18 Paraneoplastic Disorders
Introduction
Paraneoplastic neurological disorders (PND) are a heterogeneous group of disorders that can affect any part of the neuraxis, including the retina and muscle.1 Unlike other neurological complications that occur in patients with cancer, many PNDs are believed to be mediated by immune mechanisms. The current concept is that the expression of normal neuronal proteins by a cancer induces an immune response that targets the nervous system, resulting in neuronal dysfunction and/or neuronal cell death.2 These immune responses are often associated with the presence of specific antineuronal serum and cerebrospinal fluid antibodies.
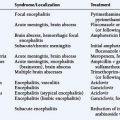
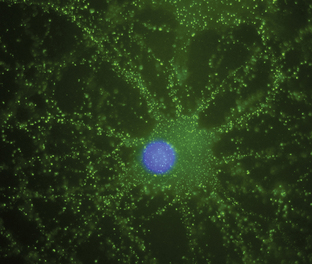
Antineuronal antibodies play a direct pathogenic role in three PNDs that affect the peripheral nervous system. These include antibodies to P/Q-type voltage-gated calcium channels (VGCC) in patients with the Lambert-Eaton myasthenic syndrome (LEMS),3 antibodies to acetylcholine receptor in patients with myasthenia gravis, and antibodies to voltage-gated potassium channels (VGKC) in some patients with peripheral nerve hyperexcitability (neuromyotonia).4 A common feature of these antibodies is that they target cell surface antigens and the associated disorders can occur without cancer; therefore, detection of these antibodies does not predict the presence of cancer. Antibodies to P/Q type VGCC are also found in a subgroup of patients with paraneoplastic cerebellar degeneration (PCD)5 and antibodies to VGKC-related proteins can be found in some patients with cancer-associated or non−cancer-associated limbic encephalitis (LE) and Morvan syndrome.6,7 In these cases, the antibodies are believed to be pathogenic, but this has not yet been proven. Similarly, there is recent evidence that antibodies to the N-methyl-D-aspartate (NMDA) receptor located on the cell surface are associated with a severe form of encephalitis and are likely pathogenic (Figure 18-1).8 An antibody-mediated immunopathogenesis is also strongly suggested for the cerebellar and stiff-person syndromes associated with antibodies to glutamic-acid decarboxylase (GAD), and the paraneoplastic stiff-person syndrome related to antiamphiphysin antibodies. These two antigens are intracellular, close to the synaptic membrane, and the patients’ antibodies appear to have a functional effect in vivo.9,10
For other PNDs, usually those that affect the central nervous system, more complex immune mechanisms appear to exist. In addition to the presence of antineuronal antibodies, PNDs of the central nervous system are associated with infiltrates of CD4+ and CD8+ T cells, microglial activation, gliosis, and variable neuronal loss.11–13 The infiltrating T cells are often in close contact with neurons undergoing degeneration, suggesting a primary pathogenic role. The interaction of B- and T-cell mechanisms and the subacute development of extensive inflammatory abnormalities and neuronal degeneration could explain the difficulty in treating these disorders as well as their poor response to plasma exchange or intravenous immunoglobulins (IVIg).
Although they are increasingly becoming recognized, significant diagnostic delays are frequent even for well-described syndromes. In a series of 50 patients with LEMS, about half of the patients were initially misdiagnosed, usually with myasthenia gravis.14 Another study noted an inverse correlation between the severity of the neurologic symptoms and the time to the diagnosis of the PND.15 For patients who develop a syndrome that is typically associated with cancer and are found to have well-characterized paraneoplastic antibodies, the diagnosis of PND is relatively straightforward. The diagnosis of PND is more difficult in patients who develop less characteristic symptoms, especially if no antibodies are found in the serum or CSF. Features that suggest a paraneoplastic origin include an acute or subacute onset, and, if the central nervous system is involved, the CSF will often suggest an inflammatory process. If the patient is known to have cancer, metastastic or other nonmetastatic complications of cancer should be ruled out. For a patient in cancer remission, a recurrence should be suspected if symptoms of PND develop. For patients without a known cancer, if a PND is suspected, a detailed search for an underlying neoplasm is mandated. Whole body FDG-PET scans may detect tumors that escape detection by other standard imaging methods.16–18 Features of individual syndromes that may aid in diagnosis (e.g., by neuroimaging) are noted in the following descriptions of individual syndromes.
Paraneoplastic Syndromes of the Brain
PARANEOPLASTIC CEREBELLAR DEGENERATION (PCD)
Paraneoplastic cerebellar degeneration is characterized by the rapid development of severe pancerebellar dysfunction that may be preceded by prodromic symptoms including dizziness, oscillopsia, blurry or double vision, nausea, and vomiting. Eventually, symptoms progress to truncal and appendicular ataxia, dysarthria, and downbeating nystagmus.19 Symptoms of brainstem dysfunction, upgoing toes, or a mild neuropathy may occur. The subacute onset of PCD differentiates it from chronic degenerative diseases involving the cerebellum. Early MRI studies are usually normal; in some patients, transient enhancement of the cerebellar cortex has been noted. MRI studies late in the course usually show cerebellar atrophy.
The tumors more frequently involved are small cell lung cancer (SCLC), cancer of the breast and ovary, and Hodgkin lymphoma.20 The paraneoplastic antibodies typically associated with prominent or pure cerebellar degeneration are anti-Yo antibodies in patients with breast and gynecologic cancers, and anti-Tr antibodies in patients with Hodgkin lymphoma. When PCD occurs in association with paraneoplastic encephalomyelitis (PEM), anti-Hu antibodies are almost always present.21 When neoplasms other than breast and gynecological tumors are involved, patients are usually anti-Yo negative. Anti-Yo antibodies have been identified in a few male patients with PCD and cancer of the salivary gland, lung, and esophagus.22,23 Patients with predominant truncal ataxia and opsoclonus or other ocular movement abnormalities may have anti-Ri antibodies, in which case the tumor is usually a breast carcinoma or, less frequently, gynecologic, bladder, or SCLC.24,25 Antibodies to P/Q-type VGCC occur in some patients with SCLC and cerebellar dysfunction, although only some of these patients develop LEMS.5 There is a group of patients, usually with SCLC, who harbor two or more antibodies, such as Zic4 and Hu or CRMP5 or all three. Patients who harbor only Zic4 antibodies are more likely to develop cerebellar dysfunction than patients with several antibodies.26
Prompt tumor control, immunosuppressive intervention, or perhaps different pathogenic mechanisms, may explain a number of single case reports describing neurologic improvement after tumor treatment, plasma exchange, IVIg, cyclophosphamide, or steroids.27–29 However, large series of patients with well-defined antibody-positive PCD show that, in general, there is only rare improvement with treatment, if any.
PARANEOPLASTIC ENCEPHALOMYELITIS
Patients with paraneoplastic encephalomyelitis (PEM) develop multifocal involvement of the nervous system, including brain, brainstem, cerebellum, or spinal cord.15,21 Many patients with PEM also have paraneoplastic sensory neuropathy. The clinical features depend on the area(s) predominantly involved, but pathology studies almost always show abnormalities (inflammatory infiltrates, neuronal loss, gliosis) in asymptomatic regions. Several syndromes have been described that may occur alone or in combination. These include cortical encephalitis, that may present as epilepsia partialis continua; limbic and/or brainstem encephalitis, which is discussed in further detail later; cerebellar gait and limb ataxia; myelitis that may cause lower or upper motor neuron symptoms, myoclonus, muscle rigidity, and spasms; and autonomic dysfunction.
