and children: a report of the Children’s Cancer Group. J Clin Oncol. 1995;13:1377-1383.
139. A.L. Albright, J.H. Wisoff, P. Zeltzer, et al. Prognostic factors in children with supratentorial (nonpineal) primitive neuroectodermal tumors: a neurosurgical perspective from the Children’s Cancer Group. Pediatr Neurosurg. 1995;22:1-7.
140. C.C. Bailey, A. Gnekow, S. Wellek, et al. Prospective randomized trial of chemotherapy given before radiotherapy in childhood medulloblastoma. International Society of Paediatric Oncology (SIOP) and the (German) Society of Paediatric Oncology (GPO): SIOP I. Med Pediatr Oncol. 1995;25:166-178.
141. P.RM. Thomas, M. Deutsch, J.L. Kepner, et al. Low stage medulloblastoma: final analysis of trial comparing standard-dose with reduced-dose neuraxis irradiation. J Clin Oncol. 2000;18:3004-3011.
142. R.J. Packer, L.N. Sutton, R. Elterman, et al. Outcome for children with medulloblastoma treated with radiation and cisplatin, CCNU and vincristine chemotherapy. J Neurosurg. 1994;81:690-698.
143. R.J. Packer, J. Goldwein, H.S. Nicholson, et al. Treatment of children with medulloblastoma with reduced dose craniospinal radiation therapy and adjuvant chemotherapy. A Childrens Cancer Group study. J Clin Oncol. 1999;17:2127-2136.
144. V.A. Levin, L.A. Rodriguez, M.S. Edwards, et al. Treatment of medulloblastoma with procarbazine, hydroxyurea, and reduced radiation doses to whole brain and spine. J Neurosurg. 1988;68:383-387.
145. R.E. Taylor, C.C. Bailey, K. Robinson, et al. Results of a randomized study of preradiation chemotherapy versus radiotherapy alone for non-metastatic medulloblastoma: the International Society of Paediatric Oncology/United Kingdom Children’s Cancer Study Group PNET-3 study. J Clin Oncol. 2003;21:1581-1591.
146. J.G. Douglas, J.L. Barker, R.G. Ellenbogen, et al. Concurrent chemotherapy and reduced-dose cranial spinal irradiation followed by conformal posterior fossa tumor bed boost for average-risk medulloblastoma: efficacy and patterns of failure. Int J Radiat Oncol Biol Phys. 2004;58:1161-1164.
147. R.J. Packer, A. Gajjar, G. Vezina, et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average risk medulloblastoma. J Clin Oncol. 2006;24:4202-4208.
148. A. Gajjar, M. Chintagumpala, D. Ashley, et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma. Lancet Oncol. 2006;7:813-820.
149. T.E. Merchant, L.E. Kun, M.J. Krasin, et al. Multi-institution prospective trial of reduced dose craniospinal irradiation (23.4 Gy) followed by conformal posterior fossa (36 Gy) and primary site irradiation (55.8 Gy) and dose-intensive chemotherapy for average-risk medulloblastoma. Int J Radiat Oncol Biol Phys. 2008;70:782-787.
150. J.P. Krischer, A. Ragab, L. Kun, et al. Nitrogen mustard, vincristine, procarbazine, and prednisone as adjuvant chemotherapy in the treatment of medulloblastoma. J Neurosurg. 1991;74:905-909.
151. J.R. Geyer, P.M. Zeltzer, J.M. Boyett, et al. Survival of infants with primitive neuroectodermal tumors or malignant ependymomas of the CNS treated with eight drugs in one day: A report of the Children’s Cancer Group. J Clin Oncol. 1994;12:1607-1615.
152. E. Kovnar, S. Kellie, M. Horowitz, et al. Preirradiation cisplatin and etoposide in the treatment of high-risk medulloblastoma and other malignant embryonal tumors of the central nervous system: a phase II study. J Clin Oncol. 1990;8:330-336.
153. M.A. Castello, A. Clerico, G. Deb, et al. High-dose carboplatinum in combination with etoposide (JET regimen) for childhood brain tumors. Am J Ped Hem-Onc. 1991;12:297-300.
154. S.J. Kellie, C.K. Wong, L.D. Pozza, et al. Activity of postoperative carboplatin, etoposide, and high-dose methotrexate in pediatric CNS embryonal tumors: results of a phase II study in newly diagnosed children. Med Pediatr Oncol. 2002;39:168-174.
155. R. Geyer. Intensive chemotherapy pilot for infants with malignant brain tumors. Proc Amer Soc Clin Oncol. 1993;12:417.
156. R.L. Heideman, E.H. Kovnar, S.J. Kellie, et al. Preirradiation chemotherapy with carboplatin and etoposide in newly diagnosed embryonal pediatric CNS tumors. J Clin Oncol. 1995;13:2247-2254.
157. B. Timmermann, R-D Kortmann, J. Kühl, et al. Role of radiotherapy in the treatment of supratentorial primitive neuroectodermal tumors in childhood: results of the prospective German Brain Tumor trials HIT 88/89 and 91. J Clin Oncol. 2002;20:842-849.
158. R-D Kortmann, J. Kühl, B. Timmerman, et al. Postoperative neoadjuvant chemotherapy before radiotherapy as compared to immediate radiotherapy followed by maintenance chemotherapy in the treatment of medulloblastoma in childhood: results of the German prospective randomized trial HIT ‘91. Int J Radiat Oncol Biol Phys. 2000;46:269-279.
159. R.E. Taylor, C.C. Bailey, K.J. Robinson, et al. Outcome for patients with metastatic (M2-3) medulloblastome treated with SIOP/KUCCSF PNET-3 chemotherapy. Europ J Cancer. 2005;41:727-734.
160. D. Strother, D. Ashley, S.J. Kellie, et al. Feasibility of four consecutive high-dose chemotherapy cycles with stem cell rescue for patients with newly diagnosed medulloblastoma or supratentorial primitive neuroectodermal tumor: results of a collaborative study. J Clin Oncol. 2001;19:2696-2704.
161. S.N. Chi, S.L. Gardner, A.S. Levy, et al. Feasibility and response to induction chemotherapy intensified with high dose methotrexate for young children with newly diagnosed disseminated medulloblastoma. J Clin Oncol. 2005;22:4881-4887.
