Chapter 50 Pediatric Epilepsy
An Overview
After a first unprovoked seizure in childhood, the risk of a recurrence is about 50 percent, and after the second the risk is about 80 percent. By convention, epilepsy may be diagnosed only after a child has had two or more unprovoked seizures [Camfield et al., 1985a; Shinnar et al., 2000]. The term unprovoked implies that there has been no closely associated concurrent illness, fever, or acute brain injury. Recurrent seizures immediately after a head injury or associated with drug intoxication do not qualify for the diagnosis of epilepsy. Some specific provoking factors leading to reflex seizures are permitted, such as seizures provoked by patterns and flashes from video terminals for children with photosensitive epilepsy. Seizures are still viewed as unprovoked if they occur with stresses related to personal activity, such as sleep deprivation or severe emotional distress, unless these stresses are extreme.
Seizure Type and Epilepsy Syndrome
The definition of epilepsy allows for a tremendous variety of disorders. The seizure types, age of onset, cause, severity, comorbid conditions, response to medication, and clinical course vary widely. There are two main ways of grouping patients to bring an ordered approach to classification, treatment, and prognosis: seizure type and epilepsy syndrome. Both concepts continue to evolve. The International League against Epilepsy defined the seizure types listed in Box 50-1 [Commission on Classification and Terminology of the International League against Epilepsy, 1981]. There is a basic distinction between seizures with a generalized onset that seem to arise from everywhere in the cortex at once, and seizures with a partial or focal onset that begin in a defined area of cortex. A proposed update in the classification of epileptic seizures suggested that the distinction between simple partial and complex partial seizures is often difficult to assess and not needed [Engel, 2001].
Box 50-1 International Classification of Seizure Type



















Unclassifiable Epileptic Seizures
(Adapted from the Commission on Classification and Terminology of the International League against Epilepsy. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia 1981;22:489.)
A given seizure type may occur with many different associations. For example, a 2-year-old child with severe mental handicap may have generalized tonic-clonic seizures that are completely resistant to medication, or a normal teenager may have the same seizure type that is completely suppressed by medication. Factors beyond seizure type allow a more comprehensive diagnosis – an epilepsy syndrome diagnosis. Epileptic syndromes may have one or more seizure types, often with a characteristic interictal electroencephalogram (EEG). Each syndrome has a typical age of onset, a defined group of causes, sometimes a clear response to specific treatments, and sometimes a defined clinical course and prognosis. As with seizure types, syndromes have been classified as localization-related (focal) or generalized, and then subdivided into idiopathic (when microdysgenesis or a biochemical etiology is likely), symptomatic (when a diffuse or localized brain abnormality is known), and cryptogenic (when a diffuse or localized brain abnormality is suspected but unproven; Table 50-1). The term cryptogenic has been discouraged, as the detection of a structural lesion is dependent on the evolving quality of brain imaging and detection of a biochemical abnormality is dependent on the evolving understanding of genomic and proteomic abnormalities. In adults, localization-related epilepsy syndromes are most common, usually symptomatic, and caused by a localized, structural abnormality. Children have similar disorders, but there are many other important syndromes. The idiopathic partial and generalized syndromes all begin in childhood, and genetic influences are significant. The generalized symptomatic syndromes of childhood account for most intractable epilepsy in children [Arzimanogolou et al., 2004; Camfield and Camfield, 2003].
Table 50-1 Proposal from the International Classification of Epilepsies and Epileptic Syndromes and Related Seizure Disorders of 1989 and the Listing of Epilepsy Syndromes Recognized in the Proposed 2001 ILAE Classification Scheme
| 1989 ILAE Classification* | 2001 ILAE Proposal† |
|---|---|
| Localization-Related (Local, Focal, Partial Epilepsies and Syndromes | |
| Idiopathic (with Age-Related Onset) | |
| Benign childhood epilepsy with centrotemporal spikes | Benign childhood epilepsy with centrotemporal spikes |
| Childhood epilepsy with occipital paroxysms | Early-onset benign childhood occipital epilepsy (Panayiotopoulos type) (new) Late-onset childhood occipital epilepsy (Gastaut type) (new) Idiopathic photosensitive occipital lobe epilepsy (new) |
| Primary reading epilepsy | Primary reading epilepsy Now called Reflex epilepsies, which also includes: |
(Diagnosis of epilepsy not now required)
* Modified from the 1989 International League Against Epilepsy (ILAE) Proposal.
† Modified from the 2001 ILAE Proposal. In this proposal there are no headings or subheadings, only a listing of epilepsy syndromes by specific syndrome name. Our format of presentation has indicated where syndromes are removed or renamed from the 1989 ILAE proposal or newly recognized in the 2001 proposal. Each syndrome was “forced” into the 1989 proposed schema.
The International League Against Epilepsy (ILAE) classified febrile seizures as a special syndrome. Febrile seizures are the most common convulsive event in the human species. Because these seizures are provoked by fever, febrile seizures do not fit well with the usual definition of epilepsy, and few children with febrile seizures later develop unprovoked seizures. The disorder merits special consideration, as reviewed in Chapter 57.