Paraneoplastic encephalomyelitis with or without PSN has been reported in association with almost all types of tumors, but the most common is lung carcinoma, particularly SCLC. The most frequently associated antibodies are anti-Hu and anti-CRMP5/CV2; antibodies to amphiphysin and Zic proteins are less frequently reported.15,26,30
All types of PEM except LE respond poorly to treatment. Stabilization or partial neurologic improvement may occur and usually correlates with tumor response to treatment. In a large series of patients with anti-Hu−associated PEM, treatment of the tumor with or without associated immunotherapy was an independent predictor of neurologic improvement or stabilization.15 The roles of plasma exchange, IVIg, and immunosuppression have not been established. Some patients with LE show marked improvement after tumor treatment and immunomodulatory therapies.31,32
LIMBIC ENCEPHALITIS
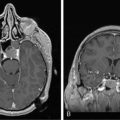
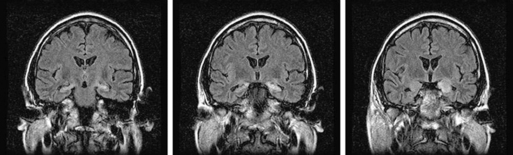
Limbic encephalitis is characterized by confusion, depression, agitation, severe short-term memory deficits, partial-complex seizures, sleep disturbances, and dementia.31 The EEG usually reveals foci of epileptic activity in one or both temporal lobes, or focal or generalized slow activity. About 80% of patients have MRI fluid-attenuated inversion recovery (FLAIR) or, in T2 sequences, hyperintense signal abnormality in the medial aspect of one or both temporal lobes (Figure 18-2). FDG-PET may show hypermetabolism in one or both temporal lobes even when the MRI is normal.33 Recent studies have shown that immune-mediated LE can be categorized into four groups based on the type and location of the target antigens.
Limbic encephalitis associated with antibodies to intracellular antigens
The main intracellular antigens related to LE are Hu, Ma2, and, less frequently, CV2/CRMP5 and amphiphysin. In these immune responses, cytotoxic T cell mechanisms are considered the main pathogenic effectors. Patients with Hu antibodies have PEM, although the disorder may initially present as a focal syndrome; the associated tumor is almost always a SCLC.15,21
Antibodies to Ma proteins are associated with limbic and brainstem encephalitis and occasionally with cerebellar symptoms; prominent hypothalamic dysfunction, hypersomnia, and cataplexy can occur.32,34 Patients less than 50 years of age with limbic dysfunction and antibodies to Ma proteins usually have an underlying germ cell tumor of the testis.35 These patients often benefit from orchiectomy and from immunotherapy that may include corticosteroids and IVIg. Overall, 35% of patients with anti-Ma2 encephalitis have neurological responses to treatment.36 One case of spontaneous neurological improvement has recently been reported.37
Anti-CV2 or CRMP5 antibodies associate with encephalomyelitis, sensorimotor neuropathy, and, more distinctively, with cerebellar ataxia, chorea, uveitis, and optic neuritis.30,38,39 The development of myelitis and optic neuritis may resemble Devic syndrome.40 SCLC and thymoma are the tumors more frequently involved. In patients with SCLC, anti-CV2/CRMP5 may coexist with anti-Hu or Zic antibodies; these patients usually have multifocal deficits or encephalomyelitis.26
Anti-N-methyl-D-aspartate (NMDA) receptor-associated encephalitis
Anti-NMDA receptor-associated encephalitis is a recently described disorder that usually affects young women.8 About 65% of patients have an underlying tumor, usually a cystic teratoma of the ovary. After prodromal symptoms that may include headache, fever, or a viral-like illness, patients develop severe psychiatric symptoms or memory loss, seizures, and decreased level of consciousness, accompanied by dyskinesias, hypoventilation, or autonomic instability. Intensive care support and ventilation may be required for several weeks or many months. Although the disorder is potentially lethal, most patients recover after immunotherapy; when a tumor is found, removal expedites recovery and decreases relapses.41 The disorder can also occur in men or women without a detectable tumor.42 Due to the location of the target antigens on the cell surface (Figure 18-1) and the dramatic response to immunotherapy, it is likely that these antibodies play a direct pathogenic role.
Encephalitis and antibodies to voltage-gated potassium channels
Recent evidence suggests that the target of these antibodies is not in fact the VGKC but other related proteins. The two main syndromes associated with these antibodies include typical LE and a lesser focal encephalitis that is associated with psychiatric symptoms, hallucinations, peripheral nerve hyperexcitability, hyperhydrosis, and other symptoms of autonomic dysfunction (Morvan syndrome). REM sleep disturbances and hyponatremia are common in both, and some patients may develop hypothermia, hypersalivation, pain, and disorders of appetite.43 About 20% of patients with antibodies to VGKC-related proteins have a tumor, often SCLC or thymoma. About 80% of patients will respond to treatment that includes corticosteroids, plasma exchange, or IVIg.
Antibodies to other cell membrane antigens
There are other antibodies to cell surface antigens that have not been fully characterized. Some of these antibodies occur along with other well-characterized immune responses, such as GAD antibodies, and the associated disorders respond differently to immunotherapy.33 It is unclear whether these novel antibodies have one or several target antigens. Tumors found in association with these antibodies include thymoma, SCLC, and Hodgkin lymphoma.
PARANEOPLASTIC OPSOCLONUS-MYOCLONUS
Opsoclonus is a disorder of eye movement characterized by spontaneous, arrhythmic, large-amplitude conjugate saccades occurring in all directions of gaze. Opsoclonus frequently associates with myoclonus and ataxia of the head, trunk, or limbs. When paraneoplastic in adults, symptoms can range from opsoclonus with mild truncal ataxia to a severe syndrome associated with encephalopathy that can lead to stupor and death. A number of associated tumors have been reported, but the most common is SCLC.44 Paraneoplastic opsoclonus-myoclonus in children usually has a subacute onset with frequent fluctuations and is accompanied by ataxia, hypotonia, and irritability.45 Almost 50% of children with paraneoplastic opsoclonus-myoclonus have neuroblastoma, and in half of the patients, the neurologic symptoms precede the diagnosis of the tumor. Children with neuroblastoma and opsoclonus have a better tumor prognosis than those without paraneoplastic symptoms.
Some adult patients, in particular those with SCLC, and 5% to 10% of children with neuroblastoma have anti-Hu antibodies.46 Patients with breast and gynecologic cancers may harbor anti-Ri antibodies;25 some of these patients develop muscle rigidity, autonomic dysfunction, and dementia. A small number of patients have been reported with other antibodies including antibodies to CRMP5/CV2, Zic2, amphiphysin, Yo, and Ma2.30,47,48 However, in many adults and children with neuroblastoma, no paraneoplastic antibodies are found.
When associated with neuroblastoma, the disorder frequently responds to treatment of the tumor, steroids, ACTH, IVIg, plasma exchange, or rituximab;49,50 however, developmental and neurologic sequelae are frequent.45 Paraneoplastic opsoclonus-myoclonus in adults may respond to immunosuppression and IVIg. Patients whose tumors are treated promptly appear to have a better prognosis than those whose tumors are not treated.51
Paraneoplastic Disorders of the Visual System
Paraneoplastic retinopathy is characterized by photosensitivity, progressive loss of vision and color perception, central or ring scotomas, and night blindness.52 The fundoscopic examination is normal or demonstrates arteriolar narrowing, and the electroretinogram (ERG) shows attenuation of photopic and scotopic responses. Paraneoplastic retinopathy associated with antibodies to recoverin is known as cancer-associated retinopathy (CAR).53 Patients with CAR usually have SCLC, but cases have been reported associated with breast or gynecological cancers. Other target antigens that have been described include the tubby-like protein, photoreceptor cell-specific nuclear receptor, and the polypyrimidine tract binding-like protein.54,55 Retinopathy in association with metastatic cutaneous melanoma is known as melanoma-associated retinopathy (MAR).56 As opposed to CAR, these patients present with acute visual loss years or months after the diagnosis of the metastatic disease. The ERG shows reduced or absent b-waves with normal dark-adapted a-waves indicating bipolar cell dysfunction. Some of these patients have antibodies that target unknown antigens in the retinal bipolar cells.57
Optic neuritis has been described in some patients with paraneoplastic syndromes of the central nervous system in association with several antibodies including anti-Hu, anti-Tr, anti-Yo, and, more frequently, anti-CV2/CRMP5.39,58 Patients present with sudden bilateral loss of vision, swollen optic discs, and field defects; the majority have SCLC.