162. S.J. Mork, A.C. Loken. Ependymoma – a follow up study of 101 cases. Cancer. 1977;40:907-915.
163. J.W. Goldwein, B.W. Corn, J.L. Finlay, et al. Is craniospinal irradiation required to cure children with malignant (anaplastic) intracranial ependymoma? Cancer. 1991;67:2766-2771.
164. N.A. Paleologos, D.R. Macdonald, N.A. Vick, et al. Neoadjuvant procarbazine, CCNU and vincristine for anaplastic and aggressive oligodendroglioma. Neurology. 1999;53:1141-1143.
165. J.C. Buckner, D. GesmeJr, J.R. O’Fallon, et al. Phase II trial of procarbazine, lomustine and vincristine as initial therapy for patients with low grade oligodendroglioma or oligoastrocytoma: efficacy and associations with chromosomal abnormalities. J Clin Oncol. 2003;21:251-255.
166. M.J. van den Bent, M.J.B. Taphoorn, A.A. Brandes, et al. Phase II study of first-line chemotherapy with temozolamide in recurrent oligodendroglial tumors: the European Organization for Research and Treatment of Cancer Brain Tumor Group Study 26971. J Clin Oncol. 2003;21:2525-2528.
167. S. Oi, K. Matsuzawa, J-U. Choi, et al. Identical characteristics of the patient populations with pineal region tumors in Japan and in Korea and therapeutic modalities. Child’s Nerv Syst. 1998;14:36-40.
168. J-U. Choi, D-S. Kim, S-S. Chung, et al. Treatment of germ cell tumors in the pineal region. Child’s Nerv Syst. 1998;14:41-48.
169. Y. Shibamoto, M. Abe, J. Yamashita, et al. Treatment results of intracranial germinoma as a function of the irradiated volume. Int J Radiat Oncol Biol Phys. 1988;15:285-290.
170. S.L. Wolden, W.M. Wara, D.A. Larson, et al. Radiation therapy for primary intracranial germ cell tumors. Int J Radiat Oncol Biol Phys. 1995;32:943-949.
171. J.C. Allen, R.C. DaRosso, B. Donahue, et al. A phase II trial of preirradiation carboplatin in newly diagnosed germinoma of the central nervous system. Cancer. 1994;74:940-944.
172. G. Calaminus, L. Andreussi, M-L. Garrè, et al. Secreting germ cell tumors of the central nervous system (CNS). First results of the cooperative German/Italian pilot study (CNS sGCT). Klin Pädiatr. 1997;209:222-227.
173. S.J. Kellie, H. Boyce, I.J. Dunkel, et al. Primary chemotherapy for intracranial nongerminomatous germ cell tumors: results of the Second International CNS Germ Cell Study Group protocol. J Clin Oncol. 2004;22:846-853.
174. P.L. Robertson, R.C. DaRosso, J.C. Allen. Improved prognosis of malignant intracranial non-germinoma germ cell tumors with multimodality therapy. J Neuro-Oncol. 1997;32:71-80.
175. B.H. Cohen, R.J. Packer, K.R. Siegel, et al. Brain tumors in children under 2 years: treatment, survival and long-term prognosis. Pediatr Neurosurg. 1993;19:171-179.
176. L.C. Strauss, T.M. Killmond, B.S. Carson, et al. Efficacy of postoperative chemotherapy using cisplatin plus etoposide in young children with brain tumors. Med Ped Oncol. 1991;19:16-21.
177. C. Dufour, J. Grill, A. Lellouch-Tubiana, et al. High-grade glioma in children under 5 years of age: a chemotherapy only approach with the BBSFOP protocol. Europ J Cancer. 2006;42:2939-2945.
178. S. Rutkowski, U. Bode, F. Deinlein, et al. Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. New Engl J Medicine. 2005;352:978-986.
179. D. Strother, J. Kepner, P. Aronin, et al. Dose-intensive (DI) chemotherapy (CT) prolongs event-free survival (EFS) for very young children with ependymoma (EP). Results of Pediatric Oncology Group (POG) study 9233. Proc Am Soc Clin Oncol. 2000;19:585a. (Abstr 2302)
180. H.K. Thorarinsdotter, B. Rood, N. Kamani, et al. Outcome in children < 4 years of age with malignant central nervous systems tumors treated with high dose chemotherapy and autologous stem cell rescue. Pediatr Blood Cancer. 2007;48:278-284.
181. G. Dhall, H. Grodman, L. Ji, et al. Outcome of children less than three years old at diagnosis with non-metastatic medulloblastoma treated with chemotherapy on the “Head Start” I and II protocols. Pediatr Blood Cancer. 2008;50:1169-1175.
182. J.F. Hirsch, D. Reiner, R. Czernichew, et al. Medulloblastoma in childhood: survival and functional results. Acta Neurochir. 1979;48:1-15.
183. R.J. Packer, L.N. Sutton, T.E. Atkins, et al. A prospective study of cognitive function in children receiving whole brain radiotherapy and chemotherapy: 2-year results. J Neurosurg. 1989;70:707-713.
184. R.K. Mulhern, L.E. Kun. Neuropsychologic function in children with brain tumors. III: interval changes in the six months following treatment. Med Ped Oncol. 1985;13:318-324.
185. D. Riva, C. Pantaleoni, N. Milani, et al. Impairment of neuropsychological functions in children with medulloblastomas and astrocytomas in the posterior fossa. Child’s Nerv Syst. 1989;5:107-110.
186. B.J. Spiegler, E. Bouffet, M.L. Greenberg, et al. Change in neurocognitive functioning after treatment with cranial radiation in childhood. J Clin Oncol. 2004;22:706-713.
187. D.M. Ris, R. Packer, J. Godlwein, et al. Intellectual outcome after reduced-dose radiation therapy plus adjuvant chemotherapy for medulloblastoma: a Children’s Cancer Group study. J Clin Oncol. 2001;19:3470-3476.
188. R.J. Packer, J.G. Gurney, J.A. Punyko, et al. Long-term neurologic and neurosensory sequelae in adult survivors of a childhood brain tumor: childhood cancer survivor study. J Clin Oncol. 2003;21:3255-3261.
189. D.A. Rottenberg, N.L. Chernik, M.D.F. Deck, et al. Cerebral necrosis following radiotherapy of extracranial neoplasms. Ann Neurol. 1977;1:339-357.
190. G. Di Chiro, E. Oldfield, D.C. Wright, et al. Cerebral necrosis after radiotherapy and/or intrarterial chemotherapy for brain tumors: PET and neuropathologic studies. Amer J Radiol. 1988;50:189-197.