The 1989 ILAE syndrome classification scheme is still in wide use, and most patients can be “forced” into one of the broad categories, but many children, especially those with symptomatic generalized epilepsy, do not meet the criteria for a more specific syndrome [Commission on Classification and Terminology of the International League Against Epilepsy, 1989]. There are many recently described epilepsy syndromes, but a newer, comprehensive syndrome classification system has been elusive. Advances in genetics have revealed that the same mutation may cause a variety of epilepsy syndromes, including partial and generalized epilepsies, even within the same family (see Chapter 52). This finding has been particularly well illustrated in large families with mutations in the gene coding for neuronal sodium channel – SCN1A.
Another evolving classification is “epilepic encephalopathy,” defined as an epilepsy syndrome where the seizures and/or interictal EEG discharge have permanent deleterious effects on brain development. Examples would be West’s syndrome with infantile spasms and hypsarrhythmia on EEG, Lennox–Gastaut syndrome with mixed generalized seizures and nearly continuous slow spike wave EEG discharge, or Landau–Kleffner syndrome with relatively mild clinical seizures but very active bitemporal epileptic EEG discharge (see Chapter 56).
A new proposal for syndrome classification in 2001 is based on a five-axis system [Engel, 2001]. Axis 1 concerns ictal phenomenology (i.e., what the seizure looks like). Axis 2 is the specific seizure type. Axis 3 is the specific epilepsy syndrome, when it can be defined; axis 4 is the cause; and axis 5 describes the degree of impairment associated with epilepsy. This proposal comes with a more comprehensive list of epilepsy syndromes, although the system is less categorical than the 1989 classification [Commission on Classification and Terminology of the International League against Epilepsy, 1989]. The 2001 proposal has not been widely accepted, although it does serve well to alert the clinician to the many aspects of the epileptic diathesis in an individual patient. Table 50-1 highlights the differences between the two classification schemes by using each to characterize some accepted epileptic syndromes. A clinician interested in epilepsy should be conversant with the 1989 and 2001 classification schemes and must anticipate additional revisions.
Incidence
In developed countries, the overall incidence of childhood epilepsy from birth to 16 years is approximately 40 cases in 100,000 children per year [Camfield et al., 1996b; Hauser et al., 1993]. The incidence in the first year of life is about 120 in 100,000. Between 1 and 10 years of age, the incidence plateaus at 40–50 cases in 100,000 children, and then drops further in the teenage years to about 20 in 100,000. The details of incidence by year of life from the Nova Scotia childhood epilepsy study are illustrated in Figure 50-1. Hauser estimates that about 1 percent of all children will have at least one afebrile seizure by age 14 years, and that 0.4–0.8 percent will have epilepsy by age 11 years [Hauser and Hesdorffer, 1990]. In less developed countries, there is suggestive evidence for a higher incidence of childhood epilepsy, possibly related to higher incidence of trauma and central nervous system infections.
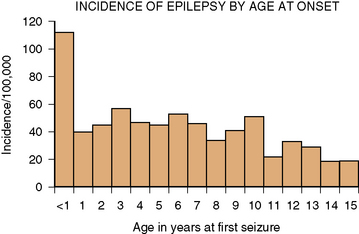
Fig. 50-1 Incidence of epilepsy by age of onset.
(From Camfield CS et al. Incidence of epilepsy in childhood and adolescents: A population based study in Nova Scotia from 1977–1985. Epilepsia 1996b;37:19.)
Epilepsy types vary in incidence. Syndromes dominated by generalized tonic-clonic or partial seizures account for 75 percent of childhood epilepsy. Syndromes dominated by absence seizures account for approximately 15 percent, and the secondary generalized epilepsies account for 10 percent. The latter group includes most of the “catastrophic” epilepsy syndromes, such as West and Lennox–Gastaut syndromes [Camfield et al., 1996b]. Prevalence data for seizure type or epilepsy syndrome are not easily collated because of various definitions of active epilepsy. When active epilepsy was defined as receiving daily antiepileptic drugs or a seizure within the past 5 years, the prevalence of epilepsy in children was 4.3–9.3 in 1000 [Hauser and Hesdorffer, 1990]. Because some epileptic syndromes rarely remit, they contribute more to prevalence of epilepsy than its incidence. The relative prevalence of symptomatic generalized epilepsies is higher than their relative incidence. This finding means that physicians who focus on newly diagnosed children will encounter a predominance of benign epilepsy syndromes. Those primarily treating chronic cases will observe a higher proportion of more malignant seizure disorders. A population-based prevalence study from Finland found that the main seizure types for each patient were focal in 43 percent, with complex partial and partial with secondary generalization most common. For 44 percent, generalized seizures were dominant, with generalized tonic-clonic seizures most common. Overall, 45 percent had localization-related epilepsy syndromes, and 48 percent had generalized syndromes [Eriksson and Koivikko, 1997].