Bilateral diffuse uveal melanocytic proliferation is a rare paraneoplastic entity in which an underlying tumor causes diffuse bilateral proliferation of melanocytes in the uveal tract, leading to bilateral visual loss.59,60 The visual symptoms precede the diagnosis of a systemic malignancy. Carcinoma of the reproductive tract in women and carcinomas of the lung and pancreas in men appear to be the more commonly-associated tumors. Patients present with abrupt bilateral visual loss and few or no findings on examination of the fundus. Nearly all patients described have had rapid cataract progression, and all have had retinal detachment.59 One case ascribed improvement in vision to treatment with external beam irradiation and subretinal fluid drainage.
In general, paraneoplastic visual loss is usually irreversible. Immunosuppression, plasma exchange, or steroids is mostly ineffective but in rare cases may result in symptom stabilization.61
Paraneoplastic Syndromes of the Spinal Cord and Dorsal Root Ganglia
PARANEOPLASTIC MOTOR NEURON SYNDROMES
A wide range of spinal cord syndromes including upper or lower motor neuron dysfunction, myelitis, myelopathy, and sensory and motor neuronopathies have been described in patients with cancer, and it is unclear if these are truly paraneoplastic or simply represent a coincidental association with cancer. Furthermore no specific paraneoplastic antibodies have been found in these patients. A recent study examined the sera of 145 patients with motor neuron disease for well characterized paraneoplastic antibodies (Hu, Yo, Ri, CV2/CRMP5, Ma2 and amphiphysin) and found only low reactivity in five sera that likely represented background activity.62 For some syndromes, such necrotizing myelopathy, the identification of nonparaneoplastic causes such as human herpesvirus has, in many instances, clarified the nature of the disorder.63
The existence of paraneoplastic motor neuron dysfunction is based on reports of patients with typical amyotrophic lateral sclerosis (ALS) who improved after treatment of the underlying tumor (usually renal cell cancer and carcinoma of the lung or breast) suggesting more than a coincidental relationship.64–66 A patient with renal cell carcinoma, neuromyotonia, and lower motor neuron syndrome had recovery of neurological deficits after tumor removal.67 For these patients the neurologic syndrome and laboratory studies are similar to those seen in typical ALS patients. A more-than-coincidental association has been suggested between lymphoproliferative disorders with motor neuron dysfunction.64,68 Patients with PEM may develop symptoms resembling motor neuron disease.21,69 These patients almost always develop signs of involvement of other areas of the nervous system, which, along with the presence of the anti-Hu antibody, helps to rule out typical ALS.
Some patients with cancer develop a subacute lower motor neuronopathy characterized by subacute, progressive, painless, and asymmetrical muscle weakness that is more prominent in the lower extremities.70 Reflexes are decreased or abolished, and, in contrast to typical ALS, bulbar muscles are usually spared, fasciculations are rare, and upper motor neuron signs are absent. Sensory symptoms, if any, are mild and transitory. The neurologic symptoms may have a benign course, independent of the activity of the neoplasm. The associated tumors are Hodgkin lymphoma and less frequently non-Hodgkin lymphoma. This disorder needs to be differentiated from the lower motor neuron syndrome that patients may develop secondary to radiation therapy of the spinal cord.71 In these patients, the distribution of muscle weakness is more distal and, although symptoms stabilize, they do not improve. Patients with Hodgkin lymphoma treated with mantle radiation may develop slowly progressive (over years) weakness and atrophy involving neck flexors and extensors and proximal muscles of the upper extremities. Characteristically, a strip of atrophy involving paraspinal muscles is also observed. Distal reflexes are usually preserved; sensation is normal. No effective therapies have been described.
A disorder with prominent upper motor neuron dysfunction that mimics primary lateral sclerosis has been reported in a few patients with breast cancer. Because no specific paraneoplastic markers have been identified, the association of these disorders may be coincidental.72
PARANEOPLASTIC STIFF-PERSON SYNDROME
This disorder is characterized by progressive muscle rigidity, stiffness, and painful spasms triggered by auditory, sensory, or emotional stimuli. Rigidity mainly involves the lower trunk and legs, but it can affect the upper extremities, shoulders and neck. Symptoms improve with sleep and general anesthetics. Electrophysiologic studies demonstrate continuous motor unit activity at rest that improves with diazepam. The paraneoplastic form of stiff-person syndrome is usually associated with breast and lung cancers and Hodgkin lymphoma. Several antibodies, indicating different immune mechanisms, have been described. The main autoantigen of the paraneoplastic form of the disorder is amphiphysin, which commonly associates with breast and lung cancer.73,74 Antibodies to GAD may occur in some patients with thymoma or cancer,75,76 but these antibodies are far more common in the nonparaneoplastic disorder.74,77 Treatment of the tumor, steroids, and drugs that enhance GABA-ergic function (diazepam, baclofen, sodium valproate, vigabatrin) usually improve symptoms. The benefit of IVIg has been demonstrated for the nonparaneoplastic disorder.78
PARANEOPLASTIC SENSORY NEURONOPATHY (PSN) OR DORSAL ROOT GANGLIONOPATHY
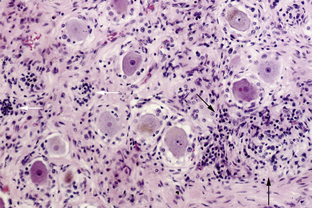
This syndrome is characterized by symmetric or asymmetric sensory deficits, painful dysesthesias, radicular pain, and decreased or absent reflexes. All modalities of sensation, including taste and hearing, can be affected. With symptom progression, the sensory deficits result in ataxia, gait difficulty, and pseudoathetoid movements. Electrophysiologic studies show decreased or absent sensory nerve potentials with normal or near-normal motor conduction velocities and normal F-wave studies.79 Some patients also have electrophysiological evidence of axonal and demyelinating neuropathy.79,80 Autopsy studies demonstrate inflammation in the dorsal root ganglia characterized by infiltrates of mononuclear cells, neuronal degeneration, and proliferation of the satellite cells (Nageotte nodules) (Figure 18-3). Almost any cancer may be found associated with PSN. In about 70% of patients, PSN precedes or associates with PEM and autonomic dysfunction and has the same immunologic and oncologic associations, mainly anti-Hu antibodies and SCLC.81 Fewer patients with PSN have antibodies to amphiphysin and CV2/CRMP5.47,82 Some patients harbor both anti-Hu and CV2/CRMP5 antibodies. The therapeutic approach focuses on prompt treatment of the tumor. Studies of patients with SCLC and anti-Hu associated PSN and PEM indicate that patients whose tumors had a complete response to therapy were more likely to have stabilization or improvement of neurological symptoms compared to patients whose tumors were not treated or did not respond well to therapy.15,83 In some patients, prompt treatment with steroids may result in partial improvement of the sensory deficits.83,84 The benefit of IVIg and plasma exchange is not proven.
Paraneoplastic Syndromes of the Nerves and Neuromuscular Junction
AUTONOMIC NEUROPATHY
Paraneoplastic autonomic neuropathy usually develops as a component of other disorders, such as LEMS and PEM. It may rarely occur as a pure or predominant autonomic neuropathy with adrenergic or cholinergic dysfunction at the preganglionic or postganglionic levels. Patients can develop several life-threatening complications, such as gastrointestinal paresis with pseudoobstruction, cardiac dysrhythmias, and postural hypotension. Other symptoms include hypoventilation, dry mouth, erectile dysfunction, anhidrosis, and sphincter dysfunction. The disorder has been reported in association with several tumors, including SCLC, cancer of the pancreas, testis, carcinoid tumors, and lymphoma. When it develops as a component of PEM, serum anti-Hu and anti-CV2/CRMP5 antibodies may be present.21 Serum antibodies to ganglionic acetylcholine receptors have been reported, but they can occur also without a cancer association.85 In some patients, treatment of the tumor may stabilize or improve the autonomic symptoms. A recent study showed that combined immunomodulatory treatment, including prednisone, mycophenolate mofetil, and plasma exchange, was effective in patients with nonparaneoplastic autoimmune autonomic ganglionopathy associated with antibodies to ganglionic acetylcholine receptors.86
PERIPHERAL NERVE HYPEREXCITABILITY (PNH)
Also known as neuromyotonia, undulating myokymia, and Isaacs syndrome, PNH is characterized by spontaneous and continuous muscle fiber activity of peripheral nerve origin triggered by voluntary muscle contraction. Patients develop cramps, stiffness, delayed muscle relaxation, and spontaneous or evoked muscle spasms. PNH is often associated with motor weakness and hyperhidrosis and, less commonly, a sensorimotor neuropathy. The electromyogram may show fibrillation, fasciculation, and doublet, triplet or multiplet single-unit discharges that have a high intraburst frequency.4 The motor discharges can continue during sleep, general anesthesia, and proximal nerve block and are abolished by blocking the neuromuscular junction. PNH can develop without cancer; when paraneoplastic, thymoma and lung cancers are more commonly involved. Patients with thymoma may also have myasthenia gravis.4 Many patients have antibodies to VGKCs that contribute to the nerve hyperexcitability.87,88 Patients with PNH and thymoma, with or without myasthenia gravis, may also harbor antibodies to acetylcholine receptors.4 Symptomatic improvement has been reported with phenytoin, carbamazepine, and plasma exchange.87,89 The cramp-fasciculation syndrome resembles PNH, but the electromyogram does not show myokymic discharges. It may occur in association with cancer (usually thymoma or lung cancer) and antibodies to VGKC.