191. W.G. Mitchell, L.S. Fishman, J.H. Miller, et al. Stroke as a late sequela of cranial irradiation for childhood brain tumors. J Child Neurol. 1991;6:128-133.
192. N.E. Anderson. Late complications of childhood central nervous system tumour survivors. Current Opinion Neurol. 2003;16:677-683.
193. N.A. Samaan, M.M. Bakdash, J.B. Caderao, et al. Hypopituitarism after external irradiation: evidence for both hypothalamic and pituitary origin. Ann Int Med. 1975;83:771-777.
194. W.P. Dickenson, D.H. Berry, L. Dickenson, et al. Differential effects of cranial radiation on growth hormone response to arginine and insulin infusion. J Pediatr. 1978;92:754-757.
195. M. Schmeigelow, U. Feldt-Rasmussen, A.K. Rasmussen, et al. A population-based study of thyroid function after radiotherapy and chemotherapy for childhood brain tumor. J Clin Endocr & Metabol. 2003;88:136-140.
196. M.T. Jennings, S. Iyengar. Pharmacotherapy of malignant astrocytomas of children and adults: current strategies and future trends. CNS Drugs. 2001;15:719-743.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
8 Pediatric Neuro-Oncology
Introduction
EPIDEMIOLOGY
The incidence of brain tumors among children (ages 0 to 14 years) is highest in the Scandinavian countries (31.4 per 106 per annum) followed by Western Europe, the United States of America, and New Zealand (24 to 27 per 106). The age-specific incidence for brain tumors is highest among children less than 5 years of age (36 per 106 per annum), then declines among those 5 to 9 years (31.9 per 106) and 10 to 14 years (24.6 per 106) to the 15-19 age group (20.2 per 106 annually).1–4
Age correlates with the relative site of origin among cancers of the brain, in a pattern that is quite different than that observed among adults. Brainstem gliomas (BSG) are more common in children between 5 and 9 years of age (5.9 per 106 per annum) than among preschoolers (4.7 per 106), adolescents of 10 to 14 years (2.8 per 106), or older teenagers (1.7 per 106 per annum). Cerebellar neoplasms are similarly distributed, occurring most frequently between 5 and 9 years of age (9.7 per 106). By comparison, the relative distribution of tumors arising in the cerebrum gradually increases following the first 5 years of life (5.8 per 106 per annum) through adolescence (15 to 19 years of age) (7 per 106).4 In contrast, during adulthood, the vast majority of primary CNS neoplasms are supratentorial in origin.
Reliable epidemiologic data confirm an increase in the incidence of children with gliomas between 1973 and 1992. This increase has been restricted to the AST (World Health Organization Grades I and II), with an average yearly rise of 3%. The trend has been more notable among females, in whom there has been an average annual increase of 3.9%. During this period, there was no change in the relative incidence of the other “classic” childhood CNS tumors, such as MBL and EPD.5
Among children and adolescents, tumors of astrocytic lineage constitute about 52% of reported cases. Other types of gliomas, such as gangliogliomas or mixed AST-OLG, contribute an additional 15.5%.4 There are two clearly defined peaks in relative incidence, observed at five years (20.7 per 106 per annum) and thirteen (19.7 per 106) years of age.3 After puberty, the incidence of AST and malignant astrocytomas (MA) falls to 12.3 per 106 annually.1
The PNETs comprise neoplasms considered “embryonal” in their pathogenetic derivation, and include the MBL, medulloepithelioma, pineoblastoma and the “cerebral neuroblastoma.” Overall, this family of cancers comprises only 3.6% of all brain tumors.6 As a group, the PNET constitute about 22% of primary CNS neoplasms occurring during childhood and adolescence.4 Their relative incidence declines throughout childhood, from the peak seen among children less than 5 years of age (9.6 per 106 per annum), through those of 5 to 9 years of age (7.4 per 106), 10 to 14 years (4 per 106), and older adolescents (2.5 per 106 annually).4 According to the National Cancer Institute’s SEER data, the incidence of MBL-PNET rose 23% between 1973-1976 and 1993-1998, that is from 4 to 4.9 per 106 person-years (PY).10
Ependymomas constitute about 9% of tumors arising within the CNS during childhood and adolescence, but represent only 3% of brain tumors overall.4,9 The relative incidence varies among age cohorts, being highest among infants, toddlers, and preschoolers (5.6 per 106 per annum), then falling to 1.1-1.6 per 106 annually among children older than 5 years of age.1 The peak age at diagnosis is during the second year of life (8.6 per 106).4
Germ cell tumors present in three principal age periods. Congenital GCTs are typically benign teratomas, and during infancy (1 month to 3 years), endodermal sinus tumors and/or malignant teratomas predominate. Adolescence and young adulthood are usually associated with endodermal sinus tumors and embryonal carcinomas, often with teratomatous elements. In contrast to the prevalence of these nongerminomatous GCTs (NG-GCT) among the young, the majority of adult GCTs are pure germinomas.8 In Japan, Taiwan, and South Korea, GCTs comprise 2.1% to 9.4% of primary intracranial neoplasms.9 This is consistently higher than the 0.4% to 3.4% reported in western series.10 In North America, origin within the brain accounts for 14% of all GCTs occurring among persons less than 20 years of age. Their relative incidence has steadily risen from 0.5 per 106 per annum in the 1970s to 1.5 per 106 in the late 1990s, which, within the industrialized countries, has suggested an environmental influence for their pathogenesis.