VASCULITIS OF THE NERVE AND MUSCLE
Patients with this disorder develop a painful symmetric or asymmetric subacute distal sensorimotor neuropathy with variable proximal weakness or, less frequently, a multiple mononeuropathy.90 It predominantly affects elderly men, and is associated with an elevated erythrocyte sedimentation rate and increased CSF protein concentration. Electrophysiological findings are compatible with axonal degeneration involving motor and sensory nerves. Lymphoma and SCLC are the main tumors involved.90 Pathology studies show axonal degeneration and T cell infiltrates involving the small vessels of the nerve and muscle.91,92 Most patients do not have paraneoplastic antibodies, although anti-Hu antibodies can be found in some patients with SCLC. Immunosuppressants (steroids and cyclophosphamide) often result in neurologic improvement.90,93
SENSORIMOTOR NEUROPATHIES
Paraneoplastic sensorimotor neuropathy may develop before or after the cancer diagnosis. The presentation is usually subacute followed by continued progression, although some patients have a relapsing and remitting course.94 The most commonly associated tumors are lung and breast cancers. There are usually no serum antineuronal antibodies, although some patients with lung cancer and thymoma may harbor CV2/CRMP5 antibodies.82 The detection of anti-Hu suggests concurrent dorsal root ganglionitis.81
An acute neuropathy identical to Guillain-Barré syndrome (GBS) has been reported in patients with lymphoma, usually Hodgkin lymphoma. In one series of 435 patients with GBS, nine developed cancer in the 6 months preceding or following the onset of the GBS.95 In general, patients with cancer and GBS appear to have higher mortality than those with GBS alone. For brachial neuritis, the differential diagnosis should include more common causes of brachial plexopathy in cancer patients, including tumor infiltration, radiation injury, ischemic neuropathy, and traumatic injury of the plexus.
About 50% of patients with osteosclerotic myeloma develop a symmetric, distal, sensorimotor neuropathy with predominant motor deficits resembling a chronic inflammatory demyelinating neuropathy. Some patients develop additional symptoms of the POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, monoclonal proteinemia and skin changes). If there is a solitary sclerotic lesion, radiation is the most effective and least toxic therapy.96 Systemic chemotherapy with or without corticosteroid therapy should be considered for patients with diffuse sclerotic lesions or those with no obvious bone lesions.
About 5% to 10% of patients with Waldenström macroglobulinemia develop a neuropathy. The neuropathy may be a distal symmetric demyelinating sensorimotor neuropathy that is often associated with IgM antibodies to myelin-associated glycoprotein or gangliosides, including GD1b and GM1. Other neuropathies include an axonal neuropathy, neuropathy associated with amyloid deposition, and a cryoglobulinemic vasculitis. In addition to treating the Waldenström macroglobulinemia, the use of plasma exchange, IVIg, chlorambucil, cyclophosphamide, fludarabine, or rituximab may result in improvement.97,98
Castleman disease, or angiofollicular lymph node hyperplasia, represents a group of lymphoproliferative disorders that are often accompanied by a marked systemic inflammatory response and acquired systemic amyloidosis. Patients may develop a painful sensorimotor neuropathy, a chronic relapsing sensorimotor neuropathy, and a predominant motor neuropathy.99 Additional symptoms indicative of POEMS syndrome are common as there is considerable overlap between the syndromes.100,101 There are reports of neurological improvement with cyclophosphamide and prednisolone or immunosuppression.102,103
Paraneoplastic Syndromes of the Neuromuscular Junction
LAMBERT-EATON MYASTHENIC SYNDROME (LEMS)
LEMS is characterized by the development of proximal muscle weakness in the lower and upper extremities.14 Symptoms usually develop gradually over a period of weeks or months but can develop acutely. The frequency of ocular symptoms including diplopia and ptosis is low at symptom presentation, but eventually about 50% of patients became affected.104 In a few reported cases, ocular symptoms were the only clinical manifestation of LEMS.105 More than 50% of patients also develop autonomic dysfunction including dry mouth, erectile dysfunction and blurring of vision.106 Reflexes are decreased or abolished but may increase after a brief muscle contraction. The diagnosis of LEMS is based on electrophysiological studies. Nerve conduction studies show small-amplitude compound muscle action potentials (CMAP). At slow rates of repetitive nerve stimulation (2 to 5 Hz) there is a decremental response, while at fast rates (20 Hz or greater) or after maximal voluntary muscle contraction, facilitation occurs with an incremental response of at least 100%.
Approximately 60% of patients with LEMS have SCLC or lymphoma; other cancers have rarely been reported. The neurologic symptoms usually precede the cancer diagnosis. LEMS can develop in association with other paraneoplastic syndromes such as PCD and PEM, and recurrence of LEMS after a remission often heralds tumor recurrence.107,108 Most patients with LEMS have serum antibodies against P/Q type VGCCs.109 When LEMS develops in association with PEM, patients often have anti-Hu antibodies. Treatment of the tumor and medication that enhances acetylcholine release (3,4-diaminopyridine, or the combination of pyridostigmine and guanidine) usually control the disorder.84,110 Plasma exchange and IVIg improve symptoms within 2 to 4 weeks but the benefit is transient.111,112 Long-term immunosuppression with prednisone or azathioprine is an alternative for patients who do not improve with 3,4-diaminopyridine.
MYASTHENIA GRAVIS
The main features of MG are weakness and fatigability of skeletal muscles that improve with rest and increase with activity. Ptosis and diplopia occur in most patients and, in about 15% of cases, symptoms remain localized to the extraocular and eyelid muscles. In contrast to LEMS, reflexes and sensation are spared. Approximately 10% of patients have thymoma or a thymic carcinoma; one third of thymoma patients develop myasthenia gravis.113 In a few instances, MG has been reported in association with other tumors, including thyroid gland tumors, SCLC, breast cancer, and lymphoma. Whether the underlying disorder is thymoma or thymic hyperplasia, about 80% to 90% of the patients have antibodies to acetylcholine receptors. About 70% of patients with symptoms restricted to the eyes also have these antibodies. A group of patients without acetylcholine receptor antibodies develop antibodies to MusK, a muscle tyrosine kinase receptor.114 Patients with MusK antibodies predominantly develop cranial and bulbar symptoms and respiratory crises. Most of these patients do not have tumors. A case with overlapping acetylcholine, MusK, and VGKC antibodies has recently been reported without an association with cancer.115 High-titer neutralizing antibodies to IL-12 and interferon-α are frequently detected in patients with MG and thymoma, but not in patients without thymoma.116 The first approach to treatment is directed at the underlying tumor. Additional therapeutic strategies, including symptomatic treatment (e.g., anticholinesterase drugs), immunomodulation (plasma exchange, IVIg), and immunosuppression (steroids, azathioprine, mycophenolate mofetil) are similar for patients with and without cancer.112
Paraneoplastic Disorders of the Muscle
DERMATOMYOSITIS
Most epidemiological studies indicate a clear association between dermatomyositis and cancer, particularly in older patients.117 The symptoms of paraneoplastic dermatomyositis are the same as those in patients without cancer. Patients usually present with the subacute onset of proximal muscle weakness. Neck flexors, pharyngeal muscles, and respiratory muscles are commonly involved, which may lead to aspiration and hypoventilation. Reflexes and sensory exam are normal. Cutaneous changes include purplish discoloration of the eyelids (heliotrope rash) with edema and erythematous lesions over the knuckles. The presence of necrotic skin ulcerations and pruritis are felt to be indicators of an underlying cancer.118,119 Life-threatening complications of dermatomyositis include respiratory muscle weakness, myocarditis, and interstitial lung disease; serum muscle enzymes are usually elevated. The electromyogram shows increased spontaneous activity (fibrillations, positive sharp waves, and complex repetitive discharges), and short duration, low-amplitude polyphasic units on voluntary activation.
When associated with cancer, the tumors more frequently involved are cancer of the breast, lung, ovary, and stomach. Less frequently associated are cancer of the pancreas, thymoma, germ cell tumors, melanoma, nasopharyngeal cancer, and lymphoma. There are no distinctive serologic markers of paraneoplastic or nonparaneoplastic dermatomysositis. Interstitial lung disease is less frequent in paraneoplastic dermatomyositis than in patients without cancer.120 After treating the tumor, the therapy of paraneoplastic dermatomyositis does not differ from cases not associated with cancer and consists of steroids and long-term immunosuppression (e.g., azathioprine).121,122 IVIg has been reported useful in refractory dermatomyositis.
ACUTE NECROTIZING MYOPATHY
This rare disorder is characterized by the acute onset of painful proximal muscle weakness with rapid generalization and involvement of respiratory and pharyngeal muscles. Serum muscle enzymes are markedly elevated and electrophysiological studies demonstrate myopathic findings. Muscle biopsy shows extensive necrosis with minimal or absent inflammation. In patients with cancer, the differential diagnosis of an acute necrotizing myopathy should include chemotherapy-induced and cytokine-induced rhabdomyolysis.123 The disorder has been reported in association with a variety of solid tumors, including carcinomas of the lung, bladder, breast, prostate, and gastrointestinal tract.124 No specific immune responses have been identified. Treatment of the tumor may result in neurologic improvement.
GENERAL TREATMENT APPROACH
For PNDs of the peripheral nervous system such as LEMS, myasthenia gravis, PNH and some types of autonomic neuropathy, the associated serum antibodies directly block the function of ion channels or membrane receptors. These disorders usually respond to plasma exchange, IVIg, and immunosuppressive therapies.125 Most patients with paraneoplastic neuropathies do not harbor antineuronal antibodies, but an immune-mediated etiology is inferred by the subacute development of symptoms, pleocytosis or increased proteins in the CSF, or presence of inflammatory infiltrates on nerve biopsy. For these disorders, and particularly those with predominant demyelinating features, plasmapheresis, IVIg, and immunosuppression can be effective. Paraneoplastic axonal neuropathies are poorly responsive to immunotherapy; treatment is largely symptomatic or supportive, along with treatment of the tumor.
In adults, there are several PNDs of the CNS that are responsive or more likely to respond to treatment of the tumor and immunomodulatory therapies. These include anti-NMDAR encephalitis and limbic encephalitis in patients without anti-Hu antibodies (some of whom may have antibodies to VGKC-related proteins)126; opsoclonus-myoclonus; limbic encephalitis in young patients with testicular tumors and anti-Ma2 antibodies; and stiff-person syndrome associated with anti-amphiphysin antibodies.36,51
For other PNDs, the first therapeutic step is the early diagnosis and treatment of the tumor.29,125 For many PNDs affecting the CNS, there is recent data that demonstrates the importance of the early institution of immunologic therapies (immunomodulation, immunosuppression) when the neurologic deficits are not fully established or still partially reversible.127,128 Since the combination of oncologic and immunosuppressive therapies may have significant toxicity, it is recommended that immunologic treatments be stratified accordingly. For patients with progressive PNDs who are receiving chemotherapy, immunosuppression or immunomodulation may include oral or intravenous corticosteroids and IVIg; anecdotal experience suggests that plasma exchange is rarely effective in PNDs of the CNS. Patients with progressive PNDs, who are not receiving chemotherapy, should be considered for more aggressive immunosuppression that may include oral or intravenous cyclophosphamide, tacrolimus, or cyclosporine.
1. L. Bataller, J.O. Dalmau. Paraneoplastic disorders of the central nervous system: update on diagnostic criteria and treatment. Semin Neurol. 2004;24:461-471.
2. J. Dalmau, H.S. Gultekin, J.B. Posner. Paraneoplastic neurologic syndromes: pathogenesis and physiopathology. Brain Pathol. 1999;9:275-284.
3. M. Motomura, I. Johnston, B. Lang, A. Vincent, J. Newsom-Davis. An improved diagnostic assay for Lambert-Eaton myasthenic syndrome. J Neurol Neurosurg Psychiatry. 1995;58:85-87.
4. I.K. Hart, P. Maddison, J. Newsom-Davis, A. Vincent, K.R. Mills. Phenotypic variants of autoimmune peripheral nerve hyperexcitability. Brain. 2002;125:1887-1895.
5. F. Graus, B. Lang, P. Pozo-Rosich, A. Saiz, R. Casamitjana, A. Vincent. P/Q type calcium-channel antibodies in paraneoplastic cerebellar degeneration with lung cancer. Neurology. 2002;59:764-766.
6. A. Vincent, C. Buckley, J.M. Schott, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain. 2004;127:701-712.
7. R. Liguori, A. Vincent, L. Clover, et al. Morvan’s syndrome: peripheral and central nervous system and cardiac involvement with antibodies to voltage-gated potassium channels. Brain. 2001;124:2417-2426.
8. J. Dalmau, E. Tuzun, H.Y. Wu, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61:25-36.
9. C. Sommer, A. Weishaupt, J. Brinkhoff, et al. Paraneoplastic stiff-person syndrome: passive transfer to rats by means of IgG antibodies to amphiphysin. Lancet. 2005;365:1406-1411.
10. M.U. Manto, M.A. Laute, M. Aguera, V. Rogemond, M. Pandolfo, J. Honnorat. Effects of anti-glutamic acid decarboxylase antibodies associated with neurological diseases. Ann Neurol. 2007;61:544-551.
11. B. Benyahia, R. Liblau, H. Merle-Béral, J.M. Tourani, J. Dalmau, J-Y Delattre. Cell-mediated auto-immunity in paraneoplastic neurologic syndromes with anti-Hu antibodies. Ann Neurol. 1999;45:162-167.
12. K. Tanaka, M. Tanaka, T. Inuzuka, R. Nakano, S. Tsuji. Cytotoxic T lymphocyte-mediated cell death in paraneoplastic sensory neuronopathy with anti-Hu antibody. J Neurol Sci. 1999;163:159-162.
13. M.L. Albert, J.C. Darnell, A. Bender, L.M. Francisco, N. Bhardwaj, R.B. Darnell. Tumor-specific killer cells in paraneoplastic cerebellar degeneration. Nat Med. 1998;4:1321-1324.
14. J.H. O’Neill, N.M. Murray, J. Newsom-Davis. The Lambert-Eaton myasthenic syndrome. A review of 50 cases. Brain. 1988;111:577-596.
15. F. Graus, F. Keime-Guibert, R. Rene, et al. Anti-Hu-associated paraneoplastic encephalomyelitis: analysis of 200 patients. Brain. 2001;124:1138-1148.
16. S. Younes-Mhenni, M.F. Janier, L. Cinotti, et al. FDG-PET improves tumour detection in patients with paraneoplastic neurological syndromes. Brain. 2004;127:2331-2338.
17. J.H. Rees, S.F. Hain, M.R. Johnson, et al. The role of 18F.fluoro-2-deoxyglucose-PET scanning in the diagnosis of paraneoplastic neurological disorders. Brain. 2001;124:2223-2231.
18. R. Linke, M. Schroeder, T. Helmberger, R. Voltz. Antibody-positive paraneoplastic neurologic syndromes: value of CT and PET for tumor diagnosis. Neurology. 2004;63:282-286.