The observed survival rates for malignant CNS tumors diagnosed between 1973 and 1999 were grim: 50.0% at 1 year, 35.0% at 2 years, 25.6% at 5 years, and only 19.5% at 10 years from diagnosis. This translates to a mortality rate of 5.3/100,000 PY for males and 3.6/100,000 PY among females.2
PROPOSED ETIOLOGIC FACTORS
There is an approximately three- to nine-fold increased risk of developing a brain tumor should a family member be so affected; however, the nature of this association is not understood.11 Allegations have been raised regarding electromagnetic waves, nitrosourea derivatives in processed meats, pesticides, and unique occupational hazards. However, no chemical agent not already known to be carcinogenic for other human tissues has been found to be uniquely associated with cancer of the brain. Neuroectodermal tissue is vulnerable to the carcinogenic effects of external-beam radiation therapy (EBRT). A strong dose-response relationship exists, as exposures higher than 2.5 Gray (Gy) are associated with a 20-fold increased carcinogenic potential.12 The latency period for postradiation neuro-oncogenesis stretches from 12 years to 15 years (range 4 to 30 years).13
CLINICAL PRESENTATION
Low-Grade and Malignant Glioma
The child afflicted with a cerebral neoplasm complains of symptoms referable to its location. Cognitive impairment (15% to 20%) and seizures, either at presentation (about one third) or during the course of the illness (50% to 70%), are common problems that bring the patient to medical attention.14 Superficial cerebral tumors may produce partial, complex partial, or secondarily generalized seizures. Symptoms of dysphasia, mental confusion, or motor deficit are predictive of significantly impaired physical, cognitive, emotional, and social function.15 Motor or sensory compromise will reflect proximity to eloquent cortex. Deeply seated neoplasms, or those situated so as to obstruct the drainage of cerebrospinal fluid (CSF), produce headache, altered mental status, and, possibly, visual obscuration with or without localizing corticospinal tract findings. The pattern of headache, such as those which awaken the patient from sleep (10% to 32%), early morning occurrence followed by vomiting, as well as the influence of position, posture, or exercise (20% to 32%), contribute to the recognition of increased intracranial pressure. Recent onset, evolution of the headache’s intensity, papilledema and/or focal deficits on exam also expedite a diagnostic investigation.16 Brainstem gliomas are recognized by the combination of multiple cranioneuropathies, corticospinal tract findings, ataxia, and/or obstructive hydrocephalus.
Medulloblastoma
Park et al.17 reviewed 144 patients treated at a major Canadian referral center between 1950 through 1980. The prediagnosis symptomatic interval was less than 6 weeks in 51%, and less than 12 weeks in 76%. The age at the time of diagnosis ranged from 7 weeks to 15 years. Eighty-one percent of patients were between 1 and 10 years of age. The clinical presentation was that of a posterior fossa mass, i.e., intracranial hypertension from hydrocephalus (Figure 8-1). Seventy-six percent had papilledema and 65% complained of headache. Recurrent vomiting occurred in 47%. Axial ataxia (62%), nystagmus (44%), and appendicular dysmetria (35%) were common neurologic signs. Hemorrhage into the primary tumor occurred in four, who presented with abrupt neurological deterioration.17
Staging evaluation is an important aspect in establishing the clinical diagnosis. Harisiadis and Chang18 stratified patients based on tumor size (“T”) and extent of metastatic spread (“M”). Spinal subarachnoid dissemination has been identified in 36% to 43% of prospectively studied patients.19 Dissemination to the bone marrow has been reported to be as high as 18% at the time of diagnosis.19 The frequency of this complication at recurrence rises to approximately 35%.20
The “atypical teratoid/rhabdoid tumor” has previously been confused with the MBL. This early childhood neoplasm of the CNS arises either supra- or infra-tentorially and is composed of rhabdoid cells and undifferentiated small cells, with epithelial, mesenchymal, and neural elements. Overall survival (OS) periods of only 11 to 13 months have been reported following surgery and EBRT.21,22
Ependymoma
Supratentorial origin is more likely to be associated with corticospinal tract deficits and increased intracranial pressure at the time of presentation. Intratentorial location is frequently identified because of cranioneuropathies, ataxia, hydrocephalus, and a briefer prodrome because of the higher grade of malignancy encountered (Figure 8-2). Spinal EPDs present with back and/or radicular pain as well as lower motor neuron signs. The overall incidence of subarachnoid dissemination with intracranial EPD is reported to be approximately 10% to 22%, but this is influenced by site of origin and tumor grade. Anaplastic infratentorial tumors have a rate approaching 30%, while the incidence of spinal dissemination among the supratentorial EPD is negligible.23
Oligodendroglioma
A series of 37 patients demonstrated that the common presenting symptoms were seizures (43%), headache (38%), motor deficits (38%), and to a lesser degree, behavioral changes (16%). The most common site of origin was the frontal lobe (43%). Low grade histopathology (WHO grades I and II) was documented in 60%, with the remainder being grade III or IV disease.24
Germ Cell Tumor
Ninety-five percent of primary intracranial GCTs arise along an axis from the suprasellar cistern (37%) to the pineal gland (48%) (Figure 8-3). In one study, germinomas were found to preferentially involve the suprasellar region (57%), while 68% of NG-GCT’s originated in the pineal recess (P < .0001). Germ-cell tumors were found in the suprasellar region in 75% of female patients; pineal involvement was more frequent (67%) among males (P = .0001). The age distribution peaked during early puberty. Nongerminomatous GCTs (24%) were more frequently diagnosed between birth and 9 years than were germinomas (11%) (P < .0001). Among patients with germinomas, 35% were reported to be symptomatic for 6 months or longer, with half of these in excess of 24 months. The prodrome was much shorter among patients with NG-GCT (P = .0007).8
Patients with germinomas, which were commonly suprasellar, presented with chiasmal visual field defects (33%), diabetes insipidus (41%), and hypothalamic-pituitary dysfunction (33%). These neuroendocrine deficits included delay or regression of sexual development (16%), hypopituitarism (16%), and growth failure (9%). Presenting symptoms and signs among the NG-GCTs, which usually localized the lesion to the pineal recess, included hydrocephalus (47%), Parinaud’s sign (34%), obtundation (26%), pyramidal tract findings (21%), and ataxia (19%). Spinal cord metastases were more prevalent among patients with germinomas (11%) and endodermal sinus tumor (23%). Abdominal and pelvic metastases developed in approximately 10% of the 106 patients who were known to have received ventriculoperitoneal shunting.8
Brain Tumors Arising During Infancy
Primary CNS neoplasms during the first 2 years of life are considered quite uncommon in individual practice. However, they actually constitute 13% of all childhood brain tumors. The ratio between malignant (47%) and low-grade tumors is nearly equal in this age-group. Symptoms and signs are related to the site of origin, which is infratentorial in most (40% to 57%).25 The most common symptom has been vomiting (43%), with early morning occurrence in only a third of infants. Headache may be suspected in 33% because of head banging and irritability. Gait ataxia (38%) and macrocephaly (19%) were other findings. The nonspecific nature of these symptoms contributes to the frequent delay in diagnosis. Tonsillar herniation has been documented in 43% at the time of diagnosis, presenting with head tilt (one third) or opisthotonic arching (one third), the latter has been misinterpreted as tonic seizure activity. As many as 10% of these children have presented as failure to thrive secondary to hypothalamic involvement, termed “the diencephalic syndrome.”25
PROGNOSTIC VARIABLES
Malignant Glioma
It has proven difficult to identify biologic determinants of survival that can be conveniently assayed in a timely manner at the time of diagnosis in order to better guide the intensity of therapy. However, some attempts have been encouraging. For example, identification of the labeling index has shown an encouraging prognostic correlation with the histopathologic grade of MG, which in the future may allow stratification of pediatric subjects in clinical trials by relative risk.26 The literature also suggests that expression of the multiple drug resistance (PgP/MDR) proteins and intratumoral hypoxia may also influence outcome.