19. K. Peterson, M.K. Rosenblum, H. Kotanides, J.B. Posner. Paraneoplastic cerebellar degeneration. I. A clinical analysis of 55 anti-Yo antibody-positive patients. Neurology. 1992;42:1931-1937.
20. S. Shams’ili, J. Grefkens, B. De Leeuw, et al. Paraneoplastic cerebellar degeneration associated with antineuronal antibodies: analysis of 50 patients. Brain. 2003;126:1409-1418.
21. J. Dalmau, F. Graus, M.K. Rosenblum, J.B. Posner. Anti-Hu—associated paraneoplastic encephalomyelitis/sensory neuronopathy. A clinical study of 71 patients. Medicine (Baltimore). 1992;71:59-72.
22. O. Felician, J.L. Renard, F. Vega, et al. Paraneoplastic cerebellar degeneration with anti-Yo antibody in a man. Neurology. 1995;45:1226-1227.
23. J. Krakauer, C. Balmaceda, J.T. Gluck, J.B. Posner, M.R. Fetell, J. Dalmau. Anti-Yo-associated paraneoplastic cerebellar degeneration in a man with adenocarcinoma of unknown origin. Neurology. 1996;46:1486-1487.
24. C. Budde-Steffen, N.E. Anderson, M.K. Rosenblum, et al. An antineuronal autoantibody in paraneoplastic opsoclonus. Ann Neurol. 1988;23:528-531.
25. F.A. Luque, H.M. Furneaux, R. Ferziger, et al. Anti-Ri: an antibody associated with paraneoplastic opsoclonus and breast cancer. Ann Neurol. 1991;29:241-251.
26. L. Bataller, D.F. Wade, F. Graus, H.D. Stacey, M.R. Rosenfeld, J. Dalmau. Antibodies to Zic4 in paraneoplastic neurologic disorders and small-cell lung cancer. Neurology. 2004;62:778-782.
27. S. Shams’ili, J. de Beukelaar, J.W. Gratama, et al. An uncontrolled trial of rituximab for antibody associated paraneoplastic neurological syndromes. J Neurol. 2006;253:16-20.
28. F. Blaes, M. Strittmatter, S. Merkelbach, et al. Intravenous immunoglobulins in the therapy of paraneoplastic neurological disorders. J Neurol. 1999;246:299-303.
29. Y.B. David, E. Warner, M. Levitan, D.M. Sutton, M.G. Malkin, J.O. Dalmau. Autoimmune paraneoplastic cerebellar degeneration in ovarian carcinoma patients treated with plasmapheresis and immunoglobulin. A case report. Cancer. 1996;78:2153-2156.
30. Z. Yu, T.J. Kryzer, G.E. Griesmann, K.K. Kim, E.E. Benarroch, V.A. Lennon. CRMP-5 neuronal autoantibody: Marker of lung cancer and thymoma related autoimmunity. Ann Neurol. 2001;49:146-154.
31. S.H. Gultekin, M.R. Rosenfeld, R. Voltz, J. Eichen, J.B. Posner, J. Dalmau. Paraneoplastic limbic encephalitis: Neurological symptoms, immunological findings, and tumor association in 50 patients. Brain. 2000;123:1481-1494.
32. M.R. Rosenfeld, J. Eichen, D. Wade, J.B. Posner, J. Dalmau. Molecular and clinical diversity in paraneoplastic immunity to Ma proteins. Ann Neurol. 2001;50:339-348.
33. B.M. Ances, R. Vitaliani, R.A. Taylor, et al. Treatment-responsive limbic encephalitis identified by neuropil antibodies: MRI and PET correlates. Brain. 2005;128:1764-1777.
34. J. Dalmau, S.H. Gultekin, R. Voltz, et al. Ma1, a novel neuronal and testis specific protein, is recognized by the serum of patients with paraneoplastic neurologic disorders. Brain. 1999;122:27-39.
35. R.M. Mathew, R. Vandenberghe, A. Garcia-Merino, et al. Orchiectomy for suspected microscopic tumor in patients with anti-Ma2-associated encephalitis. Neurology. 2007;68:900-905.
36. J. Dalmau, F. Graus, A. Villarejo, et al. Clinical analysis of anti-Ma2-associated encephalitis. Brain. 2004;127:1831-1844.
37. H. Pruss, R. Voltz, H. Gelderblom, et al. Spontaneous remission of anti-Ma associated paraneoplastic mesodiencephalic and brainstem encephalitis. J Neurol. 2008;255:292-294.
38. S. Vernino, P. Tuite, C.H. Adler, et al. Paraneoplastic chorea associated with CRMP-5 neuronal antibody and lung carcinoma. Ann Neurol. 2002;51:625-630.
39. J.C. Antoine, J. Honnorat, C. Vocanson, et al. Posterior uveitis, paraneoplastic encephalomyelitis and auto- antibodies reacting with developmental protein of brain and retina. J Neurol Sci. 1993;117:215-223.
40. F. Ducray, R. Roos-Weil, P.Y. Garcia, et al. Devic’s syndrome-like phenotype associated with thymoma and anti-CV2/CRMP5 antibodies. J Neurol Neurosurg Psychiatry. 2007;78:325-327.
41. M. Seki, S. Suzuki, T. Iizuka, et al. Neurological response to early removal of ovarian teratoma in anti-NMDAR encephalitis. J Neurol Neurosurg Psychiatry. 2008;79:324-326.
42. M.E. Novillo-Lopez, J. Rossi, J. Dalmau, J. Masjuan. Treatment-responsive subacute limbic encephalitis and NMDA receptor antibodies in a man. Neurology. 2008;70:728-729.
43. S. Jacob, S.R. Irani, Y.A. Rajabally, et al. Hypothermia in VGKC antibody-associated limbic encephalitis. J Neurol Neurosurg Psychiatry. 2008;79:202-204.
44. N.E. Anderson, M.K. Rosenblum, J.B. Posner. Paraneoplastic cerebellar degeneration: clinical-immunological correlations. Ann Neurol. 1988;24:559-567.
45. C. Russo, S.L. Cohn, M.J. Petruzzi, P.A. de Alarcon. Long-term neurologic outcome in children with opsoclonus-myoclonus associated with neuroblastoma: a report from the Pediatric Oncology Group. Med Pediatr Oncol. 1997;28:284-288.
46. B. Hersh, J. Dalmau, F. Dangond, S. Gultekin, E. Geller, P.Y. Wen. Paraneoplastic opsoclonus-myoclonus associated with anti-Hu antibody. Neurology. 1994;44:1754-1755.
47. A. Saiz, J. Dalmau, M.H. Butler, et al. Anti-amphiphysin I antibodies in patients with paraneoplastic neurological disorders associated with small cell lung carcinoma. J Neurol Neurosurg Psychiatry. 1999;66:214-217.
48. L. Bataller, M.R. Rosenfeld, F. Graus, J.J. Vilchez, N.K. Cheung, J. Dalmau. Autoantigen diversity in the opsoclonus-myoclonus syndrome. Ann Neurol. 2003;53:347-353.
49. E.D. Tate, T.J. Allison, M.R. Pranzatelli, S.J. Verhulst. Neuroepidemiologic trends in 105 US cases of pediatric opsoclonus-myoclonus syndrome. J Pediatr Oncol Nurs. 2005;22:8-19.
50. J. Bell, C. Moran, J. Blatt. Response to rituximab in a child with neuroblastoma and opsoclonus-myoclonus. Pediatr Blood Cancer. 2008;50:370-371.
51. L. Bataller, F. Graus, A. Saiz, J.J. Vilchez. Clinical outcome in adult onset idiopathic or paraneoplastic opsoclonus-myoclonus. Brain. 2001;124:437-443.
52. C.E. Thirkill. Cancer-induced, immune-mediated ocular degenerations. Ocul Immunol Inflamm. 2005;13:119-131.
53. C.E. Thirkill, R.C. Tait, N.K. Tyler, A.M. Roth, J.L. Keltner. The cancer-associated retinopathy antigen is a recoverin-like protein. Invest Ophthalmol Vis Sci. 1992;33:2768-2772.
54. T. Kikuchi, J. Arai, H. Shibuki, H. Kawashima, N. Yoshimura. Tubby-like protein 1 as an autoantigen in cancer-associated retinopathy. J Neuroimmunol. 2000;103:26-33.
55. J.G. Eichen, J. Dalmau, A. Demopoulos, D. Wade, J.B. Posner, M.R. Rosenfeld. The photoreceptor cell-specific nuclear receptor is an autoantigen of paraneoplastic retinopathy. J Neuroophthalmol. 2001;21:168-172.
56. K. Boeck, S. Hofmann, M. Klopfer, et al. Melanoma-associated paraneoplastic retinopathy: case report and review of the literature. Br J Dermatol. 1997;137:457-460.
57. A.H. Milam, C.J. Saari, S.G. Jacobson, et al. Autoantibodies against retinal bipolar cells in cutaneous melanoma-associated retinopathy. Invest Ophthalmol Visual Sci. 1993;34:91-100.
58. S.A. Cross, D.R. Salomao, J.E. Parisi, et al. Paraneoplastic autoimmune optic neuritis with retinitis defined by CRMP-5-IgG. Ann Neurol. 2003;54:38-50.
59. K.D. O’Neal, K.J. Butnor, K.R. Perkinson, A.D. Proia. Bilateral diffuse uveal melanocytic proliferation associated with pancreatic carcinoma: a case report and literature review of this paraneoplastic syndrome. Surv Ophthalmol. 2003;48:613-625.
60. W. Saito, S. Kase, K. Yoshida, et al. Bilateral diffuse uveal melanocytic proliferation in a patient with cancer-associated retinopathy. Am J Ophthalmol. 2005;140:942-945.
61. J.L. Keltner, C.E. Thirkill, P.T. Yip. Clinical and immunologic characteristics of melanoma-associated retinopathy syndrome: eleven new cases and a review of 51 previously published cases. J Neuroophthalmol. 2001;21:173-187.
62. O. Stich, B. Kleer, S. Rauer. Absence of paraneoplastic antineuronal antibodies in sera of 145 patients with motor neuron disease. J Neurol Neurosurg Psychiatry. 2007;78:883-885.
63. T. Iwamasa, Y. Utsumi, H. Sakuda, et al. Two cases of necrotizing myelopathy associated with malignancy caused by herpes simplex virus type 2. Acta Neuropathol (Berl). 1989;78:252-257.
64. P.H. Gordon, L.P. Rowland, D.S. Younger, et al. Lymphoproliferative disorders and motor neuron disease: an update. Neurology. 1997;48:1671-1678.
65. M.R. Rosenfeld, J.B. Posner. Paraneoplastic motor neuron disease. L.P. Rowland, editor. Amyotrophic Lateral Sclerosis and Other Motor Neuron Diseases. Advances in Neurology, Volume 56. New York: Raven Press. 1991:445-459.
66. B.K. Evans, C. Fagan, T. Arnold, E.J. Dropcho, S.J. Oh. Paraneoplastic motor neuron disease and renal cell carcinoma: improvement after nephrectomy. Neurology. 1990;40:960-962.
67. D. Canovas, J.M. Martinez, M. Viguera, G. Ribera. Association of renal carcinoma with neuromyotonia and involvement of inferior motor neuron. Neurologia. 2007;22:399-400.
68. E.D. Louis, A.E. Hanley, T.H. Brannagan, et al. Motor neuron disease, lymphoproliferative disease, and bone marrow biopsy. Muscle Nerve. 1996;19:1334-1337.
69. A. Verma, J.R. Berger, S. Snodgrass, C. Petito. Motor neuron disease: a paraneoplastic process associated with anti-hu antibody and small-cell lung carcinoma. Ann Neurol. 1996;40:112-116.
70. S.C. Schold, E.S. Cho, M. Somasundaram, J.B. Posner. Subacute motor neuronopathy: a remote effect of lymphoma. Ann Neurol. 1979;5:271-287.
71. C.H. Sadowsky, E. SachsJr, J. Ochoa. Postradiation motor neuron syndrome. Arch Neurol. 1976;33:786-787.
72. P.A. Forsyth, J. Dalmau, F. Graus, V. Cwik, M.K. Rosenblum, J.B. Posner. Motor neuron syndromes in cancer patients. Ann Neurol. 1997;41:722-730.
73. P. De Camilli, A. Thomas, R. Cofiell, et al. The synaptic vesicle-associated protein amphi-physin is the 128- kD autoantigen of Stiff-Man syndrome with breast cancer. J Exp Med. 1993;178:2219-2223.
74. F. Folli, M. Solimena, R. Cofiell, et al. Autoantibodies to a 128-kd synaptic protein in three women with the stiff-man syndrome and breast cancer. N Engl J Med. 1993;328:546-551.
75. L. Hernandez-Echebarria, A. Saiz, A. Ares, et al. Paraneoplastic encephalomyelitis associated with pancreatic tumor and anti-GAD antibodies. Neurology. 2006;66:450-451.
76. J.C. McHugh, B. Murray, R. Renganathan, S. Connolly, T. Lynch. GAD antibody positive paraneoplastic stiff person syndrome in a patient with renal cell carcinoma. Mov Disord. 2007;22:1343-1346.
77. P. Brown, C.D. Marsden. The stiff man and stiff man plus syndromes. J Neurol. 1999;246:648-652.
78. O.M. Vasconcelos, M.C. Dalakas. Stiff-person Syndrome. Curr Treat Options Neurol. 2003;5:79-90.
79. J.P. Camdessanche, J.C. Antoine, J. Honnorat, et al. Paraneoplastic peripheral neuropathy associated with anti-Hu antibodies. A clinical and electrophysiological study of 20 patients. Brain. 2002;125:166-175.
80. S.J. Oh, Y. Gurtekin, E.J. Dropcho, P. King, G.C. Claussen. Anti-Hu antibody neuropathy: a clinical, electrophysiological, and pathological study. Clin Neurophysiol. 2005;116:28-34.
81. J.L. Molinuevo, F. Graus, R. Rene, A. Guerrero, I. Illa. Utility of anti-Hu antibodies in the diagnosis of paraneoplastic sensory neuropathy. Ann Neurol. 1998;44:976-980.
82. J.C. Antoine, J. Honnorat, J.P. Camdessanche, et al. Paraneoplastic anti-CV2 antibodies react with peripheral nerve and are associated with a mixed axonal and demyelinating peripheral neuropathy. Ann Neurol. 2001;49:214-221.
83. S.P. Sillevis, J. Grefkens, B. De Leeuw, et al. Survival and outcome in 73 anti-Hu positive patients with paraneoplastic encephalomyelitis/sensory neuronopathy. J Neurol. 2002;249:745-753.
84. S.J. Oh, D.S. Kim, T.C. Head, G.C. Claussen. Low-dose guanidine and pyridostigmine: Relatively safe and effective long-term symptomatic therapy in Lambert-Eaton myasthenic syndrome. Muscle Nerve. 1997;20:1146-1152.
85. S. Vernino, P.A. Low, R.D. Fealey, J.D. Stewart, G. Farrugia, V.A. Lennon. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med. 2000;343:847-855.
86. C.H. Gibbons, S.A. Vernino, R. Freeman. Combined immunomodulatory therapy in autoimmune autonomic ganglionopathy. Arch Neurol. 2008;65:213-217.
87. J. Newsom-Davis, K.R. Mills. Immunological associations of acquired neuromyotonia (Isaac’s syndrome). Report of five cases and literature review. Brain. 1993;116:453-469.
88. P. Shillito, P.C. Molenaar, A. Vincent, et al. Acquired neuromyotonia: evidence for autoantibodies directed against K+ channels of peripheral nerves. Ann Neurol. 1995;38:714-722.
89. J.S. van den Berg, B.G. Van Engelen, R.H. Boerman, M.H. De Baets. Acquired neuromyotonia: superiority of plasma exchange over high-dose intravenous human immunoglobulin. J Neurol. 1999;246:623-625.
90. S.J. Oh. Paraneoplastic vasculitis of the peripheral nervous system. Neurol Clin. 1997;15:849-863.
91. K. Matsumuro, S. Izumo, F. Umehara, et al. Paraneoplastic vasculitic neuropathy: immunohistochemical studies on a biopsied nerve and post-mortem examination. J Intern Med. 1994;236:225-230.