Histopathologic Grade and Age among Malignant Astrocytomas
The Children’s Cancer Group (CCG) #943 study showed the median survival among children with anaplastic astrocytomas (AAST) was greater than 6 years; among GBM patients, overall survival averaged less than 15 months (P = .012).27 Age and histopathologic grade jointly influenced outcome. The CCG #921 study found that among children less than 24 months old, the 3-year progression-free survival (3Y-PFS) was 36%. The 3Y-PFS for patients with AAST was 44%, which was dramatically better than that for GBM (0%) (Table 8-1).28
Multidrug Resistance Phenotype
The PgP and MRP proteins are thought to function as ATP-dependent transmembrane transporters, which nonspecifically remove large molecular weight solutes from the brain and/or prevent hematogenously borne chemicals from entering the CNS.29 PgP/MDR-1-associated resistance is hypothesized to function at the interface between the blood-brain and blood-tumor barriers as well as within the neoplastic cells themselves. Despite their association with progressive anaplasia and an adverse prognosis, there is actually little evidence that the PgP/MDR1 is a major mechanism of acquired resistance among MG.29–31 Resistance to therapy appears to be a more complex phenomenon which disrupts the mechanisms of repair of genotoxic injury and/or induction of apoptosis.
The O6-methylguanine-DNA methyltransferase (MGMT) enzyme’s function is to counteract the cytotoxic effects of alkylating agents; its expression correlates inversely with survival among adult MG patients. Archival histopathologic specimens from 109 patients treated on the CCG Study #945 were analyzed, and 11% had overexpression of MGMT. The 5Y-PFS was 8.3% among these patients, in contrast to those whose tumors did not show overexpression of MGMT (5Y-PFS of 42%, P = .017). This adverse correlation was independent of age, histopathologic grade, tumor location, or extent of residual disease.32
Hypoxia and the Abrogation of Apoptosis
Intratumoral hypoxia is known to be the most significant predictor of radiotherapeutic resistance.33 Radiation therapy has only one third the effectiveness against hypoxic tumor cells that it does against well-oxygenated cells. It was previously thought that radiation-induced DNA damage required the presence of oxygen radicals. Instead, normal cells, when deprived of oxygen, will arrest in late G1 and early S phase, in concert with hypophosphorylation of the retinoblastoma gene protein, pRB, and die through induction of apoptosis.33 Arrest of the DNA-damaged cell at the G1/S interface is associated with stabilization of intact wild-type TP53 (TP53wt) expression, inhibition of replicative DNA synthesis, and possibly the initiation of repair. TP53wt appears to achieve G1/S arrest through transcriptional regulation of at least one critical downstream target, the Cdk inhibitor p21WAF1/CIP1. This protein inactivates cyclin D/Cdk4, D/Cdk6, and E/Cdk2 complex activity and directly inhibits PCNA, a regulatory subunit of DNA polymerase (the principal replicative DNA polymerase).34
Ionizing radiation also produces G1 arrest in cells with TP53wt and increases TP53 protein expression.35 In contrast, the hypoxic regions within the tumor actually create a reservoir for malignantly transformed cells, which do not arrest at the G1/S interface, are more likely to have mutations in the TP53 gene (TP53 m), and become resistant to TP53-induced apoptosis (Graeber et al., 1996).36 Stable cytoplasmic TP53wt expression has been shown to a favorable predictor for survival among AAST patients (P = .015). By comparison, such expression was not found among the GBM (P = .8), which by definition contain areas of necrosis.37 Intratumoral hypoxia thus constitutes a key mechanism of clonal selection.
Furthermore, hypoxia has also been correlated with insensitivity to carmustine (BCNU) and cisplatin, independently of the putative drug resistance genes MDR1, MRP, O6MGMT, or ERCC.38 Here too, the acquisition of resistance appears be mediated through TP53 m. For example, 86% of TP53wt positive MG cell lines have been shown to be responsive to currently available chemotherapy drugs. In contrast, there were no effective cytotoxic agents against 75% of the glioma cultures expressing TP53 m (P = .0006). Supporting this specificity, the resistance conferred by TP53 m is restricted to alkylators and platinators, which damage DNA, rather than against the microtubular toxins.39
The Molecular Basis for Multiple Models of Glioma Progression: The Tumor Suppressor Genes
There is a large literature on the nonrandom cytogenetic aberrations observed among MGs, which include gains or losses of chromosomes, as well as rearrangements. Some of these changes, such as 10q, 9p, 11p15.5-pter and 19q may be found only among AASTs and/or GBMs.40,41 Loss of heterozygosity (LOH) analysis has correlated 17p deletions with allelic loss of the tumor suppressor gene (TSG), TP53.42 Hence, it has been hypothesized that such nonrandom patterns of genetic loss can be exploited to predict the location of other as yet unknown TSGs. This in turn has generated the concept of modeling mechanisms for tumor progression.
TP53
One pathway in the evolution of the MG involves TP53 m, which occurs in 14% to 46% of adult patients and is preferentially associated with development of the glioma (often an AAST) between 18 and 45 years of age.42,43 Clonal expansion of TP53 m cells have been observed from a small subpopulation within a primary AST to become the dominant cell type within a recurrent, or “secondary” MG.44
Alternate means of TP53 wt inactivation exist as TP53 wt may be complexed by the MDM2 oncogene product. In 8% to 10% of GBM, the MDM2 gene is amplified and overexpressed, supporting the importance of TP53 wt in glial cell cycle dysregulation. A more recent study has identified that about 35% of gliomas demonstrate either TP53 m or methylation of p14(ARF), suggesting that TP53 wt is controlled by down-regulation of p14(ARF).45 Inactivation of the astrocyte’s apoptotic response may therefore be effected either by deletion of 17p, mutation of TP53, MDM2 overpression, or hypermethylation of p14(ARF), thus unmasking multiple points of vulnerability along a common pathway that permit further progression towards the endpoint of malignant transformation.45
PTEN
Another fraction of about 30% (23% to 62%) of adult GBM patients has been characterized by the association of LOH of chromosome 10 with epidermal growth factor receptor (EGFR) amplification. In contrast to the previous group, these patients (typically older individuals) appear to present de novo with a “primary” GBM. The LOH at 10q23 has been demonstrated in approximately 70% of GBMs, and therefore predicted to be the site of another glial TSG.42,43 The “Mutated in Multiple Advanced Cancers” and “Phosphatase and Tensin” gene (MMAC/PTEN) has been localized to chromosome 10q25 and shown to function as a phosphatidylinositol 3,4,5-triphosphate phosphatase, which modulates cell growth as well as apoptosis.46,47
Patients with GBM have demonstrated a significantly higher incidence of LOH at the MMAC/PTEN locus (72%) than did patients with AAST (29%)(P < .0001). The clinical relevance is that patients with LOH at the MMAC/PTEN locus have a more adverse prognosis (P < .0001), even when controlled for age at surgery and histologic grade.48 Conversely, Kaplan-Meier analysis has demonstrated significantly better survival for patients whose MG exhibited high MMAC/PTEN expression.49 These results have been interpreted to implicate alteration or loss of the MMAC/PTEN locus as a late marker of disease progression with ominous significance.
RB
About 15% to 44% of MG patients are reported to have loss of chromosome 13 as well as LOH or partial deletions of the 13q14 allele, which suggests the possibility that the RB gene is also a glioma TSG. Loss of heterozygosity at the RB 1.20 region has not been detected among low-grade gliomas, but occurred in 20% of AAST and 32% of GBM. The presence of LOH at the RB 1.20 region has also been associated with an adverse prognosis, implying that loss/inactivation of pRB contributes to disease progression.50
Investigation suggests that aberrant function of cyclin D, Cdk4, p16INK4A/MTS1, and/or pRB is critical for both the process of transformation and that of progression towards increasing anaplasia among glial neoplasms. Cyclin D1 was originally cloned from a human GBM cDNA library, where its transcript and 34 kD product were in abundant expression. Deletion of p16INK4A/MTS1 and/or amplification of its target Cdk4 occur in 50% of AAST and 85% of GBM.51,52 The coordinate deletion of both p15INK4B/MTS2 and p16INK4A/MTS1 among GBMs suggests that selection favors homozygous deletions of chromosome 9p21 as more efficient for the simultaneous inactivation of both Cdk inhibitors than independent intragenic mutations would be.51 This remarkable specificity suggests that knocking out p16INK4A/MTS1’s inhibition of cyclin D1/Cdk4-induced hyperphosphorylation of pRB marks a critical event in the progression to the most advanced grade of glioma, the GBM.52
Evidence is accumulating that cyclin D1 overexpression, absence of pRB expression, failure of pRB dephosphorylation, and/or pRB cleavage may also contribute to chemotherapeutic drug resistance as well, although the mechanism or mechanisms are not understood. Altered CDKN2/p16 function has been correlated with increased sensitivity to antimetabolite agents as opposed to the alkylators, topoisomerase inhibitors, and microtubular toxins.53
To conclude, the genetic mechanism of glial transformation in children is not understood. In one series of 20 pediatric GBMs, 25% were associated with TP53m, the majority of which presented with a brief prodromal period and were considered “primary.” Loss of p16CDKN2/INK4A/MTS1 was detected in 61% of these GBMs. Overexpression of EGFR was infrequent (11%) and was coincidental with TP53 m in one case. Of the four GBMs, which progressed from lower-grade tumors, only one contained TP53 m.54 In another series of 29 childhood MGs, 95% of cases were found to harbor an alteration in at least one member of the TP53/MDM2/p14ARF tumor suppressor pathway. Overexpression of MDM2, which downregulates TP53 transcriptional activity, was present in 65%, and loss of p14AFT, which inactivates the pathway through MDM2, was found in 10%.55
Among pediatric MGs, the presence of MMAC/PTEN mutations has also been confirmed as a marker of an adverse outcome that is independent of patient age or histopathologic grade (III vs. IV).56 In tumor specimens of 62 evaluable patients treated on the Children’s Cancer Group (CCG) #945 protocol for children with MG (vide infra), alteration of PTEN sequence was detected in just one, in conjunction with loss of chromosome 10. Deletions of PTEN without mutations were found in seven additional specimens. The PTEN alterations were more common among GBM than other histopathologic grades (P = .0056). Although 37% (14/38) of evaluable tumors had increased EGFR expression, only one exhibited amplification of the EGFR gene. This may be interpreted to indicate that pediatric MGs differ in the mechanisms of tumorigenesis from those of adulthood.57
Brainstem Glioma
Favorable prognostic features include (a) protracted symptoms, (b) origin within the optic tectum, the mesencephalon, or at the cervicomedullary junction, (c) lesions which are cystic, focal, and/or dorsally exophytic, (d) onset in adulthood, and (e) neurofibromatosis type I (NF1). By comparison, the adverse markers for disease progression are diffuse pontine infiltration, a high mitotic index, a brief symptomatic prodrome, and multiple cranioneuropathies at presentation.58
Low-Grade Astrocytoma
Low-grade glial neoplasms (LGGN), which include AST, mixed astrocytoma-oligodendroglioma (AST-OLG), and pure OLG, constitute a heterogenous group of tumors without a predictable natural history. The 5Y-OS and 10Y-OS rates for LGGN following resection and EBRT are reported to range between 40% to 70% and 11% to 50%, respectively.59 In a review of 461 AST patients, the 5Y-OS rate was 36%; that is a third of the predicted actuarial survival for a comparable population of unaffected individuals.59 Only 16% of LGGN patients of any age were alive at 15 years following diagnosis.60 Otherwise stated, the long-term survival rate among patients with LGGN is less than that of their peers suffering from acute lymphocytic leukemia.