92. D. Vincent, F. Dubas, J.J. Hauw, et al. Nerve and muscle microvasculitis in peripheral neuropathy: a remote effect of cancer? J Neurol Neurosurg Psychiatry. 1986;49:1007-1010.
93. S.J. Oh, R. Slaughter, L. Harrell. Paraneoplastic vasculitic neuropathy: a treatable neuropathy. Muscle Nerve. 1991;14:152-156.
94. J.C. Antoine, J.F. Mosnier, L. Absi, P. Convers, J. Honnorat, D. Michel. Carcinoma associated paraneoplastic peripheral neuropathies in patients with and without anti-onconeural antibodies. J Neurol Neurosurg Psychiatry. 1999;67:7-14.
95. M.C. Vigliani, M. Magistrello, P. Polo, R. Mutani, A. Chio. Risk of cancer in patients with Guillain-Barré syndrome (GBS). A population-based study. J Neurol. 2004;251:321-326.
96. F.T. Rotta, W.G. Bradley. Marked improvement of severe polyneuropathy associated with multifocal osteosclerotic myeloma following surgery, radiation, and chemotherapy. Muscle Nerve. 1997;20:1035-1037.
97. N. Latov. Prognosis of neuropathy with monoclonal gammopathy. Muscle Nerve. 2000;23:150-222.
98. R. Weide, J. Heymanns, H. Koppler. The polyneuropathy associated with Waldenstrom’s macroglobulinaemia can be treated effectively with chemotherapy and the anti-CD20 monoclonal antibody rituximab. Br J Haematol. 2000;109:838-841.
99. W.B. Bowne, J.J. Lewis, D.A. Filippa, et al. The management of unicentric and multicentric Castleman’s disease: a report of 16 cases and a review of the literature. Cancer. 1999;85:706-717.
100. A. Dispenzieri, R.A. Kyle, M.Q. Lacy, et al. POEMS syndrome: definitions and long-term outcome. Blood. 2003;101:2496-2506.
101. A. Ku, E. Lachmann, R. Tunkel, W. Nagler. Severe polyneuropathy: initial manifestation of Castleman’s disease associated with POEMS syndrome. Arch Phys Med Rehabil. 1995;76:692-694.
102. M. Donaghy, P. Hall, J. Gawler, et al. Peripheral neuropathy associated with Castleman’s disease. J Neurol Sci. 1989;89:253-267.
103. J.L. Fernandez-Torre, J.M. Polo, J. Calleja, J. Berciano. Castleman’s disease associated with chronic inflammatory demyelinating polyradiculoneuropathy: a clinical and electrophysiological follow-up study. Clin Neurophysiol. 1999;110:1133-1138.
104. M.J. Titulaer, P.W. Wirtz, A.R. Wintzen, J.J. Verschuuren. Re: Lambert-Eaton myasthenic syndrome with pure ocular weakness. Neurology. 2008;70:86-87.
105. S.A. Rudnicki. Lambert-Eaton myasthenic syndrome with pure ocular weakness. Neurology. 2007;68:1863-1864.
106. P. O’suilleabhain, P.A. Low, V.A. Lennon. Autonomic dysfunction in the Lambert-Eaton myasthenic syndrome: serologic and clinical correlates. Neurology. 1998;50:88-93.
107. P.D. Clouston, C.B. Saper, T. Arbizu, et al. Paraneoplastic cerebellar degeneration. III. Cerebellar degeneration, cancer, and the Lambert-Eaton myasthenic syndrome. Neurology. 1992;42:1944-1950.
108. W.P. Mason, F. Graus, B. Lang, et al. Small-cell lung cancer, paraneoplastic cerebellar degeneration and the Lambert-Eaton myasthenic syndrome. Brain. 1997;120:1279-1300.
109. M. Motomura, B. Lang, I. Johnston, J. Palace, A. Vincent, J. Newsom-Davis. Incidence of serum anti-P/O-type and anti-N-type calcium channel autoantibodies in the Lambert-Eaton myasthenic syndrome. J Neurol Sci. 1997;147:35-42.
110. D.B. Sanders, J.M. Massey, L.L. Sanders, L.J. Edwards. A randomized trial of 3,4-diaminopyridine in Lambert-Eaton myasthenic syndrome. Neurology. 2000;54:603-607.
111. P.G. Bain, M. Motomura, J. Newsom-Davis, et al. Effects of intravenous immunoglobulin on muscle weakness and calcium-channel autoantibodies in the Lambert-Eaton myasthenic syndrome. Neurology. 1996;47:678-683.
112. J. Newsom-Davis. Therapy in myasthenia gravis and Lambert-Eaton myasthenic syndrome. Semin Neurol. 2003;23:191-198.
113. P.W. Wirtz, M.G. Nijnuis, M. Sotodeh, et al. The epidemiology of myasthenia gravis, Lambert-Eaton myasthenic syndrome and their associated tumours in the northern part of the province of South Holland. J Neurol. 2003;250:698-701.
114. D.B. Sanders, K. El Salem, J.M. Massey, J. McConville, A. Vincent. Clinical aspects of MuSK antibody positive seronegative MG. Neurology. 2003;60:1978-1980.
115. J. Diaz-Manera, R. Rojas-Garcia, E. Gallardo, et al. Antibodies to AChR, MuSK and VGKC in a patient with myasthenia gravis and Morvan’s syndrome. Nat Clin Pract Neurol. 2007;3:405-410.
116. C. Buckley, J. Newsom-Davis, N. Willcox, A. Vincent. Do titin and cytokine antibodies in MG patients predict thymoma or thymoma recurrence? Neurology. 2001;57:1579-1582.
117. Y.H. Leow, C.L. Goh. Malignancy in adult dermatomyositis. Int J Dermatol. 1997;36:904-907.
118. G.H. Mautner, M.E. Grossman, D.N. Silvers, A. Rabinowitz, C.M. Mowad, B.L. JohnsonJr. Epidermal necrosis as a predictive sign of malignancy in adult dermatomyositis. Cutis. 1998;61:190-194.
119. E. Mahe, V. Descamps, M. Burnouf, B. Crickx. A helpful clinical sign predictive of cancer in adult dermatomyositis: cutaneous necrosis. Arch Dermatol. 2003;139:539.
120. Y.J. Chen, C.Y. Wu, J.L. Shen. Predicting factors of malignancy in dermatomyositis and polymyositis: a case-control study. Br J Dermatol. 2001;144:825-831.
121. A.A. Amato, R.J. Barohn. Idiopathic inflammatory myopathies. Neurol Clin. 1997;15:615-648.
122. M.C. Dalakas, I. Illa, J.M. Dambrosia, et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med. 1993;329:1993-2000.
123. P. Anderlini, A.C. Buzaid, S.S. Legha. Acute rhabdomyolysis after concurrent administration of interleukin-2, interferon-alfa, and chemotherapy for metastatic melanoma. Cancer. 1995;76:678-679.
124. M.I. Levin, T. Mozaffar, M.T. Al-Lozi, A. Pestronk. Paraneoplastic necrotizing myopathy: clinical and pathological features. Neurology. 1998;50:764-767.
125. M.R. Rosenfeld, J. Dalmau. Current therapies for paraneoplastic neurologic syndromes. Curr Treat Options Neurol. 2003;5:69-77.
126. L. Bataller, K.A. Kleopa, G.F. Wu, J.E. Rossi, M.R. Rosenfeld, J. Dalmau. Autoimmune limbic encephalitis in 39 patients: immunophenotypes and outcomes. J Neurol Neurosurg Psychiatry. 2007;78:381-385.
127. F. Keime-Guibert, F. Graus, A. Fleury, et al. Treatment of paraneoplastic neurological syndromes with antineuronal antibodies (Anti-Hu, anti-Yo) with a combination of immunoglobulins, cyclophosphamide, and methylprednisolone. J Neurol Neurosurg Psychiatry. 2000;68:479-482.
128. S. Vernino, B.P. O’Neill, R.S. Marks, J.R. O’Fallon, D.W. Kimmel. Immunomodulatory treatment trial for paraneoplastic neurological disorders. Neuro-oncol. 2004;6:55-62.