The literature suggests that age at presentation, seizures as the sole presenting symptom, site of origin, favorable Karnofsky status, absence of contrast enhancement on imaging, histopathologic grade (i.e., pilocytic vs. diffuse) (Figure 8-4), initial tumor dimensions, extent of resection (i.e., unresectable tumor), presence of hydrocephalus, or coexistence of NF1 are the important variables that influence quality of life as well as survival.61 The prognostic value of the labeling index has been previously reviewed.26
Site of Origin
It is accepted that the biology of LGGN arising within the hypothalamus, optic tectum, and cervico-medullary junction clearly differs from those arising within the pons and cerebrum. Consensus exists that gross total resection of an AST or mixed LGGN during childhood is adequate therapy and does not require immediate postoperative irradiation (Nishio et al., 1989).62 Univariate analysis has demonstrated that optic chiasmal origin and only partial resection are associated with a worsened prognosis.63 This has influenced the controversy regarding EBRT (vide infra).
Neurofibromatosis
The 5Y-OS and 10Y-OS for patients with optic nerve gliomas and NF1 has been reported to be 93% and 81%, respectively. This compares favorably to survival rates of 83% and 76% at 5 and 10 years, respectively, for those children without evidence of NF1. The mean time to disease progression (TDP) was significantly different between the two groups (P < .01) in favor of those with NF1, although this did not impact OS.64 However, this relative advantage does not appear to apply to patients with von Recklinghausen disease who suffer from gliomas of the cerebrum.65
Histopathologic Grade and Malignant Progression among Low-Grade Astrocytomas
The process of malignant transformation was studied among 11 of 65 children with LGGN. The median latency was 5.1 years, and the 15-year cumulative incidence estimate was 6.7 %. Overexpression of TP53 was more common after progression to higher histopathologic grade. Deletions of RB1 and/or CDKN2A were observed in 71% of the LGGN and 90% of their malignant successors. There were PTEN pathway abnormalities found in 76% of patients.66
Medulloblastoma and Primitive Neuroectodermal Tumor
Staging evaluation stratifies patients by tumor size (“T”) and extent of metastatic spread (“M”).18 Table 8-2 summarizes its prognostic significance as derived from the European Societie Internationale Oncologie Pediatrie (SIOP) and the North American CCG study #942.67,68 Stepwise analysis of survival among randomized patients demonstrated earlier age of onset to be the dominant adverse variable (P = .034). There were significant interactions between treatment and M stage, which support postoperative irradiation and chemotherapy among patients with advanced disease, i.e., high T and M stages (P = .004) (Table 8-2).68
The subsequent CCG #921 study of children with MBL (203 patients) demonstrated the significance of residual disease prior to EBRT, whether determined by extent of surgical resection (P = .023) or by evidence of metastatic tumor (M1–3) (P < .003) (Table 8-2).69,70
Pathologic Studies
A series of 330 cases studied by the Pediatric Oncology Group (POG) found significant anaplasia among 24%, which was strongly associated with an unfortunate outcome. Diffuse or extensive anaplasia was worse than focal involvement. In contrast, extensive nodularity conferred a more favorable course.71 Reevaluation of 347 MBL biopsies treated under the SIOP II trial confirmed that severe anaplasia conferred an adverse prognostic effect on 5-year progression-free survival (49.5%) relative to mild-moderate change (65.4%) (P = .001). This effect was magnified when the presence or absence of extensive apoptosis (P = .00216) was factored in.72
Drug Resistance Genes
There is evidence that the expression of the MDR1, MRP1, LRP and BCRP genes does not correlate with overall outcome among patients with MBL/PNET.73
Biologic Determinants of Survival
Established chromosome abnormalities include amplication of isochromosome 17q, novel amplicons, and losses as well as gains of chromosomes; these are known to occur in greater than 20% of MBL. Copy number abnormalities have been identified in specific regions of chromosomes 1, 8p, 10q, 11p, and 16q, which occur frequently among MBLs and may identify distinct subsets.74 Furthermore, differences in DNA copy number at chromosome regions 1p12-22.1, 9p21, 19p, and chromosome 17 have been used to segregate between MBL and supratentorial PNET. The 9p21 deletions correlated with loss of CDKN2A protein expression more frequently among the PNET (P < .001). Gains of 19p were also more evident among the PNET (P = .02), whereas gains of 17q were more common among the MBL (P = .02).75
(i) Tumor suppressor genes
Haploinsufficiency of the 17p13.3 region is the most commonly found disrupted genetic site (35-50%) among MBLs, which may indicate the site of yet another TSG(s). This chromosomal site contains the “Hypermethylated in Cancer” gene (HIC1), a transcriptional repressor, which is a frequent target of epigenetic gene silencing in MBL. HIC1 is a direct transcriptional repressor of “Atonal Homolog 1” gene (ATOH1), a proneural transcription factor essential for cerebellar development. ATOH1 is a putative target of “(Sonic) Hedgehog” (HH) signaling and its expression has been shown to be required for human MBL cell growth in vitro. The PTCH gene, an inhibitor of SHH signaling, is among the characterized TSG in MBL; however, fewer than 20% of MBLs have mutations in this gene. Recent work suggests that the HIC1 and PTCH1 TSG cooperate to silence ATOH1 expression during a critical phase of granule cell precursor differentiation in the cerebellum to contribute to the malignant progression to MBL.76
Specimens of MBL collected from 65 children treated in the SIOP/United Kingdom Children’s Cancer Group PNET3 trial were segregated into the histopathologic groups such as the large cell/anaplastic phenotype and nodular/desmoplastic variant, among others. Loss of 17p13.3 was found among 38% of samples of all variants, whereas MYCC/MYCN amplification (6%/8% respectively of MBLs) was significantly associated with the large cell/anaplastic phenotype. Loss of 9q22 coincided with the nodular/desmoplastic type. Together with metastatic disease at diagnosis, the large cell/anaplastic phenotype, 17p13.3 loss, or high frequency MYC amplification defined a high-risk group of children whose outcome was significantly worse than those without such tumor characteristics (P = .0002).77 However, the relationships between chromosome 17 lesions with anaplastic/large cell MBL and the abnormalities in the Sonic Hedgehog/PATCH (SHH/PTCH) pathway with the desmoplastic variant remain controversial.78
The SHH antagonist, cyclopamine, blocked expression of the SHH pathway targets PTCH1 and GLI1, lowered Bcl2 levels, and increased apoptosis in MBL cells in vitro. Blockade of the SHH pathway sensitized MBL cells to lovastatin, a proapoptotic agent used for lowering cholesterol levels. The combination of cyclopamine and lovastatin target pathways appear crucial for MBL cell survival.79 Agents targeting the SHH pathway are in clinical trials for the therapy of medulloblastomas.
Activation of the canonical WNT/Wingless (WNT/WG) signaling pathway occurs in up to 25% of cases of primary MBL and is associated with a favorable prognosis. Activation of WNT/WG was determined by evidence of CTNNB1 mutations and/or beta-catenin nuclear stabilization. Loss of chromosome 6 has been correlated with WNT/WG active tumors (P < .001), but few other cytogenetic aberrations including chromosome 17. In contrast, WNT/WG-negative MBLs were found to have losses of chromosomes 17p, 8, 10, and 16, with gains of chromosomes 7 and 17q. This supports the hypothesis of independent pathways of tumorigenesis among MBL, which are of potential clinical relevance.80
TP73 is a member of the TP53 TSG family that is overexpressed in a variety of tumors and mediates apoptotic responses to genotoxic stress. Biopsy samples of MBL and MBL cell lines have been reported to contain elevated levels of TP73 RNA and increased expression of the TAp73 and DeltaNp73 protein products. Overexpression of these induced apoptosis among cultured MBL cells in vitro and sensitized them, resulting in cell death upon exposure to chemotherapeutic agents. TAp73 RNA overexpression within biopsy samples was determined to correlate with a favorable PFS by Kaplan-Meier analysis.81
The 10q23.3 chromosomal region is subject to frequent allelic losses in MBL, which is the locus of the PTEN gene. Activation of the phosphoinosityl 3-kinase/ AKT (P13 k/AKT) signaling pathway appears to be associated with alterations of the PTEN gene. Proliferation of MBL cell lines has been shown to be dependent upon P13 k/AKT signaling and inhibited by a P13K antagonist as well as by AKT overexpression. Reduction of PTEN mRNA and protein expression has been found to be correlated with PTEN promoter hypermethylation in 50% of 22 MBL tissue samples. This suggests that PTEN loss or dysregulation by the P13 k/AKT signaling pathway may be an important mechanism of tumorigenesis for a subset of MBL.82 In contrast, PTEN deletion has not been found among supratentorial PNET biopsies or cell lines.83
(ii) The Putative Oncogenes.
The Duke group studied 31 MBL specimens for MYCC, MYCN and TRKC expression and correlated this with clinical outcome and histopathologic grading. The presence of MYCC mRNA was associated with shorter survival (P = .04) as well as with anaplasia. Regulation of MYCC is influenced by WNT signaling and MXI1 mutations. Nuclear translocation of beta-catenin, a marker of WNT pathway activation, was more common among the MBLs with high MYCC. No MXI1 mutations were detected in the 22 cases examined.84
Archival formalin-fixed, paraffin-embedded MBL samples from 78 patients treated on the prospective European multicenter HIT’91 protocol were studied for DNA amplification of C-MYC and N-MYC and mRNA expression of C-MYC and TRKC. TRKC and C-MYC mRNA expression were identified as independent prognostic factors on multivariate analysis. A favorable-risk group (eight patients), with a 7Y-PFS of 100%, possessed elevated TRKC and reduced C-MYC expression. The poor-risk group (15 patients) had metastatic disease, with high C-MYC and low TRKC mRNA expression. Their 7Y-PFS was only 33%. An intermediate-risk group of the remaining subjects showed a 7Y-PFS of 65%.85 Not all investigators have made similar correlations.86
Ependymoma
Pathologic Grading
The prognostic utility of pathologic grading of ependymomas has engendered a long-standing controversy, as there has been no clear distinction in 5Y-OS rates between “benign” and “malignant” EPD. However, one recent blinded review demonstrated that the PFS at 2 years following postoperative irradiation was 84% for differentiated EPD, and 32% for specimens considered anaplastic.87
Site of Origin
There is a correlation between site of origin and histopathologic grade of malignancy. Approximately two thirds of supratentorial EPDs are high-grade at diagnosis, whereas those arising in the IVth ventricle tend to have a better prognosis.88 A series of 49 patients found that 78% originated infratentorially and exhibited low grade histology.89 Those arising in the spine and cauda equina are usually low-grade and/or myopapillary.
Extent of Surgical Resection
The CCG #921 study accessioned 20 EPD and 12 anaplastic EPD patients, of whom only three had metastatic disease at diagnosis. The prognostic variables of significance included the extent of resection (gross total vs. other, P = .0001) and postoperative residual disease of < 1.5 cm2 (P < .0001), in contrast to age, staging, or treatment, emphasizing the importance of local disease control.90
The French Society of Pediatric Oncology has studied the efficacy of postoperative chemotherapy, with the intent of avoiding EBRT, among 73 children less than 5 years of age with intracranial EPD. The favorable prognostic variables included supratentorial origin (P = .0004) and complete resection (P = .0009). Patients with gross total resection demonstrated a 4Y-OS of 74% in contrast to those with residual disease (35%).91
Univariate and Multivariate Analysis
Three prognostic factors have been identified to have a significant correlation with the 5Y-PFS. These included the extent of surgical resection (P < .0001), age at diagnosis (P = .003), and the prediagnosis symptomatic interval (P = .02).23 Another multicenter retrospective study of 83 children with EPD confirmed that age of less than 3 years, identifiable postoperative residual disease, and Grade III histology were significant adverse factors for PFS in both univariate and multivariate analysis.92
Biologic Determinants of Survival
Overexpression of specific genes (YAP and LOC374491) and downregulation of others such as SULT4A1, NF-[kappa]B2, and PLEK have been implicated in determining the age of onset, relapse potential, and tumor location of pediatric EPD.93,94 The catalytic subunit of telomerase, the human telomere reverse transcriptase (hTERT), which aids in uncontrolled cell proliferation, has been shown to correlate with prognosis among 65 children with EPD. Study of 87 tumors from these patients found the presence of hTERT to be adversely correlated with 5Y-OS (41% vs. 84% in its absence).95
Oligodendroglioma
The accepted favorable prognostic variables for OLG include youth (less than 21 years), low histopathologic grade, and extent of resection.96 Multivariate analysis in a series of 51 patients aged 5 to 75 years found younger age and presentation with seizures alone to be statistically significant for a favorable outcome. Approximately one third of patients of all ages appear to be cured by aggressive treatment including surgical resection, EBRT, and chemotherapy.96
